Изобретение относится к новым имидазопиридиназолидинонам, предназначенным для применения в фармацевтической промышленности в качестве действующих веществ при изготовлении лекарственных средств.
В европейской заявке EP-A-0033094 описываются имидазо-[1,2-а] пиридины, содержащие в положении 8 арильный заместитель, представляющий собой предпочтительно радикал из числа следующих: фенил, тиенил, пиридил либо замещенный хлором, фтором, метилом, трет-бутилом, трифторметилом, метоксигруппой или цианом фенил. В качестве особенно интересных арильных радикалов в ЕР-А-0033094 названы фенил, о- либо п-фторфенил, п-хлорфенил и 2,4, 6-триметилфенил, из которых особенно предпочтительны фенил, о- либо п-фторфенил и 2,4,6-триметилфенил. В европейских заявках EP-A-0204285, EP-A-0228006, EP-A-0268989 и EP-A-0308917 описываются имидазо [1,2-а] пиридины, содержащие в положении 3 ненасыщенный алифатический радикал, прежде всего (замещенный) алкинильный радикал. В европейской заявке EP-A-0266890 описываются имидазо [1,2-а] пиридины, замещенные в положении 8 алкенильным, алкильным или циклоалкилалкильным радикалом.
Задачей настоящего изобретения является разработка новых производных имидазопиридина, обладающих противоязвенными и антисекреторными свойствами, и разработка новых лекарственных средств на их основе.
Поставленная задача решается описанными ниже более подробно соединениями, отличающимися от соединений из уровня техники прежде всего замещением в положении 8, которые обладают неожиданными и особенно положительными свойствами.
Таким образом, объектом изобретения являются имидазопиридиназолидиноны формулы I
где R0 означает (C1-C4)алкил, гидроксиметил или галоген,
R1 означает (C1-C4)алкил,
R2 означает водород или (C1-C4)алкил,
R3 означает водород или (C1-C4)алкил,
R3' означает водород, (C1-C4)алкил либо (C1-C4)алкил, замещенный одним или двумя идентичными либо различными заместителями, выбранными из группы, включающей галоген и (C1-C4)алкокси,
R4 означает водород или (C1-C4)алкил,
R5 означает водород или (C1-C4)алкил,
A означает О (кислород) или NH и
Y означает О (кислород) или CH2,
и их соли.
Кроме вышеуказанных значений, R0 может дополнительно означать тиоцианат, R2 - (C1-C4)алкокси, галоген или трифторметил.
Указанная группа заместителей в R3', когда он означает замещенный (C1-C4)алкил, дополнительно может включать (C1-C4)алкокси (C1-C4)алкокси, R4 дополнительно может обозначать (C1-C4)алкокси, галоген или трифторметил, а R5 - галоген.
(C1-C4)алкил представляет собой прямоцепочечные либо разветвленные алкильные радикалы с 1-4 атомами углерода. В качестве примеров таких радикалов можно назвать бутил, изобутил, втор-бутил, трет-бутил, пропил, изопропил, этил и прежде всего метил.
Галоген в контексте настоящего изобретения означает бром, фтор и прежде всего хлор.
(C1-C4)алкокси представляет собой атом кислорода, с которым связан один из вышеназванных (C1-C4)алкильных радикалов. В качестве примеров можно назвать метоксильный и этоксильный радикалы.
(C1-C4)алкокси-(C1-C4)алкокси представляет собой один из вышеназванных (C1-C4)алкоксильных радикалов, замещенный в свою очередь другим (C1-C4)алкоксильным радикалом. В качестве примера можно назвать метоксиэтоксильный радикал.
К солям соединений формулы I можно отнести предпочтительно все кислотно-аддитивные соли. Среди таковых следует назвать прежде всего фармакологически приемлемые соли неорганических и органических кислот, обычно применяемых в галенике. Фармакологически неприемлемые соли, которые могут образовываться в качестве первоначальных продуктов при получении соединений согласно изобретению в промышленном масштабе, с помощью методов, известных специалисту в данной области техники, могут переводиться в фармакологически приемлемые соли. В качестве таковых пригодны водорастворимые и водонерастворимые кислотно-аддитивные соли с такими кислотами, как, например, соляная кислота, бромистоводородная кислота, фосфорная кислота, азотная кислота, серная кислота, уксусная кислота, лимонная кислота, D-глуконовая кислота, бензойная кислота, 2-(4-гидроксибензоил) бензойная кислота, масляная кислота, сульфосалициловая кислота, малеиновая кислота, лауриновая кислота, яблочная кислота, фумаровая кислота, янтарная кислота, щавелевая кислота, винная кислота, эмбоновая кислота, стеариновая кислота, толуолсульфоновая кислота, метансульфоновая кислота или 3-гидрокси-2-нафтойная кислота, причем при получении солей - в зависимости от того, используют ли одно- или многоосновную кислоту, и от того, какую соль требуется получить, - кислоты применяют в эквимолярном или несколько в другом количественном соотношении.
Среди соединений формулы I следует выделить те из них, где
R0 означает (C1-C4)алкил, гидроксиметил или галоген,
R1 означает (C1-C4)алкил,
R2 означает (C1-C4)алкил,
R3 означает водород или (C1-C4)алкил,
R3' означает водород, (C1-C4)алкил либо (C1-C4)алкил, замещенный заместителем, выбранным из группы, включающей галоген и (C1-C4)алкокси,
R4 означает водород,
R5 означает водород,
A означает О (кислород) или NH и
Y означает О (кислород) или CH, и их соли.
К особенно предпочтительным соединениям формулы I относятся таковые, где
R0 означает метил, гидроксиметил, хлор или фтор,
R1 означает метил,
R2 означает (C1-C4)алкил,
R3 означает водород или (C1-C4)алкил,
R3' означает водород, (C1-C4)алкил либо (C1-C4)алкил, замещенный (C1-C4)алкоксигруппой,
R4 означает водород,
R5 означает водород, A означает O (кислород) или NH и
Y означает О (кислород) или CH
и их соли.
Предпочтительными являются далее такие соединения формулы I, где
R0 означает метил, гидроксиметил или хлор,
R1 означает метил,
R2 означает (C1-C4)алкил,
R3 означает водород или (C1-C4)алкил,
R3' означает водород, (C1-C4)алкил, (C1-C4)алкокси (C1-C4)алкил,
R4 означает водород,
R5 означает водород, A означает O (кислород) или NH
Y означает O (кислород) или CH,
и их соли.
Примеры предлагаемых согласно изобретению соединений представлены в таблице 1 (см. табл. 1 в конце описания).
К этим примерам относятся также соли представленных в таблице соединений.
Соединения формулы I в положениях, в которых присоединены заместители R3, соответственно R3', могут иметь соответственно центр хиральности. Изобретение включает в соответствии с этим в случае хиральных соединений как чистые энантиомеры и диастереомеры, так и их смеси в любом соотношении 15 компонентов, включая рацематы.
