Изобретение относится к новому ряду 4-фенилпиперазинов, 4-фенилпиперидинов и 4-фенил-1,2,3,6-тетрагидропиридинов. Имеющие действия как на центральный серотониновый 5-HT1A и допаминовый D2 рецепторы, новые соединения полезны при лечении некоторых психических и неврологических расстройств.
Предшествующий уровень техники
В публикации заявки на международный патент N WO 92/03426 раскрывается класс пиперазиновых производных, имеющих фенил, нафтил или хинолил в 4-положении и N-арил замещенную карбамоил алкильную группу или N-арил замещенную уреидо алкильную группу в 1-положении. Как заявлено, указанные соединения демонстрируют сродство к различным рецепторам, включая 5-HT2, 5-HT1A, альфа и допаминовый рецепторы.
EP A1 0376607 относится к некоторым 1-[4-(3-индолил)бутил]-4- (2-оксифенил)пиперазиновым соединениям, которые являются частичными 5-HT1A агонистами.
EP A1 0526434, среди ряда других соединений, раскрывает 1-[(4-фенилпиперазин-1-ил)-C2-6 алкил]-бензимидазол-2-оновые соединения, причем указанные соединения демонстрируют 5-HT1A агонистическую активность и 5-HT2A антагонистическую активность.
Пат. США N 3374237 раскрывает класс заявляемых 1-фенил-3-(4-фенил-1-пиперазинил-C2-4 алкил)-2-имидазолидинонов, которые могут быть полезными в качестве транквилизаторов. Никаких данных по испытанию не представлено. FR Патенты NN 1394708 и 1513604, соответственно, раскрывают сходные соединения без фенильного заместителя в 4-положениии пиперазинильной группы и указанные соединения обладают транквилизирующими и психофармакодинамическими свойствами, соответственно.
AU Патент N 15658/83 раскрывает соединения 3-(4-фенил-1- пиперазинил-C2-4алкил)гидантоина, имеющие антигипертензивные действия
WO 92/00282 относится к субгруппе соединений Пат. США N 3374237, которые представляют 1-фенил-3-[4-(4-фенил-1-пиперазинил)-1-бутил] - 2-имидазолидиноны, имеющие необязательный атом хлора в 2-положении фенильного заместителя в 1-положении кольца имидазолидинона и иметокси или этокси заместитель в 2- и/или 3-положении другого фенильного заместителя. Эти соединения показывают допаминергические действия.
Клинические исследования известных 5-HT1A частичных агонистов, таких как например, буспирон, 8-[4-[4-(2-пиримидил)-1-пиперазинил]-бутил]-8-азаспиро 4,5-декан- 7,9-дион, гепирон, 4,4-диметил-1-[4-[4-(2-пиримидил)-1-пиперазинил] бутил-2,6] -пиперидиндион, и ипсапирон, 2-[4-[4-(2-пиримидил)-1-пиперазинил] бутил] -1,2-бензотиазол-3(2H)-он- 1,1-диоксид, показали, что 5-HT1A частичные агонисты полезны при лечении расстройств, связанных со страхом, таких как генерализированное расстройство, связанное с тревогой, расстройство, вызванное паникой, и расстройство, вызванное компульсивной обсессией (Glitz, D.A., Pohl, R., Drugs 1991, 41, 11). Предклинические исследования показали, что, кроме того, полные агонисты используются при лечении вышеупомянутых родственных страху расстройств (Schihher, Human Psychopharmacol., 1991, 6, S 53).
Кроме того, существует доказательство, как клиническое, так и преклиническое, в поддержку положительного воздействия 5-HT1A агонистов при лечении депрессии, нарушений мотивационного контроля и злоупотребления алкоголем (van Hest, Psychopharmacol., 1991, 107, 474; Schipper et al., Human Psychopharmacol. , 1991, 6, S53; Cervo et al., Eur. J. Pharmacol., 1988, 158, 53; Glitz, D. A., Pohl, R., Drugs 1991, 41, 11; Grof et al., Int. Clin Psychopharmacol., 1993, 8, 167 - 172; Ansseau et al., Human Psychopharmacol., 1993, 8, 279 - 283).
5-HT1A агонисты и частичные агонисты ингибируют вызванную изоляцией агрессию у мышей-самцов, указывая на то, что эти соединения полезны при лечении агрессии (Sanchez et al., Psychopharmacology, 1993, 110, 53 - 59).
Кроме того, 5-HT1A агонисты, как сообщается, проявляют антипсихотическое действие на животных моделях (Wadenberg and Ahlenius, J. Neural. Transm., 1991, 83, 43; Ahlenius, Pharmacol. & Toxicol., 1989, 64, 3; Lowe et al., J. Med. Chem. , 1991, 34, 1860; New et al., J. Med, Chem., 1989, 32, 1147; и Martin et al. , J. Med Chem., 1989, 32, 1052) и, кроме того, недавние исследования показали, что 5-HT1A рецепторы важны в серотонинергической модуляции вызванной галоперидолом каталепсии (Hicks, Life Science 1990, 47, 1609, Wadenberg et al., Pharmacol, Biochem. & Behav. 1994, 47, 509 - 513), что дает возможность предположить, что 5-HT1A агонисты полезны при лечении экстрапирамидальных побочных эффектов (ЭПП, EPS), вызванных обычными антипсихотическими средствами, такими как галоперидол.
Подавление допаминовой (DA) сверхактивности с помощью использования лекарственных средств, блокирующих DA рецептор, является сегодня наиболее важным принципом при лечении шизофрении, в частности, позитивных симптомов ее, и других психозов. "Классические нейролептики", такие как галоперидол, цис(Z)-флупентиксол и хлорпромазин, как полагают, вызывают антипсихотическое действие посредством блокады DA рецептора. К сожалению, эти классические нейтролептики также вызывают ЭПП (EPS), что, по-видимому, коррелируется со склонностью этих соединений вызывать каталепсию у грызунов (Arnt et al., Neuropharmacology, 1981, 20, 1331 - 1334). Комбинация агонизма 5-HT1A рецептора, который может предотвращать ЭПП (EPS) у человека (ссылка выше), и блокады допаминового рецептора, которая лечит позитивные симптомы шизофрении, была бы очень целесообразна.
Кроме того, 5-HT1A агонисты проявляют нейтрозащитные свойства на модели грызунов с локальной и глобальной церебральной ишемией и, поэтому, они могут быть полезны при лечении состоящий ишемической болезни (Prehn, Eur. J. Pharm., 1991, 203, 213).
Представлены фармакологические исследования, которые показали, что 5-HT1A антагонисты полезны при лечении старческого слабоумия (Bowen et al., Trends Neur. Sci. 1992, 15, 84).
Как на животных моделях, так и при клинических испытаниях, было показано, что 5-HT1A агонисты оказывают антигипертензивные действия через центральный механизм (Saxena and Villalon, Treds Pharm. Sci. 1990, 11, 95; Gillis et al., J.Pharm. Exp. Ther. 1989, 248, 851).
Поэтому, 5-HT1A лиганды могут быть полезны при лечении сердечно-сосудистых нарушений.
Таким образом, агенты, действующие как на 5-HT1A рецептор, включая агонисты, частичные агонисты и антогонисты, так и в то же самое время блокирующие допаминовый D2 рецептор, как полагают, потенциально могут быть использованы при лечении таких состояний, в частности, при лечении психозов, и, поэтому, в них существует большая потребность.
Сущность изобретения
Было установлено, что новый ряд фенилпиперазинов, 4-фенилпиперидинов и 4-фенил-1,2,3,6-тетрагидропиридинов обладает как центральной серотонинергической 5-HT1A, так и антидопаминергической D2 активностью.
В соответствии с этим, данное изобретение относится к новым соединениям 4-фенилпиперазина, 4-фенилпиперидина и 4-фенил-1,2,3,6-тетрагидропиридина общей формулы I: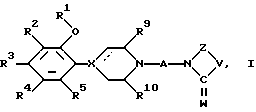
в которой A представляет спэйсерную (пространственную) группу, выбранную из разветвленного или неразветвленного C3-8 алкилена, C3-8 алкенилена и C3-8 алкинилена, и C3-7 циклоалкилена, причем указанная спэйсерная группа произвольно замещена низшим алкилом, арилом или гидрокси;
R1 представляет разветвленную C3-10 алкил, C3-10 алкенил или C3-10 алкинильную группу, циклоалк(ен)ил, циклоалк(ен)ил-низший алк(ен/ин)ил, трифторметилсульфонил, или низший алкилсульфонил, R2-R5 независимо выбирают из группы, состоящей из водорода, галоида, низшего алкила, низшей алкокси, низшей алкилтио, гидрокси, низшего алкилсульфонила, циано, низшего алкилкарбонила, фенилкарбонила, галоид замещенного фенилкарбонила, трифторметила, трифторметилсульфонилокси, циклоалкила, циклоалкил-низшего алкила, нитро, низший алкиламино, ди-низшей алкиламино и трифторметилтио;
R9 и R10 независимо являются водородом, низшим алкилом или они могут соединяться вместе, образуя при этом этиленовый или пропиленовый мостик;
W означает O или S;
V представляет O, S, CR6R7, или NR8, где R6, R7 и R8 независимо выбирают из водорода или низшего алкила или низшего алкенила, циклоалкила, циклоалкил-низшего-алкила, арил-низшего алкила или арила, или R6 и R7 связаны вместе, образуя 3-7 членное спиросоединенное кольцо;
Z представляет - (CH2)m-, причем m равно 2 или 3 или Z представляет -CH= CH-;
пунктирная линия, исходящая от X, указывает на необязательную связь и когда этой связи нет, X означает N или CH, а когда она есть X означает C; причем любая присутствующая алкил, циклоалкил или циклоалкилалкильная группа произвольно замещена одной или двумя гидрокси группами, которые, кроме того, произвольно этерифицированы алифатической или ароматической карбоновой кислотой; и любой присутствующий арильный заместитель произвольно замещен галоидом, низшим алкилом, низшей алкокси, низшей алкилтио, гидрокси, низшим алкилсульфонилом, циано, ацилом, трифторметилом, трифторметилсульфонилокси, циклоалкилом, циклоалкилалкилом или нитро; и их фармацевтически приемлемым солям кислого присоединения.
