Настоящее изобретение относится к способу получения в существенной степени безводного хлористого магния, к в существенной степени безводному хлористому магнию, полученному этим способом, и к в существенной степени безводному хлористому магнию как таковому.
Предшествующий уровень техники.
В существенной степени чистый металлический магний может быть получен электролитическим способом из хлористого магния с высвобождением газообразного хлора. Однако, при использовании гидратного хлористого магния в качестве материала, загружаемого в электролитический электролизер, эффективность работы электролизера существенно снижается в течение короткого периода времени из-за образования оксидов магния, вызывающих корродирование электродов и образование шлама, который необходимо периодически удалять из электролизера. Хлормагнивое сырье также обычно содержит загрязняющие примеси, такие как углеводороды, соли бора и соли других металлов, которые также существенно снижают эффективность работы электролитического электролизера. Поэтому желательно получать в существенной степени чистый безводный хлористый магний, соответствующий требованиям электролитического производства металлического магния.
В зависимости от температурного режима, выделенный из водных растворов хлористый магний содержит различные количества молекул кристаллизационной воды. Гидратные формы хлористого магния могут быть до определенной степени обезвожены путем нагрева. Однако гидратные формы хлористого магния имеют тенденцию к расплавлению в их собственной кристаллизационной воде с образованием сгущенного, частично обезвоженного продукта, который крайне трудно поддается дальнейшему обезвоживанию нагреванием. Кроме того, невозможно осуществить полную дегидратацию хлористого магния путем нагревания на открытом воздухе, поскольку хлористый магний, содержащий менее двух молекул кристаллизационной воды, подвергается не столько дегидратации, сколько гидролитическому расщеплению с выделением хлороводорода. С учетом изложенного, предлагаются альтернативные подходы к технологии получения безводного хлористого магния.
Безводный хлористый магний может быть получен прямым хлорированием магния и сушкой газообразным хлороводородом. Первый из этих двух способов явно неприемлем для получения безводного хлористого магния для использования его в электролитическом производстве металлического магния. Во втором из указанных способов гидратный хлористый магний формуют в гранулы, которые помещают в колонну и продувают горячим хлороводородом, удаляя все следы воды. Этот второй способ не проявил себя как достаточно эффективный, поскольку обычно в конце технологического цикла присутствуют гидратные формы хлористого магния, которые могут при дальнейшем использовании продукта преобразовываться в нежелательные оксиды магния. Кроме того, этот способ требует использования больших объемов газообразного хлороводорода, что порождает проблемы, связанные с его хранением и использованием.
Альтернативный подход к технологии получения безводного хлористого магния предусматривает формирование раствора гидратного хлористого магния в растворителе, удаление воды из полученного раствора, формирование комплекса хлористого магния взаимодействием обезвоженного раствора с осаждающим веществом и нагревом комплекса хлористого магния с получением безводного хлористого магния. Целый ряд вариантов этого общего технологического принципа изложен в патентной литературе на протяжении прошедших лет. Эти варианты объединены тем общим признаком, что в качестве осаждающего вещества используется аммиак. Далее эти способы упоминаются как аммиачные или аммонизационные способы. Аммиачные способы связаны с различными технологическими проблемами, и составителям настоящей заявки неизвестны примеры когда-либо осуществлявшегося промышленного получения безводного хлористого магния аммиачным способом.
Нужный комплекс хлористого магния, нагреваемый для получения безводного хлористого магния в аммиачном процессе, представляет собой гексааммониат хлористого магния (MgCl2•6NH3). В случаях использования этиленгликоля в качестве растворителя для получения раствора гидратного хлористого магния, как удалось убедиться создателям настоящего изобретения, в ходе осуществления аммиачного способа параллельно с гексаммониатом хлористого магния или в связи с ним могут формироваться гликолятные соединения хлористого магния. К гликолятам хлористого магния относятся тригликолят хлористого магния. (MgCl2 • 3(HOCH2 • CH2OH) и биаммиакат бигликолята хлористого магния (MgCl2 • (HOCH2 • CН2OH) • 2NH2). Создателями настоящего изобретения определены рентгенографические (XRD) характеристики и инфракрасные (по преобразованию Фурье [FTIR] (спектры гликолятных соединений хлористого магния. Подробности сведены в таблицах 1 - 4. Гликоляты хлористого магния нежелательны в аммиачном процессе, поскольку считается, что они разлагаются при нагревании и образуют кислородсодержащие соединения, загрязняющие конечный продукт - безводный хлористый магний. Попадание кислорода в электролитический электролизер, в который подается исходный продукт в процессе получения металлического магния, приводит как к сокращению срока службы графитовых анодов, обычно используемых в таких электролизерах, так и к снижению эффективности работы самого электролизера.
В патенте США N 2381995, выданном в 1942 г. на имя Mwkellog Company, аммиачный способ, в котором изоамиловый спирт используется в качестве предпочтительного растворителя для получения раствора гидратного хлористого натрия, а в качестве осаждающего вещества используется аммиак, и где в качестве комплекса хлористого магния, нагреваемого для получения безводного хлористого магния, определяется гексааммониат хлористого магния. Патентом США N 2381995 предусматривается также тщательное смешивание обезвоженного раствора хлористого магния и аммиака перед их подачей в охладитель, где предполагается осаждение гексааммониата хлористого магния.
В патенте США N 3966888, выданном в 1975 г. на имя Nalco Chemical Company, также описывается аммиачный способ. В ходе обсуждения решения по патенту США N 2381995 в патенте США N 3966888 утверждается, что решение по патенту США N 2381995:
"основывается на растворении гидратного хлористого магния в одноатомном насыщенном алифатическом спирте. Раствор этот затем нагревают в течение времени, достаточного для отгонки имеющейся в нем воды. Предполагаемый обезвоженный раствор обрабатывают аммиаком для осаждения аммиачного комплекса хлористого магния, который затем отделяют от спирта и подвергают нагреву для отгонки аммиака от комплексного соединения.
Сложности практического осуществления способа по патенту США N 2381995 убедительно доказывают специалисту в данной области, что этот способ исходно непригоден для широкомасштабного промышленного использования.
Прежде всего, при нагревании спиртового раствора гидратного хлористого магния с целью удаления из него воды невозможно удалить воду при температуре, близкой к точке кипения используемого спирта. Это в особенности верно при использовании изоамилового спирта в качестве растворителя для гидратного хлористого магния. Таким образом, хлористый магний обезвоживается неполностью. Когда предполагаемый обезвоженный хлористый магний осаждают аммиаком из спирта, как это излагается в патенте США N 2381995, образуется плотный воскоподобный осадок, содержащий большие количества захваченного спирта. Плотность и воскообразная структура осадка делают его непригодным для переработки на стандартном промышленном оборудовании с целью освобождения осадка от захваченного растворителя. Таким образом, невозможно осуществлять дальнейшую обработку осадка без сопутствующих потерь спирта в ходе технологической стадии удаления аммиака".
В патенте США N 3966888 в целом заявляется "способ получения безводного хлористого магния из гидратов хлористого магния, содержащий следующие операции:
А) растворение гидратного хлористого магния в этиленгликоле с получением этиленгликолевого раствора гидратного хлористого магния;
Б) нагревание полученного этиленгликолевого раствора гидратного хлористого магния до температуры и в течение временного интервала, достаточных для удаления всей воды из указанного раствора, с получением этиленгликолевого раствора безводного хлористого магния;
В) обработку полученного этиленгликолиевого раствора безводного хлористого магния аммиаком для образования аммиачного комплекса хлористого магния, нерастворимого в этиленгликоле, при этом температура этиленгликолевого раствора хлористого магния лежит в пределах от -15o до 50oC;
Г) отделение полученного аммиачного комплекса хлористого магния от этиленгликоля;
Д) промывку указанного аммиачного комплекса хлористого магния полярным растворителем, имеющим более низкую, чем этиленгликоль, температуру кипения, для удаления любых количеств этиленгликоля, захваченных аммиачным комплексом хлористого магния;
Е) нагревание полученного аммиачного комплекса хлористого магния до температуры и в течение временного интервала, достаточных для отгонки аммиака, с формированием, таким образом, безводного хлористого магния; и затем
Ж) извлечение безводного хлористого магния, имеющего содержание оксида магния менее 0,8% вес.".
В патенте США N 3966888 вышеизложенная операция В) более конкретно характеризуется тем, что
а) этиленгликолевый раствор безводного хлористого магния, охлажденный до температуры, лежащей в пределах от -15o до 50oC, обрабатывают, по меньшей мере, 6 молями аммиака, исходя из содержания хлористого магния в этиленгликоле; и
б) указанный этиленгликолевый раствор безводного хлористого магния охлаждают до температуры, лежащей в пределах от 0o до 25oC, перед добавлением в указанный раствор аммиака.
В описании операции В) патент США N 3966888 излагает следующее:
"Безводный этиленгликолевый раствор хлористого магния затем охлаждают до температуры от -15o до 50oC, предпочтительно в пределах от 0oC до 25oC. В этом температурном режиме раствор обрабатывают безводным аммиаком из расчета не менее 6 молей, предпочтительно 9 молей, на моль хлористого магния, содержащегося в этиленгликолевом растворе. Введение аммиака может осуществляться относительно быстро, хотя в условиях лабораторных исследований с небольшими объемами материалов введение аммиака может осуществляться на протяжении временного интервала от 1 до 2 часов.
Было установлено, что при охлаждении этиленгликолевого раствора хлористого магния до указанного температурного интервала аммиак обретает большую растворимость в этом растворе и что осадок не формируется до тех пор, пока не добавлено по меньшей мере 6 молей аммиака. После ввода большей части аммиака в гликоль начинает формироваться мелкий белый зернистый осадок, который представляет собой обезвоженный аммиачный комплекс хлористого магния".
В примере практического осуществления операции В) по патенту США N 3966888 описывается, что этиленгликолевый раствор безводного хлористого магния охлаждают до 15oC, и приблизительно 9 молей безводного аммиака подают в охлажденный раствор в течение одного часа, при этом осадок начинает формироваться после тридцати минут введения аммиака.
Патент США N 3966888 определяют также как предпочтительное содержание хлористого магния в этиленгликолевом растворе безводного хлористого магния в объеме 8 - 12 вес.%.
В 1980 году была опубликована работа доктора Рональда Дж. Аллена, сотрудника Nalco Chemical Company, озаглавленная "Новый экономичный способ получения безводного хлористого магния (далее - работа Аллена), в которой рассматривался аммиачный способ, основанный на положениях, изложенных в патенте США N 3966888. В связи с операцией В) описанного в указанном патенте способа в работе Аллена показано, что с целью выделения хлористого магния из этиленгликолевого раствора безводного хлористого магния через этот раствор пропускают с барботированием газообразный безводный аммиак. Происходит немедленное растворение аммиака, который вначале насыщает раствор, а затем формирует осадок гексааммониат хлористого магния. В работе указывается, что наиболее удобно выполнять аммонизацию в простом резервуаре путем подачи аммиака с одновременным перемешиванием, под давлением, слегка превышающим атмосферное. Отмечается, что реакция носит по существу мгновенный характер, при этом полностью отсутствует в заданных рабочих режимах унос аммиака с охлаждающей водой, используемой для отбора большей части высвобождающейся теплоты. В работе указывается, что на практике реактору позволяют разогреваться до 70oC с последующим охлаждением его в процессе реакции до конечной температуры в пределах от 15 до 30oC. Таким образом, изложенное в работе Аллена в существенной степени аналогично операции В) в патенте США N 3966888, т.е. этиленгликолевый раствор хлористого магния обрабатывается аммиаком путем помещения указанного этиленгликолевого раствора хлористого магния в емкость с последующим вводом в эту емкость аммиака. Различие между работой Аллена и изложением способа в патенте США N 3966888 состоит в том, что в работе Аллена говорится об охлаждении реактора от 70oC до 15 - 30oC в ходе процесса подачи аммиака, тогда как решением по патенту США N 3966888 оговаривается, что температура этиленгликолевого раствора хлористого магния перед введением безводного аммиака должна лежать в пределах от -15 до 50oC.
