Изобретение относится к новым соединениям, предназначенным для применения в фармацевтической промышленности в качестве действующих веществ при изготовлении лекарственных средств.
В европейской патентной заявке ЕР-А-0033094 описываются имидазо[1,2-а] пиридины, содержащие в положении 8 арильный заместитель, представляющий собой фенил, тиенил или пиридил либо фенил, замещенный на хлор, фтор, метил, трет.-бутил, трифторметил, метокси или циано. В качестве арильных радикалов, представляющих особый интерес, в упомянутой европейской заявке ЕР-А-0033094 названы фенил, о- либо п-фторфенил, п-хлорфенил и 2,4,6-триметилфенил, причем наиболее предпочтительными из них являются фенил, о- либо п-фторфенил и 2,4,6-триметилфенил.
В европейских патентных заявках ЕР-А-0204285, ЕР-А-0228006, ЕР-А- 0268989 и ЕР-А-0308917 описываются имидазо[1,2-а] пиридины, содержащие в положении 3 ненасыщенный алифатический радикал, прежде всего (замещенный) алкинильный радикал.
В европейской патентной заявке ЕР-А-0266890 описываются имидазо[1,2-а] пиридины, замещенные в положении 8 на алкенильный, алкильный либо циклоалкильный радикал.
Задачей настоящего изобретения является разработка новых соединений, обладающих противоязвенными и антисекреторными свойствами, способа их получения и лекарственного средства на их основе.
Было найдено, что подробно описанные ниже соединения, отличающиеся от известных соединений из уровня техники прежде всего замещением в положении 3 или 8, обладают неожиданными и особенно положительными свойствами.
Таким образом, объектом настоящего изобретения являются алкоксиалкилкарбаматы имидазо[1,2-а]пиридинов формулы I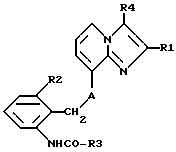
где R1 обозначает C1-C4алкил,
R2 обозначает C1-C4алкил,
R3 обозначает C1-C4алкокси-C2-C4алкокси,
R4 обозначает C1-C4алкил или гидроксиметил и
A обозначает О (кислород) или NH, и их соли.
C1-C4алкил представляет собой линейные либо разветвленные алкильные радикалы с числом атомов углерода 1-4. В качестве примеров можно назвать бутил, изобутил, втор. -бутил, трет. -бутил, пропил, изопропил, этил и в первую очередь метил.
C1-C4алкокси представляет собой атом кислорода, с которым связан один из вышеназванных C1-C4алкильных радикалов. Предпочтителен из них метоксильный радикал. C2-C4алкокси представляет собой атом кислорода, с которым связан один из C2-C4алкильных радикалов, выбранный из группы указанных выше C1-C4алкильных радикалов. C1-C4алкокси-C2-C4алкокси представляет собой C2-C4алкоксильный радикал, с которым связан C1-C4алкоксильный радикал. Предпочтителен среди них 2-метоксиэтоксильный радикал.
В качестве солей соединений формулы I могут рассматриваться предпочтительно все кислотно-аддитивные соли. Особо следует назвать фармакологически приемлемые соли неорганических и органических кислот, применяемые обычно при изготовлении средств в галеновой форме. Фармакологически неприемлемые соли, которые могут являться первичными продуктами способа, осуществляемого при получении соединений по изобретению в промышленном масштабе, переводятся затем с помощью известных специалисту методов в фармакологически приемлемые соли. В качестве таковых пригодны водорастворимые, равно как и водонерастворимые кислотно-аддитивные соли, образованные от таких кислот, как, например, соляная кислота, бромистоводородная кислота, фосфорная кислота, азотная кислота, серная кислота, уксусная кислота, лимонная кислота, D-глюконовая кислота, бензойная кислота, 2-(4-гидроксибензоил)бензойная кислота, масляная кислота, сульфосалициловая кислота, малеиновая кислота, лауриновая кислота, яблочная кислота, фумаровая кислота, янтарная кислота, щавелевая кислота, винная кислота, эмбоновая кислота, стеариновая кислота, толуолсульфоновая кислота, метансульфоновая кислота или 3-гидрокси-2-нафтойная кислота, причем для получения соответствующей соли - в зависимости от того, какую кислоту используют: одноосновную или многоосновную, и в зависимости от того, какую соль требуется получить, кислоты применяют в эквимолярном количественном соотношении либо приблизительно в таком соотношении.