Соединения формулы I согласно изобретению могут быть получены следующими способами
а) для получения соединений формулы I, в которых R0 означает гидроксиметил, соединения формулы II
где R1, R2, R3, R3', R4, R5, A и Y имеют указанные выше значения подвергают восстановлению, или
б) соединения формулы III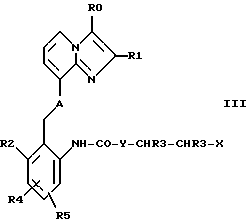
где R0, Rl, R2, R3, R3', R4, R5, A и Y имеют указанные выше значения, а X представляет собой соответствующую уходящую группу подвергают циклизации с отщеплением HX,
или при необходимости полученные соединения формулы I переводят затем в их соли, или же при необходимости из полученных солей соединений 1 высвобождают затем соединения формулы I.
Восстановление соединений формулы II проводят но методике, известной специалисту в данной области техники. Реакцию осуществляют в инертных растворителях, например в низших алифатических спиртах, например, с применением соответствующих гидридов, таких, в частности, как борогидрид натрия, при необходимости с добавками воды.
Циклизацию соединений формулы III осуществляют по методике, известной специалисту в данной области техники, например, так, как это описано в приведенных ниже примерах, в инертных растворителях и в присутствии связывающего кислоту агента (акцептора протонов). В качестве акцепторов протонов можно назвать среди прочих гидроксиды щелочных металлов, такие, как гидроксид натрия либо гидроксид калия, или гидриды металлов, такие, как гидрид натрия. Соответствующей уходящей группой является, например, атом галогена (предпочтительно хлор либо бром) или метансульфонилоксигруппа.
Выбор конкретных условий, необходимых для осуществления способа, определяет сам специалист на основе своих знаний и опыта.
Выделение и очистку соединений согласно изобретению проводят по известной методике, например отгонкой растворителя под вакуумом и перекристаллизацией полученного остатка из соответствующего растворителя, или одним из обычных методов очистки, таким как, например, колоночная хроматография на приемлемом материале-носителе.
Кислотно-аддитивные соли получают растворением свободного основания в пригодном для этой цели растворителе, например в воде, в хлорированном углеводороде, таком, как метиленхлорид или хлороформ, в низшем алифатическом спирте (этаноле, изопропаноле), в кетоне, таком, как ацетон, или в простом эфире, таком, как ТГФ или диизопропиловый эфир, содержащем требуемую кислоту или же в который добавляют затем требуемую кислоту.
Соли получают путем фильтрации, переосаждения, осаждения с помощью нерастворителя соли присоединения или путем выпаривания растворителя. Полученные соли можно путем подщелачивания, например, водным раствором аммиака, переводить в свободные основания, которые в свою очередь можно переводить в кислотно-аддитивные соли. Таким способом фармакологически неприемлемые кислотно-аддитивные соли могут быть превращены в фармакологически приемлемые кислотно-аддитивные соли.
Исходные соединения формулы II получают путем циклизации соединений формулы III, в которых R0 имеет значение CHO.
Соединения формулы III получают, например, взаимодействием соединений формулы IV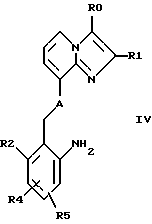
где R0, Rl, R2, R4, R5 и А имеют указанные выше значения, с соединениями Hal-CO-Y-CHR3'-CHR3-X, где R3, R3', Y и X имеют указанные выше значения, a Hal представляет собой атом галогена (предпочтительно хлор либо бром).
Исходные соединения формулы IV известны из европейских заявок EP-A-0268989 и EP-A-0308917 или их можно получить по описанной в этих публикациях методике. Так, в частности, исходные соединения IV могут быть получены по известной методике из соответствующих нитросоединений восстановлением либо гидролизом соответствующих N-ацилпроизводных.
Получение предлагаемых согласно изобретению соединений поясняется подробнее на приведенных ниже примерах. В первую очередь эти примеры служат для выборочного описания процесса и методики получения соединений формулы I, а также получения некоторых исходных соединений. Точно так же по аналогичной методике или же по методике, известной специалисту в данной области техники, с использованием обычных технологических приемов могут быть получены другие соединения формулы I, равно как и другие исходные соединения, получение которых не описано подробно.
Употребляемые сокращения означают: КТ - комнатная температура, ч - час(ы), мин - минута (ы), tпл - температура плавления, tкип - температура кипения, разл. - разложение.
Примеры
Конечные продукты
1.3- [2-(2.3-диметилимидазо[1.2-а]пиридин-8-иламинометил)-3-метилфенил] оксазолидин-2-он
а) 8-[2-(2-хлорэтоксикарбониламино)-6-метилбензиламино] -2,3-диметилимидазо [1,2-а] пиридин
К раствору 8-(2-амино-6-метилбензиламино)-2,3-диметилимидазо[1,2-а]пиридина (19,65 г, 70 ммолей) в безводном дихлорметане (600 мл) при КТ в течение приблизительно 30 мин добавляют по каплям 2-хлорэтиловый эфир хлормуравьиной кислоты (12,4 г, 81 ммоль), растворенный в дихлорметане (40 мл). Затем перемешивают в течение 16 ч при КТ, после чего экстрагируют раствором гидрокарбоната натрия (4х200 мл). После экстракции водной фазы дихлорметаном (200 мл) объединенные органические экстракты промывают водой (2х200 мл), сушат над сульфатом магния и концентрируют. Полученный неочищенный продукт (33, 9 г) непосредственно используют в последующей реакции в примере 1б.
б) 3-[2-(2,3-диметилимидазо[1,2-а}пиридин-8-иламинометил)-3-метилфенил] оксазолидин-2-он
К раствору неочищенного продукта из примера 1а (33,8 г) в безводном этаноле (800 мл) при интенсивном перемешивании порциями добавляют в течение приблизительно 30 мин гидрид натрия (3,15 г, 80%-ный в парафине). Затем продолжают перемешивание еще в течение 30 мин при КТ, после чего добавляют порциями воду (200 мл) и этанол отгоняют с помощью ротационного испарителя. Осадок отсасывают, промывают водой и сушат под вакуумом. Сырой продукт очищают хроматографией на силикагеле (система растворителей: толуол-диоксан= 4: 1). После объединения фракций с Rf=0,15 и кристаллизации из этилацетата-диизопропилового эфира выделяют указанное в заголовке соединение (13,6 г, 63%) в виде твердого вещества бежевого цвета. tпл 139-140oC.
в) Взаимодействием растворенного в ацетоне соединения, указанного в заголовке, с метансульфокислотой получают метансульфонат указанного в заголовке соединения с tпл 232-235oC.
