Настоящее изобретение относится к классу неизолированных бензопроизводных, в сильной степени связанных с рецепторами 5-HT1A и обладающих центральной серотонергической активностью 5-HT1A. Поэтому эти неизолированные бензопроизводные полезны для лечения определенных психических и неврологических заболеваний.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Ряд соединений, структурно относящихся к соединениям согласно данному изобретению, были известны из предыдущих исследований.
Так, ЕП N 0138280 и N 0185429 раскрывают очень широкий класс соединений пиперазинила, имеющих в положении 4 бициклический гетероарильный радикал и в положении 1 гетероарильную, арильную или алкилзамещенную карбамоилэтильную или карбамоилпропильную группу. Этим соединениям приписывается свойство снижать кровяное давление посредством центрального механизма. ЕП 0372657 раскрывает аналогичные производные, отличающиеся лишь тем, что они имеют несколько иные заместители в бициклическом гетероарильном радикале. Эти последние производные, как показано, демонстрируют анксиолитическое действие в моделях на животных, но не воздействуют на кровяное давление. Недавно сообщалось, что одно из соединений, охватываемых ЕП 0138280, например, 4-фтор-N-[2-(4-(2-гидроксиметил-1,4- бензодиоксан-5-ил)-пиперазин-1-ил)этил]бензамид, известное как флезиноксан, имеет высокую агонистическую 5-HT1A -активность, обладая антидепрессивным и анксиолитическим действиями (Schipper et al., Human Psychopharm., 1991, 6, S53).
ЕП N 0364327 раскрывает класс производных 4-[2-(4-нафтил- или изохинолил)пиперазин-1-ил)этил] -2-хинолона, обладающих активностью относительно рецепторов 5-HT1A и 5-НТ2. Отмечено, что эти соединения являются полными агонистами, частично агонистами или антагонистами in vivo. ЕП 0343050 описывает группу соединений б-фенил-3-[(4-нафтил или изохинолил)пиперазин-1- ил)алкил(2-4)] -1H, 3H-пиримидин-2,4-диона, обладающих активностью относительно рецепторов 5-HT1A и 5-НТ2. И еще относительно рецептора 5-HT1A в международной патентной публикации N WO 92/03426 описан класс производных пиперазина, имеющих нафтил или хинолил в положении 4 и N-арилзамещенную карбамоилалкильную группу или N-арилзамещенную уреидоалкильную группу в положении 1. Заявляется, что эти соединения обладают сродством к различным рецепторам, включая рецепторы 5-HT1A, 5-НТ2, α- рецепторы и рецепторы домамина.
ЕП N 0466585 касается 1-(бензамидоалкил)-4-(нафтил- или хинолил) пиперазинов или -тетрагидропиридинов, имеющих сродство к рецептору 5-HT1A и которые, как было найдено, имеют сильное антигипертензивное воздействие на животных.
Наконец, ЕП 0490772 A 1 раскрывает класс 1,4-бизамещенных производных пиперазина, которым приписывается антагонистическая активность 5-HT1A. Указанные производные имеют в положении 4 радикал 5-бензодиоксанила или 7-изобензофуранила и цепь низших алкилов, замещенных бициклической карбоциклической системой в положении 1.
Соединения, обладающие центральной серотонергической активностью 5-HT1A, можно разделить, пользуясь хорошо известными фармакологическими принципами, на полных агонистов, частичных агонистов и антагонистов.
Клинические изучения известных частичных агонистов 5-HT1A, таких как, например, бушпирон(8-[4-[4-(2-пиримидил)-1-пиперазинил] бутил] -8-азаспиро[4,5] декан-7,9-дион, ипсапирон (4,4-диметил-1-[4-[4-(2-пиримидил)-1-пиперазинил] бутил]-2,6-пиперидиндион) и гепирон(2-[4-[4-(2-пиримидил)1- пиперазинил] бутил]-1,2-бензотиазол-3(2Н)-он-1,1-диоксид, показали, что частичные агонисты 5-HT1A полезны для лечения тревожных состояний, таких как общее тревожное состояние, паническое состояние и компульсивные нарушения (Glitz, D. A. , Pohl, R., Drugs, 1991, 41, 11). Доклинические испытания показывают, что полные агонисты также полезны для лечения приведенных выше нарушений, вызванных состоянием тревоги (Schipper, Human Psychopharm., 1991, 6, S53).
Существуют также доказательства, как клинические, так и доклинические, в поддержку успешного воздействия частичных агонистов 5-HT1A на лечение депрессии, а также и контролируемых нарушений и злоупотребления алкоголем (van Hеst, Psychopharm. , 1992, 107, 474; Schipper et al., Human Psychopharm., 1991, 6, S53; Cervo et al., Eur. J. Pharm., 1988, 158, 53; Glitz, D.A., Pohl, R., Grugs, 1991, 41, 11).
Агонисты 5-HT1A и частичные агонисты ослабляют агрессию мужских особей мышей, индуцированную их изоляцией, что указывает на полезность этих соединений для лечения агрессии (Sanchez et al., Psychopharmacology, 1993, 110, 53 - 59).
Более того, последние исследования показали также, что рецепторы 5-HT1A являются важными для серотонергического модулирования каталепсии, вызванной галоперидолом (Hicks, Life Science, 1990, 47, 1609), и предполагается, что агонисты 5-HT1A полезны для лечения побочных эффектов, вызванных общепринятыми антипсихотическими средствами, такими как, например, галоперидол.
Агонисты 5-HT1A продемонстрировали нейропротекторные свойства при моделировании на грызунах фокальной и церебральной ишемии, и поэтому они полезны для лечения ишемических состояний (Prehn, Eur. J. Pharm., 1991, 203, 213).
Фармакологические испытания указывают на то, что антагонисты 5-HT1A полезны для лечения сенильного слабоумия (Bowen et al., Trends Neur. Sci., 1992, 15, 84).
И в опытах на животных, и при клинических испытания было показано, что агонисты 5-HT1A оказывают антигипертензивное воздействие через центральный механизм (Saxena, Villalon, Trends Pharm. Sci., 1990, 11, 95; Gillis et al., J. Pharm. Exp. Ther., 1989, 248, 851). Поэтому лиганды 5-HT1A могут быть успешно использованы для лечения кардиоваскулярных нарушений.
Следовательно, как полагают, и агонистические, и антагонистические средства, действующие на рецептор 5-HT1A, полезны для использования в терапии приведенных выше состояний, и поэтому средства эти весьма желательны.
В настоящее время установлено, что соединения определенного класса неизолированных бензопроизводных связаны с рецептором 5-HT1A с высокой степенью сродства. Более того, установлено, что эти соединения имеют широкую избирательность относительно рецептора 5-HT1A, рецептора допамина D2 и α1- адреноцептора, а также сильно различны по эффективности.
РЕЗЮМЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение обеспечивает новый класс неизолированных бензосоединений общей формулы I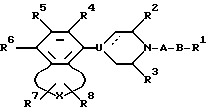
где A представляет собой 2-6-членную пространственную группу, выбранную из алкилена, алкенилена, и алкинилена, каждый из которых может представлять прямую или разветвленную цепь, или 3 - 7-членную циклоалкиленовую группу, причем указанная пространственная группа может быть замещена арилом или гидроксилом;
В представляет собой полярную бивалентную группу, выбранную из SO, SO2 и группу формулы II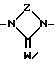
в которой W представляет собой О или S, a Z выбирают из - (CH2)n-, где n= 2 или 3, -CH=CH-, -COCH2-, -CSCH2- или 1,2-фенилен, который может быть замещен галогеном или трифторметилом;
U представляет собой N или CH; пунктирная линия обозначает факультативную связь, и если она обозначает связь, то U представляет собой C;
X выбирают из группы бивалентных 3-4-членных групп, содержащих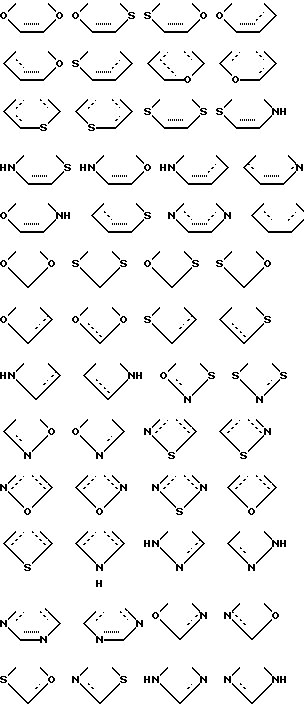
где пунктирные линии обозначают факультативные связи, образуя таким образом карбоциклическое или гетероциклическое кольцо, слившееся с бензольным кольцом;
R1 представляет собой алкил, алкенил, циклоалк(ен)ил, арил, циклоалк(ен)илалк(ен/ин)ил, арилалкил, дифенилалкил, любую алкильную группу, которая факультативно может быть замешена одной или двумя гидроксигруппами при условии, что если Z представляет собой 1,2-фенилен и U представляет собой N, то R1 выбирают из арила и замещенного арила;
R2 и R3 представляют собой независимо водород, низший алкил или они могут быть связаны вместе, образуя при этом этиленовый или пропиленовый мостик;
R4, R5 и R6 независимо выбирают из группы, содержащей водород, галоген, низший алкил, низший алкокси, гидрокси, низший алкилтио, низший алкиламино или низший диалкиламино, циано, нитро, трифторметил и трифторметилтио;
R7 и R8 независимо выбраны из группы, содержащей водород, галоген, трифторметил, низший алкил, низший алкил замещенный одной или более гидроксигруппой, арил, циан, группу -COOR9 и группу - CONR10R11, R9, R10 и R11 при этом представляют собой водород или низший алкил; причем любая из присутствующих арилгрупп может быть факультативно замещена одним или большим числом заместителей, выбранным из галогена, низшего алкила, низшего алкокси, гидрокси, низшего алкилтио, низшего алкилсульфонил, низшего алкил- или диалкиламино, циано, трифторметил или трифторметилтио;
и фармацевтически приемлемую добавку солей этих кислот.
Вторым аспектом настоящего изобретения является обеспечение фармацевтической композиции, содержащей по меньшей мере одно новое неизолированное бензопроизводное согласно настоящему изобретению, как это было определено выше, или фармацевтически приемлемую добавку соли этих кислот или пролекарство из них в терапевтически эффективном количестве и в сочетании с одним или более фармацевтически приемлемыми носителями или разбавителями.
Следующим аспектом настоящего изобретения обеспечивается использование неизолированных бензопроизводных, имеющих определенную выше формулу I, или солей их кислот или пролекарств из них для изготовления фармацевтического препарата для лечения тревожных состояний, депрессий, психоза, контролируемых нарушений, злоупотребления алкоголем, ишемии, кардиоваскулярных нарушений, побочных эффектов, вызванных общеизвестными антипсихотическими средствами и сенильного слабоумия.
Было установлено, что соединения настоящего изобретения замещают тритированный 8-гидрокси-2-дипропиламинотетралин (8-OH-DPAT) из 5- HT1A in vitro, причем большинство соединений имеет сродство выше чем 50 nM. Более того, было доказано, что настоящие соединения охватывают более широкий диапазон избирательности для рецепторов 5-HT1A по сравнению с α1- адреноцепторами и рецепторами D2. Некоторые из соединений согласно настоящему изобретению более селективны в отношении рецепторов 5-HT1A, в то время как другие соединения настоящего изобретения имеют сродство к некоторым из указанных выше местам связи. Было показано, что настоящие соединения охватывают широкий диапазон эффективностей.
Особенно интересная группа соединений демонстрирует высокое сродство и к рецептору 5-HT1A, и к рецепторам D2. С точки зрения того факта, что антагонисты D2 эффективны при лечении шизофренических нарушений (см. Lowe et al. , Med. Res. Rev., 1988, 8, 475), и так как агонисты 5-HT1A, как это было выше отмечено, могут смягчить побочные нейролептические эффекты, то такие соединения полезны для лечения шизофренических нарушений.
ДЕТАЛЬНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Некоторые соединения общей формулы I могут существовать в виде своих оптических изомеров, и такие оптические изомеры также включаются в настоящее изобретение.
Употребляемый здесь термин "алкил" относится к прямой или разветвленной C1-C20-группе, аналогично термин "алкенил" и "алкинил" означает прямую или разветвленную цепь углеводородов C2-C20, имеющую одну или более двойных или тройных связей соответственно. Термин "циклоалкил" обозначает карбоциклическое кольцо, включающее 3 - 8 атомов углерода, или бициклическое или трициклическое карбоциклическое кольцо, такое как, например, адамантил.