Создатели настоящего изобретения предприятия попытку получить безводный хлористый магний, следуя рекомендациям работы Аллена и описания к патенту США N 3966888, а также технологическим деталям, приведенным в сравнительном примере. Для этой цели этиленгликолевый раствор безводного хлористого магния был подготовлен в соответствии с рекомендациями по патенту США N 3966888. Этот раствор, который содержал 10 вес.% хлорида магния, охлаждали до 15o -20oC и в охлажденный раствор пробовали вводить 6-9 молей безводного аммиака. Однако было выявлено, что охлажденный раствор оказывался слишком вязким, чтобы позволить легкое прокачивание и диспергирование безводного аммиака. Создатели настоящего изобретения выяснили, что затруднения, обусловленные вязкостью охлажденного раствора, избегались, если температура этиленгликолевого раствора безводного хлористого магния составляла порядка 70oC перед вводом аммиака, и что выход осадка возрастал, если температуру после ввода аммиака понижали до 15oC. Однако было выявлено, что получаемый осадок не представлял собой в существенной степени желаемый гексааммониат хлористого магния, а содержал значительные количества нежелательных гликолятных соединений хлористого магния. Исходя из этого, можно считать, что способы, описанные в патенте США N 3966888 и в работе Аллена, исходно непригодны для применения в промышленном производстве безводного хлористого магния.
Раскрытие сущности изобретения
В первом своем аспекте настоящее изобретение предлагает способ получения в существенной степени безводного хлористого магния, содержащий следующие операции:
а) формирование спиртового раствора хлористого магния путем смешивания гидратного хлористого магния со спиртом, совместимым с водой;
б) обезвоживание указанного спиртового раствора хлористого магния с целью формирования безводного спиртового раствора хлористого магния;
в) формирование осадка, содержащего гексааммониат хлористого магния, подачей обезвоженного спиртового раствора хлористого магния и аммиака в реактор с неводным раствором, имеющим содержание аммиака более 7 вес.%;
г) извлечение полученного осадка из реактора;
д) промывку извлеченного осадка промывочным растворителем с целью получения промытого осадка; и
е) нагревание полученного промытого осадка для получения в существенной степени безводного хлористого магния.
Если проводить параллель между операцией (а) способа по настоящему изобретению и решением по патенту США N 3966888, в последнем в качестве спирта предусматривается этиленгликоль. В настоящем изобретении, напротив, спирт может быть выбран из группы спиртов, включающих гликоли. Указанный спирт должен быть водосовместим и способен растворять гидратный хлористый магний с формированием раствора хлористого магния. Предпочтительно в качестве спирта используется один из спиртов, в котором гидратный хлористый магний проявляет по меньшей мере умеренную растворимость. Например, спирт может быть выбран из группы, представленной метанолом, этанолом, пропанолом, бутанолом, этиленгликолем и диэтиленгликолем. В дополнение к сказанному, алкильные группы спирта могут иметь разветвленную структуру или представлять собой прямые цепи и могут содержать ненасыщенность. Указанным спиртом предпочтительно является этиленгликоль.
В операции (б) может использоваться любой удобный способ дегидратации. Могут быть, например, использованы дистилляции, мембранное разделение, молекулярные сита или сочетание этих способов, предпочтительным из которых, в целом, является способ дистилляции. Применение того или иного способа до некоторой степени зависит от спирта, использованного в операции (а). Например, если в качестве спирта в операции (а) используется метанол, дистилляция едва ли будет использоваться в качестве метода дегидратации из-за сложностей, обусловленных тем, что температура кипения метанола меньше температуры кипения воды. При использовании общепринятого метода дистилляции вода может отгоняться в стандартных дистилляционных колоннах либо при атмосферном давлении, либо под разрежением. Вакуумная дистилляция является предпочтительной, поскольку температура, необходимая для осуществления дистилляции, может быть понижена во избежание разложения спирта, используемого в процессе. Например, в качестве спирта используется этиленгликоль, предпочтительно поддерживать температурный режим процесса дистилляции ниже 150oC во избежание разложения этиленгликоля. Операция б) решения по патенту США N 3966888 предписывает удаление всей воды из этиленгликолевого раствора гидратного хлористого магния перед обработкой его аммиаком. Напротив, настоящим изобретением не оговаривается необходимость полного обезвоживания продукта в операции (б) с целью получения продукта, приемлемого по качеству для использования в операции (в). Не вдаваясь в серьезные теоретические исследования, мы может все же предположить, что это является следствием измененной методики, используемой в операции (в). Например, дегидратированный спиртовый раствор хлористого магния, полученный на операции (б), может содержать до 50000 ppm воды без заметного негативного влияния на качество в существенной степени безводного хлористого магния, получаемого в результате изложенного процесса.
В операции (в) обезвоженный спиртовый раствор хлористого магния и аммиак подают в реактор с неводным раствором, имеющим содержание аммиака более 7 вес. %. В отличие от этого, патентом США N 3966888 и работой Аллена предусматривается подача аммиака в резервуар, содержащий обезвоженный спиртовой раствор хлористого магния, а патентом США N 2381995 предусматривается тщательное смешивание аммиака и обезвоженного спиртового раствора хлористого магния перед подачей их в охлажденную емкость-кристаллизатор.
Не желая вдаваться в серьезные теоретические исследования, мы можем все же предположить, что получению гексааммониата хлористого магния реакцией взаимодействия обезвоженного спиртового раствора хлористого магния и аммиака благоприятствуют пониженные температурные режимы и повышенные концентрации растворенного аммиака, тогда как получение гликолятных соединений хлористого магния более вероятно при повышенных температурных режимах и при пониженных концентрациях растворенного аммиака.
В операции (в) желательно, чтобы, во-первых, не было контакта между обезвоженным спиртовым раствором хлористого магния и аммиаком до их ввода в реактор, и, во-вторых, чтобы в ходе формирования осадка исходно и непрерывно наличествовало достаточное количество аммиака для взаимодействия его с обезвоженным спиртовым раствором хлористого магния с целью формирования осадка, в общем и целом содержащего гексааммониата хлористого магния. Исходя из изложенного, предпочтительно, чтобы обезвоженный спиртовый раствор хлористого магния и аммиак раздельно и одновременно вводились в реактор.
Указанный неводный раствор может представлять собой жидкий аммиак, однако в этом случае необходимо, чтобы реактор представлял собой аппарат высокого давления. Поэтому предпочтительно, чтобы указанный неводный раствор представлял собой раствор из спирта, обработанного газообразный аммиаком. Для сведения к минимуму числа используемых в процессе растворителей предпочтительно, чтобы указанный неводный раствор представлял собой раствор того спирта, который смешивается с гидратным хлористым магнием в операции (а) изложенного способа.
Указанный безводный раствор содержит не менее 7 вес.% аммиака. Предпочтительно, неводный раствор содержит более 7 вес.% аммиака и, более предпочтительно, безводный раствор насыщен аммиаком. В ходе выполнения операции (в) требуемый уровень содержания аммиака в безводном растворе может достигаться регулированием скоростей подачи обезвоженного спиртового раствора хлористого магния и аммиака.
В реактор в ходе выполнения операции (в) может подаваться либо газообразный, либо жидкий аммиак. Предпочтительно, чтобы подаваемый в реактор аммиак представлял собой газообразный безводный аммиак.
Дистилляция является предпочтительным методом обезвоживания (дегидратации) спиртового раствора хлористого магния в ходе операции (б), поскольку поступающий после дистилляции на операцию (в) обезвоженный спиртовый раствор хлористого магния имеет повышенную температур. Несмотря на то, что получение гексааммониата хлористого магния является предпочтительным при пониженных температурных режимах в практическом промышленном осуществлении настоящего изобретения предпочтительно будет не охлаждать в существенной степени обезвоженный спиртовый раствор хлористого магния перед его взаимодействием с аммиаком, поскольку было установлено, что, даже если температура не понижается перед реакцией взаимодействия, удается выделить осадок, в существенной степени состоящий из гексааммониата хлористого магния. Можно предположить, что это является следствием технологических условий, в которых обезвоженный спиртовый раствор хлористого магния и аммиак взаимодействуют в соответствии с настоящим изобретением. В случае использования изложенного технологического подхода предпочтительно, чтобы температура в реакторе падала до уровня примерно ниже 45oC по завершении операции (в).
Операция (в) может осуществляться либо в цикле периодической загрузки, либо в непрерывном цикле. При использовании технологического цикла периодической загрузки предпочтительно, чтобы температура в реакторе составляла менее 45oC по завершении формирования осадка, хотя следует подразумевать, что температура в реакторе может быть и выше 45oC перед завершением формирования осадка. При использовании технологии непрерывного цикла может использоваться либо единичный реактор, либо последовательный ряд реактора. В промышленной реализации настоящего способа предпочтительным, очевидно, будет осуществление операции (в) в непрерывном технологическом цикле с использованием последовательного ряда реакторов. При задействовании непрерывного технологического цикла с использованием только одного реактора предпочтительно, чтобы температура в реакторе по завершении формирования осадка составляла менее 45oC. При задействовании непрерывного технологического цикла с использованием последовательного ряда реакторов предпочтительно, чтобы температура в последнем реакторе в цепочке по завершении формирования осадка составляла менее 45oC.
Не желая вдаваться в серьезные теоретические изыскания, создатель настоящего изобретения считают возможным предположить, что осадок, сформированный по завершении операции (в), не содержит существенных количеств гликолятных соединений хлористого магния, поскольку наличие значительного уровня содержания аммиака в реакторе, в который подаются обезвоженный спиртовый раствор хлористого магния и аммиак, приводит к почти мгновенному осаждению требуемого гексааммониата хлористого магния, нежели к формированию нежелательных гликолятных соединений хлористого магния. Кроме того, в случае формирования нежелательных гликолятных соединений хлористого магния, они предположительно подвергаются реакции твердофазного преобразования в гексааммониат хлористого магния в ходе операции (в), благодаря постоянному наличию значительного количества аммиачного реагента. В противоположность сказанному, можно предположить, что в результате операции (В) по патенту США N 3966888 формируется осадок, содержащий гликолятные соединения хлористого магния, поскольку небольшой поток аммиака вводится в сравнительно большой объем обезвоженного спиртового раствора хлористого магния. Кроме того, можно предположить, что гликолятные соединения хлористого магния, сформировавшиеся в ходе операции (В) по патенту США N 3966888, не преобразуются свободно в гексааммониат хлористого магния в заданных условиях реакции из-за отсутствия аммиака, с которым они могут взаимодействовать. Можно предположить, что все это ведет к тому, что хлористый магний, полученный в результате операции (Е) по патенту США N 3966888, имеет более низкое качество, чем хлористый магний, полученный в соответствии с первым аспектом настоящего изобретения.