Соединения данного изобретения имеет средство к 5-HT1A рецептору и допаминовому D2 рецептору in vitro и они также проявляют 5-HT1A агонистическую или антагонистическую, а также допаминергическую активность in vivo. Кроме того, у соединений отсутствует каталептическое действие или они только слабо каталептогенны у крыс, что указывает на очень низкую возможность вызвать ЭПП (EPS) у человека. Соответственно этому, соединения данного изобретения, как полагают, являются полезными в качестве лекарственных средств при лечении психоза, позитивных симптомов шизофрении, расстройств, вызванных страхом, таких как генерализированное расстройство, вызванное тревогой, расстройство, вызванное паникой, и расстройство, вызванное компульсивной обсессией, депрессии, расстройств, связанных с мотивационным контролем, злоупотребления алкоголем, агрессии, ЭПП (EPS), вызванных обычными антипсихотическими средствами, состояний ишемической болезни, старческого слабоумия и сердечно-сосудистых нарушений.
В другом аспекте, изобретение обеспечивает фармацевтическую композицию, содержащую по крайней мере один новый заявляемый, определенный выше, 4-фенилпиперазин, 4-фенилпиперидин или 4-фенил-1,2,3,6-тетрагидропиридин или их фармацевтически приемлемую соль кислого присоединения или пролекарство в терапевтически эффективном количестве или в комбинации с одним или более фармацевтически приемлемых носителей или разбавителей.
В следующем аспекте, данное изобретение обеспечивает использование предлагаемого соединения 4-фенилпиперазина, 4-фенилпиперидина или 4-фенил-1,2,3,6-тетрагидропиридина или его соли кислого присоединения или пролекарства для производства фармацевтического препарата для лечения вышеупомянутых нарушений и заболеваний.
Детальное описание изобретения
Некоторые из соединений общей формулы (I) могут существовать в виде их оптических изомеров, и такие оптические изомеры входят в объем изобретения.
Пролекарства соединений общей формулы (I) также входят в объем изобретения.
Термин циклоалкил обозначает карбоциклическое кольцо, имеющее 3-8 углеродных атомов, включительно, или бициклический или трициклический карбоцикл, такой как адамантил, и циклоалкенил обозначает соответствующие группы, содержание ненасыщенную связь.
Термин низший алкил относится к неразветвленной и разветвленной алкильным группам, имеющим от одного до шести углеродных атомов, включительно. Примерами таких групп являются метильная, этильная, 1-пропильная, 2-пропильная, 1-бутильная, 2-бутильная, 2-метил-2-пропильная и 2-метил-1-пропильная. Соответственно, термины низшая алкокси, низшая алкилтио, низший алкилсульфонил, низший алкилкарбонил и низшая алкиламино относятся к таким группам, в которых алкильная часть представляет собой низшую алкильную группу, как определено выше, такую как метокси, этокси, 1-пропокси, метилтио, этилтио, 1-пропилтио, 2-пропилтио, метилсульфонил, этилсульфонил и т.д. Аналогично, низший алкенил и алкинил, соответственно, обозначают такие группы, имеющие от двух до шести углеродных атомов, включительно. Предпочтительными низшими алкильными алкенильными и алкинильными группами являются группы, имеющие вплоть до четырех углеродных атомов.
Термин арил относится к моно- или бициклической карбоциклической или гетероциклической ароматической группе, такой как фенил, индолил, тиенил, пиримидил, оксазолил, изоксазолил, тиазолил, изотиазолил, имидазолил, бензофуранил, бензотиенил, пиридил, нафтил, и фуранил, в частности, фенил, пиримидил, индолил и тиенил.
Галоид означает фтор, хлор, бром или иод.
Ацил относится к арилкарбонильной, C1-18 алкилкарбонильной, N'-(C1-18) алкил- или N',N'-ди(C1-18) алкилкарбонильной группе.
Выражение алк(ен/ин)ил означает, что группа может быть алкильной, алкенильной или алкинильной группой.
В формуле I, A предпочтительно представляет собой -(CH2)n-группу, в которой n равно целому числу 3-8, включительно, более предпочтительно 4-6, включительно. Наиболее предпочтительно равно 4.
R1 представляет собой предпочтительно разветвленный C3-6 алкил, циклоалкил-низший алкил или трифторметилсульфонил, более предпочтительно 2-пропил, 2-метил-1-пропил, 2-бутил, 3-пентил, 2,2-диметил-1-пропил, трет-бутил, циклопропилметил, циклопентил, 2,4-диметил-3-пентил, или трифторметилсульфонил, в частности, 2-пропил, циклопентил, циклопропилметил или трифторметилсульфонил.
R2-R5 представляют предпочтительно водород, галоид или циано и более предпочтительно все они являются водородом или один из заместителей является галоидом, а другие - водородами. R9 и R10 оба представляют собой водород.
Z представляет предпочтительно -CH2CH2- или -CH=CH- и V предпочтительно обозначает N-R8, где R8 представляет низший алкил, циклоалкил, фенил, или фенил, замещенный галогеном, наиболее предпочтительно циклогексил, адамантил, изопропил или 4-фторфенил.
W предпочтительно является кислородом.
К предпочтительным соединениям относятся:
3-Циклогексил-1-[4-[4-[2-(2-пропилокси)фенил] -1-пиперазинил]- бутан-1-ил]-2-имидазолидинон.
3-(4-фторфенил)-1-[4-[4-[2-(2-пропилокси)фенил] -1-пиперазинил]- бутан-1-ил]-2-имидазолидинон.
3-Циклогексил-1-[4-[4-(2-циклопентилоксифенил)-1-пиперазинил] - бутан-1-ил]-2-имидазолидинон.
1-[4-[4-(2-Циклопентилоксифенил)-1-пиперазинил] -бутан-1-ил] - 3-(4-фторфенил)-2-имидазолидинон.
1-[3-[4-[2-(2-Пропокси)фенил] -1-пиперазинил] -1-пропил]- 3-фенил-2-имидазолидинон.
3-(4-фторофенил)-1-[4-[4-[2-(2-пропилокси)фенил] -1-пиперидинил] - бутан-1-ил]-2-имидазолидинон.
3-Циклогексил-1-[4-[4-[2-(2-пропилокси)фенил] -1-пиперидинил]- бутан-1-ил]-2-имидазолидинон.
3-Циклогексил-1-[3-[4-[2-(2-пропилокси)фенил] -1-пиперидинил]- 1-пропил] -2-имидазолидинон.
3-(2-Пропил)-1-[4-[4-[2-(2-пропилокси)фенил] -1-пиперидинил]- 1-бутил]-2-имидазолидинон.
3-Циклогексил-1-[6-[4-[2-(2-пропилокси)фенил] -1-пиперидинил] - гексан-1-ил]-2-имидазолидинон.
3-Циклогексил-1-[4-[4-(2-циклопропилметилоксифенил)-1- пиперидинил]-бутан-1-ил]-2-имидазолидинон.
3-Циклогексил-1-[4-[4-[2-(2,2-диметилпропилокси)фенил] -1- пиперидинил] -бутан-1-ил]-2-имидазолидинон.
3-Циклогексил-1-[4-[4-[2-(2-пропилокси)фенил] -1- пиперидинил]-бутан-1-ил]-1,3-дигидроимидазол-2-он.
3-(1-Адамантил)-1-[4-[4-[2-(2-пропилокси)фенил] -1- пиперидинил]-бутан-1-ил]-1,3-дигидроимидазол-2-он.
3-(4-Фторфенил)-1-[4-[4-[2-(2-трифторметилсульфонилокси)фенил] -1- пиперазинил]-бутан-1-ил]-2-имидазолидинон.
3-Циклогексил-1-[4-[4-[2-(2-трифторметилсульфонилокси)фенил] -1- пиперазинил]-бутан-1-ил]-2-имидазолидинон.
3-(4-Фторфенил)-1-[4-[4-[2-(2-трифторметилсульфонилокси)фенил] -1- пиперидинил]-бутан-1-ил]-2-имидазолидинон.
3-Циклогексил-1-[4-[4-[2-(2-пропилокси)фенил] -1,2,3,6- тетрагидропиридин-1-ил]бутан-1-ил]-2-имидазолидинон.
3-(4-Фторфенил)-1-[4-[4-[2-(2-пропилокси)фенил]-1,2,3,6- тетрагидропиридин-1-ил]бутан-1-ил]-2-имидазолидинон.
3-Циклогексил-1-[4-[4-[2-(циклопропилметокси)фенил]-1,2,3,6- тетрагидропиридин-1-ил]бутан-1-ил]-2-имидазолидинон.
3-Циклогексил-1-[4-[4-[2-(2-пропилокси)фенил] -1,2,3,6- тетрагидропиридин-1-ил]бутан-1-ил]-2,3-дигидроимидазол-2-он.
3-(1-Адамантил)-1-[4-[4-[2-(пропилокси)фенил] -1,2,3,6- тетрагидропиридин-1-ил]бутан-1-ил]-2,3,-дигидроимидазол-2-он.
3-Циклогексил-1-[4-[4-[2-(1,1-диметилэтилокси)фенил]-1- пиперидинил]-бутан-1-ил]-2-имидазолидинон.
3-Циклогексил-1-[4-[4-[2-циклопропилоксифенил] -1- пиперидинил]-бутан-1-ил]-2-имидазолидинон.
3-Циклопентил-1-[3-[4-[2-(2-пропилокси)фенил] -1- пиперидинил]-бутан-1-ил]-2-имидазолидинон.