2. 3- [2-(3-хлор-2-метилимидазо[1.2-а]пиридин-8-иламинометил) -3-метилфенил]оксазолидин-2-он
а) 8- [2-(2-хлорэтоксикарбониламино)-6-метилбензиламино] -3-хлор-2-метилимидазо[1,2-а]пиридин
По описанной в примере 1а методике из 8-(2-амино-6-метилбензиламино)-3-хлор-2-метилимидазо[1,2-а] пиридина (0,27 г) и 2-хлорэтилового эфира хлормуравьиной кислоты (0,18 г) в дихлорметане (30 мл) получают указанное в заголовке соединение в виде коричневого масла, которое непосредственно используют в последующей реакции в 2б.
б) 3-[2-(3-хлор-2-метилимидазо[1,2-а] пиридин-8-иламинометил)-3-метилфенил] оксазолидин-2-он
По описанной в примере 1б методике из неочищенного продукта 2а (0,37 г) и гидрида натрия (0,45 г, 80%-ный в парафине) в безводном этаноле (20 мл) после кристаллизации из этилацетата-петролейного эфира получают указанное в заголовке соединение (70 мг, 21%) в виде твердого вещества бежевого цвета, tпл 137-139oC.
3. 3- [2-(3-гидроксиметил-2-метилимидазо[1.2-а]пиридин-8-иламинометил) -3-метилфенил]оксазолидин-2-он
а) 8-[2-(2-хлорэтоксикарбониламино)-6-метилбензиламино]-3-формил-2- метилимидазо[1,2-а]пиридин
По описанной в примере 1а методике из 8-(2-амино-6-метилбензиламино)-3-формил-2-метилимидазо[1,2-а] пиридина (2,03. г) и 2-хлорэтилового эфира хлормуравьиной кислоты (1,01 г) в дихлорметане (120 мл) получают указанное в заголовке соединение в виде коричневого масла, которое непосредственно используют в последующей реакции в примере 3б.
б) 3-[2-(3-формил-2-метилимидазо[1,2-а]пиридин-8-иламинометил)- 3-метилфенил]оксазолидин-2-он
По описанной в примере 1б методике из неочищенного продукта 3а (0,12 г) и гидрида натрия (0,01 г, 80%-ный в парафине) в безводном этаноле (10 мл) после кристаллизации из этилацетата-диизопронилового эфира получают указанное в заголовке соединение (80 мг, 73%) в виде твердого вещества бежевого цвета, tпл 196-197oC.
в) 3- [2-(3-гидроксиметил-2-метилимидазо[1,2-а]пиридин-8-иламинометил)- 3-метилфенил]оксазолидин-2-он
Раствор 3- [2-(3-формил-2-метилимидазо[1,2-а]пиридин-8-иламинометил)-3- метилфенил] оксазолидин-2-она (0,18 г, 0,49 ммоля) и бораната натрия (19 мг, 0,5 ммоля) в безводном этаноле (20 мл) перемешивают в течение 30 мин при КТ. Затем добавляют воду (75 мл), этанол отгоняют с помощью ротационного испарителя и водный остаток экстрагируют этилацетатом (3х50 мл). Органические экстракты промывают водой (50 мл), сушат над сульфатом магния и концентрируют. После кристаллизации из диизопропилового эфира получают указанное в заголовке соединение (120 мг, 66%) в виде твердого вещества бежевого цвета, tпл 152-157oC.
4.3- [2-(3-гидроксиметил-2-метилимидазо[1,2-а]пиридин-8-илоксиметил) -3-метилфенил]оксазолидин-2-он
а) 8-[2-(2-хлорэтоксикарбониламино)-6-метилбензилокси] -3-формил-2- метилимидазо[1,2-а]пиридин
По описанной в примере 1а методике из 8-(2-амино-6-метилбензилокси)-3-формил-2-метилимидазо[1,2-а] пиридина (2,36 г) и 2-хлорэтилового эфира хлормуравьиной кислоты (1,3 г) в дихлорметане (150 мл) получают указанное в заголовке соединение в виде коричневого масла, которое непосредственно используют в реакции 4б.
б) 3-[2-(3-формил-2-метилимидазо[1,2-а]пиридин-8-илоксиметил)-3- метилфенил]оксазолидин-2-он
По описанной в примере 1б методике из неочищенного продукта 4а (1,14 г) и гидрида натрия (0,13 г, 80%-ный в парафине) в безводном этаноле (200 мл) после кристаллизации из этилацетата-диизопропилового эфира получают указанное в заголовке соединение (0,7 г, 67%) в виде твердого вещества бежевого цвета, tпл 242-244oC.
в) 3-[2-(3-гидроксиметил-2-метилимидазо[1,2-а]пиридин-8-илоксиметил) -3-метилфенил] оксазолидин-2-он
Раствор 3-[2-(3-формил-2-метилимидазо[1,2-а] пиридин-8-илоксиметил)-3- метилфенил] оксазолидин-2-она (1,39 г, 3,8 ммоля) бораната натрия (0,19 г, 4,75 ммоля) в безводном этаноле (125 мл) перемешивают в течение 1,5 ч при КТ. Затем добавляют воду (100 мл), этанол отгоняют с помощью ротационного испарителя и водный остаток перемешивают в течение 30 мин при 4oC. Осадок отфильтровывают, промывают холодной водой и сушат под вакуумом. Таким путем получают указанное в заголовке соединение (1,28 г, 91%) в виде твердого вещества бежевого цвета, tпл 190-193oC.
5. 5-хлорметил-3-[2-(2.3-диметилимидазо [1.2-а] пиридин-8-иламинометил)-3-метилфенил] оксазолидин-2-он
а) К 591 мл (1,14 моля) раствора фосгена в толуоле (1,93 моль/л) в течение 2 ч при 5-8oC по каплям добавляют смесь 150 г (1,14 моля) 1,3-дихлорпропанола и 90,2 г (1,14 моля) пиридина и перемешивают еще в течение 24 ч при 20oC. Затем пропускают поток газа N2, отфильтровывают гидрохлорид пиридина, промывают водой, сушат над сульфатом магния и после фракционной перегонки получают 102 г (47%) (2-хлор-1-хлорметилэтилового эфира) хлормуравьиной кислоты, tкип 140-145oC/18 мбар.
б) В течение 2 дней при КТ перемешивают раствор 2,0 г (7,13 ммолей) 8-(2-амино-6-метилбензиламино)-2,3-диметилимидазо [1,2-а] пиридина и 1,5 г (7,84 ммолей) полученного на стадии а) соединения в 80 мл дихлорметана. Органический раствор промывают насыщенным раствором карбоната натрия и водой, сушат сульфатом магния и концентрируют под вакуумом. Таким путем получают 3,4 г неочищенного 8-12- [(2-хлор-1-хлорметилэтокси)карбониламино]-6-метилбензиламино) -2,3-диметилимидазо[1,2-а] пиридина в виде масла, которое используют на последующей стадии.