В качестве примеров соединений можно назвать следующие: 8-{2-[(2- метоксиэтокси)карбониламино] -6-метилбензилокси}-2- метилимидазо[1,2-а]пиридин-3-метанол, 8-{2-[(2-метоксиэтокси)карбониламино]-6-метилбензиламино}-2- метилимидазо[1,2-а] пиридин-3-метанол и 8-{2-[(2-метоксиэтокси)карбониламино] -6-метилбезиламино}-2,3- диметилимидазо[1,2-а]пиридин и их соли.
Среди соединений формулы I следует выделить такие, в которых R4 обозначает гидроксиметил и A представляет собой О (кислород), a R1, R2 и R3 имеют указанные выше значения, а также соли этих соединений.
Другим объектом изобретения является способ получения соединений формулы I и их солей. Способ заключается в том, что
а) соединения формулы II,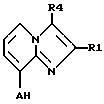
где R1, R4 и A имеют указанные выше значения, или их соли подвергают взаимодействию с соединениями формулы III,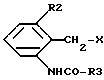
где R2 и R3 имеют указанные выше значения, а X представляет собой соответствующую реакционноспособную отщепляемую группу, либо с их солями, или
б) для получения соединений формулы 1, в которых R4 обозначает гидроксиметил, восстанавливают соединения формулы IV,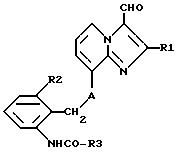
где R1, R2, R3 и A имеют указанные выше значения и затем при необходимости соединения I, полученные согласно а) или б), переводят в их соли, или затем при необходимости из полученных солей соединений I высвобождают соединения I.
Взаимодействие соединений II с соединениями III осуществляют по методике, известной специалистам. Соответствующей реакционноспособной отщепляемой группой является, например, атом галогена (предпочтительно хлор либо бром) или метансульфонилоксигруппа. Обменную реакцию проводят предпочтительно в присутствии основания (например, неорганического гидроксида, такого, как гидроксид натрия, либо неорганического карбоната, такого, как карбонат калия, либо органического азотистого основания, такого, как триэтиламин, пиридин, коллидин или 4-диметиламинопиридин), причем добавками катализаторов, таких, как иодид щелочного металла или бромид тетрабутиламмония, можно эффективно воздействовать на ход реакции.
Восстановление соединений IV осуществляют по обычной методике, хорошо известной специалистам. Реакцию проводят в инертных растворителях, например, в низших алифатических спиртах, например, с использованием соответствующих гидридов, таких, как борогидрид натрия, при необходимости с добавками воды.
Выбор тех или иных параметров реакции, требуемых для осуществления способа, не доставит квалифицированному специалисту трудностей.
Выделение и очистку веществ по изобретению осуществляют с помощью известных методов, используя для этой цели, например, отгонку растворителя под вакуумом и перекристаллизацию полученного остатка из соответствующего растворителя или применяя один из обычных методов очистки, как, например, колоночная хроматография на соответствующем носителе.
Кислотно-аддитивные соли получают путем растворения свободного основания в соответствующем растворителе, например, в хлорированном углеводороде, таком, как метиленхлорид или хлороформ, в низкомолекулярном алифатическом спирте (этаноле, изопропаноле), в кетоне, таком, как ацетон, или в простом эфире, таком, как ТГФ или диизопропиловый эфир, который содержит требуемую кислоту или в который затем вводят требуемую кислоту.
Выделение солей осуществляют путем фильтрации, переосаждения, высаживания с помощью растворителя, нерастворяющего аддитивную соль, или путем выпаривания растворителя. Полученные соли можно переводить подщелачиванием, например, водным раствором аммиака, в свободные основания, которые в свою очередь могут быть снова переведены в кислотно-аддитивные соли. Таким путем фармакологически неприемлемые кислотно-аддитивные соли могут быть превращены в фармакологически приемлемые кислотно-аддитивные соли.
