Изобретение относится к производным антибиотиков липопептидного комплекса A 1437, способу их получения и их применению в качестве лекарственного средства.
В заявке на патент ФРГ 43 19 007.3 предлагаются липопептиды с очень гомологичными аминокислотными последовательностями, однако с различными остатками жирных кислот (липидной частью), которые синтезируются Actinoplanes sp. во время ферментации и выделяются в питательную среду, а также способ выделения липопептидов из питательной среды, их очистка и применение липопептидов в качестве фармакологических биологически активных веществ, в особенности против грамположительных бактерий.
Задачей настоящего изобретения теперь является поиск производных липопептидного комплекса A 1437 с меньшей токсичностью по сравнению с природными A 1437 липопептидами.
Эта задача, согласно изобретению, решается за счет производных согласно соединению формулы (I).
Таким образом, изобретение относится:
1) к производным липопептидам A 1437 формулы (I)
где R1 обозначает ОН или NH2;
R2 обозначает линейный или разветвленный, насыщенный или ненасыщенный, алифатический ацильный остаток с 8-22 C-атомами, который может быть прерван фенильными или циклоалкильными группами или кислородом;
и их фармацевтически приемлемым солям;
2) к способу получения соединения формулы (I), отличающемуся тем, что соединение формулы (II):
где R1 имеет вышеуказанное значение; и
R3 обозначает известную из химии пептидов защитную для амино-группы группу, предпочтительно трет. -бутоксикарбонильную (BOC), бензилоксикарбонильную (Z, Cbz,), флуоренилметоксикарбонильную (Fmoc) или аллилоксикарбонильную (Alloc) защитную группу; вводят во взаимодействие с карбоновой кислотой формулы (III):
R2OH, (III),
где R2 имеет вышеуказанные значения;
или с активированным по карбонильной группе производным этой карбоновой кислоты;
3) к фармацевтической композиции, содержащей липопетидной производное согласно формуле (I), а также фармацевтический носитель;
4) к применению липопептидного производного согласно формуле (I) для получения лекарственного средства против бактериальных инфекций.
Ниже изобретение описывается подробно, в особенности в его предпочтительных вариантах осуществления. Далее, изобретение определяется содержанием пунктов формулы изобретения.
Исходные соединения [соединения формулы (II)] получают из защищенных продуктов ферментации, например, из A 1437 [формула (I), R1 = ОН; R2 = (CH3)2CH(CH2)7CH = CHCH2CO] и 9-флуоренилметилового сложного эфира хлормуравьиной кислоты при образовании соответствующего соединения с
и путем последующего ферментативного отщепления остатка жирной кислоты с помощью Actinoplanes utahensis NRRL 12052 (J. Antibiotics, 1988, 1093).
Если используют сами карбоновые кислоты формулы (III) в качестве ацилирующего средства, то целесообразно работать в присутствии конденсационного средства, например карбодиимида, как N,N'-дициклогексилкарбодиимид. Активирование карбоновых кислот формулы (II) можно осуществлять обычными в химии пептидов способами, которые, например описываются в "Chemie in unserer Leit" 27, 274-286 (1993).
В качестве активированных производных, соответственно этому, пригодны галоидангидриды кислот, например хлорангидриды кислот, ангидриды или смешанные ангидриды, например, со сложными эфирами муравьиной кислоты; азиды; активированные сложные эфиры, как п-нитрофениловый, пентафторфениловый, 4,6-диметокси-1,3,5-триазин-2-иловый сложный эфир или сложный эфир с N-гидроксисукцинимидом или 1-гидроксибензотриазолом, которые получают с карбодиимидами в качестве реагентов сочетания, или сложные тиоэфиры, например, с 2-меркаптобензотриазолом. Другими пригодными реагентами сочетания являются N, N'-карбонилдиимидазол или таковые на основе солей фосфония или урония, как, например ВОР, HBTU, PyBOP, TBTU или TOTU (O-/циано-(этоксикарбонил)метилиденамино-1,1,3,3-тетраметил/-уроний-тетрафторборат).