В ходе операции (г) осадок может извлекаться из реактора любым удобным способом. Могут, например, использоваться центрифугирование, декантация, фильтрование или сочетание таких методов. Предпочтительно, извлечение осадка осуществляется в безводной среде, которая может образовываться атмосферой из таких газов как аммиак, аргон, азот, осушенный воздух и т.п.
В ходе операции (д) осадок промывают промывочным растворителем с целью удаления следов спирта и безводного раствора, имеющихся в осадке. В качестве промывочного растворителя могут быть использованы аммиак и различные спирты, например, метанол, этанол, пропанол и бутанол; однако предпочтительно, чтобы, в случае использования в качестве промывочного растворителя спирта, этот спирт был насыщен аммиаком. Патент США N 3966888 предусматривает использование метанола в качестве промывочного растворителя. Создатели настоящего изобретения убедились, что использование только метанола в качестве промывочного растворителя приводит к повторному растворению осадка. Есть основание считать, что используемый сам по себе метанол растворяет как гликолят соединения хлористого магния, так и гексааммониат хлористого магния, в результате чего качество продукта повышается, а выход продукта уменьшается. Предполагается, что гликолятные соединения хлористогоо магния подвергаются комбинированному процессу растворения и преобразования в гексааммониат хлористого магния при промывке аммонизированным метанолом, причем есть основание считать, что растворимость гексааммониата хлористого магния в метаноле уменьшается с увеличением содержания аммиака в метаноле. Соответственно, можно сделать вывод о том, что промывка гексааммониата хлористого магния, содержащего некоторое количество гликолятных соединений хлористого магния, аммонизированным метанолом, приводит к повышению качества продукта и к увеличению выхода продукта. Промывочный растворитель предпочтительно насыщают аммиаком в температурном диапазоне примерно 3-40oC, более предпочтительно 20-30oC.
В ходе операции (е) промытый осадок нагревают с целью удаления аммиака и формирования в существенной степени безводного хлористого магния. Может использоваться любая приемлемая методика нагрева, например, нагрев в кальцинационной печи, нагрев во вращающейся печи, нагрев в псевдосжиженном слое; предпочтительным, однако, является нагрев в псевдосжиженном слое. Предпочтительно, осадок нагревать в псевдосжиженном слое до температуры выше, чем примерно 380oC, и, более предпочтительно, температура нагрева лежит в пределах от 400 до 500oC. Перед нагреванием с целью удаления аммиака предпочтительно удалить из промытого осадка метанол, если метанол использовался в промывочном растворе. Для удаления метанола может использоваться любой приемлемый метод, включая помещение промытого осадка в вакуум. Однако предпочтительно, чтобы метанол удалялся нагревом промытого осадка при температуре ниже 120oC.
Как отмечалось ранее, предпочтительно, чтобы указанный неводный раствор представлял собой раствор спирта, смешиваемый с гидратным хлористым магнием. Для оптимизации экономических показателей процесса по настоящему изобретению предпочтительно регенерировать и повторно использовать в технологическом цикле различные химикаты, используемые в процессе. Например, предпочтительно, вслед за извлечением осадка из реактора регенерировать из реактора спирт и рициклировать спирт на смешивание со свежим гидратным хлоридом магния.
В зависимости от сырьевого источника гидратного хлористого магния, вместе с гидратным хлористым магнием на операции (а) в процессе могут попадать такие соли, как хлорид кальция, хлорид калия и хлорид натрия. Эти соли не осаждаются в ходе формирования осадка гексааммониата хлористого магния в ходе операции (в) и потому будут накапливаться в ходе процесса, если спирт из реактора после извлечения осадка рециклируется на операцию (а), что является предпочтительным. Поскольку указанные соли не осаждаются в ходе операции (в), их присутствие терпимо. Однако, если концентрации указанных солей имеют тенденцию к постепенному нарастанию за счет рециклирования на операцию (а), эффективность процесса в конечном счете будет снижаться. Следовательно, предпочтительно осуществлять периодическое или непрерывное удаление указанных солей из процесса, что предпочтительно выполняется как часть операции регенерации и рециркулирования спирта из реактора после извлечения осадка из указанного реактора. Существуют различные подходы к удалению солей, обеспечивающие регенерацию и рециклирование спирта. Например, соли могут удаляться отбором части спирта, содержащего соли; однако такой прием приводит к нежелательным потерям спирта. Создателями настоящего изобретения разработаны два предпочтительных способа регенерации спирта из реактора, позволяющие удалять соли.
Первый из указанных способов предпочтительно используется в случае присутствия в спирте хлорида кальция и содержит следующие операции:
(1) удаление из извлеченного из реактора спирта любых количеств аммиака или полярного растворителя, например, метанола;
(2) смешивание раствора растворимого бикарбоната магния со спиртом, полученным на операции (1), с формированием смеси бикарбонат магния и спирт;
(3) нагревание полученной смеси бикарбоната магния и спирта с получением осадка карбоната кальция; и
(4) отделение осадка карбоната кальция.
В ходе операции (1) аммиак и полярный растворитель могут удаляться любым удобным методом. Например, спирт может подвергаться дистилляции для удаления любых количеств аммиака или полярного растворителя. Этот метод предпочтителен, поскольку он позволяет повторно использовать отогнанные аммиак и полярный растворитель.
В ходе операции (2) спирт может непосредственно смешиваться с раствором бикарбоната магния, однако предпочтительнее выпаривать достаточное количество спирта до получения спиртового раствора, содержащего 15 - 35% вес. хлористого кальция. Более предпочтительно содержание в спирте примерно 25 вес.% хлористого кальция.
Любое приемлемое молярное отношение бикарбоната магния к спирту может быть задействовано таким образом, чтобы сформировать максимальное осаждение карбоната кальция. Предпочтительно, молярное отношение бикарбоната магния к хлористому кальцию находится в пределах 0,8 - 5, более предпочтительно 0,8 - 1.
Может использоваться любой приемлемый раствор растворимого бикарбоната магния. Например, оксид магния может быть смешан с водой с последующим добавлением в смесь двуокиси углерода с целью формирования растворимого бикарбоната магния. Предпочтительно, температура в ходе добавления двуокиси углерода поддерживается в диапазоне от 5 до 25oC, более предпочтительно от 13 до 18oC. Такой раствор предпочтительно используется вскоре после его приготовления, более предпочтительно в пределах 8 часов после приготовления.
При нагревании смеси бикарбоната магния и спирта с целью осаждения карбоната кальция в операции (3) смесь предпочтительно нагревают до 60 - 120oC. Если в качестве спирта используется гликоль, температура нагрева предпочтительно составляет 90 - 100oC.
После операции (4) осадок карбоната кальция может промываться водой с целью улавливания следов спирта, захваченного осадком; при этом сам осадок отделяют от спирта и воды любым удобным методом.
Второй из указанных способов позволяет удалять из спирта, извлеченного из реактора, практически все разновидности ионов. Например этот второй способ пригоден для удаления из спирта хлористого кальция хлорида натрия, хлорида калия, бора, сульфатов и силикатов.
Указанный второй способ содержит следующие операции:
(1) удаление из спирта любых количеств аммиака или полярного растворителя, например, метанола;
(2) непрерывную подачу спирта, полученного в операции (1), в верхнюю часть выпарной колонны, в нижнюю часть которой непрерывно подается пар, с отбором из нижней части колонны водосолевого раствора, в существенной степени не содержащего спирта, и с отбором из верхней части колонны потока пара спирта и воды, не содержащих солей.
При осуществлении операции (1) аммиак и полярный растворитель могут удаляться любым удобным методом. Например, спирт может подвергаться дистилляции для удаления любых количеств аммиака или полярного растворителя. Этот метод предпочтителен, поскольку он позволяет повторно использовать отогнанные аммиак и полярный растворитель.
В ходе операции (2) спирт после операции (1) может подаваться прямо на выпарную колонну, однако предпочтительнее его нагреть и выпарить спирт, чтобы получить спиртовый раствор, имеющий 15-35 вес.% суммарного содержания солей. Более предпочтительно, суммарное содержание солей в спирте составляет примерно 25 вес.%.
Спирт предпочтительно подается в верхнюю часть выпарной колонны при температуре в диапазоне 15 - 200oC, предпочтительно 100 - 150oC. Предпочтительно также разбавить спирт водой с целью снижения вязкости раствора перед подачей его в колонну. Колонна может быть снабжена любой удобной насадкой или оборудована тарелками, как это принято в технологии дистилляции. Предпочтительно, пар должен иметь давление 1 - 15 бар (абсолютных), более предпочтительно 5 - 10 бар (абсолютных) может содержать некоторое количество перегретого пара. Давление в колонне составляет предпочтительно 1 - 15 бар (абсолютных), более предпочтительно 5 бар (абсолютных). Соотношение между скоростью подачи спирта и скоростью подачи пара находится предпочтительно в пределах 0,01-2, более предпочтительно 0,05-0,3.
Газообразный продукт, содержащий спирт и не содержащий солей, непрерывно отбирается из верхней части колонны, а водосолевой раствор содержащий небольшое количество спирта, непрерывно отбирается из нижней части колонны. Предпочтительно, содержание солей в воде составляет 5 - 40%, более предпочтительно 25 - 30%.
В дополнение к регенерированию и рециклированию отработавшего в реакторе спирта, что выполняется после извлечения из реактора осадка, предпочтительно также улавливать и рециклировать:
(1) любые количества спирта, удаленного из спиртового раствора хлористого магния дегидратацией в операции (б),
(2) смывы, получаемые в результате промывки извлеченного осадка, и
(3) аммиак и любой спирт, удаленные из промытого осадка в операции (е).
Во втором своем аспекте настоящее изобретение предлагает в существенной степени безводный хлористый магний, полученный способом в соответствии с первым аспектом настоящего изобретения.
В третьем своем аспекте настоящее изобретение предлагает в существенной степени безводный хлористый магний, содержащий менее 0,05 вес.% оксида магния и, предпочтительно, менее 40 ppm кальция.
Хлористый магний в соответствии с третьим аспектом настоящего изобретения может быть получен способом в соответствии с первым аспектом настоящего изобретения.
На чертеже дана блок-схема опытной установки для получения безводного хлористого магния, описанной в примере 10.
Сравнительный пример.
Нижеследующий пример иллюстрирует способ по патенту США N 3966888 и по работе Аллена. Пример приводится исключительно в целях сравнения.
Сравнительный пример.
В двухлитровую пятигорлую круглодонную колбу, снабженную термометром, магнитным бруском для перемешивания, насадочной дистилляционной колонной диаметром 25 мм с разделителями флегмы, конденсатором и сборником, помещали 1302 грамма этиленгликоля. Туда же добавляли 429 граммов водного раствора хлористого магния, содержащего 33,6% (весовых) хлористого магния. С помощью вакуумного насоса в колбе понижали давление до 50 мм ртутного столба. Под колбу устанавливали электронагреватель с магнитной мешалкой, и температуру медленно повышали до 150oC в течение 6 часов с целью отгонки воды. Верхнюю порцию конденсата, почти чистую воду, возвращали в режиме дефлегмации в колонну с коэффициентом обратного потока 1 с целью сокращения потерь этиленгликоля в конденсат.
По завершении отгонки воды дистилляцией этиленгликолевый раствор хлористого магния анализировали на присутствие воды титрованием по методу Карла Фишера, определяя содержание воды 300 ppm. Температуру содержимого колбы с помощью нагревателя поддерживали на уровне 150oC.