3-Циклогексил-1-[4-[4-[5-фтор-2-(2-пропилокси) фенил]-1-пиперидинил]бутан-1-ил]-2-имидазолидинон.
3-Циклогексил-1-[4-[4-[4-хлор-2-(2-пропилокси) фенил]-1-пиперидинил]бутан-1-ил]-2-имидазолидинон.
3-Циклогексил-1-[4-[4-[5-бром-2-(2-пропилокси) фенил]-1-пиперидинил]бутан-1-ил]-2-имидазолидинон.
3-Циклогексил-1-[4-[4-[5-циано-2-(2-пропилокси) фенил] -1-пиперидинил] бутан-1-ил]-2-имидазолидинон.
3-Адамантил-1-[4-[4-[2-(2-пропилокси) фенил] -1-пиперидинил]бутан-1-ил] -2-имидазолидинон.
3-Циклогексил-1-[4-[4-[2-(2,4-диметил-3-пентилокси) фенил] -1-пиперидинил]бутан-1-ил]-2-имидазолидинон.
Кислые соли присоединения изобретения представляют собой фармацевтически приемлемые соли соединений формулы 1, образованные с нетоксичными кислотами. Примерами таких органических солей являются соли с малеиновой, фумаровой, бензойной, аскорбиновой, эмбоновой (embonic), янтарной, щавелевой, бисметиленсалициловой, метансульфоновой, этандисульфоновой, уксусной, пропионовой, винной, салициловой, лимонной, глюконовой, молочной, яблочной, миндальной, коричной, цитраконовой, аспарагиновой, стеариновой, пальмитиновой, итаконовой, гликолевой, п-аминобензойной, глутаминовой, бензолсульфоновой, и теофеллин уксусной кислотами, а также 8-галотеофиллинами, например, 8-бромотеофиллином. Примерами таких неорганических солей являются соли с хлористоводородной, бромистоводородной, серной, сульфаминовой (sulfamic), фосфорной, и азотной кислотами.
Фармацевтические композиции данного изобретения или фармацевтические композиции, получаемые в соответствии с этим изобретением, могут назначаться с помощью любого подходящего приема, например, перорально, в виде таблеток, капсул, порошков, сиропов и т.д., или парентерально в виде растворов для инъекции. Для получения таких композиций могут быть использованы способы, хорошо известные в данной области, и могут быть использованы любые фармацевтически приемлемые носители, разбавители, наполнители, или другие обычно используемые в данной области техники вспомогательные вещества.
Удобно, чтобы соединения данного изобретения вводились в единичной лекарственной форме, содержащей указанные соединения в количестве от около 0,01 до 100 мг.
Суммарная суточная доза обычно находится в пределах около 0,05 - 500 мг, наиболее предпочтительно от около 0,1 до 50 мг, активного соединения изобретения.
Кроме того, изобретение относится к способу получения новых 4-фенилпиперазинов, 4-фенилпиперидинов и 4-фенил-1,2,3,6-тетрагидропиридинов формулы I, включающему:
а) взаимодействие соединения формулы II с соединением формулы III:

где R1-R5, R9, R10, X, V, W, Z, n, и пунктирная линия такие, как определено ранее, и Y - подходящая отщепляемая группа, такая как галоид, мезилат, или тозилат; или
b) восстановление амидкарбонила соединения формулы IV: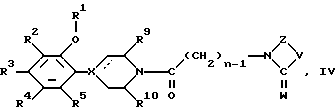
где R1-R5, R9, R10, X, V, W, Z, n, и пунктирная линия такие, как определено ранее;
или
c) взаимодействие соединения формулы V:
где R1-R5, R9, R10, X, V, W, Z, A, и пунктирная линия такие, как определено ранее, с соединением R1Y, где R1 такой, как определено ранее, и Y - подходящая отщепляемая группа, такая как галоид, мезилат или тозилат; или
d) гидроалкилирование NH группы соединения формулы VI:
где R1-R5, R9, R10, X, V, W, Z, A, и пунктирная линия такие, как определено ранее, с альдегидом R'CHO, кетоном R'' R'''CO или карбоновой кислотой R'COOH, и в этих формулах R', R'' и R''' являются группами, которые вместе с атомом азота образуют N-CH2R' и N-CHR''R''' группы, соответственно, которые входят в ранее данное определение V; или
e) восстановление двойной связи производного 1,2,3,6-тетрагидропиридина формулы VII:
где R1-R5, R9, R10, X, V, W, Z, A, и такие, как определено выше; после чего соединение формулы I выделяют в виде свободного основания или его фармацевтически приемлемой аддитивной соли кислоты.
Взаимодействие соединения формулы II с соединением формулы III согласно способу a) осуществляют в подходящем органическом растворителе, таком как ацетон, метилизобутилкетон, этанол, 2-пропанол, N-метил-2-пирролидинон, предпочтительно при повышенной температуре, например при температуре кипения растворителя, и обычно в присутствии основания (такого как карбонат калия или триэтиламин).
Восстановление согласно способу b) обычно проводят с использованием LiAlH4, AlH3, или диборана, в инертном растворителе, таком как тетрагидрофуран, диоксан, или диэтиловый эфир, при комнатной температуре или при слегка повышенной температуре.
Взаимодействие фенольного соединения формулы V согласно способу c) обычно проводят сначала генерируя фенолятный ион путем добавления сильного основания (например, трет-бутоксид калия) в инертном растворителе, таком как диэтиловый эфир, тетрагидрофуран, толуол или диметоксиэтан, предпочтительно при комнатной температуре или ниже. После чего фенолятный ион подвергают взаимодействию с соединением формулы: R1Y, при повышенной температуре, например, при температуре кипения растворителя. Трифторметилсульфонилокси производные обычно получают путем трифторирования (triflation) (смотри способы в публикации заявки на патент WO 93/11761) соответствующим образом замещенных фенолов формулы V. Ангидрид трифторсодержащей (triflic) кислоты, N-фенилтрифторметансульфонимид, и хлорангидрид трифторсодержащей (triflic) кислоты являются предпочтительными в качестве агентов трифторирования.
Гидроалкилирование соединения формулы VI согласно способу d) обычно проводят в кислых условиях, например, в уксусной кислоте, используя NaBH4, NaCNBH3 или каталитическое гидрирование (Pt или Pd в качестве катализаторов). Обычно температура комнатная или ниже.
Восстановление тетрагидропиридильной двойной связи соединения формулы VII согласно способу e) обычно проводят путем каталитического гидрирования при низком давлении (< 3 атм.) в аппарате Парра (Parr), или путем использования восстанавливающего агента, такого как диборан, в инертном растворителе, таком как тетрагидрофуран, диоксан или диэтиловый эфир.
1-Незамещенные 4-арилпиперазины формулы II (X=N) либо коммерчески доступны, либо они могут быть синтезированы из соответствующих анилинов и N', N',-бис-(2-хлорэтил)амина при кипячении с обратным холодильником в высококипящих растворителях, как например, хлорбензол, обычно в течение нескольких дней (2-3) согласно способам, описанным Martin et al., J. Med. Chem. 1989, 32 1052 - 1056.
4-Фенилпиперидины формулы II (X=CH) либо коммерчески доступны, либо их получают как описано, например, в пат. США N 2 891 066; McElvain et al., J. Amer. Chem. Soc. 1950, 72, 3134; Bally et al., Chem. Ber. 1987, 20, 2590.
Соответствующие 4-фенил-1,2,3,6-тетрагидропиридины формулы II (X=C) получают из N-защищенных 4-пиперидинов путем добавления соответствующим образом замещенных фениллитий галогенидов или фенилмагний галогенидов с последующим катализируемым кислотой водным элиминированием. Наконец, N-защищающую группу (карбамат, бензил, сульфонил, ацетил) удаляют обычным способом.
Синтезы конкретных соединений формулы II даны детально в Экспериментальном Разделе.
1-(3-Хлорпропил)-, 1-(4-хлорбутил)-, 1-(5-хлорпентил)- и 1-(6-хлоргексил)-2-имидазолидиноны или соответствующие 3-замещенные 2-имидазолидиноны получают согласно способам Perregaard et al., J. Amer. Chem. 1992, 35, 1092 - 1101 или ссылкам, цитированным там, или способам, детально описанным ниже.
Экспериментальный раздел
Ниже данное изобретение дополнительно иллюстрируется примерами, которые, однако, не следует рассматривать как ограничивающие изобретение.
Во всех примерах, температуры плавления определяют на Biichi MP-20 приборе и они некорретированы. 1H ЯМР спектры записаны на 250 Мгц Brucker AC 250 спектрометре. В качестве растворителей использовали дейтерированный хлороформ (99,8% D) и диметилсульфоксид (99,9% D). ТМС (TMS) использовали в качестве внутреннего стандарта. Значения химических сдвигов выражены в ппм-значениях. Использовали следующие аббревиатуры для множества ЯМР-сигналов: с=синглет, д=дублет, т=триплет, к=квартет, п=пентет, г=гептет, дд=двойной дублет, дт=двойной триплет, дк=двойной квартет, кв(qui)=квинтет триплетов, м=мультиплет.
Пример 1 (аналогичный способу d)
1-(4-Хлор-1-бутил)-3-циклогексил-2-имидазолидинон (1a).
Смесь 1-(4-хлор-1-бутил)-2-имидазолидинона (50 г) и циклогексанона (83,3 г) в ледяной уксусной кислоте (1000 мл) перемешивают в течение одного часа при комнатной температуре. Смесь охлаждают до 10 - 15oC и добавляют малыми порциями NaBH4 (42,4 г) в течение 5 часов. После перемешивания в течение ночи при комнатной температуре уксусную кислоту выпаривают в вакууме. Добавляют воду (500 мл) и дихлорметан (300 мл) и pH доводят до > 9 путем добавления водного разбавленного NH4OH. Органическую фазу отделяют, сушат безводным MgSO4, фильтруют и растворитель испаряют в вакууме. Остающийся сырой продукт очищают колоночной хроматографией на силикагеле (элюируют этилацетатом). Очищенное названное соединение 1a кристаллизуется при стоянии. Выход: 33 г, т. пл.: 30 - 35oC.