В формулах, включаемых в определения для X, пунктирные линии обозначают факультативную связь, например, в случае, если пунктирная линия обозначает связь, вероятно, что эта связь может быть двойной. Конечно, двойные связи не должны находиться в смежных позициях и расположение связей не должно находиться в противоречии с общепринятыми правилами, что легко понятно людям, квалифицированным в этой области.
Термин "алк(ен/ил)ил" означает, что эта группа может представлять собой алкил, алкенил или алкинил группу.
Термины "низший алкил", "низший алкокси", "низший алкилтио" и т. д. относятся к таким разветвленным или неразветвленным группам, которые включают 1 - 6 атомов углерода. Примерами таких групп являются метил, этил, 1- пропил, 2-пропил, 1-бутил, 2-бутил, 2-метил-2-пропил, 2-метил-1-пропил, метокси, этокси, 1-пропокси, метилтио, этилтио, 1-пропилтио, 2-пропилтио, метилсульфонил, этилсульфонил и им подобные.
Термин "арил" относится к карбоциклической или гетероциклической ароматической моноциклической или неизолированной бициклической группе или бифенильной группе. Примерами таких групп являются: тиенил, фурил, оксазолил, изоксазолил, тиазолил, изотиазолил, пиразолил, имидазолил, триазолил, оксадиазолил, тиадиазолил, тетразолил, бензофуранил, бензотиенил, бензизотиазолил и бензизоксазолил, индолил, фенил, пиридил, пиримидинил, пиридазинил, нафтил, хинолинил и хиназолинил; особенно фенил, тиенил, нафтил или фуранил.
В формуле I:
A является предпочтительно 2 - 6-членной алкиленовой группой.
B является предпочтительно SO, SO2 или группой формулы II, согласно данному выше определению, где W представляет собой О, a Z выбирают из -(CH2)n-, причем n=2 или 3, -CH=CH или 1,2-фенилен, который может быть замещен галогеном или трифторметилом.
X предпочтительно выбирают из группы бивалентных 3 - 4-членных групп, содержащих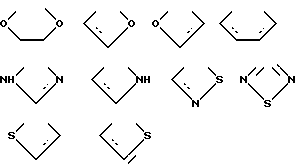
R1 предпочтительно представляет собой низший алкил; арил; циклоалкил или арил-низший арил; наиболее предпочтительно низший алкил; фенил; фенил, замещенный одним из заместителей, указанных выше; C5-C6 циклоалкил; адамантил; фенил-низший алкил, который может быть замещен одним из заместителей, указанных выше или нафтил.
R2 и R3 оба предпочтительно представляют собой водород.
R4, R5 и R6 предпочтительно выбирают независимо из группы, содержащей водород и галоген.
R7 и R8 предпочтительно независимо выбирают из группы, содержащей водород, низший алкил, арил, группу -COOR9, причем R9 представляет собой водород или низший алкил и группа -CONH2. Наиболее предпочтительно, чтобы R7 и R8 были независимо выбраны из водорода, низшего алкила, фенила, который может быть замещен одним или более из указанных выше заместителей, группы -COOR9, где R9 представляет собой водород или низший алкил, и группы -CONH2.
Соли кислот по настоящему изобретению представляют собой фармацевтически приемлемые соли соединений формулы I и нетоксичных кислот. Примерами таких органических солей являются соли малеиновой, фумаровой, бензойной, аскорбиновой, янтарной, щавелевой, бис-метиленсалициловой, метансульфоновой, этандисульфоновой, уксусной, пропионовой, винной, салициловой, лимонной, глюконовой, молочной, миндальной, коричной, цитраконовой, аспартиновой, стеариновой, пальмитиновой, итаконовой, гликоновой, п-аминобензойной, глютаминовой, бензолсульфоновой и теофиллинуксусной кислот, а также 8-галотеофиллинов, например, 8-бромотеофиллин. Примерами таких неорганических солей служат соли соляной, бромистоводородной, серной, сульфамидной, фосфорной и азотной кислот.
Фармацевтические соединения или соединения, полученные согласно настоящему изобретению, можно использовать любым подходящим способом, например, орально в виде таблеток, капсул, порошков, сиропов и т. д. или парентерально в виде растворов для инъекции. Для получения таких соединений могут быть применены известные в этой области методы и любые приемлемые фармацевтические носители, разбавители или иные добавки, обычно используемые в этой области. Обычно соединения настоящего изобретения вводятся в единичной дозировочной форме, содержание указанных соединений составляет от ~ 0.01 до 50 мг.
Общая дневная дозa обычно составляет от ~0.05 до 500 мг, а наиболее предпочтительно от ~0.1 до 20 мг активного соединения по данному изобретению.
Соединения формулы I получают:
а) взаимодействием соединения формулы III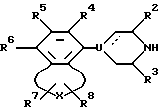
где R2-R8, U, X и пунктирные линии таковы, как это было определено выше, с соединением формулы R1-B-A-V, где R1, A и B таковы, как это было определено выше, а V представляет собой подходящую отщепляемую группу, такую как галоген, мезилат или тозилат;
b) восстановлением амида карбонила соединения формулы IV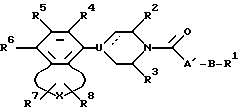
где R1-R8, B, U, X и пунктирная линия таковы, как это было определено выше, а A' - таково, что группа CH2-A' представляет собой 2 - 6-членный разветвленный алкилен или алкилен с прямой цепью, алкениленовую или алкиниленовую группу, которая может быть замещена таким арилом или гидрокси, который включает в себя определения A;
с) восстановительным алкилированием амина формулы III, которая была определена выше, с альдегидом формулы R1-B-A'-CHO, карбоновой кислотой формулы R1-B-A'-COOH или кетоном формулы R-B-A''-CO-A''', где R1, B и A' таковы, как это было определено выше, а A" и A''' такие, что группы A''-CH-A''' представляют собой 2 - 6-членную разветвленную или прямую цепь групп алкилена, алкенилена или алкинилена, которая может быть замещена арилом или гидроксилом, что включается определением для A;
d) окисление сульфидного атома серы в соединении формулы V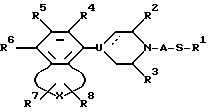
где для R1-R8, A, U, X и пунктирной линии выше были даны определения, до соответствующего сульфоксида или сульфона;
е) 1,4-присоединением амина общей формулы III, определенного выше, к α,β- ненасыщенному соединению общей формулы R12R13C=CR14-B-R1, где для R1 и В дано определение выше, а R12, R13 и R14 таковы, что R12R13C=CR14-BR1 представляет собой 2 - 6-членную алкениленовую группу с разветвленной или прямой цепью, которую можно заместить арилом или гидрокси, как это было указано в определениях для A;
f) восстановительным алкилированием NH-группы соединения общей формулы VI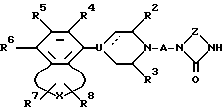
в которой для R2 - R8, A, U, X, Z и пунктирной линии дано определение выше, с альдегидом формулы R1'-CHO, карбоновой кислотой формулы R1'-COOH или кетоном формулы R1"-CO-R1'', где R1', R1'' и R1''' такие группы, что R1'-CH2 и R1''-CH2-R1''' соответственно представляют собой группы, входящие в определения R1;
g) циклизацией соединений общей формулы VII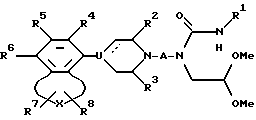
в котором R1-R8, A, U, X и пунктирная линия были определены выше;
h) арилированием NH группы соединения общей формулы VIII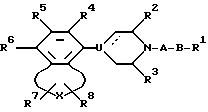
в котором A, B, R1-R8, пунктирная линия и U были определены выше, а X' такой, как это было выше определено для X, полагая, что X' обозначает гетероатомную кольцевую систему, содержащую функциональную группу NH, с помощью арилирующего агента, имеющего формулу Ar-hal, где Ar обозначает арил, как это было определено выше, a hal обозначает галоген;
i) превращением соединения общей формулы I, в которой R7 или R8 в обозначает группу -COOR9, в соответствующее соединение, в котором R7 или R8 обозначает группу -CONR10R11, где R7-R11 были определены выше;
j) обработкой соединения общей формулы I, в котором кольцевая система обозначена X, содержащего одну или более двойных связей, для восстановления одной или более указанных связей, получая таким образом соответствующую частично или полностью восстановленную кольцевую систему;
k) восстановительным отщеплением заместителей R4-R8 соединения общей формулы I, в котором один или более из этих заместителей выбраны из группы, содержащей хлор, бром или иод;
l) восстановлением двойной связи в тетрагидропиридиновом кольце соединения общей формулы I, в котором U представляет собой C, а пунктирная линия представляет собой связь, для получения соответствующего производного пиперидина;
при этом соединение формулы I выделяют в виде свободного основания или фармацевтически приемлемой соли указанной кислоты.
Взаимодействие соединения формулы III согласно методу a) удобно проводить в инертном органическом растворителе, таком как подходящий кипящий спирт или кетон, предпочтительно в присутствии основания (карбонат калия или триэтиламин) при температуре перегонки.
Реагенты формулы R1-B-A-V, в которой В представляет собой SO или SO2, получают окислением соответствующих сульфидов согласно методам, хорошо известным в этой области. Исходные сульфиды получают с помощью обычных методов, описанных в литературе.
Реагенты, в которых В представляет собой группу формулы II, в которой Z - это -(CH2)2-, a W представляет собой O, получают с помощью метода, раскрываемого в DE-OS N 2 035 370. Получение таких реагентов, в которых Z представляет собой -CH=CH- или 1,2-фенилен, описаны в примерах 5 и 12-13 соответственно.
Производные арилпиперазина формулы III удобно получать из соответствующих ариламинов согласно методу, описанному Martin et al., J. Med. Chem., 1989, 32, 1052 или методом, описанным Kruse et al., Rec. Trav. Chim Pays-Bas, 1988, 107, 303.
Исходные ариламины или доступны в коммерческом отношении, или описаны в следующей литературе.
Синтез 5-амино-1,4-бензодиоксана описан Dauksas et al., Zh. Org. Khim., 1967, 3, 1121.
Синтез 7-амино-2,3-дигидробензофурана описан в патенте США, приложение N 4302592.
Синтез этил 7-амино-2-индолил карбоксилата описан Scriven et al., J. Chem. Soc., Perkin Trans. 1, 1979, 53.
Синтез 7-аминобензофурана описан Van Wijngaarden et al., J. Med. Chem., 1988, 31, 1934.
Синтез 7-амино-2,3-дигидро-2,2-диметилбензофурана описан в Ger. Offen. DE 3526510.
Синтез 7-амино-бензо[b] тиофена описан Boswell et al., J. Heterocycl. Chem., 1968, 5, 69.
Синтез 7-аминоиндола описан в патенте США, приложение N 4506078.
Синтез 7-амино-1,2-бензотиазола описан Ricci et al., Ann. Chim. (Rome), 1963, 53, 1860.
Синтез 4-аминоиндола описан Melhado et al., J. Org. Chem., 1983, 48, 5130.
4-аминобензофуран и этил 4-амино-2-бензофуран карбоксилат получают восстановлением соответствующих нитросоединений (Andrisano et al., Gazz. Chim. Ital., 1956, 86, 1257).
7-амино-2-фенилбензофуран получают из 2-фенил-7-бензо- фуранилкарбоновой кислоты (Европейский патент, приложение N Ер 147044 A2) через перегруппировку Картиуса.
Замещенные производные различных циклических систем получают с помощью методов, аналогичных изложенным выше методам.
Пиперидин и производные 1,2,5,6-тетрагидропиридина формулы III получают известными методами, например, патент США N 2891066; McElvain et al., J. Amer. Chem. Soc., 1950, 72, 3134 или так, как это описано в примерах 10 и 11.
Восстановление согласно методу b) предпочтительно проводить в инертном органическом растворителе, таком как диэтиловый эфир или тетрагидрофуран, в присутствии литий-алюминий-гидрида при температуре перегонки.
Амиды формулы IV удобно получать обработкой соединений общей формулы III хлорангидридом подходящей карбоновой кислоты R1-B-A'COCl в присутствии основания (карбонат калия или триэтиламин). Ангидриды карбоновых кислот получают согласно стандартным методам.
Восстановительное алкилирование аминов формулы III согласно методу с) осуществляют с помощью стандартных литературных методов (см. пример 4). Альдегиды, карбоновые кислоты и кетоны формул R1-B-A'CHO, R1-B-A'COOH и R1-B-A''-CO-A''' соответственно получают согласно стандартным методам.