Исходные соединения формулы II известны в частности из европейских патентных заявок ЕР-А-0290003 и ЕР-А-0299470. Исходные соединения формулы III являются новыми. Их получают аналогично способам, известным из соответствующих публикаций, а именно следующим образом: в соединениях III, где X обозначает OH, гидроксильную группу взаимодействием с галогенирующим средством, таким, как, например, тионилхлорид, тионилбромид, фосфортрибромид либо оксалилхлорид, переводят в реакционноспособную отщепляемую группу, например, в атом галогена, или взаимодействием с метансульфонилхлоридом, при необходимости в присутствии основания, ее превращают в метансульфонилоксигруппу.
Соединения формулы IV представляют собой новые соединения и также являются объектом настоящего изобретения. Как и соединения формулы I, их получают аналогичным взаимодействием соединений II, где R4 обозначает CHO, с соединениями III, описанными выше.
Нижеследующие примеры поясняют подробнее способ получения соединений формулы I. Сокращение KT обозначает комнатную температуру, ч обозначает час(ы).
Примеры
Конечные и промежуточные продукты
1. 8-{ 2-[(2-метоксиэтокси)карбониламино]-6-метилбензилокси}- 2-метилимидазо[1,2-а]пиридин-3-карбоксальдегид
Смесь 2,0 г (11,35 ммолей) 8-гидрокси-2-метилимидазо[1,2-а]пиридин-3-карбоксальдегида, 1,2 г безводного карбоната натрия, 0,17 г (1,14 ммоля) иодида натрия и 3,3 г (12,8 ммолей) 2-метоксиэтилового эфира [2-(хлорметил)-3-метилфенил] карбаминовой кислоты в 30 мл ацетона перемешивают в течение 24 ч при КТ и выливают в 200 мл ледяной воды. Затем осадок отфильтровывают, сушат и перекристаллизовывают из толуола/диизопропилового эфира. В результате получают 3,9 г (86,5%) соединения, указанного в заголовке, с Тпл 119-120oC.
2. 8-{ 2-[(2-метоксиэтокси)карбониламино] -6-метилбензилокси} 2-метилимидазо[1,2-а] пиридин-3-метанол
Суспензию 3,7 г (9,3 ммолей) 8-{2-[(2-метоксиэтокси)карбониламино]-6-метилбензилокси} -2-метилимидазо[1,2-а] пиридин-3-карбоксальдегида в 40 мл метанола смешивают при КТ с 362 мг (9,3 ммолей) 97%-ного борогидрида натрия и перемешивают в течение 75 мин. Затем выливают в ледяную воду, экстрагируют дихлорметаном и концентрируют в ротационно-вакуумном испарителе. Оставшееся масло кристаллизуют с помощью 5 мл изопропилового спирта, 5 мл толуола и диизопропилового эфира. Таким путем получают 2,7 г (72,7%) соединения, указанного в заголовке, с Тпл 121-123oC.
3. 8-{ 2-[(2-метоксиэтокси)карбониламино] -6-метилбензиламино} -2-метилимидазо[1,2-а]пиридин-3-карбоксальдегид
Смесь из 2,0 г (11,41 ммолей) 8-амино-2-метилимидазо[1,2-а]пиридин-3-карбоксальдегида, 1,21 г (11,41 ммолей) безводного карбоната натрия, 0,17 г (1,14 ммоля) иодида натрия и 3,5 г (13,6 ммолей) 2-метоксиэтилового эфира [2-(хлорметил)-3-метилфенил]карбаминовой кислоты в 30 мл ацетона перемешивают в течение 24 ч при КТ и концентрируют в ротационно-вакуумном испарителе. Остаток смешивают со 100 мл воды, после чего экстрагируют этилацетатом, органическую фазу сушат сульфатом магния и концентрируют в вакууме. Остаток перекристаллизовывают из толуола. В результате получают 3,31 г (73%) соединения, указанного в заголовке, с Тпл 153-155oC.