В общем, взаимодействие соединений формулы (II) с карбоновой кислотой формулы (III) или ее активированным производным осуществляют в присутствии инертного растворителя, как, например дихлорметан или диметилформамид, предпочтительно в присутствии третичного основания, как, например, пиридин или этилдиизопропиламин. При применении замещенных бензоилхлоридов можно работать также в присутствии воды и добавки оснований, как пиридин или карбонат натрия. Взаимодействие можно осуществлять в температурном интервале от -20oC до +50oC, предпочтительно при -10oC - +30oC.
Отщепление защитных групп R3 при образовании соединений формулы (I) осуществляют по известным из литературы способам, например BOC-группу отщепляют с помощью трифторуксусной кислоты; Z - остаток отщепляют с помощью смеси HBr с ледяной уксусной кислотой или путем каталитического гидрирования; Alloc-группу отщепляют с помощью нуклеофильного соединения плюс Pd-катализатор; или Fmoc-группу отщепляют с помощью вторичных аминов, например пиперидина.
Предпочтительны соединения формулы (I), в которой R2 обозначает насыщенный алифатический ацильный остаток CH3(CH2)nCO; разветвленный насыщенный алифатический ацильный остаток, предпочтительно (CH3)2CH(CH2)nCO или CH3CH2CH(CH3)(CH2)nCO; ненасыщенный алифатический ацильный остаток, который может содержать одну или несколько двойных связей, причем двойная связь может находиться в транс- или цис-форме, предпочтительно
H2C = CH(CH2)nCO; (CH3)2CH(CH2)nCH = CH(CH2)nCO;
CH3(CH2CH = CH)n(CH2)nCO;
CH3(CH2)nCH = CH(CH2)nCH = CH(CH2)nCO;
CH3(CH2)nCH=CH-CO; CH3(CH2)nCH = CH(CH2)nCO;
H(CH2-C(CH3=CH CH2)nCO;
ненасыщенный алифатический остаток с одной или несколькими тройными связями, предпочтительно
HC ≡ C(CH2)nCO;
CH3(CH2)n ≡ C(CH2)nCO;
CH3(CH2)nC ≡ C-C ≡ C(CH2)nCO;
прерываемый фенильными или циклоалкильными остатками алифатический ацильный остаток, предпочтительно:
прерываемый кислородом ацильный остаток, предпочтительно:
и где "n" обозначает целые числа 0-20.
Особенно предпочтительны соединения, которые содержат линейный или разветвленный C12-C15-ацильный остаток, как, например тетрадеканоил, тридеканоил, 12-метилтридеканоил; ненасыщенный C12-C18-ацильный остаток с одной или несколькими двойными или тройными связями, как, например цис-10-пентадеценоил, транс-9-гексадеценоил,
H(CH2-C(CH3)=CHCH2)3CO; или прерываемый 1-3-я фенильными остатками и/или дополнительно кислородом, алифатический ацильный остаток, как, например:
где "n" обозначает целые числа 0 - 8.
Особенно предпочтительны соединения, которые содержат прерываемый 3 фенильными группами алифатический ацильный остаток, как, например,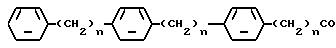
где "n" обозначает целые числа 0-2.
Далее, изобретение охватывает способ получения соединений формулы (I), который отличается тем, что соединение формулы (II):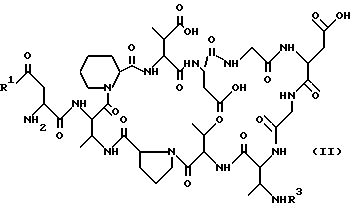
где R1 имеет вышеуказанное значение; и
R3 обозначает известную из химии пептидов защитную для амино-группы группу, как, например трет. -бутоксикарбонильная (BOC), бензилоксикарбонильная (Z, Cbz), флуоренилметоксикарбонильная (Fmoc) или аллилоксикарбонильная (Alloc) защитная группа, вводят во взаимодействие с карбоновой кислотой формулы (III):
R2OH, (III),
где R2 имеет уже указанные значения.
В качестве фармацевтически приемлемых солей соединений формулы (I) особенно пригодны соли с неорганическими и органическими кислотами, например, как соляная кислота, серная кислота, уксусная кислота, лимонная кислота, п-толуолсульфокислота; с неорганическими и органическими основаниями, как NaOH, KOH, Mg(OH)2, диэтаноламин, этилендиамин; или с аминокислотами, как аргинин, лизин, глутаминовая кислота и т.д. Их получают по стандартным методикам.
