Клиническая картина диабета характеризуется повышенными содержаниями сахара в крови. В случае инсулинзависимости от типа I диабета причиной является отмирание продуцирующих инсулин β- клеток поджелудочной железы; поэтому лечение осуществляют путем введения инсулина (заместительная терапия). Напротив, инсулиннезависимый или тип II диабета характеризуется уменьшенным действием инсулина на мышечную и жировую ткань (резистентность к инсулину) и повышенной выработкой печенью глюкозы. Причина этих нарушений обмена вещества еще далеко неясны, известная терапия с помощью сульфонилмочевин пытается компенсировать резистентность к инсулину за счет повышения аутогенного выделения инсулина, однако это не во всех случаях ведет к нормализации уровня сахара в крови и не может остановить распространение болезни; многие диабетики типа II в конце концов в результате "истощения" β- клеток становятся инсулинзависимыми и позднее, при дальнейшем развитии болезни, страдают такими заболеваниями, как катаракты, нефропатии и ангиопатии.
Поэтому желательны новые терапевтические принципы лечения диабета типа II.
Натощак концентрация глюкозы в крови определяется выработкой глюкозы печенью. Различные группы исследователей показали, что повышение содержания сахара в крови при диабете типа II коррелирует с пропорционально повышенным выделением глюкозы из печени. Глюкоза, выделяемая печенью в кровь, может образоваться как в результате разложения гликогена печени (гликогенолиз), так и также путем глюконеогенеза.
Глюкозо-6-фосфат является общим конечным продуктом как глюконеогенеза, так и гликогенолиза. Конечная стадия выделения печенью глюкозы из глюкозо-6-фосфата катализируется глюкозо-6-фосфатазой (ЕС 3.1.3.9). Глюкозо-6-фосфата представляет собой многоферментный комплекс, встречающийся в эндоплазматической сетке (ER). Этот ферментный комплекс состоит из находящейся в ER-мембране глюкозо-6-фосфаттранслоказы, локализованной на люминальной стороне эндоплазматической сетки глюкозо-6-фосфатазы и фосфатранслоказы /см. F. Ashmore и G. Wеber "The Role of Hepatic Glucose-6-phosphatase in Regulation of Carbohydrate Metabolism" в "Vitamins and Hormones" т. XVII/Harris R. S. Marrian G.F.Timann K.V.Edts 92-132 (1959): Burchell A., Waddell I.D. "The molecular basis of the hepatic microsomal glucose-6-phosphatase system," Biochim. Biophys. Acta. 1092, 129-137 (1990)/. Существующая обширная литература показывает, что при всех исследованных условиях, приводящих в опытах на животных c повышенным содержаниям глюкозы в крови, например, как в случае стрептозоцина, аллоксана, кортизона, тироидных гормонов и голодовок, активность этого многоферментного комплекса также повышается. Сверх того, многочисленные исследования указывают на то, что наблюдаемое у диабетиков типа II повышенное выделение глюкозы связано с повышенной активностью глюкозо-6-фосфатазы. Значение глюкозо-6-фосфатазной системы для нормального глюкозного гомеостаза подчеркивается, далее, гипогликемическими симптомами у пациентов с заболеванием типа 16, связанным с накоплением гликогена, при котором отсутствует транслоказная компонента глюкозо-6-фосфатной системы.
Уменьшение активности гпюкозо-6-фосфатазы благодаря пригодным биологически активным веществам (ингибиторам) должно приводить к соответственно уменьшенному гепатитному выделению глюкозы. Эти биологически активные вещества должны обладать способностью адаптировать выделение глюкозы печенью к эффективному периферическому расходу. Снижающееся благодаря этому в состоянии натощак у диабетиков типа II содержание глюкозы в крови, кроме того, должно оказывать также предохранительное действие в отношении наносимого позднее диабетом вреда.
В литературе описан ряд неспецифических ингибиторов глюкозо-6-фосфатазы, как, например, флорризин [Soodsma J.F.Legeer B. и Nordlie R.C., J. Biol. Chem 242, 1955-1960 (1967)]; 5,5'-дитио-бис-нитробензойная кислота (Wallin B. K. и Arion W. J., Biochem. Biophys. Res. Commun., 48, 694-699 (1972)]; 2,2-диизотиоцианатостильбен и 2-изотиоцианато-2'-ацетокси-стильбен [Zoccoli M.A.Karnowski M.L., J.Biol. Chem. 255, 1113-1119 (1980)]J. Однако до сих пор еще нет никаких терапевтически применимых ингибиторов глюкозо-6-фосфатазной системы.
Охарактеризованные подробнее далее производные циклогексана являются новыми, до сих пор не описанными в химической и биологической литературе соединениями.
В настоящее время заявителем найдено, что определенные сложные эфиры замещенных циклогексанкарбоновых кислот, как, например, соединение примера 14, являются ингибиторами глюкозо-6-фосфатазной системы.
Поэтому изобретение относится к производным циклогексана формулы I
где R1 обозначает CN, COOH;
R2 обозначает C1-C10-алкил (R11)n, O-C1-C10-алкил (R11)n, O-C3-C10-алкенил (R11)n, O-C3-C10-алкинил (R11)n, S-C1-C10-алкил (R11)n причем в случае необходимости R11 замещен с помощью R12, R12-фенил, причем остаток может быть одно- или многократно замещен хлором; R3, R11 и R13, обозначают алкил с 1-10 C-атомами; циклоалкил с 3-8 C-атомами в кольце; фенил, настил, пиридил, тиенил, фурил, индолил, имидазолил, хинолил или их тиено-, пиридино-, пиримидино- или бензоанеллированные производные, причем ароматический, соответственно, гетероароматический остаток может быть одно- или многократно замещен фтором, хлором, OH, CF3, CN, C1-C4-алкоксилом, фенилом, O-фенилом, причем заместители могут быть одинаковыми или разными, и также R3, R11 и R13 являются одинаковыми или разными;
R4, R5 обозначают H, OH, защищенную обычными защитными для спиртовой группы группами OH-группу, или имеют указанные для R2 значения, причем R4, R5 и R6 являются одинаковыми или разными;
R6 означает водород;
x обозначает (CH2)m;
y - кислород, (-O-);
Z обозначает - (CH2)m, серу, C3-C10-циклоалкенилен, причем 1-3 C-атома кольца могут быть заменены атомами азота; -CH=C(C1-C4-алкил), -CH= C(C1-C4-алканоил); -CH=C(R13); 
n обозначает нуль, 1 или 2;
m обозначает нуль, 1, 2, 3 или 4,
а также физиологически приемлемые соли соединений формулы I .
Предлагаемые согласно изобретению соединения формулы I, поскольку они содержат карбоксильную группу, могут образовывать соли с неорганическим или органическими основаниями. Поэтому изобретение относится также к физиологически приемлемым солям соединений формулы I.
Предлагаемые согласно изобретению соединения формулы I содержат ряд стереоцентров. Изобретение относится ко всем возможным энантиомерам и диастореомерам. Все они охватываются формулой I.
Если не указано ничего другого, то для выше- и нижеуказанного имеет значение следующее:
Указанные под обозначениями R11, R12, R13, R1, R3, Z и алкильные, алканоильные и алкоксильные остатки являются линейными или разветвленными.
Указанные под обозначением R2 алкильные, алкенильные и алкинильные группы являются линейными, разветвленными или циклическими, причем также только часть остатка может образовывать кольцо; R11 может быть замещен с помощью R12 и при n = 2 оба остатка R11 являются одинаковыми или разными. Ненасыщенные остатки одно- или многократно ненасыщены.
Предпочтительны соединения формулы I, в которых R1 обозначает CN, COOH, а остальные остатки имеют вышеуказанные значения.
Особенно предпочтительны соединения формулы I, где остатки имеют следующие значения:
обозначает CN, COOH;
R2 обозначает O-C1-C10-aлкил (R11)n (n = 0, 1, 2), причем алкильная часть линейная, разветвленная, R11 может быть замещен с помощью R12 и при n = 2 оба остатка R11 являются одинаковыми или разными; O-C3-C10-алкенил (R11)n (n = 0, 1, 2), причем алкенильная часть линейная, разветвленная, R11 может быть замещен с помощью R12 и при n = 2 оба остатка R11 являются одинаковыми или разными; O-C3-C10-алкинил (R11)n (n = 0, 1, 2), причем алкинильная часть линейная, разветвленная, R11 может быть замещен с помощью R12 и при n = 2 оба остатка R11 являются одинаковыми или разными; R3-R13 имеют вышеуказанные значения; x обозначает - (CH2)m; y - кислород; z обозначает -(CH2)m (m = 0, 1, 2, 3, 4), серу, C3-C10-циклоалкилен,  -CH=C(C1-C4-aлкил) (линейный или разветвленный).
-CH=C(C1-C4-aлкил) (линейный или разветвленный).
Предлагаемые в изобретении соединения формулы (I), поскольку они содержат карбоксильную группу, могут образовывать соли с неорганическими или органическими основаниями. Предпочтительны соли с неорганическими основаниями, особенно физиологически приемлемые соли щелочных металлов, прежде всего соли натрия и калия.
Соединения формулы (I) ингибируют глюкозо-6-фосфатазную систему печени млекопитающих. Соединения поэтому пригодны в качестве лекарственного средства. Изобретение относится поэтому также к лекарственным средствам на основе соединений формулы (I), в случае необходимости в виде физиологически приемлемых солей.
Изобретение также относится к применению соединений формулы (I), соответственно, солей, для лечения заболеваний, связанных с повышенным выделением глюкозы печенью.
Изобретение относится, кроме того, к применению соединений формулы (I), соответственно, солей, для лечения диабета типа II (инсулиннезависимого или старческого диабета).
Изобретение, далее, включает применение соединений формулы (I), соответственно, солей, для получения лекарственных средств для лечения диабета или других заболеваний, которые характеризуются повышенным выделением глюкозы из печени или повышенной активностью глюкозо-6-фосфатазной системы.
Действие предлагаемых согласно изобретению соединений на глюкозо-6-фосфатазную систему исследовалось в ферментном тесте на микросомах печени.
Для приготовления содержащей глюкозо-6-фосфату микросомной фракции используют свежую печень самцов крыс Wistar и обрабатывают как описано в литературе [Canfield W.K. и Arion W.J., J. Biol. Chem. 263, 7458-7460 (1988)]. Эта микросомная фракция может храниться при -70oC по меньшей мере 2 месяца без значительной потери активности.
Обнаружение глюкозо-6-фосфатазной активности осуществляют, как указано в литературе [Arion W. J. в Methods Enxymol, 174, Academic Press. 1989, с. 58-67] , путем определения высвобождающегося из глюкозо-6-фосфата фосфата. 0,1 мл испытуемой смеси содержит глюкозо-6-фосфат (1 ммоль/л), испытуемое вещество, 0,1 мг микросомной фракции и 100 ммоль/л HEPES-буфера [4-(2-гипроксиэтил)пиперазин-1-этансульфокислота] , pH 7,0. Реакцию инициируют путем добавки фермента. По истечении 20 минут при комнатной температуре реакцию прекращают за счет добавки 0,2 мл фосфатного реагента. Пробу инкубируют в течение 30 минут при 37oC, и затем измеряют поглощение (A) голубого цвета при 570 нм. Ингибирующую активность испытуемого вещества получают путем сравнения с контрольной реакцией, при которой не содержится никакого испытуемого вещества, по формуле
Если необходимо, ингибирующее действие испытуемого вещества определяют как функцию используемой концентрации испытуемого вещества и из нее рассчитывают концентрацию для 50%-ного ингибирования активности фермента (ИК50)
Для нижеуказанных соединений определено ИК50 значение:
Пример 1
(1S, 3R, 4R,5S)-3-[(E-)-3-/4-Гидроксифенил/пропеноил]окси-4,5- дигидрокси-1-фенилметилокси-циклогексанкарбоновая кислота. ИК50 = 190 мкмоль
Пример 2
(1S, 3R, 4R,5S)-3-[(E-)-3-/4-Гидроксифенил/пропеноил]окси-4,5- дигидрокси-1-(2-тиенилметил)окси-циклогексанкарбоновая кислота. ИК50 = 110 мкмоль
Пример 3
(1S, 3R,4R,5S)-3-[(E-)-3-/4-Гидроксифенил/пропеноил]-окси-4,5- дигидрокси-1-/2-пропинил/окси-циклогексанкарбоновая кислота. ИК50 = 560 мкмоль.
Пример 8.
(1S, 3R, 4R, 5S)-3-[(E)-3-(4-Гидроксифенил)пропеноил]окси-4,5- дигидрокси-1-пропилокси-циклогексанкарбоновая кислота. ИК50= 230 мкмоль.
Пример 4.
(1S, 3R, 4R,5S)-1-[4-Хлорфенилпропил]-окси-4,5-дигидрокси-3- /2-пиридинкарбонил/-окси-циклогексанкарбоновая кислота. ИК50=26 мкмоль.
Пример 44.
(1S, 3R, 4R,5R)-1-/4-Хлорфенилпропил/-окси-3-[(E)-3- (4-гидроксифенил)-пропил]окси-4,5-дигидрокси-циклогексанкарбоновая кислота. ИК50 = 9,3 мкмоль
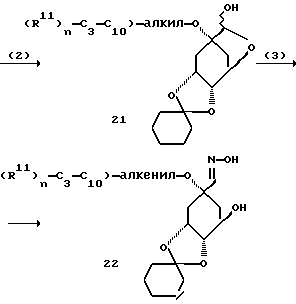
Получение предлагаемых в изобретении соединений формулы I в которых остатки: R2 обозначает O-алкил(R11)n, O-алкенил(R11)n или O-алкинил(R11)n; R4 = R5 = OH и Y обозначает O, можно осуществлять указанным в следующей схеме путем A:
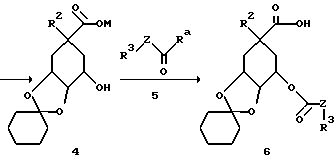