Соответствующим способом получают следующие имидазолидиноны:
1-(3-Хлор-1-пропил)-3-циклогексил-2-имидазолидинон (1b),
1-(4-Хлор-1-бутил)-3-изопропил-2-имидазолидинон (1c),
1-(6-Хлор-1-гексил)-3-циклогексил-2-имидазолидинон (1d).
Пример 2
1-(4-Хлор-1-бутил)-3-(4-фторфенил)-2-имидазолидинон (2a).
К аминоэтанолу (1100 г) в этаноле (1000 мл) добавляют 4-хлорбутанол (220 г). Смесь кипятят с обратным холодильником в течение 4 часов. После охлаждения до 10oC, добавляют метоксид натрия в метанол (380 мл). Осадок отфильтровывают и летучее вещество испаряют в вакууме. Оставшееся масло дистиллируют при пониженном давлении. 4-[N-(2-гидроксиэтил)амино]бутанол собирают при 135 - 140oC при 0,2 - 0,4 мм Hg. Выход: 106 г аминоспирта (25 г) растворяют в дихлорметане (150 мл) и добавляют по каплям при 0 - 8oC раствор 4-фторфенилизоцианата (26 г) в дихлорметане (25 мл). После кипячения в течение 2 часов растворитель выпаривают в вакууме, получая неочищенную 1-(2-гидроксиэтил)-1-(4-гидроксибутил)-3-(4- фторфенил)мочевину в виде масла. Выход 56 г. К раствору неочищенного производного мочевины и N,N-диметилформамида (ДМФ, 1 мл) в дихлорметане (250 мл) добавляют по каплям раствор тионилхлорида (40 мл) в дихлорметане (60 мл) при 10 - 20oC. Смесь кипятят с обратным холодильником в течение 3 часов. Летучее вещество испаряют в вакууме. Оставшееся вещество нагревают при 130 - 150oC в течение 2 часов. Добавляют дихлорметан (50 мл) и смесь прямо фильтруют через силикагель (элюируют смесью 1:1 этилацетата и гептана). Названное соединение 2a кристаллизуется после выпаривания растворителей. Выход: 33 г, т. пл.: 62 - 64oC.
Соответствующим способом получают следующий имидазолидинон:
1-(3-Хлор-1-пропил)-3-фенил-2-имидазолидинон (2b).
Пример 3
4-(3-Циклогексил-1,3-дигидроимидазол-2-он-1-ил)-1-бутил метансульфонат, (3a).
Смесь 4-бутиролактона (54 г) и аминоацетальдегид диметилацеталя (55 г) в тетрагидрофуране (ТГФ) (500 мл) кипятят с обратным холодильником в течение 4 ч. Добавляют дополнительную порцию аминоацетальдегид диметилацеталя (27 г) и затем кипятят с обратным холодильником в течение 2 ч. Продолжают кипячение с обратным холодильником и дополнительно добавляют две порции ацеталя с 2 ч интервалами. Концентрирование реакционной смеси в вакууме с последующей дистилляцией оставшегося масла дает N-(2,2-диметоксиэтил)-4-гидроксибутирамид в виде масла, т.к. 230oC/10 торр, выход 72 г. Масло растворяют в сухом ТГФ (800 мл) и медленно добавляют к суспензии литийалюминийгидрида в ТГФ (200 мл). После кипячения с обратным холодильником в течение 16 ч, реакционную смесь охлаждают и затем гасят водой (88 мл), 15% раствором NaOH (44 мл) и водой (220 мл). Фильтрация и удаление растворителя в вакууме дают масло, которое растворяют в метиленхлориде (500 мл) и сушат над сульфатом магния. Фильтрация и удаление растворителя в вакууме дают 4-(2,2-диметоксиэтиламино)-1-бутанол в виде масла, выход 62 г. Часть масла (20 г) растворяют в метиленхлориде (100 мл) и добавляют по каплям к раствору циклогексилизоцианата в метиленхлориде (50 мл) при 5oC. Кипячение с обратным холодильником в течение 3 ч и концентрирование в вакууме дает N-циклогексил-N'-(2,2-диметоксиэтил)-N'-(4- гидрокси-1-бутил)-мочевину в виде масла, выход 35 г. Масло растворяют в смеси ТГФ (200 мл) и 2 М хлористоводородной кислоты (200 мл) и затем кипятят с обратным холодильником в течение 24 ч. Реакционную смесь концентрируют в вакууме и затем добавляют этилацетат (300 мл) и 4 М NaOH (300 мл). Органическую фазу сушат над сульфатом магния. Удаление растворителя дает 1-циклогексил-3-(4-гидрокси-1-бутил)- 1,3-дигидроимидазол-2-он в виде масла, выход 29 г. Масло растворяют в метиленхлориде (250 мл) и добавляют триэтиламин (13 г) с последующим охлаждением до 5oC. Раствор метансульфонил хлорида (14 г) в метиленхлориде (20 мл) добавляют по каплям, затем перемешивают в течение 1,5 ч. при комнатной температуре. Реакционную смесь промывают водой, сушат над сульфатом магния и концентрируют в вакууме, получая названное соединение в виде масла, выход 36 г.
1H ЯМР (CDCl3): δ 1,05 - 1,25 (м, 1H), 1,30 - 1,50 (м, 4H), 1,60 - 2,00 (м, 9H), 3,00 (с, 3H), 3,65 (т, 2H), 3,85 - 4,05 (м, 1H), 4,25 (т, 2H), 6,20 (д, 1H), 6,25 (д, 1H).
Соответствующим способом получают следующий 1,3-дигидроимидазол-2-он:
4-[3-(1-Адамантил)-1,3-дигидроимидазол-2-он-1-ил] -1-бутил метансульфонат, (3b).
Пример 4 (способ a)
3-Циклогексил-1-[4-[4-[2-(2-пропилокси)фенил] -1-пиперазинил]- бутан-1-ил]-2-имидазолидинон, оксалат (4a).
Смесь 1-[2-(2-пропилокси)фенил] -пиперазина (5,0 г), 1-(4-хлоро-1-бутил)-3-циклогексил-2-имидазолидинона 1a (3,0 г), кристалла иодида калия и карбоната калия (4,0 г) в метил изобутил кетоне (МИБК) (100 мл) кипятят с обратным холодильником в течение 16 часов. Неорганические соли отфильтровывают, пока смесь еще горячая. После охлаждения до комнатной температуры, сырую реакционную смесь фильтруют через силикагель (элюируют смесью 1:1 этилацетата и метанола). После выпаривания растворителей, остается 3,9 г сырого продукта в виде вязкого масла. Оксалатную соль кристаллизуют из ацетона. Выход: 2,4 г, т. пл.: 133 - 134oC.
1H ЯМР (ДМСО-d6): δ 1,0 - 1,80 (м, 14H), 1,25 (г, 6H), 2,85 - 3,30 (м, 16H), 3,35 - 3,55 (м, 1H), 4,60 (г, 1H), 6,80 - 7,00 (м, 4H).
Аналогичным способом получают следующие соединения:
3-(4-Фторфенил)-1-[4-[4-[2-(2-пропилокси)фенил] -1- пиперазинил]-бутан-1-ил]-2-имидазолидинон (4b), т. пл.: 135 - 137oC (этилацетат).
1H ЯМР (CDCl3): δ 1,45 (д, 6H); 1,60 (т, 4H), 2,45 (т, 2H); 2,65 (широкий с, 4H); 3,15 (широкий с, 4H); 3,35 (т, 2H); 3,50 (т, 2H); 3,80 (т, 2H), 4,60 (г, 1H); 6,85 - 7,00 (м, 4H); 7,05 (т, 2H); 7,50 (дд, 2H).
3-Циклогенксил-1-[4-[4-(2-Циклопентилоксифенил)-1- пиперазинил]-бутан-1-ил]-2-имидазолидинон, дигидрохлорид (4c), т.пл.: 182 - 193oC (ацетон).
1H ЯМР (ДМСО-d6): δ 1,05 - 2,00 (м, 22H); 3,05 - 3,20 (м, 8H); 3,40 - 3,55 (м, 5H); 4,85 (кв, 1H); 6,85 - 7,00 (м, 4H); 4,60 (шир.с, 1H); 11,05 (шир.с, 1H).
1-[4-[4-(2-Циклопентилоксифенил)-1-пиперазинил]бутан-1-ил]-3- (4-фторфенил)-2-имидазолидинон, (4), т.пл.: 111 - 116oC (ацетон)
1H ЯМР (CDCl3): δ 1,60 - 1,95 (м, 12H); 2,40 (широкий т, 2H); 2,65 (широкий с, 4H); 3,05 (широкий с, 4H); 3,25 (широкий т, 2H); 3,45 (т, 2H); 3,75 (т, 2H); 4,80 (кв, 1H); 6,80 - 7,05 (м, 6H); 7,45 (дд, 2H).
1-[3-[4-(2-(2-пропилокси)фенил)-1-пиперазинил] -1-пропил] -3- фенил-2-имидазолидинон, (4e) т.пл.: 167 - 68oC (этанол)
1H-ЯМР (CDCl3): δ 1,25 (д, 6H); 1,75 (кв, 2H); 2,60 (т, 2H); 2,75 (м, 4H); 3,05 (м, 4H); 3,25 (т, 2H); 3,45 (т, 2H); 3,80 (т, 2H); 4,60 (г, 1H); 6,60 (с, 3H); 6,80 - 6,95 (м, 4H); 7,00 (т, 1H); 7,30 (т, 2H); 7,55 (д, 2H).
