Клиническая картина диабета характеризуется повышенными содержаниями сахара в крови. В случае инсулинзависимого или типа I диабета причиной является отмирание продуцирующих инсулин β- клеток поджелудочной железы; поэтому лечение осуществляют путем приема инсулина (заместительная терапия). Напротив, не зависящий от инсулина или тип II диабета характеризуется уменьшенным действием инсулина на мышечную и жировую ткань (резистентность к инсулину) и повышенной выработкой глюкозы печенью. Причины этих нарушений обмена веществ еще далеко не ясны. Известная терапия сульфонилмочевинами пытается компенсировать резистентность к инсулину за счет повышения аутогенного выделения инсулина, однако это не во всех случаях ведет к нормализации уровня сахара в крови и не может остановить распространение болезни; многие диабетики типа II в конце концов в результате "истощения" β- клеток становятся инсулинзависимыми и позже страдают такими недугами, как катаракты, нефропатии и ангиопатии.
Поэтому желательны новые терапевтические принципы лечения диабета типа II.
Натощак концентрация глюкозы в крови определяется выработкой глюкозы печенью. Различные группы исследователей показали, что повышение содержания сахара в крови при диабете типа II коррелирует с пропорционально повышенным выделением глюкозы из печени. Глюкоза, выделяемая печенью в кровь, может образовываться как в результате разложения гликогена печени (гликогенолиз), так и также путем гликонеогенеза.
Глюкозо-6-фосфат является общим конечным продуктом как глюконеогенеза, так и гликогенолиза. Конечная стадия выделения печенью глюкозо-6-фосфата катализируется глюкозо-6-фосфатазой (EC 3.1.3.9). Глюкозо-6-фосфатаза представляет собой многоферментный комплекс, встречающийся в эндоплазматической сетке (ER). Этот ферментный комплекс состоит из находящейся в ER-мембране глюкозо-6-фосфаттранслоказы, локализованной на люминальной стороне эндоплазматической сетки глюкозо-6-фосфатазы и фосфаттранслоказы [см. J.Ashmore и G.Weber "Роль гепатитной глюкозо-6-фосфатазы в регулировании углеводного метаболизма" в книге "Витамины и гормоны", т. XVII, (ред. Harris R.S., Marrian G. F. , Thimann K.V.) 92-132 (1959); Burchell A., Waddell I.D. "Молекулярные основы гепатитной микросомальной глюкозо-6-фосфатазной системы", Biochim. Biophys. Acta, 1092, 129-137 (1990)]. Существующая обширная литература показывает, что при всех исследованных условиях, приводящих в опытах на животных к повышенным содержаниям глюкозы в крови, например, как при использовании стрептозоцина, аллоксана, кортизона, тироидных гормонов и в случае голодовок, активность этого многоферментного комплекса также повышается. Сверх того, многочисленные исследования указывают на то, что наблюдаемое у диабетиков типа II повышенное выделение глюкозы связано с повышенной активностью глюкозо-6-фосфатазы. Значение глюкозо-6-фосфатазной системы для нормального глюкозного гомеостаза подчеркивается далее гипогликемическими симптомами у пациентов с заболеванием типа Iб, связанным с накоплением гликогена, при котором отсутствует транслоказная компонента глюкозо-6-фосфатной системы.
Уменьшение активности глюкозо-6-фосфатазы благодаря пригодным активным веществам (ингибиторам) должно приводить к соответственно уменьшенному гепатитному выделению глюкозы. Эти биологически активные вещества должны обладать способностью адаптировать выделение глюкозы печенью к эффективному периферическому расходу. Снижающееся благодаря этому в соответствии натощак у диабетиков типа II содержание глюкозы в крови, кроме того, должно оказывать также превентивное действие в отношении наносимого позднее диабетом вреда.
В литературе описан ряд неспецифических ингибиторов глюкозо-6-фосфатазы, как, например, флорхизин [Soodsma J.F., Legler B. и Nordlie R.C., J. Biol. Chem, 242, 1955-1960 (1967)], 5,5'-дитио-бис-нитробензойная кислота [Wallin B. K. и Arion W. J. , Biochem. Biophys. Res. Commun. 48, 694-699 (1972)], 2,2'-диизотиоцианатостильбен и 2-изотиоцианато-2'-ацетокси-стильбен Zoccoli M. A., Karnowski J. Biol Chem. 255, 1113-1119 (1980)]. Однако до сих пор еще нет никаких терапевтически применимых ингибиторов глюкозо-6-фосфатазной системы.
Замещенные производные циклогексана, которые более подробно описаны ниже, являются отчасти известными из химической и биологической литературы соединениями, которые могут быть выделены из многочисленных растений [R. Krasemann, Arch. Pharm. 293, 721 (1960)]. Однако о фармакологических и биохимических действиях этих сложных эфиров известно только немного. Хлорогеновая кислота, типичный представитель рассматриваемых здесь соединений, между прочим описана в качестве ингибитора липоксигеназы [M.Nishizawa и др., Chem. Pharm. Bull., 34 (3), 1419 (1986)].
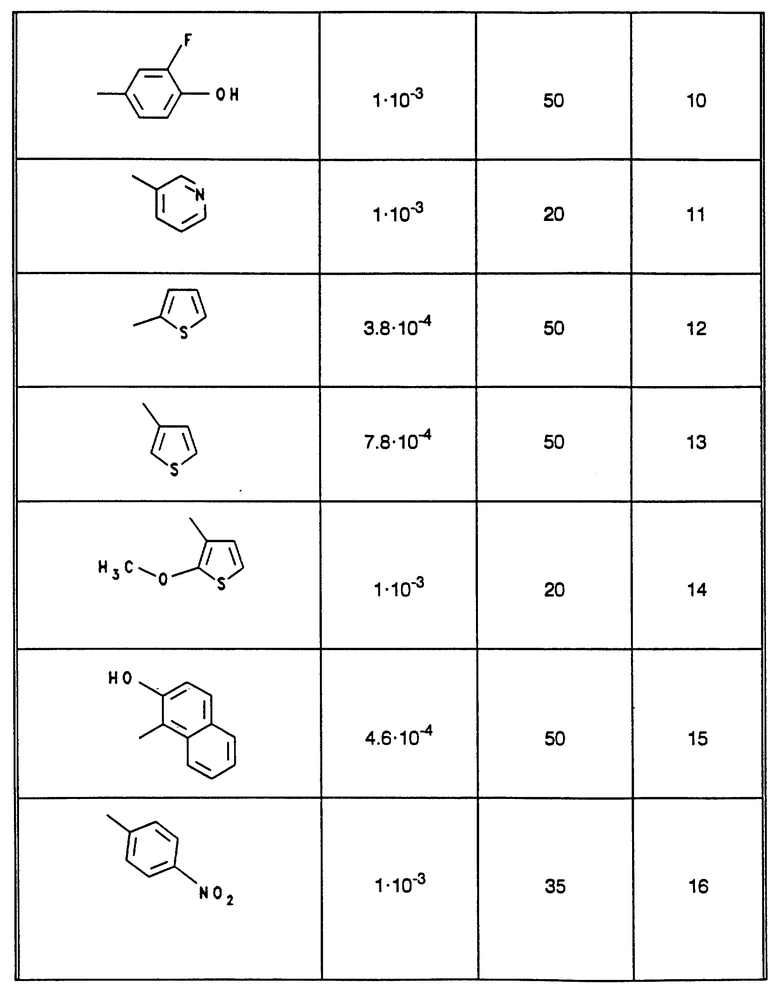
В настоящее время заявитель нашел, что определенные сложные эфиры замещенных циклогексанкарбоновых кислот, как, например, хлорогеновая кислота (N 17 из исследованных соединений), являются ингибиторами глюкозо-6-фосфатазной системы.
Поэтому изобретение относится к сложным эфирам производных циклогексана формулы I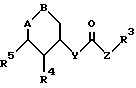
где A-B означает группу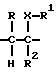
или группу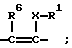
R1 означает CN, COOH, COO-(C1-C4-алкил), C1-C4-алканоил, SO3-(C1-C4-алкил), SO3H, PO(OH)2, PO(OH)(O-C1-C4-алкил) или PO(O-C1-C4-алкил)2;
R2 означает H, OH или F;
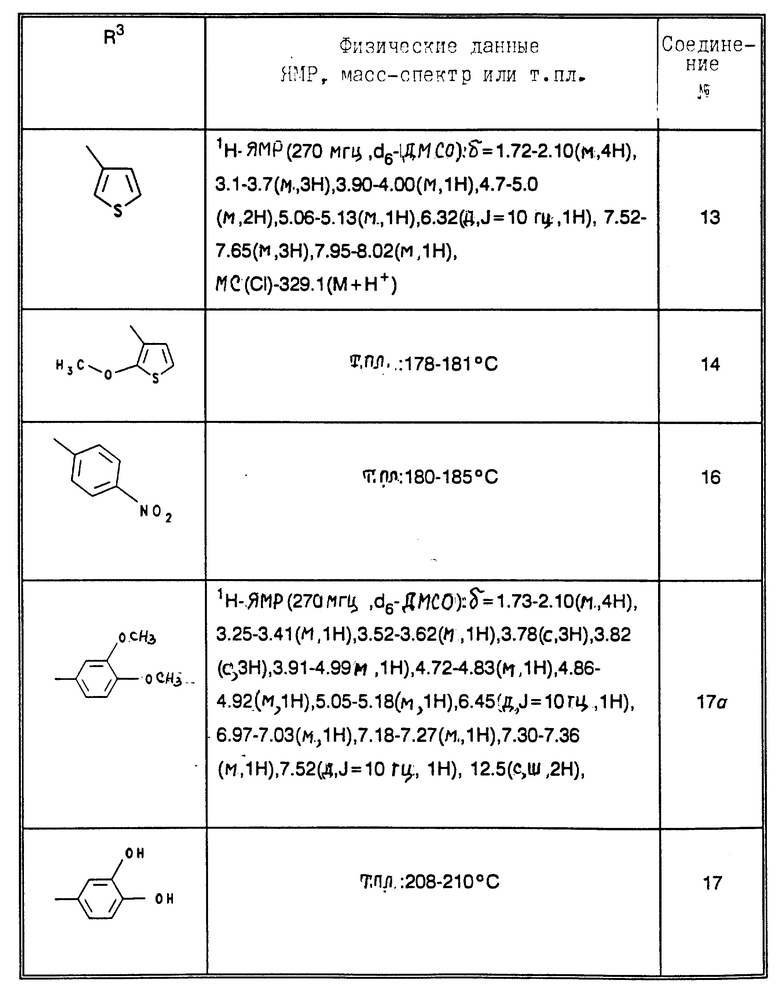
R3 означает H, фенил, нафтил, пиридил, тиенил, фурил, причем ароматический или гетероароматический остаток может быть одно- или многократно замещен фтором, хлором, бромом, иодом, OH, NO2, C1-C4-алканоилом, (C1-C4-алкоксил), C1-C4-алкилом, фенилом, фенокси, тиенилом, фурилом, пиридилом, имидазолилом или бензилокси, причем заместители являются одинаковыми или разными;
R4, R5, R6 означают H, OH, фтор, хлор, бром, C1-C4-алканоил, C1-C4-алкил, фенил, фенокси, тиенил, фурил, пиридил, имидазолил или бензилокси, причем R4, R5, R6 являются одинаковыми или разными;
X означает -(CH2)n-; -CH=CH- или -CH2OCH2-;
Y означает -(CH2)n-; O, S или NH;
Z означает -(CH2)n-; или -CH=CH- и n означает нуль, 1, 2, 3 или 4,
являющимися ингибиторами глюкозо-6-фосфатазной системы печени млекопитающих.