4. 8-{2-[(2-метоксиэтокси)карбониламино]-6-метилбензиламино} -2-метилимидазо[1,2-а]пиридин-3-метанол
2,8 г (7,06 молей) 8-{2-[(2-метоксиэтокси)карбониламино]-6- метилбензиламино} -2-метилимидазо[1,2-а] пиридин-3-карбоксальдегида восстанавливают с помощью борогидрида натрия аналогично примеру 2, метанол отгоняют в вакууме, смешивают с водой и этилацетатом и раствором гидрофосфата калия устанавливают pH 9. Затем несколько раз экстрагируют этилацетатом, сушат, концентрируют в вакууме и перекристаллизовывают из толуола/диизопропилового эфира. Таким путем получают 2,28 г (81%) соединения, указанного в заголовке, с Тпл 138-140oC.
5. 8-{2-[(2-метоксиэтокси)карбониламино]-6-метилбензиламино} -2,3-диметилимидазо[1,2-а]пиридин-изопропиловый спирт (1/1)
Смесь 3,0 г (18,6 ммолей) 8-амино-2,3-диметилимидазо[1,2-а]пиридина, 4,9 г (46,2 ммолей) безводного карбоната натрия, 0,28 г (1,86 ммоля) иодида натрия и 5,8 г (22,5 ммоля) 2-метоксиэтилового эфира [2-(хлорметил)-3-метилфенил] карбаминовой кислоты в 30 мл ацетона перемешивают в течение 20 ч при КТ. Затем фильтруют, концентрируют в вакууме, смешивают с водой и этилацетатом, разбавленной соляной кислотой устанавливают pH 6 и экстрагируют этилацетатом. Органический раствор сушат и концентрируют в вакууме. Остаток растворяют в 40 мл ацетона и смешивают с раствором 1,2 г (10,3 ммолей) фумаровой кислоты в 80 мл ацетона. Выпадение кристаллов не происходит. Тогда повторно концентрированный раствор смешивают с толуолом и изопропиловым спиртом и при температуре 0oC осаждают с помощью диизопропилового эфира 4,9 г фумарата. Полученный фумарат смешивают с 50 мл этилацетата и 10 мл воды, едким натром устанавливают pH 9 и свободное основание экстрагируют этилацетатом. После концентрирования в вакууме растворяют в толуоле/изопропиловом спирте и при температуре 0oC осаждают с помощью петролейного эфира (Ткип 40oC). В результате получают 2,2 г (26,7%) соединения, указанного в заголовке, с Тпл 85-86oC.
6. 8-{2-[(2-метоксиэтокси)карбониламино]-6-метилбензилокси} -2-метилимидазо[1,2-а]пиридин-3-метанол.
Смесь 178 мг (1,0 ммоль) 8-гидрокси-2-метилимидазо[1,2-а]пиридин-3-метанола, 117 мг (1,1 ммоля) безводного карбоната натрия, 15 мг (0,1 ммоля) иодида натрия и 283 мг (1,1 ммоля) 2-метоксиэтилового эфира [2-(хлорметил)- 3-метилфенил]карбаминовой кислоты в 5 мл ацетона перемешивают в течение 48 ч при КТ, затем перерабатывают аналогично примеру 2 и хроматографируют с помощью этилацетата/изопропилового спирта (соотношение 9:1). Таким путем получают 247 мг (62%) соединения, указанного в заголовке.
7. 8-{ 2-[(2-метоксиэтокси)карбониламино]-6-метилбензилокси} -2,3-диметилимидазо[1,2-а]пиридин.
Аналогично примеру 5 подвергают взаимодействию 2,0 г (12,4 ммолей) 8-гидрокси-2,3-диметилимидазо[1,2-а] пиридина, 3,6 г (13,9 ммолей) 2-метоксиэтилового эфира [2-(хлорметил)-3-метилфенил] карбаминовой кислоты, 0,18 г иодида натрия и 1,3 г карбоната натрия в 30 мл ацетона. В результате получают 1,07 г (22,5%) соединения, указанного в заголовке, с Тпл 107-108oC.
Исходные продукты
Аа. 2-Метоксиэтиловый эфир [2-(гидроксиметил)-3-метилфенил] карбаминовой кислоты
К раствору 33 г (0,24 моля) 2-амино-6-метилбензилового спирта и 19,4 мл (0,24 моля) пиридина в 600 мл изопропилового спирта при температуре 10oC добавляют по каплям при перемешивании и охлаждении 33,2 г (0,24 моля) 2-метоксиэтилового эфира хлормуравьиной кислоты. Перемешивание продолжают еще в течение 2 ч при 0oC, затем смешивают с водой и изопропилацетатом и несколько раз экстрагируют изопропилацетатом. Органическую фазу сушат сульфатом магния и концентрируют при 50oC в ротационно-вакуумном испарителе. Остаток хроматографируют на силикагельной колонне с помощью этилацетата. После концентрирования в вакууме получают 36 г (68%) соединения, указанного в заголовке, в виде масла.