Отдельную однолитровую пятигорлую плоскодонную колбу, снабженную верхней мешалкой, трубкой для барботирования газообразного аммиака с фильтром из пористого стекла (фритты), и термометром, помещали в охлаждаемую водяную баню. В эту колбу помещали 1000 г обезвоженного этиленгликолевого раствора хлористого магния, и колбу с ее содержимым охлаждали до 15oC, используя охлаждаемую водяную баню. Жидкость была очень вязкой и трудноперемешиваемой.
Поток безводного аммиака поступал от газового баллона через устройство регулирования давления. Высокая вязкость раствора крайне затрудняла обеспечение какого-либо потока аммиака через фильтр из пористого стекла. Чтобы преодолеть эту проблему, давление подачи аммиака было увеличено, однако это привело к взрыву барботерной трубки и фильтра с высвобождением большого объема сжатого аммиака внутри кристаллизатора. Это, в свою очередь, привело к разгерметизации крышки с сильным выбросом этиленгликолевого раствора хлористого магния и газообразного аммиака из емкости.
Анализ подтвердил, что вязкость раствора составляла около 120 mPas.
С целью преодоления затруднений с вводом аммиака при 15oC, обусловленных высокой вязкостью раствора, эксперимент был повторен с подачей аммиака в этиленгликолевый раствор хлористого магния при температуре раствора 60oC. Температуру раствора поддерживали с помощью водяной бани, оборудованной средствами нагрева и охлаждения.
Аммиак подавался в плоскодонную колбу, содержащую этиленгликолевый раствор хлористого магния, со скоростью 3,0 г в минуту. Подачу аммиака продолжали до достижения емкостью и ее содержимым постоянного веса, что указывало на насыщение раствора аммиаком, и о чем также свидетельствовало истечение аммиака через жидкостный затвор, заполненный этиленгликолем. Изменение веса содержимого колбы, включая некоторое количество сформировавшихся кристаллов, составило 149 граммов. Это свидетельство о достижении молярного соотношения аммиака к хлористому магнию, равного 7,87.
Раствору и кристаллам затем позволяли медленно остыть до 20oC в течение 16 часов при продолжающемся перемешивании. Содержимое колбы затем выгружали в перчаточной камере в атмосфере аргона и далее помещали в лабораторную корзиночную центрифугу, снабженную фильтровальной тканью из полипропилена.
Шлам затем центрифугировали в течение 10 минут.
Фильтровальную лепешку собирали и подвергали анализу. Было выявлено, что отфильтрованный осадок содержал 24,2 вес. % хлористого магния, 0,7 вес.% хлористого кальция, 7,3 вес. % аммиака и 68,5 вес.% гликоля, включая как свободный этиленгликоль, так и гликолевые лиганды любых гликолятных соединений хлористого магния. Содержание хлористого магния и хлористого кальция определялось титрованием этилендиаминтетрауксусной кислотой (EDTA), содержание аммиака определялось анализом Кьелдала, а содержание гликоля определялось жидкостной хроматографией высокого давления (HPLC). Значение содержания аммиака и хлористого магния в 7,3 вес.% и 24,2 вес.%, соответственно, свидетельствуют о величине молярного оношения аммиака к хлористому магнию во влажном кристалле, составляющей 1,7:1,0, что в значительной степени расходится с величиной отношения в 6,0:1,0, ожидаемой для чистого гексааммониата хлористого магния. Присутствие как тригликолята хлористого магния, так и биаммониата бигликолята хлористого магния подтверждалось как методами XRD, так и FTIR - спектрографией.
Описание предпочтительных вариантов осуществления изобретения.
Нижеприводимые примеры иллюстрируют предпочтительные варианты осуществления настоящего изобретения и не должны истолковываться как ограничивающие объем настоящего изобретения в той или иной степени.
Пример 1. Периодическое получение безводного хлористого магния в этиленгликоле.
В двухлитровую пятигорлую круглодонную колбу, снабженную термометром, магнитным бруском для перемещения, насадочной дистилляционной колонной диаметром 25 мм с разделителем флегмы, конденсатором и сборником, помещали 1000 граммов этиленгликоля. Туда же добавляли 535 граммов водного раствора хлористого магния, содержащего 33% (весовых) хлористого магния. С помощью вакуумного насоса в колбе понижали давление до 50 мм ртутного столба. Под колбу устанавливали электронагреватель с магнитной мешалкой и температуру медленно повышали до 150oC в течение 6 часов с целью отгонки воды. Верхнюю порцию конденсата, почти чистую воду, возвращали в режиме дефлегмации в колонну с коэффициентом обратного потока 1 с целью сокращения потерь этиленгликоля в конденсат.
По завершении отгонки воды дистилляцией обезвоженный этиленгликолевый раствор хлористого магния анализировали на присутствие воды титрованием по методу Карла Фишера, определяли содержание воды в 200 ppm. Температуру содержимого колбы с помощью нагревателя поддерживали на уровне 150oC.
Отдельную однолитровую пятигорлую плоскодонную колбу, снабженную верхней мешалкой, трубкой для барботирования газообразного аммиака и термометром, помещали в охлаждаемую водяную баню. В эту колбу помещали 100 г этиленгликоля, насыщенного безводным аммиаком при 15oC. Полученный обезвоженный этиленгликолевый раствор хлористого магния затем закачивали в плоскодонную колбу в течение 4-часового периода с одновременным барботированием безводным газообразным аммиаком через жидкое содержимое колбы. Аммиак добавлялся в количестве, достаточном для создания 20% избытка по отношению к требуемому количеству, о чем свидетельствовало истечение газообразного аммиака из колбы, фиксируемое ротаметром газового потока. Температуру содержимого колбы поддерживали на уровне 15oC в течение подачи этиленгликолевого раствора хлористого магния путем регулирования температуры водяной бани. Зернистый осадок формировался почти немедленно при вводе этиленгликолевого раствора хлористого магния в насыщенный аммиаком этиленгликоль и продолжал формироваться в ходе непрерывной подачи обезвоженного этиленгликолевого раствора хлористого магния и аммиака в течение последующих 4 часов.
Содержимое колбы затем выгружали в перчаточной камере в атмосфере аргона и далее помещали в лабораторную корзиночную центрифугу, снабженную фильтровальной тканью из полипропилена. Шлам затем центрифугировали в течение 10 минут. Отдельную порцию в 500 г метанола при 15oC насыщали безводным аммиаком и помещали в промывочное устройство. Находящиеся в центрифуге кристаллы затем промывали при вращении центрифуги струей насыщенного аммиаком метанола из промывочного устройства. В результате анализа было выявлено, что отфильтрованная лепешка содержала 46,3 вес.% хлористого магния, 49,7 вес.% аммиака, 4,0 вес.% метанола и менее 50 ppm гликоля. Содержание хлористого магния, содержание аммиака, содержание метанола и содержание гликоля определялись EDTA - титрованием, анализом Кьелдала, HPLC и HPLC, соответственно. Значение содержания аммиака и хлористого магния в 49,7 вес.% и 46,3 вес.%, соответственно, свидетельствуют о величине молярного отношения аммиака к хлористому магнию, равному 6,1:1,0 что хорошо согласуется с величиной молярного отношения 6,0:1,0, ожидаемой для чистого гексааммониата хлористого магния. Методы XRD и FTIR - спектрографией показывали отсутствие соединений гликолята хлористого магния.
Примерно 100 граммов твердого вещества выгружали из центрифуги и помещали в круглодонную 500-миллиметровую колбу в перчаточной камере в атмосфере аргона. Колба была снабжена верхней мешалкой со стеклянным импеллером (лопастное колесо), а также термопарой со стеклянной защитой. Колбу устанавливали на нагревателе и содержимое нагревали до 650oC в течение 3 часов при продувке колбы сухим аргоном во избежание проникновения любого количества водяных паров в колбу. Количественный анализ конечного твердого осадка выявил содержание менее 50 ppm аммиака и 0,04% оксида магния, тогда как остальную часть осадка составлял хлористый магний. Значение содержания аммиака, оксида магния и хлористого магния определялись анализом Кьелдала, обратным титрованием соляной кислоты/гидроксидом натрия (кислотно-основным титрованием), и EDTA-титрованием, соответственно.
Пример 2. Периодическое получение гексааммониата хлористого магния в метаноле.
В двухлитровую пятигорлую круглодонную колбу, снабженную термометром и магнитным бруском для перемешивания, помещали 733 грамма в существенной степени безводного метанола. В течение 30 минут в метанол вводили 100 граммов безводного хлористого магния. Под колбу устанавливали электронагреватель с магнитной мешалкой, и температуру поднимали до 35oC с целью растворения хлористого магния в метаноле. Это осуществлялось за 30 минут, а небольшое количество остающегося нерастворимого материала отфильтровывали с помощью лабораторного пресс-фильтра. Чистый раствор возвращали в круглодонную колбу, температуру в которой поддерживали на уровне 35oC с помощью нагревателя.
Отдельную однолитровую пятигорлую плоскодонную колбу, снабженную верхней мешалкой, трубкой для барботирования газообразного аммиака и термометром, помещали в нагреваемую водяную баню. В эту колбу помещали 100 г метанола, насыщенного аммиаком при 35oC. Затем в плоскодонную колбу в течение 4 часов закачивали полученный метаноловый раствор хлористого магния при одновременном барботировании безводного газообразного аммиака через жидкое содержимое колбы. Аммиак добавляли в количестве, достаточном для создания 20% избытка по отношению к требуемому количеству, о чем свидетельствовало истечение газообразного аммиака из колбы, фиксируемые ротаметром газового потока. Температуру содержимого колбы поддерживали на уровне 35oC в течение подачи метанолового раствора хлористого магния в насыщенный аммиаком метанол путем регулирования температуры водяной бани. Зернистый осадок формировался почти немедленно при вводе метанолового раствора хлористого магния и продолжал формироваться в ходе непрерывной подачи метанолового раствора хлористого магния и аммиака в течение последующих 4 часов.
Содержимое колбы выгружали из нее в перчаточной камере в атмосфере аргона и помещали в лабораторную корзиночную центрифугу, снабженную фильтровальной тканью из полипропилена. Шлам затем центрифугировали в течение 10 минут. В результате анализа было выявлено, что отфильтрованная лепешка содержала 42,0% вес. хлористого магния, 44,2% вес. аммиака и 13,8% вес. метанола. Величины содержания хлористого магния, содержания аммиака и содержания метанола определялись EDTA титрованием, анализом Кьелдала и HPLC, соответственно. Значение содержания аммиака и хлористого магния в 44,2% вес. и 42,0% вес. , соответственно, свидетельствуют о величине молярного отношения аммиака к хлористому магнию, равной 5,9:1,0, что хорошо согласуется с величиной молярного отношения 6,0: 1,0, ожидаемой для чистого гексааммониата хлористого магния. Методы XRD и FTIR - спектроскопия показали отсутствие гликолятных соединений хлористого магния.
Пример 3. Периодическое получение гексааммониата хлористого магния в диэтиленгликоле.