Предпочтительно использование таких соединений формулы (I), в которых остатки имеют следующее значение:
R1 означает COOH, COO-(C1-C4-алкил), PO(OH)2, PO(OH)(O-C1C4-алкил) или PO(O-C1-C4)-алкил)2;
R2 означает H или OH;
R3 означает H, фенил, нафтил, пиридил, тиенил, фурил, причем ароматический или гетероароматический остаток может быть одно-, двух- или трехкратно замещен фтором, хлором, бромом, иодом, NO2, OH, C1-C4-алканоилом, C1-C4-алкоксилом, C1-C4-алкилом, фенилом, фенокси, тиенилом, фурилом, пиридилом, имидазолилом или бензилокси, причем заместители являются одинаковыми или разными;
R4, R5 и R6 означает H, OH, фтор, хлор, бром, C1-C4-алканоил, C1-C4-алкил, фенил, тиенил, фурил, пиридил, имидазолил или бензилокси, причем R4, R5, R6 являются одинаковыми или разными;
X означает -(CH2)n-; -CH=CH- или -CH2OCH2-;
Y означает -(CH2)n-; O, S или NH;
Z означает -(CH2)n- или -CH=CH- и
n = 0, 1, 2, 3 или 4.
Предпочтительно применение таких соединений формулы (I), в которых остатки имеют следующее значение:
R1 означает COOH или COO-(C1-C4-алкил);
R2 означает H или OH;
R3 означает H, фенил, нафтил, пиридил, тиенил, фурил, причем ароматический или гетероароматический остаток может быть одно-, двух- или трехкратно замещен фтором, хлором, OH, NO2, C1-C4-алканоилом, C1-C4-алкилом, фенилом, фенилокси или бензилокси, причем заместители являются одинаковыми или разными;
R4, R5, R6 означают H или OH, причем R4, R5, R6 являются одинаковыми или разными;
X означает -(CH2)n- и
n = 0, 1 или 2;
Y означает кислород или NH;
Z означает -(CH2)n-, с
n = 0 или 2,
или -CH=CH-.
Имеющиеся в соединениях формулы (I) алкильные, алкоксильные и алканоильные остатки являются линейными или разветвленными.
Далее изобретение относится к применению соединений формулы (I) для лечения заболеваний, связанных с повышенной активностью глюкозо-6-фосфатазной системы.
Изобретение также относится к применению соединений формулы (I) для лечения заболеваний, связанных с повышением выделением глюкозы печенью.
Кроме того, изобретение относится к применению соединений формулы (I) для лечения диабета типа II (инсулиннезависимого или старческого диабета).
Далее изобретение охватывает применение соединений формулы (I) для приготовления лекарств для лечения диабета и других заболеваний, характеризующихся повышенным выделением глюкозы из печени или повышенной активностью глюкозо-6-фосфатазной системы.