Окисление серы согласно методу d) осуществляют с использованием хорошо известного окислительного агента, например м-хлорбензойной кислоты, перекиси водорода или пероксимоносульфата калия. Сульфоксиды предпочтительно получают при использовании м- хлорбензойной кислоты согласно стандартным методам. Сульфоны предпочтительно получают при использовании перекиси водорода в ледяной уксусной кислоте согласно стандартным методам.
Сульфиды формулы V получают или методом a), используя реагент R1-S-A-V, или методом b), используя соединения формулы IV, где B определяется как S, или методом с), используя альдегиды формулы R1-S-A'-CHO, или карбоновые кислоты формулы R1-S-A'-COOH, или кетоны формулы R1-S-A''-CO-A'''. Все указанные выше реагенты получают согласно стандартным методам.
Присоединение аминов к α,β- ненасыщенным соединениям согласно методу е) удобно осуществлять в инертном растворителе, таком как метиленхлорид при комнатной температуре. Ненасыщенные соединения формулы R12R13C=CR14-B-R получают стандартными методами.
Циклизация согласно методу g) проводится в этаноле в присутствии соляной кислоты. Исходные соединения общей формулы VII получают алкилированием аминов формулы III ацетонитрилом с последующим восстановлением цианогруппы до соответствующего первичного амина. Моноалкилирование 2-бромацетальдегидиметилацеталем и последующее присоединение изоцианатов дает соединение VII.
Арилирование согласно методу h) наиболее удобно проводить, применяя широко известную реакцию Ульмана. Арилирующие реагенты Ar-hal доступны в коммерческом отношении, а превращение эфиров согласно методу i) хорошо описано в литературе.
Восстановление двойных связей согласно методу j) удобно проводить при каталитическом гидрировании в спирте, используя платиновый катализатор, или при обработке цианоборогидридом натрия в трифторуксусной кислоте (см. пример 9), или при гидрировании дибораном или предшественником диборана, таким как триметиламин или диметилсульфидный комплекс в тетрагидрофуране или диоксане при температурах от 0oC до температуры перегонки с последующим каталитическим кислотным гидролизом промежуточных производных борана.
Удаление заместителей галогена согласно методу k) и восстановление двойной связи согласно методу l) удобно осуществлять при каталитическом гидрировании в спирте в присутствии палладиевого катализатора или при обработке формиатом аммония в спирте при повышенной температуре в присутствии палладиевого катализатора.
ПРИМЕРЫ
Далее изобретение иллюстрируется примерами, которые не следует рассматривать как ограничительные.
ПРИМЕР 1
1-(1,4-бензодиоксан-5-ил)-4-(3-циклогексилсульфонил-1-пропил) пиперазин, оксалат, 1a.
К суспензии трет-бутоксида калия (100 г) в толуоле (600 мл) по каплям добавили циклогексилтриол (100 г). После перемешивания в течение получаса при комнатной температуре по каплям добавили 3- бром-1-пропанол (100 г). Смесь перемешивали в течение 3 часов при 60oC, а затем вылили в 2 М раствор гидроксида натрия (1 л). Провели разделение фаз и органическую фазу промыли 2 М гидроксидом натрия (500 мл). После удаления в вакууме растворителя было получено бесцветное масло (120 г) 3-циклогексилтио-1- пропанола, которое было достаточно чистым для его последующего использования на следующей стадии.
К раствору 3-циклогексилтио-1-пропанола (60 г) в ледяной уксусной кислоте (250 мл) добавили при 10oC перекись водорода (35% в воде, 210 мл) с последующей дефлегмацией в течение 2 часов. После охлаждения смесь вылили на лед, а затем провели экстракцию этилацетатом (1 л). Органическую фазу несколько раз промывали 1 М гидроксидом натрия. После удаления растворителя было получено масло, которое обрабатывали в течение 1 часа 1 М гидроксидом натрия (600 мл) при температуре перегонки. После экстракции этилацетатом, высушивания органической фазы над сульфатом магния и удаления в вакууме растворителя было получено бесцветное масло (37 г) 3-циклогексилсульфонил-1-пропанола, которое было использовано на следующей стадии без дальнейшей очистки.
Раствор 3-циклогексилсульфонил-1-пропанола (37 г) и триэтиламина (30 мл) в метиленхлориде (400 мл) обрабатывали, прикапывая при - 5oC метансульфонилхлорид (15 мл). После перемешивания в течение 2 часов при комнатной температуре смесь промыли водой и высушивали над сульфатом магния. Удалив в вакууме растворитель, получили вязкое масло (49 г) 3-циклогексилсульфонил- 1-пропил метансульфоната.
Смесь 3-циклогексилсульфонил-1-пропил метансульфоната (8.5 г), 1-(1,4-бензодиоксан-5-ил-пиперазина (5.4 г) и карбоната калия в метилизобутилкетоне (200 мл) подвергали дефлегмации в течение 20 ч. После фильтрования и удаления в вакууме растворителя получили масло, которое было очищено колоночной хроматографией (силикагель, элюент: этил/метанол/триэтиламин= 96:2:2). Соединение, указанное в заглавии, кристаллизовали как оксалатную соль из ацетона при добавлении щавелевой кислоты. Выход 8.1 г, температура плавления 162-64oC. 1H ЯМР ( δ, ДМСО): 1.05-1.45 (m, 6H), 1.00-1.90 (m, 2H), 1.95-2.10 (m, 4H), 2.90-3.20 (m, 13H), 4.15-4.30 (m, 4H), 6.45-6.60 (m, 2H), 6.75 (d, 1H).
Аналогичным способом были получены:
1-(1,4-бензодиоксан-5-ил)-4-(3-фенилсульфонил-1-пропил)пиперазин, гидрохлорид, 1b, температура плавления 184-96oC.
1H ЯМР (δ, ДМСО): 2.00-2.20 (m, 2H), 3.00-3.25 (m, 6H), 3.30-3.60 (m, 6H), 4.15-4.30 (m, 4H), 6.45-6.60 (m, 2H), 6.75 (t, 1H), 7.60-7.80 (m, 3H), 7.95 (d, 2H), 8.00 (b, 2H).
1-(3-циклогексилсульфонил-1-пропил)-4-(2,3-дигидробензофуран-7- ил)пиперазин, малеат, 1c, температура плавления 166-68oC. 1H ЯМР (δ, ДМСО): 1.05-1.50 (m, 5H), 1.60-1.70 (m, 1H), 1.75-1.90 (m, 2H), 1.95-2.20 (m, 4H), 3.00-3.40 (m, 17H), 4.50 (t, 2H), 6.05-7.80 (s. 2H), 6.65-6.80 (m, 2H), 6.90 (d, 1H).
1-(2,3-дигидробензофуран-7-ил)-4-(3-метилсульфонил-1- пропил)пиперазин, малеат, 1d, температура плавления 150-51oC. 1H ЯМР (δ, ДМСО): 2.00-2.20 (m, 2H), 3.05 (s, 3H), 3.00-3.50 (m, 16H), 4.55 (t, 3H), 6.10 (s, 2H), 6.65-6.85 (m, 2H), 6.90 (d, 1H).
1-(1,4-бензодиоксан-5-ил)-4-(3-изопропилсульфонил-1- пропил)пиперазин, фумарат, 1e, температура плавления 166-67oC. 1H ЯМР (δ, ДМСО): 1.25 (d, 6H), 1.80-2.00 (m, 2H), 2.50-2.65 (m, 6H), 2.90-3.05 (m, 4H), 3.05-3.15 (m, 2H), 3.30 (h, 1H), 4.15-4.30 (m, 4H), 6.50 (t, 2H), 6.60 (s, 2H), 6.70 (t, 1H).
1-[3-(1-адамантил)сульфонил-1-пропил]-4-(1,4-бензоикоксан-5- ил)пиперазин. 1f, температура плавления 143-44oC. 1H ЯМР (δ, CDCl3): 1.65-1.85 (m, 6H), 2.00-2.25 (m, 11H), 2.55 (t, 2H), 2.60- 2.70 (m, 4H), 2.90-3.00 (m, 2H), 3.00-3.15 (m, 4H), 4.20-4.25 (m, 2H), 4.25-4.35 (m, 2H), 6.50-6.60 (m, 2H), 6.80 (l, 1H).
ПРИМЕР 2
1-[3-[4-(l, 4-бензодиоксан-5-ил)-1-пиперазинил] -1- пропил]-3-фенил-2-имидазолидинон, гидрохлорид, 2a.
Смесь 1-(1,4-бензодиоксан-5-ил)-1-пиперазина (1.5 г), 1-(3- хлор-1-пропил)-3-фенил-2-имидазолидинона (1.4 г), карбоната калия (3 г) и иодида калия (0.1 г) в метилизобутилкетоне подвергалась дефлегмации в течение 20 ч. После фильтрования и удаления в вакууме растворителя было получено вязкое масло, которое отделили хроматографией на колонке (силикагель, элюент этилацетат/метанол/триэтиламин=15:4:1). Соединение, указанное в заглавии, было выделено в виде масла, которое кристаллизовалось как гидрохлоридная соль из ацетона при добавлении соляной кислоты. Выход 1.9 г, температура плавления 229-32oC. 1H ЯМР (δ, ДМСО): 1.95-2.15 (m, 2H), 3.00-3.25 (m, 6H), 3.30 (t, 2H), 3.40-3.65.(m, 4H), 3.70-4.00 (m, 4H), 4.15-4.30 (m, 4H), 6.45-6.70 (m, 2H), 6.75 (t, 1H), 7.00 (t, 1H), 7.30 (t, 2H), 7.60 (d, 2H), 11.30 (b, 1H).
Аналогичным способом были получены:
1-[2-[4-(1,4-бензодиоксан-5-ил)-1-пиперазинил] этил-3- циклопентил-2-имидазолидинон], гидрохлорид, 2b, температура плавления 266-68oC. 1H ЯМР (δ, CDCl3): 1.45-1.95 (m, 8H), 3.00-3.30 (m, 4H), 3.35-3.60 (m, 8H), 3.60-3.85 (m, 4H), 4.15-4.35 (m, 5H), 6.50 (d, 1H), 6.65 (d, 13H), 6.80 (d, 1H), 12.30 (b, 1H).
1-[2-[4-(1,4-бензодиоксан-5-ил)-1-пиперазинил] этил] -3- фенил-2-имидазолинон, гидрохлорид, 2c, температура плавления 288-290oC. 1H ЯМР (δ, ДМСО): 3.00-3.75 (m, 10H), 3.85 (t, 2H), 4.10-4.35 (m, 4H), 4.50-4.75 (m, 4H), 6.45-6.70 (m, 2H), 6.75 (t, 1H), 7.00 (t, 3H), 7.35 (t, 2H), 7.60 (b, 2H), 10.95 (b, 1H).
1-[2-[4-(1,4-бензодиоксан-5-ил)-1-пиперазинил] этил] -3- циклогексил-2-имидазолидинон, фумарат, 2d, температура плавления 103-14oC. 1H ЯМР (δ, ДМСО): 0.95-1.15 (m, 1H), 1.15-1.45 (m, 4H), 1.45-1.65 (m, 3H), 1.65-1.80 (m, 2H), 2.60 (t, 2H), 2.65-2.80 (m, 4H), 2.90-3.05 (m, 4H), 3.15-3.35 (m, 6H), 3.40- 3.55 (m, 1H), 4.15-4.30 (m, 4H), 6.4-6.55 (m, 2H), 6.60 (s, 2H), 6.70 (t, 1H), 7.90 (b, 1H).
1-[4-[4-(1,4-бензодиоксан-5-ил)-1-пиперазинил] 1-бутил]-3- циклогексил-2-имидазолидинон, гидрохлорид, 2e, температура плавления 212-22oC. 1H ЯМР (δ, ДМСО): 0.95-1.15 (m, 1H), 1.15-1.40 (m, 4H), 1.40-1.65 (m, 5H), 1.65-1.85 (m, 4H), 3.00-3.25 (m, 8H), 3.25 (m, 4H), 3.40-3.60 (m, 5H), 4.15-4.30 (m, 4H), 6.45-6.60 (m, 2H), 6.75 (t, 1H), 8.00 (b, 1H), 11.40 (b, 1H).
1-циклопентил-3-[2-[4-(2,3-дигидробензофуран-7-ил)-1-пиперазинил] этил] -2-имидазолидинон, гидрохлорид, 2f, температура плавления 200-2oC. 1H ЯМР ( ДМСО): 1.40-1.80 (m, 8H), 3.00-3.80 (m, 18H), 4.00-4.15 (m, 1H), 4.50 (t, 2H), 6.65-6.85 (m, 2H), 6.90 (t, 2H), 11.05 (b, 1H).
