Клиническая картина диабета характеризуется повышенным содержанием сахара в крови. Причиной, вызываемого недостатком инсулина диабета, или диабета типа 1, является отмирание продуцирующих инсулин β- клеток поджелудочной железы; поэтому лечение осуществляют путем введения инсулина (заместительная терапия). Инсулинонезависимый диабет, или диабет типа II, напротив, характеризуется уменьшенным воздействием инсулина на мышечную и жировую ткань (резистентность к инсулину) и повышенным продуцированием глюкозы печенью. Причины этих нарушений обмена веществ еще неясны. При обычном лечении с помощью сульфонилмочевин компенсируют резистентность к инсулину за счет повышения аутогенного выделения инсулина, однако, это не во всех случаях приводит к нормализации уровня сахара в крови и не замедляет прогрессирование заболевания; многие диабетики типа II, за счет "исчерпания" β- -клеток становятся инсулинозависимыми и страдают от позднее возникающих заболеваний, как катаракта, нефропатия и ангиопатия.
Вследствие этого желательны новые подходы к лечению диабета типа II.
Концентрация глюкозы в крови в состоянии натощак определяется продуцированием глюкозы печенью. Было показано, что в случае диабета типа II повышение содержания сахара в крови коррелируют с пропорционально повышенным выделением глюкозы из печени. Выделяемая печенью в кровь глюкоза может образовываться как за счет разрушения гликогена печени (гликогенолиз), так и благодаря глюконеогенезу.
Глюкозо-6-фосфат представляет собой конечный продукт как глюконеогенеза, так и гликогенолиза. Конечная стадия выделения печенью глюкозы из глюкозо-6-фосфата катализируется глюкозо-6-фосфатазой (ЕС 3.1.3.9). Глюкозо-6-фосфатаза представляет собой находящийся в эндоплазматической сетке (ER) многоферментный комплекс. Этот ферментный комплекс состоит из находящейся в ER-мебране глюкозо-6-фосфаттранслоказы, локализованной на люминальной стороне эндоплазматической сетки глюкозо-6-фосфатазы и фосфат-транслоказы (CM.:Ashmore J. и Weber G.,"The Role of Hepatic Glucose-6-phosphatase in the Regulation of Carbohydrate Metabolism в Vitamins and Hormones, т. XVII (Harris R.S., Marrian G.F.,Thimann K.V. издатели), 92-132 (1959); Burchell A., Vaddell I.D.,"The molecular basis of the hepatic microsoma glucose-6-phosphatase system, "Biochem. Biophys. Asta 1092, 129-137 (1990)). Имеющаяся обширная литература показывает, что при всех исследованных условиях, которые в опытах на животных приводят к повышенным содержаниям сахара в крови, например, в случае использования стрептозотоцина, аллоксана, кортизона, тироидных гормонов и голоданий, активность этого многоферментного комплекса также повышена. Показано, что наблюдаемое в случае диабетиков типа II повышенное продуцирование глюкозы связано с повышенной активностью глюкозо-6-фосфатазы.
Значение глюкозо-6-фосфатазной системы для нормального гомеостаза глюкозы, подчеркивается гипогликемическими симптомами у пациентов с заболеванием типа 16, связанным с накоплением гликогена, при этом отсутствует транслоказная компонента глюкозо-6-фосфатсистемы.
Снижение активности глюкозо-6-фосфатазы за счет исследования пригодных биологически активных веществ (ингибиторов) должно приводить к уменьшенному выделению глюкозы печенью. Эти биологически активные вещества должны обеспечить продуцирование глюкозы печенью для эффективного периферического потребления. Благодаря этому в состоянии натощак у диабетиков типа II снижается содержание глюкозы в крови, сверх того, оказывается превентивное воздействие в отношении возникающих вследствие диабета заболеваний.
В литературе описывается ряд неспецифических ингибиторов глюкозо-6-фосфатазы, например, флоррицин [(Soodsma J. Legler В. nHerdlie R.C., J.Biol. Chem. 242, 1955-1960 (1967)] ; 5,5'-дитио-бис-2-нитро-бензойная кислота [(Wallin В. К. и Arion W.J., Biochem. Biophys., Res.Commun., 48, 694-699 (1972)] ; 2,2'-ди-изотиоцианато-стильбен и 2-изотиоцианато-2'-ацетокси-стильбен [(Zoccoli M. A. и Karnowski M.L., J.Bio. Chem., 255, 1113-1119 (1980)] . Первые терапевтически применимые ингибиторы глюкозо-6-фосфатазной системы предложены в европейских заявках 93114260.8 и 93114261.6.
Охарактеризованные ниже подробнее циклогексановые производные представляют собой новые, до сих пор не описанные в литературе соединения.
Авторами показано, что сложные эфиры определенных производных циклогексанола, например, соединение согласно примеру 4, являются очень хорошими ингибиторами глюкозо-6-фосфатазной системы.