Действие соединений согласно изобретению на глюкозо-6-фосфатазную систему исследовалось в ферментном тесте на микросомах печени.
Для приготовления микросомной фракции, содержащей глюкозо-6-фосфатазу, используют свежую печень самцов крыс Wistar и обрабатывают как описано в литературе [Canfield W.K. и Arion W.J., J. Biol.Chem. 263, 7458-7460 (1988)] . Эта микросомная фракция может храниться при -70oC по крайней мере 2 месяца без значительной потери активности.
Обнаружение глюкозо-6-фосфатазной активности, как указано в литературе (Arion W.J. в Methods Enzymol 174, Acаdemic Press, 1989, с. 58 - 67), осуществляют путем определения фосфата, высвобождаемого из глюкозо-6-фосфата. 0.1 мл испытуемой смеси содержит глюкозо-6-фосфат [1 ммоль/л], испытуемое вещество 0.1 г микросомной фракции и 100 ммоль/л HEPES-буфера [4-(2-гидроксиэтил)пиперазин-1-этансульфокислоты], pH=7.0. Реакцию инициируют путем добавки фермента. По истечении 20 мин при комнатной температуре реакцию прекращают за счет добавления 0.2 мл фосфатного реагента. Пробу инкубируют в течение 30 мин при 37oC и затем измеряют поглощение (A) голубого цвета при 570 нм. Ингибирующую активность испытуемого вещества получают путем сравнения с контрольной реакцией, которая не содержит никакого испытуемого вещества, по формуле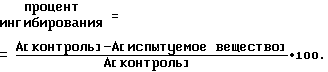
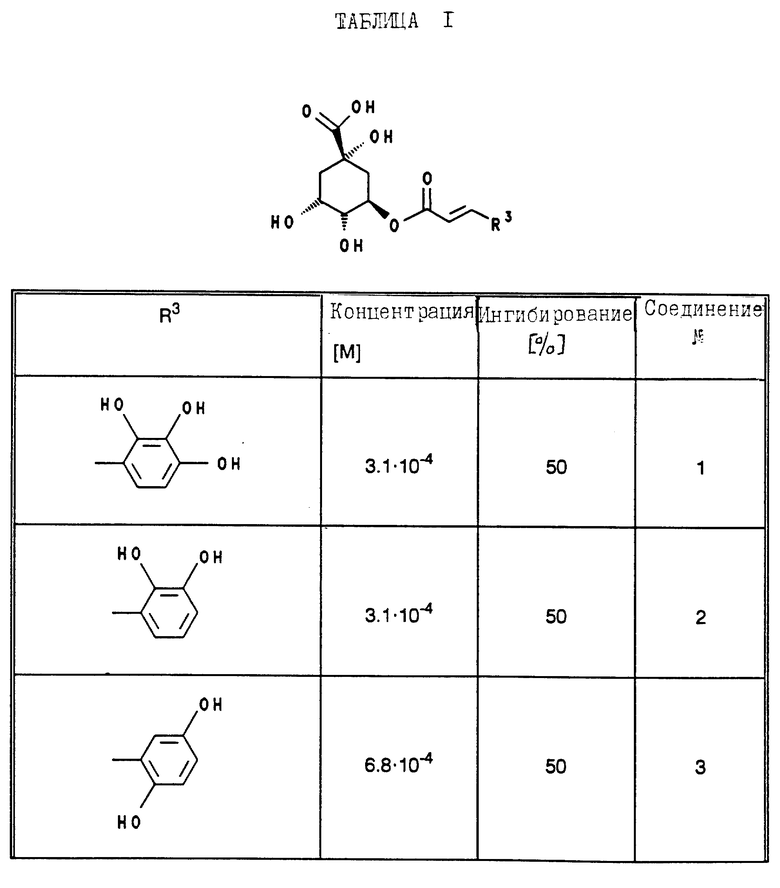
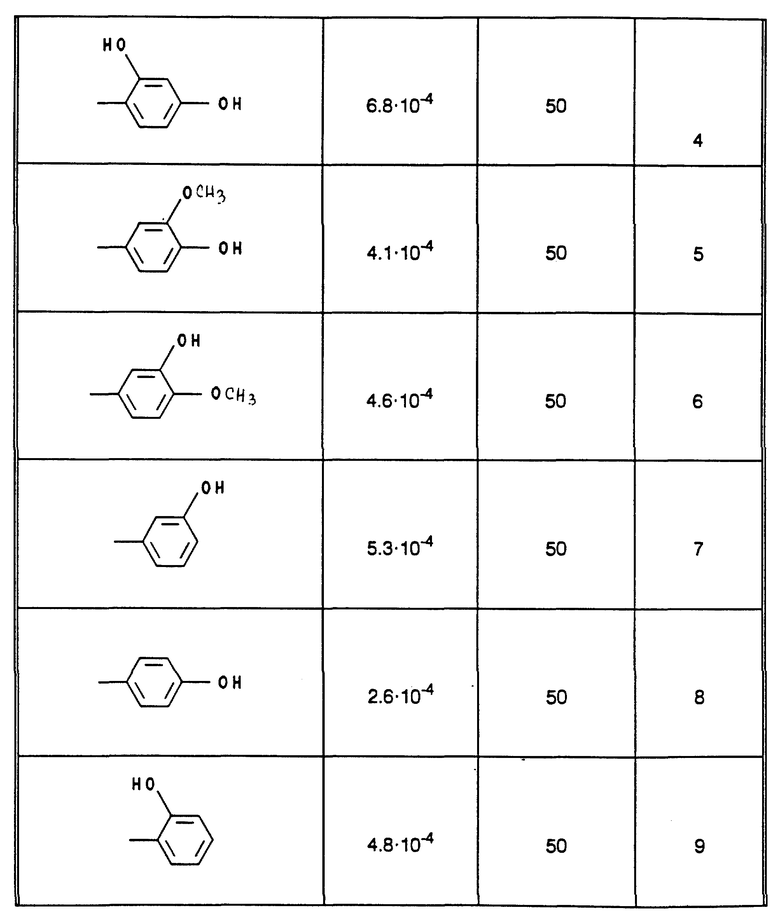
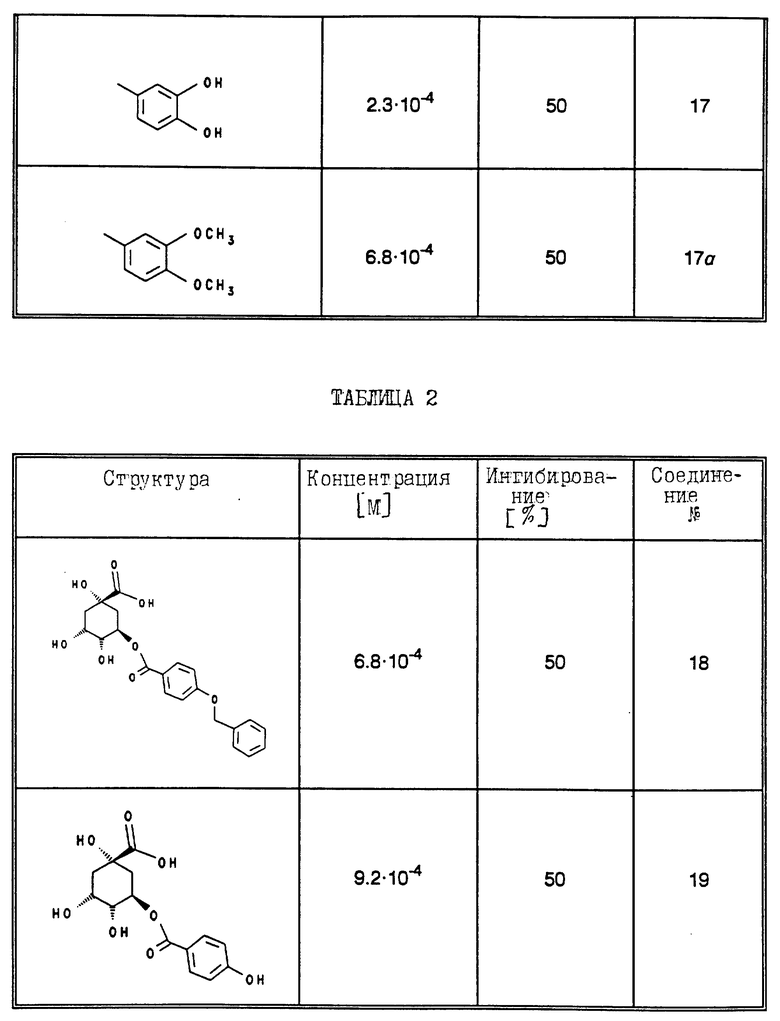
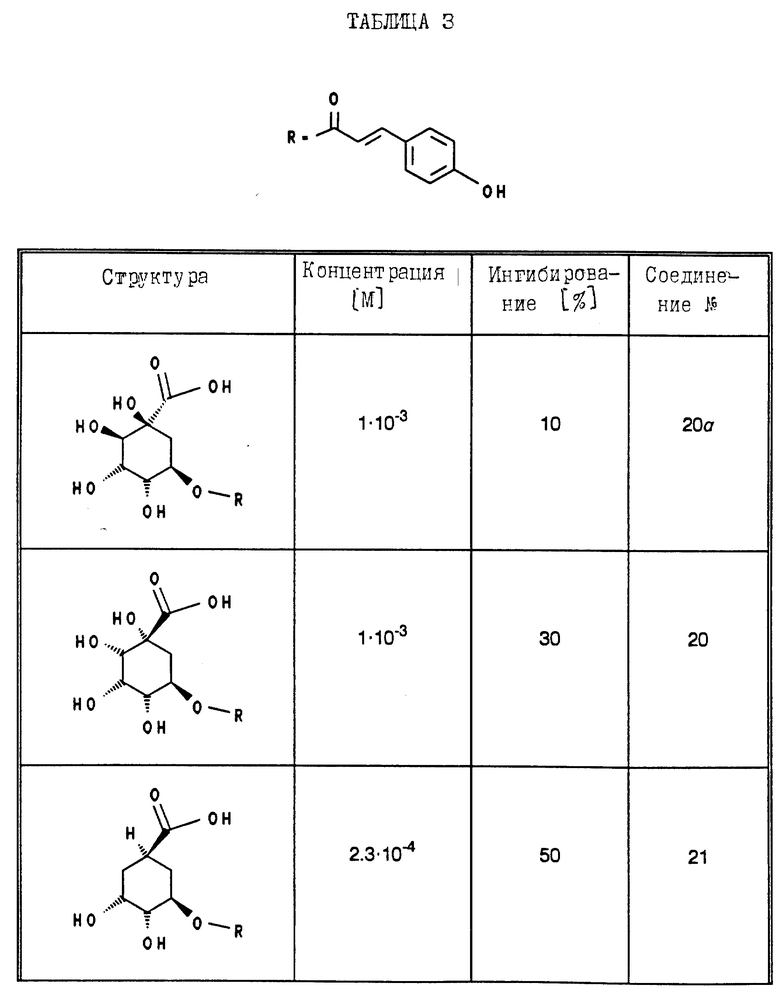
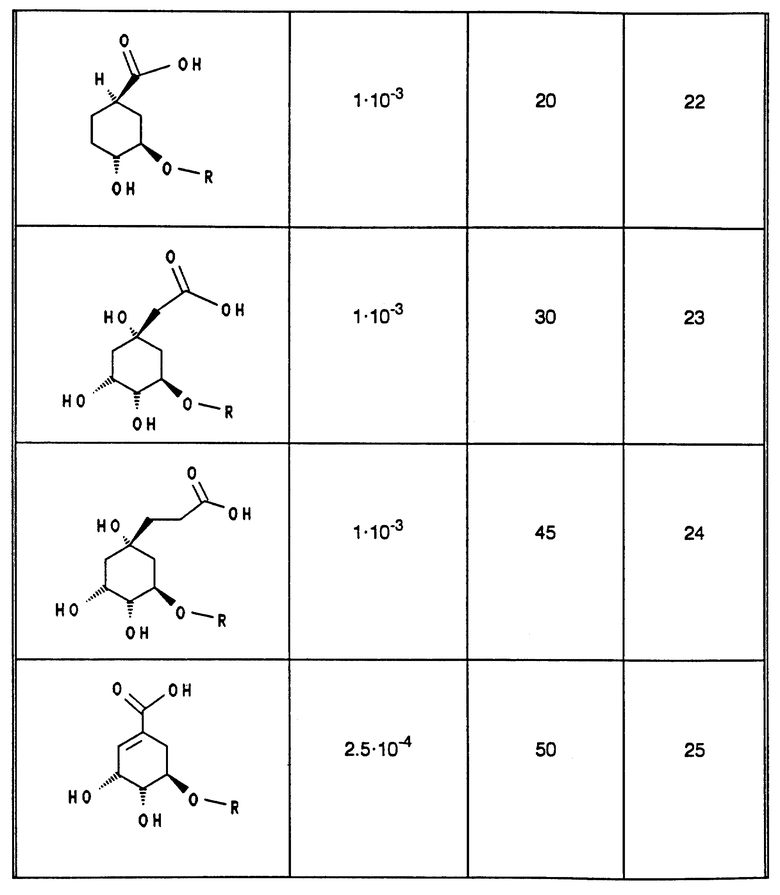
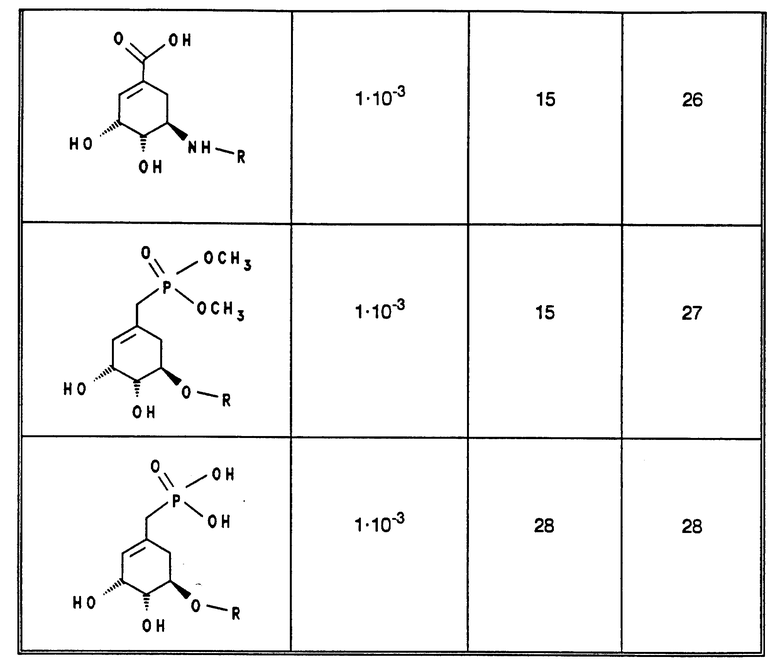
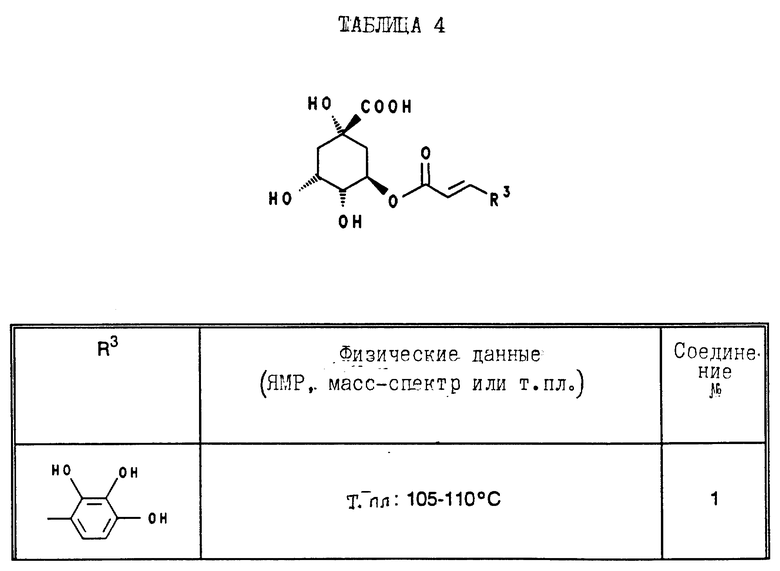
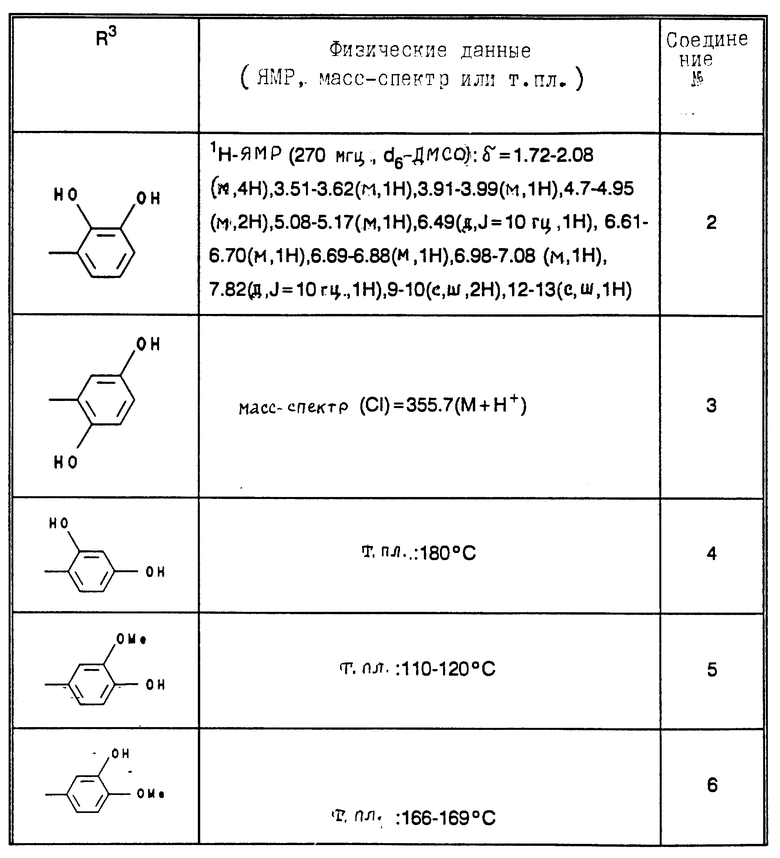
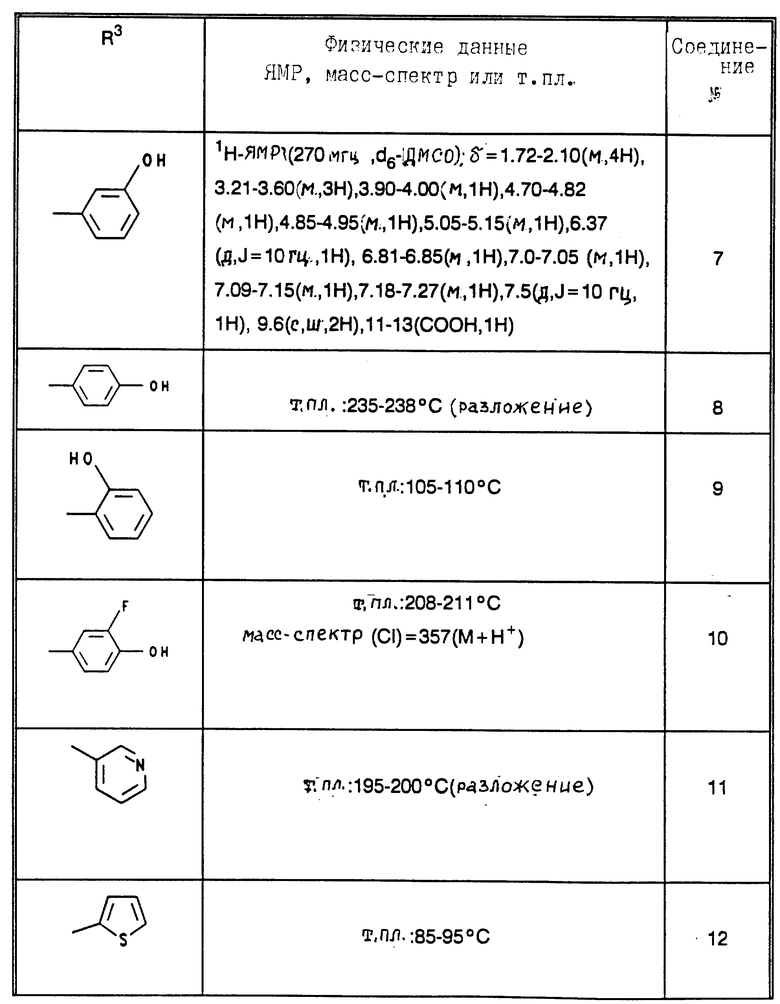
В табл. 1 - 3 (см. в конце описания) приведены в качестве примера величины ингибирования, полученные для ряда соединений формулы (I). Исследованные соединения частично известны из литературы. Получение описано в примерах осуществления.
В лекарствах согласно настоящему изобретению, получаемых обычными способами, кроме соединений формулы (I) могут содержаться также фармакологически приемлемые добавки, такие как разбавители и/или носители. Под этим нужно понимать физиологически приемлемые вещества, которые после смешения с биологически активным веществом переводят его в пригодную для приема форму. Предпочтительно оральное введение.