Одно или несколько соединений из предлагаемых согласно изобретению липопептидов, соответственно, их солей, на основании своего ценного фармакологического свойства пригодны для использования в качестве лекарственного средства.
Предлагаемые согласно изобретению вещества обладают фармакологической активностью, в частности антибиотической активностью против грамположительных бактерий, особенно предпочтительно против MRSA- и гликопептид-резистентных штаммов.
В случае резистентных к пенициллину, соответственно, метициллину штаммов (MRSA-штаммы), которые проявляют резистентности к другим антибиотикам, зачастую только гликопептиды, как ванкомицин или тейкопланин, обладают терапевтически достаточной активностью. Однако в возрастающей степени резистентные штаммы выступают также против этих антибиотиков (FEMS Microbiol. Lett. 98 (1992), 109-116).
Одно или несколько соединений из предлагаемых согласно изобретению липопептидов обладают превосходной активностью также против этих вызывающих проблему микроорганизмов.
Изобретение также относится к фармацевтическим композициям, включающим одно или несколько соединений из предлагаемых согласно изобретению липопептидов, соответственно, их солей.
Одно или несколько соединений из предлагаемых согласно изобретению липопептидов, предпочтительно одно или несколько соединений с тремя фенильными радикалами в ацильном остатке R2, в принципе могут вводиться как таковые. Предпочтительно применение в смеси с пригодными вспомогательными веществами, носителями или разбавителями. В качестве носителей в случае лекарственных средств для животных можно применять обычные кормовые смеси, соответственно, в случае лекарственных средств для людей - любые фармакологически приемлемые носители и/или вспомогательные вещества.
Предлагаемые согласно изобретению лекарственные средства в общем вводят орально или парентерально, однако также в принципе возможно ректальное (кишечное) применение. Пригодными твердыми или жидкими галеновыми формами композиций являются, например грануляты, порошки, таблетки, драже, (микро)капсулы, свечи, сиропы, эмульсии, суспензии, аэрозоли, капли или растворы для инъекций в ампулах, а также препараты с пролонгированным выделением биологически активного вещества, при приготовлении которых обычно находят применение носители и добавки и/или вспомогательные средства, как наполнители, связующие, покрывные средства, способствующие набуханию средства, придающие скользкость вещества или смазки, вкусовые вещества, подслащивающие вещества или агенты растворения.
В качестве обычно применяемых носителей или вспомогательных веществ следует назвать, например карбонат магния, диоксид титана, лактозу, маннит и другие сахара, тальк, молочный белок, желатину, крахмалы, витамины, целлюлозу и ее производные, животные или растительные масла, полиэтиленгликоли и растворители, как, например стерильная вода, спирты, глицерин и многоатомные спирты.
В качестве разбавителей следует упомянуть, например полигликоли, этанол и воду. Буферные вещества представляют собой, например, органические соединения, как, например N,N'-дибензилэтилендиамин, диэтаноламин, этилендиамин, N-метилглюкамин, N-бензилфенетиламин, диэтиламин, трис(гидроксиметил) аминометан; или органические соединения, как, например фосфатный буфер, бикарбонат натрия, карбонат натрия.
Также можно применять биологически активные вещества как таковые, без носителей или разбавителей, в пригодной форме. Пригодные дозы соединений формулы (I) или их фармацевтически приемлемых солей составляют примерно 0,4 г, предпочтительно 0,5 г - максимально 20 г в день для взрослого с весом тела примерно 75 кг. Можно вводить разовые дозы или многоразовые дозы, причем разовая доза может содержать биологически активное вещество в количестве примерно 50-1000 мг.
В случае необходимости, дозировочные единицы для орального введения можно микрокапсулировать, чтобы замедлить выделение или растянуть выделение в течение более продолжительного времени, как, например за счет покрытия или введения биологически активного вещества в форме частиц в пригодные полимеры, воски или т.п.
Предпочтительно фармацевтические композиции готовят и вводят в дозировочных единицах, причем каждая единица в качестве составной части содержит определенную дозу одного или нескольких соединений из предлагаемых согласно изобретению липопептидов. В случае твердых дозировочных единиц, как таблетки, капсулы или свечи, эта доза может составлять примерно вплоть до 200 мг, однако предпочтительно примерно 0,1-100 мг, а в случае растворов для инъекций в ампулах - вплоть до примерно 200 мг, предпочтительно, однако, примерно 0,5-100 мг в день.