ДМСО): 1.40-1.80 (m, 8H), 3.00-3.80 (m, 18H), 4.00-4.15 (m, 1H), 4.50 (t, 2H), 6.65-6.85 (m, 2H), 6.90 (t, 2H), 11.05 (b, 1H).
1-[3-[4-(2,3-дигидробензофуран-7-ил)-1-пиперазинил] -1-пропил] -3- фенил-2-имидазолидинон, гидрохлорид, 2g, температура плавления 225-28oC. 1H ЯМР (δ, ДМСО): 1.95-2.10 (m, 2H), 2.95-3.40 (m, 12H), 3.40-3.70 (m, 6H), 3.80 (t, 2H), 4.50 (t, 2H), 6.65-6.80 (m, 2H), 6.90 (d, 1H), 7.00 (t, 1H), 7.35 (t, 2H), 7.60 (d, 2H), 11.20 (b, 1H).
4-[4-[2-(3-фенилимидазолидин-2-он-1-ил)этил] -1- пиперазинил]- 2,1,3-бензотиадиазол, малеат, 2h, температура плавления 182-83oC. 1H ЯМР (δ, ДМСО): 3.20-3.95 (m. 18H), 6.10 (s, 2H), 6.90-7.10 (m, 2H), 7.35 (t, 2H), 7.55-7.70 (m, 4H).
1-[2-[4-(2,3-дигидробензофуран-7-ил)-1-пиперазинил] этил] - 3-(4-фторфенил)-2-имидазолидинон, фумарат, 2i, температура плавления 188-90oC. 1H ЯМР (δ, ДМСО): 2.55-2.70 (m, 6H), 2.95-3.15 (m, 4H), 3.10 (t, 2H), 3.35 (t, 2H), 3.55 (t, 2H), 3.80 (t, 2H), 4.50 (t, 2H), 5.10 (b, 2H), 6.60 (s, 2H), 6.65 (d, 1H), 6.75 (t, 1H), 6.80 (d, 1H), 7.15 (t, 2H), 7.50-7.60 (m, 2H).
Этил 7-[4-[2-(3-фенил-2-имидазолидин-2-он-1-ил)этил] -1- пиперазинил]-2-индолил карбоксилат, фумарат, 2j, температура плавления 202-4oC. 1H ЯМР (δ, ДМСО): 1.35 (t, 3H), 2.70 (t, 2H), 2.75-2.90 (m, 4H), 2.95-3.15 (m, 4H), 3.40 (t, 2H), 3.60 (t, 2H), 3.00 (t, 2H), 4.35 (q, 2H), 7.60 (s, 2H), 6.80 (d, 1H), 6.95- 7.05 (m, 2H), 7.15 (d, 1H), 7.25-7.40 (m, 2H), 7.60 (d, 2H).
1-[2-[4-(1-нафтил)-1-пиперазинил] этил]-3-фенил-2-имидазодинон, фумарат, 2k, температура плавления 176-80oC. 1H ЯМР (δ, ДМСО): 1.35 (t, 2H), 2.65-2.90 (m, 4H), 2.95-3.15 (m, 4H), 3.40 (t, 2H), 3.55 (t, 2H), 3.80 (t, 2H), 6.60 (s, 2H), 7.00 (t, 1H), 7.10 (d, 1H), 7.30 (t, 2H), 7.40 (t, 1H), 7.45-7.65 (m, 5H), 7.89-7.95 (m, 1H), 8.05-8.20 (m, 1H).
1-[2-[4-(1,4-бензодиоксан-5-ил)-1-пиперазинил]этил]-3-этил-2- имидазодинон, гидрохлорид, 2l, температура плавления 250-52oC. 1H ЯМР (δ, ДМСО): 1.05 (t, 3H), 2.95-3.70 (m, 18H), 4.15-4.30 (m, 4H), 6.50 (d, 1H), 6.55 (d, 1H), 6.25 (t, 1H), 10.65 (b, 1H).
1-[2-[4-бензофуран-7-ил-1-пиперазинил] этил] -3-фенил-2- имидазолидинон, полуфумарат, 2m, температура плавления 175-76oC. 1H ЯМР (δ, ДМСО): 2.60 (t, 2H), 2.65-2.75 (m, 4H), 3.20-3.35 (m, 4H), 3.40 (t, 2H), 3.60 (t, 2H), 3.80 (t, 2H), 3.75 (s, 1H), 6.65 (d, 1H), 6.90 (s, 1H), 7.00 (t, 1H), 7.05- 7.25 (m, 2H), 7.30 (t, 2H), 7.60 (d, 1H), 7.95 (s, 1H).
1-[2-[4-(2,3-дигидро-2,2-диметилбензофуран-7-ил)-1-пиперазинил] этил]-3-фенил-2-имидазолидинон, дигидрохлорид, 2n, температура плавления 220-30oC. 1H ЯМР (δ, ДМСО): 1.40 (s, 6H), 3.00 (s, 2H), 3.10-3.45 (m, 6H), 3.50-3.75 (m, 8H), 3.85 (t, 2H), 6.65-6.80 (m, 2H), 6.85 (d, 1H), 7.00 (t, 1H), 7.35 (t, 2H), 7.60 (d, 2H), 9.35 (b, 1H), 11.30 (b, 1H).
1-[2-[4-(1,4-бензодиоксан-5-ил)-1-пиперазинил] этил] -3--изопропил-2- имидазолидинон, гидрохлорид, 2o, температура плавления 228-30oC. 1H ЯМР (δ, ДМСО): 1.05 (d, 6H), 2.95-3.65 (m, 16H), 3.90 (h, 1H), 4.15-4.30 (m, 4H), 6.50 (d, 1H), 6.60 (d, 1H), 6.25 (d, 1H), 10.95 (b, 1H).
1-циклопентил-3-[2-[4-(2,3-дигидро-2,2-диметилбензофуран-7-ил)- 1-пиперазинил] этил] -2-имидазолидинон, дигидрохлорид, 2p, температура плавления 185-95oC. 1H ЯМР (δ, ДМСО): 1.45 (s, 6H), 1.45-1.75 (m, 8H), 3.00 (s, 2H), 3.10-3.40 (m, 10H), 3.50 (t, 2H), 3.55-3.70 (m, 4H), 4.00-4.15 (m, 1H), 6.70-6.80 (m, 2H), 6.35 (d, 1H), 7.35 (b, 1H), 11.30 (b, 1H).
1-адамантил-3-[2-[4-(1,4-бензодиоксан-5-ил)-1-пиперазинил] этил] -2-имидазолидинон, гидрохлорид, 2q, температура плавления 246-48oC. 1H ЯМР (δ, ДМСО): 1.55-1.65 (m, 6H), 1.90-2.10 (m, 9H). 2.96-3.60 (m, 16H), 4.15-4.30 (m, 4H), 6.50 (d, 1H), 6.55 (d. 1H), 6.75 (t, 1H), 10.85 (b, 1H).
1-[2-[4-бензофуран-4-ил-1-пиперазинил] этил] -3-фенил-2- имидазолидинон, сесквифумарат, 2r, температура плавления 207-9oC. 1H ЯМР (δ, ДМСО): 2.65 (t, 2H), 2.70-2.80 (m, 4H), 3.10-3.20 (m, 4H), 3.40 (t, 2H), 3.55 (t, 2H), 3.80 (t, 2H), 6.60 (s, 3H), 6.65-6.70 (m, 1H), 6.95 (m, 2H), 7.10-7.20 (m, 2H), 7.30 (t, 2H), 7.55 (d,2H), 7.90 (s, 1H).
1-[2-[4-бензофуран-4-ил-1-пиперазинил] этил] -3-циклопентил-2- имидазолидинон, дигидрохлорид, 2s, температура плавления 237-39oC. 1H ЯМР (δ, ДМСО): 1.40-1.80 (m, 8H), 3.15- 3.45 (m, 10H), 3.55 (t, 2H), 3.55-3.75 (m, 4H), 4.00-4.20 (m, 1H), 4.45 (b, 1H), 6.75 (dd, 1H), 7.10 (d, 1H.), 7.20-7.30 (m, 2H), 8.00 (s, 1H), 11.20 (b, 1H).
1-[2-[4-бензо[b] тиофен-7-ил-1-пиперазинил] этил] -3-фенил-2- имидазолидинон, 2t, температура плавления 136-38oC. 1H ЯМР (δ, CDCl3): 2.70 (t, 2H), 2.70-2.85 (m, 4H), 3.15-3.35 (m, 4H), 3.50 (t, 2H), 3.55 (t, 2H), 3.80j (t, 2H), 6.90 (d, 1H), 7.00 (t, 1H), 7.20-7.45 (m, 5H), 7.45-7.65 (m, 3H).
1-циклопентил-3-[2-[-4-(7-индолил)-1-пиперазинил] этил] -2-имидазолидинон, 2u, температура плавления 188-89oC. 1H ЯМР (δ, CDCl3): 1.40-1.90 (m, 8H), 2.60 (t, 2H), 2.65- 2.75 (m, 4H), 3.05-3.15 (m, 4H), 3.20-3.45 (m, 6H), 4.25 (p, 1H), 6.50-6.55 (m, 1H), 6.80 (d, 1H), 7.05 (t, 1H), 7.10-7.20 (m, 1H), 7.35 (d, 1H), 8.40 (b, 1H).
1-[2-[4-(7-индолил)-1-пиперазинил] этил]-3-фенил-2-имидазолидинон, фумарат, 2v, температура плавления 215-16oC. 1H ЯМР (δ, ДМСО): 2.70 (t, 2H), 2.75- 2.85 (m, 4H), 3.00-3.15 (m, 4H), 3.40 (t, 2H), 3.55 (t, 2H), 3.80 (t, 2H), 6.35-6.40 (m, 1H), 6.60 (s, 2H), 6.65 (d, 1H), 6.90 (t, 1H), 7.00 (t, 1H), 7.15-7.35 (m, 4H), 7.60 (d, 2H).
1-[2-[4-(1,2-бензизотиазол-7-ил)-1-пиперазинил] этил] -3-фенил-2- имидазолидинон, гидрохлорид, 2х, температура плавления 237-44oC. 1H ЯМР (δ, ДМСО): 3.10-3.80 (m, 14H), 3.85 (t, 2H), 7.00 (t, 1H), 7.20 (d, 1H), 7.30 (t, 2H), 7.50 (t, 1H), 7.60 (d, 2H), 7.90 (d, 1H), 9.15 (s, 1H), 11.25 (b, 1H).
1-циклопентил-3-[2-[4-(4-индолил)-1-пиперазинил]этил]-2- имидазолидинон, дигидрохлорид, 2y, температура плавления 214-20oC. 1H ЯМР (δ, ДМСО): 1.50-1.80 (m, 8H), 3.20-3.60 (m, 1H), 3.60-3.80 (m, 4H), 3.95-4.20 (m, 1H), 6.60j (s, 1H), 6.70 (d, 1H), 7.00 (t, 1H), 7.20 (d, 1H), 7.35 (s, 1H), 11.30 (b, 1).
1-[2-[4-(4-индолил)-1-пиперазинил] этил] -3-фенил-2-имидазолидинон, дигидрохлорид, 2z, температура плавления 233-38oC. 1H ЯМР (δ, ДМСО): 3.25-3.50 (m, 8H), 3.60 (t, 2H), 3.60-3.74 (m, 4H), 3.85 (t, 2H), 5.00 (b, 2H), 6.50 (2, 1H), 6.60 (d, 1H), 6.95-7.00 (m, 2H), 7.15 (d, 1H), 7.25-7.40 (m, 3H), 7.60 (d, 2H), 11.20 (b, 1H).
1-[2-[4-бензо[b] тиофен-7-ил-1-пиперазинил] этил] -3-циклопентил- 2-имидазолидинон, гидрохлорид, 2aa, температура плавления 264-67oC. 1H ЯМР (δ, ДМСО): 1.40-1.75 (m, 8H), 3.20-3.45 (m, 10H), 3.50 (t, 2H), 3.60-3.75 (m. 4H), 4.10 (p, 1H), 7.05 (d, 1H), 7.40 (t, 1H), 7.50 (d, 1H), 7.60 (d, 1H), 7.75 (d, 1H), 11.30 (b, 1H).