Аб. 2-Метоксиэтиловый эфир [2-(хлорметил)-3-метилфенил] карбаминовой кислоты
К раствору 18,0 г (0,075 моля) указанного выше соединения в 80 мл толуола при температуре 17-20oC по каплям добавляют при перемешивании и охлаждении 9,4 г (0,079 моля) тионилхлорида и оставляют на ночь при КТ. Затем охлаждают на ледяной бане, растирают и в результате получают 11,2 г (57,7%) соединения, указанного в заголовке, с Тпл 100-102oC. После концентрирования маточного раствора и кристаллизации из толуола/петролейного эфира (Ткип 40oC), получают еще 4,8 г (24,7%) осадка с аналогичной температурой плавления.
Б. 8-гидрокси-2-метилимидазо[1,2-а]пиридин-3-карбоксальдегид
4,77 г (0,02 моля) 8-бензилокси-2-метилимидазо[1,2-а]пиридина перемешивают в смеси Вильсмайера, содержащей 20 мл диметилформамида и 2,3 мл хлорокиси фосфора, в течение 2,5 ч при температуре 60oC и смесь разделяют по обычной методике с помощью ледяной воды и гидрокарбоната калия. Таким путем получают 8-бензилокси-2-метилимидазо[1,2-а] пиридин-3-карбоксальдегид с Тпл 105-106oC (из диизопропилового эфира). Это соединение аналогично тому, как это описано у Kaminski и др., в Journ. Med. Chem. 28, 876 (1985), Methode Н. , дебензилируют до соединения, указанного в заголовке, с Тпл 251-252oC.
Промышленная применимость
Соединения формулы I и их соли обладают ценными фармакологическими свойствами, обеспечивающими их промышленную применимость. Прежде всего они обладают ярко выраженной способностью ингибировать секрецию кислоты желудочного сока и отличным защитным действием по отношению к желудку и кишечнику у теплокровных. При этом наряду с хорошей растворимостью в водной среде соединения по изобретению отличаются высокой избирательностью действия, сравнительно продолжительной длительностью действия, хорошей энтеральной эффективностью, отсутствием существенных побочных эффектов и широким терапевтическим спектром.
Под понятием "защита желудка и кишечника" в контексте изобретения имеются в виду предупреждение и лечение заболеваний желудочно-кишечного тракта, прежде всего желудочно-кишечных воспалительных заболеваний и поражений (как, например, язвенная болезнь желудка, язва двенадцатиперстной кишки, гастрит, раздражение желудка, вызванное повышенной кислотностью или действием определенных лекарств), которые могут быть обусловлены, например, микроорганизмами (например, Helicobacter pylori), бактериальными токсинами, медикаментами (например, некоторыми противовоспалительными средствами и антиревматическими средствами), химикалиями (например, этанолом), кислотой желудочного сока или стрессовыми ситуациями. Соединения по изобретению обладают, кроме того, также собственным действием против зародыша Helicobacter pylori.
По своим отличным противоязвенным и антисекреционным свойствам, проходившим экспериментальную проверку на различных моделях, соединения по изобретению неожиданным образом заметно превосходят соединения, известные из уровня техники. Благодаря этим свойствам соединения формулы I и их фармакологически приемлемые соли могут исключительно эффективно применяться в медицине и ветеринарии, причем свое применение они могут находить в первую очередь для лечения и/или профилактики заболеваний желудка и кишечника, равно как и для лечения остеопороза.
Объектом изобретения являются поэтому соединения по изобретению, обладающие способностью ингибировать секрецию кислоты желудочного сока.
Изобретение включает далее применение соединений по изобретению для изготовления лекарственных средств, обладающих способностью ингибировать секрецию кислоты желудочного сока, которые предназначены для лечения и/или профилактики вышеназванных заболеваний.