Пригодными твердыми и жидкими галеновыми формами композиций являются, например, таблетки, драже, порошки, капсулы, сиропы, эмульсии, суспензии, капли, а также препараты с пролонгированным высвобождением биологически активного вещества. В качестве часто используемого носителя, соответственно разбавителя, следует назвать, например, различные сахара и виды крахмалов, производные целлюлозы, карбонат магния, желатину, животные и растительные масла, полиэтиленгликоли, воду или другие пригодные растворители, a также содержащие воду буферы, которые за счет добавления солей можно делать изотоническими. Кроме того, в случае необходимости могут найти применение поверхностно-активные вещества, красители и вкусовые вещества, стабилизаторы, а также консерванты в качестве других добавок в лекарственные композиции согласно изобретению.
Препараты преимущественно можно получать в виде дозировочных единиц, особенно в виде таблеток и капсул. Каждая дозировочная единица, особенно для орального применения, может содержать до 500 мг, предпочтительно, однако, от 10 до 200 мг, активнoй составной части. Однако также можно использовать лежащие выше или ниже этих значений дозировочные единицы, которые в случае необходимости перед приемом можно делить. При необходимости дозировочные единицы для орального приема можно микрокапсулировать, для того чтобы, например, замедлить выделение. Контролируемого выделения достигают, например, также путем покрытия или внедрения частицеобразного материала в пригодные полимеры, воски или т.п.
Исследуемые соединение синтезируют как описано ниже.
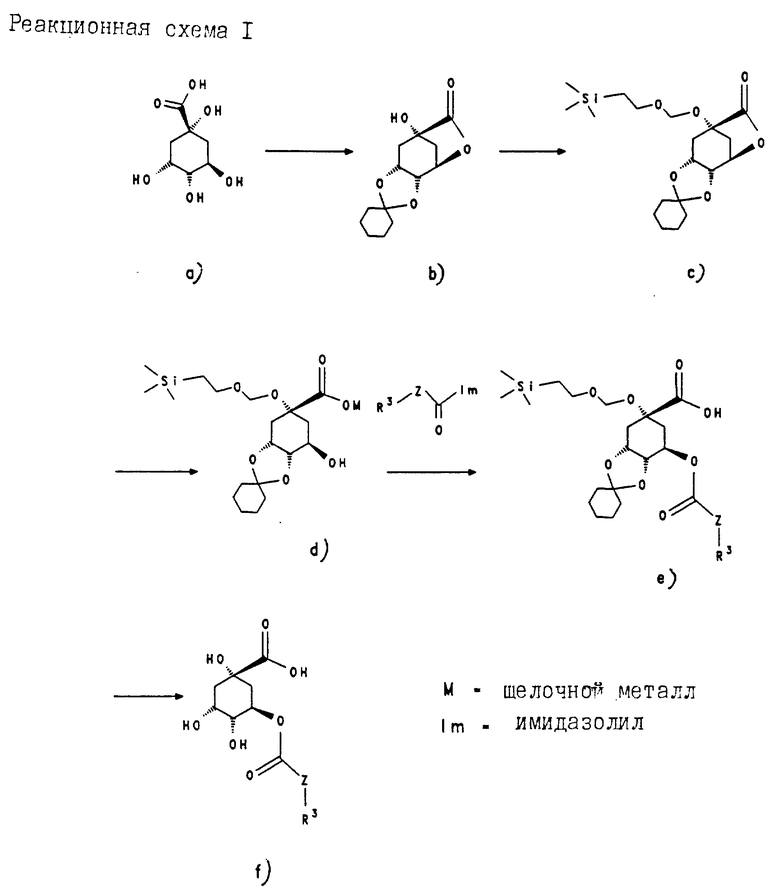
Получение соединения (b) из соединения (a) (соединения a-f см. в конце описания. Реакционная схема 1).
163.3 г (0.85 моль) соединения (a) [Fischer, Dangschat, Chem. Ber. 65, 1009 (1932)] суспендируют в 186 мл (1.8 моль) циклогексанона и добавляют 0.5 мл концентрированной серной кислоты. Затем медленно нагревают до температуры нагревательной бани 200oC и отгоняют азеотроп воды с циклогексаноном. После прекращения отгонки азеотропа светло-коричневый реакционный раствор перемешивают еще 2 ч при температуре бани 200oC. Затем реакционный раствор охлаждают до 70oC и добавляют к нему 10 г гидрокарбоната натрия. После этого смешивают с 700 мл этилацетата, органическую фазу промывают водой насыщенным раствором хлорида натрия. Затем органическую фазу концентрируют в вакууме. Светло-желтый остаток кристаллизуют из смеси изопропанола с водой (1:1) и получают 142.1 (75%) лактона (b) в виде бесцветных кристаллов; т.пл. 140-141oC.