в) К раствору полученного на стадии б) соединения в 30 мл этанола при 20oC по каплям добавляют раствор 0,48 г 85%-ного гидроксида калия в 4 мл этанола и перемешивают еще в течение 5 ч. Затем концентрируют под вакуумом, смешивают с водой, экстрагируют дихлорметаном и высушенный сульфатом магния органический слой концентрируют под вакуумом. Остаток смешивают с 5 мл этанола и охлаждают на ледяной бане. Таким путем получают 1,2 г указанного в заголовке соединения, tпл 141-143oC.
6. Гидрохлорид 3-[2-(2,3-диметилимидазо[1.2-а]пиридин-8-иламинометил)-3-метилфенил] -5-метилоксазолидин-2-она
а) Аналогично примеру 5а из 2-хлор-1-пропанола и фосгена получают (2-хлор-1-метилэтиловый эфир) хлормуравьиной кислоты, tкип 55-56oC/17 мбар.
б) Аналогично примеру 5б подвергают взаимодействию 2,0 г (7,13 ммолей) 8-(2-амино-6-метилбензиламино)-2,3-диметилимидазо[1,2-а] пиридина с 1,23 г (7,85 ммолей) полученного на стадии а) соединения, получая в результате 3,1 г неочищенного 8-{2- [(2-хлор-1-метилэтокси) карбониламино]-6-метилбензил-амино]-2,3-диметилимидазо[1,2-а] пиридина в виде аморфной массы.
в) К 2,9 г полученного на стадии б) соединения в 15 мл этанола при КТ по каплям добавляют раствор 0,48 г 85%-ного КОН в 15 мл этанола и перемешивают в течение последующих 12 ч. Затем концентрируют под вакуумом, смешивают с водой, экстрагируют дихлорметаном и высушенный сульфатом магния органический слой концентрируют под вакуумом. Остаток смешивают с 0,3 мл концентрированной соляной кислоты и ацетоном, получая в результате 1,5 г указанного в заголовке соединения, tпл 274-275oC (разл.).
7. 3-[2-(2.3-диметилимидазо [1.2-а]пиридин-8-иламинометил)-3-метилфенил] -4-метилоксазолидин-2-он
а) Аналогично примеру 5б из (2-хлорпропилового эфира) хлормуравьиной кислоты и 8-(2-амино-6-метилбензиламино)-2,3-диметилимидазо[1,2-а] пиридина получают 8-[2-(2-хлорпропоксикарбониламино)-6-метилбензиламино] -2,3-диметилимидазо[1,2-а]пиридин.
б) Из полученного в а) соединения и раствора гидроксида калия в этаноле аналогично примеру 5в получают указанное в заголовке соединение.
8. 3- [2-(2.3-диметилимидазо [1.2-а]пиридин-8-иламинометил)-3-метилфенил] -5-метоксиметилоксазолидин-2-он
а) Аналогично примеру 5б из (1-хлорметил-2-метоксиэтилового эфира) хлормуравьиной кислоты (см. Helv. Chim. Acta 44, 105, 1961) и 8-(2-амино-6-метилбензиламино)-2,3-диметилимидазо[1,2-а] пиридина получают 8-[2-(1- хлорметил-2-метоксиэтоксикарбониламино)-6-метилбензиламино] -2,3-диметилимидазо[1,2-а]пиридин.
б) Из полученного в а) соединения и раствора гидроксида калия в этаноле аналогично примеру 5в получают указанное в заголовке соединение.
9. 1-[2-(2.3-диметилимидазо [1.2-а] пиридин-8-иламинометил)-3-метилфенил] пирролидин-2-он
а) 8-[2-(4-хлорбутириламино)-6-метилбензиламино] -2,3-диметилимидазо[1,2-а]пиридин
К раствору 2,2 г (7,85 ммолей) 8-(2-амино-6-метилбензиламино)-2,3-диметилимидазо[1,2-а]пиридина и 0,63 мл (7,85 ммолей) пиридина в 30 мл дихлорметана при 0-5oC при перемешивании добавляют по каплям 0,88 мл (7,85 ммолей) 4-хлорбутирилхлорида и продолжают перемешивание еще в течение 2 ч при 0-20oC. Затем смешивают с водой и гидрокарбонатом калия до pH 7, экстрагируют встряхиванием, органический слой сушат сульфатом магния и концентрируют под вакуумом. Остаток перекристаллизовывают из толуола и петролейного эфира (50/70oC). Таким путем получают 2,6 г ( 86%) указанного в заголовке соединения, tпл 138-140oC.
б) 1-[2-(2,3-диметилимидазо[1,2-а]пиридин-8-иламинометил)- 3-метилфенил] пирролидин-2-он
К раствору 2,4 г (6,24 ммолей) полученного в а) соединения в 60 мл этанола при КТ добавляют по каплям раствор 0,41 г (6,25 ммолей) 85%-ного КОН в 15 мл этанола и перемешивают еще в течение 4 ч. Затем концентрируют под вакуумом до объема приблизительно 30 мл, разбавляют водой и несколько раз экстрагируют путем встряхивания с этилацетатом. Органический слой сушат с помощью сульфата магния, концентрируют под вакуумом и остаток кристаллизуют диизопропиловым эфиром. Таким путем получают 2,0 г (92%) указанного в заголовке соединения, tпл 138-141oC.
10. 1-[2-(3-хлор-2-метилимидазо[1.2-а] пиридин-8 -иламинометил)-3-метилфенил] пиролидин-2-он
а) 8-[2-(4-хлорбутириламино)-6-метилбензиламино] -3-хлор-2-метилимидазо [1,2-а]пиридин
По описанной в примере 9а методике взаимодействием 8-(2-амино-6-метилбензиламино)-3-хлор-2-метилимидазо [1,2-а] пиридина с 4-хлорбутирил-хлоридом после кристаллизации из этилацетата-циклогексанона получают указанное в заголовке соединение в виде порошка бежевого цвета. Выход 66%; tпл 127-130oC.
б) 1-[2-(3-хлор-2-метилимидазо[1,2-а] пиридин-8-иламинометил)- 3-метилфенил] пирролидин-2-он
По описанной в примере 9б методике взаимодействием полученного в а) соединения с гидроксидом калия в этаноле и последующей кристаллизацией из диизопропилового эфира получают указанное в заголовке соединение в виде порошка бежевого цвета. Выход 77%; tпл 265-267oC.
