Изобретение относится к ряду терапевтических соединений, конъюгированных с одной-тремя ацильными группами жирных кислот. Терапевтические соединения выбраны из следующей группы:
1. кортикостероновое семейство лекарственных средств;
2. опиоиды и антагонисты опиоидов;
3. антивирусные нуклеозиды, такие как АЗТ;
4. циклоспорины и родственные им циклопептиды;
5. фолатные антагонисты, в том числе фолиевая кислота и аналоги фолиевой кислоты;
6. предшественники катехоламинов, такие как ДОПА и Допамин, и катехоламины, такие как адреналин, норадреналин и производные;
7. алкилирующие агенты, содержащие карбоксильную группу, такие как хлорамбуцил и мелфалан.
В частности, данное изобретение относится к изменению фармакокинетического профиля и способам доставки этих терапевтических соединений путем конъюгирования их с одним-тремя ацильными производными жирных кислот.
1. Кортикостероновое семейство лекарственных средств
Среди наиболее часто используемых терапевтических агентов находится кортикостероновое семейство лекарственных средств, основанное на природно-встречающихся гормонах, продуцируемых корковым веществом надпочечника. Существуют две основные группы кортикостероновых гормонов с перекрывающимися активностями: глюкокортикоиды - нормальное биологическое действие заключается в регуляции метаболизма углеводов, обладают противовоспалительной активностью при высоких уровнях; минералокортикоиды - связаны с метаболизмом воды и минеральных соединений.
В основе кортикостеронов, как природных, так и синтетических, лежит молекула холестерина, и, в целом, они имеют общие структурные признаки представленной ниже структуры: а) гидроксиацетил в положении 17 (-CO-CH2OH); b) кетогруппа в положении 3 (=O); с) двойная связь между атомами 4 и 5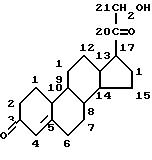
Эти группы обычно являются немодифицированными в активных аналогах гормонов, за исключением гидроксильной части (альтернативно описываемой как гидроксил в положении 21) гидроксиацетила в положении 17, например ацетата гидрокортизона.
Примером глюкокортикоида является гидрокортизон (кортизол или 17-гидроксикортикостерон)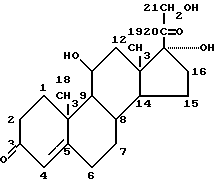
а минералокортикоидом является альдостерон.
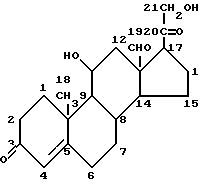
Особый интерес в одном из аспектов данного изобретения представляет противовоспалительное действие глюкокортикоидов (как природных гормонов, так и синтетических лекарственных средств), не ограничивающими примерами которых являются: кортизон, гидрокортизон, флудрокортизон, преднизон, преднизолон, метилпреднизолон, триамцинолон, дексаметазон, бетаметазон, параметазон, флуоцинолон.
Данное изобретение показало, что члены этого семейства могут быть соединены с одним-тремя ацильными производными жирных кислот. Заявители считают, что такие новые конъюгированные соединения превосходят по их свойствам неконъюгированное терапевтическое средство. Кроме того, предполагается, что эти новые соединения будут способствовать пероральной, трансдермальной, внутрисуставной, интраназальной и/или внутриглазной доставке этих лекарственных средств.
В соответствии с первым аспектом данное изобретение состоит в соединении следующей формулы: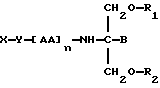
где X является членом кортикостеронового семейства гормонов или лекарственных средств и связан с Y через гидроксильную группу,
Y является спейсерной группой,
AA является аминокислотой; n = 0-5;
B является H или CH2OR3,
R1, R2 и R3 одинаковы или различны и представляют собой каждый водород, метил, этил или ацильную группу жирной кислоты, при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой жирной кислоты.
Во втором аспекте данное изобретение состоит в соединении следующей формулы:
X - Y - [AA]n - NH - CH2 - CH2O - R4,
где X является членом кортикостеронового семейства гормонов или лекарственных средств и соединен с Y через гидроксильную группу,
Y является спейсерной группой,
AA является аминокислотой; n = 0-5 и
R4 является ацильной группой жирной кислоты.
В третьем аспекте данное изобретение состоит в способе пролонгирования или изменения активности члена кортикостеронового семейства гормонов или лекарственных средств, предусматривающем введение соединения в форме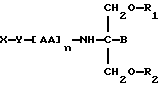
где X является членом кортикостеронового семейства гормонов или лекарственных средств и соединен с Y через гидроксильную группу,
Y является линкерной группой,
AA является аминокислотой; n = 0-5,
B является H или CH2O-R3,
R1, R2 и R3 одинаковы или различны и представляют собой каждый водород, метил, этил или ацильную группу жирной кислоты, при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой жирной кислоты.
В четвертом аспекте данное изобретение состоит в способе пролонгирования или изменения активности члена кортикостеронового семейства гормонов или лекарственных средств, предусматривающем введение соединений в форме
X - Y - [AA]n - NH - CH2 - CH2O - R4,
где X является членом кортикостеронового семейства гормонов или лекарственных средств и соединен с Y через гидроксильную группу,
Y является спейсерной группой,
AA является аминокислотой; n = 0-5 и
R4 является ацильной группой жирной кислоты.
Жирная кислота может быть насыщенной или ненасыщенной.
Как отмечалось выше, X соединен через гидроксильную группу с линкером Y. Как правило, эта гидроксильная группа находится в положении 17 или 21, однако она может быть в других положениях, например в положении 16.
Линкеры Y для связывания соединений с гидроксильной группой с аминогруппой Триса (когда B обозначает CH2O-R3) или промежуточной аминокислоты (AA, если она присутствует), применимые в данном изобретении, включают:
а) линкер с карбоксильной группой к соединению и карбоксильной группой к Трису (или аминокислоте, если она присутствует), такой как дикарбоновая кислота, через ангидрид, например янтарный ангидрид, малеиновый ангидрид;
b) линкер с карбоксильной группой к соединению и альдегидной группой к Трису (или аминокислоте, если она присутствует), такой как глиоксиловая кислота (в присутствии восстанавливающего агента, например NaBH4);
c) линкер группу с карбоксильной группой к соединению и галогенидной группой к Трису (или аминокислоте, если она присутствует), такой как хлоруксусная кислота;
d) линкер группу с карбоксильной группой к соединению и группой N=C=O к Трису (или аминокислоте, если она присутствует), такой как этилизоцианатоацетат.
X может быть любым из членов кортикостеронового семейства соединений, однако в настоящее время предпочтительно, чтобы X представлял собой гидрокортизон или кортизон.
В следующих предпочтительных вариантах этого аспекта данного изобретения Y является дикарбоновой кислотой, AA не присутствует или является глицином или аланином и связывание имеет место в положении 21.
Как должно быть понятно, R1, R2 и R3 представляют собой водород или ацильную группу жирной кислоты. Специалистам в данной области также понятно, что при R1, R2 и R3 возможны иные замещения, чем метил или этил. Основным требованием является, чтобы по меньшей мере один из R1, R2 и R3 был ацильной группой жирной кислоты.
В случае, когда каждый из R1, R2 и R3 является ацильной группой жирной кислоты, предпочтительно, чтобы ацильные группы жирных кислот имели углеродную цепь из 3-18 атомов, более предпочтительно из 10-18 атомов углерода.
Специалистам в данной области понятно, что подобные модификации могут быть сделаны с некоторыми членами других классов стероидных гормонов (или их аналогов), таких как мужские и женские половые гормоны, при гидроксильных группах, расположенных в различных положениях в молекуле.
Данное изобретение обеспечивает также терапевтические композиции, содержащие соединение первого или второго аспекта данного изобретения и фармацевтически приемлемый носитель. Композиция может, кроме того, включать неконъюгированный член семейства кортикостероновых гормонов или лекарственных средств.
Терапевтическую композицию можно вводить любым подходящим способом, как будет понятно специалистам в данной области. Такие способы включают трансдермальный, внутрисуставной, пероральный, интраназальный и внутриглазной способы.
2. Опиоиды и антагонисты опиоидов
Морфин является классическим примером опиатного аналгетического средства, которое действует на рецепторы ЦНС для природно-встречающихся опиоидных пептидов, энкефалинов и эндорфинов, имитируя их действие. Он представляет собой сильнодействующее наркотическое лекарственное средство, используемое для снятия или ослабления сильной боли, связанной с такими состояниями, как сердечный приступ, рак, колика, вызванная камнями в почках или желчном пузыре, после хирургии и при серьезных ожогах. Он имеет короткий биологический полупериод существования (в организме) и в норме доставляется перорально или при помощи инъекции. Подобные опиоидные аналгетики или антагонисты включают гидроморфон, оксиморфон, леворфанол, леваллорфан, кодеин, налмефен, налорфин, налоксон, бупренорфин, буторфанол и налбуфин.
Морфин имеет структуру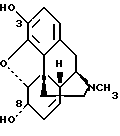
Модификация гидроксильных групп в положении 3 или 6 липофильными группами изменяет скорость всасывания и распределение морфина особенно в ЦНС.
Заявители показали, что морфин и родственные опиоидные аналгетики или антагонисты ("семейство морфина") могут быть присоединены с образованием эфиров при гидроксиле в положении 3 через спейсеры к одному-трем ацильным производным жирных кислот. Полагают, что такие новые конъюгированные соединения превосходят по их действию неконъюгированный терапевтический агент. Авторы также считают, что подобная связь может иметь место через гидроксильную группу в положении 6.
В соответствии с пятым аспектом данное изобретение состоит в соединении следующей формулы: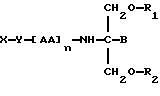
где X является членом семейства морфина и соединен с Y через гидроксильную группу, например гидроксильную группу в положении 3 или 6,
Y является линкерной группой,
AA является аминокислотой; n = 0-5,
B является H или CH2O-R3,
R1, R2 и R3 одинаковы или различны и представляют собой каждый водород, метил, этил или ацильную группу жирной кислоты, при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой жирной кислоты.
В шестом аспекте данное изобретение состоит в соединении следующей формулы:
X - Y - [AA]n - NH - CH2 - CH2O - R4,
где X является членом семейства морфина и соединен с Y через гидроксильную группу в положении 3 или 6,
Y является спейсерной группой,
AA является аминокислотой; n = 0-5 и
R4 является ацильной группой жирной кислоты.
В седьмом аспекте данное соединение состоит в способе пролонгирования или изменения активности члена семейства морфина, предусматривающем введение соединения в форме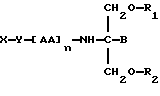
где X является членом семейства морфина и соединен с Y через гидроксильную группу, например гидроксильную группу в положении 3 или 6,
Y является линкерной группой,
AA является аминокислотой; n = 0-5,
B является H или CH2O-R3,
R1, R2 и R3 одинаковы или различны и представляют собой каждый водород, метил, этил или ацильную группу жирной кислоты, при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой жирной кислоты.
В восьмом аспекте данное изобретение состоит в способе пролонгирования или изменения активности члена семейства морфина, предусматривающем введение соединения в форме
X - Y - [AA]n - NH - CH2 - CH2O - R4,
где X является членом семейства морфина и соединен с Y через гидроксильную группу в положении 3 или 6,
Y является спейсерной группой,
AA является аминокислотой; n = 0-5 и
R4 является ацильной группой жирной кислоты.
Жирная кислота может быть насыщенной или ненасыщенной.
Линкеры Y для связывания соединений с гидроксильной группой с аминогруппой Триса (когда B обозначает CH2O-R3) или промежуточной аминокислоты (AA, если она присутствует), применимые в данном изобретении, включают:
а) линкер с карбоксильной группой к соединению и карбоксильной группой к Трису (или аминокислоте, если она присутствует), такой как дикарбоновая кислота, через ангидрид, например янтарный ангидрид, малеиновый ангидрид;
b) линкер с карбоксильной группой к соединению и альдегидной группой к Трису (или аминокислоте, если она присутствует), такой как глиоксиловая кислота (в присутствии восстанавливающего агента, например NaBH4);
с) линкер с карбоксильной группой к соединению и галогенидной группой к Трису (или аминокислоте, если она присутствует), такой как хлоруксусная кислота;
d) линкер с карбоксильной группой к соединению и группой N=C=O к Трису (или аминокислоте, если она присутствует), такой как этилизоцианатоацетат.
В предпочтительном варианте данного изобретения X представляет собой морфин, модифицированный в положении 3 или 6, Y представляет собой дикарбоновую кислоту и AA не присутствует или является глицином или аланином.
Как отмечалось выше, R1, R2 и R3 представляют собой каждый водород или ацильную группу жирной кислоты. Специалистам в данной области также понятно, что возможны замещения, иные, чем метил или этил, в R1, R2 и R3. Основным требованием является, чтобы по меньшей мере один из R1, R2 и R3 обозначал ацильную группу жирной кислоты.
В случае, когда R1, R2 и R3 являются ацильными группами жирных кислот, предпочтительно, чтобы они представляли собой одну и ту же группу. Также предпочтительно, чтобы ацильные группы жирных кислот имели углеродную цепь из 3-18 атомов, более предпочтительно 10-18 атомов углерода.
Данное изобретение обеспечивает также терапевтические композиции, содержащие соединение пятого и шестого аспекта данного изобретения и фармацевтически приемлемый носитель. Кроме того, композиция может содержать неконъюгированный член семейства морфина.
Терапевтическую композицию можно вводить любым подходящим способом, как будет понятно специалистам в данной области. Такие способы включают местное нанесение, локальную инъекцию, внутрибрюшинный и внутривенный способы инъекции.
3. Антивирусные нуклеозиды
Как отмечалось выше, в одном аспекте данное изобретение относится к терапевтическим конъюгатам АЗТ (азидотимидину или зидовудину) и другим антивирусным нуклеозидам (например, ацикловиру, ганцикловиру, видарабину, идоксуридину, трифуридину, валацикловиру, фамцикловиру) и включает антивирусные агенты, связанные через линкерную группу (группы) с одной-тремя ацильными группами, и способы с применением этих соединений. В частности, данное изобретение касается изменения фармакокинетики и/или способа доставки и нацеливания этих лекарственных средств при связывании их с одним-тремя ацильными производными жирных кислот.
АЗТ является примером антиретровирусного лекарственного средства. Он активен против вируса иммунодефицита человека (ВИЧ) и других ретровирусов млекопитающих. Это лекарственное средство представляет собой аналог тимидина, который превращается в трифосфатное производное обычными клеточными ферментами. В этой форме он ингибирует обратную транскрипцию вируса (РНК-зависимый синтез ДНК). Цепи ДНК терминируются включением модифицированного тимидина. АЗТ широко рекомендован для СПИДа и, поскольку он имеет короткий биологический полупериод существования, его следует вводить каждые 4 часа. Его применение связано со многими побочными эффектами от тошноты до подавления образования новых клеток крови и связанных с этим состояний.
АЗТ имеет структуру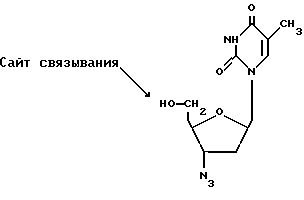
Данное изобретение показало, что АЗТ и подобные лекарственные средства (далее называемые "антивирусными нуклеозидами") могут быть связаны с одним-тремя ацильными производными жирных кислот. Заявители считают, что такие новые соединения будут улучшать доставку, поглощение, полупериод существования в организме и нацеливание в клетке лекарственных средств после перорального, интраназального, трансдермального, внутриглазного и других способов доставки. Кроме того, может быть улучшено распределение лекарственного средства в теле с увеличением процента лекарственного средства, доставляемого в ЦНС и в лимфоциты в лимфатической системе.
В соответствии с девятым аспектом данное изобретение относится к соединению следующей формулы: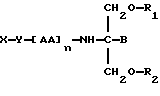
где X является антивирусным нуклеозидом и соединен с Y через гидроксильную группу,
Y является спейсерной группой,
AA является аминокислотой; n = 0-5,
B является H или CH2O-R3,
R1, R2 и R3 одинаковы или различны и представляют собой каждый водород, метил, этил или ацильную группу жирной кислоты, при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой жирной кислоты.
В десятом аспекте данное изобретение относится к соединению следующей формулы:
X - Y - [AA]n - NH - CH2 - CH2O - R4,
где X является антивирусным нуклеозидом и соединен с Y через гидроксильную группу,
Y является спейсерной группой,
AA является аминокислотой; n = 0-5 и
R4 является ацильной группой жирной кислоты.