В двухлитровую пятигорлую круглодонную колбу, снабженную термометром, магнитным бруском для перемешивания, насадочной дистилляционной колонной диаметром 25 мм с разделителем флегмы, конденсатором и сборником, помещали 1300 граммов этиленгликолята. Туда же добавляли 200 граммов водного раствора хлористого магния, содержащего 32% (весовых) хлористого магния. С помощью вакуумного насоса в колбе понижали давление до 50 мм ртутного столба. Под колбу устанавливали электронагреватель с магнитной мешалкой и температуру медленно повышали до 170oC в течение 6 часов с целью отгонки воды. Верхнюю порцию конденсата, почти чистую воду, возвращали в режиме дефлегмации в колонну с коэффициентом обратного потока 1 с целью сокращения потерь этиленгликоля в конденсат.
По завершении отгонки воды дистилляцией обезвоженный этиленгликолевый раствор хлористого магния анализировали на присутствие воды титрованием по методу Карла Фишера, определяя содержание воды в 200 ppm. Затем содержимому колбы позволяли остыть до комнатной температуры.
Отдельную однолитровую пятигорлую плоскодонную колбу, снабженную верхней мешалкой, трубкой для барботирования газообразного аммиака и термометром, помещали в охлаждаемую водяную баню. Колба была оборудована боковым переливным отводом. В эту колбу помещали 800 г диэтиленгликоля, насыщенного безводным аммиаком при 15oC и вводили заправочные кристаллы гексааммониата хлористого магния. Полученный обезвоженный диэтиленгликолевый раствор хлористого магния затем закачивали в плоскодонную колбу в течение 5 часов с одновременным барботированием безводным газообразным аммиаком через жидкое содержимое колбы. Получаемый шлам переливался через боковой отвод колбы и собирался в герметизированный двухлитровой колбе. Аммиак добавляли в количестве, достаточном для создания 20% избытка по отношению к требуемому количеству, о чем свидетельствовало истечение газообразного аммиака из колбы, фиксируемое ротаметром газового потока. Температуру содержимого колбы поддерживали на уровне 15oC в течение подачи обезвоженного диэтиленгликолевого раствора хлористого магния путем регулирования температуры водяной бани. Мелкокристаллический осадок формировался почти немедленно при вводе обезвоженного диэтиленгликолевого раствора хлористого магния в насыщенный аммиаком диэтиленгликоль и продолжал формироваться в ходе непрерывной подачи обезвоженного диэтиленгликолевого раствора хлористого магния и аммиака в течение последующих 5 часов.
Содержимое колбы выгружали из нее в перчаточной камере в атмосфере аргона и помещали в лабораторную корзиночную центрифугу, снабженную фильтровальной тканью из полипропилена. Шлам затем центрифугировали в течение 10 минут и промывали метанолом, насыщенным аммиаком. Анализ показал, что отфильтрованная лепешка содержала 39,1 вес.% хлористого магния, 40,3 вес.% аммиака, 0,7 вес. % диэтиленгликоля, и остаток составляла промывочная жидкость. Значение содержания диэтиленгликоля определялись EDTA-титрованием, анализом Кьелдала и HPLC, соответственно. Величины содержания аммиака и хлористого магния 40,3% вес. и 39,1% вес., соответственно, свидетельствуют о величине молярного отношения аммиака к хлористому магнию 5,8:1,0, что хорошо согласуется с величиной молярного отношения 6,0:1,0, ожидаемой для чистого гексааммониата хлористого магния.
Пример 4. Одностадийная непрерывная кристаллизация гексааммониата хлористого магния.
В двадцатилитровую трехгорлую круглодонную колбу, снабженную дистиллятором вихревого слоя (B/R Instrument Corp), помещали 12 кг этиленгликоля и 6,6 кг водного раствора хлористого магния (33% вес.).
С помощью вакуумного насоса давление в колбе понижали до 50 мм ртутного столба. Под колбу устанавливали электронагреватель с магнитной мешалкой и температуру медленно повышали до 150oC в течение 6 часов с целью отгонки воды. Верхнюю порцию конденсата, почти чистую воду, возвращали в режиме дефлегмации в колонну с коэффициентом обратного потока 2 с целью сокращения потерь этиленгликоля в конденсат.
По завершении отгонки воды дистилляцией обезвоженный этиленгликолевый раствор хлористого магния анализировали на содержание воды титрованием по методу Карла Фишера, определяя содержание воды в 300 ppm. Температуру содержимого колбы поддерживали на уровне 150oC с помощью электронагревателя.
Двухлитровый кристаллизатор оборудовали электронными средствами контроля верхнего и нижнего уровней, предназначенными для задействования перистальтического насоса, который обеспечивал перенос кристаллического шлама из кристаллизатора в емкость для сбора продукта примерно с 10-минутными интервалами. Исходная жидкость, представляющая собой обезвоженный этиленгликолевый раствор хлористого магния, непрерывно закачивалась в кристаллизатор. Кристаллизатор представлял собой двухлитровый стеклянный сосуд, снабженный в верхней части стандартной фланцевой горловиной из притертого стекла. Крышка кристаллизатора была выточена из нержавеющей стали марки 316 и содержала узел механической герметизации для мешалки, а также каналы для ввода исходного материала, трубки для барботирования аммиака, чехла для термопары, каналы для вывода продукта и для отвода избытка аммиака. Корпус кристаллизатора подвешивали за крышку сосуда и опускали в пятилитровый стакан, в который поступала охлажденная вода для поддерживания требуемого температурного режима кристаллизации. Газообразный аммиак поступал в сосуд через ротаметр под нижним импеллером из двух четырехлопастных пропеллерных мешалок, приводимых во вращение верхним лабораторным приводом.
Поток охлажденной воды, поступающей к кристаллизатору, регулировали для поддержания кристаллизатора в рабочем температурном режиме в 30oC.
Перед началом подачи в кристаллизатор исходного материала, кристаллизатор наполняли этиленгликолем, насыщенным аммиаком, и вводили в него затравочный кристалл гексааммониата хлористого магния.
Обезвоженный этиленгликолевый раствор хлористого магния затем закачивали в кристаллизатор со скоростью 2,5 кг/час. В кристаллизатор подавали избыток аммиака, о чем свидетельствовало истечение газообразного аммиака из кристаллизатора, фиксируемое ротаметром газового потока. В течение пяти часов работы кристаллизатора содержание аммиака в этиленгликоле в кристаллизаторе обычно составлял 12% вес.
Содержимое емкости для сбора шламообразного продукта перекачивали в корзиночную центрифугу диаметром 335 мм, снабженную фильтровальной тканью из полипропилена. В ходе загрузки центрифуги скорость ее вращения удерживали на уровне 1250 об/мин, а затем увеличивали до 1780 об/мин в ходе отжима жидкой фазы. Отдельно, 8 кг метанола при 20oC насыщали безводным аммиаком в стеклянном сосуде. Этот раствор затем закачивали на кристаллы в центрифуге, вращающейся со скоростью 1780 об/мин. После завершения промывки кристаллы центрифугировали дополнительно 10 минут для уменьшения содержания в них метанола.
Образцы отцентрифугированных промытых кристаллов анализировали, определяли содержание в них 47,8 вес.% хлористого магния, 50,1 вес.% аммиака и 348 ppm хлористого кальция. Величины содержания хлористого магния и хлористого кальция определялись EDTA-титрованием, а содержание аммиака определяли анализом Кьелдала. Значения содержания аммиака и хлористого магния 50,1 вес.% и 47,8 вес.%, соответственно, свидетельствовали о величине молярного отношения аммиака к хлористому магнию 5,9: 1,0, что хорошо согласуется с величиной молярного отношения 6,0: 1 ожидаемой для чистого гексааммониата хлористого магния. XRD и FTIR-спектроскопия свидетельствовали об отсутствии гликолятных соединений хлористого магния.
Пример 5. Двухстадийная непрерывная кристаллизация гексааммониата хлористого магния.
В двадцатилитровую трехгорлую круглодонную колбу, снабженную дистиллятором вихревого слоя (B/R Instrument Corp), помещали 12 кг этиленгликоля и 6,6 кг водного раствора хлористого магния (33% вес.).
С помощью вакуумного насоса давление в колбе понижали до 50 мм ртутного столба. Под колбу устанавливали электронагреватель с магнитной мешалкой и температуру медленно повышали до 150oC в течение 6 часов с целью отгонки воды. Верхнюю порцию конденсата, почти чистую воду, возвращали в режиме дефлегмации в колонну с коэффициентом обратного потока 2 с целью сокращения потерь этиленгликоля в конденсат.
По завершении отгонки воды дистилляцией обезвоженный этиленгликолевый раствор хлористого магния анализировали на содержание воды титрованием по методу Карла Фишера, определяли содержание воды в 300 ppm. Температуру содержимого колбы поддерживали на уровне 150oC с помощью электронагревателя.
Два двухлитровых кристаллизатора соединяли последовательно; каждый из кристаллизаторов оборудовали электронными средствами контроля верхних и нижних уровней, предназначенными для задействования перистальтических насосов, которые обеспечивали перенос кристаллического шлама из первого кристаллизатора во второй кристаллизатор и из второго кристаллизатора - в емкость для сбора продукта примерно с 10-минутными интервалами. Исходная жидкость, представляющая собой обезвоженный этиленгликолевый раствор хлористого магния, непрерывно закачивалась в первый кристаллизатор. Каждый из кристаллизаторов представлял собой двухлитровый стеклянный сосуд, снабженный в верхней части стандартной фланцевой горловиной из притертого стекла. Крышки кристаллизаторов были выточены из нержавеющей стали марки 316 и содержали соответственно каждая узел механической герметизации для мешалки, а также каналы для ввода исходного материала, трубки для барботирования аммиака, чехлы для термопары, каналы для вывода продукта и для отвода избытка аммиака. Корпуса кристаллизаторов подвешивали за крышки сосудов и опускали в пятилитровые стаканы, в которые поступала охлажденная вода для поддерживания требуемого температурного режима кристаллизации. Газообразный аммиак поступал соответственно в каждый из сосудов через ротаметр под нижним импеллером из двух четырехлопастных пропеллерных мешалок, приводимых во вращение верхними лабораторными приводами.
Поток охлажденной воды, поступающей к кристаллизаторам, регулировали для поддержания кристаллизатора в рабочем температурном режиме 59oC, и рабочего температурного режима во втором кристаллизаторе 39oC.
Перед началом подачи в первый кристаллизатор исходного материала оба кристаллизатора наполняли этиленгликолем, насыщенным аммиаком, и вводили в него затравочные кристаллы гексааммониата хлористого магния.
Обезвоженный этиленгликолевый раствор хлористого магния затем закачивали в первый кристаллизатор со скоростью 3,2 кг/час. В каждый из кристаллизаторов подавали избыток аммиака, о чем свидетельствовало истечение газообразного аммиака из каждого кристаллизатора, фиксируемое ротаметрами газового потока. В течение пяти часов работы кристаллизаторов в последовательном цикле уровень аммиака в этиленгликоле в кристаллизаторах варьировали между 7.1% вес. и 11,8% вес.
Содержимое емкости для сбора шламообразного продукта перекачивали в корзиночную центрифугу диаметром 335 мм, снабженную фильтровальной тканью из полипропилена. В ходе загрузки центрифуги скорость ее вращения удерживали на уровне 1250 об/мин, а затем увеличивали до 1780 об/мин в ходе отжима жидкой фазы. Отдельно, 8 кг. метанола при 20oC насыщали безводным аммиаком в стеклянном сосуде. Этот раствор затем закачивали на кристаллы в центрифуге, вращающейся со скоростью 1780 об/мин. После завершения промывки кристаллы центрифугировали дополнительно 10 минут для уменьшения содержания в них метанола.