Пример 5
1-(Трет-бутилоксикарбонил)-4-(2-гидроксифенил)пиперидин (5a).
2-Метоксибензальдегид (200 г) и этил ацетоацетат (400 г) смешивают и охлаждают до 5oC. Добавляют пиперидин (25 мл) и смесь перемешивают в течение ночи при комнатной температуре. На следующий день добавляют трет-бутоксид калия (25 г). Через 1,5 часа смесь целиком отверждается и ее выдерживают в течение 2 дней. Добавляют этанол (2000 мл) и осадок отфильтровывают, промывают этанолом и окончательно сушат в вакууме. Выход: 326 г. Весь полученный таким образом продукт (325 г) добавляют малыми порциями в течение 1 часа к раствору гидроксида калия (260 г) в воде (320 мл), поддерживая температуру 80 - 90oC. После перемешивания еще в течение двух часов при 80oC, добавляют воду (2000 мл) и диэтиловый эфир (1000 мл). После перемешивания органическую фазу отделяют и отбрасывают. К оставшемуся водному раствору добавляют лед и концентрированную хлористоводородную кислоту до pH<1. После перемешивания в течение 45 мин., высадившийся продукт отфильтровывают и сушат. Выход 3-(2-метоксифенил)-1,5-пентандикарбоновой кислоты: 137 г, т.пл.: 183 - 185oC. Смесь пентандикарбоновой кислоты (97 г) и мочевины (28 г) нагревают при 160 - 165oC в течение 2 часов. После охлаждения до 70oC добавляют этанол (150 мл). Высадившийся 4-(2-метоксифенил)-2,6-пиперидиндион отфильтровывают и затем сушат. Выход: 66 г. Т.пл.: 127 - 129oC. К суспензии LiAlH4 (30 г) в сухом ТГФ (1000 мл) малыми порциями (в сумме 65 г) добавляют пиперидиндион, при этом температура постепенно повышается до температуры кипения смеси. После кипячения с обратным холодильником в течение 2,5 часов, смесь охлаждают ниже 10oC и осторожно добавляют разбавленный водный (4М) раствор NaOH (60 мл). Осадившиеся неорганические соли отфильтровывают. Растворитель выпаривают и остающееся масло растворяют в дихлорметане, сушат (безв. Na2SO4), фильтруют и дихлорметан выпаривают, получая 56 г 4-(2-метокфенил) пиперидина в виде масла. Весь пиперидин растворяют в смеси 48% водной бромистоводородной кислоты (400 мл) и 33% раствора бромистого водорода в уксусной кислоте (400 мл). Раствор кипятят с обратным холодильником в течение 19 часов. Избыток бромистоводородной и уксусной кислот выпаривают в вакууме, получая гидробромид 4-(2-гидроксифенил)пиперидина в виде вязкого масла. Гидробромид (45 г) растворяют в смеси воды (300 мл) и ТГФ (150 мл). Добавляют малыми порциями карбонат калия (80 г). Добавляют по каплям раствор ди-трет-бутилдикарбоната (40 г) в ТГФ (150 мл). Смесь перемешивают на протяжении ночи. Водную фазу отделяют и промывают диэтиловым эфиром (2х100 мл). Объединенные органические фазы сушат (безв. MgSO4) и растворители выпаривают. Оставшийся продукт перемешивают с диизопропиловым эфиром и высадившийся продукт отфильтровывают. Выход названного фенольного производного 5a:24 г, т. пл.: 187 - 189oC.
Пример 6 (способ a)
3-(4-фторфенил)-1-[4-[4-[2-(2-пропилокси)фенил] -1-пиперидинил]- бутан-1-ил]-2-имидазолидинон, оксалат (6a).
К раствору 1-(трет-бутилоксикарбонил)-4-(2-гидроксифенил)- пиперидина (5a) (5 г) в сухом ТГФ (100 мл) добавляют трет-бутоксид калия (2,2 г). После перемешивания в течение 5 минут добавляют 2-иодопропан (9 г) и смесь кипятят с обратным холодильником в течение 3 часов. Дополнительно добавляют 1,1 г трет-бутоксида калия и кипячение с обратным холодильником продолжают в течение ночи. После охлаждения до комнатной температуры, неорганические соли отфильтровывают и растворители выпаривают в вакууме. Оставшееся масло очищают путем фильтрации через силикагель (элюируют смесью 7:3 гептана и этилацетата). Выход: 4,5 г 1-(трет-бутилоксикарбонил)-4-[2-(2- пропилокси)фенил] пиперидина, который весь растворяют в смеси дихлорметана (90 мл) и трифторуксусной кислоты (40 мл). После перемешивания в течение одного часа при комнатной температуре, все летучие вещества испаряют в вакууме. К оставшемуся маслу добавляют этилацетат (200 мл) и воду (200 мл). pH доводят до >10 путем добавления разбавленного водного NH4OH. Органическую фазу отделяют и промывают соляным раствором (2х50 мл). Обработка органической фазы, как указано выше, дает 2,6 г 4-[2-(2-пропилокси)фенил]пиперидина. Смесь выделенного таким образом 1-незамещенного пиперидина (2,5 г), 1-(4-хлор-1-бутил)-3-(4- фторфенил)-2-имидазолидинона 2a (3,0 г), карбоната калия (1,6 г) и иодида калия (0,5 г) в МИБК (M1BK) (50 мл) кипятят с обратным холодильником на протяжении ночи. Неорганические соли отфильтровывают и МИБК выпаривают в вакууме. Оставшийся сырой продукт очищают колоночной хроматографией на силикагеле (элюируют 4% триэтиламина в этилацетате). Очищенное названное соединение 6a кристаллизуют из этилацетата. Выход: 2,6 г, т.пл.: 130 - 131oC.
1H ЯМР (CDCl3): δ 1,45 (д, 6H); 1,60 (т, 4H); 1,70 - 1,90 (м, 4H); 2,00 - 2,15 (м, 2H); 2,40 (т, 2H); 2,90 - 3,05 (м, 3H); 3,35 (т, 2H); 3,45 (т, 2H); 3,75 (т, 2H); 4,55 (г, 1H); 6,80 - 6,90 (м, 2H); 7,0 (т, 2H); 7,10 (дт, 1H); 7,15 (дд, 1H); 7,50 (дд, 2H).
Аналогичным путем получают следующие соединения:
3-Циклогексил-1-[4-[4-[2-(2-пропилокси)фенил]-1-пиперидинил] бутан-1-ил] -2-имидазолидинон, оксалат (6), т.пл.: 138 - 141oC (ацетон).
1H ЯМР (ДМСО-d6): δ 0,95 - 1,20 (м, 1H); 1,20 - 1,40 (м, 4H); 1,30 (д, 6H); 1,40 - 1,80 (м, 9H); 1,80 - 2,00 (м, 4H); 2,90 -3,20 (м, 7H); 3,25 (с, 4H); 3,40 - 3,60 (м, 3H); 4,65 (г, 1H); 6,90 (т, 1H); 7,00 (д, 1H); 7,10 - 7,25 (м, 2H).
3-Циклогексил-1-[3-[4-[2-(2-пропилокси)фенил] -1-пиперидинил] -1-пропил] имидазолидинон, оксалат, (6с), т.пл.: 136 - 39oC (ацетон).
1H ЯМР (ДМСО-d6): δ 0,95 - 1,45 (м, 5H); 1,30 (д, 6H); 1,50 - 1,65 (м, 3H); 1,65 - 2,00 (м, 8H); 2,85 - 3,15 (м, 7H); 3,25 (с, 4H); 3,40 - 3,60 (м, 3H); 4,60 (г, 1H); 6,90 (т, 1H); 7,00 (д, 1H); 7,05 - 7,25 (м, 2H).
3-(2-Пропил)-1-[4-[4-[2-(2-пропилокси)фенил] -1-пиперидинил]-1-бутил]-2- имидазолидинон, оксалат, (6d), т.пл.: 82 - 84oC (ацетон).
1H ЯМР (ДМСО-d6): δ 1,05 (д, 6H); 1,25 (д, 6H); 1,40 - 1,55 (м, 2H); 1,55 - 1,75 (м, 2H); 1,80 - 2,00 (м, 4H); 2,90 - 3,15 (м, 7H); 3,15 - 3,30 (м, 4H); 3,40 - 3,60 (м, 2H); 3,90 (г, 1H); 4,60 (г, 1H); 6,90 (т, 1H); 7,00 (д, 1H); 7,05 - 7,25 (м, 2H).
3-Циклогексил-1-[6-[4-[2-(2-пропилокси)фенил] -1-пиперидинил] -1-гексил] -2-имидазолидинон, оксалат, (6e), т.пл.: 77 - 79oC (ацетон).
1H ЯМР (ДМСО-d6): δ 0,95 - 1,50 (м, 14H); 1,25 (д, 6H); 1,50 - 1,80 (м, 6H); 1,80 - 2,00 (м, 4H); 2,80 - 3,15 (м, 7H); 3,15 - 3,25 (м, 4H); 3,35 - 3,60 (м, 3H); 4,60 (г, 1H); 6,90 (т, 1H); 7,00 (д, 1H); 7,05 - 7,25 (м, 2H).
3-Циклогексил-1-[4-[4-[2-(циклопропилметокси)фенил] -1- пиперидинил] -бутан-1-ил]-2-имидазолидинон, оксалат, (6f), т.пл.: 186 - 193oC (этилацетат).
1H ЯМР (ДМСО-d6): δ 0,25 - 0,40 (м, 2H); 0,60 - 0,70 (м, 2H); 0,95 - 1,15 (м, 1H); 1,20 - 1,45 (м, 5H); 1,50 - 1,90 (м, 9H); 1,95 - 2,10 (м, 2H); 2,10 - 2,35 (м, 2H); 2,85 (т, 2H); 3,05 - 3,25 (м, 3H); 3,20 (т, 2H); 3,25 - 3,35 (м, 4H); 3,55 - 3,85 (м, 3H); 3,80 (д, 2H); 6,80 (д, 1H); 6,90 (т, 1H); 7,10 - 7,25 (м, 2H).