В одиннадцатом аспекте данное изобретение относится к способу пролонгирования или изменения активности антивирусного нуклеозида, предусматривающему введение антивирусного нуклеозида в форме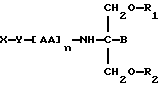
где X является антивирусным нуклеозидом и соединен с Y через гидроксильную группу,
Y является линкерной группой,
AA является аминокислотой; n = 0-5,
B является H или CH2O-R3,
R1, R2 и R3 одинаковы или различны и представляют собой каждый водород, метил, этил или ацильную группу жирной кислоты, при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой жирной кислоты.
В двенадцатом аспекте данное изобретение относится к способу пролонгирования или изменения активности антивирусного нуклеозида, предусматривающему введение антивирусного нуклеозида в форме
X - Y - [AA]n - NH - CH2 - CH2O - R4,
где X является антивирусным нуклеозидом и соединен с Y через гидроксильную группу,
Y является спейсерной группой,
AA является аминокислотой; n = 0-5 и
R4 является ацильной группой жирной кислоты.
Жирная кислота может быть насыщенной или ненасыщенной.
Линкеры Y для связывания соединений с гидроксильной группой с аминогруппой Триса (когда В обозначает CH2O-R3) или промежуточной аминокислоты (AA, если она присутствует), применимые в данном изобретении, включают:
а) линкер с карбоксильной группой к соединению и карбоксильной группой к Трису (или аминокислоте, если она присутствует), такой как дикарбоновая кислота, через ангидрид, например янтарный ангидрид, малеиновый ангидрид;
b) линкер с карбоксильной группой к соединению и альдегидной группой к Трису (или аминокислоте, если она присутствует), такой как глиоксиловая кислота (в присутствии восстанавливающего агента, например NaBH4);
с) линкер с карбоксильной группой к соединению и галогенидной группой к Трису (или аминокислоте, если она присутствует), такой как хлоруксусная кислота;
d) линкер с карбоксильной группой к соединению и группой N=C=O к Трису (или аминокислоте, если она присутствует), такой как этилизоцианатоацетат.
В предпочтительном варианте данного изобретения X представляет собой АЗТ, ацикловир, ганцикловир, видарабин, идоксуридин, трифлуридин, ddI, ddC, ddA или рибавирин, однако в настоящее время в качестве X предпочтителен АЗТ. Предпочтительно также, чтобы Y был дикарбоновой кислотой и AA не присутствовала или представляла собой глицин или аланин.
Как отмечалось выше, R1, R2 и R3 обозначают водород или ацильную группу жирной кислоты. Специалистам в данной области также понятно, что возможны замещения, иные, чем метил или этил, в R1, R2 и R3. Основным требованием является, чтобы по меньшей мере один из R1, R2 и R3 обозначал ацильную группу жирной кислоты.
В случае, когда R1, R2 и R3 являются ацильными группами жирных кислот, предпочтительно, чтобы они представляли собой одну и ту же группу. Также предпочтительно, чтобы ацильные группы жирных кислот имели углеродную цепь из 3-18 атомов, более предпочтительно 10-18 атомов углерода.
Данное изобретение обеспечивает также терапевтические композиции, содержащие соединение девятого или десятого аспектов данного изобретения и фармацевтически приемлемый носитель. Кроме того, такая композиция может содержать неконъюгированный антивирусный нуклеозид.
Терапевтическая композиция может вводиться любым подходящим способом, как будет понятно специалистам в данной области. Такие способы включают пероральный, интраназальный, трансдермальный и внутриглазной способы.
4. Циклоспорины и родственные им циклопептиды
Циклоспорины представляют собой семейство близкородственных циклических пептидов, которые проявляют сильную иммуносупрессивную активность. Циклоспорины используют интенсивно (часто в комбинации с глюкокортикоидами, такими как преднизолон) в трансплантации органов для предотвращения отторжения. По-видимому, циклоспорины действуют обратимо на хелперные T-лимфоциты путем ингибирования продуцирования интерлейкинов и интерферонов и/или ингибирования связывания интерлейкина с рецепторами на T-лимфоцитах-киллерах ("убийцах"), снижая тем самым клеточно-опосредованную ответную реакцию на чужеродные клетки трансплантируемых ткани или органа. Исследования на животных показали, что циклоспорины ингибируют ряд иммунных ответных реакций, в том числе кожную аллергическую реакцию замедленного типа, индуцированный адъювантом Фрейнда артрит и зависимое от T-клеток образование антител, что открывает возможность их применения в более широком диапазоне приложений, чем это практикуется в настоящее время, например местное применение циклоспорина могло бы быть благотворным в лечении псориаза и/или артрита.
Структура соединений циклоспоринового семейства показана ниже, причем циклоспорины A, B, C, D и G отличаются только в боковой цепи R.
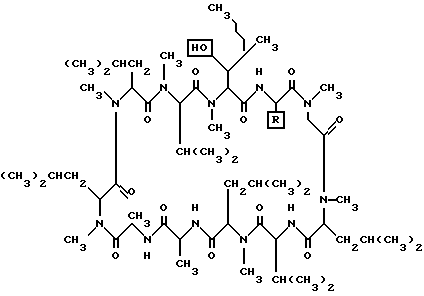
Заявители предполагают, что члены этого семейства могут быть соединены с одним-тремя ацильными производными жирных кислот. Это могло бы достигаться связыванием с постоянной гидроксильной группой или через связывание с различными R. Например, циклоспорин C имеет боковую цепь треонина в положении R, которая могла бы использоваться в качестве точки связывания. Заявители считают, что такие новые соединения будут улучшать доставку, поглощение, полупериод существования в организме и/или способ доставки членов циклоспоринового семейства лекарственных средств. Кроме того, можно предполагать, что эти новые соединения будут способствовать пероральной, трансдермальной, интраназальной, парентеральной и/или внутриглазной доставке этих лекарственных средств путем облегчения их транспорта через липофильные мембраны.
Согласно тринадцатому аспекту данное изобретение относится к соединению следующей формулы: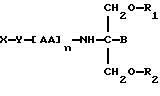
где X является членом циклоспоринового семейства лекарственных средств и соединен с Y через гидроксильную группу,
Y является спейсерной группой,
AA является аминокислотой; n = 0-5,
B является H или CH2O-R3,
R1, R2 и R3 одинаковы или различны и представляют собой каждый водород, метил, этил или ацильную группу жирной кислоты, при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой жирной кислоты.
В четырнадцатом аспекте данное изобретение относится к соединению следующей формулы:
X - Y - [AA]n - NH - CH2 - CH2O - R4,
где X является членом циклоспоринового семейства лекарственных средств и соединен с Y через гидроксильную группу,
Y является спейсерной группой,
AA является аминокислотой; n = 0-5 и
R4 является ацильной группой жирной кислоты.
В пятнадцатом аспекте данное изобретение относится к способу пролонгирования или изменения активности лекарственного средства, являющегося членом циклоспоринового семейства, предусматривающему введение соединения в форме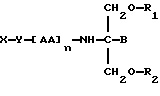
где X является членом циклоспоринового семейства лекарственных средств и соединен с Y через гидроксильную группу,
Y является линкерной группой,
AA является аминокислотой; n = 0-5,
B является H или CH2O-R3,
R1, R2 и R3 одинаковы или различны и представляют собой каждый водород, метил, этил или ацильную группу жирной кислоты, при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой жирной кислоты.
В шестнадцатом аспекте данное изобретение относится к способу пролонгирования или изменения активности лекарственных средств, являющихся членами циклоспоринового семейства, предусматривающему введение соединения в форме
X - Y - [AA]n - NH - CH2 - CH2O - R4,
где X является членом циклоспоринового семейства лекарственных средств и соединен с Y через гидроксильную группу,
Y является спейсерной группой,
AA является аминокислотой; n = 0-5 и
R4 является ацильной группой жирной кислоты.
В случае, когда X является членом циклоспоринового семейства лекарственных средств, он может быть соединен с Y через постоянный гидроксил этого семейства или через гидроксильную группу треонина в качестве боковой цепи циклоспорина C. Альтернативно, в противоположность связыванию только через гидроксил, специфические новые аналоги могут быть получены с рядом реакционноспособных боковых цепей в этом положении.
Жирная кислота может быть насыщенной или ненасыщенной.
Линкеры Y для связывания соединений с гидроксильной группой с аминогруппой Триса (когда B обозначает CH2O-R3) или промежуточной аминокислоты (AA, если она присутствует), применимые в данном изобретении, включают:
а) линкер с карбоксильной группой к соединению и карбоксильной группой к Трису (или аминокислоте, если она присутствует), такой как дикарбоновая кислота, через ангидрид, например янтарный ангидрид, малеиновый ангидрид;
b) линкер с карбоксильной группой к соединению и альдегидной группой к Трису (или аминокислоте, если она присутствует), такой как глиоксиловая кислота (в присутствии восстанавливающего агента, например NaBH4);
с) линкер с карбоксильной группой к соединению и галогенидной группой к Трису (или аминокислоте, если она присутствует), такой как хлоруксусная кислота;
d) линкер с карбоксильной группой к соединению и группой N=C=O к Трису (или аминокислоте, если она присутствует), такой как этилизоцианатоацетат.
Предпочтительно X является членом циклоспоринового семейства, предпочтительно циклоспорином C. Y предпочтительно является дикарбоновой кислотой, а AA не присутствует или представляет собой глицин или аланин.
Как отмечалось выше, R1, R2 и R3 обозначают водород или ацильную группу жирной кислоты. Специалистам в данной области также понятно, что возможны замещения, иные, чем метил или этил, в R1, R2 и R3. Основным требованием является, чтобы по меньшей мере один из R1, R2 и R3 обозначал ацильную группу жирной кислоты.
В случае, когда R1, R2 и R3 являются ацильными группами жирных кислот, предпочтительно, чтобы они представляли собой одну и ту же группу. Также предпочтительно, чтобы ацильные группы жирных кислот имели углеродную цепь из 3-18 атомов, более предпочтительно 10-18 атомов углерода.
Данное изобретение обеспечивает также терапевтические композиции, содержащие соединение тринадцатого или четырнадцатого аспектов данного изобретения и фармацевтически приемлемый носитель. Кроме того, такая композиция может содержать неконъюгированный член циклоспоринового семейства лекарственных средств или родственных циклопептидов.
Такая терапевтическая композиция может вводиться любым подходящим способом, как будет понятно специалистам в данной области. Такие способы включают в себя пероральный, трансдермальный, интраназальный, парентеральный и внутриглазной способы.
5. Фолатные антагонисты, в том числе метотрексат, фолиевая кислота и аналоги фолиевой кислоты
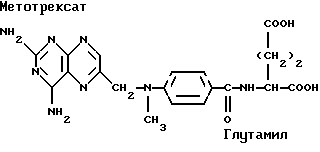
Метотрексат, антиметаболит, является примером лекарственных средств, относящихся к семейству фолатных антагонистов. Он снижает пролиферацию новых клеток, действуя как конкурентный ингибитор редуктазы фолиевой кислоты, предотвращая тем самым превращение витамина фолиевой кислоты в его активную форму, фолиновую кислоту. Метотрексат прописывают для лечения раков, а также его используют для уменьшения пролиферации эпителиальных клеток в лечении псориаза, который не чувствителен к другим способам лечения. Было обнаружено, что низкая доза метотрексата эффективна в остановке прогрессирования и ослаблении симптомов ревматоидного артрита, предположительно вследствие ингибирования воспалительной клеточно-опосредованной реакции.
Заявители показали, что члены семейства метотрексата могут быть соединены с одним-тремя ацильными производными жирных кислот. Они полагают, что такие новые соединения будут улучшать доставку, поглощение, устойчивость во внутрисуставных зонах, биологический полупериод существования и/или способ доставки и распределение в ЦНС этих лекарственных средств. Кроме того, заявители считают, что эти новые соединения будут способствовать их пероральной, интраназальной, трансдермальной, внутриопухолевой, парентеральной, внутрисуставной и/или внутриглазной доставке.
Согласно семнадцатому аспекту данное изобретение относится к соединению формулы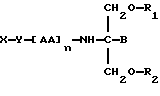
где X является членом семейства фолатных антагонистов и соединен с Y через карбоксильную группу,
Y является необязательной спейсерной группой,
AA является аминокислотой; n = 0-5,
B является H или CH2O-R3,
R1, R2 и R3 одинаковы или различны и представляют собой каждый водород, метил, этил или ацильную группу жирной кислоты, при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой жирной кислоты.
В восемнадцатом аспекте данное изобретение относится к соединению следующей формулы:
X - Y - [AA]n - NH - CH2 - CH2O - R4,
где X является членом семейства фолатных антагонистов и соединен с Y через карбоксильную группу,
Y является необязательной спейсерной группой,
AA является аминокислотой; n = 0-5 и
R4 является ацильной группой жирной кислоты.
В девятнадцатом аспекте данное изобретение относится к способу пролонгирования или изменения активности члена семейства фолатных антагонистов, предусматривающему введение соединения в форме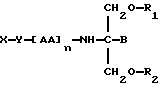
где X является членом семейства фолатных антагонистов и соединен с Y через карбоксильную группу,
Y является необязательной спейсерной группой,
AA является аминокислотой; n = 0-5,
B является H или CH2O-R3,
R1, R2 и R3 одинаковы или различны и представляют собой каждый водород, метил, этил или ацильную группу жирной кислоты, при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой жирной кислоты.
В двадцатом аспекте данное изобретение относится к способу пролонгирования или изменения активности члена семейства фолатных антагонистов, предусматривающему введение соединения в форме
X - Y - [AA]n - NH - CH2 - CH2O - R4,
где X является членом семейства фолатных антагонистов и соединен с Y через карбоксильную группу,
Y является необязательной спейсерной группой,
AA является аминокислотой; n = 0-5 и
R4 является ацильной группой жирной кислоты.
Жирная кислота может быть насыщенной или ненасыщенной.
Линкеры Y для связывания соединений (таких как метотрексат) с карбоксильной группой с аминогруппой Триса (когда B обозначает CH2O-R3) или промежуточной аминокислоты (AA, если она присутствует), применимые в данном изобретении, включают:
а) линкер с аминокислотой к соединению и карбоксильной группой к Трису (или аминокислоте, если она присутствует), такой как аминокислота или антибиотик;
b) линкер с аминогруппой к соединению и группой сульфокислоты к Трису (или аминокислоте, если она присутствует), такой как 2-аминоэтансульфоновая кислота (таурин);
с) линкер с гидроксильной группой к соединению и карбоксильной группой к Трису (или аминокислоте, если она присутствует), такой как гликолевая кислота, молочная кислота и т.д.;
d) линкер с гидроксильной группой к соединению и группой сульфокислоты к Трису (или аминокислоте, если она присутствует), такой как 2-гидроксиэтансульфоновая кислота (изэтионовая кислота);
e) линкер с гидроксильной группой к соединению и реакционноспособной галогенидной группой к Трису (или аминокислоте, если она присутствует), такой как 2-хлорэтанол;
f) другие примеры потенциально пригодных линкеров между соединением с реакционноспособной карбоксильной группой и аминогруппой Триса (или аминокислоты, если она присутствует) включают семейства соединений, примерами которых являются п-гидроксибензальдегид, 2-хлоруксусная кислота, 1,2-дибромэтан и этиленоксид.
В предпочтительном варианте данного изобретения X является метотрексатом, Y отсутствует, является аминокислотой, гликолевой кислотой, 3-гидроксипропионовой кислотой или молочной кислотой, AA не присутствует или является глицином или аланином и связь представляет собой амидную связь или эфирную связь, предпочтительно, с γ-карбоксилом глутамила метотрексата.
Как отмечалось выше, R1, R2 и R3 представляют собой каждый водород или ацильную группу жирной кислоты. Специалистам в данной области также понятно, что возможны замещения, иные, чем метил или этил, в R1, R2 и R3. Основным требованием является, чтобы по меньшей мере один из R1, R2 и R3 обозначал ацильную группу жирной кислоты.
В случае, когда R1, R2 и R3 являются ацильными группами жирных кислот, предпочтительно, чтобы они представляли собой одну и ту же группу. Также предпочтительно, чтобы ацильные группы жирных кислот имели углеродную цепь из 3-18 атомов, более предпочтительно 10-18 атомов углерода.
Данное изобретение обеспечивает также терапевтические композиции, содержащие соединение семнадцатого или восемнадцатого аспектов данного изобретения и фармацевтически приемлемый носитель. Кроме того, такая композиция может содержать неконъюгированный член семейства фолатных антагонистов.
Терапевтическая композиция может вводиться любым подходящим способом, как будет понятно специалистам в данной области. Такие способы включают пероральный, интраназальный, трансдермальный, внутриопухолевый, парентеральный, внутрисуставной и внутриглазной способы доставки.
6. Предшественники катехоламинов и катехоламины
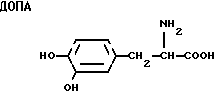
ДОПА является предшественником катехоламинов, важной фармакологически активной группы соединений, включающей в себя адреналин, норадреналин и допамин; нейротрансмиттерные амины, которые действуют как адренергические стимуляторы и сосудосуживающие агенты. ДОПА и аналоги (далее называемые семейством ДОПА) уменьшают акинезию в случае болезни Паркинсона, возможно, путем снижения уровней допамина в мозгу.