Получение соединения (c) из соединения (b):
38.14 г (0.15 моль) гидроксилактона (b) растворяют в 180 мл дихлорметана. Добавляют 53.0 мл (0.3 моль) диизопропилэтиламина. К этому раствору при комнатной температуре прикапывают 45.0 мл (0,254 моль) триметилсилилэтилоксиметилхлорида и перемешивают 6 ч при температуре кипения с обратным холодильником. Затем реакционный раствор вносят в насыщенный раствор хлорида аммония и экстрагируют этилацетатом. Объединенные органические фазы экстрагируют охлажденным примерно до 6oC 1 н раствором гидросульфата калия и сушат над сульфатом натрия. После концентрирования в вакууме получают светло-желтый остаток, который кристаллизуют из смеси гептана с этилацетатом (6 : 1). Получают 57.0 г (98%) соединения (c); т.пл. 100-102oC.
Получение соединения (d) из соединения (c).
1.38 г (3.6 моль) соединения (с) растворяют в 8 мл диоксана. После добавления 0.4 мл воды прикапывают при комнатной температуре 3.8 мл 1 н раствора гидроксида натрия. Реакционную смесь перемешивают в течение 2 ч и затем концентрируют в вакууме. Получают 1.3 г (85%) соединения (d) в виде аморфного твердого вещества.
1H-ЯМР (270 мГц, d6-ДМСО) δ, м.д. = 0.01 (с., 9H); 0.72-0.89 (м., 2H), 1.21-1.62 (м. , 10H); 1.65-1.78 (м, 1H); 1.82-1.92 (м., 1H); 1.94-2.08 (м., 2H); 3.38-3.63 (м, 3H); 3.82-3.88 (м., 1H); 4.18-4.47 (м., 2H); 7.80-7.90 (м., 1H).
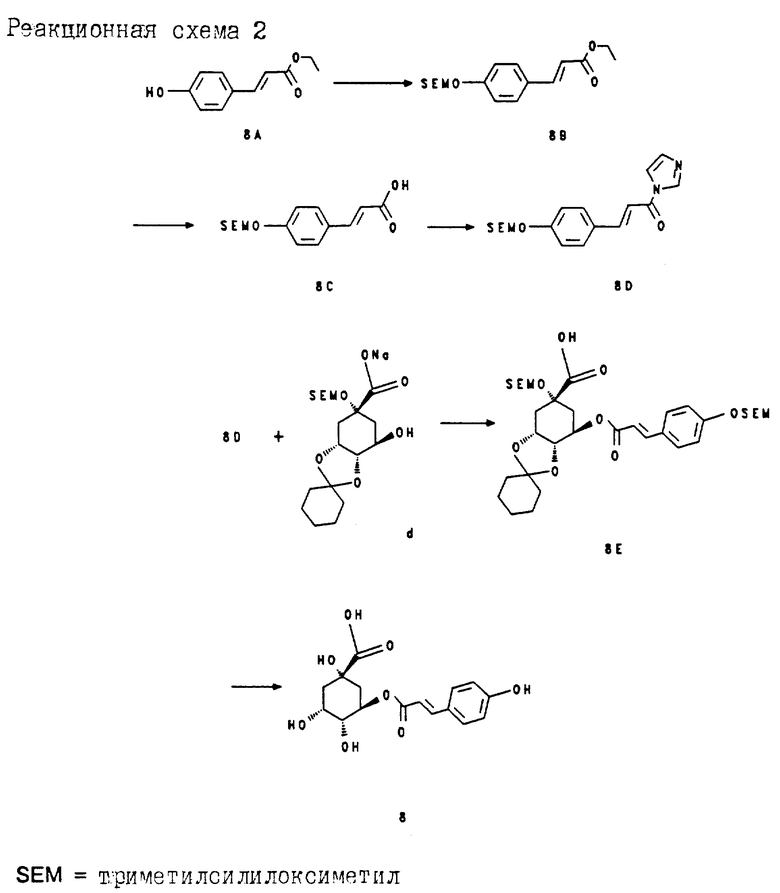
Стадии (d), (e) и (f) описывают на примере получения соединения 8.
Получение соединения 8C (из соединения 8A через соединение 8B) (соединения 8A-8E cм. в конце описания. Реакционная схема 2).
10.0 г (0.052 моль) сложного эфира п-гидроксикоричной кислоты (8A) растворяют в 60 мл безводного дихлорметана. Добавляют 27 мл (0,156 моль) диизопропилэтиламина и при комнатной температуре и в атмосфере аргона прикапывают 19.5 мл (0.11 моль) триметилсилилэтоксиметилхлорида. Перемешивают 4 ч при комнатной температуре и затем реакционный раствор выливают в охлажденный льдом раствор хлорида аммония. Экстрагируют этилацетатом, объединенные органические фазы промывают последовательно охлажденным льдом 1 н раствором гидросульфата калия и насыщенным раствором хлорида натрия. После высушивания органической фазы над сульфатом натрия концентрируют в вакууме. Получают 16.8 г простого эфира 8B, который без дальнейшей очистки растворяют в 600 мл диоксана и при комнатной температуре смешивают со 160 мл (0.8 моль) 5 н раствора гидроксида натрия. Спустя 24 ч метанол отгоняют в вакууме и водную суспензию натриевой соли соединения 8C подкисляют 2 н соляной кислотой до pH 4. Кислота 8C осаждается почти количественно и ее можно отсасывать и промывать водой. Получают 16.02 г соединения 8C; т.пл. 93-96oC.
Получение соединения 8E из соединений 8C и (d) (соответствует стадии (e) на схеме 1).
a) 7.95 г (27 ммоль) соединения 8C растворяют в 35 мл безводного диметилформамида. При комнатной температуре прикапывают раствор 4.54 (27 ммоль) карбонилдиимидазола в 35 мл безводного диметилформамида. Затем этот раствор нагревают в течение 1 ч при 60-70oC, причем можно наблюдать выделение CO2.
б) К раствору 8.92 г (0.021 моль) натриевой соли соединения (d) в 50 мл безводного диметилформамида добавляют при комнатной температуре и в атмосфере аргона 0.75 г (0.025 моль) 80%-ного гидрида натрия. Эту суспензию перемешивают в течение 1 ч при комнатной температуре и затем добавляют к ней при 0-5oC полученный согласно п.(а) раствор имидазолида 8D. Раствор перемешивают 2.5 ч при 0-5oC и после этого реакционную смесь выливают в насыщенный раствор хлорида аммония. Путем добавки 1 н раствора гидросульфата калия смесь подкисляют до pH 4 и экстрагируют этилацетатом. Объединенные органические фазы промывают последовательно насыщенным раствором хлорида аммония, водой и насыщенным раствором хлорида натрия. Органическую фазу сушат над сульфатом натрия, концентрируют в вакууме и маслянистый остаток хроматографируют на силикагеле (растворитель: этилацетат/н-гептан/ледяная уксусная кислота = 20 : 60 : 1). Получают 10.3 г (78%) соединения 8E в виде бесцветного масла.
1H-ЯМР (270 мГц, CDCl3): δ, м.д. = 0.02 (с., 9H). 0.05 (с., 9H); 0.91-1.03 (м. , 4H); 1.5-1.78 (м., 10H); 1.91-2.05 (м., 1H); 2.28-2.42 (м., 2H); 2.57-2.63 (м. , 1H); 3.68-3.90 (м., 4H); 4.14-4.20 (м., 1H); 4.42-4.52 (м., 1H); 4.91-4.96 (м., 1H); 5.11-5.18 (м., 1H); 5.24 (с., 2H); 5.21-5.34 (м., 1H); 6.32 (д., J = 10 Гц, 1H); 7.02-7.08 (м., 2H); 7.42-7.5 (м., 2H); 7.65 (д., J = 10 Гц, 1H); 13 (с., ш., COOH, 1H).
Получение соединения 8 из соединения 8E (соответствует стадии (f) на схеме 1).
5.02 г (7.4 ммоль) соединения 8E растворяют в 130 мл диоксана и при комнатной температуре и перемешивании смешивают с 95 мл (0.19 моль) 2 н соляной кислоты. Перемешивают в течение 20 ч при комнатной температуре. По окончании реакции в прозрачном растворе устанавливают pH 3-4 с помощью 2 н раствора гидроксида натрия и концентрируют в вакууме. Твердый остаток перемешивают в смеси этилацетата с метанолом (3 : 1) и отфильтровывают нерастворимый хлорид натрия. Фильтрат снова концентрируют и остаток хроматографируют на силикагеле (этилацетат/метанол/вода/ледяная уксусная кислота = 100/10/10/5). Получают 1.95 г (70%) соединения 8; т.пл. 235-238oC.
Приведенные в табл. 4 в качестве примеров соединения получают вышеописанным способом. При этом синтез соединений, содержащих гидроксильные группы в остатке R3 общей формулы (I), за счет соответствующих операций с защитными группами отличается от прочих синтезов, при которых эти операции не нужны.
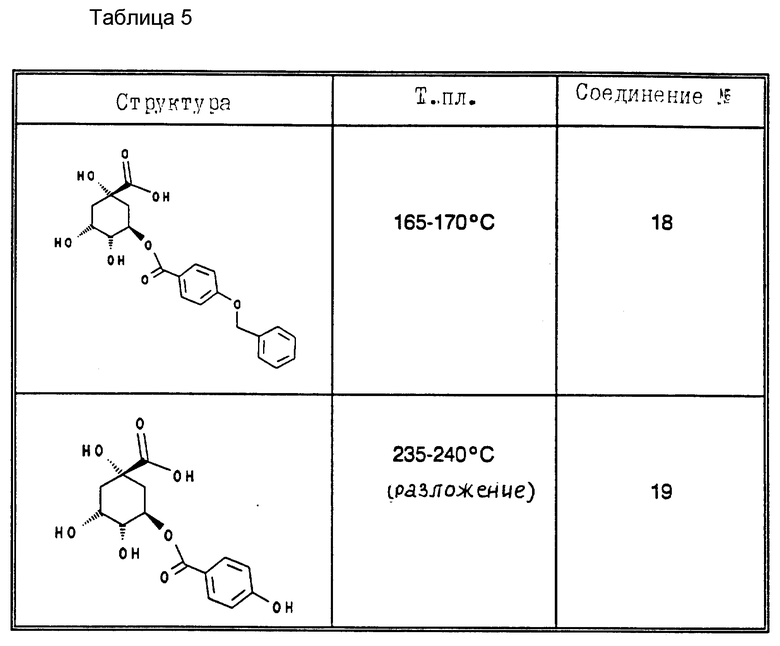
В табл. 4 и 5 (см. в конце описания) представлены физические характеристики синтезированных в качестве примеров соединений.