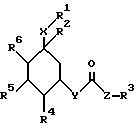
Поэтому изобретение относится к сложным эфирам циклогексанола формулы (1):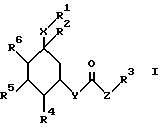
где R1 обозначает CONHSO2R14, где R14 является C1-C10 алкилом; бензтиазолилом, замещенным C1-C4алкоксилом; фенилом, незамещенным или замещенным фтором и/или нитро-группой; нафтилом, незамещенным или замещенным NR8R9-группой; тетразолилом; тиазолилом; или группу формулы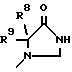
где R8 и R9 обозначают водород или C1-C4- алкил,
R2 обозначает O-C1-C10алкил(R11), R11 является C3-циклоалкилом или C1-C10 алкилом, который в свою очередь может быть замещен фенилом или хлорфенилом, n = 0,1 или R1 и R2 образуют кольцо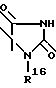
где R16 обозначает C1-C10 алкил (R11);
R3 обозначает бензимидазолил,
Z обозначает CH=C(R13), где R13 - фенил, незамещенный или замещенный ОН;
R4, R5, R6 обозначает водород, ОН, причем R4, R5, R6 являются одинаковыми или разными;
Y является -O-;
X обозначает (CH2)m, где m = 0.
Предлагаемые согласно изобретению соединения формулы (I), если они содержат карбоксильную группу, могут образовывать соли с неорганическими или органическими основаниями.
Поэтому изобретение также относится к физиологически приемлемым солям соединений формулы (I).
Предлагаемые согласно изобретению соединения формулы (I) содержат ряд стереоцентров. Изобретение относится ко всем возможным энантиомерам и диастереомерам, соответствующим формуле (I). В приведенном тексте заместители имеют следующие значения:
Указанные значения для R1, R3, R8, R9, R11, R13 и R16 алкильные и алкоксильные остатки являются линейными или разветвленными. Указанные значения для R2 и R14 алкильные группы являются линейными, разветвленными или циклическими, причем также только одна часть остатка может образовывать кольцо.
Защитными спиртовыми группами являются группы, образующие замещенные простые эфиры, как метоксиметил, метилтиометил, трет-бутилтиометил, бензилоксиметил, п-метоксибензилоксиметил, трет-бутоксиметил, силоксиметил, 2-метоксиэтоксиметил, 1-этокси-этил, аллил, бензил, п-метоксибензил, 3,4-диметоксибензил, о-нитробензил, п-нитробензил, п-галогенбензил, 2,6-дихлор-бензил, п-цианбензил, п-фенил-бензил, 2- и 4-пиколил.
Защитными группами для аминокислоты являются:
а) образующие карбаматы группы, как метил и этил; 9-флуоренилметил; 9-(2-сульфо)- флуоренилметил; 9-(2,7-ди бром) флуоренилметил; 2,7-ди-трет-бутил-[9-(10,10-диоксо-10,10,10,10-тетрагидротиоксантил) 7-метил; 4-метоксифенацил; 2,2,2-трихлорэтил; 2-триметилсилилэтил; 2-фенилэтил; 1-(1-адамантил)-1-метилэтил; 1-диметил-2-галогенэтил; 1,1-диметил-2, 2-дибромэтил; 1, 1-диметил-2,2,2-трихлорэтил; 1-метил-1-(4-бифенил)-этил; 1- (3,5-ди-трет-бутилфенил)-1-метилэтил; 2-(2'- и 4'-пиридил)-этил; 2-(N,N-дициклогексилкарбоксамидо)-этил; трет-бутил; 1-адамантил; винил; аллил; 1-изопропилаллил; циннамил; 4-нитроциннамил; 8-хинолил; N-гидроксипиперидинил; алкилтио; бензил; п-метоксибензил; п-нитробензил; п-бромбензил; п-хлорбензил; 2,4-дихлорбензил; 4-метилсульфинилбензил; 9-антрилметил; и дифенилметил, трет-амил, S-бензил-тио-группа; п-цианобензил, циклобутил, циклогексил, циклопентил, циклопропилметил, п-дециклобензил, диизопропилметил, 2,2-диметоксикарбонилвинил, о-(N,N-диметил-карбоксамидо)-бензил, 1,1-диметил-3-(N,N-диметилкарбоксамидо) пропил, 1,1-диметилпропинил, ди-(2-пиридил) метил, 2-фуранилметил, 2-йодэтил, изоборнил, изобутил, изоникотинил, п-(п'-метоксифенилазо) бензил, 1-метилциклобутил, 1-метилциклогексил, 1-метил-1-циклопропилметил, 1-метил-1-(3,5-диметоксифенил)этил, 1-метил-1-(п-фенилазофенил)-этил, 1-метил-1-фенилэтил, 1-метил-1-(4-пиридил)-этил, фенил, п-(фенилазо) бензил, 2,4,6-три-трет-бутилфенил, 4-(триметиламмоний) бензил и 2,4,6-триметилбензил;
б) образующие производные мочевины группы, как фенотриазинил-(10)-карбонильные, N'-п-толуолсульфониламинокарбонильные и N'-фениламинотиокарбонильные производные;
в) образующие амиды группы, как N-формил, N-ацетил, N-хлорацетил, N-трихлорацетил, N-трифторацетил, N-фенилацетил, N-3-фенилпропионил, N-пиколиноил, N-3-пиридилкарбоксил, N-бензоилфенилаланил, N-бензоил и N-п-фенил-бензоил.
Предпочтительные соединения формулы (I), в которых:
R1 означает CONHSO2R14,
R2 означает O-C1-C10алкил(R11), n = 1, причем алкильная часть является линейной или разветвленной, или R1 и R2 вместе образуют кольцо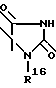
Предлагаемые согласно изобретению предпочтительные соединения формулы (I), если они содержат карбоксильную группу, также могут образовывать соли с неорганическими или органическими основаниями. Предпочтительны соли с неорганическими основаниями, особенно физиологически приемлемые соли щелочных металлов, прежде всего соли натрия и калия.