Вводимая суточная доза зависит от веса тела, возраста, пола и состояния млекопитающего. При необходимости, однако, также можно вводить более высокие или более низкие суточные дозы. Введение суточной дозы может осуществляться как путем одноразовой дачи в форме единичной дозировочной единицы или, однако, в виде нескольких более маленьких дозировочных единиц, так и путем многократной дачи разделенных на части доз в определенные интервалы.
Предлагаемые согласно изобретению лекарственные средства получают тем, что одно или несколько соединений из предлагаемых согласно изобретению липопептидов вместе с обычными носителями, а также в случае необходимости добавками и/или вспомогательными веществами доводят до пригодных, соответственно, пригодной формы введения.
Особенно предпочтительные соединения формулы (I) с 3-я фенильными радикалами в ацильном остатке R2 (например, соединения 55, 56), сверх того, обладают особенно благоприятными токсикологическими свойствами. Так, в стандартном месте на гемолиз они практически не показывают никакого намека на гемолиз, в то время как все испытуемые соединения с линейными или разветвленными алифатическими ацильными остатками, включая природные вещества, проявляют значительную активность, составляющую 16-25% (см. ниже).
Гемолитическая активность ин витро 2)
Пример - Гемолиз
A 14371) X XF3CO2H - 17,5
1 - 19,6
6 - 16,5
7 - 25,7
8 - 19,3
9 - 22,8
14 - 22,9
49 - 0,0
55 - 0,5
56 - 0,4
1) Продукт ферментации формулы (I) (R1 = ОН;
R2 = (CH3)2CH(CH2)7CH = CHCH2CO).
2) Для измерения гемолитической активности применяют свежеотобранную, венозную кровь макак-резус. Кровь собирают в гепаринизированные пробирки и разделяют на аликвоты по 200 мкл на 12 полиэтиленовых пробирок.
Одну аликвоту смешивают с 200 мкл дистиллированной воды, и она служит в качестве 100%-ного стандарта; другую аликвоту смешивают с 200 мкл физиологического раствора хлорида натрия (0,9% NaCl) (0%-ный стандарт).
В прочие пробирки распределяют по 200 мкл разбавлений вещества в физиологическом растворе хлорида натрия до 1600; 800; 400; 200; 100; 50; 25; 12,5; 6,25 и 3,125 мг/л. Все пробирки осторожно встряхивают и затем инкубируют в течение 3-х часов при 37oC. Затем доливают 100%-ный стандарт с помощью 5 мл дистиллированной воды, а остальные доливают по 5 мл физиологического раствора хлорида натрия и центрифугируют в течение 5 минут при 700 g.
Гемолиз определяют путем измерения абсорбции надосадочной жидкости в спектрофотометре при длине волны 540 нм. Абсорбцию стандарта с полным гемолизом (дистиллированная вода) принимают за 100%. Абсорбции разбавлений испытуемых препаратов и 0%-ного стандарта измеряют, и в процентах указывают максимально индуцируемый гемолиз.
Следующие примеры осуществления для получения соединений согласно изобретению служат для дальнейшего пояснения изобретения.
В нижеследующих примерах изобретение поясняется далее. Данные в процентах относятся к весу. Соотношения компонентов в смеси в случае жидкостей относятся к объему, если не указано ничего другого.
Чистоту продуктов реакции определяют с помощью аналитической ВЭЖХ (Merck, Дармштадт, LiChrospher (R) 100 RP-8, 125 x 4 мм, элюирующая система: вода + трифторуксусная кислота, pH 2,5; 0,1% октансульфонат натрия/ацетонитрил; детектирование с помощью УФ при 220 нм); структуру доказывают с помощью Electrospray-масс-спектроскопии (BIO-Q-MS).
Для упрощения ниже для соединения формулы (I)
с R2 = водород используют обозначение A 1437-циклопептид.
Пример 1.