[соединение 7 = формула /1/; R2 = O-C1- C10-алкил/R11/n; O-C3-C10-алкенил/R11/n или O-C3-C10-алкинил/R11/n; R4= R5=OH; R6=H; Y=O; X= /CH2/m c m = нуль; R1=COOH; Z, R3, R11 и n имеют указанное по формуле /I/ значение].
Способ A отличается тем, что известное из литературы, получаемое из соединения 1 соединение 2 депротонируют с помощью сильного основания, как трет. -бутилат калия, гидрид натрия или гидрид калия, и для введения R2 вводят во взаимодействие с соответствующими галогенидами, сложными эфирами трифторсульфокислоты, сложными эфирами метилсульфокислоты или сложными эмирами п-толуолсульфокислоты, предпочтительно в полярном апротонном растворителе, как диметилформамид, диметилсульфоксид или тетрагидрофуран, причем образуется соединение 3. В качестве растворителя предпочтительно применение диметилформамида, а в качестве основания - гидрида натрия.
Реакцию 2 __→ 3 осуществляют при температурах от -20oC до температуры кипения используемого растворителя. Предпочтителен температурный интервал от -10oC до 60oC, особенно при 0-30oC.
Предпочтительный вариант реакции 2 __→ 3/ осуществляют в диметилформамиде в присутствии гидрида натрия или гидрида калия при температурах 0-60oC. При этом реакцию предпочтительно осуществляют в отсутствие влажности в атмосфере защитного газа (азот или аргон).
Необходимые для превращения соединения 2 в соединение 3 исходные материалы, которые соответствуют остатку R2, можно получать по известному специалисту стандартному способу. При этом речь идет о структурах типа R2-B с указанным для способа А ограничением (разумеется, без связывающего атома кислорода). "B", например, обозначает удаляемую группу, как Cl, Br, йод или OSO2R (R=CH3 фенил, толил, CF3).
Вместо циклогексилиденовой защитной группы в соединении 2, соответственно, соединении 3 также можно применять другие, отщепляющиеся в мягких кислых условиях защитные группы, как изопропилиден- или бензилиден-ацетали, а также трет. -бутиловый, метоксиметиловый, 1-этоксиэтиловый или тетрагидропираниловый простой эфир; простые силильные эфиры, как триметилсилильный или трет. -бутилдиметилсилильный, или карбонаты, как бензилоксикарбонил- и трет. -бутоксикарбонил-производные, которые общеизвестны из химии пептидов и стероидов. Получение таких защищенных соединений также следует из соединения 1.
Следующей стадией способа A является гидролиз лактона 3 по щелочной соли 4 с помощью гидроксидов щелочных металлов, как гидроксид лития, гидроксид натрия или гидроксид калия. Реакцию предпочтительно осуществляют в протонных или апротонных растворителях, как низшие спирты, тетрагидрофуран или диоксан; предпочтительно применение диоксана.
Реакцию превращения соединения 3 в соединение 4 осуществляют при температурах от -20oC до температуры кипения используемого растворителя. Предпочтителен температурный интервал от -10oC по 60oC, особенно 0-30oC.
Следующая стадия представляет собой реакцию превращения соединения 4 в соединение 6, при которой остаток R3-Z-C(O)- присоединяется к соединению 4. Для этой цели соединение 4 в апротонном органическом растворителе, как, например, тетрагидрофуран, диметилформамид, дихлорметан, пиридин или диметилсульфоксид, вводят во взаимодействие с соединением R3-Z-C(O)-Ra (соединение 5), причем Ra может обозначать, например, хром, бром, OC(O)-C1-C4-алкил, имидазолил, триазолил или тетраазолил, причем особенно предпочтительны имидазолил и триазолил. Особенно предпочтительно осуществление реакции в диметилформамиде в присутствии основания, как, например, гидрид натрия, гидрид калия, 4-пиалкиламинопирипин или трет.-амины, особенно, однако, гидрид натрия.
Реакцию превращения соединения 4 в соединение 6 осуществляют при температурах от -20oC по температуры кипения используемого растворителя. Предпочтителен температурный интервал от -10oС до 60oC, особенно 0-30oC.
Соединение R3-Z-C(O)-Ra (5) можно получать известными специалисту стандартными способами.
Предпочтительный вариант реакции превращения соединения 4 в соединение 6 состоит во введении во взаимодействие соединения 4 с гидридом натрия в диметилформамиде и путем последующей добавки раствор R3-Z-C(O)-имидазола (соединение 5) в диметилформамиде при 0-20oC, предпочтительно в отсутствие влажности в атмосфере защитного газа (аргон или азот).
Отщепление защитной группы при реакции превращения соединения 6 в соединение 7 осуществляют общеизвестным образом, например, путем обработки разбавленными неорганическими кислотами, как, например, соляная кислота, пли сильными органическими кислотами, как, например, трифторуксусная кислота, в инертных органических растворителях, как простые циклические эфиры, необязательно в присутствии воды, при температурах от -20oC до температуры кипения растворителя, предпочтительно при 0-30oC.
Полученные согласно изобретению соединения формулы (I), поскольку они содержат карбоксильную группу, могут образовывать соли с неорганическими или органическими основаниями. Поэтому также предпочтительным такие соли с неорганическими основаниями, особенно физиологически приемлемые соли щелочных металлов, прежде всего соли натрия и калия.
Из солей щелочных металлов соединений формулы (I) с карбоксильной группой можно получать указанные сложные эфиры. Для этого соединение 7 в инертном органическом растворителе, как тетрагидрофуран, диметилсульфоксид, предпочтительно диметилформамид, при от -10oC до 60oC, вводят во взаимодействие, например, с C1-C4-алкилгалогенидом, предпочтительно с C1-C4-алкилиодидом, бензилбромидом, или C1-C4-алканоил-O-CH(R7)-Br, или C1-C4-алканоил-O-CH(R7)-J, с получением соединений формулы (I) со сложноэфирной группой в качестве R1 и с X = (CH2)m при m = O с указанными в способе A деталями.
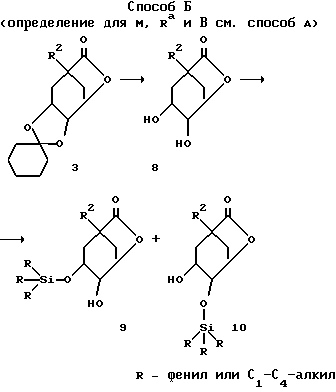


(соединения 17 и 18 = формула (I) с R2 = O-C1-C10-алкил (R11)n; O-C3-C10-алкенил (R11)n или O-C3-C10-алкинил(R11)n; R4 имеет указанное для R2 значения и R5 = OH, соответственно, R4 = OH и R5 имеет указанное для R2 значение; R6 = H; Y = O; X = (CH2)m с "m" = нуль: R1 = COOH; Z, R3 и R11, а также "n" имеют указанное в формуле (I) значение).
Для того, чтобы изменять остатки R4 и R5, применяют способ Б. При этом необходимо отщеплять циклогексилиденовую защитную группу в соединении 3 для получения соединения 8. Это можно осуществлять согласно любому из известных специалисту стандартных способов. При этом предпочтителен гидролиз соединения 3 в инертных органических растворителях, как низшие спирты, в присутствии сильных органических кислот, как сульфокислоты, например, n-толуолсульфокислота или трифторуксусная кислота.
Реакцию превращения соединения 3 в соединение 8 осуществляют, например, при температурах от -20oC для температуры кипения используемого растворителя. Предпочтителен температурный интервал от -10oC по +60oC, особенно при 20-50oC. Особенно предпочтительно превращение соединения 3 в соединение 8 в изопропаноле в присутствии п-толуолсульфокислоты при 40oC.
Характерной для способа Б стадией является дифференцирование обеих свободных гидроксильных групп в соединении 8. Для этого соединение 8 вводят во взаимодействие с пространственно затрудненными триалкилсилилгалогенидами, как, например, трет.-бутилдиметилсилилхлорид, трет.-бутилдифенилсилилхлорид или триизопропилсилилхлорид, в инертном органическом растворителе, особенно в диметилформамиде, при температурах от -10oC до 40oC, в присутствии основания, особенно имидазола, с получением соединений 9 и 10, которые можно разделять хроматографически.
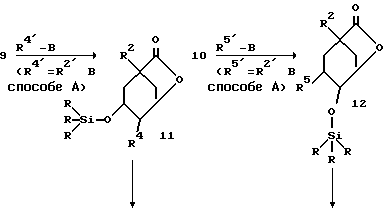
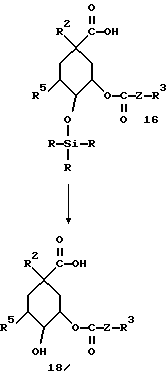
Соединения 9 и 10 можно вводить во взаимодействие аналогично превращениям, осуществляемым от соединения 2 до соединения 7 из способа А, так что указанные для формулы (I) вариации остатков R4 и R5 возможны согласно этому способу.
Способ В (Определение для Ra см. Способ А)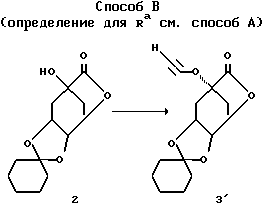