Заявители показали, что ДОПА могут быть соединены с одним-тремя ацильными производными жирных кислот. Они считают, что такие новые соединения будут улучшать доставку ДОПА через желудочно-кишечный тракт и гематоэнцефалический барьер и улучшать полупериод существования ДОПА в организме. Кроме того, заявители считают, что эти новые соединения будут способствовать пероральной, трансдермальной, интраназальной, парентеральной и/или внутриглазной доставке этого лекарственного средства.
Согласно двадцать первому аспекту данное изобретение относится к соединению следующей формулы: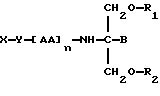
где X является членом семейства ДОПА и соединен с Y через карбоксильную группу или аминогруппу,
Y является необязательной спейсерной группой,
AA является аминокислотой; n = 0-5,
В является H или CH2O-R3,
R1, R2 и R3 одинаковы или различны и представляют собой каждый водород, метил, этил или ацильную группу жирной кислоты, при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой жирной кислоты.
В двадцать втором аспекте данное изобретение относится к соединению следующей формулы:
X - Y - [AA]n - NH - CH2 - CH2O - R4,
где X является членом семейства ДОПА и соединен с Y через карбоксильную группу или аминогруппу,
Y является необязательной спейсерной группой,
AA является аминокислотой; n = 0-5 и
R4 является ацильной группой жирной кислоты.
В двадцать третьем аспекте данное изобретение относится к способу пролонгирования или изменения активности члена семейства ДОПА, предусматривающему введение его в форме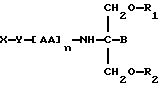
где X является членом семейства ДОПА и соединен с Y через карбоксильную группу или аминогруппу,
Y является необязательной спейсерной группой,
AA является аминокислотой; n = 0-5,
B является H или CH2O-R3,
R1, R2 и R3 одинаковы или различны и представляют собой каждый водород, метил, этил или ацильную группу жирной кислоты, при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой жирной кислоты.
В двадцать четвертом аспекте данное изобретение относится к способу пролонгирования или изменения активности члена семейства ДОПА, предусматривающему введение его в форме
X - Y - [AA]n - NH - CH2 - CH2O - R4,
где X является членом семейства ДОПА и соединен с Y через карбоксильную группу или аминогруппу,
Y является необязательной спейсерной группой,
AA является аминокислотой; n = 0-5 и
R4 является ацильной группой жирной кислоты.
Линкеры Y для связывания соединений (таких как ДОПА) с карбоксильной группой с аминогруппой Триса (когда В обозначает CH2O-R3) или промежуточной аминокислоты (AA, если она присутствует), применимые в данном изобретении, включают:
а) линкер с аминокислотой к соединению и карбоксильной группой к Трису (или аминокислоте, если она присутствует), такой как аминокислота или антибиотик;
b) линкер с аминогруппой к соединению и группой сульфокислоты к Трису (или аминокислоте, если она присутствует), такой как 2-аминоэтансульфоновая кислота (таурин);
c) линкер с гидроксильной группой к соединению и карбоксильной группой к Трису (или аминокислоте, если она присутствует), такой как гликолевая кислота, молочная кислота и т.д.;
d) линкер с гидроксильной группой к соединению и группой сульфокислоты к Трису (или аминокислоте, если она присутствует), такой как
2-гидроксиэтансульфоновая кислота (изэтионовая кислота);
e) линкер с гидроксильной группой к соединению и реакционноспособной галогенидной группой к Трису (или аминокислоте, если она присутствует), такой как 2-хлорэтанол;
f) другие примеры потенциально пригодных линкеров между соединением с реакционноспособной карбоксильной группой и аминогруппой Триса (или аминокислоты, если она присутствует) включают семейства соединений, примерами которых являются п-гидроксибензальдегид, 2-хлоруксусная кислота, 1,2-дибромэтан и этиленоксид.
Нелимитирующие примеры линкеров Y для связывания соединений (таких как ДОПА) с аминогруппой Триса (когда B=CH2O-R3) или промежуточной аминокислоты (если она присутствует), применимые в данном изобретении, включают такие бифункциональные соединения, как:
а) линкер с карбоксильной группой к соединению и карбоксильной группой к Трису (или аминокислоте, если она присутствует), такой как дикарбоновая кислота, через ангидрид, например янтарный ангидрид, малеиновый ангидрид и т. д. Подобным образом можно использовать соединения с двумя группами сульфокислоты или двумя реакционноспособными галогенидными группами.
b) линкер с карбоксильной группой к соединению и группой сульфокислоты к Трису (или аминокислоте, если она присутствует), такой как гидроксиэтансульфоновая кислота (изэтионовая кислота), или с группой сульфокислоты к соединению и карбоксильной группой к Трису или промежуточной аминокислоте (если она присутствует);
c) линкер с карбоксильной группой к соединению и реакционноспособной галогенидной группой к Трису (или аминокислоте, если она присутствует), такой как 2-хлорэтанол, или с реакционноспособной галогенидной группой к соединению и карбоксильной группой к Трису или промежуточной аминокислоте (если она присутствует);
d) линкер с реакционноспособной галогенидной группой к соединению и группой сульфокислоты к Трису или аминокислоте (если она присутствует) или с группой сульфокислоты к соединению и реакционноспособной галогенидной группой к Трису или промежуточной аминокислоте (если она присутствует).
В предпочтительном варианте данного изобретения X является ДОПА, Y отсутствует, является аминокислотой, гликолевой кислотой, 3-гидроксипропионовой кислотой или молочной кислотой, AA не присутствует или является глицином или аланином и связь представляет собой амидную связь или эфирную связь с карбоксильной группой.
Как отмечалось выше, R1, R1 и R3 обозначают водород или ацильную группу жирной кислоты. Специалистам в данной области также понятно, что возможны замещения, иные, чем метил или этил, в R1, R2 и R3. Основным требованием является, чтобы по меньшей мере один из R1, R2 и R3 обозначал ацильную группу жирной кислоты.
В случае, когда R1, R2 и R3 являются ацильными группами жирных кислот, предпочтительно, чтобы они представляли собой одну и ту же группу. Также предпочтительно, чтобы ацильные группы жирных кислот имели углеродную цепь из 3-18 атомов, более предпочтительно 10-18 атомов углерода.
Данное изобретение обеспечивает также терапевтические композиции, содержащие соединение двадцать первого или двадцать второго аспектов данного изобретения и фармацевтически приемлемый носитель. Кроме того, такая композиция может содержать неконъюгированный член семейства ДОПА.
Терапевтическая композиция может вводиться любым подходящим способом, как будет понятно специалистам в данной области. Такие способы включают пероральный, трансдермальный, интраназальный, парентеральный и внутриглазной способы доставки.
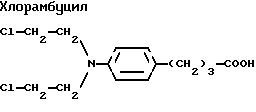
7. Алкилирующие агенты, содержащие группу карбоновой кислоты
Примером этого семейства соединений является хлорамбуцил. Он является бифункциональным алкилирующим агентом и действует как цитотоксическое лекарственное средство путем сшивания цепей ДНК и предотвращения в результате этого репликации клеток. В настоящее время он применяется для лечения болезни Ходжкина, некоторых форм отличающейся от болезни Ходжкина лимфомы, некоторых лейкозов, рака яичников и молочной железы.
Заявители показали, что хлорамбуцил и подобные лекарственные средства могут быть соединены в одним-тремя ацильными производными жирных кислот. Они считают, что такие новые соединения будут улучшать доставку, поглощение, биологический полупериод существования и/или способ доставки этих лекарственных средств. Кроме того, они считают, что эти новые соединения будут способствовать их пероральной, интраназальной, трансдермальной, парентеральной, внутриопухолевой и/или внутриглазной доставке.
Согласно двадцать пятому аспекту данное изобретение относится к соединению следующей формулы: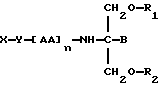
где X является членом семейства хлорамбуцила и соединен с Y через карбоксильную группу,
Y является необязательной спейсерной группой,
AA является аминокислотой; n = 0-5,
B является H или CH2O-R3,
R1, R2 и R3 одинаковы или различны и представляют собой каждый водород, метил, этил или ацильную группу жирной кислоты, при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой жирной кислоты.
В двадцать шестом аспекте данное изобретение относится к соединению следующей формулы:
X - Y - [AA]n - NH - CH2 - CH2O - R4,
где X является членом семейства хлорамбуцила и соединен с Y через карбоксильную группу,
Y является необязательной спейсерной группой,
AA является аминокислотой; n = 0-5 и
R4 является ацильной группой жирной кислоты.
В двадцать седьмом аспекте данное изобретение относится к способу пролонгирования или изменения активности соединения, являющегося членом семейства хлорамбуцила, предусматривающему введение соединения в форме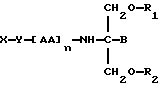
где X является членом семейства хлорамбуцила и соединен с Y через карбоксильную группу,
Y является необязательной спейсерной группой,
AA является аминокислотой; n = 0-5,
B является H или CH2O-R3,
R1, R2 и R3 одинаковы или различны и представляют собой каждый водород, метил, этил или ацильную группу жирной кислоты, при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой жирной кислоты.
В двадцать восьмом аспекте данное изобретение относится к способу пролонгирования или изменения активности соединения, являющегося членом семейства хлорамбуцила, предусматривающему введение соединения в форме
X - Y - [AA]n - NH - CH2 - CH2O - R4,
где X является членом семейства хлорамбуцила и соединен с Y через карбоксильную группу,
Y является необязательной спейсерной группой,
AA является аминокислотой; n = 0-5 и
R4 является ацильной группой жирной кислоты.
Жирная кислота может быть насыщенной или ненасыщенной.
Линкеры Y для связывания соединений (таких как хлорамбуцил) с карбоксильной группой с аминогруппой Триса (когда B обозначает CH2O-R3) или промежуточной аминокислоты (AA, если она присутствует), применимые в данном изобретении, включают:
а) линкер с аминокислотой к соединению и карбоксильной группой к Трису (или аминокислоте, если она присутствует), такой как аминокислота или антибиотик;
b) линкер с аминогруппой к соединению и группой сульфокислоты к Трису (или аминокислоте, если она присутствует), такой как 2-аминоэтансульфоновая кислота (таурин);
c) линкер с гидроксильной группой к соединению и карбоксильной группой к Трису (или аминокислоте, если она присутствует), такой как гликолевая кислота, молочная кислота и т.д.;
d) линкер с гидроксильной группой к соединению и группой сульфокислоты к Трису (или аминокислоте, если она присутствует), такой как 2-гидроксиэтансульфоновая кислота (изэтионовая кислота);
e) линкер с гидроксильной группой к соединению и реакционноспособной галогенидной группой к Трису (или аминокислоте, если она присутствует), такой как 2-хлорэтанол;
f) другие примеры потенциально пригодных линкеров между соединением с реакционноспособной карбоксильной группой и аминогруппой Триса (или аминокислоты, если она присутствует) включают семейства соединений, примерами которых являются п-гидроксибензальдегид, 2-хлоруксусная кислота, 1,2-дибромэтан и этиленоксид.
В предпочтительном варианте данного изобретения X является хлорамбуцилом, Y отсутствует, является аминокислотой, гликолевой кислотой, 3-гидроксипропионовой кислотой или молочной кислотой, AA не присутствует или является глицином или аланином и связь является амидной связью или эфирной связью с карбоксильной группой.
Как отмечалось выше, R1, R2 и R3 являются водородом или ацильной группой жирной кислоты. Специалистам в данной области также понятно, что возможны замещения, иные, чем метил или этил, в R1, R2 и R3. Основным требованием является, чтобы по меньшей мере один из R1, R2 и R3 обозначал ацильную группу жирной кислоты.
В случае, когда R1, R2 и R3 являются ацильными группами жирных кислот, предпочтительно, чтобы они представляли собой одну и ту же группу. Также предпочтительно, чтобы ацильные группы жирных кислот имели углеродную цепь из 3-18 атомов, более предпочтительно 10-18 атомов углерода.
Данное изобретение обеспечивает также терапевтические композиции, содержащие соединение двадцать пятого или двадцать шестого аспектов данного изобретения и фармацевтически приемлемый носитель. Кроме того, такая композиция может содержать неконъюгированный член семейства хлорамбуцила.
Терапевтическая композиция может вводиться любым подходящим способом, как будет понятно специалистам в данной области. Такие способы включают пероральный, интраназальный, трансдермальный, парентеральный, внутриопухолевый и внутриглазной способы доставки.
Для более ясного понимания характера данного изобретения далее будут обсуждаться предпочтительные формы со ссылками на соответствующие примеры.
Используемые аббревиатуры:
АЗТ - 3'-Азидо-3'-дезокситимидин;
ДЦК - 1,3-Дициклогексилкарбодиимид;
ДXM - Дихлорметан;
ДЦМ - N,N'-Дициклогексилмочевина;
ДИЭА - N,N'-Диизопропилэтиламин;
ДМАП - 4-Диметиламинопиридин;
ДМФ - Диметилформамид;
ДОПА - 3-(3,4-дигидроксифенил)-аланин;
ДСК - N,N'-Диcукцинилимидилкapбoнaт;
EtOAc - Этилацетат;
Gly - Глицин;
GTP1 - Глицин-Трис-Монопальмитат;
GTP2 - Глицин-Трис-Дипальмитат;
GTP3 - Глицин-Трис-Трипальмитат;
HOSu - N-Гидроксисукцинимид;
ВЭЖХ - Высокоэффективная жидкостная хроматография;
17-б-Гидрокортизон, 17-Бутират-Гидрокортизон;
MeOH - метанол;
Мор - Морфин;
MTX - Метотрексат;
ЯМР - Ядерный магнитный резонанс;
Suc - Янтарная кислота;
ТМДМС - трет-Бутилдиметилсилил;
ТФК - Трифторуксусная кислота;
ТГФ - Тетрагидрофуран;
ТСХ - Тонкослойная хроматография;
TPTU - Тетрафторборат O-(1,2-Дигидро-2-оксо-1-пиридил)-N,N,N',N'-тетраметилурония;
Трис - 2-Амино-2-гидроксиметил-1,3-пропандиол;
TSTU - Тетрафторборат O-(N-Сукцининимидил)-N,N,N',N'- тетраметилурония;
Z - Бензилоксикарбонил.
Аналитическая ВЭЖХ
Выполненная на оборудовании Waters HPLC с использованием колонок C18 с обращенной фазой (Radial Pak).
Система I - Для соединений, не связанных с жирной кислотой.
Буфер A - 0,1% ТФК в воде.
Буфер B - смесь 80% ацетонитрил : 20% вода, содержащая 0,1% ТФК.
Программа градиента от 30% B до 100% B в течение 5 мин, поддерживаемая до 8 мин; скорость 2 мл/мин.
Времена удерживания - RtI
Система II - Для гидрофобных соединений, обычно содержащих часть молекулы, являющуюся жирной кислотой.
Буфер A - смесь 50% ацетонитрил : 50% вода, содержащая 0,1% ТФК.
Буфер B - смесь 50% ацетонитрил : 50% ТГФ, содержащая 0,1% ТФК.
Программа градиента от 20% B до 100% B в течение 5 мин, поддерживаемая до 8 мин, скорость 2 мл/мин.
Времена удерживания - RtII
Получение Gly-Триса
Соединение заголовка получали гидрированием раствора Z-Gly-Триса в этаноле при давлении 40 Па в гидрогенизаторе Парра в присутствии палладия на угле (10%). Удаление группы Z наблюдали при помощи ВЭЖХ. Катализатор удаляли фильтрованием и промывали этанолом. Выпаривание растворителя дало соединение заголовка с 95% выходом. Получение Z-Gly-Триса описано в Whittaker R.G., Hayes P. J. and Bender V.J. (1993). Peptide Research 6; 125 and Australian Patent N 649242.
Пример 1
Синтез конъюгата Гидрокортизон-Suc-Gly-Трис-(Пальмитат)n: где n = 1, 2 или 3.
I. Гидрокортизон-Сукцинат
К раствору Гидрокортизона (3,65 г, 10 ммоль) в ацетонитриле (450 мл) добавляли янтарный альдегид (1,65 г, 15 ммоль) и ДИЭА (1,7 мл, 10 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 36 ч. Анализ ВЭЖХ реакционной смеси показал 93% соединения заголовка. Растворитель выпаривали и остаток повторно растворяли в этилацетате и промывали водой. Этилацетат выпаривали при пониженном давлении и остаток растирали в диэтиловом эфире с получением 4,4 г белого порошка с выходом 95%. RtI 5,94 мин.