Соединения формулы (I) ингибируют фермент глюкозо-6-фосфатазы печени млекопитающих. Поэтому соединения пригодны в качестве фармацевтического препарата. Изобретение относится также к фармацевтическому препарату на основе соединений формулы (I), в случае необходимости в виде физиологически приемлемых солей.
Изобретение включает также применение соединений формулы (I) или их солей для лечения заболеваний, которые связаны с повышенной активностью фермента глюкозо-6-фосфатазы.
Изобретение включает также применение соединений формулы (I), соответственно, их солей, для лечения заболеваний, которые связаны с повышенным продуцированием глюкозы печенью.
Кроме того, изобретение включает применение соединений формулы (I), а также их солей, для лечения диабета типа II (инсулиннезависимого или старческого диабета).
Изобретение, далее, относится к применению соединений формулы (I), соответственно, их солей, для получения лекарственных средств для лечения диабета и других заболеваний, которые характеризуются повышенным выделением глюкозы из печени или повышенной активностью фермента глюкозо-6-фосфатазы.
Действие предлагаемых согласно изобретению соединений на фермент глюкозо-6-фосфатазы исследовано в ферментном тесте на микросомах печени.
Для приготовления содержащей глюкозо-6-фосфатазу микросомной фракции используют свежую печень самцов крыс Wistar и обрабатывают как описано в литературе [Canfield W.K. и Arion W.J., J. Biol.Chem. 263, 7458-7460 (1988)] . Эту микросомную фракцию можно хранить при -70 С, по меньшей мере, в течение 2-х месяцев без значительной потери активности.
Активность глюкозо-6-фосфатазы определяли согласно Arion W.J. (Methods buzymol 174, Academic Press, 1989, с. 58-67) путем определения высвобождающегося фосфата из глюкозо-6-фосфата. Испытуемая смесь (0,1 мл) содержит глюкозо-6-фосфат (1 ммоль/л), испытуемое вещество, 0,1 мг микросомной фракции и 100 ммоль/л HEPES -буфера [4-(2-гидроксиэтил) пиперазин-1-этансульфокислота], pH 7,0. Реакцию инициируют путем добавки фермента. По истечении 20 минут при комнатной температуре реакцию прекращают добавлением 0,2 мл фосфатного реагента. Пробу инкубируют в течение 30 минут при 37oC и затем измеряют абсорбцию (A) синего красителя при 570 нм. Ингибирующую активность исследуемого вещества определяют путем сравнения с контрольным образцом, который не содержит исследуемого вещества, по формуле: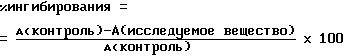
В случае необходимости, ингибирующее действие исследуемого вещества определяют как функцию используемой концентрации вещества и из нее рассчитывают концентрацию для 50%-ного ингибирования активности фермента (IC50).
Для нижеуказанных соединений определено следующее IC50-значение:
Соединение - IC50 (мк моль)
4 - 0,02
9 - 0,3
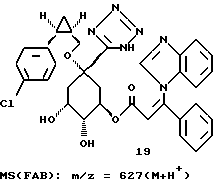
19 - 0,8
Следующим объектом изобретения является фармацевтический препарат, который содержит эффективное количество одного или нескольких соединений формулы (I), согласно изобретению, и/или их фармакологически приемлемых солей.
Лекарственные препараты получают известными для специалиста способами. В качестве лекарственных средств предлагаемые в изобретении фармакологически активные соединения используются либо как таковые, либо предпочтительно в комбинации с пригодными фармацевтическими вспомогательными веществами в форме таблеток, драже, капсул, свечей, эмульсий, суспензий, гранулятов, порошков, растворов или препаратов с пролонгированным выделением биологически активного вещества, причем содержание биологически активного вещества предпочтительно составляет 0,1-95%.
Вспомогательные вещества, пригодные для рецептуры лекарственного средства, специалисту известны. Наряду с растворителями, гелеобразователями, основами суппозиториев, вспомогательными веществами для таблеток и другими носителями биологически активных веществ, можно применять, например, антиоксиданты, диспергаторы, эмульгаторы, антивспениватели, улучшающие вкус вещества, консерванты, агенты растворения или красители.
Биологически активные вещества можно применять топически, орально, парентерально или внутривенно, причем предпочтительный способ введения зависит от излечиваемого заболевания. Предпочтительно оральное введение.
Для оральной формы применения, активные соединения смешивают с пригодными для этой цели добавками, как носители, стабилизаторы или инертные разбавители, и обычными способами доводят до пригодной формы введения, как таблетки, драже, разъемные капсулы, водные, спиртовые или масляные суспензии или водные, спиртовые или масляные растворы. В качестве инертных носителей можно применять, например, гуммиарабик, магнезию, карбонат магния, фосфат калия, молочный сахар, глюкозу или крахмал; в особенности кукурузный крахмал. При этом препарат можно получать как в виде сухого, так и в виде мокрого гранулята. В качестве масляных носителей или растворителей принимают во внимание растительные или животные масла, как подсолнечное масло или рыбий жир.
Для подкожного или внутривенного введения, активные соединения или их физиологически приемлемые соли, в желательном случае вместе с обычными для этой цели веществами, как агенты растворения, эмульгаторы или другие вспомогательные вещества, переводят в раствор, суспензию или эмульсию, в качестве растворителей принимают во внимание, например, воду, физиологический раствор хлорида натрия или спирты, например, как этанол, пропанол, глицерин; наряду с ними также растворы сахара, как растворы глюкозы или маннита, или также смесь различных растворителей.