Образцы отцентрифугированных промытых кристаллов анализировали, определяли содержание в них 51,4% вес. хлористого магния, 49,3% вес. аммиака и 348 ppm хлористого кальция. Величины содержания хлористого магния и хлористого кальция определялись EDTA-титрованием, а содержание аммиака определяли анализом Кьелдала. Значения содержания аммиака и хлористого магния 50,1% вес. и 47,8% вес., соответственно, свидетельствовали о величине молярного отношения аммиака к хлористому магнию 5,4: 1,0, что довольно неплохо согласуется с величиной молярного отношения 6,0:1,0, ожидаемой для чистого гексааммониата хлористого магния. XRD и FTIR - спектроскопия свидетельствовали об отсутствии гликолятных соединений хлористого магния.
Пример 6. Влияние уровня содержания воды в обезвоженном гликолевом растворе хлористого магния на уровень содержания безводного хлористого магния и оксида магния.
В двухлитровую пятигорлую круглодонную колбу, снабженную термометром, магнитным бруском для перемешивания, насадочной дистилляционной колонной диаметром 25 мм с разделителем флегмы, конденсатором и сборником, помещали 2000 граммов этиленгликоля. Туда же добавляли 535 граммов водного раствора хлористого магния, содержащего 33% вес. хлористого магния. С помощью вакуумного насоса в колбе понижали давление до 50 мм ртутного столба. Под колбу устанавливали электронагреватель с магнитной мешалкой, и температуру медленно повышали до 150oC в течение 6 часов с целью отгонки воды. Верхняя порция конденсата вначале представляла собой почти чистую воду, однако, по завершении дистилляции она представляла собой чистый этиленгликоль. Чистый этиленгликоль затем добавляли в обезвоженный этиленгликолевый раствор хлористого магния для корректировки хлористого магния до 15% вес.
По завершении отгонки воды дистилляцией и корректировки уровня содержания хлористого магния обезвоженный этиленгликолевый раствор хлористого магния анализировали на присутствие воды титрованием по методу Карла Фишера, определяли содержание воды в 320 ppm. Температуру содержимого колбы поддерживали на уровне 150oC с помощью электронагревателя.
Отдельную однолитровую пятигорлую плоскодонную колбу, снабженную верхней мешалкой, трубкой для барботирования газообразного аммиака и термометром, помещали в охлаждаемую водяную баню. В эту колбу помещали 100 г. этиленгликоля, насыщенного безводным аммиаком при 30oC. Полученный обезвоженный этиленгликолевый раствор хлористого магния затем закачивали в плоскодонную колбу в течение 4 часов с одновременным барботированием безводным газообразным аммиаком через жидкое содержимое колбы. Аммиак добавляли в количестве, достаточном для создания 20% избытка по отношению к требуемому количеству, о чем свидетельствовало истечение газообразного аммиака из колбы, фиксируемое ротаметром газового потока. Температуру содержимого колбы поддерживали на уровне 30oC в течение подачи обезвоженного этиленгликолевого раствора хлористого магния путем регулирования температуры водяной бани. Зернистый осадок формировался почти немедленно при вводе обезвоженного этиленгликолевого раствора хлористого магния в насыщенный аммиаком этиленгликоль и продолжал формироваться в ходе непрерывной подачи обезвоженного этиленгликолевого раствора хлористого магния и аммиака в течение последующих 4 часов.
Содержимое колбы выгружали из нее в перчаточную камеру в атмосфере аргона и помещали в лабораторную корзиночную центрифугу, снабженную фильтровальной тканью из полипропилена. Шлам затем центрифугировали в течение 10 минут. Отдельную порцию безводного метанола в 500 граммов, с содержанием 290 ppm воды, при 15oC насыщали аммиаком и помещали в промывочное устройство. Находящиеся в центрифуге кристаллы затем промывали при вращении центрифуги струей насыщенного аммиаком метанола из промывочного устройства в течение 2 минут репульпированием в 500 граммах насыщенного при 15oC аммиаком метанола, и затем повторно центрифугировали. XRD-анализ образцов, промытых репульпированием и отцентрифугированных кристаллов свидетельствовал об отсутствии гликолятных соединений хлористого магния.
Около 20 граммов твердого осадка выгружали из центрифуги в перчаточной камере в атмосфере аргона и помещали в 200-миллилитровую круглодонную колбу. Колба была снабжена верхней и мешалкой со стеклянным импеллером, а также термопарой со стеклянной защитой. Колбу устанавливали на нагревателе и содержимое нагревали до 450oC в течение 3 часов при продувке колбы сухим аргоном во избежание проникновения любого количества водяных паров в колбу. Количественный анализ конечного твердого осадка выявил содержание в осадке 0,22% вес. оксида магния.
Весь цикл эксперимента повторяли еще пять раз с корректированием содержания воды в обезвоженном этиленгликолевом растворе хлористого магния в различных экспериментах до 960 ppm, 5140 ppm, 9830 ppm, и 50560 ppm. Полученный в результате прокаленный хлористый магний содержал 0,25%, 0,21%, 0,19%, 0,35% и 0,28% оксида магния, соответственно. Эти результаты свидетельствовали о небольшой зависимости между концентрацией прокалочной окиси магния и концентрацией воды в обезвоженном этиленгликолевом растворе хлористого магния до уровня содержания воды в 50560 ppm. Количественные FTIR-анализы прокаленных продуктов указывали на присутствие небольших количеств производного пиридина, которое при кислотно-основном титровании в определении содержания оксида магния оценивалось как оксид магния, что приводило к неверно завышенной оценке оксида магния. XRD-анализ свидетельствовал также о присутствии силикатов магния, образовавшихся в результате реакции безводного хлористого магния с натриево-силикатным стеклом кальцинатора при повышенных температурах. Эти силикаты также оценивались как оксид магния при кислотном титровании. Последующие эксперименты показали, что соединения пиридина разрушались при 650oC, и что истинный уровень содержания оксида магния составлял менее 0,1% при использовании кальцинатора из инертного кварцевого стекла.
Пример 7. Влияние уровня содержания воды в обезвоженном гликолевом растворе хлористого магния на уровни содержания безводного хлористого магния и оксида магния.
В двухлитровую пятигорлую круглодонную колбу, снабженную термометром, магнитным бруском для перемешивания, насадочной дистилляционной колонной диаметром 25 мм с разделителем флегмы, конденсатором и сборником, помещали 2000 граммов этиленгликоля. Туда же добавляли 535 граммов водного раствора хлористого магния, содержащего 33% вес. хлористого магния. С помощью вакуумного насоса в колбе понижали давление до 50 мм ртутного столба. Под колбу устанавливали электронагреватель с магнитной мешалкой и температуру медленно повышали до 150oC в течение 6 часов с целью отгонки воды. Верхняя порция конденсата вначале представляла собой почти чистую воду, однако, по завершении дистилляции она представляла собой чистый этиленгликоль. Чистый этиленгликоль затем добавляли в обезвоженный этиленгликолевый раствор хлористого магния для корректировки концентрации хлористого магния до 15% вес.
По завершении отгонки воды дистилляцией и корректировки уровня содержания хлористого магния обезвоженный этиленгликолевый раствор хлористого магния анализировали на присутствие воды титрованием по методу Карла Фишера, определяя содержание воды в 55 ppm. Температуру содержимого колбы поддерживали на уровне 150oC с помощью электронагревателя.
Отдельную однолитровую пятигорлую плоскодонную колбу, снабженную верхней мешалкой, трубкой для барботирования газообразного аммиака и термометром, помещали в охлаждаемую водяную баню. В эту колбу помещали 100 г этиленгликоля, насыщенного безводным аммиаком при 35oC. Полученный обезвоженный этиленгликолевый раствор хлористого магния затем закачивали в плоскодонную колбу в течение 4 часов с одновременным барботированием безводным газообразным аммиаком через жидкое содержимое колбы. Аммиак добавляли в количестве, достаточном для создания 20% избытка по отношению к требуемому количеству, о чем свидетельствовало истечение газообразного аммиака из колбы, фиксируемое ротаметром газового потока. Температуру содержимого колбы поддерживали на уровне 35oC в течение подачи обезвоженного этиленгликолевого раствора хлористого магния путем регулирования температуры водяной бани. Зернистый осадок формировался почти немедленно при вводе обезвоженного этиленгликолевого раствора хлористого магния в насыщенный аммиаком этиленгликоль и продолжал формироваться в ходе непрерывной подачи обезвоженного этиленгликолевого раствора хлористого магния и аммиака в течение последующих 4 часов.
Содержимое колбы выгружали из нее в перчаточной камере в атмосфере аргона и помещали в лабораторную корзиночную центрифугу, снабженную фильтровальной тканью из полипропилена. Шлам затем центрифугировали в течение 20 минут. Отдельную порцию безводного метанола в 500 граммов при 15oC насыщали безводным аммиаком и помещали в промывочное устройство. Находящиеся в центрифуге кристаллы затем промывали при вращении центрифуги струей насыщенного аммиаком метанола из промывочного устройства. XRD-анализ образцов, промытых и отцентрифугированных кристаллов свидетельствовали об отсутствии гликолятных соединений хлористого магния.
Около 20 граммов твердого осадка выгружали из центрифуги и помещали в пробирку из кварцевого стекла диаметром 20 мм и длиной 400 мм. Горловину пробирки герметизировали от контакта с атмосферой, и для мягкой продувки верхней части объема пробирки использовали аргон, осушенный пропуском через последовательный ряд пробирок, содержащих молекулярные сита в 3A. Выходящий из пробирки газ пропускали через два жидкостных затвора, заполненных безводным этиленгликолем, во избежание обратного подсасывания влажного воздуха.
Кварцевую пробирку вертикально устанавливали в лабораторную печь и температуру в печи повышали до 650oC в течение 4 часов. Количественный анализ конечного твердого осадка с помощью кислотно-основного титрования выявил содержание в осадке менее 0,02% оксида магния.
Весь цикл эксперимента повторяли еще два раза с корректированием содержания воды в обезвоженном этиленгликолевом растворе хлористого магния до 300 ppm в одном случае и 500 ppm во втором случае. Полученный в результате прокаленный хлористый магний содержал 0.08% хлористого магния и 0.07% оксида магния, соответственно. Эти эксперименты показывали, что при уровнях содержания воды до 500 ppm в обезвоженном этиленгликолевом растворе хлористого магния возможно получение безводного хлористого магния, содержащего менее 0,1% оксида магния.
Пример 8. Удаление кальция из рециклируемого гликоля.
В двухлитровую трехгорлую круглодонную колбу, снабженную термометром, магнитным бруском для перемешивания и конденсатором, и содержащую некоторое количество хлористого магния, помещали 900 граммов этиленгликоля, содержащего хлористый кальций и некоторое количество хлористого магния. С помощью вакуумного насоса в колбе понижали давление до 50 мм ртутного столба, и этиленгликоль выпаривали из смеси при 150oC в течение 5 часов. По завершении выпаривания оставалось 100 г раствора, который анализировали EDTA-титрованием, определяя содержание 171 г/кг хлористого кальция и 46 г/кг хлористого магния в этиленгликоле. Температуру этого раствора поддерживали на уровне 100oC.