1-циклогексил-3-[4-[4-(2,3-дигидро-2,2-диметилбензофуран-7-ил)- 1-пиперазинил] -1-бутил]-2-имидазолидинон, дигидрохлорид, 2bb, температура плавления 196-203oC. 1H ЯМР (δ, ДМСО): 1.20-1.65 (m, 10H), 1.40 (s, 6H), 1.65-1.80 (m, 4H), 3.00 (s, 2H), 3.00-3.20 (m, 8H), 3.30-3.25 (m, 6H), 3.40-3.55 (m, 2H), 3.60-3.65 (m, 1H), 6.70-6.80 (m, 2H), 6.85 (d, 1H), 7.60 (b, 1H), 11.30 (b, 1H).
Этил [4-[4-[2-(3-циклопентил-2-имидазолидинон-1-ил)этил]-1- пиперазинил] -2-бензофуранил] карбоксилат, гидрохлорид, 2cc, температура плавления 198-201oC. 1H ЯМР (δ, ДМСО): 1.35 (t, 3H), 1.40-1.75 (m, 8H), 3.25-3.75 (m, 16H), 4.00-4.15 (s, 1H), 4.35 (q, 2H), 6.80 (d, 1H), 7.30 (d, 1H), 7.40 (t, 1H), 7.95 (s, 1H).
1-[4-[4-(2,3-дигидро-2,2-диметилбензофуран-7-ил)-1-пиперазинил] - 1-бутил] -3-(4-фторфенил)-2-имидазолидинон, 2dd, температура плавления 158-60oC. 1H ЯМР (δ, CDCl3): 1.50 (s, 6H), 1.55-1.65 (m, 4H), 2.45 (t, 2H), 2.55-2.70 (m, 4H), 3.00 (s, 2H), 3.10-3.20 (m, 4H), 3.30 (t, 2H), 3.45 (t, 2H), 3.80 (t, 2H), 6.65-6.70 (m, 1H), 6.75 (d, 2H), 7.00 (t, 2H), 7.40-7.55 (m, 1H).
1-[2-[4-(1,4-бензодиоксан-5-ил)-1-пиперазинил] этил] -3-t- бутил-2-имидазолидинон, гидрохлорид, 2ee, температура плавления 229-31oC. 1H ЯМР (δ, ДМСО): 1.30 (s, 90H), 34.00-3.60 (m, 16H), 4.20-4.30 (m, 4H), 6.45-6.60 (m, 2H), 6.75 (t, 1H).
1-[3-[4-(2,3-дигидро-2,2-диметилбензофуран-7-ил)-1-пиперазинил] - 1-пропил] -3-фенил-2-имидазолидинон, фумарат, 2ff, температура плавления 183-85oC. 1H ЯМР (δ, ДМСО): 1.40 (s, 6H), 1.75 (hep, 2H), 2.50 (t, 2H), 2.60-2.70 (m, 4H), 2.95 (s, 2H), 3.00-3.15 (m, 4H), 3.25 (t, 2H), 3.45 (t, 2H), 3.80 (t, 2H), 6.60 (s, 2H), 6.65 (d, 1H), 6.70 (t, 1H), 6.75 (d, 1H), 7.00 (t, 1H), 7.30 (t, 2H), 7.55 (d, 2H).
1-адамантил-3-[4-[4-(2,3-дигидро-2,2-диметилбензофуран-7-ил)- 1-пиперазинил]-1-бутил]-2-имидазолидинон, 2gg, температура плавления 125-27oC. 1H ЯМР (δ, CDCl3): 1.50 (s, 6H), 1.50-1.55 (m, 3H), 1.65-1.70 (m, 6H), 2.00-2.10 (m, 9H), 2.40 (t, 2H), 2.55-2.65 (m, 4H), 3.00 (s, 2H), 3.10-3.20 (m, 8H), 3.30 (t, 2H), 6.70 (t, 1H), 6.75 (d, 2H).
1-[4-[4-(5-хлор-2-фенилбензофуран-7-ил)-1-пиперазинил] - 1-бутил]-3-циклогексил-2-имидазолидинон, дигидрохлорид, 2hh, температура плавления 198-200oC. 1H ЯМР (δ, ДМСО): 1.00-1.85 (m, 14H), 3.10 (t, 2H), 3.15-3.70 (m, 14H), 4.00- 4.10 (m, 1H), 2.95 (b, 2H), 6.85 (s, 1H), 7.30 (s, 1H), 7.40 (s, 1H), 7.45 (t, 1H), 7.50 (t, 2H), 7.95 (d, 2H).
1-[2-[4-(5-хлор-2-фенилбензофуран-7-ил)-1-пиперазинил]-этил]-3- циклопентил-2-имидазолидинон, фумарат, 2ii, температура плавления 155-57oC. 1H ЯМР (δ, ДМСО): 1.40-1.70 (m, 8H), 2.55 (t, 2H), 2.65-2.75 (m, 4H), 3.30-3.45 (m, 10 H), 4.00-4.15 (m, 1H),. 6.60 (s, 2H), 6.70 (s, 1H), 7.20 (s, 1H), 7.35 (s, 1H), 7.45 (t, 1H), 7,50 (t, 2H), 7.90 (d, 2H).
1-[4-[4-(2,3-дигидро-2,2-диметилбензофуран-7-ил)-1-пиперазинил] - 1-бутил] -3-(1-нафтил)-2-имидазолидинон, фумарат, 2jj, температура плавления 220-21oC. 1H ЯМР (δ, ДМСО): 1.40 (s, 6H), 1.55-1.65 (m, 4H), 2.55 (t, 2H), 2.65-2.75 (m, 4H), 2.95 (s, 2H), 3.05-3.15 (m, 4H), 3.25 (t, 2H), 3.60 (t, 2H), 3.80 (t, 2H), 6.60 (s, 2H), 6.65 (d, 1H), 6.70 (t, 1H), 6.80 (d, 1H), 7.45 (d, 1H), 7.45-7.60 (m, 3H), 7.85-8.00 (m, 3H).
1-циклогексил-3-[3-[4-(2, 3-дигидро-2, 2-диметилбензофуран- 7-ил)-1-пиперазинил]-1-пропил]-2-имидазолидинон, оксалат, 2kk, температура плавления 191-92oC. 1H ЯМР (δ, ДМСО): 1.00-1.90 (m, 10H), 1.40 (s, 6H), 2.90-3.00 (m, 4H), 3.10 (t, 2H), 3.15-3.30 (m, 10H), 3.40-3.50 (m, 1H), 4.10 (b, 2H), 6.65 (d, 1H), 6.70 (t, 1H), 6.80 (d, 1H).
1-[4-[4-(2, 3-дигидро-2,2-диметил-5-фторбензофуран-7-ил)-1- пиперазинил] -1-бутил] -3-(4-фторфенил)-2-имидазолидинон, оксалат, 2ll, температура плавления 126-27oC. 1H ЯМР (δ, ДМСО): 1.45 (s, 6H), 1.50-1.65 (m, 4H), 2.40 (t, 2H), 2.55-2.65 (m, 4H), 2.95 (s, 2H), 3.05-3.20 (m, 4H), 3.30 (t, 2H), 3.95 (t, 2H), 3.80 (t, 2H), 6.30-6.50 (m, 2H), 7.00 (t, 2H), 7.40-7.55 (m, 2H).
1-циклогексил-3-[4-[4-(2, 3-дигидро- 2,2-диметил-5-фтор-бензофуран-7-ил)-1-пиперазинил] -1-бутил] -2- имидазолидинон, оксалат, 2mm, температура плавления 125-35oC. 1H ЯМР (δ, ДМСО): 1.00-1.80 (m, 14H), 1.40 (s, 6H), 2.95 (s, 2H), 3.00-3.50 (m, 17H), 6.50 (dd, 1H), 6.65 (dd, 1H).
1-циклопентил-3-[6-[4-(2,3-дигидро-2,2-диметилбензофуран-7-ил)- 1-пиперазинил] -1-гексил] -2-имидазолидинон, оксалат, 2nn, температура плавления 132-34oC. 1H ЯМР (δ, ДМСО): 1.15-1.75 (m, 14H), 1.40 (s, 6H), 2.95 (s, 2H), 2.95-3.10 (m, 4H), 3.15-3.45 (m, 12H), 4.00-4.15 (m, 1H), 6.65 (d, 1H), 6.75 (t, 1H), 6.85 (d, 1H).
1-[2-[4-(5-хлор-2,3-дигидро-3,3-диметил)-7-бензофуранил)-1- пиперазинил] -этил] -3-циклопентил-2-имидазолидинон, оксалат, 2oo, температура плавления 104-7oC. 1H ЯМР (CDCl3) δ, : 1.25 (s, 6H), 1.40-1.75 (m, 8H), 3.00 (t, 2H), 3.05- 3.15 (m, 4H), 2.20-3.35 (m, 8H), 3.40 (t, 2H), 4.00-4.15 (m, 1H), 4.25 (s, 2H), 6.70 (d, 1H), 6.90 (d, 1H).
1-[6-[4-(5-хлор-2,3-дигидро-3,3-диметил)-7-бензофуранил)-1- пипepaзинил] -1-гекcил]-3-циклoпeнтил-2-имидaзoлидинон, оксалат, 2pp, температура плавления 125-27oC. 1H ЯМР (CDCl3) δ, : 1.25 (s, 6H), 1.20-1.75 (m, 16H), 2.95 (t, 2H), 3.00 (t, 2H), 3.10-3.40 (m, 12H), 4.00-4.15 (m, 1H), 4.25 (s, 2H), 6.70 (d, 1H), 6.90 (d, 1H).
1-[3-[4-(7-хлор-2, 3-дигидро-3,3-диметил)-4-бензофуранил)-1- пиперазинил] -1-пропил] -3-циклогексил-2-имидазолидинон, оксалат, 2qq, температура плавления 123-33oC. 1H ЯМР (CDCl3) δ, : 0.95-1.50 (m, 5H), 1.44 (s, 6H), 1.50-1.65 (m, 3H), 1.65-1.90 (m, 4H), 2.85-3.30 (m, 18H), 3.35-3.50 (m, 1H), 6.45 (d, 1H), 7.10 (d, 1H).
ПРИМЕР 3
1-(1,4-бензодиоксан-5-ил)-4-(3-циклогексилтио-1- пропил)-пиперазин S оксид, оксалат, 3a.
Раствор 1-(1,4-бензодиоксан-5-ил)-4-(3-циклогексилтио-1- пропил)пиперазина (7 г) в тетрагидрофуране (70 мл) охладили до 0oC, затем порциям добавляли м-хлорпербензойную кислоту (6.4 г), сохраняя при этом температуру 0oC. После перемешивания в течение 3 часов при указанной температуре добавили водный карбонат натрия (20% раствор, 100 мл). Фазы разделили и водную фазу экстрагировали метиленхлоридом. Cобранные органические фазы концентрировали в вакууме, а полученное в результате масло очищали колоночной хроматографией (элюент: этилацетат/метанол/диэтиламин=88:8:4). Соединение, указанное в заглавии, кристаллизовали как оксалатную соль из смеси ацетон/метанол при добавлении щавелевой кислоты. Выход 1.5 г, температура плавления 113-15oC. 1H ЯМР (δ, ДМСО): 1.00-1.50 (m, 6H), 1.55-2.20 (m, 7H), 2.55-2.95 (m, 4H), 2.95-3.35 (m, 8H), 4.15-4.35 (m, 4H), 6.50 (d, 1H), 6,55 (d, 1H), 6.75 (t, 1H).
ПРИМЕР 4
1-[3-[4(1,4-бензодиоксан-5-ил)-1-пиперазинил] -1- пропил]-3-бензил-2-имидазолидинон, гидрохлорид, 4a.
Раствор 1-[3[4(1,4-бензодиоксан-5-ил)-1-пиперазинил] -1- пропил]-2-имидазолидинона (полученного из 1-(1,4-бензодиоксан-5- ил)-пиперазина и 1-(3-хлор-1-пропил)-2-имидазолидинона согласно методу, описанному в примере 2) (2.5 г) и бензальдегида (2.3 г) в ледяной уксусной кислоте (30 мл) обрабатывали порциями борогидрида натрия (0.6 г), сохраняя при этом температуру 10oC. После перемешивания в течение 40 мин при комнатной температуре добавили дополнительное количество бензальдегида (2.3 г) и борогидрида натрия (0.6 г) и смесь перемешивали при комнатной температуре в течение 16 часов. После удаления в вакууме растворителя получили тяжелое масло, которое очищали колоночной хроматографией (элюент этилацетат/этанол/триэтиламин = 10:1:1). Соединение, указанное в заглавии, было выделено как вязкое масло, которое кристаллизовали в виде гидрохлорида из смеси ацетон/эфир при добавлении эфирного раствора сухого HCl. Выход 2.8 г, температура плавления 181-91oC 1H ЯМР (δ, ДМСО): 1.90-2.10 (m, 2H), 3.00-3.25 (m, 10H), 3.30 (t, 2H), 3.35-3.65 (m, 4H), 4.20 (s, 4H), 4.25 (s, 2H), 6.50 (d, 1H), 6.55 (d, 1H), 6.75 (t, 1H), 7.00 (b, 2H), 7.20-7.40 (m, 5H)
Аналогичным способом были получены:
1-[3-[4-(1,4-бензодиоксан-5-ил)-1-пиперазинил] -1-пропил] -3-этил-2- имидазолидинон, гидрохлорид, 4b, температура плавления 240-43oC. 1H ЯМР (δ, ДМСО): 1.00 (t, 3H), 1.85-2.05 (m, 2H), 2.95-3.35 (m, 14H), 3.35-3.65 (m, 4H), 4.25 (s, 4H), 6.35 (b, 2H), 6.50 (d, 1H), 6.55 (d, 1H), 6.75 (t, 1H).