Тридеканоильное производное A 1437- циклопептида (соединение формулы (I), R1 = HO; R2 = CH3(CH2)11CO)
Способ сочетания с TOTU:
а) Активирование тридекановой кислоты:
113 мг (0,527 моль) Тридекановой кислоты растворяют в 3,75 мл N,N-диметилформамида (ДМФ), добавляют 172,5 мг (0,526 ммоль) TOTU и 1,25 г раствора этилдиизопропиламина (0,5 ммоль) в ДМФ (0,4 ммоль/г). Раствор выдерживают 1 час при комнатной температуре.
б) Сочетание:
348 мг (0,264 ммоль) Fmoc-производного формулы (II) (R1 = OH; R3 = флуоренилметоксикарбонил; пример 69) суспендируют в 7,2 мл безводного ДМФ и добавляют 2,9 г активного раствора а) (0,25 ммоль) на ледяной бане. Образовавшийся коричневатый раствор перемешивают 1,5 часа при комнатной температуре.
в) Отщепление Fmoc-защитной группы:
Раствор б) охлаждают до 10oC, добавляют 6 мл пиперидина и перемешивают 1 час при комнатной температуре. Затем разбавляют с помощью 250 мл воды и подвергают сушке вымораживанием.
г) Очистка:
Остаток после сушки вымораживанием суспендируют в 100 мл смеси воды с ацетонитрилом (5:1), с помощью 1,5 мл 2н HCl устанавливают pH 2,0, и прозрачный раствор хроматографируют через 90 г RP18 - силикагеля (Merck, арт. 9303) с помощью смеси воды (+0,01% CF3COOH) с ацетонитрилом. Последовательность элюирования: 500 мл смеси в соотношении компонентов 3:1; 500 мл смеси в соотношении компонентов 2:1; 600 мл смеси в соотношении компонентов 1:1.
Целевое соединение находится во фракции с соотношением компонентов 1:1 (УФ-детектирование, 220 нм).
Выход = 267 мл (78% от теории). Чистота 72%.
Сырой продукт снова хроматографируют через Buchi-колонку среднего давления (250 г. RP18; элюирование с помощью смеси воды (+0,01% CF3COOH) с ацетонитрилом (в соотношении 3:2). Фракции продукта подвергают сушке вымораживанием.
Выход = 130 мг; чистота: 96%.
C58H93N13O20 (1292,5) MS: 1293.
Пример 2.
4-Октилбензоильное производное A 1437-циклопептида (соединение формулы (I); R1 = HO;
Способ с хлорангидридом кислоты:
а) Сочетание
6,6 мг (0,005 ммоль) Fmoc-производного формулы (II) (пример 69) растворяют в 200 мг смеси пиридина с водой (9:1) и при -20oC добавляют 25 мг (0,1 ммоль) 4-октилбензоилхлорида. Раствор перемешивают 4 часа при комнатной температуре. После добавки 2 мл диоксана растворитель удаляют в вакууме, и остаток растворяют в 0,2 мл ДМФ.
б) Отщепление Fmoc-защитной группы
К раствору а) добавляют 0,2 мл пиперидина и оставляют стоять в течение 1 часа при комнатной температуре. Раствор разбавляют с помощью 5 мл воды и подвергают сушке вымораживанием.
в) Очистка:
Остаток после сушки вымораживанием хроматографируют через 10 г RP18-силикагеля с помощью смеси воды (+0,01% CF3COOH) с ацетонитрилом. Последовательность элюирования: 80 мл смеси с соотношением компонентов 3:1; 80 мл смеси с соотношением компонентов 1:1.
Фракцию продукта в смеси с соотношением компонентов 1:1 подвергают сушке вымораживанием.
Выход = 4,6 мг (70% от теории); чистота: 85%.