3-Циклогексил-1-[4-[4-[2-(2,2-диметил-1-пропилокси)фенил] - 1-пиперидинил] бутан-1-ил] -2-имидазолидинон, оксалат, (6g), т. пл. : 162 - 168oC (этилацетат/ацетон).
1H ЯМР (ДМСО-d6): δ 1,05 (с, 9H); 1,15 - 1,85 (м, 14H); 1,80 - 2,05 (м, 4H); 2,85 - 3,15 (м, 7H); 3,20 (с, 4H); 3,35 - 3,60 (м, 3H); 3,65 (с, 2H); 6,85 - 7,00 (м, 2H); 7,05 - 7,25 (м, 2H).
Аналогичным способом, но применяя мезилаты (3a) и (3b) вместо хлоридов в качестве алкилирующих агентов, получают следующие производные:
3-Циклогексил-1-[4-[4-[2-(2-пропилоксил)фенил] - 1-пиперидинил]бутан-1-ил]-1,3-дигидроимидазол-2-он, оксалат, (6h), т.пл.: 68 - 71oC (ацетон).
1H ЯМР (ДМСО-d6): δ 1,05 - 1,55 (м, 5H); 1,30 (д, 6H); 1,55 - 1,85 (м, 9H); 1,85 - 2,00 (м, 4H); 2,90 - 3,20 (м, 5H); 3,40 - 3,60 (м, 4H); 3,75 (дт, 1H); 4,65 (г, 1H); 6,50 (д, 1H); 6,55 (д, 1H); 6,90 (т, 1H); 7,00 (д, 1H); 7,10 - 7,25 (м, 2H).
3-(1-Адамантил)-1-[4-[4-[2-(2-пропилокси)фенил] -1-пиперидинил] бутан-1-ил]-1,3-дигидроимидазол-2-он, оксалат, (6i), т.пл.: 116 - 122oC (этилацетат/ацетон).
1H ЯМР (ДМСО-d6): δ 1,30 (д, 6H); 1,50 - 1,75 (м, 10H); 1,75 - 2,10 (м, 4H); 2,00 - 2,20 (м, 9H); 2,85 - 3,20 (м, 5H); 3,35 - 3,60 (м, 4H); 4,65 (г, 1H); 6,50 (с, 2H); 6,90 (т, 1H);
7,00 (д, 1H); 7,05 - 7,25 (м, 2H).
Пример 7 (способ а)
3-(4-фторфенил)-1-[4-[4-[2-(2-трифторметилсульфонилокси)фенил] -1- пиперидинил]бутан-1-ил]-2-имидазолидинон, (7а)
Раствор
1-(трет-бутилоксикарбонил)-4-(2-гидроксифенил)пиперидина (5а) (9 г) и триэтиламина (7 мл) в дихлорметане (90 мл) охлаждают до 5oC и добавляют по каплям раствор ангидрида трифторметансульфокислоты (10 мл) в дихлорметане (15 мл). После перемешивания в течение одного часа при комнатной температуре, добавляют воду (200 мл). Органическую фазу отделяют и производят обработку, как ранее описано. Выход неочищенного 1-(трет-бутилоксикарбонил)-4-(2-трифторметилсульфонилоксифенил) пиперидина: 14 г. 1-(трет-бутилоксикарбонил) N-защищающую группу отщепляют, как в Примере 5, получая 9 г неочищенного 4-(2-трифторметилсульфонилоксифенил)пиперидина. Смесь таким образом выделенного 1-незамещенного пиперидина (5,5 г), 1-(4-хлоро-1-бутил)-3-(4-фторфенил)-2-имидазолидинона (2а) (4,0 г), и иодида калия (0,5 г) в МИБК (М1ВК) (80 мл) кипятят с обратным холодильником на протяжении ночи. МИВК выпаривают в вакууме. Оставшееся масло растворяют в этилацетате (100 мл) и воде (100 мл) и pH доводят до > 9 путем добавления разбавленного водного NH4OH. Органическую фазу отделяют и производят обработку, как описано ранее. Оставшийся неочищенный продукт очищают колоночной хроматографией на силикагеле (элюируют 4% триэтиламином в смеси 9:1 этилацетата и этанола). Очищенное названное соединение 7а кристаллизуют из смеси 1:1 диэтилового и диизопропилового эфира.
Выход: 2,0 г, т.пл.: 77-79oC.
1H ЯМР (CDCL3): δ 1,55 - 1,70 (м, 4H); 1,70 - 1,85 (м, 4H); 2,05 (дт, 2H); 2,40 (т, 2H); 2,80 - 2,95 (м, 1H); 3,05 (д, 2H); 3,35 (т, 2H); 3,45 (т, 2H); 3,80 (т, 2H); 7,00 (т, 2H); 7,20 - 7,45 (м, 4H); 7,50 (дд, 2H).
Аналогичным способом получают трифторметилсульфонилокси производные:
3-Циклогексил-1-[4-[4-[2-(2-трифторметилсульфонилокси)фенил] -1- пиперазинил] бутан-1-ил] -2-имидазолидинон, гидрохлорид, (7b), т.пл.: 153 - 154oC (ацетон).
1H ЯМР (ДМСО-d6): δ 1,00 - 1,75 (м, 14H); 3,00 - 3,55 (м, 17H); 7,25 - 7,50 (м, 4H); 11,20 (с, 1H).
3-(4-Фторфенил)-1-[4-[4-[2-(2-трифторметилсульфонилокси)фенил] -1- пиперазинил]бутан-1-ил]-2-имидазолидинон, (7c), т.пл.: 68 - 70oC (диизопропиловый эфир).
1H ЯМР (ДМСО-d6): δ 1,55 - 1,65 (м, 4H); 2,45 - 2,55 (м, 2H); 2,65 (широкий с, 4H); 3,05 (т, 4H); 3,35 (т, 2H); 3,50 (т, 2H); 3,80 (т, 2H); 7,00 - 7,20 (м, 5H); 7,35 (дт, 1H); 7,50 (дд, 2H).
Пример 8
4-[2-(2-пропилокси)фенил]-1,2,3,6-тетрагидропиридин (8a).
2-Бромфенол (10 г), 2-бромпропан (7,1 г), карбонат калия (12 г) и кристалл иодида калия кипятят с обратным холодильником в МИВК (100 мл) в течение 6 часов. Неорганические соли отфильтровывают и МИВК испаряют в вакууме. Добавляют охлажденную льдом воду и диэтиловый эфир (200 мл) и доводят pH до > 10 путем добавления разбавленного водного NaOH. Органическую фазу отделяют и продолжают обработку как описано выше. Выход 2-бромфенил 2-пропилового эфира (11 г) в виде масла. К диэтиловому эфиру (38 мл), охлажденному ниже -10oC, добавляют 1,6 М раствор н-бутиллития в гексане (31 мл). Полученный раствор охлаждают до -50oC и добавляют по каплям весь вышеупомянутый 2-бромфенил эфир в диэтиловом эфире (20 мл). После перемешивания еще в течение 20 минут при -50oC, по каплям при -50oC добавляют раствор 1-бензил-4-пиперидона (9,7 г) в диэтиловом эфире (25 мл). Смеси дают возможность постепенно нагреться до -10oC и выливают в разбавленную водную хлористоводородую кислоту. Органическую фазу отбрасывают и добавляют по каплям разбавленный водный NH4OH до pH > 9. Экстракция диэтиловым эфиром (2 х 200 мл) и обработка органической фазы, как указано выше, дает 1-бензил-4-гидрокси-4-[2-(2-пропилокси)фенил] пиперидин (13,7 г) в виде масла. Весь гидроксипиперидин кипятят с обратным холодильником в трифторуксусной кислоте (100 мл) в течение 2,5 часов. Лед (1000 г) и диэтиловый эфир (300 мл) добавляют и доводят pH до > 9 путем добавления разбавленного водного NH4OH. После экстракции несколько раз (3 х 200 мл) диэтиловым эфиром, объединенные органические фазы подвергают обработке, как описано ранее. Неочищенный продукт очищают колоночной хроматографией на силикагеле (элюируют 4% триэтиламином в смеси 3:1 гептана и этилацетата). Выход: 4,2 г в виде масла. К полученному таким образом 1-бензил-4-[2-(2-пропилокси)фенил] -1,2,3,6-тетрагидропиридину в 1,1,1-трихлорэтане (40 мл) добавляют по каплям раствор 2,2,2-трихлорэтил хлороформиата (2,2 мл) в трихлорэтане (10 мл) при температуре кипения смеси. После кипячения в течение 1,5 часов, растворитель испаряют. Неочищенный продукт фильтруют через силикагель (элюируют смесью этил-ацетат/гептан 1:3), получая 4,5 г очищенного производного 2, 2,2-трихлорэтил карбамата в виде масла. Весь карбамат растворяют в уксусной кислоте (40 мл). Добавляют воду и при 40 - 50oC малыми порциями добавляют Zn порошок (8 г) в течение 10 минут. После перемешивания в течение 2 часов при 50oC, неорганические соли отфильтровывают и растворители испаряют в вакууме. Добавляют лед и этилацетат и доводят pH > 9 путем добавления разбавленного водного NH4OH. Органическую фазу отделяют и обрабатывают, как описано выше, получая 265 г названного соединения 8a в виде масла.
Пример 9
3-Циклогексил-1-[4-[4-[2-(2-пропилокси)фенил] -1,2,3,6-тетрагидропиридин-1-ил] бутан-1-ил]-2-имидазолидинон оксалат (9а).