1-[3-[4(1, 4-бензодиоксан-5-ил)-1-пиперазинил] -1-пропил] -3- циклогексил-2-имидазолидинон, гидрохлорид, 4c, температура плавления 189-200oC. 1H ЯМР (δ, ДМСО): 0.95-1.50 (m, 5H), 1.50-1.65 (m, 3H), 1.65-1,85 (m, 2H), 1.90-2.10 (hep, 2H), 3.00-3.35 (m, 12H), 3.35-3.60 (m, 5H), 4.15-4.30 (m, 4H), 6.45-6.60 (m, 2H), 6.75 (t, 1H).
ПРИМЕР 5
1-[3-[4(1,4-бензодиоксан-5-ил)-1-пиперазинил] -1- этил]-1,3-дигидро-3-(4-фторфенил)-2-имидазолон, гидрохлорид, 5a.
Раствор 1-(1,4-бензодиоксан-5-ил)пиперазина (11 г) и триэтиламина (7 мл) в N-метил-2-пирролидоне по каплям обрабатывали хлорацетонитрилом (4.5 г). После перемешивания в течение 2 часов при 100oC смесь вылили на лед и экстрагировали этилацетатом. Органическую фазу промывали водой, высушили над сульфатом магния, отфильтровали и концентрировали в вакууме. Продукт, 1-(1,4-бензодиоксан-5-ил)-4-цианометилпиперазин, получили в виде масла (17.4 г), которое было в достаточной степени чистым для использования на следующей стадии.
Суспензию литий алюминий хлорида (8.2 г) в сухом эфире (170 мл) обрабатывали, прикапывая раствор алюминий хлорида (8.2 г) в эфире (170 мл) при охлаждении. После перемешивания в течение получаса при комнатной температуре по каплям добавили раствор 1-(1,4- бензодиоксан-5-ил)-4-цианометилпиперазина (9.4 г) в сухом тетрагидрофуране (250 мл) при 15oC. После дефлегмации в течение 1.5 часа смесь охладили и по каплям добавили концентрированный раствор гидроксида натрия (40 мл). После фильтрования и удаления в вакууме растворителя получили масло, которое растворили в метиленхлориде и высушили над сульфатом магния. После удаления в вакууме растворителя получили 1-(2-амино-1-этил)-4-(1,4- бензодиоксан-5-ил)пиперазина (9.1 г) в виде вязкого масла. Смесь 1-(2-амино-1-этил)-4-(1,4-бензодиоксан-5-ил)пиперазина (9.1 г), бромацетальдегида диметилацетата (6.5 г), иодида калия (0.5 г) и карбоната калия (4.8 г) в 1,4-диоксане (200 мл) подвергали дефлегмации в течение 16 часов. Добавили воду, а затем экстрагировали этилацетатом. После концентрирования в вакууме органической фазы, оставшееся масло поместили в колонку с силикагелем (элюент этилацетат/метанол=1:3). Продукт, 1-(1,4- бензодиоксан-5-ил)-4-[2-(2,2-диметокси-1-этиламино)-1- этил] пиперазин, получили в виде масла (4.7 г). Раствор 1-(1,4- бензодиоксан-5-ил)-4-[2-(2,2-диметокси-1-этиламино)- 1-этил]пиперазина (2.3 г) и 4-фторфенилизоцианат (0.9 г) в метиленхлориде (100 мл) подвергали дефлегмации в течение 2 часов. После удаления в вакууме растворителя получили масло, которое очищали на колонке с силикагелем (элюент этилацетат/метанол=3:1). Продукт, 1-(1,4-бензодиоксан-5-ил)-4-[2-N-(2, 2-диметокси-1-этил)-N-(4-фторфениламинокарбонил)-(амино)-1- этил)пиперазин, был получен как твердое вещество (2.5 г).
Раствор 1-(1,4-бензодиоксан-5-ил)-4-[2-N-(2,2-диметокси-1- этил)-N-(4-фторфениламинокарбонил)амино)-1-этил)пиперазина (2.5 г) и 3М соляная кислота (2.5 мл) в этаноле (50 мл) перемешивали в течение 72 часов при комнатной температуре. Соединение, указанное в заглавии, собрали при фильтровании в виде гидрохлорида. Выход 1.2 г, температура плавления 301-5oC. 1H ЯМР (δ, ДМСО): 3.00-3.60 (m, 10H), 4.05 (t, 2H), 4.20-4.35 (m, 4H), 6.55 (t, 2H), 6.75 (t, 1H), 6.80 (d, 1H), 7.00 (d, 1H), 7.25 (t, 2H), 7.65-7.80 (m, 2H).
Аналогичным образом также были получены:
1-[3-[4(1,4-бензодиоксан-5-ил)-1-пиперазинил] -этил] -1,3-дигидро- 3-фенил-2-имидазолон, гидрохлорид 5b, температура плавления 295-300oC. 1H ЯМР (δ, ДМСО): 3.00-3.60 (m, 10H), 4.05 (t, 2H), 4.20-4.30 (m, 4H), 6.50 (t, 2H), 6.70 (t, 1H), 6.80 (d, 1H), 7.00 (d, 1H), 7.20 (t, 1H), 7.45 (t, 2H), 7.70 (d, 2H).
ПРИМЕР 6
1-(2-циклогексилсульфонил-1-этил)-4-(2,3-дигидробензо-фуран-7-ил) пиперазин, малеат, 6a.
Раствор 2-пиклогексилсульфонилэтанола (22 г) и триэтиламина (30 мл) в метиленхлориде (200 мл) обрабатывали, прикапывая раствор метансульфонилхлорида (15 мл) в метиленхлориде (100 мл) при 10oC. После перемешивания в течение 2 часов при комнатной температуре смесь промыли водой, высушили над сульфатом магния и концентрировали в вакууме, получив в результате продукт, циклогексилвинилсульфон, в виде масла (19 г).
Раствор циклогексилвинилсульфона (2.4 г) и 1-(2,3-дигидробензофуран-7-ил-пиперазин (2.5 г) в метиленхлориде (50 мл) перемешивали при комнатной температуре в течение 16 часов. После удаления в вакууме растворителя получили масло, которое поместили в колонку с силикагелем (элюент: этилацетат/метанол/диэтиламин 97: 2:1). Соединение, указанное в заглавии, получили в виде масла, которое кристаллизовали как соль малеата из ацетона при добавлении малеиновой кислоты. Выход 3.4 г, температура плавления 178-79oC. 1H ЯМР (δ, ДМСО): 1.00-1.50 (m, 5H), 1.60-1.70 (m, 1H), 1.75-1.90 (m, 2H), 2.00-2.15 (m, 2H), 3.00-3.35 (m, 13H), 3.45 (t, 2H), 4.50 (t, 2H), 6.10 (s, 2H), 6.55 (d, 1H), 6.75 (t, 1H), 6.85 (d, 1H).
ПРИМЕР 7
1-циклопентил-3-[2-[4-[1-(4-фторфенил)-4-индолил] - 1-пиперазинил]этил] -2-имидазолидинон, оксалат, 7a.
Смесь 2y (1.3 г), 4-фториодобензола (2.0 г), медного порошка (0.2 г) и карбоната калия (0.8 г) в N-метилвинилпирролидоне (20 мл) перемешивали в течение 5 часов при 170oC. После охлаждения реакционную смесь отфильтровали и добавили воду (200 мл) с последующей экстракцией дихлорметаном (2 х 100 мл). После удаления в вакууме растворителя и очистки тонкослойной хроматографией (силикагель, этилацетат/триэтиламин 95:5) получили свободное основание в виде твердого вещества (0.8 г). Указанная в заглавии оксалатная соль кристаллизовалась при добавлении щавелевой кислоты к этанольному раствору этого основания. Выход 0.7 г, температура плавления 210-12oC. 1H ЯМР (δ, ДМСО): 1.40-1.75 (m, 8H), 3.10 (t, 2H), 3.20-3.45 (m, 16H), 4.05-4.15 (m, 1H), 6.65 (d, 1H), 6.70 (dd, 1H), 7.06-7.15 (m, 2H), 7.40 (t, 2H), 7.55-7.65 (m, 3H).
ПРИМЕР 8
4[4-2-(3-циклопентил-2-имидазолидинон-1-ил)этил-1-пиперазинил] - 2-бензофуранилкарбоксамид, гидрохлорид, моногидрат, 8a.
Раствор 2cc (1.0 г) в смеси концентрированного аммония (50 мл) и тетрагидрофурана (25 мл) выдерживали в течение 48 часов при 50oC. После экстракции эфиром (3 х 50 мл), высушивания над сульфатом магния и удаления растворителя в вакууме было получено свободное основание в виде масла. При добавлении эфирного раствора HCl к этанол/гептановому раствору основания была получена указанная в заглавии гидрохлоридная соль. Выход 0.5 г, температура плавления 166-70oC. 1H ЯМР (δ, ДМСО): 1.40-1.75 (m, 8H), 3.20-3.85 (m, 16H), 4.05-4.15 (m, 1H), 6.80 (d, 1H), 7.26 (d, 1H), 7.35 (t, 1H), 7.65 (b, 1H), 7.80 (s, 1H), 8.10 (b, 1H), 11.15 (b, 1H).
ПРИМЕР 9
1-циклопентил-3-[2-[4-(7-индолинил)-1-пиперазинил] этил] -2- имидазолидинон, 9a.
Раствор 2u (1.3 г) в трифторуксусной кислоте при комнатной температуре обрабатывали в течение 3 часов порциями цианоборогидрида натрия (0.6 г). После дополнительного перемешивания в течение 0.5 часа смесь вылили на лед, а затем провели экстракцию этилацетатом (3 х 100 мл). После удаления в вакууме растворителя и очистки хроматографией (силикагель, этилацетат/триэтиламин 96:4) было получено соединение, указанное в заглавии, в виде кристаллического вещества. Выход 0.2 г, температура плавления 130-32oC. 1H ЯМР (δ, CDCl3): 1.40-1.85 (m, 8H), 2.55 (t, 2H), 2.55-2.70 (m, 4H), 2.95-3.05 (m, 4H), 3.05 (t, 2H), 3.20-3.45 (m, 6H), 3.55 (t, 2H), 4.25 (hep, 1H), 6.65-6.75 (m, 1H).
ПРИМЕР 10
1-циклогексил-3-[4-[4-(2,3-дигидро-2,2-диметилбензофуран-7-ил)- 1,2,3,6-тетрагидропирид-1-ил]-1-бутил]-2-имидазолидинон, оксалат, 10a.
Смесь 2,3-дигидро-2,2-диметилбензофурана (25 г) и тетраметилэтилендиамина (46 г) в гептане (250 мл) обрабатывали при комнатной температуре 1.6 М BuLi в гексане (250 мл). После перемешивания в течение 1.5 часа при 30-40oC смесь охладили до -40oC и при этой температуре по каплям добавляли 1-бензил-4- пиперидинон (32 г). В течение 3 часов допускалось нагревание реакционной смеси до комнатной температуры, а затем ее резко охладили водой. После концентрирования в вакууме к реакционной смеси добавили дихлорметан (500 мл) и провели промывку водой (3 х 500 мл). Удалив в вакууме растворитель, получили масло, которое очищали тонкослойной хроматографией (силикагель, гептан/этилацетат/триэтиламин 50: 48: 2). Добавлением гептана к очищенному маслу был получен 7-(1-бензил-4-гидрокси-4-пиперидинил)-2, 3-дигидро-2,2-диметилбензофуран в виде твердого вещества (11 г).