В качестве фармацевтических препаратов для топического или локального применения пригодны глазные капли, которые содержат активное соединение в водном или масляном растворе. Для введения в нос пригодны аэрозоли и пульверизаторы, а также грубодисперсные пудры, которые вводят путем быстрых ингаляций в носовые отверстия, и прежде всего капли в нос, которые содержат активные соединения в водном или масляном растворе.
Дозировка вводимого биологически активного вещества формулы (I) и частота введения зависят от интенсивности действия и длительности действия используемого соединения согласно изобретению, кроме того, также от рода и силы излечиваемого заболевания, как также от пола, возраста, веса и индивидуальной предрасположенности излечиваемого млекопитающего. В среднем рекомендуемая суточная доза предлагаемого согласно изобретению соединения составляет примерно 1- 500 мг для млекопитающего весом 75 кг, в первую очередь для человека, предпочтительно примерно 10 - 250 мг, причем можно осуществлять при необходимости введение нескольких доз в день и в случае необходимости доза может быть также меньше или больше.
Получение предлагаемых согласно изобретению соединений формулы (I) поясняется примерами. Комнатная температура обозначает температуру 20 - 25oC.
Пример 1.
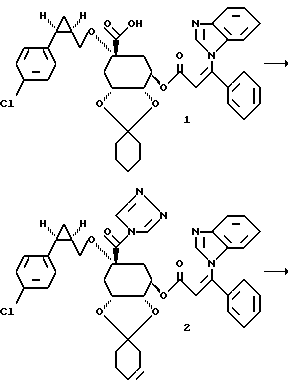
Получение соединения 2 из соединения 1.
3,7 г (0,054 моля) карбоновой кислоты 1 (получение см.: европейская заявка на патент N 93114261.6; реакционная схема способа A, структурный элемент 68B) растворяют в 36 мл безводного диметилформамида и при комнатной температуре в атмосфере аргона смешивают с 1,81 г (0,011 моля) N,N'-карбонил-ди-(1,2,4-триазола) и в течение 1,5 часов нагревают при 50 - 60oC. После охлаждения 0,15 М раствор соединения 2 без дальнейшей обработки используют в следующей стадии.
Получение соединения 3 из соединения 2.
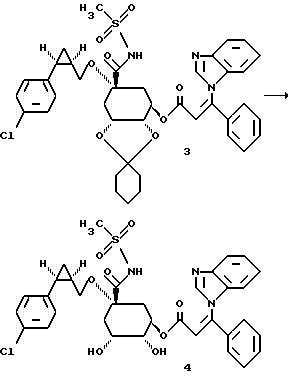
0,057 г (0,006 моля) метансульфонамида растворяют в 3 мл безводного диметилформамида и при комнатной температуре добавляют 0,02 г (0,0066 моля) гидрида натрия (80%-ного в масле). Суспензию перемешивают в течение 45 минут при 50 - 60oC. Затем при этой температуре прикапывают 3,1 мл (0,00047 моля) 0,15 М раствора триазолида формулы 2. Реакционную смесь перемешивают в течение 1 часа при 60oC. Затем реакционную смесь добавляют в насыщенный раствор хлорида аммония, причем продукт формулы 3 осаждается в виде аморфного твердого вещества. Осадок отсасывают, промывают дистиллированной водой и высушивают таким образом полученное твердое вещество в течение 3-х часов при 40oC и давлении 10-2 торр над хлоридом кальция. Получают 0,248 г соединения 3.
Получение соединения 4 из соединения 3.
0,24 г (0,000316 моля) циклогексилиденкеталя формулы 3 вносят в 10 мл диоксана и при комнатной температуре и интенсивном перемешивании добавляют 1,6 мл (0,0032 моля) 2М соляной кислоты. Прозрачный раствор перемешивают 2 часа при 50 - 60oC. Затем реакционный раствор охлаждают до 10 - 20oC и доводят до pH 3 с помощью 1 М раствора гидроксида натрия, разбавляют с помощью 20 мл дистиллированной воды и реакционную смесь концентрируют в вакууме до тех пор, пока не будет более отгоняться никакого диоксана. При перемешивании с водой медленно выкристаллизовывается осадок, который отсасывают и промывают водой. После высушивания при 40oC в высоком вакууме получают 0,18 г соединения 4 в виде бесцветного твердого вещества.