Отдельную однолитровую плоскодонную колбу снабжали трехгорлой крышкой и верхней мешалкой с импеллером из нержавеющей стали, а также трубкой для барботирования диоксида углерода. Этот аппарат помещали в охлаждаемую водяную баню, и в колбу вводили 500 граммов деионизированной воды, которую охлаждали до 15oC. Затем через воду барботировали диоксид углерода и в течение двух часов в смесь воды с диоксидом углерода вводили 15,8 грамма тонкодисперсного порошка оксида магния. Оксид углерода подавали со скоростью 250 миллилитров в минуту для обеспечения его избытка по отношению к требуемому количеству. В ходе подачи оксида магния температуру в жидкости тщательно удерживали на уровне 150oC. Полученную в результате жидкость анализировали, определяя содержание в ней 14,3 г/кг магния (бикарбоната магния).
К 90 граммам концентрированного раствора хлористого кальция/хлористого магния в этиленгликоле добавляли 253 грамма полученного бикарбоната магния в течение 30 минут. Осадок формировался сразу же при вводе бикарбоната магния. Температуру смеси поддерживали на уровне 100oC в течение всего периода подачи бикарбоната магния и в течение дополнительных 15 минут после окончания подачи бикарбоната магния. Содержимое колбы, представляющее собой смесь твердого осадка карбоната кальция и хлористого магния, этиленгликоля и воды в растворе, помещали в воронку Бюхнера, снабженную фильтровальной бумагой. Твердый осадок легко отфильтровывался и затем его промывали 50 граммов воды.
Фильтрат анализировали с использованием атомносорбционной спектрграфии, убеждаясь, что 91% кальция, содержавшегося в концентрированном этиленгликолевом растворе хлористого кальция/хлористого магния, выпал в осадок.
Пример 9. Паровая десорбция солей из рециклиуемого гликоля.
В двухлитровую трехгорлую круглодонную колбу, снабженную магнитным бруском для перемешивания, термометром, и конденсатором, помещали 1700 граммов этиленгликоля, содержащего хлористый кальций и некоторое количество хлористого магния. С помощью вакуумного насоса в колбе понижали давление до 50 мм ртутного столба и этиленгликоль выпаривали из смеси при 150oC в течение 6 часов. Пробу полученного в результате раствора анализировали, определяли содержание в нем 7,2% хлористого магния, 24,6% хлористого кальция и 68,2% этиленгликоля. Величины содержания хлористого магния, хлористого кальция и этиленгликоля определялись EDTA-титрованием, EDTA-титрованием, и HPLC, соответственно.
Пробу в 200 граммов концентрированного этиленгликолевого раствора соли разбавляли 132 граммами воды для снижения вязкости раствора. Эту разбавленную смесь удерживали в температурном режиме 120oC и закачивали в верхнюю часть колонны диаметром 25 мм и высотой 400 мм, снабженной насадкой Sultzer, со скоростью 55 граммов в час. Давление в колонне поддерживали на уровне в 5 бар, используя регулятор давления, и выходящий из верхней части колонны газ конденсировали и собирали в сборнике. Жидкость из нижней части колонны отводили в автоклавный резервуар, из которого можно было осуществлять отбор проб.
Одновременно с подачей этиленгликоль-водного раствора солей в верхнюю часть колонны пар с давлением в 9 бар подавали в нижнюю часть колонны со скоростью 795 граммов в час. Тест-цикл продолжался в течение 6 часов, и анализы раствора, полученного из нижней части колонны, показали содержание в нем 5,6% хлористого магния, 20,1% хлористого кальция, 76,1% воды и лишь 700 ppm этиленгликоля. Величины содержания хлористого магния, хлористого кальция и этиленгликоля определялись EDTA-титрование, титрованием по методу Карла Фишера и HPLC, соответственно.
Пример 10. Опытная установка для получения безводного хлористого магния.
Для демонстрации способа по настоящему изобретению в более широком масштабе была задействована опытная рециркулировочная установка непрерывного действия.
33%-ный Раствор хлористого магния в воде (10) закачивали со скоростью потока в 53 грамма в минуту в двухлитровый стеклянный смеситель (11) вместе с рециклированным этиленгликолем (12), закачивали со скоростью потока в 100 граммов в минуту. Оба потока смешивались с помощью верхней мешалки с импеллером. Смесь этиленгликоля, хлористого магния и воды (13) затем закачивали в последовательную цепочку дегидратационных дистилляционных колонн. Дистилляционные колонны (14), (15) и (16) работали в технологических режимах, известных специалистам в технике дистилляции, обеспечивая отделение воды от этиленгликоля и хлористого магния. Энергообеспечение колонн осуществлялось с использованием паровых змеевиков в ребойлерах, а воду (17) отводили из верхней части колонны и конденсировали, возвращая некоторое количество конденсата в насадку верхней части колонны для содействия разделению воды и этиленгликоля. Дегидратационные дистилляционные колонны (14), (15) и (16) функционировали в режимах последовательного нарастающего вакуума с целью контроля температурных режимов ребойлеров на уровне до 150oC с целью предотвращения распада этиленгликоля, при одновременном уменьшении размеров колонн, обусловленных требованиями высоковакуумной дегидратации. Продукт (18) из нижней части колонны (14) подавали на колонну (15), а продукт (19) колонны (15) подавали на колонну (16). Колонна (14) работала в режиме атмосферного давления, тогда как колонны (15) и (16) работали в режимах пониженного давления.
Продукт (20) из нижней части колонны (16) имел температуру приблизительно 150oC и, по результатам количественного анализа, содержал 150 ppm воды, 15% вес. хлористого магния и этиленгликоль - остальное.
Обезвоженный этиленгликолевый раствор хлористого магния (20) непрерывно закачивали со скоростью приблизительно 117 граммов в минуту в двадцатилитровый кристаллизатор (21) при одновременной раздельной подаче безводного газообразного аммиака (22) со скоростью 34 грамма в минуту. Кристаллизатор (21) представлял собой плоскодонный резервуар, снабженный четырьмя внутренними отражательными перегородками и охлаждающей водяной рубашкой. Содержимое кристаллизатора перемешивалось с помощью верхней мешалки, снабженной четырехлопастным импеллером, вращавшимся со скоростью 750 об/мин. Обезвоженный этиленгликолевый раствор хлористого магния (20) подавался на поверхность содержимого кристаллизатора, тогда как аммиак (22) поступал в шлам кристаллизации через барботажную трубку, размещенную под импеллером. Поток охлаждающей воды регулировали для удержания температуры шлама на уровне 30oC. Количественный анализ маточного раствора кристаллизатора показывал содержание аммиака в 10% вес.
Кристаллический шлам (23) откачивали из кристаллизатора (21) с постоянной скоростью с целью поддерживания стабильного уровня шлама в кристаллизаторе (21). Шлам (23) выгружали в пятнадцатилитровый напорный фильтр (24), снабженный полипропиленовым сетчатым мешочным фильтром. Периодическое повышение давления подкачкой сухого азота (25) отфильтровывало маточный раствор кристаллизатора от кристаллов. После 4 часов подачи потока (23) к фильтру (24) поток переключали на второй идентичный фильтр (не показан), и приблизительно 8 килограммов кристаллов гексааммониата хлористого магния промывали четырьмя четырехлитровыми порциями метанола, содержащего 20% вес. аммиака (26). После промывки кристаллы обычно содержали менее 0,2% вес. этиленгликоля, и XRD-анализы свидетельствовали об отсутствии каких-либо гликолятных соединений хлористого магния.
Промытые кристаллы гексааммониата хлористого магния (27) вручную выгружали из фильтра (24) и помещали в бункер-накопитель для непрерывной подачи кристаллов на двухстадийную кальцинацию в псевдосжиженном слое (показан только один кальцинатор) (28). Рециклированный газообразный аммиак (29) использовался для псевдоожижения содержимого кальцинатора, а нагрев обеспечивался электрическими нагревательными элементами. Аппараты (28) были оборудованы переточными трубами, которые позволяли кристаллическому материалу непрерывно проходить из одного аппарата (28) в следующий аппарат (28).
В первом псевдосжиженном слое (28) температуру удерживали на уровне 120oC, чтобы обеспечить выпаривание метанола из влажных кристаллов. Второй псевдосжиженный слой (28) работал при 450oC с целью прокаливания сухих кристаллов до безводного хлористого магния с высвобождением газообразного аммиака (30) для непрерывного его рецикла в кристаллизаторе (21). Прокаленный кристаллический материал (31) перетекал из второго кальцинатора (28) и собирался в инертной атмосфере азота в двадцатилитровую колбу, изолированную от внешней атмосферы. Обычная скорость потока прокаленного кристаллического материала составляла 17 граммов в минуту. Прокаленные кристаллы анализировали, определяя содержание в них кальция в среднем менее 40 ppm.
Растворители (32), представленные этиленгликолем и метанолом, содержащим аммиак и небольшое количество растворимого хлористого магния, непрерывно закачивали со скоростью примерно 180 граммов в минуту в колонны регенерации растворителей (33) и (34). Первая колонна (33) работала при атмосферном давлении с температурой ребойлера 150oC. Продукт (35) из нижней части колонны (33) непрерывно закачивался в колонну глубокого вакуума (34) с температурой ребойлера 130oC для обеспечения полной регенерации метанола и аммиака. Колонны (33) и (34) были сконструированы и работали по технологическим принципам, изложенным ранее для гидратационных колонн. Насыщенный аммиаком жидкий метанол собирался в конденсаторах и рециклировался в напорные фильтры (24) для повторного использования, тогда как продукты (12) из нижней части колонны глубокого вакуума (34) рециклировались в стеклянный смеситель (11) для повторного использования.