C60H89N13O20 (1312,5) MS: 1313
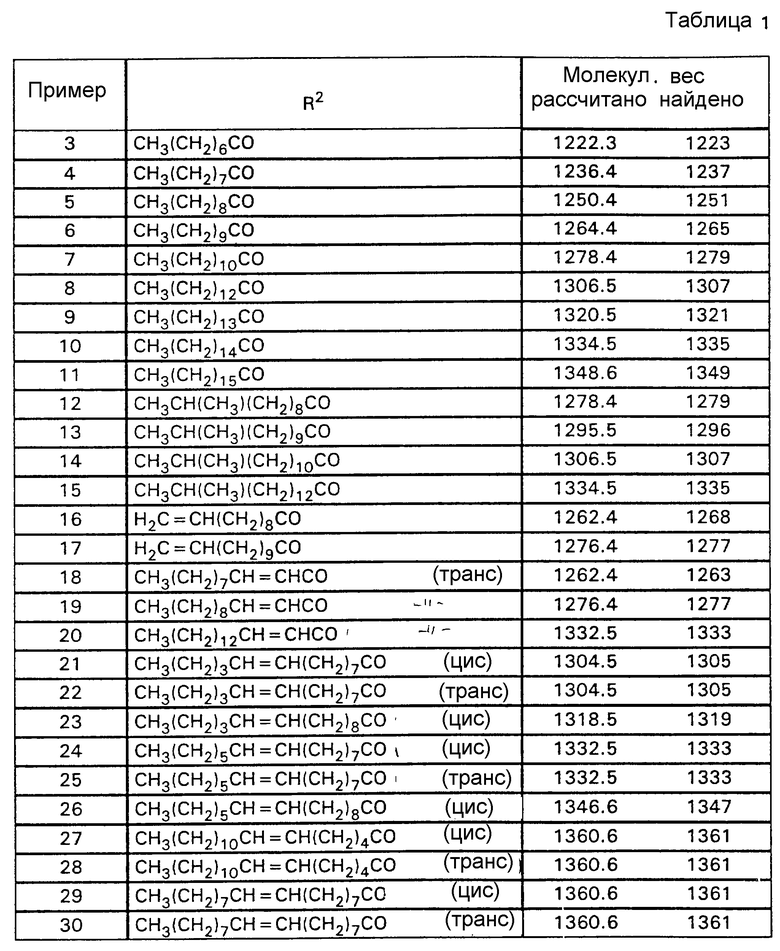
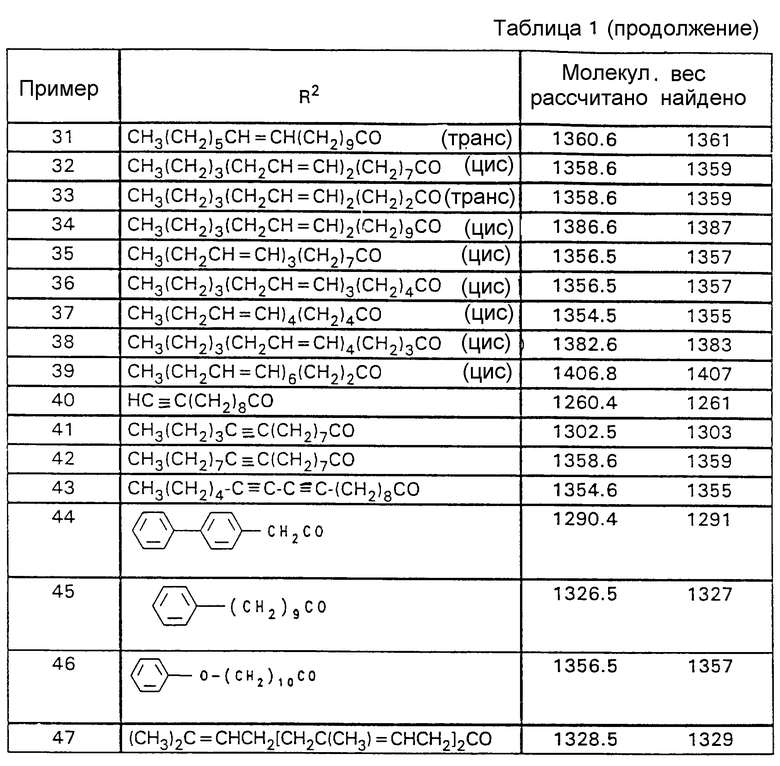
Аналогично примеру 1 получают нижеследующие соединения формулы (I), где R1 обозначает HO и которые содержат указанные в табл. 1 заместители R2.
Выходы составляют 60-85% от теории; чистота составляет 75-98%.
Аналогично примеру 2 получают нижеследующие соединения формулы (I), где R1 обозначает HO и которые содержат указанные в табл. 2 заместители.
Выходы составляют 70 - 85% от теории; чистота составляет 80 - 98%.