Таким образом синтезируют следующие соединения формулы (I):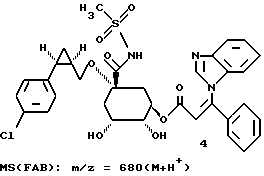
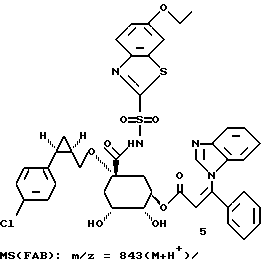
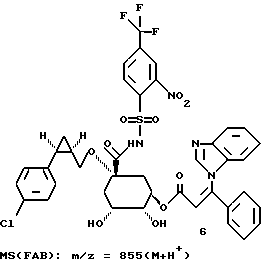
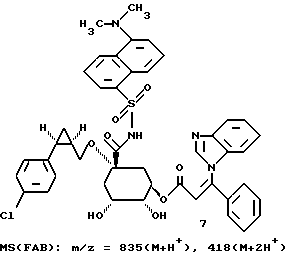
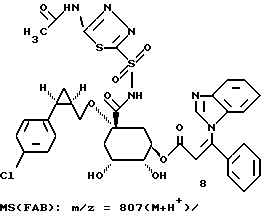
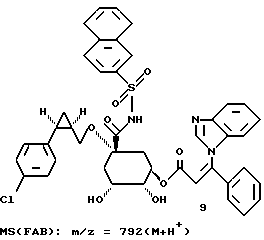
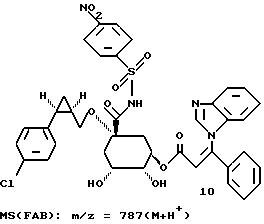
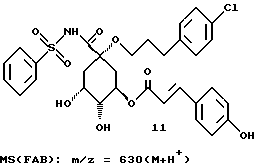
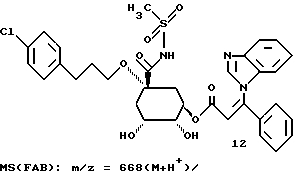
Получение соединения 14 из соединения 13.
5,0 г (0,012 моля) лактона формулы 13 (получение см. европейская заявка на патент N 93114261.6, реакционная схема способа A, структурный элемент 68B) растворяют в 80 мл безводного толуола и при -78oC в атмосфере аргона прикапывают 10 мл (0,012 моля) 1,2 М раствора диизобутилалюминийгидрида в гексане. Спустя 1 час, при -50oC, гидролизуют c помощью насыщенного раствора хлорида аммония. Экстрагируют c помощью этилацетата, объединенные органические фазы промывают насыщенным раствором хлорида натрия и высушивают над сульфатом магния. Органическую фазу концентрируют в вакууме и таким образом полученный лактол формулы 14 без дальнейшей очистки используют в следующей стадии.
Получение соединения 15 из соединения 14.
4,6 г (0,011 моля) лактола формулы 14 и 0,761 г (0,011 моля) гидроксиламин-гидрохлорида растворяют в 50 мл метанола. Добавляют 750 мл (0,014 моля) гидрокcида калия. Этот раствор перемешивают в течение 1 часа при комнатной температуре. Затем раствор смешивают с 300 мл простого метил-трет-бутилового эфира, промывают водой и насыщенным раствором хлорида натрия и органическую фазу концентрируют в вакууме, после высушивания над сульфатом магния. Остаток очищают путем хроматографии на силикагеле (элюирующее средство: этилацетат/н-гептан = 1:2). Получают 3,6 г окcима формулы 15 в виде бесцветного масла.
Получение соединения 16 из соединения 15.
20,0 г (0,046 моля) оксима формулы 15 вводят в 200 мл безводного дихлорметана и добавляют 23,0 г (0,14 моля) N,N'-карбонилдиимидазола. Происходит сильное газовыделение. Спустя 14 часов выдерживания при комнатной температуре, к реакционному раствору добавляют 100 мл метанола и следующие 4 часа кипятят с обратным холодильником, для обработки раствор выпаривают досуха на роторном испарителе и обрабатывают простым метил-трет-бутиловым эфиром. Органическую фазу промывают смесью воды с 0,1 М раствором гидросульфата калия, сушат над сульфатом магния и концентрируют в вакууме. Остаток очищают путем хроматографии на силикагеле (размер зерен силикагеля 35 - 78 мкм, элюирующая система: этилацетат/н-гептан = 1:5, к концу долю н-гептана снижают до соотношения 1:3). Получают 12,9 г нитрила формулы 16 в виде бесцветного масла.
Получение соединения 17 из соединения 16.
12,9 г (0,0286 моля) нитрила формулы 16 растворяют в 250 мл безводного толуола и нагревают при 110oC. Через три дня в интервал времени 24 часа, смотря по обстоятельствам, добавляют 5,89 г (0,0286 моль) триметилазида олова. Затем реакционный раствор концентрируют в вакууме и остаток смешивают, при интенсивном перемешивании, с 50 мл 10 М раствора гидроксида натрия и 20 мл тетрагидрофурана. Образовавшуюся натриевую соль соединения 17 отсасывают, после чего суспендируют в дистиллированной воде и подкисляют с помощью 2 М уксусной кислоты. Экстрагируют этилацетатом, объединенные органические фазы сушат над сульфатом магния и концентрируют в вакууме. Получают 7,7 г тетразола формулы 17.
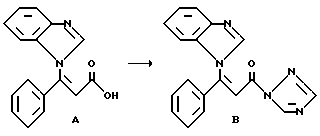
Получение исходного соединения B из соединения A.
274 мг (0,001 моля) карбоновой кислоты формулы A (получение см.: европейская заявка на патент N 93114261.6, способ I ), в атмосфере аргона и при комнатной температуре, растворяют в 20 мл диметилформамида и добавляют 180,4 мг (0,0011 моль) N,N'-карбонил-ди-(1,2,4-триазола). Реакционный раствор перемешивают в течение 1 часа при 60oC. Полученный раствор соединения B без дальнейшей обработки используют в следующей стадии.
Получение соединения 18 из соединения 17.