Промышленная применимость
В существенной степени безводной хлористый магний, полученный способом в соответствии с первым аспектом настоящего изобретения, и хлористый магний, полученный в соответствии с третьим аспектом настоящего изобретения, могут использоваться в электролизном производстве металлического магния.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОКУСКОВАНИЯ МЕЛКОЗЕРНИСТОГО ТИТАНСОДЕРЖАЩЕГО МИНЕРАЛА | 1989 |
|
RU2080396C1 |
| ДИЦИАНОВЫЕ ФУМИГАНТЫ И СПОСОБ ФУМИГАЦИИ С ИСПОЛЬЗОВАНИЕМ ДИЦИАНА | 1995 |
|
RU2194390C2 |
| СПОСОБ НАНЕСЕНИЯ КРАСИТЕЛЯ НА КЕРАТИНОВЫЕ ВОЛОКНА (ВАРИАНТЫ) | 1993 |
|
RU2128257C1 |
| СПОСОБ МОДИФИКАЦИИ, ПО МЕНЬШЕЙ МЕРЕ, ЧАСТИ ПОВЕРХНОСТИ ПОЛИМЕРА | 1996 |
|
RU2163246C2 |
| ФУМИГАНТ, ФУМИГАНТНАЯ КОМПОЗИЦИЯ И СПОСОБ ОКУРИВАНИЯ | 1993 |
|
RU2141762C1 |
| МАГНИЕВОЕ ЛИТЬЕ ПОД ДАВЛЕНИЕМ | 1998 |
|
RU2212980C2 |
| СПОСОБ ФУМИГАЦИИ ПРОДУКТОВ ФОСФИНОМ И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 1990 |
|
RU2088097C1 |
| СПОСОБ ПОЛУЧЕНИЯ ФОСФИНА, УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ И СИСТЕМА БЕЗОПАСНОСТИ ДЛЯ ПРОИЗВОДСТВА ФОСФИНА | 1991 |
|
RU2087415C1 |
| СПОСОБ И УСТРОЙСТВО ДЛЯ ОТЛИВКИ МЕТАЛЛИЧЕСКИХ СЛИТКОВ | 1996 |
|
RU2182859C2 |
| КОНЪЮГАТЫ ТЕРАПЕВТИЧЕСКОГО СОЕДИНЕНИЯ С ЖИРНОЙ КИСЛОТОЙ | 1996 |
|
RU2166512C2 |
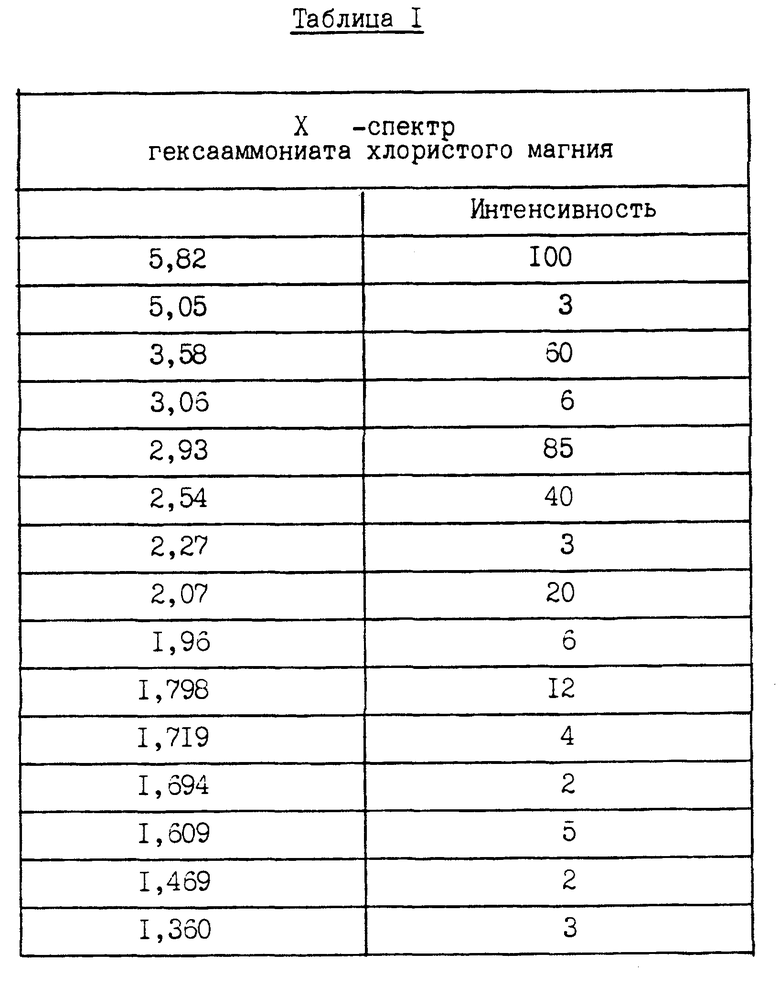
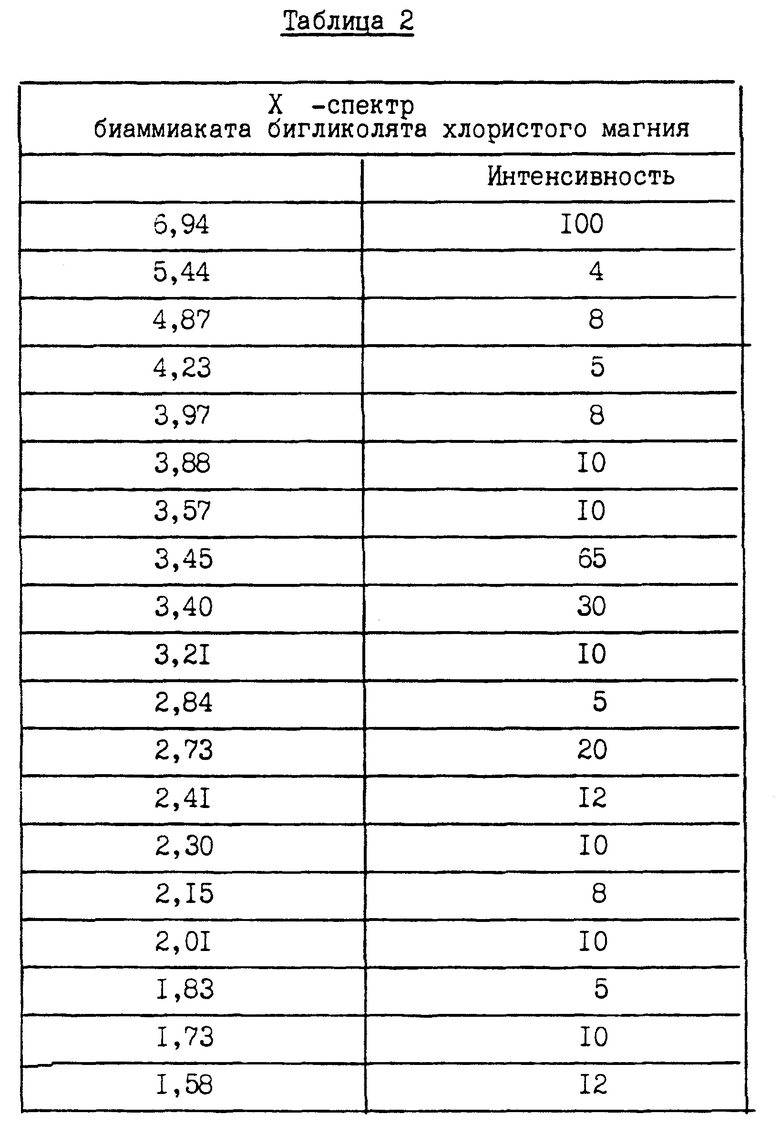
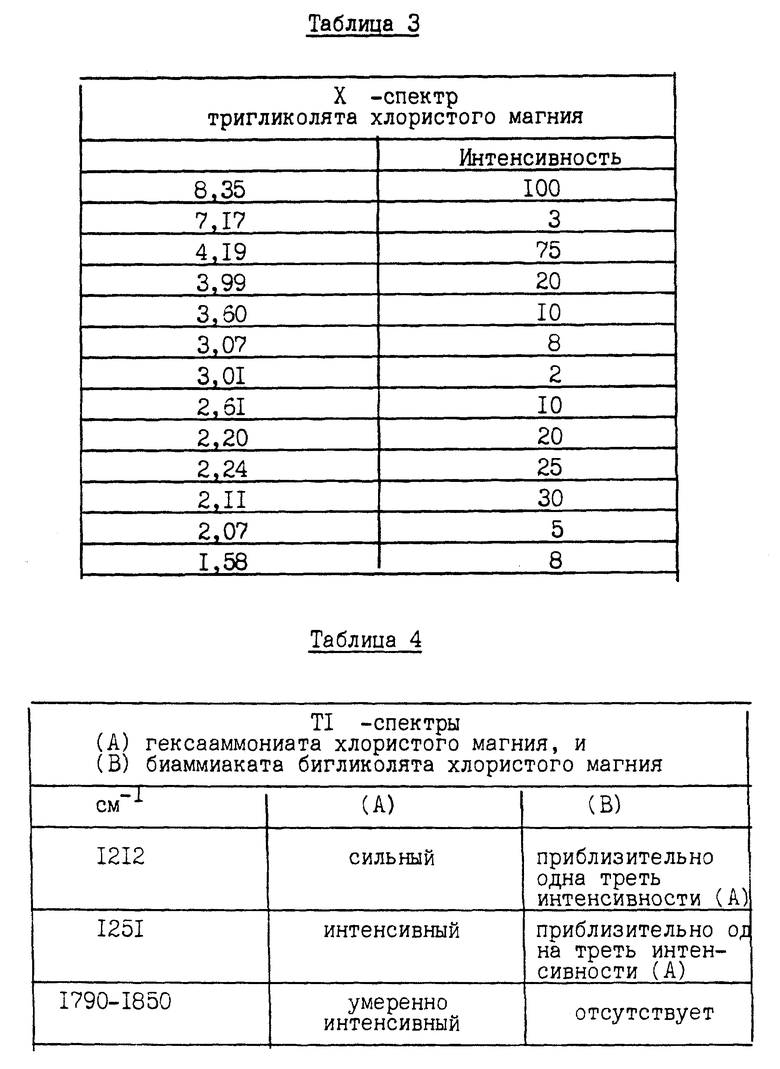
Изобретение относится к безводному хлористому магнию и к способу получения в существенной степени безводного хлористого магния. Заявленный в существенной степени безводный хлористый магний содержит менее 40 частиц на миллион кальция и менее 0,05 вес.% оксида магния. В существенной степени безводный хлористый магний получают способом, включающим получение спиртового раствора хлористого магния, обезвоживание полученного раствора хлористого магния, получение осадка, содержащего гексааммониат хлористого магния посредством введения этого раствора и аммиака в реактор, содержащий неводный раствор с содержанием аммиака более 7 вес.%, или подачей обезвоженного этиленгликолевого раствора хлористого магния в реактор, содержащий насыщенный аммиаком этиленгликоль. После промывки извлеченного осадка его нагревают до получения в существенной степени безводного хлористого магния. Способ позволяет получить в существенной степени безводный хлористый магний, не содержащий нежелательных гликолятных соединений хлористого магния и может быть использован в электролизном производстве металлического магния. 3 с. и 31 з. п. ф-лы, 4 табл., 1 ил.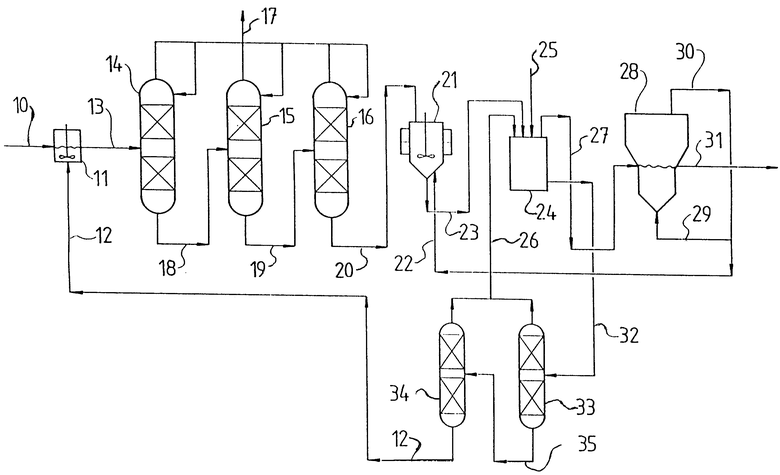
| US 3966888 A, 1975 | |||
| СПОСОБ СЖИЖЕНИЯ ХЛОРМАГНИЕВОГО СЫРЬЯ | 0 |
|
SU237845A1 |
| Способ обезвоживания хлормагниевого сырья | 1988 |
|
SU1742212A1 |
| US 3983224 A, 1976 | |||
| 0 |
|
SU287001A1 | |
| Способ получения электродов на основе TiS для электрохимических накопителей энергии с неорганическим водным Mg-ионным электролитом | 2019 |
|
RU2713401C1 |
| DE 1960232 B2, 1977 | |||
| Способ получения безводного хлористого магния | 1973 |
|
SU480645A1 |