Аналогично примеру 1 (соединения 59-66) или примеру 2 (соединения 67 и 68) получают нижеследующие соединения формулы (I), где R1 обозначает NH2 и которые имеют указанные в табл. 3 заместители R2.
Выходы составляют 75 - 85% от теории; чистота составляет 80 - 98%.
Получение исходных соединений
Пример 69. 9-Флуоренилметилоксикарбонильное производное A 1437
(R1 = HO; R2 = (CH3)2CH(CH2)7CH = CHCH2CO;
10 г (7,67 ммоль) A 1437 (формула (I), R1 = HO; R2 = (CH3)2CH(CH2)7-CH= CH2CO) и 3,24 г (38,35 ммоль) бикарбоната натрия растворяют в смеси из 920 мл воды и 640 мл ацетона.
Затем, при контроле за pH-значением, при pH 8,5 в течение 100 минут прикапывают раствор 2,97 г (11,5 ммоль) 9-флуоренилметилового сложного эфира хлормуравьиной кислоты в 240 мл ацетона. При этом реакционный раствор нагревается до 27oC.
Перемешивают дополнительно в течение 1 часа при комнатной температуре. После удаления ацетона в вакууме водный раствор подвергают сушке вымораживанием. Бесцветный остаток дважды перемешивают с метилендихлоридом, беря каждый раз по 500 мл метилендихлорида, для удаления низкомолекулярных примесей.
Выход = 12,2 г; MS: 1526,7.
Пример 70. 9-Флуоренилметилоксикарбонильное производное A 1437-циклопептида
(формула (II); R1 = HO; 
Смесь из 10 г продукта из примера 69 и 300 г влажного мицелия из Actinoplanes utahensis в 1 л стерильного калийфосфатного буфера (100 ммоль, pH 7,2, 50 ммоль ЭДТК, 0,02% азида натрия) перемешивают в течение 48 часов при 32oС. Затем биомассу отделяют путем центрифугирования, для фиксирования продукта раствор фильтруют через 500 г MCl-геля (фирма Mitsubishi), и продукт элюируют смесью воды с метанолом (1:1). Элюат концентрируют для удаления метанола, и водный раствор хроматографируют через 500 г RP18 с помощью воды (+0,05% трифторуксусной кислоты) с ацетонитрилом (2:1). Фракции продукта концентрируют в вакуума и подвергают сушке вымораживанием.
Выход = 6 г; MS: 1318,4.
Пример 71. 4-[(2-/4-(2-Фенилэтил)/-фенилэтил)] бензойная кислота
Раствор 22,9 г метилового эфира 4-бромметилбензойной кислоты в 1000 мл толуола смешивают с 33,9 г трифенилфосфина и кипятят с обратным холодильником. Спустя 7 часов, взаимодействие полностью заканчивается. Оставляют охлаждаться, и продукт отсасывают. Выход = 47,6 г.
Стадия 2:
58,9 г Продукта стадии 1 суспендируют в 500 мл безводного тетрагидрофурана, охлаждают до 0oC и смешивают со 120 мл 1M раствора бис-триметилсилиламида лития в тетрагидрофуране. Спустя 1 час стояния при комнатной температуре, еще раз охлаждают до 0oC и добавляют 19,3 г стильбен-4-альдегида. Затем перемешивают 2,5 часа при 50oC, охлаждают до 0oC, и осадившееся твердое вещество отсасывают. Остаток дополнительно промывают с помощью 0,5 л ТГФ.
Органическую фазу разбавляют с помощью 750 мл этилацетата и промывают с помощью 750 мл насыщенного раствора хлорида аммония. Водную фазу экстрагируют с помощью 750 мл этилацетата, органическую фазу сушат над сульфатом натрия и концентрируют. Сырой продукт используют в ближайшей стадии.
Выход = 49,9 г.
Стадия 3:
26,7 Сырого продукта из стадии 2 вместе с 5 г палладия на активном угле (10% Pd) суспендируют в 1000 мл метанола. Гидрируют 3 часа при комнатной температуре при нормальном давлении. Катализатор отфильтровывают горячим, раствор концентрируют в вакууме, и продукт очищают путем хроматографии на силикагеле с помощью смеси гептана с этилацетатом (10:1).
Выход = 7,4 г.