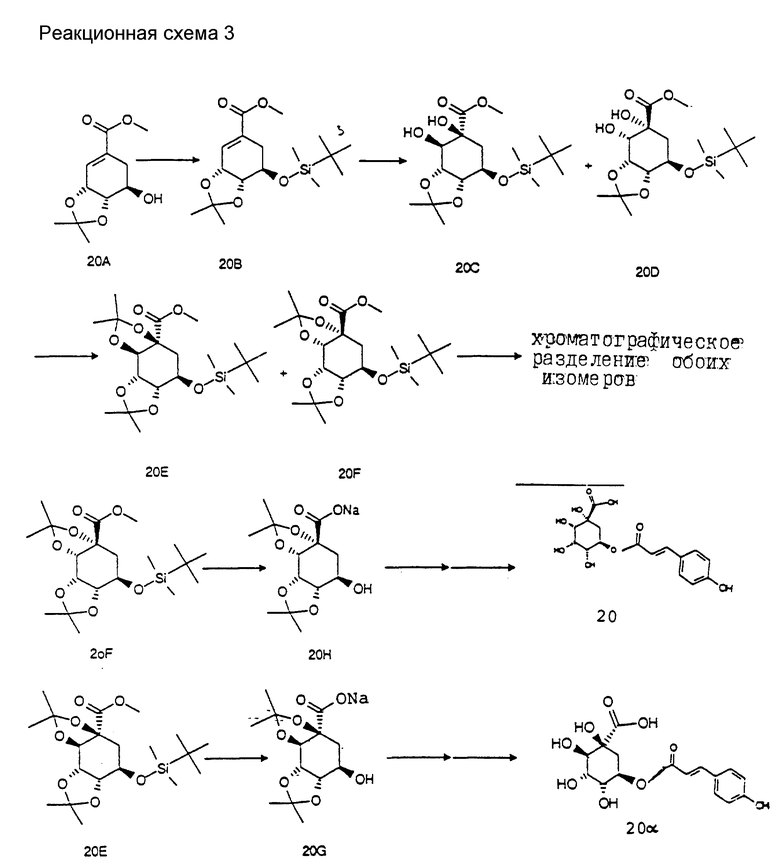
Получение соединения 20, соответственно 20α (соединения 20A-20α приведены в конце описания. Реакционная схема 3).
Получение соединения 20B из соединения 20A.
4.0 г (17.5 ммоль) известного соединения 20A [S.A.Bowles и др., Tetrahedron 46, 3981 (1990)] растворяют в 30 мл безводного диметилформамида. Добавляют 1.61 г (23.7 ммоль) имидазола, а также 2.64 г (12.5 ммоль) трет.-бутилдиметилсилилхлорида. Через 12 ч при 25oC реакционный раствор смешивают с 200 мл насыщенного раствора хлорида аммония и экстрагируют порциями с помощью 300 мл метил-трет. -бутилового эфира. Объединенные органические фазы промывают водой и насыщенным раствором хлорида натрия и сушат над сульфатом магния. Получают 5.4 г (90%) соединения 20B в виде бесцветного масла.
1H-ЯМР (270 мГц, CDCl3): δ, м.д. = 0.06 (с., 3H); 0.09 (с., 3H); 0.76 (с. , 9H); 1.39 (с., 6H); 2.23 - 2.40 (м., 1H); 2.48 - 2.62 (м., 1H); 3.76 (с., 3H); 4.0 - 4.12 (м., 2H); 4.66 - 4.72 (м., 1H); 6.80 - 6.86 (м., 1H).
Получение соединения 20C, соответственно 20D из соединения 20B:
5.4 г (15.8 ммоль) соединения 20B растворяют в 100 мл трет.-бутанола. Добавляют 1.9 г (25.3 ммоль) триметиламин-N-оксида, а также 20 мл воды. Затем добавляют 100 мг (0.4 ммоль) тетраоксида осмия в виде комплекса с 2.0 г поливинилпиридина и перемешивают 14 ч при температуре кипения. После этого отфильтровывают катализатор, концентрируют фильтрат и остаток хроматографируют на силикагеле (растворитель: этилацетат/н-гептан = 1:1). Получают 2.5 г (42%) смеси соединений 20C/20D в соотношении 3 : 1 в виде бесцветного масла.
Смесь обоих изомеров 20C/20D:
1H-ЯМР (270 мГц, CDCl3): δ, м.д. = 0.08 - 0.14 (м., 6H); 0.88 - 0.92 (м. , 9H); 1.38 - 1.40 (м., 3H); 1.51 - 1.55 (м., 3H); 1.80 - 2.0 (м., 1H); 2.28 - 2.48 (м., 1H); 3.61 - 4.52 (м., 7H).
Получение соединений 20E и 20F из соединений 20C, соответственно 20D.
2.5 г (6.6 моль) смеси соединений 20C/20D в соотношении 3 : 1 растворяют в 60 мл безводного дихлорметана. Добавляют 5 мл 2,2-диметоксипропана, а также 200 мг пиридиний-п-толуолсульфоната. Реакционный раствор кипятят в течение 6 ч и затем раствор концентрируют в вакууме. Остаток, смесь из соединений 20E и 20F, разделяют на силикагеле (растворитель: этилацетат/н-гептан = 3:1) и получают в целом 2.4 г (87%) соединений 20E и 20F, смотря по обстоятельствам, в виде бесцветного масла.
1H-ЯМР (270 мГц, CDCl3): δ, м.д. = 0.08 (с., 3H); 0.09 (с., 3H); 0.90 (с. , 9H); 1.34 (c., 3H); 1.39 (с., 3H); 1.45 (с., 3H); 1.50 (с., 3H); 1.72 (дд. J = 13.5 Гц, J = 12 Гц, 1H); 2.19 (дд., J = 4.0 Гц, J = 14.5 Гц, 1H); 3.81 (с., 3H); 3.81 - 3.92 (м., 1H); 4.05 - 4.11 (м., 1H); 4.42 - 4.48 (м., 1H); 4.68 - 4.70 (м., 1H).
Получение соединения 20G из соединения 20E.
1.4 г (3.4 ммоль) соединения 20E растворяют в 30 мл диоксана. Прикапывают 2 мл 6 н раствора гидроксида натрия. Спустя 2 ч реакционный раствор концентрируют, смешивают с 20 мл этилацетата и выливают в 200 мл насыщенного раствора хлорида аммония. Эту смесь подкисляют с помощью 1 н раствора гидросульфата калия до pH 5 и органическую фазу промывают насыщенным раствором хлорида натрия и сушат над сульфатом натрия. После концентрирования маслянистый остаток растворяют в 15 мл безводного ТГФ и добавляют 3.0 г (9.5 ммоль) тетрабутиламмоний фторида (тригидрат), а также 0.5 мл триэтиламина. После этого раствор нагревают 12 ч при 60oC. Затем раствор концентрируют и остаток очищают на силикагеле (растворитель: этилацетат/н-гептан/ледяная уксусная кислота = 30:10:1. Выделяют 600 мг (54%) соединения 20G в виде бесцветного масла.
Получение соединения 20H из соединения 20F.
Соединение 20H из соединения 20F получают аналогично получению соединения 20G из соединения 20E.
Получение соединения 20 из соединения 20H, соответственно соединения 20α из соединения 20G.
Синтез соединения 20, соответственно соединения 20α, осуществляют аналогично методикам синтезов d - f (как описано для соединения 8).
Соединение 20; т.пл. 275oC (разложение).
Cоединение 20α; т.пл. 165-175oC (разложение).
Получение соединения 21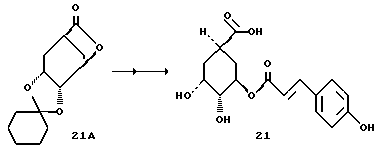
Известный из литературы лактон 21A [S.Hanessian, Tetrahedron, 45, 6623 (1989)] превращают в соединение 21 аналогично методикам синтезов d - f (как описано для соединения 8); т.пл. 227-229oC.
Получение соединения 22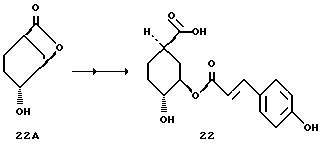
Известное соединение 22A [S. Mills и др., Tetrahedron. Let. 29, 281 (1988)] превращают в соединение 22 аналогично методикам синтезов d - f (как описано для соединения 8); т.пл. 204 - 206oC.
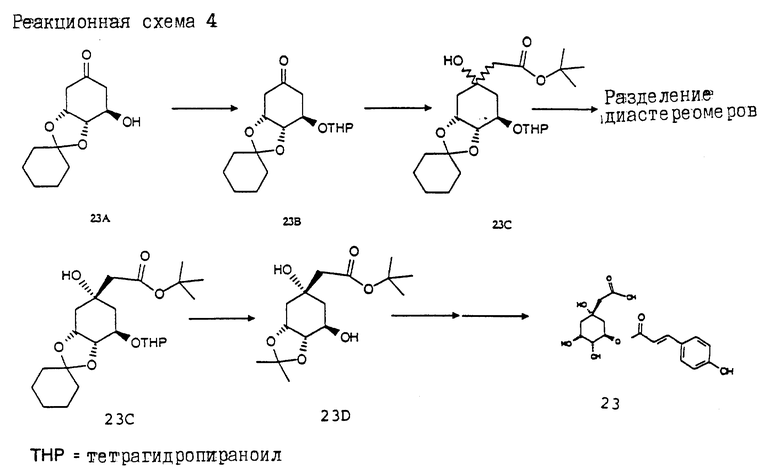
Получение соединения 23 (см. в конце описания. Реакционная схема 4).
Получение соединения 23B из соединения 23A.