Смесь 4-[2-(2-пропилокси)фенил]-1,2,3,6-тетрагидропиридина (8а) (2,3 г), 1-(4-хлоро-1-бутил)-3-циклогексил-2-имидазолидинона (1а) (1,25 г), карбоната калия (1,6 г) и кристалла иодида калия в МИВК (60 мл) кипятят с обратным холодильником на протяжении ночи. Неорганические соли отфильтровывают и растворитель испаряют в вакууме. Неочищенное названное соединение 9а очищают колоночной хроматографией на силикагеле (элюируют 4% триэтиламином в смеси 3: 1 этилацетата и гептана). Выход: 1 г в виде масла. Оксалатную соль кристаллизуют из 2-пропанола, т.пл. 131-133oC.
Аналогичным способом получают тетрагидропиридинильные производные:
3-(4-Фторфенил)-1-[4-[4-[2-(2-пропилокси)фенил] -1,2,3,6-тетрагидропиридин- 1-ил] бутан-1-ил] -2-имидазолидинон оксалат (9b), т. пл. 150-2oC (ацетон).
1H ЯМР (ДМСО-d6): δ 1,25 (д, 6H), 1,45 - 1,65 (м, 2H), 1,65 - 1,80 (м, 2H), 2,60 - 2,80 (м, 2H), 3,10 (т, 2H), 3,20 (т, 2H), 3,20 - 3,35 (м, 2H), 3,45 (т, 2H), 3,65 - 3,90 (м, 4H), 4,60 (г, 1H), 5,65 - 5,80 (м, 1H), 6,90 (т, 1H), 7,00 (д, 1H), 7,05 - 7,20 (м, 3H), 7,25 (т, 1H), 7,50 - 7,65 (м, 2H).
3-Циклогексил-1-[4-[4-[2-(циклопропилметокси)фенил] -1,2,3,6 -тетрагидропиридин-1-ил]-2-имидазолидинон оксалат (9c), бесцветное масло.
1H ЯМР (CDCl3): δ 0,25 - 0,45 (м, 2H), 0,50 - 0,75 (м, 2H), 0,90 - 1,00 (м, 1H), 1,00 - 1,95 (м, 14H), 2,50 (т, 2H), 2,55 - 2,75 (м, 4H), 3,05 - 3,15 (м, 2H), 3,20 (т, 2H), 3,25 (с, 4H), 3,30 (дт, 1H), 3,80 (д, 2H), 5,75 - 5,80 (м, 1H), 6,80 (д, 1H), 6,90 (т, 1H), 7,10 - 7,30 (м, 2H).
Аналогичным способом, но применяя мезилаты (3a) и (3b) вместо хлоридов в качестве алкилирующих агентов, получают производные:
3-Циклогексил-1-[4-[4-[2-(2-пропилокси)фенил] -1,2,3,6-тетрагидропиридин-1-ил] бутан-1-ил]-2,3-дигидроимидазол-2-он (9d), бесцветное масло.
1H ЯМР (CDCl3): δ 1,10 - 1,50 (м, 5H), 1,30 (д, 6H), 1,50 - 1,75 (м, 5H), 1,75 - 2,00 (м, 4H), 2,45 (т, 2H), 2,50 - 2,60 (м, 2H), 2,60 - 2,68 (м, 2H), 3,05 - 3,15 (м, 2H), 3,65 (т, 2H), 3,90 - 4,05 (м, 1H), 4,50 (г, 1H), 5,70 - 5,75 (м, 1H), 6,20 (д, 1H), 6,25 (д, 2H), 6,90 (т, 2H), 7,10 - 7,25 (м, 2H).
3-(1-Адамантил)-1-[4-[4-[2-(2-пропилокси)фенил] -1,2,3,6-тетрагидропиридин-1-ил] бутан-1-ил]-2,3-дигидроимидазол-2-он оксалат (9e), т.пл.: 104 - 9oC (этилацетат/ацетон).
1H ЯМР (ДМСО-d6): δ 1,25 (д, 6H), 1,50 - 1,75 (м, 10H), 2,00 - 2,20 (м, 9H), 2,60 - 2,80 (м, 2H), 3,00 - 3,20 (м, 2H), 3,30 (т, 2H), 3,50 (т, 2H), 3,70 - 3,85 (м, 2H), 4,60 (г, 1H), 5,70 - 5,80 (м, 1H), 6,50 (с, 2H), 6,90 (т, 1H), 7,00 (д, 1H), 7,15 (д, 1H), 7,25 (т, 1H).
Пример 10 (способ c)
3-Циклогексил-1-[4-[4-[2-(1,1-диметилэтокси)фенил] -1-пиперидинил] -бутан-1-ил]- 2-имидазолидинон оксалат. (10a).
Раствор 3-циклогексил-1-[4-[4-(2-гидроксифенил)-1-пиперидинил] -бутан-1-ил] -2- имидазолидинона (1 г, полученный из 4-(2-гидроксифенил)-пиперидина (описано в Примере 5) и (1a) по способу, описанному в Примере 4), в сухом метиленхлориде (10 мл) охлаждают до -20oC. Изобутилен (5 мл, конденсирован при -25oC) добавляют в атмосфере азота и затем добавляют трифторметансульфокислоту (0,4 мл). Смесь перемешивают в течение 3 ч. при -20oC и затем добавляют триэтиламин (2 мл). После нагревания до комнатной температуры реакционную смесь концентрируют в вакууме, затем добавляют 2М аммиака и экстрагируют метиленхлоридом. Органическую фазу сушат над сульфатом магния и концентрируют в вакууме, получая масло, которое очищают флеш хроматографией (силикагель, элюент: этилацетат/MeOH/ триэтиламин 97:2:1). Полученное бесцветное масло растворяют в смеси этилацетата и ацетона. Добавление щавелевой кислоты дает названное соединение, (10a), в виде кристаллического вещества. Выход: 0,8 г, т.пл.: 147 - 153oC.
1H ЯМР (ДМСО-d6): δ 0,95 - 2,10 (м, 18H), 1,35 (с, 9H), 2,85 - 3,15 (м, 7H), 3,25 (с, 4H), 3,35 - 3,60 (м, 3H), 6,90 - 7,25 (м, 4H).
Фармакология
Соединения формулы I были испытаны согласно хорошо отработанным и надежным фармакологическим способом для определения активности по отношению к 5-HT1A и D2 рецептору, соответственно, и для каталептогенных действий. Испытания проводили так, как описано ниже.
Ингибирование 3H-8-OH-DPAT связывания с серотониновыми 5-HT1A рецепторами в головном мозге крысы in vitro.
С помощью этого способа определяют in vitro ингибирование лекарственными средствами связывания 5-HT1A агонист 3H-8-OH-DPAT (1 нМ) с 5-HT1A рецепторами в мембранах из головного мозга крысы минус мозжечек. Таким образом, это является тестом на сродство к 5-HT1A рецептору. Тест проводят, как описано Hyttel et al., Drug Dev. Res. 1988, 15 389-404.
Ингибирование 3H-спироперидольного связывания с допаминовыми D2 рецепторами в головном мозге крысы in vitro.
С помощью этого способа определяют in vitro ингибирование лекарственными средствами связывания D2 антагониста 3H-спироперидола (0,5 нМ) с D2 рецепторами в мембранах из полосатого тела (corpus straitum) крысы. Таким образом, это является тестом на сродство к допаминоваму D2 рецептору. Этот способ описан детально J. Hyttel et al., J. Neurochem., 1985, 44, 1615. Результаты испытаний по связыванию даны в таблице 1.
Каталептогенное действие
Оценка каталептогенных действий соединений данного изобретения проводилась согласно способу Sanchez C. et al., Drug Dev. Res 1991, 22, 239-250.
Примеры испытанных соединений представлены в таблице 2.
Кроме того, соединения данного изобретения были испытаны в отношении своей способности ингибировать 5-МеО-DMT- вызванный 5-HT синдром у крыс. 5-Метокси-N, N-диметилтриптамин (5-MeO DMT) представляет собой 5-HT1A агонист. Частичные 5-HT1A агонисты, такие как буспирон, ингибируют характерный 5-HT синдром, продуцированный 5-MeO-DMT. Таким образом, указанный тест является тестом для определения антагонистических воздействий испытываемого соединения на 5-HT1A рецепторы in vivo. Испытание проводят, как описано Smith L.M., Peroutka S.J., Pharmacol. Biochem. Behav., 1986, 24, 1513-1519. Соединения данного изобретения активны в этой тестовой модели.
Соединения, кроме того, были испытаны в Метилфенидатном тесте, описанном Pederson and Christensen, in Acta Pharmacol. et Toxicol. 31 488-496 (1972) и в перголидном тесте по вращению (Pergolide Rotation Test), описанном Arnt. J. and Hyttel, J. , J.Neural. Transm. 1986, 67, 225-240. Обе эти тестовые модели являются in vivo тестами на андидопаминергическую активность. Некоторые из соединений изобретения также демонстрируют эффекты в этих тестовых моделях.
Как видно из вышесказанного, соединения данного изобретения показывают сродство как к 5-HT1A, так и допаминовому D2 рецепторам in vivo. Кроме того, они показывают агонистическое действие на 5-HT1A рецептор in vivo и в то же время они не имеют каталептогенных действий. Наконец, некоторые из соединений показывают, кроме того, допамин D2 антагонистические действия in vivo. Таким образом, соединения имеют комбинированные действия на указанные два рецептора, т.е. действуют как агонисты, частичные агонисты или антагонисты, и блокируют допаминовый D2 рецептор, не проявляя каталептогенность. Лекарственные средства, имеющие такие свойства, полезны при лечении психических нарушений, как упомянуто ранее. В частности, предполагают, что они являются полезными при лечении психоза, включая положительный симптомы шизофрении.
Примеры приготовления лекарственного средства.
Фармацевтические формуляции изобретения могут быть получены обычными для данной области способами.