Изобретение включает далее применение соединений по изобретению для лечения и/или профилактики вышеназванных заболеваний.
Другим объектом настоящего изобретения являются лекарственные средства, содержащие одно или несколько соединений формулы I и/или их фармакологически приемлемые соли.
Лекарственные средства изготавливают с помощью хорошо известных и доступных специалисту методов. В качестве лекарственных средств соединения по изобретению, обладающие фармакологическим действием (т.е. действующие вещества), применяют либо как таковые, либо предпочтительно в комбинации с соответствующими фармацевтическими вспомогательными веществами и/или наполнителями, причем они могут изготавливаться в виде таблеток, драже, капсул, суппозиториев, пластырей (например, в виде чрескожно действующей терапевтической системы), эмульсий, суспензий или растворов, содержание действующих веществ в которых составляет при этом предпочтительно от 0,1 до 95 %, а благодаря выбору вспомогательных веществ и наполнителей можно получать средства в галеновой форме, точно соответствующей действующему веществу и/или желаемому наступлению действия (например, в форме пролонгированного действия или в форме, устойчивой к действию желудочного сока).
Выбор вспомогательных веществ, соответственно носителей для изготовления требуемых препаративных форм для специалиста, обладающего опытом в данной области, трудностей не представляет. Наряду с растворителями, загустителями, основными веществами для суппозиториев, вспомогательными веществами для таблеток и другими носителями действующих веществ могут применяться, например, покрытия для таблеток, антиоксиданты, диспергаторы, эмульгаторы, антивспениватели, вкусовые добавки, консерванты, вещества, способствующие растворимости, красители или прежде всего промоторы для проницаемости и комплексообразователи (например, циклодекстрины).
Действующие вещества могут назначаться для орального, парентерального или чрескожного введения.
Было установлено, что для достижения положительного терапевтического эффекта при лечении человека суточная доза при оральном введении, при необходимости назначаемая в виде нескольких, предпочтительно от 1 до 4, разовых доз, должна содержать действующее вещество, соответственно действующие вещества в количестве от приблизительно 0,01 до приблизительно 20, предпочтительно 0,05-5, прежде всего 0,1-1,5 мг/кг веса тела. Для парентерального введения могут назначаться препараты примерно с той же дозировкой действующих веществ или (в частности при внутривенном введении), как правило, с более низкой дозировкой. Выбор необходимой оптимальной дозировки, равно как и формы введения действующих веществ в каждом отдельном случае компетентный специалист в данной области определяет сам.
При применении соединений по изобретению и/или их солей для лечения указанных выше заболеваний фармацевтические композиции могут содержать в своем составе также один либо несколько фармацевтически активных ингредиентов, относящихся к другим группам лекарственных средств, такие, как антацидные средства, например, гидроксид алюминия, алюминат магния; транквилизаторы, такие, как бензодиазепины, например, диазепам; спазмолитические средства, как, например, биэтамиверин, камилофин; антихолинэргические средства, как, например, оксифенциклимин, фенкарбамид; местно-анестезирующие средства, как, например, тетракаин, прокаин; при необходимости также ферменты, витамины или аминокислоты.
В этой связи следует подчеркнуть особое значение соединений по изобретению при их применении в сочетании с другими медикаментозными средствами, ингибирующими секрецию кислоты, к каковым относятся, например, H2-блокаторы (например, циметидин, ранитидин), ингибиторы H+/К+- аденозинтрифосфотазы (например, омепразол, пантопразол), или, далее, с так называемыми периферийными антихолинэргическими средствами (как, например, пирензепин, телензепин), а также с гастриновыми антагонистами с целью дополнительного и даже сверхдополнительного усиления основного действия и/или исключения либо снижения побочных эффектов, или, далее, в сочетании с субстанциями, обладающими антибактериальным действием (как, например, цефалоспорины, тетрациклины, налидиксиновая кислота, пенициллины или также висмутовые соли), для борьбы с Helicobacter pylori.
Фармакология
Отличное защитное действие соединений по изобретению по отношению к желудку и их способность ингибировать секрецию кислоты желудочного сока подтверждают результаты экспериментальных исследований, проведенных на моделях животных. Соединения по изобретению, прошедшие экспериментальную проверку на представленной ниже модели, получили порядковые номера, соответствующие нумерации этих соединений в примерах.