II. Гидрокортизон-Suc-OSu
К раствору Гидрокортизон-сукцината (4,0 г, 8,65 ммоль) в ацетонитриле (120 мл) добавляли ДСК (4,43 г, 17,3 ммоль) в 30 мл ДМФ и ДИЭА (1 мл). После 30 мин образовался белый осадок и анализ ВЭЖХ показал образование нового пика при 8,7 мин при 90%. Осадок отфильтровывали с получением 4 г соединения заголовка (с чистотой 100% согласно ВЭЖХ). Фильтрат выпаривали и остаток растирали в ацетонитриле и диэтиловом эфире с получением еще 0,6 г соединения заголовка. Общий выход 95%, RtI 6,7 мин.
III. Гидрокортизон-Suc-Gly-Трис
К раствору Гидрокортизон-Suc-OSu (3,9 г, 7 ммоль) в 20 мл ДМФ добавляли Gly-Трис (1,78 г, 10 ммоль) в 20 мл ДМФ и реакционную смесь перемешивали при комнатной температуре. Соединение заголовка (RtI 4,98 мин) образовалось с выходом 69% согласно анализу ВЭЖХ после 4 ч. Растворитель удаляли при пониженном давлении и остаток повторно растворяли в 50 мл этилацетата и промывали 100 мл воды. Водную фазу выпаривали до 20 мл и соединение заголовка экстрагировали 200 мл этилацетата (3 раза) с получением 2,4 г соединения заголовка с чистотой 95% согласно анализу ВЭЖХ; выход 56%, RtI 4,98 мин.
IV. Гидрокортизон-Suc-Gly-Трис-(Пальмитат)n: где n = 1, 2 или 3
К раствору Гидрокортизон-Suc-Gly-Триса (2,4 г, 3,85 ммоль) в 100 мл ДХМ и 10 мл ДМФ добавляли каталитическое количество ДМАП и реакционную смесь охлаждали до 0oC. К реакционной смеси добавляли через капельную воронку ДЦК (2,08 г, 10,02 ммоль) в 20 мл ДХМ. Реакционную смесь перемешивали при 0oC в течение 30 мин и затем при комнатной температуре. После 4 ч реакции образовалась смесь соединения заголовка с 1, 2 или 3 пальмитиновыми кислотами (36%, 29%, 3,2% соответственно). Растворитель выпаривали и остаток повторно растворяли в ДХМ. ДЦМ отфильтровывали и фильтрат выпаривали досуха. Остаток повторно растворяли в смеси 1:1 ацетонитрила и ТГФ и разделяли при помощи препаративной ВЭЖХ с применением колонки C18 (40 X 100 мм) с получением 800 мг Гидрокортизон-Suc-Gly-Трис-Монопальмитата, 1700 мг Гидрокортизон-Suc-Gly-Трис-Дипальмитата и 230 мг Гидрокортизон-Suc-Gly-Трис-Трипальмитата. Каждый конъюгат очищали дополнительно на колонке с диоксидом кремния с применением градиента от смеси этилацетат:петролейный эфир (60:40) до смеси этилацетат:метанол (90:10).
Пример 2
Синтез конъюгата 17-бутират-(17-б)-Гидрокортизон-21-Suc-Gly- Трис-(Пальмитат)n: где n = 1, 2 или 3
I. 17-бутират-(17-б)-Гидрокортизон-21-Сукцинат
К раствору 17-б-Гидрокортизона (2,16 г, 5 ммоль) в ацетонитриле (80 мл) добавляли янтарный ангидрид (1,25 г, 12,5 ммоль) и ДИЭА (0,34 мл, 5 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 40 ч. Анализ ВЭЖХ реакционной смеси показал до 98% соединения заголовка. Растворитель выпаривали и остаток повторно растворяли в этилацетате и промывали водой. Фазу этилацетата выпаривали при пониженном давлении и остаток растирали в диэтиловом эфире с получением 2,6 г белого порошка с выходом 95%.
II. 17-б-Гидрокоотизон-21-Suc-OSu
К раствору 17-б-Гидрокортизон-21-сукцината (1,5 г, 2,8 ммоль) в ацетонитриле (40 мл) добавляли ДСК (2,16 г, 7 ммоль) в 30 мл ДМФ и ДИЭА (0,47 мл). После 1 ч реакции при комнатной температуре образовалось 85% соединения заголовка согласно анализу ВЭЖХ (чистота 100% согласно ВЭЖХ). Растворители выпаривали и следующую стадию проводили без дополнительной очистки.
III. 17-б-Гидрокортизон-21-Suc-Gly-Трис
К 20 мл раствора 17-б-Гидрокортизон-21-Suc-OSu (2,8 ммоль) в ДМФ добавляли Gly-Трис (1,5 г, 8,4 ммоль) в 20 мл ДМФ и реакционную смесь перемешивали при комнатной температуре. Соединение заголовка (RtI 6,09 мин) получали с выходом 80% при помощи анализа ВЭЖХ после 5 ч. Растворитель выпаривали при пониженном давлении и остаток повторно растворяли в 20 мл смеси вода/ацетонитрил 50:50 и очищали препаративной ВЭЖХ (Waters Prep4000 с применением колонки C18) с получением 0,75 г соединения заголовка.
IV.17-б-Гидрокортизон-21-Suc-Gly-Трис-(Пальмитат)n: где n = 1, 2 или 3
Пальмитиновую кислоту (0,57 г, 2,22 ммоль) и каталитическое количество ДМАП добавляли к раствору 17-б-Гидрокортизон-21-Suc- Gly-Триса (0,51 г, 0,74 ммоль) в 20 мл ДХМ и реакционную смесь охлаждали до 0oC. К реакционной смеси добавляли по каплям ДЦК (0,45 г, 2,22 ммоль) в 10 мл ДХМ. Реакционную смесь перемешивали при 0oC в течение 30 мин и затем при комнатной температуре. После 2 ч образовалась смесь соединения заголовка с 1, 2 или 3 пальмитатными группами (7%, 47%, 46% соответственно, согласно ВЭЖХ). ДЦМ отфильтровывали и фильтрат выпаривали досуха. Остаток повторно растворяли в смеси 1:1 ацетонитрила и ТГФ. ВЭЖХ показала, что раствор содержал смесь соединений заголовка в соотношении 7:16:69; моно-, ди-, трипальмитат. Смесь разделяли препаративной ВЭЖХ с применением колонки C18 (40 X 100 мм) с получением 850 мг 17-б-Гидрокортизон-21-Suc-Gly-Трис-Трипальмитата, 150 мг 17-б-Гидрокортизон-21-Suc-Gly-Трис-Дипальмитата и 120 мг Монопальмитата. Трипальмитат-конъюгат имел чистоту 100% после препаративной ВЭЖХ, тогда как Моно- и Дипальмитат-конъюгаты требовали дополнительной очистки хроматографией на диоксиде кремния с применением градиента от этилацетат:петролейный эфир (60:40) до этилацетат:метанол (90:10).
ВЭЖХ-анализ показал, что соединения заголовка имеют высокую чистоту и не содержат исходного лекарственного средства и других производных.
Пример 3
Синтез Морфин-Suc-Gly-Ди- и Трипальмитата
Данное изобретение продемонстрировало, что фенольная гидроксигруппа в положении C-3 морфина может быть успешно связана с Gly-Трис-Дипальмитатом или Gly-Трис-Трипальмитатом через янтарную кислоту (Suc) в качестве соединительной группы, без защиты второй гидроксигруппы в положении C-6. Синтез Мор-Suc-Gly- Трис-Дипальмитата включал в себя две стадии с общим выходом 54%.
I. Получение Морфин-Сукцината
Триэтиламин (9,584 мл, 69,45 ммоль) добавляли по каплям к суспензии сульфата морфина (9,286 г, 27,78 ммоль) в сухом ДМФ (140 мл) при 0oC в атмосфере азота. После перемешивания в течение 10 мин к реакции добавляли порциями янтарный ангидрид (2,781 г, 27,78 ммоль). Реакцию наблюдали при помощи ТСХ (50% EtOH/H2O) и при помощи аналитической ВЭЖХ (RtI Мор-Suc было 5,06 мин). Реакция завершалась в пределах периода 24-48 ч и продукт осаждался. Осадок отфильтровывали и промывали небольшим объемом холодного ДМФ и ТГФ. Как 1H-ЯМР, так и ВЭЖХ показали, что продукт имел достаточную чистоту для следующей реакции (Мор-Suc 6,11 г, выход 57%).
Предварительный 1H-ЯМР показал, что сукцинилирование имело место при фенольном гидроксиле в положении C-3.
II. Получение Морфин-Suc-Gly-Трис-Дипальмитата
Общий способ получения Мор-Suc-Gly-Трис-(Пальмитата)n
К суспензии Мор-Suc (0,860 г, 2,23 ммоль) в сухом ТГФ (44 мл) при 0oC в атмосфере азота добавляли ДЦК (0,506 г, 2,45 ммоль) и N-гидроксисукцинимид (0,308 г, 2,68 ммоль). Полученную смесь медленно нагревали до комнатной температуры, затем кипятили с обратным холодильником в течение ночи. Реакцию наблюдали при помощи ВЭЖХ (Система I); время удерживания активного эфира, Мор-Suc-OSu, было 5,45 мин. После завершения реакции смесь охлаждали до 0oC и осадок отфильтровывали и промывали сухим ДХМ. Фильтрат добавляли непосредственно к Gly-Трис-Дипальмитату (1,459 г, 2,23 ммоль) при комнатной температуре с интенсивным перемешиванием. Аминолиз Мор-Suc-OSu наблюдали при помощи ВЭЖХ (Система II) и ТСХ (10% MeOH/ДХМ). Как ВЭЖХ, так и ТСХ показали, что реакция завершалась после перемешивания в течение ночи. Растворитель удаляли в вакууме и остаток повторно растворяли в ДХМ и промывали водой несколько раз до pH 7. Органическую фазу сушили (MgSO4) и выпаривали с получением светло-желтого твердого вещества. Неочищенный продукт очищали флэш-хроматографией (диоксид кремния, 10% MeOH/ДХМ), что дало соединение заголовка (2,16 г) с превосходным выходом (94,7%).
III. Получение Морфин-Suc-Gly-Трис-Трипальмитата
Согласно общему способу, описанному выше, Мор-Suc (0,101 г, 0,26 ммоль) успешно связывали с GTP3 с получением Мор-Suc-GTP3 (0,215 г) с выходом 65,6%. Продукт очищали флэш-хроматографией на оксиде алюминия с элюцией 10% MeOH/EtOAc.
Согласно ВЭЖХ (Система II) как Мор-Suc-GTP2, так и Мор-Suc-GTP3 имели высокую чистоту и не содержали исходное лекарственное средство.
Пример 4
Синтез АЗТ Gly-Трис-(Пальмитата)n: где n = 1, 2 или 3
Обзор схемы синтеза
АЗТ реагировал с янтарным ангидридом с образованием АЗТ-янтарной кислоты. Она реагировала по методу ДЦК/HOSu со смесью Gly-Трис-Моно-, Ди- и Трипальмитата (GTPn), которую получали каталитическим гидрированием Z-GTPn. Колоночную хроматографию на силикагеле и препаративную ВЭЖХ использовали для выделения конечных соединений.
I. АЗТ-янтарная кислота
АЗТ (1,068 г, 4 ммоль), янтарный ангидрид (0,440 г, 4,4 ммоль) и ДМАП (0,015 г) взвешивали в колбу на 25 мл, снабженную конденсатором. Добавляли ДМФ (10 мл) и смесь перемешивали в течение 5 мин и погружали в масляную баню, нагретую предварительно до 90oC, и выдерживали в течение 1,5 ч, пока ВЭЖХ-анализ не показал завершение реакции. ДМФ выпаривали под вакуумом (< 40oC) и остаток использовали сразу для следующей реакции. Остаток может быть очищен хроматографией на силикагеле и продукт может быть выделен с выходом > 87% с чистотой 97%, если требуется.
II. АЗТ-Suc-Gly-Трис-(Пальмитаты)n: где n = 1, 2 и 3
АЗТ-янтарную кислоту (1,468 г, 4 ммоль) растворяли в ДХМ (30 мл) и добавляли HOSu (0,552 г, 4,8 ммоль). Смесь перемешивали в течение 5 мин, затем добавляли ДЦК (0,988 г, 4,8 ммоль) в виде одной порции. Перемешивание продолжали в течение 1 ч, пока ВЭЖХ-анализ не показал завершение реакции. Реакционную смесь фильтровали и нерастворимую ДЦМ промывали ДХМ (35 мл). Объединенный фильтрат собирали в реакционном сосуде, затем добавляли GTPn (2,5 г) и перемешивали в течение ночи. Реакцию наблюдали при помощи ВЭЖХ. Добавляли дополнительно GTPn, пока весь активный эфир OSu не был использован. Реакционную смесь выпаривали в вакууме. Продукт очищали колоночной хроматографией на диоксиде кремния с применением смеси гексан/этилацетат и смеси этилацетат/метанол в качестве элюента для разделения АЗТ-Suc-Gly-Трис-Моно-, Ди- и Трипальмитата. Продукты очищали дополнительно препаративной ВЭЖХ (колонка C18).
Пример 5
Способ 1 для получения циклоспорин-Suc-Gly-Трис-(Пальмитата)n: где n = 1, 2 или 3
I. Циклоспорин-Suc
Соединение заголовка может быть получено из циклоспоринов A, B, D и G реакцией их с янтарным ангидридом в присутствии триэтиламина в ДМФ. Боковая цепь треонина в циклоспорине C должна была быть защищена перед проведением этой реакции.
II. Циклоспорин-Suc-Gly-Трис
Соединение заголовка может быть получено реакцией циклоспорин-Suc с Gly-Трисом в присутствии ДЦК и HOSu.
III. Циклоспорин-Suc-Gly-Трис-Моно-, Ди- и Трипальмитаты
Соединение заголовка может быть получено реакцией циклоспорин-Suc-Gly-Триса с пальмитиновой кислотой при молярном соотношении 1 к 2 в присутствии ДЦК. Затем три соединения заголовка могут быть разделены препаративной ВЭЖХ или гель-хроматографией на силикагеле с элюцией органическими растворителями. В случае циклоспорина С защита боковой цепи должна быть удалена перед очисткой.
Способ 2 для получения Циклоспорин-Suc-Gly-Трис- (Пальмитата)n; где n = 1, 2 или 3
I. Получение бензилового эфира Suc-Gly-Трис-TriOTBDMS
Suc-монобензиловый эфир может быть получен реакцией янтарного ангидрида с бензиловым спиртом в присутствии сильного основания. Оставшаяся немодифицированная карбоксигруппа может затем реагировать с Gly-Трисом в присутствии ДЦК и HOSu с получением Bzl-Suc-Gly-Триса. Три гидроксильные группы Триса могут быть затем защищены реакцией с TBDMS-хлоридом в присутствии имидазола с получением соединения заголовка.
II. Циклоспорин-Suc-Gly-Трис-Моно-, Ди- и Трипальмитата
Бензиловый эфир Bzl-Suc-Gly-Трис-TriOTBDMS может быть удален при помощи водорода в присутствии палладия на угле для генерирования незащищенной карбоксильной группы, которая может реагировать с гидроксильной группой циклоспорина (A, B, I и G; как описано выше, защита боковой цепи требуется для циклоспорина C) в присутствии ДЦК с образованием эфирной связи. Три защищенных TBDMS гидроксила Триса в этом соединении могут быть деблокированы действием уксусной кислоты и реагировать с пальмитиновой кислотой в присутствии ДЦК с получением смеси соединений заголовка. Затем три соединения заголовка могут быть разделены препаративной ВЭЖХ или гель-хроматографией на силикагеле.
Пример 6
Синтез Метотрексат-Gly-Трис-Ди- и Трипальмитата
Обзор схемы синтеза
Изобретенный способ включал удаление глутамильной части метотрексата (МТХ) ферментативным расщеплением Карбоксипептидазой G и заменой ее глутаминовой кислотой, которая имела две селективно этерифицированные карбоксильные группы (α-трет-бутил; γ-метил). Селективное удаление метилового эфира обеспечило в дальнейшем место для присоединения Gly-Триса, который затем пальмитировали, и дипальмитатное производное (и небольшое количество трипальмитата) выделяли хроматографией на диоксиде кремния и/или экстракцией.
I. [[(2,4-Диамино-6-птеридинил)метил]метиламино]бензойная кислота
Соединение заголовка получали отщеплением карбоксипептидазой G глутаминовой кислоты от метотрексата, как описано в литературе (J. Med. Chem. 24, 1450-1455 (1981)). Выход был практически количественным.