3,0 г (0,00652 моля) соединения 17 в атмосфере аргона растворяют в 30 мл безводного диметилформамида и при комнатной температуре добавляют 0,70 г (0,023 моля) гидрида натрия (80%-ная дисперсия в масле). Спустя 1 час прикапывают 157 мл (0,0078 моля) 0,5 М раствора соединения B в диметилформамиде и снова перемешивают в течение 1 часа при комнатной температуре. Затем реакционный раствор смешивают с насыщенным раствором хлорида аммония и экстрагируют этилацетатом. Объединенные органические фазы промывают насыщенным раствором хлорида натрия, сушат над сульфатом магния и концентрируют в вакууме. Сырой продукт очищают с помощью хроматографии (размер зерен силикагеля: 35-70 мкм; элюирующая система: этилацетат/н-гептан/метанол/ледяная уксусная кислота = 30: 10: 2: 1). Получают сложный эфир формулы 18 в виде аморфного твердого вещества.
Получение соединения 19 из соединения 18.
3,8 г (0,0053 моля) циклогексилидена формулы 18 обрабатывают 150 мл диоксана и при перемешивании смешивают с 10 мл (0,02 моля) 2 М соляной кислоты. Этот раствор нагревают 2 часа при 60oC, затем устанавливают pH-значение реакционного раствора равным 3 с помощью 18 мл 1 М раствора гидроксида натрия и растворитель удаляют на роторном испарителе. Остаток обрабатывают этилацетатом и выделившийся осадок отфильтровывают. Фильтрат концентрируют в вакууме и остаток очищают путем хроматографии на силикагеле (размер зерен силикагеля: 35-70 мкм; элюирующая система: этилацетат/метанол/вода/ледяная уксусная кислота = 4:1:1:0,5 ). Получают 2,5 г соединения 19 в виде бесцветного аморфного твердого вещества.
Таким образом синтезируют следующие соединения формулы (I):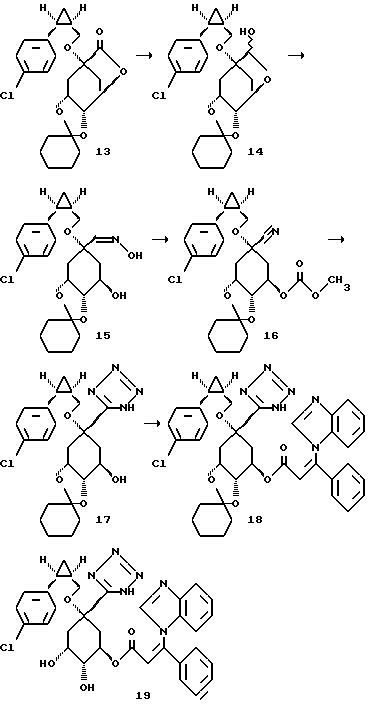
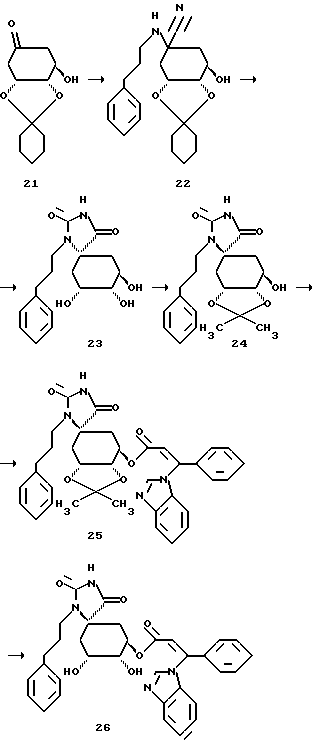
Получение соединения 22 из соединения 21.
2,26 г (0,01 моля) известного из литературы кетона формулы 21 (см. F.C. Barrier и др. , Helv. Chim Acta, 66, 296 (1983)) и 4,29 г (0,025 моля) 3-фенилпропиламингидрохлорида, в атмосфере аргона, вносят в 5 мл метанола и 3 мл дистиллированной воды. Смесь охлаждают до 0oC и прикапывают раствор из 1,63 г (0,025 моля) цианида калия в 4 мл дистиллированной воды. Реакционную смесь перемешивают 4 часа при 0oC и 1 час при комнатной температуре и затем при перемешивании вносят в смесь воды со льдом и трижды экстрагируют этилацетатом. Объединенные органические фазы трижды промывают дистиллированной водой и один раз насыщенным раствором хлорида натрия, сушат над сульфатом магния и концентрируют в вакууме. Получают 5,0 г сырого продукта формулы 22, который без дальнейшей очистки используют в следующей стадии.
Получение соединения 23 из соединения 22.
3,7 г (0,01 моля) цианосоединения формулы 22 растворяют в 8 мл ледяной уксусной кислоты и при перемешивании и при комнатной температуре смешивают с раствором 1,62 г (0,02 моля) цианата калия в 4 мл дистиллированной воды. Реакционный раствор перемешивают в течение 75 минут при комнатной температуре и затем выливают на смесь воды со льдом, экстрагируют дважды этилацетатом и объединенные органические фазы промывают один раз дистиллированной водой и один раз насыщенным раствором хлорида натрия. После высушивания органической фазы над сульфатом магния, ее концентрируют в вакууме, таким образом полученный маслянистый остаток растворяют в 4 мл диоксана и этот раствор при перемешивании смешивают с 10 мл 2М соляной кислоты. После перемешивания в течение 1 часа при 55oC, реакционную смесь выливают в смесь воды со льдом и экстрагируют трижды этилацетатом. Органические фазы промывают трижды водой и один раз насыщенным раствором хлорида натрия, сушат над сульфатом магния и концентрируют в вакууме. Маслянистый остаток очищают с помощью хроматографии на силикагеле (размер зерен силикагеля 35-70 мкм; элюирующее средство: этилацетат/н-гептан/метанол/ледяная уксусная кислота = 20:10:2: 1) и получают 0,36 г продукта формулы 23.