20.0 г (88.4 ммоль) известного из литературы соединения 23A [J.C.Barriere и др., Helv.Chim.Acta 66, 296 (1983)] растворяют в 200 мл безводного дихлорметана и при 25oC смешивают с 14.9 г (176.8 ммоль) дигидропирана и 200 мг пиридиний-п-толуолсульфоната. Этот раствор перемешивают 12 ч при комнатной температуре. Затем добавляют 500 мл этилацетатa и промывают органическую фазу раствором гидрокарбоната натрия и насыщенным раствором хлорида натрия. Органическую фазу сушат над сульфатом магния и концентрируют в вакууме. Получают 26.0 г (95%) соединения 23B в виде бесцветного твердого вещества; т.пл. 55 - 58oC.
Получение соединения 23C из соединения 23B.
3.66 г (36 ммоль) диизопропиламина растворяют в 100 мл безводного тетрагидрофурана. В атмосфере аргона и при -20oC прикапывают 25 мл 1.5 М раствора н-бутиллития в гексане. Реакционный раствор оставляют нагреваться до 0oC и после этого снова охлаждают до -60oC. При этой температуре медленно прикапывают 4.1 г (35,3 ммоль) трет.-бутилового эфира уксусной кислоты, растворенного в 20 мл безводного тетрагидрофурана. Раствор перемешивают 30 мин при -60oC и затем при -60oC прикапывают 10.0 г (32.2 ммоль) соединения 23B, растворенного в 30 мл безводного тетрагидрофурана. После перемешивания в течение 1 ч при той же температуре реакционную смесь гидролизуют насыщенным раствором гидрокарбоната натрия. Экстрагируют этилацетатом и объединенные органические фазы промывают насыщенным раствором хлорида натрия и сушат над сульфатом магния. После концентрирования получают 11.9 г (87%) соединения 23C в виде светло-коричневого масла.
Получение соединения 23D из соединения 23C.
11.9 г (27.9 ммоль) соединения 23C растворяют в 200 мл метанола. Добавляют 1.8 г пиридиний-п-толуолсульфоната, кипятят с обратным холодильником в течение 1 ч и затем реакционный раствор концентрируют. Остаток растворяют в 200 мл безводного дихлорметана и добавляют 8.6 г (93.5 ммоль) диметоксипропана. Спустя 72 ч при комнатной температуре раствор концентрируют в вакууме и остаток очищают путем хроматографии на силикагеле (растворитель: этилацетат/н-гептан = 1:1). Получают 6.6 г (82%) соединения 23D.
1H-ЯМР (270 мГц, CDCl3) δ, м.д. = 1.35 (с., 3H); 1.47 (с., 9H); 1.53 (с. , 3H); 1.9 - 2.12 (м., 1H); 2.22 - 2.32 (м., 1H); 2.43 (с., 1H); 3.87 - 3.94 (м., 1H); 4.12 - 4.25 (м., 1H); 4.35 - 4.45 (м., 1H).
Получение соединения 23 из соединения 23D.
Аналогично методикам d - f (как описано для соединения 8) получают соединение 23 в виде бесцветного твердого вещества; т.пл. 85 - 92oC.
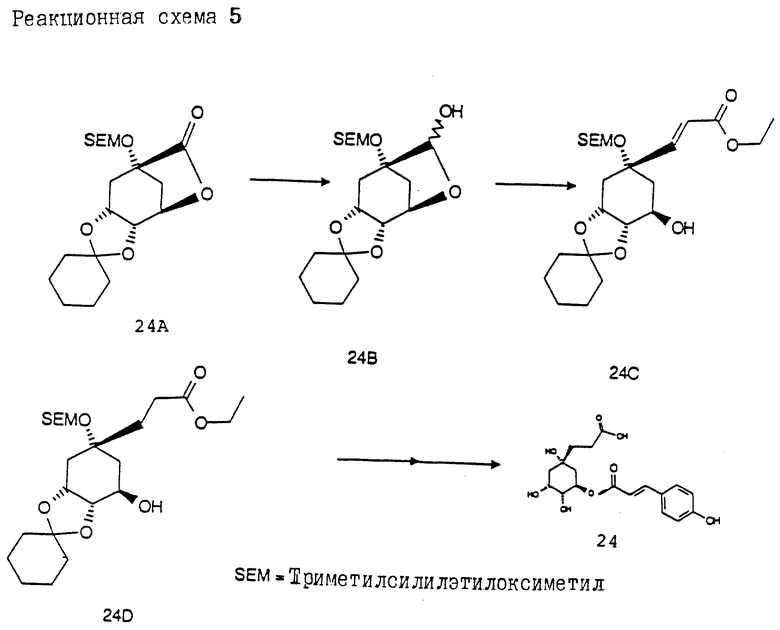
Получение соединения 24 (см. в конце описания. Реакционная схема 5).
Получение соединения 24B из соединения 24A.
15.0 г (39 ммоль) соединения 24A [J.R. Falck,. J. Org. Chem. 54, 5851 (1989)] растворяют в 200 мл безводного толуола. При -70oC прикапывают 38 мл (43 ммоль) 1.2 М раствора диизобутилалюминийгидрида в гексане. В течение 2 ч оставляют нагреваться до 0oC и реакционную смесь гидролизуют с помощью 10 мл насыщенного раствора гидрокарбоната натрия. Затем последовательно добавляют 10 мл 1 н раствора гидроксида натрия и 10 мл воды. Реакционную смесь при энергичном перемешивании смешивают с 50 г сульфата магния и 50 г сульфата натрия. Перемешивают 30 мин при комнатной температуре, твердый осадок отсасывают и фильтрат концентрируют. Получают 12.9 г (85%) соединения 24B в виде бесцветного масла, которое кристаллизуется при 0oC; т.пл. 20-25oC.
Получение соединения 24C из соединения 24B:
К суспензии 0.9 г (29.9 ммоль) 80%-ного гидрида натрия в 200 мл безводного тетрагидрофурана, в атмосфере аргона и при 0oC, прикапывают 7.5 (33.5 ммоль) триэтилового эфира фосфоноуксусной кислоты. Реакционную смесь оставляют медленно нагреваться до комнатной температуры и затем прозрачный коричневатый раствор охлаждают до -30oC. Прикапывают 7.7 г (19.9 ммоль) соединения 24B, растворенного в 20 мл безводного тетрагидрофурана. Этот раствор перемешивают в течение 2 ч при температуре от -20 до -30oC и после этого смешивают со 100 мл насыщенного раствора хлорида натрия. Экстрагируют этилацетатом, промывают насыщенным раствором хлорида натрия и объединенные органические фазы сушат над сульфатом магния. После концентрирования в вакууме остаток очищают на силикагеле (растворитель : этилацетат/н/гептан = 1:1) и получают 7.5 г (82%) соединения 24C в виде бесцветного масла.
1H-ЯМР (200 мГц, CDCl3): δ, м.д. = 0.01 (с., 9H); 0.85 - 1.0 (м., 2H); 1.1 - 1.85 (м., 15H); 2.1 - 2.25 (м., 2H); 2.35 - 2.5 (м., 1H); 3.42 - 3.9 (м., 3H); 4.1 - 4.4 (м., 4H); 4.65 - 4.8 (м., 2H); 5.92 (д., J = 15 Гц, 1H).
Масс-спектр (FAB): 463.3 (M + Li+).
Получение соединения 24D из соединения 24C.
1.0 г (2.2 ммоль) соединения 24C растворяют в 50 мл этилацетата. Добавляют 100 мг Rh/Al2O3 [5% Rh]. Встряхивают 3 ч в атмосфере водорода при 25oC и нормальном давлении. Катализатор отфильтровывают и фильтрат концентрируют в вакууме. Получают 0.95 г (94%) соединения 24D в виде бесцветного твердого вещества.
Получение соединения 24 из соединения 24D.
Аналогично методикам синтезов d - f (как описано для соединения 8) из соединения 24D получают соединение 24; т.пл. 172oC (H2O).
Получение соединения 25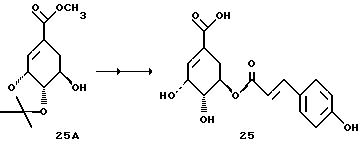
Аналогично методикам синтезов d - f (как описано для соединения 8) из известной из литературы предстадии 25A [J.L. Pawlak и др., J. Org. Chem. 52, 1765 (1987)] получают соединение 25; т.пл. 75-80oC (вспенивание).
Получение соединения 26 из соединения 26A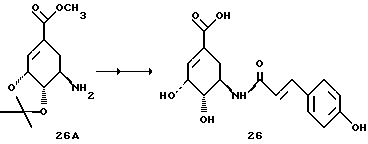
Аналогично методикам синтезов d - f (как описано для соединения 8) из известной из литературы предстадии 26A получают соединение 26 в виде бесцветного аморфного твердого вещества.
1H-ЯМР (270 мГц, d6-ДМСО): δ, м.д. = 1.95 - 2.14 (м., 1H); 2.55 - 2.70 (м. , 1H); 3.62 - 3.76 (м., 1H); 4.08 - 4.26 (м., 2H); 4.55 - 4.75 (м., 1H); 4.9 - 5.1 (м., 1H); 6.48 (д., J = 10.0 Гц, 1H); 6.63 - 6.72 (м., 1H); 6.75 - 6.88 (м. , 2H); 7.29 - 7.46 (м., 3H); 7.89 (д., J = 5 Гц, 1H); 9.70 - 10.0 (1H); 12.2 - 12.6 (1H).
Масс-спектр (Cl): 225.2 (M + H+).
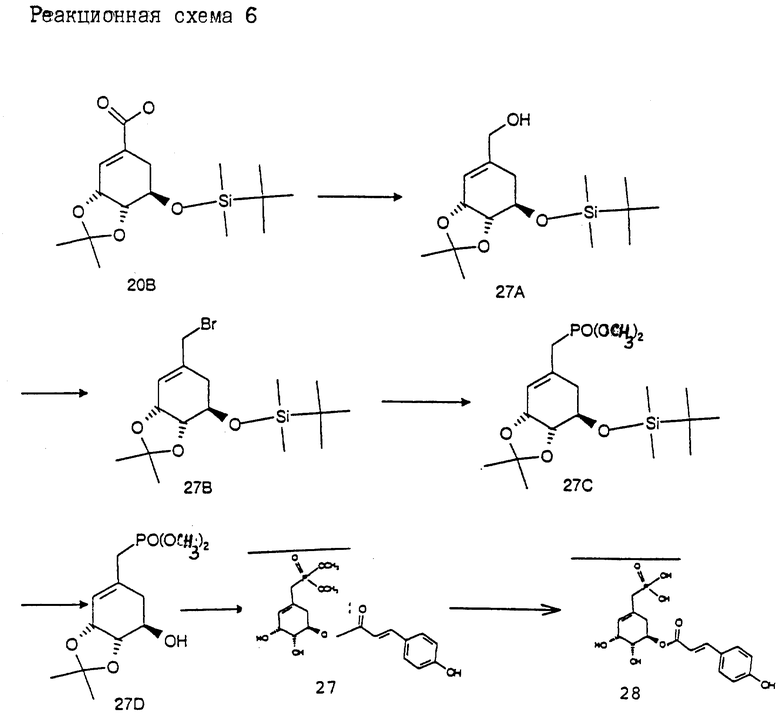
Получение соединения 27 из соединения 28 (см. в конце описания. Реакционная схема 6).