II. -α-трет-Бутил-γ-метил-L-глутамат
Соединение заголовка получали из γ-метил-L-глутамата в соответствии с литературой (Justus Liebigs Ann. Chem. 646, 134 (1961)). Выходы сильно различались, но обычно находились в пределах 30-50%. Соединение II, полученное в виде масла, использовали непосредственно в следующих реакциях связывания.
III. α-трет-Бутил-γ-метил-метотрексат
Соединение заголовка получали из I и II согласно литературе (J. Med. Chem. 24, 1450-1455 (1981)). Выходы, как правило, были 50-60%.
IV. α-трет-Бутил-метотрексат
Соединение заголовка получали гидролизом III при помощи Ba(OH)2 согласно литературе (J. Med. Chem. 24, 1450-1455 (1981)). Соединение IV обычно получали достаточно чистым, не требующим очистки ионообменной хроматографией, как показано при помощи 1H-ЯМР. Выходы, как правило, находились в пределах 75-80%.
V. Метотрексат-α-трет-бутил-γ-Gly-Трис
К IV (100 мг, 0,20 ммоль) в смеси ДМФ/ацетонитрил (1:1, 0,22 ммоль) добавляли HOSu (21 мг, 0,22 ммоль) с последующим добавлением ДЦК (45 мг, 0,22 ммоль). Реакционную смесь перемешивали при комнатной температуре и наблюдали ВЭЖХ (Система I). Получение OSu-эфира завершалось на > 85% после 5 ч согласно ВЭЖХ-анализу, что указывало на то, что остальная часть была IV вместе с некоторым количеством продуктов распада. Добавляли раствор Gly-Триса (полученного гидрированием Z-Gly-Триса в ДМФ на 10% Pd/C (1,5 экв. в 3 мл ДМФ)) и реакцию сопровождали ВЭЖХ. Реакция завершалась в пределах 30-60 мин. Затем реакционную смесь разбавляли H2O (3-5 мл), давали ей стоять в течение 10 мин и ДЦМ отфильтровывали. Растворитель удаляли из фильтрата и остаток очищали препаративной ВЭЖХ с получением соединения заголовка в виде ярко-желтого твердого вещества после удаления растворителя лиофилизацией. Выход - 30 мг, 23%. В большем масштабе с использованием 800 мг IV получали выход V 69%. ВЭЖХ-анализ показал чистоту продукта > 97%.
VI. Метотрексат-α-трет-бутиловый эфир-γ-Gly-Трис(пальмитат)n: где n = 2 или 3
К суспензии V (650 мг, 0,97 ммоль) в ДХМ (50 мл) добавляли достаточное количество ДМФ для солюбилизации (5-10 мл) и затем ДМАП (30 мг), пальмитиновую кислоту (500 мг, 2 экв.) и ДЦК (400 мг, 2 экв.). Смесь перемешивали при комнатной температуре в течение 36 ч, после чего ее разбавляли ДХМ (80 мл) и охлаждали во льду в течение 10 мин. Смесь фильтровали и фильтрат экстрагировали 0,01 М HCl (100 мл) и затем солевым раствором (2 x 100 мл). После высушивания (MgSO4) и удаления растворителя остаток наносили на колонку из диоксида кремния и элюировали последовательно ДХМ, смесью ДХМ/5% MeOH, смесью ДХМ/10% MeOH, что дало Три- и Дипальмитаты VI в виде желтых полос, которые вытекали из колонки. Фракции, которые были чистыми при анализе ТСХ (ДХМ/30% изопропанол), объединяли и растворитель удаляли с получением соединений заголовка в виде ярко-желтых твердых веществ.
VII. Метотрексат-γ-Gly-Трис-Дипальмитат
К VI (200 мг, 0,17 ммоль) добавляли ТФК (5 мл). Смесь перемешивали при комнатной температуре в течение 20-30 мин, после чего растворитель удаляли выпариванием. Остаток распределяли между ДХМ (50 мл) и H2O (50 мл), добавляли ТФК к водной фазе до pH > 7. После этого добавляли уксусную кислоту до pH 3-4. Органическую фазу собирали и промывали H2O (50 мл), сушили (Na2SO4) и растворитель удаляли. Продукт, который был чистым согласно ТСХ (бутанол/уксусная кислота/вода 4:2:1), промывали этанолом и сушили с получением продукта заголовка в виде твердого вещества золотого цвета.
Пример 7
Синтез L-ДОПА-Gly-Трис-(Пальмитата)n; где n = 1, 2 или 3
Обзор схемы синтеза
Две фенольные гидроксильные группы ДОПА, а также аминогруппу защищали защитными группами и получали активный эфир Z3-ДОПА. Образование активного эфира полностью защищенного соединения было наилучшим с использованием TPTU, возможно, вследствие структурно заторможенной природы этого соединения. Он реагировал с Gly-Трис-Дипальмитатом с образованием Z3-ДОПА-Gly-Трис- Дипальмитата.
Альтернативный синтез состоял в пальмитировании Z3-ДОПА-Gly-Триса (полученного из Z3-ДОПА и Gly-Триса по способу с применением активного эфира). Гидрирование этих продуктов дало желаемые ДОПА-Gly-Трис-Дипальмитат и ДОПА-Gly-Трис-Трипальмитат с хорошими выходами.
I. N,O',O'-Трикарбобензокси-L-ДОПА(Z3-ДОПА)
Z3-ДОПА получали по способу Felix et al., 1974 (J. Med. Chem. 17. 422-426). Соединение заголовка синтезировали добавлением L-ДОПА (7 г, 35,5 ммоль) к предварительно охлажденному раствору 1 М NaOH (35,5 мл) и воды (74 мл) при -10oC под током азота в 3-горлой колбе. Раствор интенсивно перемешивали, добавляя по каплям одновременно 1 М NaOH (100 мл) и раствор карбобензоксихлорида (20,2 г, 116,5 ммоль) в диэтиловом эфире (100 мл) в течение периода 1 ч при -10oC. Перемешивание продолжали при -10oC в течение 1 ч, затем 1 ч при 0oC и наконец при 20oC в течение 2 ч. Осажденные соли натрия собирали фильтрованием, промывали диэтиловым эфиром и водой и распределяли между диэтиловым эфиром и 1 М лимонной кислотой (по 100 мл). Эфирный слой промывали водой, сушили (Na2SO4) и растворитель удаляли в вакууме с получением неочищенного продукта заголовка в виде масла (18 г, выход 84,6%).
Дальнейшая очистка продукта заголовка препаративной ВЭЖХ (колонка C18 с обращенной фазой) дала хроматографически чистый продукт заголовка, RtII 4,77 мин.
II. Z3-ДОПА-Gly-Трис-Дипальмитат
Активный эфир Z3-ДОПА получали добавлением раствора TPTU (1,68 г, 5,62 ммоль) в ацетонитриле (30 мл) к перемешиваемому раствору Z3-ДОПА (1,68 г, 2,8 ммоль) в ацетонитриле (20 мл) и ДИЭА (550 мкл до pH 8,50). Реакцию сопровождали ВЭЖХ и наблюдением при 300 нм. После 10 мин реакция завершалась и растворитель и основание выпаривали в вакууме. Остаток повторно растворяли в ДХМ, повторно выпаривали и эту процедуру повторяли дважды для гарантии полного удаления основания. Раствор остатка в ДХМ (30 мл) добавляли к перемешиваемому раствору GTP2 (2,0 г, 3 ммоль) в ДХМ (15 мл) в атмосфере азота в темноте.
ВЭЖХ с мониторингом при 280 нм показала, что реакция завершалась за 30 мин с образованием единственного продукта. Реакционную смесь разбавляли ДХМ (120 мл), промывали водой (3 x 120 мл) и сушили (Na2SO4). Слой ДХМ содержал продукт заголовка, который дал один пик при ВЭЖХ (RtII 8,70 мин). Растворитель удаляли в вакууме и остаток повторно промывали несколько раз холодным ацетонитрилом с получением продукта заголовка в виде белого пушистого осадка, 3,40 г, с выходом 98%. ВЭЖХ, ТСХ и ЯМР-спектроскопия показали, что продукт заголовка был высокоочищенным.
Гидрирование аликвоты этого продукта в этаноле в присутствии каталитических количеств 10% палладия на угле в течение 5 ч дало L-ДОПА-GTP2, RtII 8,05 мин с максимумом поглощения при 285 нм. Выход 785 мг, 83,5%.
Анализ ВЭЖХ показал, что продукт имел чистоту 99% и не содержал исходного лекарственного средства.
III. Получение Z3-ДОПА-Gly-Триса
Раствор Z3-ДОПА (800 мг, 1,33 ммоль) в сухом ацетонитриле (10 мл) реагировал с раствором TPTU (1 г, 3,4 ммоль) в ацетонитриле (5 мл) и ДИЭА (350 мкл, до pH 8,3) и образование активного эфира наблюдали ВЭЖХ в Системе II при 300 нм. Реакция завершалась через 10 мин с образованием активного эфира с выходом 89% согласно ВЭЖХ, RtII 5,19 мин. Повторяющееся выпаривание растворителя и ДИЭА из ДХМ дало маслянистый остаток, который повторно растворяли в ацетонитриле (15 мл) и добавляли по каплям к раствору Gly-Триса (1 г, 5,6 ммоль) в свежеотогнанном сухом ДМФ (5 мл) с перемешиванием. Реакцию наблюдали при помощи ВЭЖХ при 300 нм и при 280 нм, что показало образование продукта заголовка в обеих системах ВЭЖХ, I и II (Rt 7,11 и 3,05 мин соответственно).
Растворители удаляли в вакууме и продукт заголовка очищали препаративной ВЭЖХ с получением 655 мг Z3-ДОПА-Gly-Трис (выход 60,4%). Структура продукта была подтверждена ЯМР-спектроскопией.
IV. Z3-ДОПА-GIy-Трис-Ди- и Трипальмитат
Раствор Z3-ДОПА-Gly-Трис (600 мг, 1 ммоль) в ДХМ (10 мл) взаимодействовал с пальмитиновой кислотой (312 мг, 1,22 ммоль) и ДЦК (250 мг, 1,2 ммоль) как описано выше. Реакция сопровождалась ВЭЖХ с дополнительным добавлением пальмитиновой кислоты в виде аликвот по 10 мг, в случае необходимости. После добавления дополнительных 40 мг пальмитиновой кислоты получали Z3-ДОПА-Gly-Трис-Моно- и Дипальмитат в соотношении 1:1, как определено при помощи ВЭЖХ. После удаления ДЦМ фильтрованием растворитель удаляли в вакууме и смесь разделяли препаративной ВЭЖХ с применением колонки C18.
Во время процедуры обработки большая часть продукта превращалась в формы Ди- и Трипальмитата, которые разделяли с получением 260 мг дипальмитата и 520 мг трипальмитата и рассчитывали в процентах по результатам ВЭЖХ смеси (21% P2, 35% P3).
L-ДОПА-GTP2 и GTP3 получали гидрированием в этаноле в присутствии катализатора Pd/C с получением продуктов заголовка.
Пример 8
Синтез Хлорамбуцил-Gly-Трис-(Пальмитата)n; где n = 1, 2 или 3
Соединения заголовка получали путем получения активного эфира хлорамбуцила и взаимодействием его со смесью Gly-Трис-Моно-, Ди- и Трипальмитата (GTPn), полученной гидрированием Z-GTPn. Полученные продукты очищали колоночной хроматографией и препаративной ВЭЖХ.
I. Хлорамбуцил-Gly-Трис-(Пальмитат)n; где n= 1, 2 или 3
Хлорамбуцил (0,913 г, 3 ммоль) и HOSu (414 мг, 3,6 ммоль) взвешивали в колбу на 50 мл с магнитным стержнем для перемешивания. ДХМ (15 мл) добавляли и раствор перемешивали в течение 5 мин. Добавляли ДЦМ (742 мг, 3,6 ммоль) в ДХМ (15 мл) в течение 5 мин. Аналитическая ВЭЖХ показала завершение реакции после 1 ч.
Раствор фильтровали и остаток промывали ДХМ (10 мл). Объединенный фильтрат собирали в колбе, перемешивали и добавляли твердый GTPn (4 г). Перемешивание продолжали в течение ночи и добавляли свежий GTPn (300 мг). ВЭЖХ после 2 ч показала отсутствие эфира хлорамбуцил-OSu, что свидетельствовало о завершении реакции.
Смесь выпаривали под вакуумом (< 40oC). Продукт хроматографировали через силикагель с применением смеси гексан:этилацетат с последующей препаративной ВЭЖХ для получения высокоочищенных продуктов.
II. Хлорамбуцил-Gly-Трис-Монопальмитат
К раствору хлорамбуцила (0,972 г, 3,2 ммоль) в ДХМ (32 мл) добавляли ДЦК (0,726 г, 3,5 ммоль) и HOSu (0,387 г, 3,3 ммоль) порциями при 0oC. Полученную смесь перемешивали при комнатной температуре в течение ночи. После завершения реакции согласно ВЭЖХ (Система I) осадок (ДЦМ) отфильтровывали и промывали сухим ДХМ (10 мл). К фильтрату добавляли Gly-Трис-Монопальмитат (1,208 г, 2,9 ммоль) порциями с интенсивным перемешиванием при комнатной температуре в течение ночи. По завершении реакции полученную смесь разбавляли ДХМ, затем промывали H2O несколько раз. Органическую фазу сушили (MgSO4) и выпаривали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали флэш-хроматографией (80% этилацетат/гексан, 2,5% MeOH/этилацетат) с получением соединения заголовка (0,950 г) в виде светло-желтого полутвердого вещества с выходом 46,6%.
III. Хлорамбуцил-Gly-Трис-Трипальмитат
К раствору хлорамбуцила (0,240 г, 0,789 ммоль) в ДХМ (16 мл) при 0oC добавляли ДЦК (0,171 г, 0,828 ммоль) и ДМАП (0,005 г, 0,039 ммоль) с последующим добавлением Gly-Трис-Трипальмитата (0,740 г, 0,828 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. Осадок отфильтровывали и фильтрат промывали 5% водным раствором уксусной кислоты, затем H2O три раза до pH 7. Органическую фазу сушили (MgSO4), затем концентрировали в вакууме. Неочищенный продукт очищали флэш-хроматографией (30%, 40% этилацетат/гексан), затем перекристаллизовывали из смеси этилацетат/гексан с получением соединения заголовка (0,45 г) в виде белого твердого вещества с выходом 48,2%.
Пример 9
Противовоспалительное действие Гидрокортизона и конъюгатов Гидрокортизона с жирной кислотой в модели UVB (УФС)
Противовоспалительное действие Гидрокортизона и конъюгатов Гидрокортизона с жирными кислотами в модели UVB
Используемые лекарственные средства
Гидрокортизон приобретали из Sigma Chemicals, Lot N 13H0525, H-4001. Гидрокортизон-Suc-GTP2 и Гидрокортизон-Suc-GTP3 и 17-б-Гидрокортизон-Suc-GTP3 синтезировали как описано выше.
Эритемный УФС-тест
Во всех экспериментах использовали самок бесшерстных мышей-альбиносов штамма i/b Skh-1, ранее не подвергавшихся экспонированию под УФС-светом. Средний возраст был 12 недель, и мышей распределяли по ящикам в группы по три (средний вес 30 г). Для индукции эритемы (воспаления) применяли источник света FS40 с одной УФС-трубкой. Мышей экспонировали до 15 мин на УФС-свете эквивалентном экспозиции с 2 МЭД (минимальными эритемными дозами). Облучаемые мыши либо получали обработку после облучения, либо их обрабатывали предварительно в течение нескольких дней перед экспозицией с УФС-светом путем равномерного диспергирования 100 мкл раствора Гидрокортизона, Гидрокортизон-Suc-GTP2 или Гидрокортизон-Suc-GTR3 в этаноле (носителе) на спинки мышей при помощи пипеттора Гильсона. Мышей оставляли на 24 ч и измерения кожных складок выполняли с применением ручного микрометра.
Экспериментальная часть
В качестве начальной части этого исследования испытывали действие Гидрокортизона на вызываемое УФС воспаление. Группу мышей Skh-1 облучали 2 МЭД УФС и затем намазывали местно Гидрокортизоном в этаноле. Увеличение нетто-толщины кожных складок (NSET) определяли после 24 ч. Построенная концентрационная кривая определяла оптимальную дозу Гидрокортизона, необходимую для наилучшей противовоспалительной защиты от экспозиции с 2 МЭД УФС. Доза 0,5 мг/мышь дала почти 100% защиту от эритемы и отека и ее использовали в последующих экспериментах, проводимых для изучения эффектов конъюгатов Гидрокортизона.