Например: таблетки могут быть получены путем смешения активного ингредиента с обычными адъювантами и/или разбавителями и затем прессования смеси в обычной таблетирующей машине. Примерами адъювантов или разбавителей являются кукурузный крахмал, картофельный крахмал, тальк, стеарат магния, желатин, лактоза, смолы, и т.п. Некоторые другие адъюванты или добавки, обычно используемые для таких целей, такие как красители, отдушки, консерванты и т. д. , могут быть использованы при условии, что они совместимы с активными ингредиентами.
Растворы для инъекций могут быть получены путем растворения активного ингредиента и возможных добавок в части раствора для инъекции, предпочтительно стерильной воде, доведения раствора до требуемого объема, стерилизации раствора и заполнения подходящих ампул или флаконов. Может быть добавлена любая подходящая добавка, обычно используемая в данной области, такая как тонизирующие агенты, консерванты, антиоксиданты, и т.д.
Типичными примерами рецептов формуляции изобретения являются следующие:
1) Таблетки, содержащие 5,0 мг Соединения 4a в расчете на свободное основание:
Соединение 4a - 5,0 мг
Лактоза - 60 мг
Кукурузный крахмал - 30 мг
Гидроксипропилцеллюлоза - 2,4 мг
Микрокристаллическая целлюлоза - 19,2 мг
Кроскармелоза Натрий Тип A - 2,4 мг
Стеарат магния - 0,84 мг
2) Таблетки, содержащие 0,5 мг Соединения 4a в расчете на свободное основание:
Соединение 4a - 0,5 мг
Лактоза - 46,9 мг
Кукурузный крахмал - 23,5 мг
Повидон - 1,8 мг
Микрокристаллическая целлюлоза - 14,4 мг
Кроскармелоза Натрий Тип A - 1,8 мг
Стеарат магния - 0,63 мг
3) Сироп, содержащий на миллилитр:
Соединение 4b - 25 мг
Сорбит - 500 мг
Гидроксипропилцеллюлоза - 15 мг
Глицерин - 50 мг
Метил-парабен - 1 мг
Пропил-парабен - 0,1 мг
Этанол - 0,005 мл
Отдушка - 0,05 мг
Сахарин натрий - 0,5 мг
Вода - до 1 мл
4) раствор для инъекции, содержащий на мл:
Соединение 6b - 0,5 мг
Сорбит - 5,1 мг
Уксусная кислота - 0,08 мг
Вода для инъекции - до 1 мл
| название | год | авторы | номер документа |
|---|---|---|---|
| ДИМЕРНЫЕ СОЕДИНЕНИЯ 4-ФЕНИЛПИПЕРИДИНА, 4-ФЕНИЛ-1,2,3,6-ТЕТРАГИДРОПИРИДИНА ИЛИ 4-ФЕНИЛПИПЕРАЗИНА ИЛИ ДИМЕРНЫЕ СОЕДИНЕНИЯ СПИРОЦИКЛИЧЕСКИХ ПИПЕРИДИНОВ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2129552C1 |
| ПРОИЗВОДНЫЕ 4-АРИЛ-1-(ИНДАНМЕТИЛ ИЛИ ДИГИДРОБЕНЗОТИОФЕНМЕТИЛ)ПИПЕРИДИНА,- -ТЕТРАГИДРОПИРИДИНА ИЛИ ПИПЕРАЗИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2142458C1 |
| КОНДЕНСИРОВАННОЕ БЕНЗОСОЕДИНЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2141959C1 |
| СОЕДИНЕНИЯ 3-ЗАМЕЩЕННОГО 1-АРИЛИНДОЛА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1994 |
|
RU2139287C1 |
| СПОСОБ ПОЛУЧЕНИЯ ФАРМАЦЕВТИЧЕСКОЙ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ СОСТОЯНИЙ БЕСПОКОЙСТВА, ПРОИЗВОДНЫЕ ПИПЕРИДИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ СОСТОЯНИЙ БЕСПОКОЙСТВА ИЛИ ЭПИЛЕПСИИ | 1992 |
|
RU2142952C1 |
| 1-ФЕНИЛ-БЕНЗИМИДАЗОЛЬНЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ РАССТРОЙСТВА ИЛИ ЗАБОЛЕВАНИЯ, ЧУВСТВИТЕЛЬНОГО К МОДУЛЯЦИИ ГАМК-РЕЦЕПТОРНОГО КОМПЛЕКСА ЦЕНТРАЛЬНОЙ НЕРВНОЙ СИСТЕМЫ | 1997 |
|
RU2194699C2 |
| ПРОИЗВОДНЫЕ ТРАНС-ИЗОМЕРОВ 1-ПИПЕРАЗИН-1,2-ДИГИДРОИНДЕНА | 1993 |
|
RU2114106C1 |
| НОВЫЕ АРИЛПИПЕРАЗИНИЛОВЫЕ СОЕДИНЕНИЯ | 2004 |
|
RU2339627C2 |
| НОВОЕ СОЕДИНЕНИЕ, ПРОЯВЛЯЮЩЕЕ ИНГИБИТОРНУЮ АКТИВНОСТЬ В ОТНОШЕНИИ PARP | 2012 |
|
RU2606635C2 |
| ПЕРВИЧНЫЕ КАРБОКСАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ BТK | 2014 |
|
RU2708395C2 |
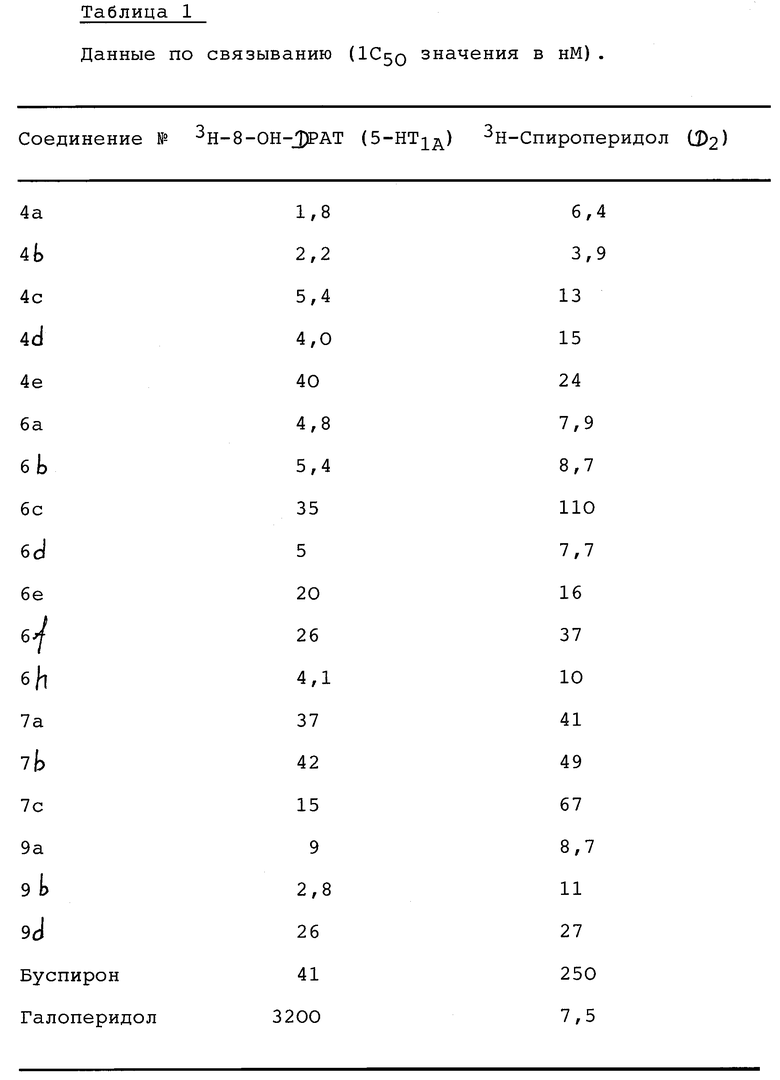

Соединение 4-фенилпиперазина, 4-фенилпиперидина и 4-фенил-1,2,3,6-тетрагидропиридина формулы I, где А - С3-8алкилен, R1 - С3-10алкил, циклоалкил, циклоалкил-низший алкил, трифторметилсульфонил или алкилсульфонил; W - O; V - NR2, R2 - Н, низший алкил, адамантил или фенил, возможно замещенный галогеном; Z - (СН2)m, m = 2; или Z представляет -СН=СН-; пунктирная линия указывает на необязательную связь и, когда связи нет, Х - N или СН, а когда связь есть, Х означает С, и его фармацевтически приемлемая аддитивная соль кислоты, имеет действие на центральный серотониновый 5-HT1A и допаминовый D2 рецепторы. Новые соединения полезны при лечении некоторых психических и неврологических расстройств. 2 с. и 4 з.п.ф-лы, 2 табл.

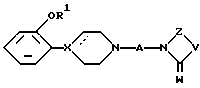
в которой A - разветвленный или неразветвленный C3-8алкилен;
R1 - разветвленный C3-10алкил, циклоалкил, циклоалкил-низший алкил, трифторметилсульфонил или низший алкилсульфонил;
W представляет O;
V представляет NR2, R2 выбирают из водорода, низшего алкила, циклоалкила, циклоалкил-низшего алкила, адамантила или фенила, возможно замещенного галоидом;
Z представляет -(CH2)m, причем m равен 2, или Z представляет -CH=CH-;
пунктирная линия, исходящая от X, указывает на необязательную связь и, когда связи нет, X означает N или CH, а когда связь есть, X означает C,
и его фармацевтически приемлемая аддитивная соль кислоты.
| Способ получения 1-бензимидазолилалкил 4-замещенных пиперидинов или их солей | 1976 |
|
SU663306A3 |
| EP 534904, 1993. | |||