Соединение 7'= формула (I) с R2 = O - C3-алкил (R11)n с "n" - 1 и R11 обозначает ароматический остаток, как указано в формуле (I); R4, R5 = OH; R6 = H; Y = O; X = (CH2)m с "m" = нуль и R1 = COOH; Z и R3 имеют указанное в формуле (I) значение, причем соединения с R2 = O-C3-алкенил(R11)n, соответственно, O-C3-алкинил(R11)n получают тогда, когда не осуществляют последующего гидрирования до соединения 3''', соответственно, 3IV).
Альтернативный А способ для ряда предлагаемых согласно изобретению соединений формулы (I) представляет собой способ В. Для R2 = O-C3-алкинил(R11) может служить промежуточная стадия 3', при которой R2 соответствует 2-пропинилокси. При этом соединение 3' (R2 = 2-пропинилокси) в инертном органическом растворителе, как, например, толуол, бензол или н-гептан, при катализе с помощью комплекса палладия и галогенида меди-(1), особенно иодида меди-(1), вводят во взаимодействие с арилгалогенидом, особенно арилбромидом или арилиодидом, с получением соединения 3" (R2=R11-2-пропинилокси). В этом случае нужно добавлять основание, как, например, первичные, вторичные или третичные амины, особенно триэтиламин. Необязательно основание также одновременно может служить в качестве растворителя и необязательно можно отказываться от добавки другого органического растворителя. Предпочтителен температурный интервал 20-90oC, особенно 60-80oC.
В качестве палладиевого комплекса может служить, например, получаемый in situ из дихлорида палладия и трифенилфосфина комплекс дитрифенилфосфинпалладийдихлорид или получаемый таким же образом из ацетата палладия-(II) комплекс дитрифенилфосфинпалладийдиацетат, предпочтительно дитрифенилфосфинпалладий дихлорид.
Из соединения 3" (R2 = R11-2-пропинилокси) благодаря катализаторам гидрирования можно целевым путем получать соединение 3" (R2 = R11-2-пропенилокси) или соединение 3IV (R2 = R11-2-пропилокси). Реакции осуществляют в этаноле или пиридине в атмосфере водорода при нормальном давлении.
Реакцию соединения 3" (R2 = R11-2-пропинилокси) с целью получения соединения 3" (R2 = R11-2-пропенилокси) осуществляют с помощью катализатора палладий-на-сульфате бария при температурах от 0oC до температуры кипения используемого растворителя. В качестве растворителя предпочтителен пиридин и предпочтителен температурный интервал 20-50oC, особенно 20-30oC.
Реакцию превращения соединения 3''' [R2 = (R11)-2- пропинилокси] в соединение 3IV [R2 = (R11)-пропилокси] осуществляют с помощью палладия-на-угле в качестве катализатора в этаноле при температурах от -20oC вплоть до температуры кипения используемого растворителя. Предпочтителен температурный интервал 20-50oC, особенно 20-30oC.
Дальнейшие реакции превращения соединения 3'' в соединение 3IV, а также соединения 4' в соединение 7', т.е. в соединения общей формулы (I), подробно описаны в способе А.
Способ Г (определение для Ra см. в способе А):
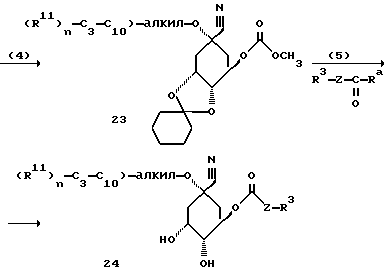
[соединение 24 = формула (I) с R1 = CN; X = (CH2)m с "m" = нуль; R2 = O-C3-C10-алкил-/R11/n с "n" = нуль или I; R4, R5 = OH; R6 = H, Y = O и R3, R11 и Z имеют указанное в формуле (I) значение].
Осуществление способа Г соответственно стадиям способа (1)-(5) пояснено в примере 68.
Способ D (определение для Ra см. способ А):

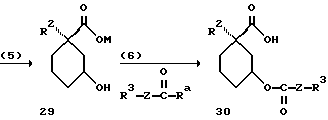
[соединение 30 = формула (I) с R2 = C1-C10-алкил (R11)n; R4, R5, R6=H; Y= O; X=(CH2)m с "m"=нуль; R1=COOH; Z, R3, R11 и "n" имеют указанное в формуле (I) значение].
Способ D пригоден для получения описанного в примере 70 соединения. В этом примере можно видеть пригодные реакционные условия.
Способ E (определение для Ra см. способ А)
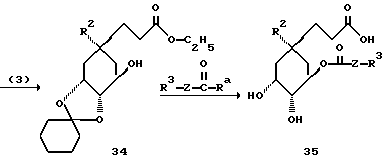 [соединение 35 - формула (I) с R1= COOH; X= (CH2)m с "m"=3; R2 = O-C1-C10-алкил(R11)n; O-C3-C10-алкенил(R11)n или O-C3-C10-алкинил-(R11)n; R4, R5=OH; R6=H и Z, R3, R11 и "n" имеют указанное в формуле (I) значение].
[соединение 35 - формула (I) с R1= COOH; X= (CH2)m с "m"=3; R2 = O-C1-C10-алкил(R11)n; O-C3-C10-алкенил(R11)n или O-C3-C10-алкинил-(R11)n; R4, R5=OH; R6=H и Z, R3, R11 и "n" имеют указанное в формуле (I) значение].
Способ пояснен в примере 63.
Исходные соединения для способов А-E известны, соответственно, их можно получать аналогично известным из литературы способам или их получают описанными в заявке способами.
Получение соединения 5 (см. способ А), которое используется, например, при синтезе соединений примеров 68, 78, 82, 96, 97, 98, осуществляют целесообразнее по способам I, II, III, как описано ниже.