Исходные соединения
A1. 3-хлор-2-метил-8-пивалоиламиноимидазо[1.2-а]пиридин
В ледяном уксусе (20 мл) растворяют 5,0 г (18,6 ммолей) гидрохлорида 2-метил-8-пивалоиламиноимидазо[1,2-а] пиридина, полученного из 8-амино-2-метилимидазо [1,2-а] пиридина и пивалоилхлорида, tпл 229-230oC, и при 15oC медленно пропускают газообразный хлор до момента завершения реакции, определяемого с помощью ТХ (приблизительно 20 мин). Затем растворитель отгоняют, остаток растворяют в этилацетате-воде (соответственно по 30 мл), насыщенным раствором гидрокарбоната натрия устанавливают основное значение pH и экстрагируют. Затем повторно экстрагируют этилацетатом (3х30 мл). Объединенные органические экстракты промывают водой (50 мл), сушат над сульфатом магния и концентрируют. Остаток очищают посредством хроматографии на силикагеле (система растворителей: толуол-диоксан= 9:1). После концентрирования фракций с Rf=0,2 получают указанное в заголовке соединение (4,1 г, 83%) в виде бесцветного твердого вещества, tпл 117-118oC.
A2. 8-амино-3-хлоримидазо[1,2-а]пиридин
Суспензию 3-хлор-2-метил-8-пивалоиламиноимидазо[1,2-а] пиридина (4,0 г, 15 ммолей) в 60%-ной серной кислоте (25 мл) нагревают в течение 1 ч при 100oC. После охлаждения до КТ добавляют воду (100 мл) и 10H едким натром доводят pH до 10. Затем экстрагируют этилацетатом (3х50 мл). Объединенные органические экстракты промывают водой (50 мл), сушат над сульфатом магния и концентрируют. Остаток растворяют в кипящем толуоле, осветляют силикагелем и кристаллизуют. Указанное в заголовке соединение выделяют в виде твердого вещества бежевого цвета. Выход 1,9 г (70%), tпл 126-127oC.
Б1. 8-(6-метил-2-нитробензиламино)-2.3-диметилимидазо[1.2-а]пиридин
К раствору 8-амино-2,3-диметилимидазо [1,2-а] пиридина (14,7 г) и 6-метил-2-нитробензилхлорида (18,6 г) в 100 мл ацетона добавляют при КТ 15,0 г иодида натрия и 31,0 г карбоната натрия, после чего нагревают с обратным холодильником в течение 6 ч до кипения. После охлаждения раствора до КТ и концентрирования остаток растворяют в смеси 200 мл этилацетата и 200 мл воды и органическую фазу отделяют. После проведения последующей трехкратной экстракции порциями по 100 мл этилацетата соответственно объединенные органические экстракты сушат над сульфатом магния и затем концентрируют до объема 80 мл. 12,1 г указанного в заголовке соединения кристаллизуются в виде твердого вещества, слегка окрашенного в желтый цвет. Маточный раствор концентрируют. После хроматографической очистки остатка на силикагеле (система растворителей: толуол-диоксан=6:1) получают дополнительно 14 г кристаллического продукта. После перекристаллизации обеих фракций из этилацетата получают 21,5 г (76%) указанного в заголовке соединения с tпл 160-162oC.
Б2. 8-(6-метил-2-нитробензилокси)-2,3-диметилимидазо [1,2-а] пиридин
Суспензию 8-гидрокси-2,3-диметилимидазо [1,2-а] пиридина (7,46 г, 46 ммолей), 6-метил-2-нитробензилхлорида (9,35 г, 50,4 ммолей), карбоната натрия (11,24 г, 105 ммолей) и иодида натрия (7,63 г, 50,4 ммолей) в ацетоне (750 мл) в течение 8 ч нагревают с обратным холодильником. После охлаждения до КТ добавляют воду (200 мл) и ацетон отгоняют с помощью ротационного испарителя. Затем водный остаток перемешивают в течение 30 мин при 4oC. Далее отфильтровывают светлокоричневый осадок, который промывают водой и сушат под вакуумом. Последующую очистку проводят посредством хроматографии на силикагеле (система растворителей: толуол-диоксан= 5:1). Фракции с Rf=0,2 концентрируют и перекристаллизовывают из этилацетата-циклогексана. Указанное в заголовке соединение выделяют в виде желтого твердого вещества. Выход 8,31 г (58%), tпл 168-171oC.
Б3. 3-хлор-2-метил-8-(6-метил-2-нитробензиламино)имидазо[1.2-а] пиридин
Исходя из 8-амино-3-хлоримидазо[1,2-а] пиридина (9,26 г), 6-метил-2-нитробензилхлорида (10,5 г), карбоната натрия (13,7 г) и иодида натрия (8,55 г) в ацетоне (380 мл), по описанной в примере Б1 методике после хроматографии на силикагеле (система растворителей: толуол-диоксан=20:1) и кристаллизацией из этилацетата-циклогексана получают 10,6 г (63%) указанного в заголовке соединения с tпл 142-144oC.
Б4. 8-(2-трет-бутоксикарбониламино-6-метилбензиламино)-2.3-диметилимидазо [1.2-а]пиридин
Исходя из 8-амино-2,3-диметилимидазо[1,2-а]пиридина (4,8 г), 2-трет-бутоксикарбониламино-6-метилбензилхлорида (9,2 г), иодида натрия (5,5 г) и карбоната натрия (8,0 г) в ацетоне (250 мл), по методу, аналогичному описанному в примере Б1, после хроматографии на силикагеле (система растворителей: толуол-диоксан= 20: 1) и перекристаллизации из диизопропилового эфира получают 7,1 г (62%) указанного в заголовке соединения с tпл 149-152oC.
Б5. 8-(2-трет-бутоксикарбониламино-6-метилбензилокси)-2,3-диметилимидазо [1.2-а]пиридин
Исходя из 8-гидрокси-2,3-диметилимидазо[1,2-а] пиридина (1,6 г), 2-трет-бутоксикарбониламино-6-метилбензилхлорида (3,1 г), иодида натрия (1,8 г) и карбоната натрия (2,7 г) в ацетоне (350 мл), по методу, аналогичному описанному в примере Б1, после хроматографии на силикагеле (система растворителей: толуол-диоксан= 5:1) и перекристаллизации из циклогексана получают 3,0 г (78%) указанного в заголовке соединения с tпл 128-131oC.
Б6. 8-(2-трет-бутоксикарбониламино-6-метилбензиламино)-3-формил- 2-метилимидазо[1.2-а]пиридин
Исходя из 8-амино-3-формил-2-метилимидазо[1,2-а] пиридина (4,0 г) 2-трет-бутоксикарбониламино-6-метилбензилхлорида (7,0 г), иодида натрия (4,1 г) и карбоната натрия (6,1 г) в ацетоне (250 мл), по методу, аналогичному описанному в примере Б1, после хроматографии на силикагеле (система растворителей: толуол-диоксан= 9:1) и перекристаллизации из диизопропилового эфира получают 7,3 г (81%) указанного в заголовке соединения с tпл 210-212oC.