Испытание ингибирующего секрецию действия на перфундированном желудке крыс
В нижеследующей таблице представлено влияние соединений по изобретению после внутривенного введения на стимулированную пентагастрином секрецию кислоты перфундированного желудка крыс in vivo.
Методика
У наркотизированных крыс (крыса линии CD, женская особь, 200-250 г; 1,5 г/кг, i. m. уретан) после трахеотомии разрезом по центральной линии эпистральной области вскрывали живот и введенный трансорально один ПХВ-катетер фиксировали в пищеводе, а другой в привратнике таким образом, что концы мягких трубок входили в просвет желудка. Катетер, выходящий из привратника, через боковое отверстие с правой стороны живота выходил наружу.
После тщательного промывания (приблизительно 50-100 мл) желудок при температуре 37oC непрерывно спринцевали теплым физиологическим раствором NaCl (0,5 мл/мин, pH 6,8-6,9; прибор типа Braun-Unita I). В собранном (с помощью мерного цилиндра) через каждые 15 мин эффлюате определяли значение pH (измеритель кислотности pH-Meter 632, стеклянный электрод ЕА 147; ⊘ = 5 мм , прибор Metrohm), а путем титрования свежеприготовленной 0,01H NaOH до pH 7 (прибор Dosimat 655 Metrohm) определяли выделенную HCl.
Стимулирование желудочной секреции осуществляли длительным внутривенным вливанием 1 мкг/кг (=1,65 мл/ч) i.v. пентагастрина (в левую бедренную вену) приблизительно через 30 мин после завершения операции (т.е. после определения двух предварительных фракций). Испытуемые субстанции вводили внутривенно в объеме жидкости 1 мл/кг через 60 мин после начала длительного внутривенного вливания пентагастрина.
Температуру тела подопытных животных с помощью инфракрасного облучения и грелки (автоматическое, бесступенчатое регулирование через ректальный температурный датчик) поддерживали постоянной на уровне 37,8-38oC.
Соединения формулы I обладают низкой токсичностью. Так, например, для соединения из примера 2 значения LD50 составляют:
Мыши.р.о. - > 1000мг/кг
Крысы р.о. - > 1000мг/кг
Мыши в/в - около 80 мг/кг
Крысы в/в - около 20 мг/кг.
Получение составов лекарственного средства может быть проиллюстрировано следующими примерами:
а) Таблетки с содержанием 40 мг действующего вещества
20 кг 8-{2-[(2-метоксиэтокси)карбониламино]-6-метилбензилокси}-2- метилимидазо[1,2-а] пиридин-3-метанола, 40 кг молочного сахара, 26 кг маисового крахмала и 3 кг поливинилпирролидона смачивают в 20 литрах воды и затем гранулируют пропусканием через сита с размером ячеек 1,25. Гранулят высушивают в псевдоожиженном слое до получения продукта с влажностью 50- 60% и затем смешивают с 8 кг натрийкарбоксиметилцеллюлозы, 2 кг талька и 1 кг стеарата магния. Готовый гранулят таблетируют в таблетки по 200 мг диаметром 8 мм.
б) Капсулы с содержанием 30 мг действующего вещества
300 г 8-{2-[(2-метоксиэтокси)карбониламино]-6-метилбензилокси}-2- метилимидазо[1,2-а] пиридин-3-метанола, 695 г микрокристаллической целлюлозы и 5 г аморфной кремниевой кислоты подвергают тонкому измельчению, тщательному смешиванию и инкапсулированию с образованием твердых желатиновых капсул 4 размера.
в) Капсулы с содержанием 10 мг действующего вещества
100 г 8-{2-[(2-метоксиэтокси)карбониламино]-6-метилбензиламино}-2- метилимидазо[1,2-а] пиридин-3-метанола, 895 г микрокристаллической целлюлозы и 5 г аморфной кремниевой кислоты подвергают тонкому измельчению, тщательному смешиванию и инкапсулированию с образованием твердых желатиновых капсул 4 размера.