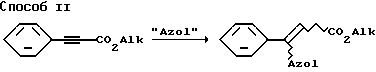

"Alk" обозначает C1-C4-алкил; "Azol" обозначает R13 в значении: имидазолил, индолил, пиперазинил, тетразолил, триазолил или их тиено-, пиридино-, пиримидине- или бензо-анеллированные производные:
Способ I.
Получение β- азол-замещенного метилового эфира коричной кислоты.
Смесь 50 г метилового эфира 2,3-дибром-3-фенилпропановой кислоты 100 мл триэтиламина и 500 мл толуола кипятят в течение 1 часа, затем охлаждают по комнатной температуры и отфильтровывают. Фильтрат выпаривают в вакууме и таким образом полученную α- бромкоричную кислоту используют далее без очистки. К суспензии 4,7 г aH (80%-ная в минеральном масле) в 100 мл безводного ДМФ при перемешивании прикапывают 0,1 моля азольного производного, растворенного в 150 мл безводного ДМФ. Температуру смеси при этом поддерживают ниже 35oC за счет охлаждения льдом. По окончании добавления перемешивают N 1 час при комнатной температуре. Вышеполученную α- бромкоричную кислоту растворяют в 200 мл безводного ДМФ и при перемешивании и при охлаждении льдом прикапывают раствор натриевой соли азота. После перемешивания в точение 2 часов при комнатной температуре добавляют 10,8 мл ледяной уксусной кислоты, смесь вносят при перемешивании в 1,5 л ледяной воды, экстрагируют многократно этилацетатом и органические фазы промывают водой. Органические фазы сушат, выпаривают в вакууме и остаток очищают путем колоночной хроматографии на силикагеле [растворитель: н-гептан/этилацетат] или перекристаллизации.
Способ II.
Получение β- азол-замещенного этилового эфира коричной кислоты
Смесь из 20 г этилового эфира фенилпропиоловой кислоты, 0,11 моль азольного производного и 15 мл безводного ДМФ перемешивают при комнатной температуре в атмосфере аргона. Добавляют на кончике шпателя aH (80%-ный в минеральном масле). Когда закончится выделение водорода, нагревают до 100-150oC (температура бани) и за протеканием реакции следят с помощью ТСХ (растворитель: н-гептан/этилацетат). По окончании реакции охлаждают до комнатной температуры, концентрируют в вакууме и остаток перекристаллизуют из н-гептана или разбавляют небольшим количеством смеси н-гептана с этилацетатом и очищают путем колоночной хроматографии на силикагеле [растворитель: н-гептан/этилацетат].
Способ III.
Получение β- азол-замещенных коричных кислот из β- азол-замещенных сложных эфиров коричной кислоты.
В растворе 0,77 г NaOH в 50 мл воды и 10 мл метанола суспендируют 6,4 ммоль β- азол-замещенного метилового или этилового эфира коричной кислоты и смесь перемешивают при комнатной температуре до тех пор, пока ТСХ но покажет полного превращения [растворитель: н-гептан/этилацетат] и пока но образуется прозрачный раствор. Этот раствор концентрируют в вакууме, разбавляют примерно 50 мл воды и при охлаждении льдом с помощью 2 н HCl устанавливают в нем pH 2-3. Если осаждается твердое вещество, то его отсасывают и высушивают в вакууме. В другом случае многократно экстрагируют с помощью CH2Cl2, органические фазы сушат, выпаривают в вакууме и остаток очищают путем перекристаллизации или хроматографии на силикагеле [растворитель: н-гептан/этилацетат/ледяная уксусная кислота].
Следующим предметом изобретения являются лекарственные средства, которые содержат одно или несколько предлагаемых согласно изобретению соединений формулы (I) и/или их фармаколически приемлемых солей.
Лекарственные средства получают само по себе известными, доступными специалисту способами. В качестве лекарственных средств использует предлагаемые согласно изобретению, фармакологически активные соединения (= биологически активное вещество) либо сами по себе, либо предпочтительно в комбинации с пригодными фармацевтическими вспомогательными веществами в форме таблеток, драже, капсул, свечей, эмульсий, суспензий, гранулятов, порошков, растворов или препаратов с пролонгированным выделением биологически активного вещества предварительно составляет 0,1-95%.
Какие вспомогательные вещества пригодны для желательной формулировки лекарственных средств, известно специалисту на основании его знания. Наряду с растворителями, гель-образователями, основами свечей, вспомогательными веществами для таблеток и другими носителями биологически активных веществ, можно применять, например, антиоксиданты, диспергаторы, эмульгаторы, антивспениватели, улучшающие вкус вещества, консерванты, агенты растворения или красители.
Биологически активные вещества можно вводить топически, орально, парентерально или внутривенно, причем предпочтительный род введения зависит от излечиваемого заболевания.
Предпочтительно оральное применение.
Для оральной формы применения активные соединения смешивают вместе с пригодные для этой цели добавками, как носители, стабилизаторы или инертные разбавители, и обычными способами переводят в пригонные формы для приема, как таблетки, драже, разъемные капсулы, водные, спиртовые или масляные суспензии или водные, спиртовые или масляные растворы. В качестве инертных носителей можно применять, например, гуммиарабик, магнезию, карбонат магния, фосфат калия, молочный сахар, глюкозу или крахмал, в особенности кукурузный крахмал. При этом приготовление можно осуществлять как в виде сухого, так и также мокрого гранулята. В качестве масляных носителей или растворителей принимают во внимание растительные или животные масла, как подсолнечное масло или рыбий жир.
Для подкожного или внутривенного ведения активные соединения или их физиологически приемлемые соли, в желательном случае вместе с обычными для этой цели веществами, как агенты растворения, эмульгаторы или другие вспомогательные вещества, растворяют, суспендируют, или эмульгируют. В качестве растворителей принимают во внимание, например, воду, физиологический раствор хлорида натрия или спирты, например, этанол, пропанол, глицерин, наряду с ними также растворы сахаров, как растворы глюкозы или маннита, или также смесь из различных растворителей.
В качестве фармацевтических препаратов для топического и локального применения пригодны глазные капли, которые содержат активное соединение в водном или масляном растворе. Для введения в нос пригодны аэрозоли и разбрызгиватели, а также грубые порошки, которые вводят за счет быстрых ингаляций через носовые отверстия и прежде всего капли в нос, которые содержат активные соединения в водном или масляном растворе.
Дозировка вводимого биологически активного вещества формулы (I) и частота приема зависят от силы действия и длительности действия используемого согласно изобретению соединения; кроме того, также от рода и интенсивности излечиваемого заболевания, как и от пола, возврата, веса и индивидуальной предрасположенности излечиваемого млекопитающего. В среднем рекомендуемая суточная доза предлагаемого согласно изобретение соединения составляет для млекопитающего весом примерно 75 кг, в первую очередь для человека, около 10-500 мг, предпочтительно примерно 25-250 мг, причем прием можно осуществлять при необходимости в виде нескольких доз в день.
Нижеследующие примеры должны иллюстрировать настоящее изобретение, однако, на ограничивая его объема охраны. Комнатная температура составляет примерно 18-25oC.
Пример 1
Получение 1,5-лактона 1L-1(OH),3,4,-O-циклогексилиден-5-тетрагидроксициклогексанкарбоновой кислоты (соединение 2) из D-хинной кислоты (соединение I).
163,3 г (0,85 моль) соединения 1 суспендируют в 186 мл (1,8 моль) циклогексанона. Добавляют 0,5 мл концентрированной сорной кислоты. Затем медленно нагревают до температуры нагревательной бани 200oC и отгоняют азеотроп воды с циклогексаном. После того, как никакого азеотропа более не отгоняется, светлокоричневый реакционный раствор перемешивают следующие 2 часа при температуре бани 200oC. После этого реакционный раствор охлаждают по 70oC и добавляют 10 г гидрокарбоната натрия. Затем смешивают с 700 мл этилацетата, органическую фазу промывают водой и насыщенным раствором хлорида натрия. После этого органическую фазу концентрируют в вакууме. Светложелтый остаток кристаллизуют из смеси изопропанола с водой в соотношении 1:1 и получают 142,1 г (75%) лактона 2 в виде бесцветных кристаллов.
Т. пл. 140-141oC.
Получение (1R, 2R, 3R, 5R)-1,2-O-циклогексилиден-5-фенилметилокси-3,5-лактонил- циклогексан-1,- 2-диола [соединение 3, R2=O-CH2C6H5]
0,81 г (28 ммоль) гидрида натрия (80%-ный в минеральном масле) в атмосфере аргона суспендируют в 14 мл безводного диметилформамида, и при 0oC прикапывают 7,1 г (28 ммоль) спирта 2, растворенного в 16 мл безводного диметилформамида. Затем перемешивают 1 час при 25oC и после этого снова при 0oC добавляют 3,5 мл бензилбромида. Реакционную смесь оставляют перемешиваться 4 часа при комнатной температуре и затем смешивают при 0oC с насыщенным раствором хлорида аммония. Экстрагируют этилацетатом, объединенные органические фазы промывают насыщенным раствором хлорида натрия и сушат над сульфатом натрия. После концентрирования в вакууме остаток перекристаллизуют из смеси н-гептана с метил-трет.-бутиловым эфиром (5:1). Получают 7,18 г (75%) простого бензилового эфира 3 (R2=O-CH2C6H5) в виде бесцветных кристаллов.
Т.пл. 122-126oC.
Получение натриевой соли (1S,3R,4R,5S)-3-гидрокси-4,5-O-циклогексилиден-1- фенилметоксициклогексанкарбоновой кислоты (соединение 4); R2=O-CH2C6H5) из соединения 3 (R2=O-CH2C6H5)
3,1 г (9 ммоль) Лактона 3 (R2=O-CH2C6H5) растворяют в 20 мл диоксана и при комнатной температуре добавляют 9,5 км 1 н. раствора гидроксида натрия. Эмульсию перемешивают 4 часа при комнатной температуре и затем концентрируют в вакууме; и бесцветней остаток сушат в течение 24 часов при 60oC в высоком вакууме над гидроксидом калия. Получают 3,32 г (96%) натриевой соли 4 (R2= O-CH2C6H5) в виде бесцветного твердого вещества.
Т. пл. 276-279oC (разложение).
Получение (1S, 3R,4R,5S)-3-[(E)-3-[4-(триметилсилилэтоксиметоксифенил)] пропеноил] окси-4,5-O-циклогексилиден-1-фенил-метилокси- циклогексанкарбоновой кислоты 6 (R2=O-CH2C6H5) из соединения 4 (R2=O-CH2C6H5)
а) Получение имидазолида (E-)-3-(-триметилсилилэтоксиметоксифенил)-2-пропеновой кислоты 5 (Ra=имидазолил)
а) - 1,62 г (5,5 ммоль) (Триметилсилилэтоксиметоксифенил)-пропеновой кислоты 5 [Ra=OH, Z = CH=CH; R3 (защищенный) = 4-(триметилсилилэтоксиметоксифенил)] растворяют в 10 мл безводного диметилформамида. При комнатной температуре прикапывают раствор 0,92 г (5,5 ммоль) карбонилдиимидазола, растворенного в 10 мл безводного диметилформамид. Затем этот раствор нагревают в течение 1 часа при 60-70oC, причем наблюдается выделение CO2.
б) - 2,1 г (5,5 - ммоль) соединения 4 (R2=O-CH2C6H5) растворяют в 20 мл безводного диметилформамида. В атмосфере аргона при 25oC добавляют 165 мг (5,5 ммоль) гидрида натрия (80%-ный в минеральном масле). Перемешивают в течение 1 часа при 25oC. Затем при 0oC прикапывают полученный в п. а) раствор соединения 5. Спустя 3 часа при 0-5oC реакционную смесь добавляют к насыщенному раствору хлорида аммония, экстрагируют этилацетатом и объединенные органические фазы промывают насыщенным раствором хлорида натрия, а затем сушат над сульфатом-магния. После концентрирования в вакууме остаток хроматографируют на силикагеле (растворитель:этилацетат/н-гептан/ледяная уксусная кислота = 20:60:1). Получают 2.5 г (71%) сложного эфира 6 [R2=O-CH2C6H5 ; Z = CH=CH; R3 (защищен) = 4-(триметилсилилэтоксиметоксифенил)] в виде бесцветного масла.
Получение (1S,3R,4R,5S)-3-[(E)-3-(4-гидроксифенил)-2-пропеноил]- окси-4,5-дигидрокси-1-фенилметилокси-циклогексанкарбоновой кислоты (соединение 7) [R3 = 4-гидроксифенил; Z = CH=CH, R2=фенилметилокси) из соединения 6
2,7 г (7,0 ммоль) соединения 6 растворяют в 130 мл диоксана и при комнатной температуре и при перемешивании смешивают с 95 мл (0,19 моль) 2 н. соляной кислоты. Перемешивают 20 часов при комнатной температуре. По окончании реакции в прозрачном растворе с помощью 2 н. раствора гидроксида натрия устанавливают pH 3-4 и концентрируют в вакууме. Твердый остаток при нагревании перемешивают в этилацетате и нерастворимый хлорид натрия отфильтровывают. Фильтрат концентрируют снова и остаток перемешивают с метил-трет.-бутиловым эфиром. Остаток отсасывают и сушат в высоком вакууме. Получают 2,0 г (70%) (1S,3R,4R,5S)-3- [(E)-3-(4-гидроксифенил)-2-пропеноил]окси-4,5-дигидрокси-1- фенилметилокси-циклогексанкарбоновой кислоты (7) в виде бесцветного твердого вещества.
Т. пл. 208-212oC.
Пример 2
(1S, 3R, 4R,5S)-3-[(E)-3-(4-Гидроксифенил)-2-пропеноил]-окси-4,5- дигидрокси-1-(2-тиенилметил)окси-циклогексанкарбоновая кислота
Т. пл. 140oC.
Пример 3
(1S, 3R, 4R,5S)-3-[(E)-3-(4-Гидроксифенил)-2-пропеноил]-окси-4,5- дигидрокси-1-(2-пропинил)окси-циклогексанкарбоновая кислота.
Т. пл. 197oC.
Пример 4
(1S, 3R, 4R,5S)-1-(4-Хлорфенилпропил)-окси-4,5-дигидрокси-3- (2-пиридинкарбонил)-окси-циклогексанкарбоновая кислота.
Т. пл. 128-130oC.
Пример 5
(1S, 3R,4R,5S)-1-(4-Хлорфенилпропил)окси-3-[(E)-3- (4-гидроксифенил)-2-пропеноил]окси-4,5-дигидрокси- циклогексанкарбоновая кислота.
Т. пл. 215-219oC.
Пример 6
(1S, 3R,4R,5S)-1-Метокси-3-[(E)-3-(4-Гидроксифенил)-2- пропеноил]-окси-4,5-дигидрокси-циклогексанкарбоновая кислота.
Т. пл. 242-243oC.
Пример 7
(1S, 3R, 4R,5S)-1-Этокси-3-[(E)-3-(4-гидроксифенил)-2-пропеноил]- окси-4,5-дигидроксициклогексанкарбоновая кислота.
Т. пл. 227-228oC.
Пример 8
(1S, 3R, 4R, 5S)-3-[(E)-3-(4-Гидроксифенил)-2-пропеноил]окси-4,5- дигидрокси-1-пропилоксициклогексанкарбоновая кислота.
Т. пл. 221oC.
Пример 9
(1S, 3R, 4R,5S)-1-(3-Фенилпропил)окси-3-[(E)-3-(4-гидроксифенил)- 2-пропеноил]-окси-4,5-дигидрокси-циклогексанкарбоновая кислота.
Т. пл. 203oC.
Пример 10
(1S, 3R, 4R,5S)-1-(4-Хлорфенилметил)окси-3-[(E)-3- (4-гидроксифенил)-2-пропеноил]-окси-4,5-дигидрокси- циклогексанкарбоновая кислота
Т. пл. 211oC.
Пример 11
(1S, 3R,4R,5S)-1-(4-Метилфенилметил)окси-3-[(E)-3- (4-гидроксифенил)-2-пропеноил]окси-4,5-дигидрокси-циклогексанкарбоновая кислота.
Т. пл. 198oC.
Пример 12
(1S,3R,4R,5S)-1-(4-Трифторметилфенилметил)окси-3-[(E)-3-(4- гидроксифенил)-2-пропеноил]окси-4,5-дигидрокси- циклогексанкарбоновая кислота.
Т. пл. 195-200oC.
Пример 13
(1S,3R,4R,5S)-(4-Бифенилметил)окси-3-[(E)-3-(4-гидроксифенил)- -2-пропеноил]окси-4,5-дигидрокси-циклогексанкарбоновая кислота.
Т. пл. 222oC.
Пример.14
(1S, 3R, 4R,5S)-1-(1-Нафтилметил)окси-3-[(E)-3-(4-гидроксифенил)- 2-пропеноил]окси-4,5-дигидрокси-циклогексанкарбоновая кислота.
Т. пл. 165-170oC.
Пример 15
(1S, 3R, 4R,5S)-1-(2-Нафтилметил)окси-3-[(E)-3-(4-гидроксифенил)- 2-пропеноил]окси-4,5-дигидроксициклогексанкарбоновая кислота.