Б7. 8-(2-трет-бутоксикарбониламино-6-метилбензилокси)-3-формил-2- метилимидазо[1.2-а]пиридин
Исходя из 3-формил-8-гидрокси-2-метилимидазо [1,2-а] пиридина (2,4 г), 2-трет-бутоксикарбониламино-6-метилбензилхлорида (4,2 г), иодида натрия (2,5 г) и карбоната натрия (3,7 г) в ацетоне (400 мл), по методу, аналогичному описанному в примере Б1, после перекристаллизации из диизопропилового эфира-этилацетата получают 4,4 г (80%) указанного в заголовке соединения с tпл 189-191oC.
В1. 8-(2-амино-6-метилбензиламино)-2,3-диметилимидазо[1.2-а]пиридин
Метод A
Раствор 8-(6-метил-2-нитробензиламино)-2,3-диметилимидазо[1,2-а]пиридина (61 г) в метаноле (5,5 л) гидрируют в присутствии 15 г палладия на активированном угле (5%-ной) в качестве катализатора при КТ и при атмосферном давлении в течение 1,5 ч. После отфильтровывания катализатора и концентрирования остаток растворяют в кипящем этилацетате (2,7 л). После охлаждения до КТ выделяют 51 г (82%) указанного в заголовке соединения с tпл 206-208oC. Метод Б
6,7 г 8-(2-трет-бутоксикарбониламино-6-метилбензиламино)-2,3-диметилимидазо [1,2-а]пиридина при 25-30°С порциями добавляют к смеси трифторуксусной кислоты (30 мл) и анизола (3 мл). После 30-минутного перемешивания при КТ раствор выливают в 100 мл ледяной воды, после чего смешивают с 75 мл 6H едкого натра. Осадок отфильтровывают и очищают посредством хроматографии на силикагеле (система растворителей: толуол-диоксан = 8:1). После перекристаллизации из этилацетата получают 3,1 г (62%) указанного в заголовке соединения с tпл 206-208oC.
В2. 8-(2-амино-6-метилбензиламино)-3-формил-2-метилимидазо[1,2-а]пиридин
Исходя из 8-(2-трет-бутоксикарбониламино-6-метилбензиламино)-3-формил-2- метилимидазо[1,2-а] пиридина (3,6 г), трифторуксусной кислоты (15 мл) и анизола (5 мл), но методике, описанной в примере В1 (метод Б), после хроматографии на силикагеле (система растворителей: толуол-диоксан=9:1) и кристаллизации из этилацетата-циклогексана получают 2,3 г (76%) указанного в заголовке соединения с tпл 230-234oC.
В3. 8-(2-амино-6-метилбензилокси)-3-формил-2-метилимидазо[1.2-а] пиридин
Исходя из 8-(2-трет-бутоксикарбониламино-6-метилбензилокси)-3-формил-2- метилимидазо [1,2-а] пиридина (5,0 г) и трифторуксусной кислоты (40 мл), по методу, аналогичному описанному в примере В1 (метод Б), получают 3,57 г (96%) указанного в заголовке соединения с tпл 144-150oC (разл.).
В4. Гидрохлорид 8-(2-амино-6-метилбензиламино)-3-хлор-2-метилимидазо [1.2-а] пиридина
Раствор 8-(2-нитро-6-метилбензиламино)-3-хлор-2-метилимидазо[1,2-а] пиридина (2,0 г, 6 ммолей) в метаноле (175 мл) и диоксане (175 мл) подвергают взаимодействию с катализатором платина на угле (5%-ный) и гидрируют в течение 2 ч при КТ и при атмосферном давлении. По истечении 2 ч добавляют 2H соляную кислоту (5 мл) и повторно гидрируют в течение 1 ч в тех же условиях. Затем катализатор отфильтровывают, pH фильтрата 2H едким натром доводят до 8,5 и растворитель отгоняют с помощью ротационного испарителя. Остаток растворяют в кипящем этилацетате (400 мл). После охлаждения до КТ добавляют диизопропиловый эфир (250 мл) и для полного завершения кристаллизации перемешивают в течение 30 мин при 4oC. Затем осадок отсасывают, промывают диизопропиловым эфиром и сушат под вакуумом. Указанное в заголовке соединение (1,66 г, 92%) выделяют в виде твердого вещества бежевого цвета, tпл 243-246oC.
Промышленная применимость
Соединения формулы I и их соли обладают ценными фармакологическими свойствами, которые обеспечивают возможность их промышленной применимости. Прежде всего они проявляют ярко выраженный ингибирующий эффект в отношении секреции желудочной кислоты и отличное действие по защите желудка и кишечника у теплокровных. При этом соединения согласно изобретению отличаются высокой избирательностью действия, сравнительно длительной продолжительностью действия, хорошей энтеральной эффективностью, отсутствием существенных побочных явлений и широким терапевтическим спектром.
Под понятием "защита желудка и кишечника" в этой связи имеются в виду предупреждение и лечение заболеваний желудочно-кишечного тракта, прежде всего воспалений и поражений желудочно-кишечного тракта (таких, например, как язва желудка, язва двенадцатиперстной кишки, гастрит, гиперацидный либо медикаментозный раздраженный желудок), которые могут быть вызваны, например, микроорганизмами (в частности бактериями Helicobacter pylori), бактериальными токсинами, медикаментами (например, определенными противовоспалительными средствами и антиревматическими средствами), химикалиями (например, этанолом), желудочной кислотой или стрессовыми состояниями. Соединения согласно изобретению обладают при этом также проприоцептивным действием против микроорганизма Helicobacter pylori.
По своим отличным свойствам соединения по изобретению, проходившие экспериментальную проверку на различных моделях, где определялись их противоязвенные и антисекреторные свойства, неожиданно значительно превосходили соединения, известные из уровня техники. Благодаря этим свойствам соединения формулы I и их фармакологически приемлемые соли пригодны для высокоэффективного применения в медицине и ветеринарии, причем в первую очередь их можно использовать для лечения и/или профилактики заболеваний желудка и/или кишечника.
Еще одним объектом изобретения в соответствии с этим являются соединения по изобретению, предназначенные для применения при лечении и/или профилактике указанных выше заболеваний.
Изобретение включает далее применение предлагаемых соединений для изготовления лекарственных средств, предназначенных для лечения и/или профилактики указанных выше заболеваний.
Изобретение включает далее применение предлагаемых соединений для лечения и/или профилактики указанных выше заболеваний.
Другим объектом изобретения является лекарственное средство, предназначенное для предупреждения и лечения заболеваний желудочно-кишечного тракта, вызванных бактериями Helicobacter pylori или повышенной секрецией желудочной кислоты, содержащее одно либо несколько соединений формулы I и/или его фармакологически приемлемую соль.