Стадия 4:
1,98 г Продукта из стадии 3 суспендируют в 60 мл этанола и смешивают с раствором 508 мг KOH в 10 мл воды. Раствор кипятят с обратным холодильником в течение 1,5 часов. Этанол удаляют в вакууме, остаток обрабатывают 500 мл этилацетата и 200 мл воды, и в растворе устанавливают pH 2 с помощью 2н HCl.
Дополнительно перемешивают 0,5 часа, фазы разделяют, и водную фазу экстрагируют еще раз с помощью 200 мл этилацетата. Органические фазы объединяют, сушат над сульфатом натрия и концентрируют в вакууме.
Выход: 1,86 г целевого соединения.
Хлорангидрид кислоты:
1,23 г Продукта из стадии 4 суспендируют в 10 мл тионилхлорида. Затем кипятят с обратным холодильником до прекращения выделения газа. После охлаждения концентрируют в вакууме и дважды испаряют, добавляя по 5 мл толуола. Выход: 1,35 г светло-серого кристаллического соединения.
Пример 72. 4-[2-(Бифенил-4-ил)этил] бензойная кислота
Стадия 1:
Аналогично стадии 2 примера 71, 6,4 г фосфонийбромида (стадия 1 из примера 71) вводят во взаимодействие с 1,82 г бифенил-4-альдегида.
Выход = 5,8 г.
Стадия 2:
5,8 г Продукта стадии 1 гидрируют аналогично стадии 3 примера 71, и продукт очищают путем хроматографии.
Выход = 970 мг.
Стадия 3:
950 мг Продукта стадии 2 омыляют аналогично стадии 4 примера 71.
Выход = 880 мг.
Стадия 4:
850 мг Продукта стадии 3 вводят во взаимодействие с тионилхлоридом, аналогично стадии 5 примера 71, с получением хлорангидрида кислоты.
Выход = 909 мг.


Описываются новые липопептидные производные формулы (I), где R1 - OH или NH2, R2 - линейный или разветвленный, насыщенный или ненасыщенный алифатический C8-C22-ацильный остаток, который может быть прерван фенильными группами или кислородом, и их фармацевтически приемлемые соли. Они являются производными антибиотиков липопептидного комплекса А 1437; описывается также способ их получения и фармацевтическая композиция на основе соединений формул (I). 3 с. и 4 з.п. ф-лы, 3 табл.

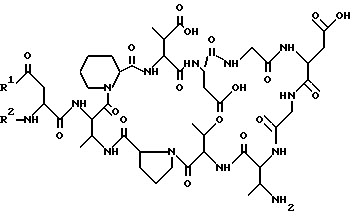
где R1 - OH или NH2;
R2 - линейный или разветвленный, насыщенный или ненасыщенный алифатический C8 - C22 - ацильный остаток, который может быть прерван фенильными группами или кислородом,
и их фармацевтически приемлемые соли.
прерываемый кислородом ацильный остаток, предпочтительно
где n = 0 - 20, целые числа.
где n = 0 - 8, целое число.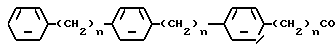
где n = 0, 1, 2.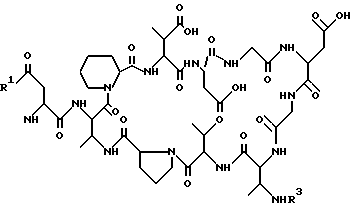
где R1 имеет указанное значение;
R3 обозначает известную из химии пептидов защитную аминогруппу, предпочтительно трет-бутоксикарбонильную (BOC), бензилоксикарбонильную (Z, Cbz), флуоренилметоксикарбольную (Fmoc) или аллилоксикарбонильную (Alloc) защитную группу,
вводят во взаимодействие с карбоновой кислотой формулы (III)
R2OH,
где R2 имеет указанные в п.1 значения,
или с активированным по карбонильной группе производным этой карбоновой кислоты.
| Способ получения органорастворимых производных хитозана | 1989 |
|
SU1823878A3 |
| US 4350627 A, 1982 | |||
| ДАТЧИК ФАЗЫ ЗВУКОВЫХ ЧАСТОТ | 0 |
|
SU337731A1 |
| Устройство для градуировки однокомпонентных точечных месдоз | 1973 |
|
SU448355A1 |
| Способ возведения изометрических сооружений | 1973 |
|
SU494515A1 |