Получение соединения 27A из соединения 20B.
6.0 г (17.5 ммоль) соединения 20B растворяют в 100 мл безводного толуола. При -20oC прикапывают 29.2 мл 1.2 М раствора диизобутилалюминийгидрида в гексане. В течение 1 ч оставляют нагреваться до 25oC, снова охлаждают до 0oC и осторожно прикапывают 20 мл смеси метанола с водой в соотношении 9 : 1. Затем прикапывают 30 мл насыщенного раствора хлорида аммония и реакционную смесь перемешивают 30 мин при 25oC. После этого экстрагируют этилацетатом и объединенные фазы промывают насыщенным раствором хлорида натрия, сушат над сульфатом магния и концентрируют в вакууме. Остаток очищают путем хроматографии на силикагеле (этилацетат/н-гептан = 1:3). Получают 3.5 г (63%) соединения 27B в виде бесцветного масла.
1H-ЯМР (270 мГц, CDCl3) δ, м.д. = 0.08 (с., 3H); 0.11 (с., 3H); 0.89 (с. , 9H); 1.39 (с., 3H); 1.46 (с., 3H); 1.97 - 2.09 (м., 1H); 2.19 - 2.30 (с., 2H); 3.88 - 3.92 (м., 1H); 3.98 - 4.09 (м., 4H); 4.62 - 4.68 (м., 1H); 5.76 - 5.82 (м., 1H);.
Получение соединения 27B из соединения 27A.
К раствору 2.9 г (16.2 ммоль) N-бромсукцинимида в 100 мл безводного дихлорметана при 0oC прикапывают 1.43 мл (19.6 ммоль) диметилсульфида. Спустя 5 мин охлаждают до -20oC и прикапывают 3.4 г (10.8 ммоль) соединения 27A, растворенного в 20 мл безводного дихлорметана. Затем светло-желтую суспензию медленно нагревают до 25oC и перемешивают 3 ч. После этого смешивают со 100 мл насыщенного раствора хлорида аммония и экстрагируют с помощью 500 мл этилацетата. Объединенные органические фазы промывают насыщенным раствором хлорида натрия и сушат над сульфатом магния. После концентрирования остаток очищают путем хроматографии на силикагеле (растворитель: этилацетат/гептан = 1:3) и получают 3.7 г (98%) соединения 27B в виде бесцветного масла.
1H-ЯМР (270 мГц, CDCl3): δ, м.д. = 0.09 (с., 3H); 0.10 (с., 3H); 0.89 (с. , 9H); 1.38 (с., 3H); 1.41 (с., 3H); 2.09 - 2.21 (м., 1H); 2.35 - 2.45 (м. , 1H); 3.92 (с., 2H); 3.97 - 4.05 (м., 2H); 4.38 - 4.65 (м., 1H); 5.83 - (м., 1H).
Масс-спектр (Cl): 377.1 (M + H+).
Получение соединения 27C из соединения 27B.
3.0 г (7.6 ммоль) соединения 27B в 42 мл триметилфосфита нагревают 6 ч при 90oC. Потом отгоняют в вакууме избыточный фосфит и остаток очищают путем хроматографии на силикагеле (растворитель: этилацетат/метанол = 5:1). Получают 3.0 г (93%) соединения 27C в виде бесцветного масла.
Получение соединения 27D из соединения 27C.
3.0 г (7.4 ммоль) соединения 27C растворяют в 50 мл метанола. Добавляют 1 мл 1 н соляной кислоты. Через 24 ч реакционный раствор нейтрализуют 1 н раствором гидроксида натрия и концентрируют в вакууме досуха. Остаток обрабатывают 50 мл безводного дихлорметана, добавляют 5 мл диметоксипропана, а также 0.5 г пиридиний-п-толуолсульфоната и нагревают 4 ч при 40oC. Затем раствор выливают в насыщенный раствор гидрокарбоната натрия и экстрагируют 500 мл этилацетата. Объединенные органические фазы промывают насыщенным раствором хлорида натрия, сушат над сульфатом магния и концентрируют в вакууме. Остаток очищают путем хроматографии на силикагеле (растворитель: этилацетат/метанол = 10:1) и получают 1.5 г (70%) соединения 27D в виде бесцветного масла.
Получение соединения 27 из соединения 27D.
Аналогично методикам синтезов e - f (как описано для соединения 8) из соединения 27D получают соединение 27.
1H-ЯМР (200 мГц, d6-ДМСО): δ, м.д. = 2.05 - 2.22 (м., 1H); 2.55 - 2.8 (м., 1H); 3.4 - 3.55 (м., 1H); 3.6 (с., 3H); 3.65 (с., 3H); 4.05 - 4.15 (м., 1H); 4.3 - 4.4 (м., 1H); 4.6 - 4.8 (м., 3H); 5.05 - 5.15 (м., 1H); 5.55 - 5.68 (м. , 1H); 6.3 - 6.45 (м., 1H); 6.37 - 6.45 (м., 2H); 7.5 - 7.7 (м., 3H); 10.0 (с., 1H).
Масс-спектр (Cl): 399 (M+); 381 (M+ - H2O).
Получение соединения 28 из соединения 27.
135 мг (0.34 ммоль) соединения 27 растворяют в 10 мл безводного ацетонитрила. При 0oC прикапывают 155 мг (1 ммоль) триметилсилилбромида. Перемешивают 30 мин и затем добавляют 5 мл воды. Смешивают с 1 н раствором гидроксида натрия примерно до pH 5 и концентрируют в вакууме. Остаток очищают путем хроматографии на силикагеле RP-8 (растворитель: вода/метанол = 4:1) и получают 23 мг (18%) соединения 28 в виде бесцветного твердого вещества; т. пл. 180-185oC.
Фармацевтические препараты получают общеизвестными способами.
Пример 1. Таблетки.
Таблетки, пригодные для орального введения и содержащие нижеуказанные составные части, получают известным образом путем гранулирования биологически активных и вспомогательных веществ и последующего прессования в таблетки.
Составные части (на таблетку) - Вес (мг)
соединение формулы (I) (например, соединение 17) - 50
молочный сахар - 100
кукурузный крахмал - 30
тальк - 3
коллоидный диоксид кремния - 3
стеарат магния - 2
Пример 2. Капсулы.
Капсулы, пригодные для орального введения, содержат нижеуказанные составные части и получаются известным образом путем смешения биологически активных и вспомогательных веществ и внесения полученной смеси в желатиновые капсулы.
Составные части (на таблетку) - Вес (мг)
соединение формулы (I) (например, соединение 21) - 50
молочный сахар - 100
кукурузный крахмал - 30
тальк - 3
коллоидный диоксид кремния - 3
стеарат магния - 22
Предложено средство для ингибирования глюкозо-6-фосфатазной системы печени млекопитающих. Средство представляет собой сложные эфиры производных циклогексана общей формулы I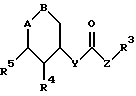
где A-B, R3, R4, R5, Y и Z имеют указанные значения. Изобретение расширяет арсенал средств указанного назначения. 2 з.п. ф-лы, 5 табл.
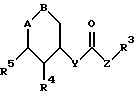
где А - В обозначает группу 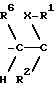 или группу
или группу 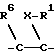
R1 обозначает CN, COOH, COO-(C1 - C4-алкил), С1 - С4-алканоил, SO3-(C1 - C4-алкил), SO3H,PO(OH)2, PO(OH)(O-C1 - C4-алкил) или РО(O-С1 - С4-алкил)2;
R2 обозначает Н, ОН или F;
R3 обозначает Н, фенил, нафтил, пиридил, тиенил, фурил, при этом ароматический или гетероароматический остаток может быть одно- или многократно замещен фтором, хлором, бромом, иодом, ОН, NO2, C1 - C1-алканоилом, С1 - С4-алкокси, С1 - С4 -алкилом, фенилом, фенокси, тиенилом, фурилом, пиридилом, имидазолилом или бензилокси, при этом заместители являются одинаковыми или разными;
R4, R5 и R6 обозначают Н, ОН, F, Cl, Br, C1 - C4-алканоил, С1 - С4-алкил, фенил, фенокси, тиенил, фурил, пиридил, имидазолил или бензилокси, при этом R4, R5, R6 являются одинаковыми или разными;
Х обозначает -(СН2)n-, -CH=CH- или -СН2-О-СН2;
Y обозначает -(СН2)n-, 0, или NH;
Z обозначает -(СН2)n-, -СН-СН-;
n обозначает нуль, 1,2,3 или 4.
Y обозначает -(СН2)n-, О, S или NH;
Z обозначает -(СН2)n-, -СН=СН-;
n обозначает нуль, 1, 2, 3 или 4.
| Способ получения производных тиазолидиндиона или их солей с щелочными металлами | 1987 |
|
SU1556540A3 |
| The Merck Index, Rahway, N.Y., 1989, p | |||
| Катодная трубка Брауна | 1922 |
|
SU330A1 |
| Haslam E | |||
| et al | |||
| The Shikimate Pathway | |||
| Part II | |||
| Conformational Analysis of (-)-Qunic Acid and Its Derivatives by proton Magnetic Resonance spectroscopy | |||
| Journal of The Chemical society | |||
| C(Organic), 1971, N 8, p | |||
| Насос для горючей жидкости для двигателей внутреннего горения | 1924 |
|
SU1496A1 |