г) Ампулы с содержанием 10 мг действующего вещества
Растворяют 3,16 г 8-{2-[(2-метоксиэтокси)карбониламино]-6- метилбензилокси} -2,3-диметилимидазо[1,2-а]пиридина в растворе 165,5 г маннита и 0,5 г карбоната натрия в 1300 мл дистиллированной воды и стерильно фильтруют. Полученный раствор разливают по 5 мл в 15 мл ампулы и лиофилизуют. Лиофилизат может быть вновь переведен в водный раствор с 10 мл воды.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИМИДАЗОПИРИДИНАЗОЛИДИНОНЫ, ИХ СОЛИ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 1995 |
|
RU2138501C1 |
| ЗАМЕЩЕННЫЕ АМИНОАЛКИЛАМИНОПИРИДИНЫ И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 1994 |
|
RU2135492C1 |
| ИМИДАЗОПИРИДИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ПОЛУЧЕНИЯ ФАРМАЦЕВТИЧЕСКОЙ КОМПОЗИЦИИ | 1994 |
|
RU2136682C1 |
| ЗАМЕЩЕННЫЕ АРИЛАЛКИЛТИОАЛКИЛТИОПИРИДИНЫ ДЛЯ БОРЬБЫ С БАКТЕРИЯМИ HELICOBACTER | 1995 |
|
RU2139286C1 |
| ТИОПИРИДИНЫ ДЛЯ БОРЬБЫ С БАКТЕРИЯМИ HELICOBAKTER | 1995 |
|
RU2142459C1 |
| ПИРИДИЛТИОСОЕДИНЕНИЯ ДЛЯ БОРЬБЫ С БАКТЕРИЯМИ HELICOBACTER | 1995 |
|
RU2149872C1 |
| ДИГИДРОБЕНЗОФУРАНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1995 |
|
RU2138498C1 |
| ЗАМЕЩЕННЫЕ АРИЛТИОАЛКИЛТИОПИРИДИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ И СПОСОБ ПОЛУЧЕНИЯ ЛЕКАРСТВЕННОГО СРЕДСТВА. | 1994 |
|
RU2154062C2 |
| ПОЛИПЕПТИДЫ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ ЛЕГОЧНО-ПОВЕРХНОСТНО АКТИВНОГО ВЕЩЕСТВА, И ФАРМКОМПОЗИЦИЯ | 1995 |
|
RU2145611C1 |
| КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ РЕСПИРАТОРНОГО ДИСТРЕСС-СИНДРОМА НОВОРОЖДЕННЫХ И ОСТРОГО РЕСПИРАТОРНОГО ДИСТРЕСС-СИНДРОМА, СОДЕРЖАЩИЕ ПО КРАЙНЕЙ МЕРЕ, ОДИН ГЛЮКОКОРТИКОСТЕРОИД В СОЧЕТАНИИ С ЛЕГОЧНЫМ ПОВЕРХНОСТНО-АКТИВНЫМ ВЕЩЕСТВОМ | 1995 |
|
RU2157222C2 |

Алкоксиалкилкарбаматы имидазо [1, 2-а]пиридинов формулы 1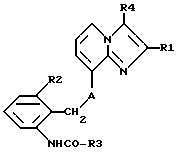
где R1, R2 - С1-4алкил; R3 - С1-4алкокси-С1-4алкокси; R4 -С1-4алкил или гидроксиметил и А - кислород или NH, или их соли, могут найти применение для лечения и/или профилактики заболеваний желудка и кишечника. 2 с. и 5 з. п. ф-лы, 1 табл.
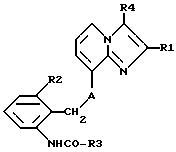
где R1 обозначает C1-4 алкил;
R2 обозначает C1-4 алкил;
R3 обозначает С1-4алкокси-С2-4алкокси;
R4 обозначает C1-4 алкил или гидроксиметил;
А обозначает О (кислород) или NН,
и их соли.
| ПОПЕРЕЧНЫЙ ЭЛЕВАТОР К КОРНЕКЛУБНЕПЛОДОУБОРОЧНЫМ МАШИНАМ | 0 |
|
SU261004A1 |
| Устройство для отсчета и записи на расстоянии показаний стрелочных приборов | 1958 |
|
SU117992A1 |