Полученное твердое вещество растворили в трифторуксусной кислоте (150 мл) и подвергали дефлегмации в течение 1 часа. Смесь вылили на лед, а затем подщелочили концентрированной NaOH. Проведя экстракцию дихлорметаном (3 х 100 мл) и удалив в вакууме растворитель, получили масло, которое очищали тонкослойной хроматографией (элюент: этилацетат/гептан/триэтиламин 50:48:2), что привело к 7-(1-бензил-1,2,3,6-тетрагидропирид-4-ил)-2, 3-дигидро-2,2-диметилбензофурану в виде масла (5.0 г).
Продукт растворили в трихлорэтане (15 мл) и добавляли по каплям к этилхлорформиату (20 мл) при температуре перегонки. После дефлегмации в течение 1 часа летучие вещества удалялись в вакууме, при этом был получен сырой 7-(1-этокси-карбонил-1,2,3, 6-тетрагидропирид-4-ил)-2,3-дигидро-2,2-диметилбензофуран в виде масла (4.5 г). Сырой продукт растворили в этаноле (50 мл) и добавили твердый KOH (3 г). После дефлегмации в течение 20 часов смесь вылили в воду, а затем экстрагировали этилацетатом. Органическую фазу высушивали над сульфатом магния, а растворитель удаляли в вакууме, после этого был получен сырой 2,3-дигидро-2,2-диметил-7-(1,2,3,6- тетрагидропирид-4-ил)-бензофуран в виде масла (2.9 г). Сырой продукт был в достаточной степени чистым для его использования на заключительной стадии.
Полученный продукт алкилировали 1-циклогексил-3-(4- хлор-1-бутил)-2-имидазолидиноном (4.5 г) согласно методу, описанному в примере 2, при этом было получено свободное основание соединения, указанного в заглавии, в виде масла (2.7 г). Оксалатную соль кристаллизовали при добавлении щавелевой кислоты к ацетоновому раствору основания. Температура плавления 132-35oC. 1H ЯМР (δ, ДМСО): 0.95-1.80 (m, 14H), 1.40 (s, 6H), 2.65-2.75 (m, 2H), 2.95 (s, 2H), 3.00-3.10 (m, 5H), 3.20-3.25 (m, 4H), 3.25-3.35 (m, 3H), 3.40-3.50 (m, 1H), 6.30 (m, 1H), 6.80 (t, 1H), 7.10 (t, 2H).
ПРИМЕР 11
1-циклогексил-3-[4-[4-(2,3-дигидро-2,2-диметилбензофуран-7-ил)- 1-пиперидинил]-1-бутил]-2-имидазолидинон, оксалат, 11a.
Смесь 10a, оксалата (1.0 г) и 5% Pd/C (0.2 г) в этаноле (20 мл) выдерживали в течение 36 часов при давлении водорода в 4 атм. После фильтрования, удаления в вакууме растворителя и добавления ацетона/эфира было получено соединение, указанное в заглавии, в виде кристаллического твердого вещества. Выход 0.5 г, температура плавления 150-54oC. 1H ЯМР (δ, ДМСО): 0.95-2.05 (t, 18H), 1.40 (s, 6H), 2.80-3.10 (m, 8H), 3.15-3.25 (m, 4H), 3.35-3.50 (m, 3H), 6.75 (t, 1H), 6.90 (d, 1H), 7.05 (d, 1H).
ПРИМЕР 12
1-[2-[4-(1,4-бензодиоксан-5-ил)-1-пиперазинил] этил] -3-(4- фторфенил)-2(3H)-бензимидазолон, 12a.
Смесь 1-(2-гидроксиэтил)бензимидазолона (J. Davoll, J. Chem. Soc., 1960, 308) (9 г), 4-фториодобензола (23 г), карбоната калия 8.0 г), иодида меди (I) (1 г) и окиси цинка (0.5 г) в N-метил-2- (3H)-пирролидоне (100 мл) выдерживали в течение 4.5 часов при 155oC. После охлаждения была добавлена вода (500 мл) при последующей экстракцией этилацетатом (3 х 200 мл). Органическую фазу промыли водой и насыщенным раствором хлорида кальция, а потом высушивали над сульфатом магния. После удаления в вакууме растворителя было получено масло, которое очищали хроматографически (силикагель, этилацетат), получив при этом 1-(4-фторфенил)-3-(2-гидроксиэтил)- 2(3H)-бензимидазолон (2 г) в виде твердого вещества с температурой плавления 124-26oC.
Масло растворили в дихлорметане (60 мл) и добавили тионилхлорид (10 мл) и диметилформамид (0.5 мл), затем проводили в течение 16 часов дефлегмацию. Удаление в вакууме летучих соединений дало 1-(2-хлорэтил)-3-(4-фторфенил)-2(3H)- бензимидазолон (2 г) в виде масла.
Полученный хлорид обрабатывали 1-(1,4-бензодиоксан-5-ил)-пиперазином (2.4 г) согласно методу, описанному в примере 2, получив при этом соединение, указанное в заглавии, в виде кристаллического вещества, выход 1.7 г, температура плавления 161-62oC. 1H ЯМР (δ, CDCl3): 2.55-2.65 (m, 4H), 2.70 (t, 2H), 2.85-2.95 (m 4H), 4.05 (t, 2H), 4.15-4.25 (m, 4H), 6.35-6.50 (m, 2H), 6.750 (t, 1H), 6.95-7.20 (m, 3H), 7.30 (d, 1H), 7.40 (t, 2H), 7.55-7.65 (m, 2H).
ПРИМЕР 13
1-[4-[4-(1,4-бензодиоксан-5-ил)-1-пиперазинил] -1-бутил] -3- (4-фторфенил)-2(3H)-бензимидазолон, 13a.
Раствор 1-(4-фторфенил)-3-(1-пропен-2-ил)-2(3H)-бензимидазолона (полученного арилированием 1-(1-пропен-2-ил)-2(3H)- бензимидазолона (J. Davoll, J. Chem. Soc., 1960, 308) согласно методу, описанному в примере 12), (5 г) в этаноле (100 мл) обработали концентрированной соляной кислотой (50 мл) при комнатной температуре. После перемешивания в течение 1.5 часа была добавлена вода (150 мл). Полученный в результате осадок собирали при фильтровании и высушили. Выход 1-(4- фторфенил)-2(3H)-бензимидазолона составил 4 г, температура плавления 209-10oC.
4 г продукта растворили в тетерагидрофуране (100 мл), а потом в течение 5-10 мин добавили трет-бутоксид калия (3.0 г).
После перемешивания в течение 10 мин добавили 1,4- дибромбутан (15 мл) с последующим нагреванием в течение 5 часов до 50oC. После фильтрования и удаления растворителя оставшееся масло очищали колоночной хроматографией (силикагель, гептан, гептан/этилацетат 1:1). Продукт, 1-(4-бром-1-бутил)-3-(4- фторфенил)-2-имидазолидинон (5.0 г), был получен в виде масла. При обработке масла (2.5г) 1-(1,4-бензодиоксан-5-ил)пиперазином (2.5 г) согласно методу, описанному в примере 2, было получено соединение, указанное в заглавии, в виде кристаллического вещества. Выход 1.9 г, температура плавления 145-47oC. 1H ЯМР (δ, CDCl3): 1.55-1.75 (m, 2H), 1.80-1.95 (m, 2H), 2.45 (t, 2H), 2.55-2.70 (m, 4H), 3.00-3.15 (m, 4H), 4.00 (t, 2H), 4.20-4.40 (m, 4H), 6.45-6.60 (m, 2H), 6.75 (t, 1H), 7.00-7.30 (m, 6H), 7.45-7.55 (m, 2H).
ПРИМЕР 14
1-циклопентил-3-[2-[4-(2-фенилбензофуран-7-ил)-1- пиперазинил]этил]-2-имидазолидинон, оксалат. 14a.
Смесь 211 (1.1 г), 5% Pd/C, ледяной уксусной кислоты (2 мл) и этанола (100 мл) выдерживали в течение 72 часов при давлении водорода 4 атм. После фильтрования и удаления в вакууме растворителя было получено масло, которое растворили в этилацетате (15 мл). При добавлении щавелевой кислоты было получено соединение, указанное в заглавии. Выход 0.5 г, температура плавления 182-83oC. 1H ЯМР (δ, ДМСО): 0.96-1.80 (m, 8H), 2.95-3.15 (m, 4H), 3.15-3.35 (m, 8H), 3.40-3.60 (m, 4H), 6.80 (d, 1H), 7.15 (t, 1H), 7.25 (d, 1H), 7.35-7.45 (m, 2H), 7.50 (t, 2H), 7.95 (d, 2H).
1-циклопентил-3-[2-[4-(2,3-дигидро-3,3- диметил)-7-бензофуранил)-1-пиперазинил] этил] -2-имидазолидинон, оксалат, 14b, температура плавления 94-98oC. 1H ЯМР (δ CDCl3): 1.25 (s, 6H), 1.40-1.75 (m, 8H), 3.00 (t, 2H), 3.05-3.35 (m, 12H), 3.40 (t, 2H), 4.00-4.15 (m, 1H), 4.20 (s, 2H), 6.65-6.75 (m, 1H), 6.75-6.85 (m, 2H).
1-циклопентил-3-[6-[4-(2,3-дигидро-3,3-диметил)-7-бензофуранил)- 1-пиперазинил] -1-гексил] -2-имидазолидинон, оксалат, 14c, температура плавления 128-31oC. 1H ЯМР (δ CDCl3): 1.25 (s, 6H),-1.20-1.75 (m, 16H), 2.95-3.10 (m, 4H), 3.15-3.40 (m, 12H), 3.95-4.10 (m, 1H), 4.20 (s, 2H), 6.65- 6.75 (m, 1H), 6.75-6.90 (m, 2H).
1-циклопентил-3-[3-[4-(2,3-дигидро-2,2-диметил)-4-бензофуpaнил)- 1-пиперазинил] -1-пропил] -2-имидазолидинон, оксалат, 14d, температура плавления 181-83oC. 1H ЯМР (δ CDCl3): 0.95-1.45 (m, 5H), 1.35 (s, 6H), 1.50-1.65 (m, 3H), 1.65-1.90 (m, 4H), 2.80-3.00 (m, 4H), 3.00-3.3 (m, 14H), 3.40-3.55 (m, 1H), 6.35 (d, 1H), 6.40 (d, 1H), 7.00 (t, 1H).
ФАРМАКОЛОГИЯ
Для определения сродства к рецептору 5-HT1A и для определения эффективности соединений относительно этого рецептора соединения формулы I испытывались согласно разработанным и надежным фармакологическим методам. Испытания эти описаны ниже.
Ослабление связывания 3H-8-OH-DPAT с рецепторами 5-HТ1A серотонина в крысином мозгу in vitro
С помощью этого метода определяли ослабление лекарственными средствами связывания 5-HT1A агониста 3H-8-OH-DPAT (1 nM) с рецепторами 5-HT1A в мембранах из мозжечка крысиного мозга in vitro. Следовательно, этот тест является тестом на сродство к рецептору 5-HT1A. Эту оценку можно осуществить так, как это описано Hyttel et al., Drug Dev. Res., 1988, 15, 389-404.
Антагонизм свойств различительного стимула, индуцированного у крыс с помощью 3H-8-OH-DPAT
Этот тест использовали для определения антагонистического воздействия испытуемых соединений in vivo. Родственный метод описан Tricklebank, M.D., et al. , Eur. J. Pharmacol., 1987, 133, 47 -56; Arnt, J. Pharmacology & Toxicology, 1989, 64, 165.
ПРОЦЕДУРА
Мужских особей крыс обучали различать 8-OH-DPAT (0.4 мг/кг, 15 минутная предварительная обработка) и физиологический раствор в рабочих камерах, оборудованных двумя рукоятками для "ответа". Между этими рукоятками была помещена емкость, в которую в качестве вознаграждения помещали воду (0.1 мл). Крыс не допускали к воде в течение по меньшей мере 24 часов, работа велась в фиксированном режиме (FR), конечное значение FR=32.