Лекарственные средства изготавливают с помощью обычных методов, известных специалисту в данной области техники. В качестве лекарственных средств предлагаемые согласно изобретению фармакологически эффективные соединения (являющиеся действующими веществами) применяют либо индивидуально, либо предпочтительно в комбинации с пригодными для этой цели фармацевтическими вспомогательными агентами и/или наполнителями в виде таблеток, драже, капсул, суппозиториев, пластырей (например, в форме чрескожно действующей терапевтической системы), эмульсий, суспензий или растворов, причем содержание действующих веществ составляет предпочтительно от 0,1 до 95% и причем благодаря соответствующему выбору вспомогательных агентов и наполнителей с учетом особенностей действующего вещества и/или требуемого эффекта можно получить готовые для применения галеновые препараты (например, форму пролонгированного действия или форму, устойчивую к действию желудочного сока).
Выбор вспомогательных агентов, соответственно наполнителей, пригодных для требуемой лекарственной композиции, определяет сам специалист на основе своих знаний и опыта. Наряду с растворителями, гелеобразователями, основными компонентами суппозиториев, вспомогательными веществами для таблеток и другими наполнителями действующих веществ могут использоваться также, например, антиоксиданты, диспергаторы, эмульгаторы, антивспениватели, корригенты вкуса, консерванты, агенты растворимости, красители или прежде всего промоторы, улучшающие проницаемость, и комплексообразователи (в частности циклодекстрины).
Действующие вещества могут вводиться орально, парентерально или чрескожно.
Как правило, в медицине для достижения необходимого эффекта оказалось целесообразным действующее вещество, соответственно действующие вещества при оральном введении назначать в суточной дозировке от приблизительно 0,01 до приблизительно 20, предпочтительно от 0,05 до 5, прежде всего 0,1 до 1,5 мг/кг веса тела, разбивая при необходимости суточную дозу на несколько разовых доз, преимущественно от 1 до 4. При парентеральном лечении могут назначаться аналогичные или же (прежде всего при внутривенном введении действующих веществ), как правило, более низкие дозировки. Определение необходимой в каждом конкретном случае оптимальной дозировки, равно как и метода введения действующих веществ может без труда проводить каждый специалист на основе своих знаний и опыта.
При применении соединений по изобретению и/или их солей для лечения вышеназванных заболеваний фармацевтические композиции могут содержать в своем составе также один либо несколько фармакологически активных ингредиентов из других групп лекарственных средств, таких, как антацидные средства, например, гидроксид алюминия, алюминат магния; транквилизаторы, такие, как бензодиазепины, например, диазепам; спазмолитические средства, такие, например, как биетамиверин, камилофин; антихолинергические средства, такие, например, как оксифенциклимин, фенкарбамид; местно-анестезирующие средства, такие, например, как тетракаин, прокаин; при необходимости также ферменты, витамины или аминокислоты.
В этой связи следует выделить в первую очередь возможность сочетания соединений согласно изобретению с фармацевтическими средствами, подавляющими секрецию кислоты, такими, например, как H2-блокаторы (в частности циметидин, ранитидин), ингибиторы H+/K+-АТФ-азы (в частности омепразол, пантопразол), или далее с так называемыми периферийными антихолинергическими средствами (как, например, пирензепин, телензенин), а также с антагонистами гастрина, что имеет целью усилить суммарное или сверхсуммарное основное действие и/или устранить либо снизить побочные эффекты, или далее возможность сочетания с веществами, обладающими антибактериальной эффективностью (такими, например, как цефалоспорины, тетрациклины, налидиксиновая кислота, пенициллины или также соли висмута), для борьбы с бактериями Helicobacter pylori.
Фармакология
Высокая эффективность соединений согласно изобретению по защите желудка и их ингибирующее действие в отношении секреции желудочной кислоты может быть подтверждена результатами исследований, проводившихся в экспериментах на моделях животных. Соединения по изобретению, проходившие экспериментальную проверку на представленной ниже модели, имеют те же порядковые номера, что и соединения, указанные в примерах.
Испытание подавляющего секрецию действия на перфундированном желудке крысы
В приведенной таблице 2 представлено in vivo воздействие соединений по изобретению после внутривенного введения на стимулированную пентагастрином секрецию кислоты на перфундированном желудке крысы.
Методика
У наркотизированных крыс (крысы линии CD, женские особи, вес 200-250 г; 1,5 г/кг внутримышечно уретан) после трахеометрии разрезом по средней линии вскрывали верхнюю брюшную часть живота и через рот вводили в пищевод ПВХ-катетер, а другой катетер в привратник, фиксируя их таким образом, чтобы концы трубок входили в просвет желудка. Катетер, ведущий из привратника, выходил через боковой разрез в правой стенке живота наружу.
После тщательного промывания (примерно 50-100 мл) через желудок непрерывно пропускали имевший температуру 37oC физиологический раствор NaCl (0,5 мл/мин, pH 6,8-6,9; фирма Braun-Unita 1). В собираемом через каждые 15 мин эффлуате (истекающая жидкость) определяли значение pH (pH-метр 632, стеклянный электрод EA 147; диаметр 5 мм, Metrohm, а также выявляли титрованием с помощью свежеприготовленной 0,01 H NaOH до pH 7 (измерительный прибор Dosimat 665 Metrohm) выделенную HCl.
Стимуляцию желудочной секреции осуществляли длительным внутривенным вливанием 1 мкг/кг (соответствует 1,65 мл/ч) пентагастрина (в левую бедренную вену) по истечении приблизительно 30 мин после окончания операции (т.е. после определения двух предварительных фракций). Испытуемые субстанции вводили внутривенно с объемом жидкости 1 мл/кг через 60 мин после начала длительного вливания пентагастрина.
Температуру тела подопытных животных путем инфракрасного излучения и использования грелок (автоматические, бесступенчатое регулирование через ректальный температурный датчик) поддерживали постоянной на уровне 37,8-38oC.
В таблице указана дозировка, обеспечивающая максимальное подавление секреции кислоты порядка 100%.
Определение общего уровня токсичности в отношении наиболее предпочтительных соединений, в частности, соединения примера 1 дает следующие показатели токсичности LD50 для этого соединения:
мыши (пероральное введение) > 750 мг/кг
крысы (пероральное введение) > 500 мг/кг,
мыши (внутривенное введение) около 80 мг/кг)
крысы (внутривенное введение) около 70 мг/кг.
Примеры получения лекарственного средства:
а) Таблетки, содержащие 40 мг действующего вещества: 20 кг 3-[2-(2,3-диметилимидазол[1,2-а] пиридин-8-иламинометил)-3-метилфенил]оксазолидин-2-она, 40 кг молочного сахара, 26 кг кукурузного крахмала и 3 кг поливинилпирролидона увлажняют 20 л воды и пропускают через специальное сито с диаметром отверстий 1,25 мм с целью получения гранул. Полученные гранулы просушивают во вращающемся сушильном аппарате до уровня содержания влаги 50-60%, после чего смешивают с 8 кг натрийкарбоксиметилцеллюлозы, 2 талька и 1 кг стеарата магния. Готовый гранулят формуют в таблетки по 200 мг и диаметром 8 мм.