Получение соединения 24 из соединения 23.
0,36 г (0,00106 моля) соединения 23 растворяют в 1,09 мл (0,0106 моля) диметоксипропана и 20 мл безводного дихлорметана. Добавляют 26 мг (10 мол. %) пиридин-пара-толуолсульфоната. Смесь нагревают 45 минут при 40oC. Затем реакционный раствор вносят в насыщенный раствор гидросульфата натрия, экстрагируют этилацетатом и высушивают объединенные органические фазы над сульфатом магния. Остаток очищают путем хроматографии на силикагеле (размер зерен силикагеля: 35-70 мкм, элюирующая система: этилацетат/н-гептан = 2:1) и получают 0,24 г в виде бесцветного твердого вещества.
Получение соединения 25 из соединения 24.
230 мг (0,0006 моля) гидрокси-соединения формулы 24 растворяют в 10 мл безводного диметилформамида и при комнатной температуре и в атмосфере аргона добавляют 55 мг (0,00184 моля) гидрида натрия (80%-ная дисперсия в масле). Спустя 30 минут выдерживания при комнатной температуре прикапывают 22 мл 0,5 М раствора соединения B в диметилформамиде. Спустя следующие 30 минут выдерживания при этой температуре получают прозрачный раствор, который смешивают с насыщенным раствором хлорида аммония, причем осаждается продукт формулы 25 в виде аморфного твердого вещества. Его отсасывают и высушивают в вакууме. Получают 310 мг соединения 25.
Получение соединения 26 из соединения 25.
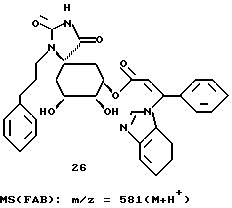
290 мг (0,00047 моля) соединения 25 растворяют в 30 мл диоксана и при комнатной температуре и интенсивном перемешивании смешивают с 4 мл (0,008 моля) 2М соляной кислоты. После перемешивания в течение 2-х часов при 50oC реакционный раствор охлаждают до 10 - 20oC и с помощью 1 М раствора гидроксида натрия устанавливают в нем pH 3. Раствор концентрируют в вакууме и маслянистый остаток обрабатывают изопропанолом, отфильтровывают осадок соли и фильтрат снова концентрируют в вакууме. Остаток перемешивают с простым метил-трет-бутиловым эфиром и аморфный осадок отсасывают. После высушивания в вакууме получают 140 мг соединения 26 (целевой продукт формулы (I)).
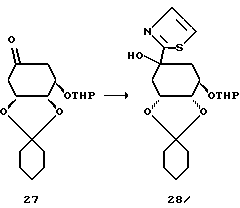
Получение соединения 28 из соединения 27.
26,4 г (14,5 мл; 0,161 моля) 2-бромтиазола, в атмосфере аргона, растворяют в 500 мл безводного диэтилового эфира и при -78oC прикапывают 107,5 мл н-бутиллития в гексане (1,5 М раствор). Перемешивают 30 минут при -76oC и затем прикапывают раствор 25,0 г (0,081 моля) кетона формулы 27 (см. европейскую заявку на патент N 92114260.8, формульная схема 4, соединение 23В) в 50 мл безводного тетрагидрофурана. Реакционный раствор оставляют нагреваться до -30oC в течение 30 минут. После этого реакционный раствор вносят в раствор хлорида аммония, экстрагируют этилацетатом и объединенные органические фазы промывают насыщенным раствором хлорида натрия и высушивают над сульфатом натрия. Органическую фазу концентрируют в вакууме и остаток очищают путем хроматографии на силикагеле (элюирующее средство: этилацетат/гептан = 1: 2; размер зерен: 35-70 мкм). Получают 23,3 г (77%) соединения 28 в виде вязкого масла.
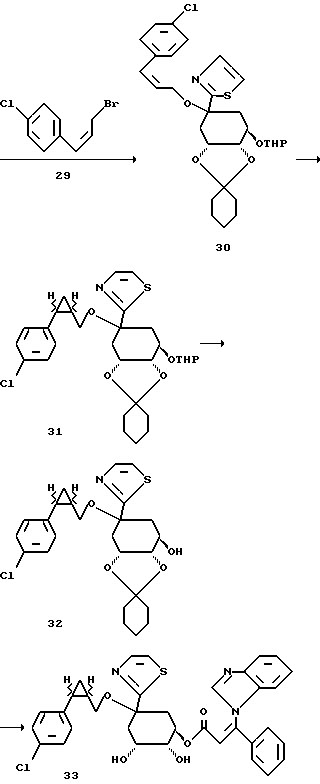
Получение соединения 30 из соединения 28.