Т.пл. 198oC.
Пример 16
(1S, 3R,4R,5S)-1-(3-Метоксифенилметил)окси-3-[(E)-3-(4-гидроксифенил) -2-пропеноил]окси-4,5-дигидрокси-циклогексанкарбоновая кислота.
Т. пл. 189-191oC.
Пример 17
(1S, 3R, 4R,5S)-1-(4-Фторфенилметил)окси-3-[(E)-3- (4-гидроксифенил)-2-пропеноил]окси-4,5-дигидрокси- циклогексанкарбоновая кислота.
Т. пл. 214oC
Пример 18
(1S, 3R,4R,5S)-1-(4-Цианофенилметил)окси-3-[(E)-3- (4-гидроксифенил)-2-пропеноил]окси-4,5-дигидрокси- циклогексанкарбоновая кислота
Т. пл. 238-241oC.
Пример 19
(1S, 3R, 4R,5S)-1-[3-(3-Метоксифенил)пропил]-окси-3-[(E)-3-(4- гидроксифенил)-2-пропеноил]окси-4,5-дигидрокси-циклогексанкарбоновая кислота.
Т. пл. 208-210oC
Пример 20
(1S, 3R, 4R,5S)-1-[(E)-3-(4-Хлорфенил)-2-пропенил]окси-3-[(E)-3- (4-гидроксифенил)-2-пропеноил]окси-4,5-дигидрокси- циклогексанкарбоновая кислота.
Т. пл. 170-173oC.
Пример 21
(1S, 3R,4R,5S)-1-[(3-Хлорфенил)пропил]окси-3-[(E)-3- (4-гидроксифенил)-2-пропеноил]-окси-4,5-дигидрокси- циклогексанкарбоновая кислота.
Т. пл. 211oC
Пример 22
(1S, 3R, 4R, 5S)-1-(4-Фенилбутил)окси-3-[(E)-3-(4-гидроксифенил)- 2-пропеноил]-окси-4,5-дигидрокси-циклогексанкарбоновая кислота.
Т. пл. 217oC.
Пример 23
(1S, 3R,4R,5S)-1-(3,3-Дифенилпропил)окси-3-[(E)-3- (4-гидроксифенил)-2-пропеноил]-окси-4,5-дигидрокси- циклогексанкарбоновая кислота.
Т. пл. 155-160oC.
Пример 24 Натриевая соль
(1S,3R,4R,5S)-1-[3-(4-трет.-Бутилфенил)метил]окси-3-[(E)-3- (4-гидроксифенил)-2-пропеноил]окси-4,5-дигидрокси- циклогексанкарбоновой кислоты.
Т. пл. 80-90oC.
Пример 25
(1S, 3R, 4R, 5S)-1-[3-(4-Хлор-2-метокси-фенил)пропил]окси-3-[(E)- 3-(4-гидроксифенил)-2-пропеноил]окси-4,5-дигидрокси- циклогексанкарбоновая кислота.
Т. пл.190-194oC.
Пример 26
(1S,3R,4R,5S)-1-[3-(5-Хлор-2-метоксифенил)пропил]окси-3-[(E)- 3-(4-гидроксифенил)-2-пропеноил]окси-4,5-дигидрокси- циклогексанкарбоновая кислота.
Т. пл. 215-218oC.
Пример 27
(1S,3R,4R,5S)-1-[3-(4-Хлорфенил)-2-пропинил]окси-3-[(E)-3-(4- гидроксифенил)-2-пропеноил]окси-4,5-дигидрокси-циклогексанкарбоновая кислота.
Т. пл. 233-234oC.
Пример 28
(1S, 3R, 4R,5S)-1-[3,3-ди(4-Хлорфенил)пропил]окси-3-[(E)-3-(4- гидроксифенил)-2-пропеноил]окси-4,5-дигидрокси-циклогексанкарбоновая кислота.
Т. пл. 122-126oC.
Пример 29
(1S,3R,4R,5S)-1-[(E)-3-(2-Хлорфенил)-2-пропенил]окси-3-[(E)-3- (4-гидроксифенил)-2-пропеноил]окси-4,5-дигидрокси- циклогексанкарбоновая кислота.
Т. пл. 192-196oC.
Пример 30
(1S,3R,4R,5S)-1-(4-феноксибутил)окси-3-[(E)-3-(4-гидроксифенил)- 2-пропеноил]окси-4,5-дигидрокси-циклогексанкарбоновая кислота
Т. пл. 194-195oC.
Пример 31
(1S,3R,4R,5S)-1-[3-(3,4-Дихлорфенил)пропил]окси-3-[(E)-3- (4-гидроксифенил)-2-пропеноил]окси-4,5-дигидрокси- циклогексанкарбоновая кислота.
Т. пл. 213-215oC.
Пример 32
(1S, 3R, 4R,5S)-1-[4-Хлорфенил)бутил]окси-3-[(E)-3- (4-метоксифенил)-2-пропеноил]окси-4,5-дигидрокси-циклогексанкарбоновая кислота.
Т.пл. 77-82oC.
Пример 33
(1S, 3R, 4R, 5S)-1-[3-(4-Хлорфенил)пропил] окси-3-[(E)-3- (4-метоксифенил)-2-пропеноил]окси-4,5-дигидрокси- циклогексанкарбоновая кислота.
Масс-спектр: m/e = 505 (M + H+).
Пример 34
(1S, 3R, 4R, 5S)-1-[3-(4-Хлорфенил)пропил] -окси-3-[(E)-3- (2-метоксифенил)-2-пропеноил]окси-4,5-дигидрокси- циклогексанкарбоновая кислота.
Масс-спектр: m/e = 505 (M + H+).
Пример 35
(1S, 3R,4R,5S)-1-[4-[3-(2-Этоксикарбонилтиенил)окси/бутил]окси- 3-[(E)-3-(4-гидроксифенил)-2-пропеноил] -окси-4,5-дигидрокси- циклогексанкарбоновая кислота.
Т. пл. 144-147oC.
Пример 36
(1S, 3R, 4R,5S)-1-[3-(2-Тиенил)пропил]окси-3-[(E)-3- (4-гидроксифенил)-2-пропеноил]окси-4,5-дигидрокси- циклогексанкарбоновая кислота.
Т. пл. 211-213oC
Пример 37
(1S, 3R, 4R,5S)-1-[(2-Тиенил)-метил]окси-3-[(E)-3- (4-гидроксифенил)-2-пропеноил]окси-4,5-дигидрокси- циклогексанкарбоновая кислота.
Т. пл. 140oC (разложение).
Пример 38
(1S, 3R, 4R, 5S)-1-[(2-Тиенил)метил]окси-3-[(E)-3- (3-метокситиенил)-2-пропеноил]окси-4,5-дигидрокси- циклогексанкарбоновая кислота.
Масс-спектр: m/e = 455 (M + H+).
Пример 39
(1S, 3R,4R,5S)-1-[3-(3-Тиенил)метил]окси-3-[(E)-3- (4-гидроксифенил)-2-пропеноил]окси-4,5-дигидрокси- циклогексанкарбоновая кислота.
Т. пл. 156-160oC.
Пример 40
(1S,3R,4R,5S)-1-[3-(2-(5-Хлор-тиенил))пропил]окси-3-[(E)-3- (4-гидроксифенил)-2-пропеноил)окси-4,5-дигидрокси- циклогексанкарбоновая кислота.
Т. пл. 215-218oC.
Пример 41
(1S,3R,4R,5S)-1-[4-(3,5-Диметилдитиено(3,2-в: 3',2'-е) пиридинил)-бутил] окси-3-[(E)-3-(4-гидроксифенил)-2-пропеноил] окси- 4,5-дигидрокси-циклогексанкарбоновая кислота.
Т. пл. 240-244oC.
Пример 42
(1S, 3R, 4R,5S)-1-[(3,5-Диметилдитиено)3,2-б:3',2'-е(-пиридинил)- метил] окси-3-[(E)-3-(4-гидроксифенил)-2-пропеноил] окси-4,5- дигидрокси-циклогексанкарбоновая кислота.
Т. пл. 240-244oC.
Пример 43
(1S, 3R,4R,5S)-1-[3-(3-Тиенил)пропил]-окси-3-[(E)-3-(4- гидроксифенил)-2-пропеноил]окси-4,5-дигидрокси-циклогексанкарбоновая кислота.
Т.пл. 211-213oC.
Пример 44
(1S, 3R, 4R, 5S)-1-(4-Хлорфенилпропил)окси-3-[(E)-3-(4- гидроксифенил)пропил]окси-4,5-дигидрокси-циклогексанкарбоновая кислота.
Т. пл. 157-159oC.
Ряд других соединений получают по способу В.
1. Алкилирование.
30,0 г (0,118 моль) Лактона 2 растворяют в 200 мл безводного диметилформамида. В атмосфере аргона и при комнатной температуре добавляют 5,3 г (0,176 моль) гидрида натрия (80%-ный в минеральном масле). Спустя 1,5 часа охлаждают до 0-10oC и в течение 30 минут прикапывают 20 мл (0,265 моль) пропаргилбромида. Раствор медленно окрашивается в темный цвет. Спустя 1 час (ТСХ-контроль) реакционную смесь концентрируют наполовину. Добавляют раствор хлорина аммония. Экстрагируют этилацетатом, органическую фазу промывает насыщенным раствором хлорида натрия и сушат над сульфатом магния. После концентрирования в вакууме остаток фильтруют через 1 кг силикагеля [растворитель: этил-ацетат/н-гептан = 1:5]. Получают 30,0 г (87%) простого пропаргилового эфира 3'(R2 = пропинилокси) в виде вязкого масла.
2-я стадия: Сопряжение
24,0 г (0,082 ммоль) простого пропаргилового эфира 3'(R2= пропинилокси) растворяют в 150 мл безводного толуола и 50 мл безводного триэтиламина. В атмосфере аргона последовательно добавляют 0,354 г (0,002 ммоль) дихлорида палладия. 1,05 г (0,004 ммоль) трифенилфосфина, 19,55 г (0,082 моль) 4-хлор-иодбензола и 0,050 г (0,0003 ммоль) иодида меди-(1). Реакционный раствор медленно нагревают до 80oC и реакционную смесь оставляют стоять в течение 4 часов при этой температуре. Затем охлаждают до комнатной температуры, отфильтровывают образовавшийся триэтиламмонийгидробромид и осадок промывают этилацетатом. Фильтрат концентрируют в вакууме и вязкий маслянистый остаток очищают путем хроматографии на 1 кг силикагеля [растворитель: этилацетат/н-гептан = 1:5; для нанесения на силикагель остаток растворяют в небольшом (! ! ) количестве этилацета]. Получают 23,0 г (69%) простого фенилпропинилового эфира 3" (R2 = 3-(3-хлорфенил)-2-пропинилокси), который можно перекристаллизовывать из метилциклогексана. Т. пл. 79oC.
Получение Алкена 3'' (R2= 3-(4-хлорфенил)-2-пропенилокси) из Алкина 3''[R2=3-(4-хлорфенил)-2-пропинилокси]:
12,0 г (29,8 ммоль) Алкина 3''/R2 = 3-(4-хлорфенил)-2- пропинилокси] растворяют в 300 мл пиридина и добавляют 3,0 г палладия-на-сульфате бария (10% палладия). Суспензию встряхивают 4 часа при 25oC в атмосфере водорода. По окончании поглощения водорода катализатор отфильтровывают и пиридиновый раствор концентрируют в вакууме. Получают 11,2 г (93%) алкена 3''[R2 = 3-(4-хлорфенил)пропенилокси) в виде бесцветного твердого вещества. Т. пл. 155-157oC.
Дальнейшие реакционные стадии осуществляют аналогично способу А (стадии 3-7).
Получение алкала 3IV (R2= 3-(4-хлорфенил)пропилокси) из алкина 3''' (R2 = 3-(4-хлорфенил)пропинилокси):
6,0 г (14,9 ммоль) Алкина 3'''(R2= 3-(4-хлорфенил)-2- пропинилокси) растворяют в 50 мл смеси этанола с этилацетатом (1:4) и добавляют 1,0 г родия на оксиде алюминия (5% родия). Реакционную смесь встряхивают атмосфере водорода при комнатной температуре в течение примерно 15 часов. Затем катализатор отфильтровывают и фильтрат концентрируют в вакууме. Получают 6,05 г (100%) алкана 3IV (R2= 3-(4-хлорфенил)пропилокси) в виде бесцветного масла.
Также таким образом полученные алканы 3 вводят во взаимодействие далее согласно способу А (стадии 3-7).
Пример 45.
(1S, 3R, 4R, 5S)-1-[3-(4-Фторфенил)пропил] окси-3-[(E)-3- (4-гидроксифенил)-2-пропеноил]окси-4,5-дигидрокси- циклогексанкарбоновая кислота.
Т. пл. 140-170oC.
Пример 46
(1S,3R,4R,5S)-1-[(Z)-3-(4-Хлорфенил)-2-пропенил]-окси-3-[(E)- 3-(4-гидроксифенил)-2-пропеноил]окси-4,5-дигидрокси- циклогексанкарбоновая кислота.
Т. пл. 208-209oC.
Пример 47
(1S,3R,4R,5S)-1-[(Z)-3-(5-Пиримидил)-2-пропенил]-окси-[(E)- 3-(4-метоксифенил)-2-пропаноил]окси-4,5-дигидрокси- циклогексанкарбоновая кислота.
Т пл. 75-78oC.
Пример 48
(1S,3R,4R,5S)-1-[(IZ)-3-(5-Пиримидил)-2-пропенил]окси-3-[(E)-3- (4-метоксифенил)-2-пропеноил]окси-4,5-O-циклогексилиден- циклогексанкарбоновая кислота.
Т. пл. 165-167oC.
Пример 49
(1S,3R,4R,5S)-1-[(Z)-3-(2-Нафтил)-2-пропенил]окси-3-[(E)-3- (4-метоксифенил)-2-пропеноил]окси-4,5-дигидрокси- циклогексанкарбоновая кислота.
Т. пл. 146-149oC.
Пример 50
(1S, 3R, 4R,5S)-I-[(Z)-3-(3-Трифторметилфенил)-2-пропенил]окси- 3-[(E)-3-(4-гипрокси)-2-пропеноил] окси-4,5-дигидрокси- циклогексанкарбоновая кислота.
Т. пл. 187-190oC.
Пример 51 Метиловый эфир
(1S, 3R, 4R, 5S)-1-[3-(4-Хлорфенил)пропил] окси-3-[(E)-3-(4- метоксифенил)-2-пропеноил]-окси-4,5-дигидрокси-циклогексанкарбоновой кислоты.
Масс-спектр: m/e = 505 (M + H+).
Пример 52
(1S,3R,4R,5S)-1-[3-(4-Хлорфенил)пропил]окси-3-[(E)-3-фенил-2- пропеноил] окси-4,5-дигидрокси-циклогексанкарбоновая кислота.
Масс-спектр: m/e = 475 (M + H+).
Пример 53
(1S, 3R, 4R, 5S)-1-[3-(4-Хлорфенил)пропил] окси-3-[(E)-3-(3,4- дихлорфенил-2-пропеноил]окси-4,5-дигидрокси-циклогексанкарбоновая кислота.
Масс-спектр: m/e = 543 (M + H+).
Пример 54
(1S, 3R,4R,5S)-1-[3-(4-Хлорфенил)пропил]окси-3-[(E)-2-фенил-1- циклопропилкарбоноил]окси-4,5-дигидрокси-циклогексанкарбоновая кислота
Масс-спектр: m/e = 489 (M + H+).
Пример 55,
(1S, 3R, 4R, 5S)-1-[3-(4-Хлорфенил)пропил] окси-3-[3- (4-гидроксифенил)пропил]окси-4,5-дигидрокси-циклогексанкарбоновая кислота
Масс-спектр: m/e = 493 (M + H+).
Пример 56
(1S, 3R, 4R, 5S)-1-[3-(4-Хлорфенил)пропил] окси-3-[3- (4-метоксифенил)-пропоил]окси-4,5-дигидрокси-циклогексанкарбоновая кислота
Масс-спектр: m/e = 507 (M + H+).
Пример 57
(1S, 3R, 4R,5S)-1-[2-(4-Хлорфенил)-1-циклопропиленметил]окси-3- [(E)-3-(4-гидроксифенил)-2-пропеноил] окси-4,5-дигидрокси- циклогексанкарбоновая кислота.
Т. пл. 195-199oC.
Примеры вариации остатков R4 и R5 согласно способу Б:
Получение лактондиола 8 [R2 = -O-CH-CH=CH-(п-хлорфенил), цис) из соединения 3 [R2= -O-CH-CH=CH-(п-хлорфенил), цис]
11,0 г (27,7 ммоль) Лактона 3 = [R2= -O-CH-CH=CH-(п-хлорфенил), цис] растворяют в изопропаноле. Добавляют 40 мл 2 н. соляной кислоты. Реакционный раствор оставляют стоять в течение 48 часов при комнатной температуре и затем нейтрализуют с помощью 1 н. раствора гидроксида натрия, концентрируют в вакууме и остаток хроматографируют на силикагеле. Получают 7:8 г (90%) лактондиола 8 [R2= -O-CH-CH= CH-(п-хлорфенил), цис] в виде бесцветного твердого вещества. Т. пл. 117-120oC.
Получение 5-трет. -бутилметилсилилокси-соединения 9 [R2= -O-CH-CH=CH-(п-хлорфенил), цис] из лактондиола 8 [R2= -O-CH-CH=CH-(п-хлорфенил), цис]:
5,0 г (15,4 ммоль) лактондиола 8 [R2= -O-CH-CH=CH-(п-хлорфенил), цис] и 4,15 г (61,9 ммоль) имидазола растворяют в 50 мл безводного диметилформамида. При 0oC добавляют 3,9 г (26 ммоль) трет.-бутилдиметилсилилхлорида. Спустя 4 часа реакционную смесь смешивают с насыщенным раствором хлорида аммония и экстрагируют метил-трет.-бутиловым эфиром. Объединенные органические фазы сушат над сульфатом магния и концентрируют в вакууме. Остаток очищают на силикагеле [растворитель:этилацетат] н-гептан= 1:3. Получают 5,7 г (84%) простого силилового эфира 9 [R2= -O-CH-CH=CH-(п-хлорфенил), цис] в виде бесцветного твердого вещества. Т. пл. 71oC.
Когда реакцию осуществляют при комнатной температуре, то таким образом также можно получать смесь обоих простых силиловых эфиров 9 и 10, которую можно разделять хроматографически на силикагеле с помощью смеси вышеуказанных растворителей.
Получение 3,3-ди-(4-хлорфенил)-2-пропенилового простого эфира II [R2= -O-CH-CH= CH-(п-хлорфенил), цис] из соединения 9 [R2= -O-CH-CH=CH-(п-хлорфенил), цис]:
1,0 г (2,3 ммоль) спирта 9 [R2= -O-CH-CH=CH-(п-хлорфенил), цис] растворяют в 20 мл безводного диметилформамида. В атмосфере аргона и при комнатной температуре прикапывают 150 мг (5 ммоль) гидрида натрия (80%-ный в минеральном масле) и перемешивают в течение 1 часа. Затем охлаждают по 0oC и добавляют 0,85 г (3,2 ммоль) 3,3-ди-(4-хлорфенил)-2-пропенилбромида, растворенного в 5 мл безводного диметилформамида, и реакционную смесь оставляют нагреваться по комнатной температуры. Спустя 14 часов реакционную смесь смешивают с насыщенным раствором хлорида аммония и экстрагируют простым метил-трет. -бутиловым эфиром. Объединенные органические фазы сушат над сульфатом магния и концентрируют в вакууме. Остаток очищают на силикагеле [растворитель: этилацетат/н-гептан = 1: 3]. Получают 0,5 г (84%) простого эфира II [R2= -O-CH-CH=CH-(п-хлорфенил), цис] в виде бесцветного масла.
Соединение II аналогично способу А превращают в соединение примера 58:
Пример 58
(1S,3R,4R,5S)-1- [(Z)-3-(4-Хлорфенил)-2-пропенил]окси-3-[(E)-3- -(4-гидроксифенил)пропоил] окси-4-[3,3-ди-(4-хлорфенил)-2-проденил]окси- 5-дигидрокси-циклогексанкарбоновая кислота.
Т. пл. 157-161oC.
Аналогично примеру 58 также синтезируют соединение примера 59:
Пример 59: Натриевая соль
(1S, 3R, 4R,5S)-1-[(Z)-3-(4-Хлорфенил)-2-пропонил]окси-3-[(E)- 3-(4-гидроксифенил)пропоил]окси-4-фенилметилокси-5-дигидрокси- циклогексанкарбоновой кислоты.
1H-ЯМР (270 МГц d6 -ДМСО): δ , т.л. = 1,85-2,3 (м, 3H); 3,3-3,5 (м, 2H); 4,05-4,70 (м, 6H). 5,2-5,38 (м, 1H), 5,82-5,93 (м, 1H), 6,3 (д, J = 10,0 гц, 1H), 6,42-6,5 (м, 1H), 6,75-6,85 (м, 2H), 7,2-7,55 (м, 12H), II (1H).
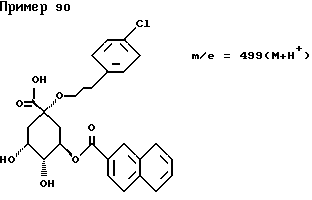
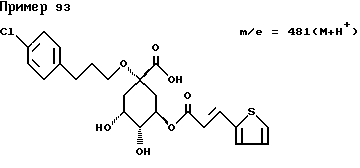
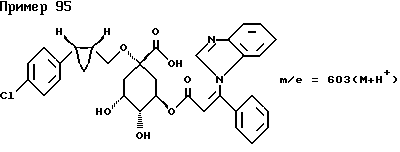
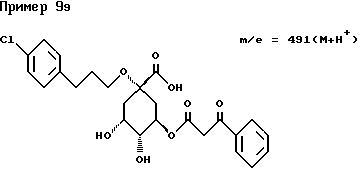
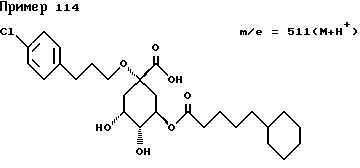
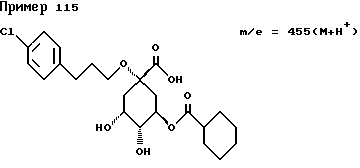
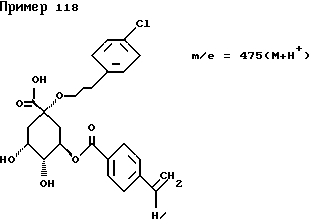
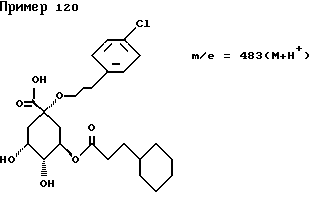
Аналогичным образом, как описано в примере I, получают следующие соединения нижеприведенных примеров:


Пример 63
1) 30,0 г (73,7 ммоль) соединения 63А: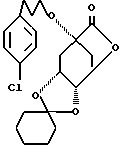
в 250 мл безводного толуола, в атмосфере аргона и при от -50oC до -60oC, смешивают с 68 мл 1,2 М раствора диизобутилалюминийгидрида. Спустя 1 час при -60oC прикапывают 50 мл смеси метанола с водой (9:1). Реакционную смесь нагревают по 0oC. Затем реакционную смесь вносят в 1 н. раствор гидросульфата калия (pH примерно 4), экстрагируют этилацетатом и органическую фазу сушат над сульфатом натрия. После концентрирования получают 30,0 г соединения 63В:
которое без очистки вводят во взаимодействие далее.
2) 19,8 г (88,1 ммоль) триэтилфосфонацетата растворяют в 200 мл безводного тетрагидрофурана. В атмосфере аргона и при 0-5oC порциями добавляют 2,65 г 80%-ного гидрида натрия. Спустя 20 минут получают прозрачный коричневатый раствор, к которому при от -40oC до -50oC прикапывают 30,0 г (73,4 ммоль) соединения 63B, растворенного в 100 мл безводного тетрагидрофурана. Спустя 4 часа при от -20oC до -30oC реакционную смесь вносят в насыщенный раствор хлорида аммония, экстрагируют этилацетатом и объединенные органические фазы сушат над сульфатом натрия и концентрируют в вакууме. После хроматографической очистки остатка на силикагеле получают 22,3 г соединения 63C:
в виде бесцветного масла.
3) Соединение 63C согласно известным специалисту способам превращают в соединение 63D: и аналогично примеру 1 (стадии "б" и 6 __→ 7 превращают далее в соединение 63 формулы:
и аналогично примеру 1 (стадии "б" и 6 __→ 7 превращают далее в соединение 63 формулы:
Аналогично примеру 1 получают соединения следующих примеров:



Пример 68
1) 15 мл 2М раствора диэтилцинка в толуоле вносят, при 0oC, в 150 мл водного дихлорметана и в атмосфере аргона прикапывают 10,4 г (59,2 ммоль) хлориодметана и перемешивают при 0-5oC в течение 50 минут. Затем прикапывают 6,0 г (14 8 ммоль) олефина 68A:
растворенного в 50 мл безводного дихлорметана. Реакционный раствор оставляют нагреваться по 25oC в точение 2-х часов и после этого гидролизуют с помощью насыщенного раствора хлорида аммония, экстрагируют этилацетатом и концентрируют в вакууме. Получают 5,5 г (91%) циклопропанового производного 68B формул: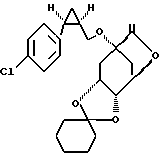

(смесь в соотношении 3:1 обоих возможных диастереомеров), которые можно разделять путем кристаллизации из изопропанола.
2) 2,0 г (4,8 ммоль) Лактона 68B в атмосфере аргона растворяют в 50 мл водного толуола и при -60oC прикапывают 4,1 мл 1,2 М раствора диизобутилалюминий гидрида в толуоле. Перемешивают 2 часа при -60oC и реакционный раствор гидролизуют с помощью 10 мл воды. Эту смесь смешивают с насыщенным раствором хлорида аммония, экстрагируют этилацетатом, сушат над сульфатом магния и концентрируют в вакууме. Получают 2,0 г (99%) лактола 68C формулы:
в виде бесцветного масла.
3) 2,0 г (4,76 ммоль) соединения 68C растворяют в 50 мл метанола. При 25oC прикапывают раствор 5,1 г гидроксиамингидрохлорида и 5,0 г гидроксида калия в 50 мл метанола. После перемешивания в течение 2 часов при 25oC реакционный раствор вносят в воду и экстрагируют простым метил-трет.-бутиловым эфиром. Объединенные органические фазы сушат над сульфатом магния и концентрируют в вакууме. Сырой продукт 68D вводят во взаимодействие далее без очистки.

4) 2,1 г (4,8 ммоль) Соединения 68D вносят в 50 мл водного дихлорметана и при 25oC добавляют 4,6 г (12,3 ммоль) карбонилдиимидазола. Затем нагревают 3 часа при 40oC и по окончании выделения CO2 добавляют 100 мл безводного метанола и снова нагревают 4 часа при 40oC. После этого концентрируют в вакууме, остаток обрабатывают простым метил-трет.-бутиловым эфиром, промывают 0,1 н/ раствором гидросульфата калия и органическую базу сушат над сульфатом магния. После концентрирования органической базы остаток очищают путем хроматографии на силикагеле [растворитель: этилацетат/н-гептан = 1:4] и получают 1,3 г соединения 68E формулы:
в виде бесцветного масла.
5) Аналогично превращению соединения 4 в соединение 6 согласно примеру 1 из соединения 68E получают соединение 68 формулы:
Пример 69
Аналогично примеру 1 получают соединение 69: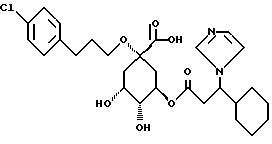
Пример 70
Стадия 1: Метиловый эфир 1-[4-(4-хлорфенил)-бутил]-циклогекс-3-ен-карбоновой кислоты 70B из соединения 70A:
85 ммоль (11,9 мл) безводного диизопропиламина растворяют в 200 мл безводного ТГФ и в атмосфере защитного газа (азот или аргон) охлаждают на охлаждающей бане со смесью сухого льда с ацетоном. К полученному раствору при хорошем перемешивании добавляют 80 ммоль (50 мл) 1,6 М раствора н-бутиллития в гексане. Перемешивают дополнительно 10 минут и затем прикапывают 75 ммоль (10,5 г) метилового эфира циклогекс-3-ен-1-карбоновой кислоты 70A:
(продажное соединение), растворенного в 10 мл ТГФ так, чтобы внутренняя температура не превышала -65oC. После этого перемешивает 30 минут при от -70oC до -80oC и затем прикапывают 74 ммоль (21,8 г) 4-/4-хлорфенил/-бутилиодида, растворенного в 25 мл ТГФ, так, чтобы внутренняя температура не превышала -35oC. Потом перемешивают 3 часа при от -70oC до -80oC и охлаждающую баню удаляют. После того, как внутренняя температура достигает 10oC, реакционный раствор при перемешивании вносят в 400 мл насыщенного раствора хлорина аммония, экстрагируют 3 раза простым метил-трот.-бутиловым эфиром, объединенные экстракты промывают 3 раза водой и 2 раза насыщенным раствором хлорида натрия, сушат над сульфатом натрия и растворитель удаляют в вакууме. Сырой продукт очищают путем флэш-хроматографии на силикагеле [растворитель: этилацетат/н-гептан = 1:9 по объему]. Продукт 70B осаждается в виде белого, низкоплавящегося воска Масс-спектр: m/e = 307 (M+H+).

Стадия 2: Натриевая соль [4-(4-хлорфенил)-бутил]-циклогекс-3-ен-1-карбоновой кислоты 70 из соединения 70B:
22,7 г метилового эфира 1[4-(4-хлорфенил)-бутил]-циклогекс-3-ен-1-карбоновой кислоты 70B растворяют в 100 мл метанола + 100 мл диоксана. К полученному раствору добавляют раствор 8 г гидроксида натрия в 50 мл воды и кипятят с обратным холодильником в течение 16 часов в атмосфере защитного газа. Охлажденный реакционный раствор смешивают с 200 мл воды, 100 мл толуола и 100 мл н-гептана и хорошо перемешивают. Осадившийся продукт отсасывают, дополнительно промывают небольшим количеством холодной воды и смесью н-гептана с толуолом (1:1 по объему) и сушат в вакууме. Получают соединение 70C формулы:
в виде бесцветных блестящих чешуек, которые но плавятся вплоть до 240oC. Полученная из натриевой соли 70C путем подкисления концентрированной соляной кислотой свободная кислота плавится при 86-87oC.
Стадия 3: 1-[4-(4-Хлорфенил)-бутил]-4-экзо-иод-6-оксабицикло/3.2.1/-октан-7- он(70D) из соединения 70C:
22,2 г натриевой соли 1-[4-(4-хлорфенил)-бутил]циклогекс-3- ен-1-карбоновой кислоты 70C суспендируют в растворе 22 г гидрокарбоната натрия и 68 г иодида калия в 350 мл воды. К полученной суспензии добавляют 175 мл простого метил-трет. бутилового эфира и 20 г йода и перемешивают 16 часов при комнатной температуре в атмосфере защитного газа. Реакционный раствор порциями смешивают с 10%-ным водным растворов бисульфита натрия, по тех пор, пока не исчезнет йодная окраска, и трижды экстрагируют этилацетатом. Экстракты промывают дважды насыщенным раствором хлорида натрия, сушат над сульфатом натрия и выпаривают в вакууме. Получают соединение 70D:
представляющее собой слегка желтоватое твердое вещество с т. пл. 84-86oC.
Стадия 4. 1-[4-(4-Хлорфенил)-бутил] -6-оксабицикло/3.2.1/-октан-7-он (соединение 70E) из соединения 70D:
2 г 1-[4-(4-Хлорфенил)-бутил]-4-экзо-иод-6-оксабицикло(3.2.1)- октан-7-она (70D) растворяют в 20 мл безводного простого метил-трет.-бутилового эфира. К раствору, в атмосфере защитного газа, добавляют примерно 0,1 1М раствора триэтилборана в ТГФ и затем прикапывают 1,35 мл гидрида трибутилолова. Дополнительно перемешивают 2 часа при комнатной температуре, затем добавляют раствор 5 г фторида калия в 50 мл воды и в течение 30 минут интенсивно перемешивают. Выделившийся осадок отсасывают, фильтрат экстрагируют трижды простым метил-трет.-бутиловым эфиром, экстракты промывают по два раза водой и насыщенным раствором хлорида натрия, сушат над сульфатом натрия и растворитель отгоняют в вакууме. Сырой продукт очищают путем флэш-хроматографии на силикагеле (растворитель: этилацетат/н-гептан = 1:3 по объему). Получают соединение 70E:
в виде бесцветного масла, которое застывает в неплавящийся воск. Масс-спектр: m/e = 293 (M+H+).
Стадии 5 и 6 осуществляют аналогично примеру 1. Получают соединение 70 формулы:
Аналогично примеру 1 получают соединения следующих примеров:

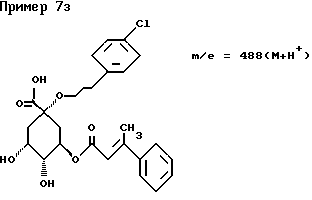

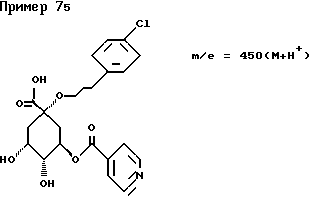
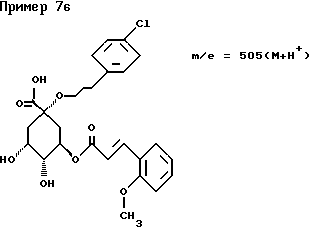


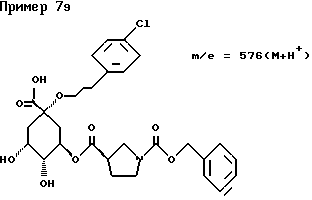
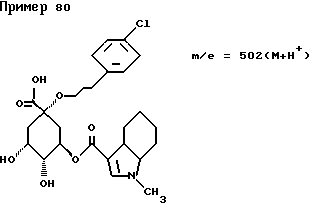
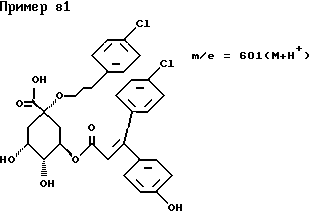





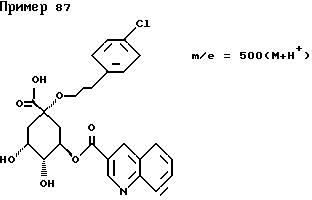













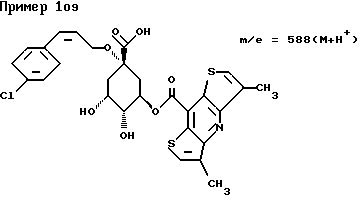
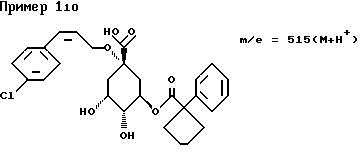
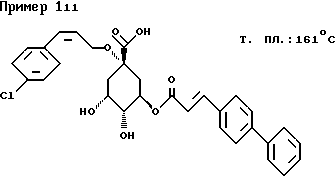
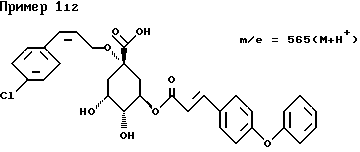




 д
д
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ЦИКЛОГЕКСАНКАРБОНОВОЙ КИСЛОТЫ, СМЕСЬ ИХ ИЗОМЕРОВ ИЛИ ОТДЕЛЬНЫЕ ИЗОМЕРЫ И ФИЗИОЛОГИЧЕСКИ ПЕРЕНОСИМЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2140416C1 |
| ПРОИЗВОДНЫЕ КАЛИХЕАМИЦИНА И ИХ КОНЪЮГАТЫ "АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО" | 2018 |
|
RU2732568C1 |
| МЕТАЛЛОЦЕНЫ С БЕНЗОКОНДЕНСИРОВАННЫМИ ПРОИЗВОДНЫМИ ИНДЕНИЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ, СПОСОБ ПОЛУЧЕНИЯ ОЛЕФИНОВОГО ПОЛИМЕРА И КАТАЛИЗАТОР ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ | 1992 |
|
RU2098423C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИОЛЕФИНОВ | 1992 |
|
RU2078771C1 |
| СПОСОБ ТОРМОЖЕНИЯ РЕТРОВИРУСНЫХ ПРОТЕАЗ | 1991 |
|
RU2028155C1 |
| НОВЫЕ ИНГИБИТОРЫ РАССАСЫВАНИЯ КОСТЕЙ И АНТАГОНИСТЫ РЕЦЕПТОРА ВИТРОНЕКТИНА | 1997 |
|
RU2195460C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПИРРОЛИДИН-3,4-ДИКАРБОКСАМИДА | 2005 |
|
RU2379288C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИОЛЕФИНОВ | 1992 |
|
RU2111211C1 |
| РЕАКЦИИ МАКРОЦИКЛИЗАЦИИ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И ДРУГИЕ ФРАГМЕНТЫ, ПРИГОДНЫЕ В ПОЛУЧЕНИИ АНАЛОГОВ ХАЛИХОНДРИНА B | 2014 |
|
RU2710545C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПЕПТИДОВ ИЛИ ИХ ФИЗИОЛОГИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ | 1990 |
|
RU2047621C1 |

Производные циклогексана формулы I, где R1 - CN или COOH, R2 - C1-10-алкил(R11)n, O-C1-C10-алкил(R11)n, O-C3-C10-алкенил(R11)n, O-C3-C10 алкинил(R11)n, S-C1-C10-алкил(R11)n, причем R11 может быть замещен R12; R12, R3, R11, R13 - C1-C10-алкил, C3-C8-циклоалкил, фенил, нафтил, пиридил, тиенил, имидазолил, фурил, индолил, хинолин или их тиено- или бнзоанеллированные производные, причем ароматический или гетероароматический остаток может быть замещен фтором, хлором, OH, CF3, CN, C1-C4-алкоксилом, фенилом или O-фенилом, R4, R5 - H, OH, обычная для гидроксила защитная группа или принимают значения, указанные для R2; R6 - H, Y - (CH2)m, Z - (CH2)m, S, - CH = C - (C1-C4-алканоил), - CH = C (R13), - CH2- CO-, C3-C10-циклоалкилен, причем 1 - 3 атома кольца могут быть замещены атомами азота, -C≡C- или -CH = C (C1-C4-алкилом), n = 0, 1, 2, m = 0, 1, 2, 3, 4, а также их физиологически приемлемые соли. Соединения I ингибируют глюкозо-6-фосфатазную систему печени млекопитающих, что должно приводить к уменьшенному гепатитному выделению глюкозы, и могут быть использованы для получения лекарственных средств для лечения диабета.

4 з.п. ф-лы, 1 табл.

где остатки имеют следующие значения:
R1 означает CN или COOH;
R2 означает C1 - C10-алкил (R11)n, или O - C1 - C10-алкил (R11)n, или O - C3 - C10-алкенил (R11)n, или O - C3 - C10-алкинил (R11)n, или S - C1 - C10-алкил (R11)n, причем в случае необходимости R11 может быть замещен, смотря по обстоятельствам, с помощью R12;
R12 - фенил, причем остаток может быть одно- или многократно замещен хлором;
R3, R11, R13 означают C1 - C10-алкил или циклоалкил с 3 - 8 атомами в кольце, или фенил, или нафтил, или пиридил, или тиенил, или имидазолил, или фурил, или индолил, или хинолил, или их тиено- или безоанеллированные производные, причем ароматический или гетероароматический остаток может быть одно- или многократно замещен фтором или хлором, или -OH, или -CF3, или -CN, или C1 - C4-алкоксилом, или фенилом, или O-фенилом, причем заместители являются одинаковыми или разными и R3, R11 и R13 являются одинаковыми и разными;
R4, R5 означают -H или -OH, или обычную для гидроксила защитную группу, или имеют значения, указанные для R2;
R6 - водород;
R4, R5 и R6 являются одинаковыми или разными;
Y - O-;
X означает -(CH2)m;
Z означает -(CH2)m, или -S-, или -CH = C - (C1 - C4-алканоил), или -CH = C(R13), или CH2 - CO-, или C3 - C10-циклоалкилен, причем 1 - 3 атома кольца могут быть замещены атомами азота или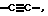 или -CH = C - (C1 - C4-алкил);
или -CH = C - (C1 - C4-алкил);
n = 0, 1, 2;
m = 0, 1, 2, 3 или 4,
а также физиологически приемлемые соли соединений формулы I.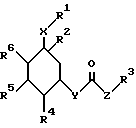
где остатки имеют следующие значения:
R1 означает -CN или -COOH;
R2 означает C1 - C10-алкил (R11)n, или O - C1 - C10-алкил (R11)n, или O - C3 - C10-алкенил (R11)n, или O - C3 - C10-алкинил (R11)n, или S - C1 - C10-алкил (R11)n, причем в случае необходимости R11 может быть замещен, смотря по обстоятельствам, с помощью R12;
R12 - фенил, одно- или многократно замещенный хлором;
R3, R11, R13 означают C1 - C4-алкил, или фенил, или нафтил, или пиридил, или тиенил, или имидазолил, или фурил, или индолил, или хинолил, или их тиено- или бензоаннелированные производные, причем ароматический, соответственно гетероароматический остаток, может быть одно- или многократно замещен фтором или хлором, или -OH, или CF3, или CN, или C1 - C4-алкоксилом, или C1 - C4-алкилом, или фенилом, или O-фенилом, причем заместители являются одинаковыми или разными и R3, R11 и R13 являются одинаковыми или разными;
R4, R5 означают H или OH, или защищенную обычной спиртовой группой OH-группу, или имеют указанные для R2 значения;
R6 - водород;
R4, R5, R6 являются одинаковыми или разными;
Y означает -O-;
X означает -(CH2)m-;
Z означает -(CH2)m-, -S-, или -CH = C - (C1 - C4-алканоил), -CH = C(R13) или CH2CO, или C3 - C10-циклоалкилен, или или -CH = C - (C1 - C4-алкил);
или -CH = C - (C1 - C4-алкил);
n = 0, 1 или 2;
n = 0, 1, 2, 3 или 4,
а также физиологически приемлемые соли соединений формулы (I). или -CH = C(C1 - C4-алкил).
или -CH = C(C1 - C4-алкил).
| Doodsma Z.F., hegeer B., etc | |||
| Y Biol | |||
| Chem., v.242, p.1955 - 1960, 1967 | |||
| Wallin B.K., etc., Biochem Biophys Res | |||
| Commun., v.48, 694 - 699, 1972 | |||
| Zoccoli M.A., Karnowski, etc | |||
| Y Biol | |||
| Chem., v.255, p.1113 - 1119, 1980 | |||
| Способ получения циклогексанкарбоновой кислоты | 1978 |
|
SU704452A3 |
| Способ получения (4,2,0)бициклооктановых производных, или их фармацевтически приемлемых нетоксичных солей, или фармацевтически приемлемых нетоксичных сложных эфиров | 1986 |
|
SU1500153A3 |