После введения 8-OH-DPAT ответная реакция усиливается только "лекарственной" рукояткой, в то время как реакция на противоположную рукоятку остается без последствий. После введения физиологического раствора ответная реакция усиливается на рукоятке, противоположной "лекарственной" рукоятке. Испытания с лекарством и физиологическим раствором чередуются случайным образом по дням. Различительная точность выражается в процентах "лекарственного" ответа и вычисляется как число правильных ответов, умноженное на 100 и деленное на сумму правильных и неправильных ответов до получения первого вознаграждения. Время до первого вознаграждения также фиксируют как меру времени реакции. При получении стабильной точности (средняя правильная реакция = 90%; индивидуальная реакция крыс составляла при этом по крайней мере 75% от правильной реакции); испытания проводят в промежутках между днями обучения. Испытуемое соединение вводят в подходящее время перед введением 8-OH-DPAT и испытание начинают спустя 15 минут после введения 8-OH-DPAT. Испытания оканчивают, когда на каждую рукоятку сделано 32 "ответа" или по истечении 20 минут. Крысы не получают вознаграждения и имеют свободный доступ к воде в течение 20-30 минут после испытания. Воздействия оцениваются в процентах ослабления лекарственной реакции. В данные анализа включаются только результаты, полученные от крыс, сделавших по меньшей мере 10 "откликов" на каждую рукоятку. Более того, включаются результаты, в которых по меньшей мере половина крыс давала "ответы".
Процентное ослабление лекарственной реакции для каждой из доз испытуемого соединения использовали для вычисления величины ЭД50 методом логарифмического анализа.
Распространение свойств различительного стимула, вызванных 8- OH-DPAT у крыс
Этот тест использовали для определения in vivo агонистического воздействия рецептора 5-HT1A испытуемых соединений. Родственный метод описан Tricklebank, M.D., supra; Arnt, J. Pharmacology & Toxicology, 1989, 64, 165.
ПРОЦЕДУРА
Процедура была такой же самой, которая описана выше для антагонистического теста, только испытуемое соединение заменили 8- OH-DPAT и введение осуществляли подкожно за 30-45 минут до начала теста.
Лекарственную реакция в процентах, полученную для каждой из доз испытуемого соединения, использовали для вычисления величины ЭД50 методом логарифмического анализа.
Ослабление 5-HТ-синдрома у крыс, вызванного 5-MEO-DMT
Так называемый синдром 5-HТ представляет собой типичную картину поведения, вызванного агонистами 5-HТ, воздействующими на рецепторы 5-HT1 и 5-HT1A (Smith, L. M., и Peroutka, S.J., Pharmacol. Biochem. & Behaviour, 1986, 24, 1513; Tricklebank, M et al., Eur. J. Pharmacol., 1985, 117, 15). Этот тест служит для определения антагонистических воздействий испытуемого соединения на рецепторы 5-HT1A in vivo для определения способности ослаблять 5-HТ синдром, вызванный 5-MeO-DMT.
ПРОЦЕДУРА
Использовались мужские особи крыс весом 170-240 г. Испытуемое соединение вводили подкожно перед подкожным введением 5 мг/кг 5-MeO-DMT. Каждую дозу вводили 4 крысам. Ежедневно в испытания включали контрольную группу, которую обрабатывали предварительно физиологическим раствором. Спустя 10, 15 и 20 минут наблюдали, имеют ли крысы серотонический (5-HТ) синдром. Были зафиксированы следующие симптомы: 1) перебирание передними лапами ("игра на фортепиано"), 2) покачивание головой и 3) отведение задних лап. Более того, отмечалась вялая подвижность. Каждую часть синдрома отмечали следующим образом: ярко выраженное воздействие (2 балла), слабый синдром (1 балл) и никакого воздействия (0 баллов). Добавлялись баллы, полученные в течение 3 периодов наблюдения. Таким образом, для 4 крыс максимально возможным является 24 балла. Влияние испытуемого соединения выражается в процентном ослаблении относительно контрольной группы.
Процентное ослабление синдрома "игры на фортепиано" использовали как "ответ", а ЭД50 вычисляли методом логарифмического анализа.
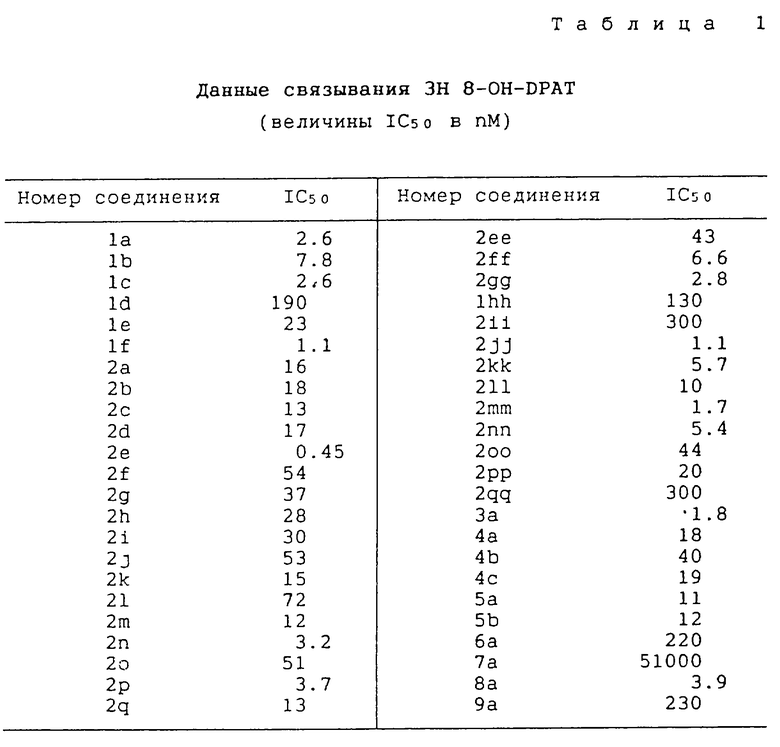
Результаты испытаний приведены в таблицах 1-3.
Из таблицы 1 видно, что большинство соединений настоящего изобретения связаны с рецептором 5-HT1A при сродстве, сравнимом с упоминаемыми соединениями, такими как бушпирон, гепирон и флезиноксан.
Из таблицы 2 видно, что соединения настоящего изобретения включают в себя и агонистов, и антагонистов, согласно определению, используя 8-OH-DPAT как модель раздражителя.
Из таблицы 3 видно, что соединения настоящего изобретения являются антагонистами в тесте ослабления 5-MeO-DMT.
Далее, было испытано сродство соединений настоящего изобретения относительно адреноцепторов α1 и рецептора допамина D2 путем определения их способности ослаблять связывание 3H-празосина с адреноцепторами α1 (Hyttel J et al., J. Neurochem., 1985, 44, 1615; Skarsfeldt, T. et al., Eur. J. Pharmacol. , 1986, 125, 323) и связывания 3H-спироперидола с рецепторами D2 (Hyttel et al., J.Neurochem., 1985, 44, 1615).
Некоторые соединения настоящего изобретения демонстрируют высокую избирательность относительно рецептора 5-HT1A, в то время как остальные соединения настоящего изобретения имеют смешанные профили связи. Соединения определенного класса в пределах настоящего изобретения демонстрируют как высокое сродство к рецепторам 5-HT1A, так и к рецепторам D2. Все указанные типы соединений результативны для лечения различных заболеваний.
Из приведенных выше таблиц 1, 2 и 3 видно, что настоящие соединения обладают высоким сродством к рецептору 5-HT1A. Более того, видно, что среди этих соединений имеются такие, которые действуют как частичные агонисты, и их эффективность колеблется от средней до низкой. В особенности отмечается, что некоторые соединения демонстрируют антагонистические эффекты в тесте 5-MeO- DMT и очень низкую эффективность в тесте 8-OH-DPAT раздражителя. Далее, некоторые соединения демонстрируют как высокое сродство к рецептору 5-HT1A и рецептору допамина D2, так и высокую эффективность в тесте 8-OH-DPAT раздражителя.
ПРИМЕРЫ ПРИГОТОВЛЕНИЯ ЛЕКАРСТВЕННЫХ СРЕДСТВ
Фармацевтические лекарственные составы согласно настоящему изобретению могут быть получены способами, удобными в этой области.
Например, таблетки можно получить, смешивая активный ингредиент с обычными адъювантами и/или разбавителями с последующим прессованием смеси в подходящей таблетировочной машине. Примеры адъювантов или разбавителей включают: кукурузный крахмал, томатный крахмал, тальк, стеарат магния, желатин, лактозу, смолы и т.п. Можно использовать также любые иные адъюванты или добавки, используемые обычно для этих целей, такие как красители, ароматизаторы, консерванты и т.д. при условии, что они совместимы с активными ингредиентами.
Растворы для инъекций могут быть получены растворением активного ингредиента и возможных добавок в части растворителя для инъекций, предпочтительно в стерильной воде, доведением раствора до требуемого объема, стерилизацией раствора и заполнением подходящих ампул или емкостей. Могут быть дополнительно введены любые подходящие добавки, которые удобно использовать в этой области, такие как тонизирующие агенты, консерванты, антиоксиданты и т. д. Типичные примеры рецептов лекарственных средств согласно настоящему изобретению приведены ниже.
1) Таблетки, содержащие 5.0 мг соединения la в расчете на свободное основание:
Соединение 1a - 5.0 мг
Лактоза - 60 мг
Кукурузный крахмал - 30 мг
Гидроксипропилцеллюлоза - 2.4 мг
Микрокристаллическая целлюлоза - 19.2 мг
Кроскармеллоз натрия тип A - 2.4 мг
Стеарат магния - 0.84 мг
2) Таблетки, содержащие 0.5 мг соединения If в расчете на свободное основание:
Соединение 2b - 0.5 мг
Лактоза - 46.9 мг
Кукурузный крахмал - 23.5 мг
Повидон - 1.8 мг
Микрокристаллическая целлюлоза - 14.4 мг
Кроскармеллоз натрия тип А - 1.8 мг
Стеарат магния - 0.63 мг
3) Сироп, содержащий в 1 мл:
Соединение 2bb - 2.5 мг
Сорбитол - 500 мг
Гидроксипропилцеллюлоза - 15 мг
Глицерол - 50 мг
Метил-парабен - 1 мг
Пропил-парабен - 0.1 мг
Этанол - 0.005 мл
Ароматизатор - 0.05 мг
Сахарин - 0.5 мг
Вода - до 1 мл
4) Раствор для инъекций, содержащий в 1 мл:
Соединение 2e - 0.5 мг
Сорбитол - 5.1 мг
Уксусная кислота - 0.08 мг
Вода для инъекций - до 1 мла

Описывается новое конденсированное бензосоединение общей формулы I,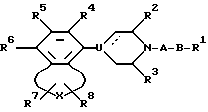
где значения А, В, R1 - R8 указаны в п.1 формулы, обладающее избирательной активностью относительно рецептора 5-НТ1А. Описывается также фармацевтическая композиция на основе вышеуказанного соединения. 2 с. и 5 з.п. ф-лы, 3 табл.
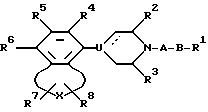
где А - алкилен, содержащий 2 - 6 атомов углерода;
В -полярная бивалентная группа, выбираемая из SO, SO2 или группы формулы II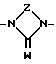
в которой W представляет собой О;
Z выбирают из группы - (CH2)h-, где h = 2 или 3, -CH=CH-, 1,2-фенилен, который может быть замещен галогеном или трифторметилом;
U представляет собой N или CH, пунктирная линия обозначает факультативную связь, и, если она обозначает связь, то U представляет собой С;
X выбирают из бивалентных 3-4 членных групп, содержащих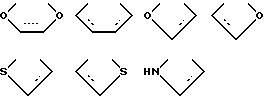
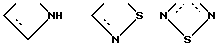
R1 представляет алкил, циклоалкил, фенил, фенил, замещенный галогеном, нафтил, адамантил, циклоалкилалкил, фенилалкил, дифенилалкил, причем любая алкильная группа может быть факультативно замещена одной или двумя гидроксигруппами, при условии, когда Z - 1,2-фенилен и U - это N, R1 выбирают из фенила или фенила, замещенного галогеном;
R2, R3 представляют собой независимо водород или низший алкил;
R4, R5 и R6 независимо выбирают из группы, состоящей из водорода, галогена, низшего алкила, низшего алкокси, гидрокси, низшего алкилтио, низшего алкиламино или низшего диалкиламино, циано, нитро, трифторметила и трифторметилтио;
R7 и R8 независимо выбирают из группы, состоящей из водорода, галогена, трифторметила, низшего алкила, фенила, фенила, замещенного галогеном, группы COOR9 и группы CONR10R11, причем R9, R10 и R11 - водород или низший алкил,
или его фармацевтически приемлемые соли.
| Adv | |||
| Heterocyclic | |||
| Chem., 14, 1972, 99 | |||
| Способ получения производных 1,4-дигидропиридина | 1983 |
|
SU1205762A3 |
| Грохот | 1987 |
|
SU1456253A1 |