Конъюгаты сукцината гидрокортизона с жирной кислотой и их защитное действие против индуцируемого УФС отека
Испытывали длительность сохранения нанесенных местно конъюгатов сукцината гидрокортизона с жирной кислотой. Использовали концентрации гидрокортизона 0,5% и 1% (масса/объем) (эквивалентно 0,5 мг и 1,0 мг на мышь соответственно). Результаты представлены ниже. Тестируемые конъюгаты наносили за 5 дней и 3 дня перед экспозицией с УФС. В день 0 конъюгаты наносили после экспозиции с УФС во избежание возможности поглощения УФ (эффекта солнечного экрана).
Результаты экспериментов с 0,5 мг/мышь даны в Таблице 1.
Результаты
Три формы конъюгатов Гидрокортизон-сукцината, Гидрокортизон-Suc-Gly-Трис-Дипальмитат (HC-Suc-GTP2), Гидрскортизон-Suc-Gly-Трис-Трипальмитат (HC-Suc-GTP3) и 17-б HC-Suc-GTP3, имели активность, равноценную приблизительно 50% активности гидрокортизона в защите против индуцируемого УФС отека при нанесении в День 0, после экспозиции на УФС. Также наблюдали, что при нанесении этих конъюгатов на кожу мышей за три дня перед экспозицией на УФС все еще наблюдалось защитное действие, присутствующее с обоими конъюгатами, и оно было слегка лучше, чем в случае одного Гидрокортизона. Наблюдали сглаживание (задержку) ответной реакции с этими конъюгатами, что указывало на измененный профиль доставки.
При более высокой концентрации 1 мг/мышь защитные эффекты конъюгатов гидрокортизона усиливались (таблица 2) с большей защитой со всеми тестируемыми соединениями при Дне 3. Опять наблюдали задержку в действии конъюгатов гидрокортизона, что позволяет предполагать более замедленный, равномерный профиль доставки.
Выводы
Эффективность конъюгатов Гидрокортизон-сукцината с жирной кислотой определяли в мышиной УФС-модели воспаления. Общебиологические открытия заключаются в том, что конъюгированные с жирной кислотой формы синтезированного гидрокортизона схожи с гидрокортизоном по их биологическому действию, измеренному по их защитному действию против индуцируемых УФС-излучением эритемы и отека. Важно то, что, по-видимому, конъюгаты с жирными кислотами имеют измененный профиль доставки к эпидермису, что предполагает задержку в достижении полной активности в День 0.
Эти результаты показывают, что может существовать некоторое преимущество в использовании конъюгированных с жирной кислотой молекул по сравнению с гидрокортизоном в смысле улучшенной компартментализации в случае трансдермальной доставки. Эта измененная доставка может также снижать побочные действия, возникающие вследствие даун-регуляции (отрицательной регуляции) оси H-P-A продолжительным использованием гидрокортизона.
Пример 10
Тест контактной повышенной чувствительности
В этом тесте использован способ, описанный в "Current Protocols in Immunology" (Vol 1, Section 4.2 Eds Coligan et al. (1991) NIH). Сенсибилизатором был оксазолон. Вкратце, способ заключался в следующем.
УФС-облучение. Мышей облучали одной флуоресцентной трубкой FL40SE UVB (Oliphant) в отражательной обшивке, обеспечивающей УФС-радиацию 2,6 • 104 Вт/см2 при измерении на расстоянии мишени при помощи радиометра (International Light IL1700 Radiometer) с детектором УФС, чувствительным между 250-315 нм. Мыши получали 0,1179 Дж/см2 радиации УФС, что составляло 1 минимальную эритемную дозу (МЭД), как определено выше, в каждый из 3 последовательных дней при неограниченном экспонировании с удаленными крышками проволочных клеток. Кумулятивная доза приводила к умеренной эритеме без кожных волдырей. Эритему оценивали количественно как компонент отека этой реакции путем измерения толщины среднедорзальной кожной складки пружинным микрометром (Merser, St. Albans, UK) при 24 ч после первой экспозиции на УФС, т.е. непосредственно перед третьей экспозицией, когда реакция оказалась максимальной.
Индукция системной (общей) контактной повышенной чувствительности
(аллергии).
Тестируемых мышей экспонировали спинкой к УФС-радиации 1 МЭД (или без УФС-радиации) в один и тот же час в дни 1, 2 и 3. В дни 8 и 9 мышей (облученных и необлученных) сенсибилизировали местным нанесением на кожу живота 0,1 мл 3% (м/об.) оксазолона (Sigma Chemical Co., St. Louis, MO), свежеприготовленного в этаноле. В день 15 измеряли толщину уха при предварительной сенсибилизации при помощи пружинного микрометра и затем мышей провоцировали нанесением 5 мкл свежеприготовленного 3% раствора в этаноле на обе поверхности каждого наружного уха. Толщину уха опять измеряли периодически в промежутке времени 16-24 ч и регистрировали максимальную толщину уха. Нетто-опухание уха рассчитывали как разность между средней толщиной уха при предварительном введении оксазолона и при пост-введении оксазолона для каждой группы обработки. Статистическую значимость различий в нетто-опухании уха между группами обработки оценивали при помощи t-критерия Стъюдента.
Мышей намазывали носителем, 0,5 мг/мышь HC, 0,5 мг/мышь HC-Suc-GTP2, 1 мг/мышь 17-б-HC-Suc-GTP3 или 17-б-HC, в пределах 10 минут после экспозиции на УФС-свету. Всего использовали 3 МЭД неэкранированного УФС-света (Таблица 3).
Результаты
Все тестированные пробы обнаружили защитное действие против индуцированной УФС иммуносупрессии, проявляющейся в увеличении толщины уха как результата системного ответа на сенсибилизатор. По-видимому, нет различий между исходными лекарственными средствами или конъюгатами в их способности изменять иммуносупрессию, вызываемую УФС. Однако важно то, что конъюгаты, по-видимому, точно так же активны в продуцировании системного ответа, как и исходные лекарственные средства.
Пример 11
АЗТ
Биологические активности АЗТ и конъюгатов АЗТ-жирная кислота тестировали на цитотоксичность и активность против ВИЧ.
I. Цитотоксичность
Цитотоксичность АЗТ и конъюгатов АЗТ-жирная кислота (АЗТ-Suc-GTP2, P2 и P2) тестировали in vitro с клетками JURKAT (острого T-клеточного лейкоза человека) и мононуклеарными клетками периферической крови (PBMS). Клетки инкубировали с лекарственным средством и определяли жизнеспособность при помощи колориметрического теста пролиферации.
Способы
a) Условия клеточной культуры
Клетки JURKAT засевали в 96-луночные планшесты при плотности 1,6 • 104 клеток/200 мкл полной среды RPMI 1640±лекарственное средство и культивировали в течение 64 ч.
Мононуклеарные клетки периферической крови человека (PBMS) выделяли из трех здоровых доноров центрифугированием на Ficoll-Paque (Pharmacia). Клетки засевали в 96-луночные планшеты при плотности 2 • 108 клеток/200 мкл полной среды RPMI±лекарственное средство и культивировали в течение 40 ч.
b) Доставка лекарственного средства
Лекарственные средства растворяли в этаноле и добавляли к культуральным средам. Конечная концентрация этанола в средах была не более 1% (об./об.).
c) Тест на жизнеспособность
Жизнеспособность определяли при помощи MTS-теста (CellTiter 96ТМ Aqueous Non-Radioactuve Cell Proliferation Assay) из Promega. Реагент (40 мкл) добавляли в каждую лунку и величины поглощения при 490 нм определяли через 1-4 ч. Действие лекарственного средства на жизнеспособность клеток сравнивали с культурами, не содержащими лекарственного средства (жизнеспособность 100%). Лунки, не содержащие клеток, использовали для контроля фонового поглощения (жизнеспособность 0%). Каждую концентрацию тестировали по меньшей мере в 3 лунках. Концентрации, необходимые для 50% ингибирования клеточного роста (IC50), рассчитывали при помощи линейной регрессии.
Результаты
АЗТ-Suc-GTP1 был в 40 раз более токсичным в отношении клеток JURKAT, чем исходное лекарственное средство (Таблица 4). Конъюгат был также более цитотоксичным в отношении PBMS, чем АЗТ.
II. Активность против ВИЧ
Также тестировали активности против ВИЧ АЗТ-Suc-GTP1 и АЗТ-Suc-GTP2. Инфицированные клетки инкубировали в течение 5 дней с соединением. Соединения растворяли в ДМСО и использовали 5-кратные разведения. Измеряли следующие параметры: образование синцитиев, образование вирусного антигена gp120, выживание клеток. В инфицированных клетках также измеряли цитотоксичность.
Использовали две клеточные линии:
C8166 (T-лимфобластоидные клетки человека), инфицированные ВИЧ-1 MN
JM (полувзрослые T-клетки человека из лимфобластоидного лейкоза), инфицированные ВИЧ-1 J4550 (?)
АЗТ малоактивен в клетках JM, возможно, вследствие недостаточного фосфорилирования, и его использовали для подтверждения способа действия соединений.
Результаты
Конъюгат АЗТ-Suc-GTP1 был в 50 раз более цитотоксичен в отношении обеих клеточных линий, чем АЗТ (Таблица 5); это согласуется с данными для клеток PBMS и JURKAT (Таблица 4). Цитотоксичность конъюгата P2 также наблюдали в отношении клеток JM и C8166 (конъюгат в 25 раз более цитотоксичен, чем АЗТ).
Вирус в линии клеток JM был в 12,5 раз более чувствителен к АЗТ-Suc-GTP1, чем к АЗТ (Таблица 6). Это увеличение токсичности в отношении вируса не отражалось в характере ВИЧ-инфекции: образовании синцитиев или профилях роста инфицированных клеток; этот эффект наблюдали только в детектировании антигена. Линия клеток JM не метаболизирует АЗТ в такой степени, в какой его метаболизируют клетки C8166, но большая доставка АЗТ через конъюгат P1 может приводить к более высоким уровням трифосфата, образуемого в клетках.
Обсуждение
АЗТ-Suc-GTP1 более токсичен, чем АЗТ, в отношении клеток PBMS, JURKAT, C8166 и JM, что предполагает увеличенное клеточное поглощение. Клеточные киназы превращают АЗТ в АЗТ-трифосфат, который ингибирует обратную транскриптазу (ОТ) ВИЧ и при концентрациях, в 100 раз более высоких, ингибирует клеточную α-ДНК-полимеразу. По-видимому, конъюгирование увеличивает клеточное поглощение, увеличивая тем самым цитотоксичность (Таблица 5). В инфицированных клетках JM АЗТ-Suc-GTP1 был более сильным в снижении антигена gp120, продуцируемого ВИЧ, однако вследствие цитотоксичной природы этого соединения клетки не возвращались к норме и не обнаруживали нормальный рост.
Это очень положительные результаты в том смысле, что они согласуются с тем, что конъюгаты поглощаются клетками и метаболизируются с высвобождением АЗТ, который действует обычным для него образом. Их цитотоксичность (которая может быть благотворным действием при нацеливании), хотя и выше цитотоксичности немодифицированного АЗТ, находится на достаточно низком уровне, чтобы позволить их применение.
Эти обнаруженные результаты могут позволить обратиться к основным недостаткам применяемой в настоящее время терапии АЗТ, а именно таким недостаткам, как быстрое выведение, низкая биодоступность и слабая доставка в лимфатическую систему, которая является первичным местом загрузки вирусом. Цитируя Charman and Porter (1996 Advanced Drug Delivery Reviews in press), можно отметить, что "основные благоприятные возможности, связанные с липофильными пролекарствами, представляют собой потенциал для (i) обхода метаболизма в печени после перорального дозирования и (ii) нацеливания лекарственных средств в лимфатическую систему". Кроме того, липофильное лекарственное средство, такое как АЗТ-Suc-GTP1, должно действовать с замедленным профилем высвобождения. Эти особенности должны приводить к более низким требующимся общим дозам АЗТ с возможностью ослабления побочных действий, несмотря на общую более цитотоксичную природу конъюгатов в сравнении с АЗТ.
Пример 12
Хлорамбуцил
Цитотоксичность хлорамбуцила и конъюгатов хлорамбуцила с жирной кислотой (CGTP1-P2 и P3) тестировали in vitro с клетками JURKAT (острого T-клеточного лейкоза человека). Клетки инкубировали с лекарственным средством и определяли жизнеспособность при помощи колориметрического теста пролиферации. Цитотоксичность в отношении других клеточных линий также тестировали (клеток PC3 рака простаты человека, клеток B16 меланомы мышей и мононуклеарных клеток периферической крови человека).
Способы
а) Условия клеточной культуры
Клетки JURKAT высевали в 96-луночные планшеты при плотности 1,6 • 104 клеток/200 мкл полной среды RPMI± лекарственное средство и культивировали в течение 64 ч.
Разведение 1:3 конфлюентных клеток PC3 в колбе на 75 мл высевали в 96-луночный планшет и культивировали в течение 48 ч в полной среде RPMI. Среду удаляли и в лунки добавляли полную RPMI±лекарственное средство. Клетки культивировали далее в течение 72 ч.
Разведение 1:8 конфлюентных клеток B16 в колбе на 75 мл высевали в 96-луночный планшет и культивировали в течение 48 ч в полной среде EMEM. Среду удаляли и в лунки добавляли полную EMEM±лекарственное средство. Клетки продолжали культивировать в течение 48 ч.
Одноядерные клетки периферической крови человека (PBMS) выделяли из одного здорового донора центрифугированием на Ficoll-Paque (Pharmacia). Клетки высевали в 96-луночные планшеты при плотности 2 • 105 клеток/200 мкл полной RPMI±лекарственное средство и культивировали в течение 40 ч.
b) Доставка лекарственного средства
Испытывали два типа доставки:
i) Этанол. Лекарственные средства растворяли в этаноле и добавляли в культуральные среды. Конечная концентрация этанола в среде была не более 1% (об. /об. ). Эту доставку использовали для экспериментов с типами клеток JURKAT, PBMS, PC3 и B16.
ii) Покрытие планшетов. Лекарственные средства растворяли в этаноле и аликвоты (до 40 мкл) добавляли к 90% (об./об.) изопропанолу. Затем раствор (50 мкл) добавляли в лунки 96-луночного планшета и растворитель выпаривали нагреванием при 37oC. Затем добавляли среду ± клетки. Эту доставку использовали для некоторых экспериментов с клетками JURKAT.
c) Тест на жизнеспособность
Жизнеспособность определяли при помощи MTS-теста (CellTiter 96ТМ Aqueous Non-Radioactive Cell Proliferation Assay) из Promega. Реагент (40 мкл) добавляли в каждую лунку и определяли величины поглощения при 490 нм через 1-4 ч. Действие лекарственного средства на жизнеспособность клеток сравнивали с культурами, не содержащими лекарственного средства (жизнеспособность 100%). Лунки, не содержащие клеток, использовали для контроля фонового поглощения (жизнеспособность 0%). Каждую концентрацию тестировали по меньшей мере в 3 лунках. Концентрации, необходимые для 50% ингибирования роста контрольных клеток (IC50), рассчитывали методом линейной регрессии.
Результаты
a) Цитотоксичность лекарственного средства
Конъюгат CGTP1 был более цитотоксичен относительно всех типов клеток, чем хлорамбуцил (таблица 7).
С клетками JURKAT наблюдали 15-кратное увеличение активности. Увеличение токсичности CGTP1 в отношении нормальных клеток (PBMS) было не столь большим (приблизительно 5-кратным). Клетки JURKAT, по-видимому, более устойчивы к хлорамбуцилу, чем PBMS. CGTP1 цитотоксичен к обоим типам клеток при сходных концентрациях. Конъюгат с GTP1 был также более токсичен в отношении типов клеток PC3 и B16.
CGTP1 был более цитотоксичен в отношении клеток JURKAT, чем эквимолярная смесь хлорамбуцила и GTP1. GTP1 не был активным. Это свидетельствует о том, что конъюгирование хлорамбуцила с GTP1 необходимо для наблюдаемой увеличенной цитотоксичности (Таблицы 7, 8, 9).
b) Влияние носителя
Оба носителя (этанол и изопропанол) не были токсичными для клеток в исследованном диапазоне доз. Доставка соединений посредством покрытия планшетов увеличивала величины IC50 хлорамбуцила и CGTP1 (Таблица 8), однако этот способ доставки обнаруживал активность с CGTP2 и CGTP3 (Таблица 9), которую не наблюдали при доставке этих соединений к культурам в этаноле. Этот эффект может быть обусловлен пониженной растворимостью P2- и P3-конъюгатов в водных тестах in vitro.
Обсуждение
Увеличенная биологическая активность P1-конъюгата хлорамбуцила была продемонстрирована с различными типами клеток. CGTP2 и CGTP3 также обнаружили улучшенную цитотоксичность в отношении клеток JURKAT.
Пример 13
Морфин
Биологическую активность морфина (Mo) и его конъюгатов с жирной кислотой тестировали в антиноцицептивном тесте с использованием мышей. Испытывали различные дозы и продолжительность болеутоляющего действия.