15,0 г (0,038 моля) спирта формулы 28 растворяют в 250 мл безводного диметилформамида и при 0 - 10oC добавляют 1,5 г (0,05 моля) гидрида натрия. Перемешивают 1,5 часа при 20oC, затем охлаждают до 0oC и прикапывают 13,2 г (0,057 моля) цис-3-(4-хлорфенил)-пропенилбромида (29), растворенные в 30 мл безводного диметилформамида. Реакционный раствор оставляют нагреваться до комнатной температуры и перемешивают 2 часа при этой температуре. Затем реакционный раствор добавляют в насыщенный раствор хлорида аммония, экстрагируют этилацетатом и объединенные органические фазы промывают насыщенным раствором хлорида натрия. После высушивания над сульфатом натрия, органическую фазу концентрируют в вакууме и очищают путем хроматографии на силикагеле (элюирующее средство: этилацетат/н-гептан = 1:2; размер зерен: 35-70 мкм). Получают 19,0 г тиазола формулы 30 в виде вязкого масла.
Получение соединения 31 из соединения 30.
56,6 мл 1 М раствора диэтилцинка в толуоле при 0oC и в атмосфере аргона прикапывают к 250 мл безводного дихлорметана и затем при 0oC добавляют 9,0 мл (0,125 моля) хлор-йод-метана. Реакционный раствор перемешивают 30 минут при той же температуре и после этого прикапывают 17,0 г (0,031 моля) олефина формулы 30, растворенные в 30 мл безводного дихлорэтана. Оставляют медленно нагреваться до комнатной температуры. Спустя 2 часа реакционный раствор вносят в насыщенный раствор хлорида аммония, экстрагируют этилацетатом и объединенные органические фазы промывают насыщенным раствором хлорида натрия. После высушивания органической фазы над сульфатом натрия концентрируют ее в вакууме и остаток перемешивают с простым метил-трет-бутиловым эфиром. Осадок отфильтровывают (в качестве побочной реакции происходит метилирование азота в тиазольном кольце) и фильтрат снова концентрируют. Получают 4,2 г (24%) соединения 31 в виде вязкого масла.
Получение соединения 32 из соединения 31.
4,2 г (0,008 моля) соединения 31 растворяют в 100 мл метанола и 150 мл дихлорметана и при комнатной температуре добавляют 0,7 г (0,003 моля) пиридиний-п-толуолсульфоната. Прозрачный раствор выдерживают 14 часов при комнатной температуре, смешивают с 20 мл 1 н раствора гидрокарбоната натрия и эту смесь концентрируют до тех пор, пока остается только водная фаза. Ее экстрагируют этилацетатом, и объединенные органические фазы промывают насыщенным раствором хлорида натрия, высушивают над сульфатом натрия, и концентрируют в вакууме. Остаток очищают путем хроматографии на силикагеле (элюирующее средство: этилацетат/н-гептан = 1:1; размер зерен: 35-70 мкм). Получают 1,82 г (51%) соединения 32 в виде бесцветного масла.
Получение соединения 33 из соединения 32.
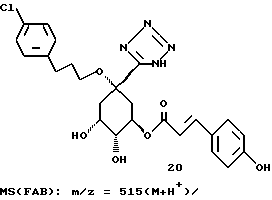
Аналогично описанному в примере 2 получению соединения 19 из соединения 17, из соединения 32 через 2 стадии получают соединение 33, соответствующее формуле (I), в виде аморфного твердого вещества. Масс-спектр (FAB): m/z = 542 (M + H+).
Описываются новые сложные эфиры циклогексанола формулы I, где значения Х, Y, Z, R1 - R6 указаны в п.1 формулы. Соединения фармакологически эффективны и поэтому могут применяться в качестве лекарственного средства, в особенности для лечения диабета и других заболеваний, которые характеризуются повышенным выделением глюкозы печенью или повышенной активностью глюкозо-6-фосфатаза-системы. 2 с. и 4 з.п.ф-лы.
где R1 обозначает CDNHSO2R14, где R14 является С1 - C10 - алкилом, бензтиазолилом, замещенным С1 - C4 - алкоксилом, фенилом, незамещенным или замещенным фтором и/или нитрогруппой, нафтилом, незамещенным или замещенным NR8R9 - группой, тетразолилом, тиазолилом,
или группы формулы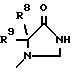
где R8 и R9 обозначают водород или С1 - C4 - алкил;
R2 обозначает О-С1-C10 - алкил(R11); R11 является С3 - циклоалкилом, С1 - C10 - алкилом, который в свою очередь может быть замещен фенилом или хлорфенилом;
n = 0, 1, или R1 и R2 образуют кольцо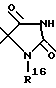
где R16 обозначает C1-C10 - алкил (R11)n; R3 обозначает бензимидазолил; Z обозначает CH=C(R13), где R13 - фенил, незамещенный или замещенный OH;
R4, R5, R6 обозначает водород, ОН, причем R4, R5, R6 являются одинаковыми или разными;
Y является -О-;
X обозначает (CH2)m, где m = 0,
а также их физиологически приемлемые соли.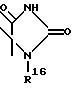
3. Соединения по п.1, обладающие ингибирующей активностью в отношении фермента глюкоза-6-фосфатазы.
| СПОСОБ ИСПЫТАНИЯ МАТЕРИАЛОВ И СОЕДИНЕНИЙ НА ПРОЧНОСТЬ | 0 |
|
SU381959A1 |
| US 5015653 A, 14.05.91 | |||
| УСТРОЙСТВО ДЛЯ ИСПЫТАНИЯ БУТЫЛОК НА РАЗРЫВ | 0 |
|
SU180838A1 |
| Способ получения оксотиазолидиновых соединений | 1986 |
|
SU1545941A3 |