б) Капсулы, содержащие 30 мг действующего вещества: 300 г 3-[2-(2,3-диметилимидазол[1,2-а] пиридин-8-иламинометил)-3- метилфенилоксазолидин-2-она, 695 г микрокристаллической целлюлозы и 5 г аморфной кремниевой кислоты растирают в порошок, тщательно перемешивают и помещают в желатиновые капсулы, имеющие размер 4.
в) Капсулы, содержащие 10 мг действующего вещества: 100 г 3- [2- (2,3-диметилимидазол [1,2-а]пиридин-8-иламинометил) -3- метилфенил]оксазолидин-2-она, 895 г микрокристаллической целлюлозы и 5 г аморфной кремниевой кислоты растирают в порошок, тщательно перемешивают и помещают в желатиновые капсулы, имеющие размер 4.
г) Ампулы, содержащие 10 мг действующего вещества: Растворяют 3,16 г метансульфоната 3-[2-(2,3-диметилимидазол[1,2-а] пиридин- 8-иламинометил)-3-метилфенил] оксазолидин-2-она в растворе, содержащем 165,5 г маннита и 0,5 г карбоната натрия в 1300 мл дистиллированной воды, доливают дистиллированную воду до метки 1500 мл и фильтруют. Полученный раствор дозируют по 5 мл в 15 мл ампулы и подвергают лиофилизации. Лиофилизат можно смешивать с 10 мл воды для получения нужной концентрации.
| название | год | авторы | номер документа |
|---|---|---|---|
| АЛКОКСИАЛКИЛКАРБАМАТЫ ИМИДАЗО[1,2-A]ПИРИДИНОВ И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 1994 |
|
RU2139288C1 |
| ЗАМЕЩЕННЫЕ АМИНОАЛКИЛАМИНОПИРИДИНЫ И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 1994 |
|
RU2135492C1 |
| ДИГИДРОБЕНЗОФУРАНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1995 |
|
RU2138498C1 |
| ИМИДАЗОПИРИДИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ПОЛУЧЕНИЯ ФАРМАЦЕВТИЧЕСКОЙ КОМПОЗИЦИИ | 1994 |
|
RU2136682C1 |
| ПИРИДИЛТИОСОЕДИНЕНИЯ ДЛЯ БОРЬБЫ С БАКТЕРИЯМИ HELICOBACTER | 1995 |
|
RU2149872C1 |
| ТИОПИРИДИНЫ ДЛЯ БОРЬБЫ С БАКТЕРИЯМИ HELICOBAKTER | 1995 |
|
RU2142459C1 |
| ЗАМЕЩЕННЫЕ АРИЛАЛКИЛТИОАЛКИЛТИОПИРИДИНЫ ДЛЯ БОРЬБЫ С БАКТЕРИЯМИ HELICOBACTER | 1995 |
|
RU2139286C1 |
| ЗАМЕЩЕННЫЕ АРИЛТИОАЛКИЛТИОПИРИДИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ И СПОСОБ ПОЛУЧЕНИЯ ЛЕКАРСТВЕННОГО СРЕДСТВА. | 1994 |
|
RU2154062C2 |
| КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ РЕСПИРАТОРНОГО ДИСТРЕСС-СИНДРОМА НОВОРОЖДЕННЫХ И ОСТРОГО РЕСПИРАТОРНОГО ДИСТРЕСС-СИНДРОМА, СОДЕРЖАЩИЕ ПО КРАЙНЕЙ МЕРЕ, ОДИН ГЛЮКОКОРТИКОСТЕРОИД В СОЧЕТАНИИ С ЛЕГОЧНЫМ ПОВЕРХНОСТНО-АКТИВНЫМ ВЕЩЕСТВОМ | 1995 |
|
RU2157222C2 |
| ПОЛИПЕПТИДЫ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ ЛЕГОЧНО-ПОВЕРХНОСТНО АКТИВНОГО ВЕЩЕСТВА, И ФАРМКОМПОЗИЦИЯ | 1995 |
|
RU2145611C1 |

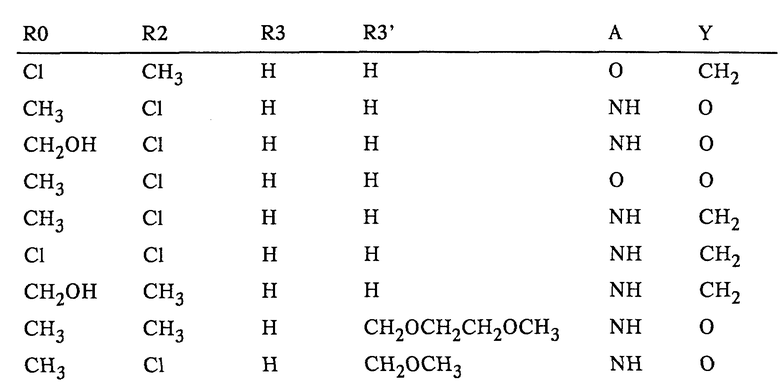

Имидазопиридиназолидиноны формулы I, где R0 - (C1-C4)алкил, гидроксиметил или галоген; R1 - (С1-C4)алкил; R2, R3, R4, R5 -водород или (С1-C4)алкил; R3' - водород, (С1-C4)алкил или (С1-C4)алкил, замещенный 1-2 галогенами или (С1-C4)алкокси; А - О или NH; Y -О или СН2, и их соли, могут быть использованы для предупреждения и лечения заболеваний желудочно-кишечного тракта. 2 с. и 4 з.п.ф-лы, 2 табл.

где R0 означает (С1-С4)алкил, гидроксиметил или галоген;
R1 означает (С1-С4)алкил;
R2 означает водород или (С1-С4)алкил;
R3 означает водород или (С1-С4)алкил;
R3' означает водород, (С1-С4)алкил либо (С1-С4)алкил, замещенный одним или двумя идентичными либо различными заместителями, выбранными из группы, включающей галоген и (С1-С4)алкокси;
R4 означает водород или (С1-С4)алкил;
R5 означает водород или (С1-С4)алкил;
А означает O (кислород) или NН;
Y означает O (кислород) или СН2,
и их соли.
| ПОПЕРЕЧНЫЙ ЭЛЕВАТОР К КОРНЕКЛУБНЕПЛОДОУБОРОЧНЫМ МАШИНАМ | 0 |
|
SU261004A1 |
| Устройство для отсчета и записи на расстоянии показаний стрелочных приборов | 1958 |
|
SU117992A1 |