Способы
Для этих экспериментов использовали штамм Quackenbush мышей (самцов 5-недельного возраста с живым весом приблизительно 30 г). Животные имели свободный доступ к пище и воде перед экспериментом и во время эксперимента. Животных делили на группы из 12 и индивидуально взвешивали. Рассчитывали средний вес для каждой группы. Затем мышей помещали на металлический блок, нагретый до 50oC, и измеряли время, требующееся для того, чтобы животные обнаружили признаки дискомфорта (базовая линия, контроль). Затем мышей инъецировали внутрибрюшинно (ip) различными дозами морфина или конъюгата морфин-жирная кислота. Морфин готовили в водном растворе, а конъюгаты морфин-жирная кислота готовили в виде эмульсии, содержащей 10% глицерилмоноолеат, 21% Miglyol, 6% этанол, 24% Твин 80 (33%) и 39% H2O. 0,2 мл соответствующего раствора инъецировали в каждую мышь. В различные точки времени после инъекции измеряли ответные реакции на нагретой пластинке. В каждом эксперименте испытывали 3 дозы морфина и конъюгата морфин-жирная кислота и 2 временные точки. Одну мышь использовали для каждой обработки и весь эксперимент повторяли по меньшей мере 3 раза. Все ответные реакции животных регистрировали съемкой и в некоторых случаях поведение мышей оценивалось независимыми людьми, которые не знали, какую обработку получили мыши. Членов группы дающих оценку людей просили оценить в баллах поведение мышей согласно следующему списку ответов:
0 = возбужденная, встревоженная мышь
1 = обнаруживающая легкий дискомфорт
2 = невозбужденная или не испытывающая дискомфорта, но еще настороженная
3 = спокойная, послушная, совершенно не испытывающая беспокойства
Результаты этой комиссии усреднялись.
Координацию мышей после обработки оценивали также помещением обработанных животных на Rotor-Rod (вращаемый стержень) при 1,6 об/мин. Оценивали способность животных удерживаться на стержне.
Результаты
Эксперимент 1. Влияние морфина и морфин-Suc-GTP2 (5, 10 и 20 мг морфина на кг) на мышей через 1 ч и 4 ч после инъекции.
Четыре повторности этого эксперимента анализировали при помощи дисперсионного анализа. Не было значимого различия между различными испытанными дозами среди лекарственных средств. Однако при объединении результатов различных доз каждого лекарственного средства были обнаружены различия (Таблица 10).
Результаты анализировали при помощи дисперсионного анализа:
p = 0,13 для временных ответов; p = 0,11 для оценок поведения.
Как данные временных ответных реакций, так и данные оценок поведения показывают, что конъюгат морфин-Suc-GTP2 имеет более продолжительное болеутоляющее действие, чем неконъюгированный морфин. Мышей, обработанных конъюгатом морфин-Suc-GTP2, тестировали также на координацию. Через 1 ч и 4 ч после инъекции они были способны оставаться на Rotor-Rod, также как обработанные морфином животные. Это указывает на то, что морфин-Suc-GTP2 не влияет на координацию тестируемых животных и оказывает истинное болеутоляющее действие на них.
Эксперимент 2. Временной ход активности морфина и конъюгата морфин-Suc-GTP2 при 5 мг Mo/кг.
Получено указание на медленное высвобождение морфина из конъюгата морфин-Suc-GTP2, так как была низкая активность через 1 ч после инъекции, но ее увеличение при 4 и 6 часах (Таблица 11). Морфин обнаруживает максимальную активность при 1 ч, которая уменьшается со временем.
Эксперимент 3. Влияние морфина и Mo-Suc-GTP3 (1 и 10 мг Mo/кг) на мышей при 4 и 6 ч после инъекции.
Mo-Suc-GTP3 обнаруживает также более продолжительное действие, чем морфин (Таблица 12).
Обсуждение
Конъюгат морфина с GTP2 обладал болеутоляющей активностью и не влиял на двигательную активность у мышей. Морфин-GTP2 имеет профиль доставки с медленным высвобождением и более продолжительное болеутоляющее действие, чем исходное лекарственное средство, морфин. Морфин-Suc-GTP2 обнаруживал сходное действие.
Пример 14
L-ДОПА
Биологические активности L-ДОПА и L-ДОПА-GTP2 испытывали в модели болезни Паркинсона на животных, состояния, обусловленного низкими уровнями нейротрансмиттера допамина в мозгу. Мышей предварительно обрабатывали резерпином, который истощает допамин и делает животных кататоническими. Измеряли способность L-ДОПА и L-ДОПА-GTP2 возвращать к норме из этого состояния.
Способы
Для этих экспериментов использовали штамм мышей Quackenbush (самцов 5-недельного возраста с живым весом приблизительно 30 г). Животные имели свободный доступ к пище и воде перед экспериментом и во время эксперимента. Мышей инъецировали внутрибрюшинно (ip) резерпином (5 мг/кг) и спустя 4 ч тестировали на кататонию. Мышь считали кататонической, если она была способна удерживаться на пробке диаметром 4,5 см в течение 5 мин. Затем кататонических животных инъецировали внутрибрюшинно L-ДОПА или L-ДОПА-GTP2, суспендированными в Miglyol (200-400 мкл/мышь) непосредственно перед использованием, и наблюдали.
Результаты
Эксперимент 1. Антирезерпиновая активность L-ДОПА.
После введения L-ДОПА кататоническим мышам все животные становились активными при дозах 364 мг/кг или более (Таблица 13). Уровень активности был высоким по сравнению с нормальными мышами. Животные имели подергивающуюся походку и обнаруживали токсичные эффекты L-ДОПА, такие как поднятие на задние лапы, слюноотделение и скачки.
Эксперимент 2. Антирезерпиновая активность L-ДОПА и L-ДОПА-GTP2.
L-ДОПА превращал кататонических мышей в очень активных (Таблица 14), как наблюдали ранее в Эксперименте 1. Животные, которые получали только Miglyol и низкие дозы L-ДОПА-GTP2 (9,1 и 91 мг ДОПА на кг), не возвращались к норме. Мыши, которые получали более высокие дозы L-ДОПА-GTP2 (364 и 637 мг ДОПА на кг), обнаружили признаки возвращения к норме при 7 и 5 ч после введения соответственно. Эти животные передвигались медленно и спорадически и не обнаруживали высокого уровня активности и токсических эффектов, которые вызывала обработка L-ДОПА. Эти результаты показывают, что L-ДОПА-GTP2 был способен к образованию допамина в мозгу кататонических мышей. Предполагается медленное высвобождение на основании времени, необходимого для появления ответной реакции, и характера этой реакции (медленные движения при 5-8 ч в противоположность сверхактивной мыши при 20 мин).
Обсуждение
L-ДОПА-GTP2 обладает способностью возвращать к норме (обращать) индуцированную резерпином кататонию у мышей. Показан эффект медленного высвобождения после введения L-ДОПА-GTP2 и не наблюдаются токсические эффекты, наблюдаемые с L-ДОПА.
Пример 15
Модель опухолевой цитотоксичности: Испытание метотрексата и хлорамбуцила и их конъюгатов с жирными кислотами.
Протокол
Группы черных мышей C57 получали инициирующую дозу 2 • 105 меланомных клеток B16 в виде интрадермальной инъекции. Эти клетки суспендировали в MEM (минимальной поддерживающей среде) без сыворотки. Живот зажимали и инъецировали 100 мкл + MEM. Спустя 8-10 дней после инъекции в месте инъекции были видны пятна растущих опухолей B16. Опухоли измеряли микрометром и фотографировали непосредственно перед инъекцией лекарственного средства (8-10 день). Цитотоксичные лекарственные средства и их конъюгаты с жирной кислотой растворяли или суспендировали в смеси 4:1 соевое масло:этилолеат. Раствор 10 мг/мл или 20 мг/мл каждого исходного лекарственного средства или его конъюгата с жирной кислотой, приготовленного с той же самой молярной концентрацией, что и раствор исходного лекарственного средства, инъецировали в ту же зону, где находились опухоли, при дозе 0,5 мг или 1 мг. Затем опухоли фотографировали, измеряли и повторно инъецировали каждые 2-3 дня.
Скорости роста опухолей показаны в Таблице 15 и статистический анализ действия лекарственного средства и конъюгатов лекарственного средства представлен в Таблице 16.
Единицы являются статистически модифицированным объемом опухоли; цифры в скобках являются средним квадратичным отклонением. Все результаты являются значимыми при 95% уровне, за исключением того, что MTX не отличается значимо от контроля. Все другие значимо отличаются от контроля, но не друг от друга.
Эти результаты показывают:
1. Нет значимого различия между мышами, обработанными носителем и метотрексатом (MTX).
2. Существует значимое различие между носителем/MTX и конъюгатами MTXGTP2 при двух различных концентрациях, но нет различия между двумя группами, обработанными конъюгатами.
3. Имеется значимое различие между группой, обработанной носителем, и группами, обработанными хлорамбуцилом, CGTP2 (0,5 мг) и CGTP2 (1,0 мг).
4. Нет различия между группой с хлорамбуцилом и двумя группами с конъюгатом или между этими группами.
5. Высокую токсичность наблюдали в тест-группе с хлорамбуцилом (3 из 7 мышей погибли); не наблюдали токсичности в любой другой тест-группе.
6. Хотя нет видимого преимущества в цитотоксичности конъюгатов хлорамбуцила по сравнению со свободным хлорамбуцилом, более низкая токсичность конъюгатов могла бы предоставлять терапевтическое преимущество.
Заключение
Данное изобретение показало, что нестероидные противовоспалительные лекарственные средства при модификации их присоединением одного-трех ацильных производных жирных кислот проявляют пролонгированную активность по сравнению с немодифицированным исходным лекарственным средством при трансдермальном применении (см. International Patent Application N PCT/AU 94/00440). В данной заявке заявители показали, что другие терапевтические агенты при подобной модификации являются биологически активными и обнаруживают измененную активность в сравнении с немодифицированными лекарственными средствами.
Специалистам в данной области будет понятно, что могут быть сделаны многочисленные изменения и/или модификации этого изобретения, показанного в характерных вариантах, без отхода от сущности или объема данного изобретения, описанных в широком плане. Данные варианты, следовательно, должны рассматриваться во всех отношениях как иллюстративные и не ограничивающие.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОНЪЮГАТЫ: ТЕРАПЕВТИЧЕСКОЕ СОЕДИНЕНИЕ - ЖИРНАЯ КИСЛОТА | 1994 |
|
RU2137755C1 |
| ДОСТАВКА НУКЛЕИНОВЫХ КИСЛОТ | 1995 |
|
RU2160278C2 |
| СПОСОБ НАНЕСЕНИЯ КРАСИТЕЛЯ НА КЕРАТИНОВЫЕ ВОЛОКНА (ВАРИАНТЫ) | 1993 |
|
RU2128257C1 |
| СПОСОБ МОДИФИКАЦИИ, ПО МЕНЬШЕЙ МЕРЕ, ЧАСТИ ПОВЕРХНОСТИ ПОЛИМЕРА | 1996 |
|
RU2163246C2 |
| СПОСОБ ФУМИГАЦИИ ПРОДУКТОВ ФОСФИНОМ И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 1990 |
|
RU2088097C1 |
| УСОВЕРШЕНСТВОВАННАЯ ФУМИГАЦИЯ МЕЛКИХ СЫПУЧИХ ПРОДУКТОВ | 1994 |
|
RU2135046C1 |
| МАГНИЕВОЕ ЛИТЬЕ ПОД ДАВЛЕНИЕМ | 1998 |
|
RU2212980C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФОСФИНА, УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ И СИСТЕМА БЕЗОПАСНОСТИ ДЛЯ ПРОИЗВОДСТВА ФОСФИНА | 1991 |
|
RU2087415C1 |
| ФУМИГАНТ, ФУМИГАНТНАЯ КОМПОЗИЦИЯ И СПОСОБ ОКУРИВАНИЯ | 1993 |
|
RU2141762C1 |
| СПОСОБ И УСТРОЙСТВО ДЛЯ СМЕШИВАНИЯ | 1998 |
|
RU2216393C2 |


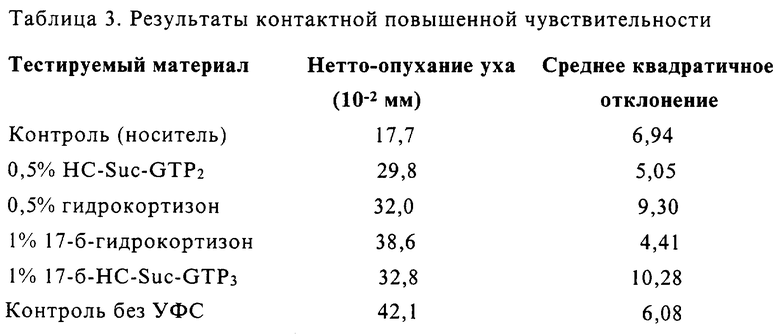

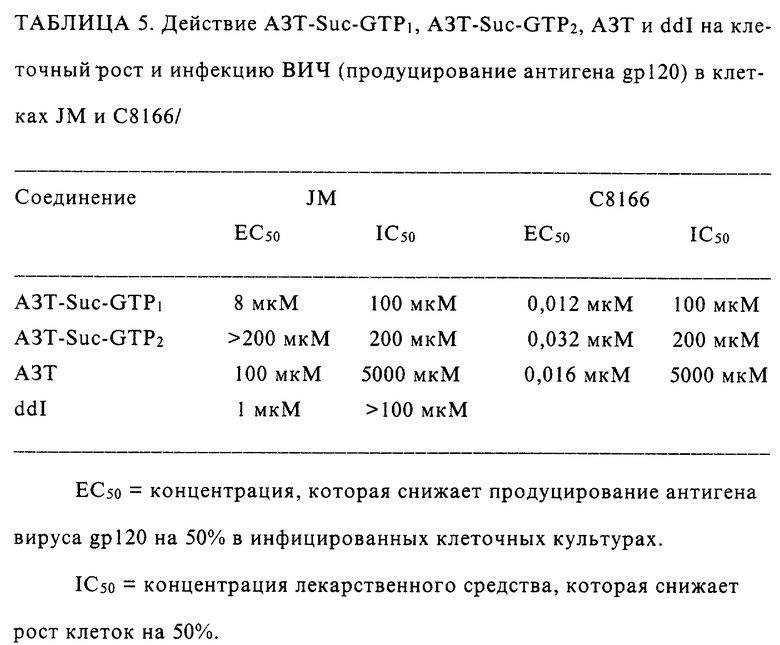

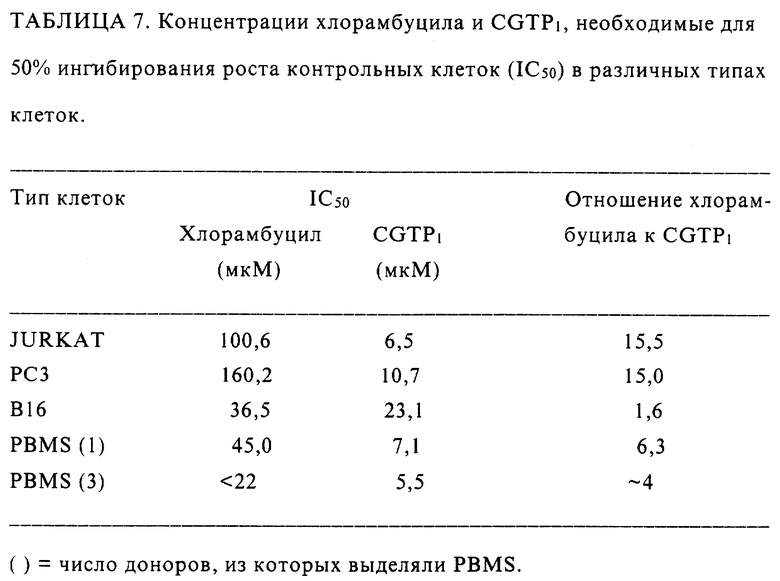

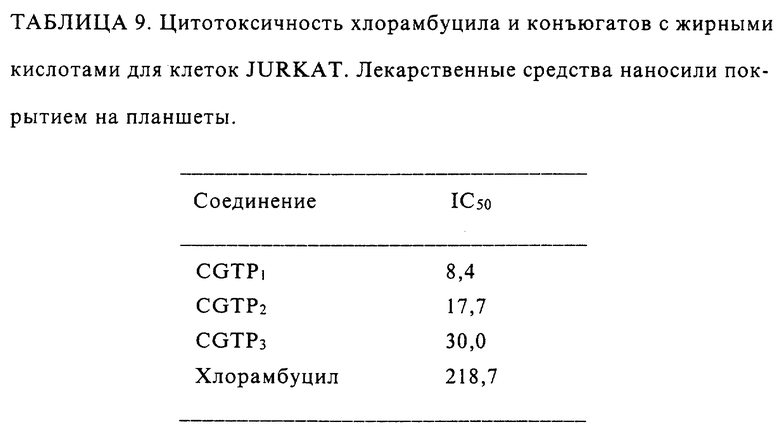





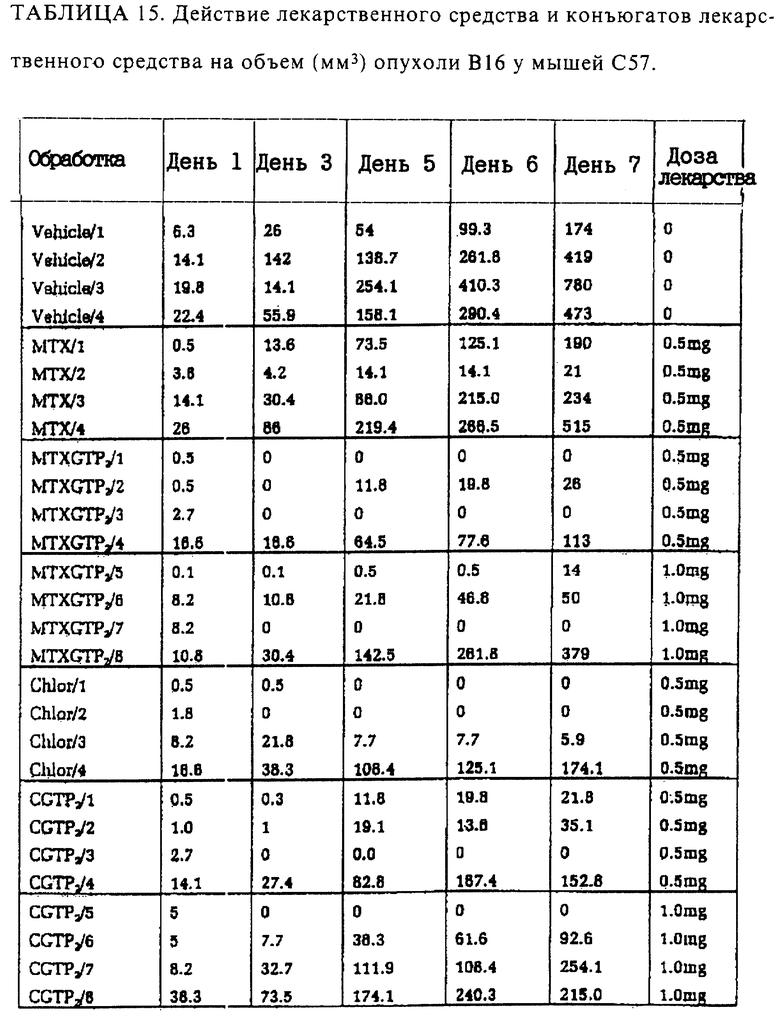
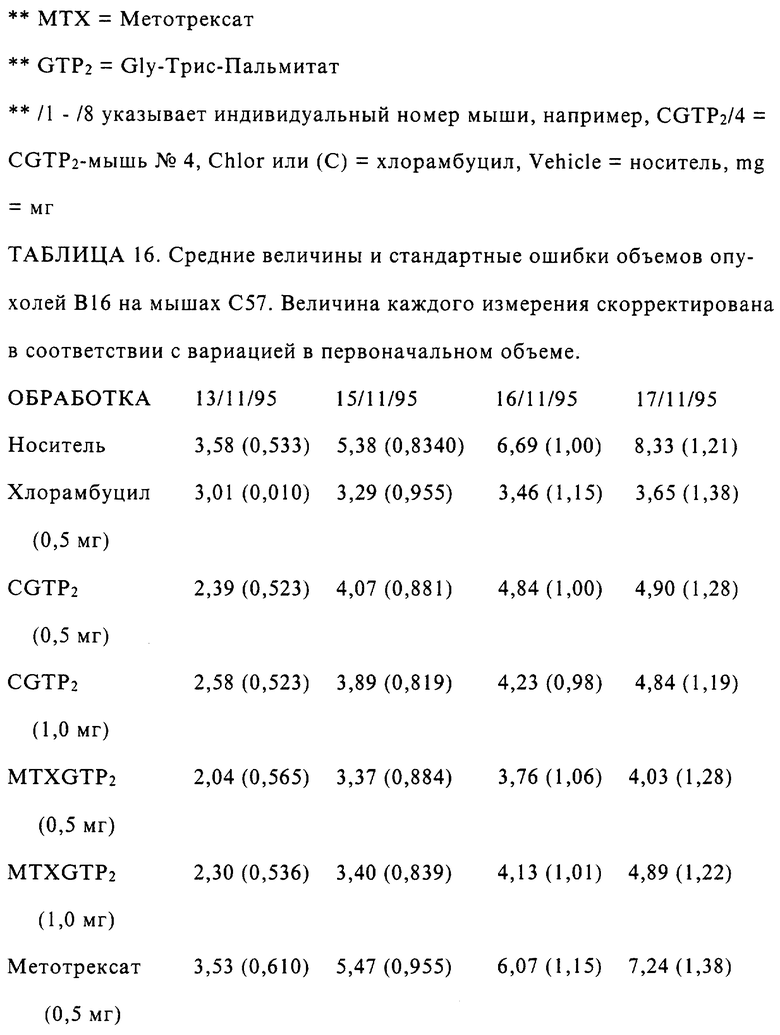
Описываются новые соединения общей формулы I, где X является членом кортикостеронового семейства гормонов и лекарственных средств и соединен с Y через гидроксильную группу; Y - остаток линкера, выбираемый из a) дикарбоновой кислоты или ангидрида; b) соединения, имеющего карбоксильную группу и альдегид; c) соединения, имеющего карбоксильную группу и галогенидную группу, d) соединения, имеющего карбоксильную группу и группу N=C=O; AA - аминокислота; n = 0 - 5; B - H или CH2O-R3; D - H или CH2O-R1; F - R2 или R4; R1, R2 и R3 одинаковы или различны и представляют собой каждый водород, метил, этил или ацильную группу жирной кислоты, при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой жирной кислоты; R4 - ацильная группа жирной кислоты. Новые соединения формулы I будут улучшать доставку, поглощение, полупериод существования в организме и нацеливание в клетке лекарственных средств после перорального, интраназального и других способов доставки. 14 с. и 20 з.п. ф-лы, 16 табл.
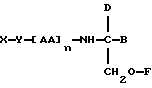
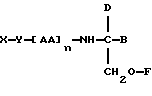
где X является членом кортикостеронового семейства гормонов и лекарственных средств и соединен с Y через гидроксильную группу;
Y - остаток линкера, выбираемый из а) дикарбоновой кислоты или ангидрида, b) соединения, имеющего карбоксильную группу и альдегид, с) соединения, имеющего карбоксильную группу и галогенидную группу, d) соединения, имеющего карбоксильную группу и группу N=C=O;
AA - аминокислота;
n = 0 - 5;
B - H или CH2O-R3;
D - H или CH2O-R1;
F - R2 или R4;
R1, R2 и R3 одинаковы или различны и представляют собой каждый водород, метил, этил или ацильную группу жирной кислоты при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой жирной кислоты;
R4 - ацильная группа жирной кислоты.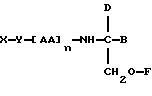
где X является членом кортикостеронового семейства гормонов и лекарственных средств и соединен с Y через гидроксильную группу;
Y - спейсерная группа;
AA - аминокислота;
n = 0 - 5;
B - H или CH2O-R3;
D - H или CH2O-R1;
F - R2 или R4;
R1, R2 и R3 одинаковы или различны и представляют собой каждый водород, метил, этил или ацильную группу жирной кислоты при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой жирной кислоты;
R4 - ацильная группа жирной кислоты.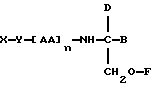
где X является членом семейства морфина и соединен с Y через гидроксильную группу, например гидроксильную группу в положении 3 или 6;
Y представляет собой остаток линкера, выбираемый из а) дикарбоновой кислоты или ангидрида, b) соединения, имеющего карбоксильную группу и альдегид, с) соединения, имеющего карбоксильную группу и галогенидную группу, d) соединения, имеющего карбоксильную группу и группу N=C=O;
AA - аминокислота;
n = 0 - 5;
B - H или CH2O-R3;
D - H или CH2O-R1;
F - R2 или R4;
R1, R2 и R3 одинаковы или различны и представляют собой каждый водород, метил, этил или ацильную группу жирной кислоты при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой жирной кислоты;
R4 - ацильная группа жирной кислоты.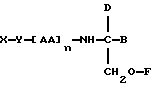
где X является членом семейства морфина и соединен с Y через гидроксильную группу в положении 3 или 6;
Y - линкерная группа;
AA - аминокислота;
n = 0 - 5;
B - H или CH2O-R3;
D - H или CH2O-R1;
F - R2 или R4;
R1, R2 и R3 одинаковы или различны и представляют собой каждый водород, метил, этил или ацильную группу жирной кислоты при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой жирной кислоты;
R4 - ацильная группа жирной кислоты.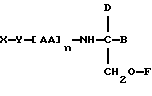
где X является антивирусным нуклеозидом и соединен с Y через гидроксильную группу;
Y - остаток линкера, выбираемый из а) дикарбоновой кислоты или ангидрида, b) соединения, имеющего карбоксильную группу и альдегид, с) соединения, имеющего карбоксильную и галогенидную группу, d) соединения, имеющего карбоксильную группу и группу N=C=O;
AA - аминокислота;
n = 0 - 5;
B - H или CH2O-R3;
D - H или CH2O-R1;
F - R2 или R4;
R1, R2 и R3 одинаковы или различны и представляют собой каждый водород, метил, этил или ацильную группу жирной кислоты при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой жирной кислоты;
R4 - ацильная группа жирной кислоты.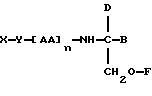
где X является антивирусным нуклеозидом и соединен с Y через гидроксильную группу;
Y - линкерная группа;
AA - аминокислота;
n = 0 - 5;
B - H или CH2O-R3;
D - H или CH2O-R1;
F - R2 или R4;
R1, R2 и R3 одинаковы или различны и представляют собой каждый водород, метил, этил или ацильную группу жирной кислоты при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой жирной кислоты;
R4 - ацильная группа жирной кислоты.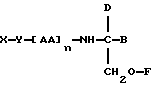
где X является членом циклоспоринового семейства лекарственных средств и соединен с Y через гидроксильную группу;
Y - остаток линкера, выбираемый из а) дикарбоновой кислоты или ангидрида, b) соединения, имеющего карбоксильную группу и альдегид, с) соединения, имеющего карбоксильную и галогенидную группы, d) соединения, имеющего карбоксильную группу и группу N=C=O;
AA - аминокислота;
n = 0 - 5;
B - H или CH2O-R3;
D - H или CH2O-R1;
F - R2 или R4;
R1, R2 и R3 одинаковы или различны и представляют собой каждый водород, метил, этил или ацильную группу жирной кислоты при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой жирной кислоты;
R4 - ацильная группа жирной кислоты.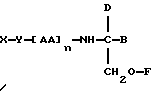
где X является членом циклоспоринового семейства лекарственных средств и соединен с Y через гидроксильную группу;
Y - спейсерная группа;
AA - аминокислота;
n = 0 - 5;
B - H или CH2O-R3;
D - H или CH2O-R1;
F - R2 или R4;
R1, R2 и R3 одинаковы или различны и представляют собой каждый водород, метил, этил или ацильную группу жирной кислоты при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой жирной кислоты;
R4 - ацильная группа жирной кислоты.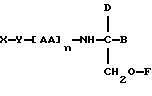
где X является членом семейства фолатных антагонистов и соединен с Y через карбоксильную группу;
Y отсутствует или выбирается из а) соединения с аминогруппой и карбоксильной группой, b) соединения с аминогруппой или группой сульфокислоты, с) соединения с гидроксильной или карбоксильной группой, d) соединения с гидроксильной группой и группой сульфокислоты, е) соединения с гидроксильной группой и реактивной галогенидной группой, f) соединения с гидроксильной и альдегидной группами, g) соединения, имеющего две реактивные галогенидные группы, и h) алкиленоксида;
AA - аминокислота;
n = 0 - 5;
B - H или CH2O-R3;
D - H или CH2O-R1;
F - R2 или R4;
R1, R2 и R3 одинаковы или различны и представляют собой каждый водород, метил, этил или ацильную группу жирной кислоты при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой жирной кислоты;
R4 - ацильная группа жирной кислоты.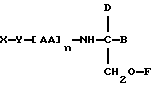
где X является членом семейства фолатных антагонистов и соединен с Y через карбоксильную группу;
Y является необязательной спейсерной группой;
AA - аминокислота;
n = 0 - 5;
B - H или CH2O-R3;
D - H или CH2O-R1;
F - R2 или R4;
R1, R2 и R3 одинаковы или различны и представляют собой каждый водород, метил, этил или ацильную группу жирной кислоты при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой жирной кислоты;
R4 - ацильная группа жирной кислоты.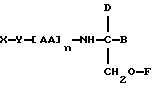
где X является членом семейства ДОПА и соединен с Y через карбоксильную или аминогруппу;
Y отсутствует или выбирается из а) соединения с аминогруппой и карбоксильной группой, b) соединения с аминогруппой и группой сульфокислоты, с) соединения с гидроксильной и карбоксильной группами, d) соединения с гидроксильной группой и группой сульфокислоты, е) соединения с гидроксильной группой и реактивной галогенидной группой, f) соединения с гидроксильной и альдегидной группами, g) соединения, имеющего две реактивные галогенидные группы и h) алкиленоксида, когда X связан через карбоксильную группу, и а) дикарбоновой кислоты, ангидрида, группы дисульфокислоты или соединения, имеющего две реактивные галогенидные группы, b) соединения с карбоксильной группой и группой сульфокислоты, с) соединения с карбоксильной группой и реактивной галогенидной группой, d) соединения с реактивной галогенидной группой и группой сульфокислоты, когда X связан через аминогруппу;
AA - аминокислота;
n = 0 - 5;
B - H или CH2O-R3;
D - H или CH2O-R1;
F - R2 или R4;
R1, R2 и R3 одинаковы или различны и представляют собой каждый водород, метил, этил или ацильную группу жирной кислоты при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой жирной кислоты;
R4 - ацильная группа жирной кислоты.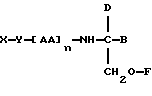
где X является членом семейства ДОПА и соединен с Y через карбоксильную группу или аминогруппу;
Y является необязательной спейсерной группой;
AA - аминокислота;
n = 0 - 5;
B - H или CH2O-R3;
D - H или CH2O-R1;
F - R2 или R4;
R1, R2 и R3 одинаковы или различны и представляют собой каждый водород, метил, этил или ацильную группу жирной кислоты при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой жирной кислоты;
R4 - ацильная группа жирной кислоты.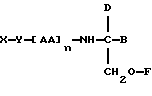
где X является членом семейства хлорамбуцила и соединен с Y через карбоксильную группу;
Y отсутствует или выбирается из а) соединения с аминогруппой и карбоксильной группой, b) соединения с аминогруппой или группой сульфокислоты, с) соединения с гидроксильной или карбоксильной группами, d) соединения с гидроксильной группой и группой сульфокислоты, е) соединения с гидроксильной группой и реактивной галогенидной группой, f) соединения с гидроксильной и альдегидной группами, g) соединения, имеющего две реактивные галогенидные группы и h) алкиленоксида;
AA - аминокислота;
n = 0 - 5;
B - H или CH2O-R3;
D - H или CH2O-R1;
F - R2 или R4;
R1, R2 и R3 одинаковы или различны и представляют собой каждый водород, метил, этил или ацильную группу жирной кислоты при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой жирной кислоты;
R4 - ацильная группа жирной кислоты.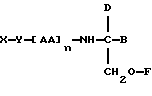
где X является членом семейства хлорамбуцила и соединен с Y через карбоксильную группу;
Y является необязательной спейсерной группой;
AA - аминокислота;
n = 0 - 5;
B - H или CH2O-R3;
D - H или CH2O-R1;
F - R2 или R4;
R1, R2 и R3 одинаковы или различны и представляют собой каждый водород, метил, этил или ацильную группу жирной кислоты при условии, что по меньшей мере один из R1, R2 и R3 является ацильной группой жирной кислоты;
R4 - ацильная группа жирной кислоты.
| Способ получения сложных эфиров аминокислот или их физиологически совместимых солей | 1989 |
|
SU1836380A3 |
| DE 4310516 A1, 11.08.1994 | |||
| US 3686238 A, 22.08.1972 | |||
| Огнетушитель | 0 |
|
SU91A1 |
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |

