Изобретение относится к новому производному пиразола и его солям, к способу его получения и фармацевтической композиции, на его основе.
В литературе уже описаны многочисленные производные пиразола; более конкретно, в европейской заявкe EP-A-268554 и патенте ФРГ DE-A-3910248 заявлены пиразолы, обладающие гербицидными свойствами, в европейской заявке EP-A-430186 и японской заявке JP-A-3031840 заявлены соединения, используемые в фотографии, а в европейской заявке AP-A-418845 заявлены пиразолы, обладающие противовоспалительной, анальгетической и антитромботической активностями.
Теперь найдено, что N-пиперидино-3-пиразолкарбоксамид обладает очень хорошим сродством к рецептору каннабиноидов и является полезным в терапевтических областях, в которых проявляет свое действие конопля.
Δ9 - THC является основным компонентом активного экстракта Cannabis sativa (Tuner, 1985, ln Marijuana, 84, Ed. Harvey, DY, IRZ, Press, Oxford).
Действие каннабиноидов возникает благодаря взаимодействию со специфическими рецепторами с высоким сродством, имеющимися на центральном и периферическом уровне (Devane et al., Molecular Pharmacology, 1988, 34, 605-613, - центральный, Hyе et al., The Journal of Pharmacology Experimental Therapeutic, 1985, 234, 784-791; Kaminski et al., 1992, Molecilar Pharmacology, 42, 736-842; Munro et al., Nature 1993, 365, 61 - 65).
Изучение характеристик этого рецептора дало возможность разработать синтетические специфические лиганды, такие как агонисты WIN 55212-2 (J. Pharmacol. Exp. Ther., 1993, 264, 1352-1362) или CP 55,940 (J. Pharmacol. Exp. Ther, 1988, 247, 1046-1051).
Один из объектов изобретения относится к N-пиперидино-5-(4-хлорфенил)-1-(2,4-дихлорфенил)-4-метилпиразол-3-карбоксамиду общей формулы
и к его фармацевтически приемлемым солям и их сольватам.
Фармацевтически приемлемые соли соединения формулы I означают соли присоединения кислот, такие как хлоргидрат, бромгидрат, сульфат, кислый сульфат, первичный фосфат, метансульфонат, метилсульфат, малеат, оксалат, фумарат, 2-нафталинсульфонат, глюконат, гликонат, цитрат, изетионат, паратолуолсульфонат.
Другой объект изобретения относится к способу получения соединения I и его солей и их сольватов, отличающемуся тем, что обрабатывают функциональное производное 5-(4-хлорфенил)-1-(2,4-дихлорфенил)-4-метилпиразол-3-карбоновой кислоты формулы
1-аминопиперидином в органическом растворителе и в присутствии основания, и при необходимости превращают полученное таким образом соединение в одну из его солей или в один из его сольватов.
В качестве функционального производного кислоты формулы II можно использовать хлорангидрид кислоты, ангидрид, смешанный ангидрид, C1-C4-алкиловый сложный эфир, в котором алкил является прямым или разветвленным, активный сложный эфир, например, п-нитрофениловый сложный эфир или свободную кислоту, при необходимости, активированную, например N,N-дициклогексилкарбодиимидом или гексафторфосфатом бензотриазол N-оксотрис (диметиламино) фосфония (ВОР).
Так, в способе согласно изобретению можно вводить в реакцию хлорангидрид 5-(4-хлорфенил)-1-(2,4-дихлорфенил)-4-метилпиразол-3-карбоновой кислоты, полученный при взаимодействии тионилхлорида с кислотой формулы II, с 1-аминопиперидином в таком растворителе, как дихлорметан, в инертной атмосфере при температуре, лежащей между 0oC и комнатной температурой, в присутствии такого основания, как триэтиламин.
Вариант способа заключается в приготовлении смешанного ангидрида кислоты формулы II при взаимодействии этилхлорформиата с кислотой формулы II в присутствии такого основания, как триэтиламин, и во взаимодействии его с 1-аминопиперидином в таком растворителе, как дихлорметан, в инертной атмосфере при комнатной температуре в присутствии основания, такого как триэтиламин.
Выделяют полученное таким образом соединение формулы I в виде свободного основания или его соли или сольвата по традиционным методикам.
Соединение формулы I может быть выделено в виде одной из его солей, например, хлоргидрата или оксалата, в этом случае свободное основание может быть приготовлено нейтрализацией указанной соли минеральным или органическим основанием, таким как гидроксид натрия или аммония, триэтиламина или карбонат или бикарбонат щелочного металла, такого как карбонат или бикарбонат натрия или калия, и превращен в другую соль, например, метансульфонат, фумарат или 2-нафталинсульфонат.
При получении соединения I в виде свободного основания солеобразование проводят при обработке выбранной кислотой в органическом растворителе. Путем обработки свободного основания, растворенного, например в эфире, таком как диэтиловый эфир, или в ацетоне, раствором кислоты в том же растворителе получают соответствующую соль, которую выделяют по классическим методикам.
Кислота формулы II, использованная в качестве исходного соединения в способе настоящего изобретения, может быть получена по классическим способам. Некоторые из этих способов детально проиллюстрированы ниже в примерах, названных "приготовление".
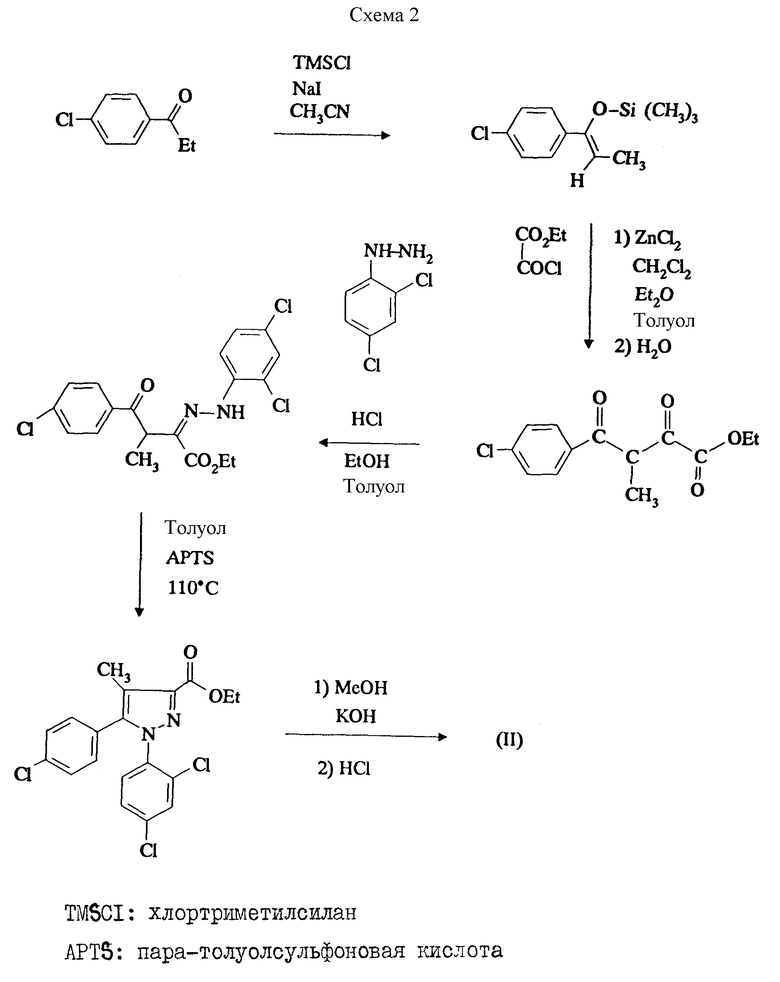
Приготовления 1 и 2 являются близкими. Их осуществляют по реакционной схеме 1 (см. в конце описания), где ZiHMDS: литиевая соль гексаметилдисилазана, APTS: паратолуолсульфоновая кислота.
Первую стадию проводят согласно методике, описанной в J. Heterocyclic. Chem. 1989, 26, 1389.
Приготовление 3 проводят по схеме 2 (см. в конце описания).
Первую стадию проводят по E.S. SCHWEIZER, J. Org. Chem. 1987, 52, 1324 - 1332. Вторую стадию проводят по K.E. TIRPAK et al., J. Org. Chem. 1982, 47, 5099-5102.
Другой реактив, используемый для способа настоящего изобретения, 1-аминопиперидин, является коммерческим продуктом.
Соединение формулы I обладает очень хорошим сродством in vitro к рецепторам центральных каннабиноидов в экспериментальных условиях, описанных Devane et al., Molecular Pharmacology, 1988, 34, 605-613.
Более конкретно, соединение настоящего изобретения как таковое или в форме одной из его фармацевтически приемлемых солей является мощным и селективным антагонистом рецепторов центральных каннабиноидов, имеющих Ki примерно 2 нМ. Оно является в 500-1000 раз более активным на центральном рецепторе, чем на периферическом рецепторе, является активным при оральном введении и проходит гематоэнцефалический барьер.
Хорошее проникновение соединений настоящего изобретения в центральную нервную систему, а также его антагонистическая природа подтверждаются результатами на модели антагонизма, гипотермии, вызванной агонистом рецепторов каннабиноидов. Например, соединение настоящего изобретения антагонизирует гипотермию, вызванную WIN 55212-2 у мышей при DE50 0,3 мг/кг и.п. и 0,4 мг/кг орально; в этом тестe (Pertwee R.G. в Marijnana, 84, Ed. Harvey, D.Y., Oxford IRL Press, 1985, 263 - 277), соединение показало длительность действия 8-10 ч после орального введения дозы 3 мг/кг.
Кроме того, соединение I, введенное одно подкожно, улучшает мнемонические способности крысы в тесте на центральную память (A. Perio et al., Psychopharmacology, 1989, 97, 262 - 268).
Благодаря этим замечательным свойствам, а именно его большому сродству при селективности для центрального рецептора и его способности проникать через гемотоэнцефалитический барьер, соединение I как таковое или в виде фармацевтически приемлемой соли или сольвата может быть использовано в качестве активного начала лекарств, предназначенных для борьбы с болезнями центральной нервной системы у млекопитающих.
Токсичность соединения I совместима с его использованием в качестве психотропного лекарства, например, для лечения расстройств, относящихся к внешнему поведению, состояний тревоги, расстройств настроения, рвоты, мнемонических расстройств, расстройств способности к познаванию, невропатий, мигрени, стресса, болезней психосоматического происхождения, эпилепсии, дискинезий или болезни Паркинсона.
Соединение I согласно изобретению также может быть использовано в качестве лекарства для лечения расстройств аппетита, например, в качестве анорексигена, для лечения шизофрении, состояний бреда, психотических расстройств в общем, а также расстройств, связанных с использованием психотических веществ. Кроме того, соединения I согласно изобретению могут быть использованы в качестве лекарства для противораковой химиотерапии.
Применение соединения согласно изобретению в качестве лекарства для лечения расстройств аппетита, состояний тревоги, расстройств настроения, шизофрении, психотических расстройств, мнемонических расстройств, расстройств способности к познаванию, и дискинезий, а также его применение в противораковой терапии составляют последующий аспект настоящего изобретения.
Соединения согласно изобретению обычно вводят единичными дозами.
Указанные единичные дозы предпочтительно составляют фармацевтические композиции, в которых активное начало смешано с фармацевтическим экципиентом.
Итак, согласно другому из аспектов настоящее изобретение относится к фармацевтическим композициям, содержащим в качестве активного начала соединение формулы I или одну из его фармацевтически приемлемых солей, или один из их сольватов.
Соединение формулы I и его фармацевтически приемлемые соли могут быть использованы в дневных дозах от 0,01 до 100 мг на 1 кг массы тела обрабатываемого млекопитающего, предпочтительно в дневных дозах от 0,1 до 50 мг/кг. Для людей доза может варьировать предпочтительно от 0,5 до 4000 мг в день, более конкретно от 2,5 до 1000 мг в зависимости от возраста больного или типа лечения: профилактического или оздоровительного.
В фармацевтических композициях настоящего изобретения для орального, сублингвального, подкожного, внутримышечного, внутривенного, трансдермического, локального или ректального введения активное начало может быть введено в единичной форме введения, в смеси с классическими фармацевтическими носителями животным и людям. Соответствующие единичных формы введения представляют собой формы для орального введения, такие как таблетки, желатиновые капсулы с лекарством, порошки, гранулы и растворы или суспензии для орального введения, сублингвальные и оральные формы введения, аэрозоли, импланты, формы для подкожного введения, внутримышечного, внутривенного, через нос или через глаза и формы ректального введения.
В фармацевтических композициях настоящего изобретения активное начало обычно формулируют в единичных дозах, содержащих 0,5-1000 мг, целесообразно 1-500 мг, предпочтительно 2-200 мг активного начала на единичную дозу для ежедневного введения.
При приготовлении твердой композиции в форме таблеток можно добавить к активному началу в микронизированной форме смачивающий агент, такой как лаурилсульфат натрия, и перемешать все с фармацевтическим носителем, таким как оксид кремния, крахмал, лактоза, стеарат магния, тальк или аналогичные добавки. Можно покрывать таблетки сахарозой, различными полимерами или другими соответствующими материалами, или же обрабатывать таким образом, что они будут обладать пролонгированной или замедленной активностью и будут высвобождать непрерывно желаемoе количество активного начала.
Композицию в виде желатиновых капсул, получают смешиванием активного начала с разбавителем, таким как гликоль или сложный эфир глицерина, и полученную смесь вводят в мягкие или твердые желатиновые капсулы.
Препарат в вида сиропа или эликсира может содержать активное начало вместе с подслащивателем, предпочтительно некалорийным, с метилпарабеном, пропилпарабеном в качестве антисептика, а также содержать агент, придающий вкус, и подходящий краситель.
Порошки и гранулы, диспергирующиеся в воде, могут содержать активное начало в смеси с диспергаторами, смачивателями или суспендирующими агентами, например поливинилпирролидоном, а также с подслащивателями или корректорами вкуса.
Для ректального введения прибегают к суппозиториям, которые готовят со связующими, плавящимися при ректальной температуре, например маслом какао или полиэтиленгликолями.
Для парентерального введения используют водные суспензии, изотонические солевые растворы или стерильные растворы для инъекций, которые содержат диспергирующие и/или солюбилизирующие агенты, фармакологически приемлемые, например пропиленгликоль или полиэтиленгликоль.
Так, для приготовления водного раствора для внутривенных инъекций можно использовать сорастворитель: спирт, такой как этанол, гликоль, такой как полиэтиленгликоль или пропиленгликоль, и поверхностно-активное гидрофильное вещество, такое как Твин 80R. Для приготовления масляного раствора для внутримышечных инъекций можно солюбилизировать активное начало триглицеридом или сложным эфиром глицерина.
Для трансдермального введения можно использовать полоски в многослойной форме или емкости, в которых активное вещество находится в спиртовом растворе.
Активное начало может выпускаться также в виде микрокапсул или микросфер, возможно с одним или несколькими носителями или вспомогательными добавками.
Активное начало также может быть представлено в форме комплекса с циклодекстрином, например α-, β- или γ- циклодекстрином, 2-оксипропил-β-циклодекстрином или метил-β-циклодекстрином.
Среди форм с пролонгированным выделением, используемых в случае лечения хроников, можно использовать импланты. Они могут быть приготовлены в виде масляной суспензии или в виде суспензии микросфер в изотонической среде.
Следующие примеры иллюстрируют изобретение, не ограничивая его.
Точки плавления или разложения продуктов, F, были измерены в капиллярной трубке на приборе Тоттоли; в некоторых случаях используют дифференциальный калориметрический анализ (DSC) для измерения температуры плавления.
В приготовлении и в примерах используют следующие сокращения:
ТГФ: тетрагидрофуран
Эфир: диэтиловый эфир
Эфир изо: диизопропиловый эфир
EtOH: этанол
AcEOt: этилацетат
MeOH: метанол
ДХМ: дихлорметан
KOH: едкий кали, гидроксид калия
AcOH: уксусная кислота
HCl: соляная кислота
NaCl: хлорид натрия
TA: комнатная температура
DSC: дифференциальный калориметрический анализ
P: точка плавления
Для интерпретации спектров ЯМР используют следующие сокращения:
С: синглет
д: дублет
т: триплет
О: квадруплет
м: мультиплет или массив.
Приготовление 1.
A) Литиевая соль этил-4-(4-хлорфенил)-3-метил-4-оксидо-2-оксобут-3-еноата.
Прибавляют в атмосфере азота 125 мл 1М раствора литиевой соли гексаметилдисилазана в ТГФ к 500 мл эфира. Охлаждают до -78oC и прибавляют по каплям раствор 21 г 4-хлорпропиофенона в 100 мг эфира. После 45 мин перемешивания быстро прибавляют 19,2 мл этилоксалата и оставляют на 16 ч при перемешивании, давая нагреться до комнатной температуры. Фильтруют образовавшийся осадок, промывают эфиром и сушат в вакууме. Получают 12,6 г целевого продукта.
B) Этиловый эфир 5-(4-хлорфенил)-1-(2,4-дихлорфенил)-4-метилпиразол-3-карбоновой кислоты.
К раствору 12,6 г литиевой соли, полученной на предшествующей стадии, в 70 мл EtOH прибавляют 9,8 г 2,4-дихлорфенилгидразина и оставляют на 16 ч при перемешивании при комнатной температуре, фильтруют образовавшийся осадок, промывают EtOH, потом эфиром и сушат в вакууме. Получают 12,6 г гидразона, который растворяют в 100 мл AcOH. Кипятят с обратным холодильником в течение 24 ч, потом выливают реакционную смесь в 500 мл ледяной воды. Экстрагируют AcOEt, промывают водой, насыщенным раствором NaCl, сушат над сульфатом магния и выпаривают в вакууме. Получают целевой продукт после кристаллизации из эфира изо, м = 9,6 г, F = 124oC.
C) 5-(4-хлорфенил)-1-(2,4-дихлорфенил)-4-метилпиразол-3- карбоновая кислота.
К раствору 9,6 г сложного эфира, полученного на предшествующей стадии, в 70 мл MeOH прибавляют раствор 3,3 г KOH в 70 мл воды и нагревают при кипячении с обратным холодильником в течение 3 ч. Выливают реакционную смесь в 200 мл ледяной воды, подкисляют до pH 1, прибавляют 10%-ный раствор HCl, фильтруют образовавшийся осадок, промывают водой и сушат в вакууме. Получают 8,8 г целевой кислоты, F = 211oC.
Приготовление 1 может быть упрощено при использовании условий, описанных в приготовлении 2 ниже.
Приготовление 2.
A) Литиевая соль этил-4-(4-хлорфенил)-3-метил-4-оксидо-2-оксобут-3-еноата.
В атмосфере азота в реакторе растворяют 2008 г литиевой соли гексаметилдисилазана в 10,1 л метилциклогексана. Медленно прибавляют при 20oC ± 5oC раствор 1686 г 4-хлорпропиофенона в 4 л метилциклогексана. После 4,5 ч перемешивания прибавляют при 20oC ± 5oC в течение 35 мин 1607 г этилоксалата. Оставляют на 17 ч при перемешивании при той же температуре. Фильтруют образовавшийся твердый осадок, промывают метилциклогексаном и сушат в вакууме (масса полученного продукта 1931 г).
B) 5-(4-хлорфенил)-1-(2,3-дихлорфенил)-4-метилпиразол-3-карбоновая кислота.
1. В атмосфере азота в реакторе растворяют 1921 г литиевой соли, полученной на предшествующей стадии, в 11 л этанола. Прибавляют сразу при 20oC ± 5oC 1493 г хлоргидрата 2,4-дихлорфенилгидразина. Перемешивают в течение 1 ч, потом прибавляют 2,88 л деионизированной воды и продолжают перемешивание в течение 17 ч при 20oC ± 5oC. Фильтруют образовавшийся осадок, промывают 80%-ным (об/об. ) этанолом и сушат в вакууме (масса полученного гидразона 2280 г), F = 140oC.
2. В атмосфере азота в реакторе растворяют 2267 г гидразона в 11,3 л толуола. Прибавляют 201,6 г паратолуолсульфоновой кислоты и кипятят с обратным холодильником в течение 3 ч. Охлаждают до 20oC ± 5oC и удаляют паратолуолсульфоновую кислоту экстракцией деионизированной водой. К толуольному раствору прибавляют 120,75 г бензилтриэтиламмоний хлорида и 636 г NaOH, растворенных в 1180 мл деионизированной воды. При интенсивном перемешивании нагревают 4 ч при 68oC ± 3oC, потом нейтрализуют гидроксидом натрия и подкисляют реакционную смесь 1500 мл HCl (d = 1,19). Охлаждают до 20oC ± 5oC, фильтруют образовавшийся осадок, промывают толуолом, деионизированнной водой и сушат в вакууме (масса полученного продукта 1585 г), F = 210oC.
Приготовление 3.
A) 1-(4-хлорфенил)-1-триметилсилоксипропен.
В атмосфере азота при 20oC ± 3oC медленно прибавляют 13,47 г хлортриметилсилана к 12,55 г триэтиламина. Продолжают прибавление реагентов, прибавляя 16,86 г 4-хлорпропиофенона (эндотермическая смесь), потом раствор 18,58 г иодида натрия в 125 мл ацетонитрила, поддерживая температуру 18oC ± 2oC. Затем нагревают в течение 3 ч при 40oC ± 5oC. Удаляют ацетонитрил при пониженном давлении и прибавляют к твердому остатку 150 мл толуола. Отгоняют при пониженном давлении 50 мл растворителя, чтобы удалить остаточный ацетонитрил. Экстрагируют минеральные продукты 100 мл ледяной воды, промывают органическую фазу 100 мл ледяной воды и сушат под сульфатом магния. Удаляют толуол при пониженном давлении и заканчивают удаление растворителей при 60oC при пониженном давлении 1 мбар в течение 15 ч (масса полученного масла 22,70 г).
ЯМР при 200 МГц (CDCl3) : 0,13 ппм : с : 9H; 1,7 ппм; д : 3H; 5,28 ппм; к : 1H; 7,21 - 7,39 ппм : м : 4H.
B) Этил-3-(4-хлорбензоил)-3-метилпируват.
В атмосфере азота суспендируют 10 г безводного хлорида цинка в 50 мл толуола. Удаляют остаточную воду азеотропной отгонкой при атмосферном давлении в течение 1 ч. Охлаждают до 20oC ± 3oC, прибавляют 20 мл толуола и 11,5 мл этилового эфира. При 0oC ± 2oC медленно прибавляют 17,0 г этилxлopглиоксилата, разбавленного 20 мл дихлорметана. При этой же температуре прибавляют в течение 1,5 ч 14,5 г продукта, полученного на предшествующей стадии. Дают нагреться до комнатной температуры, потом нагревают до 45oC в течение 4 ч. Промывают органическую фазу бикарбонатом натрия, водой и сушат над сульфатом магния. Удаляют растворители при пониженном давлении (масса полученного масла 17,6 г).
ЯМР при 200 МГц (CDCl3) : 1,25 ппм : 1 : 3H; 1,35 ппм : д : 3H; 4,20 ппм; к : 2H; 4,93 ппм : к : 1H; 7,45 - 7,90 ппм : 4H.
C) Этиловый эфир 5-(4-хлорфенил)-1-(2,4-дихлорфенил)-4-метилпиразол-3-карбоновой кислоты.
К 17,6 г соединения, полученного на предшествующей стадии, прибавляют 13,3 г хлоргидрата 2,4-дихлорфенилгидразина и перемешивают 18 ч при 20oC ± 3oC. Прибавляют без выделения гидразона 0,56 г паратолуолсульфоновой кислоты и отгоняют тройной азеотроп (вода, этанол, толуол). Поддерживают кипение толуола с обратным холодильником в течение часа. Охлаждают реакционную смесь до 20oC ± 3oC, отфильтровывают нерастворимый продукт, потом промывают толуольный раствор 2 раза по 100 мл воды. Удаляют растворители при пониженном давлении (сырое масло используют таким, как оно есть, на следующей стадии).
D) 5-(4-хлорфенил)-1-(2,4-дихлорфенил)-4-метилпиразол-3-карбоновая кислота.
К раствору масла, полученного на предыдущей стадии, в 100 мл MeOH прибавляют 8,1 г KOH в таблетках. После 1 ч при 25oC ± 3oC удаляют растворители при пониженном давлении. Обрабатывают остаток 200 мл воды и 40 мл толуола. Нагревают до 60oC ± 3oC, декантируют и экстрагируют при этой температуре водную фазу 3 раза 40 мл толуола. Прибавляют к водной фазе соляную кислоту до pH 1,5. Фильтруют образовавшиeся белые кристаллы, промывают водой и эфиром изо и сушат в вакууме (масса полученного продукта 9,9 г), F = 210oC.
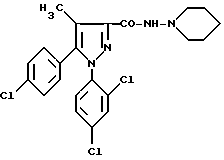
Пример 1.
N-Пиперидино-5-(4-хлорфенил)-1-(2,4-дихлорфенил)-4- метилпиразол-3-карбоксамид.
A) Хлорангидрид 5-(4-хлорфенил)-1-(2,4-дихлорфенил)-4-метилпиразол-3-карбоновой кислоты
К суспензии 8,8 г кислоты, полученной на стадии C приготовления 1, в 90 мл толуола прибавляют 5 мл тионилхлорида и кипятят с обратным холодильником в течение 3 ч. Выпаривают в вакууме досуха реакционную смесь, обрабатывают остаток 90 мл толуола и снова выпаривают в вакууме. Получают 8,0 г целевого хлорангидрида, который используют как таковой на следующей стадии.
B) N-Пиперидино-5-(4-хлорфенил)-1-(2,4-дихлорфенил)-4-метилпиразол-3-карбоксамид.
Охлаждают до 0oC раствор 2,8 мл 1-аминопиперадина, 3,6 мл триэтиламина в 100 мл ДХМ и прибавляют по каплям раствор 8,0 г хлорангидрида кислоты, полученного в предшествующей стадии, в 80 мл ДХМ. Перемешивают 3 ч и дают нагреться до комнатной температуры, потом выливают в 200 мл ледяной воды. Экстрагируют ДХМ, промывают водой, насыщенным раствором NaCl, сушат над сульфатом магния и выпаривают в вакууме. Хроматографируют на оксиде кремния, элюируя смесью AcOEt (толуол /10/90/, об/об.). Получают целевой продукт после кристаллизации из эфира изо, м = 5,9 г, F = 148oC.
Можно также получить соединение примера 1, используя рабочие условия, более приемлемые для промышленного варианта.
В атмосфере азота и суспензии 1568,6 г кислоты, полученной на стадии B приготовления 2, в 14,1 л метилциклогексана прибавляют после нагревания до 83oC ± 3oC раствор 554,5 г тионилхлорида в 1,57 л метилциклогексана. Перемешивают 3 ч при 83oC ± 3oC, потом повышают температуру реакционной смеси в течение 2 ч до температуры кипения метилциклогексана, удаляя все время избыток тионилхлорида отгонкой. Охлаждают до 10oC ± 3oC и медленно прибавляют раствор 452,9 г 1-аминопиперидина и 457,5 г триэтиламина в 3,14 л ТГФ. Перемешивают 17 ч, давая возможность нагреться до температуры 20oC ± 5oC, потом последовательно промывают органическую фазу при 20oC ± 5oC деионизированной водой и 4%-ной уксусной кислотой в воде, заканчивают промывки органической фазы при 70oC ± 3oC 1,5%-ным раствором NaOH, потом деионизированной водой и удаляют ТГФ и воду азеотропной перегонкой при атмосферном давлении. Дают остыть до 20oC ± 5oC. Кристаллизуют целевой продукт, фильтруют образовавшийся осадок, промывают метилциклогексаном и сушат в вакууме (масса полученного продукта 1627 г).
DSC : эндотермический пик, центральный при 155,5oC.
Пример 2.
N-пиперидино-5-(4-хлорфенил)-1-(2,4-дихлорфенил)-4-метилпиразол-3-карбоксамид (сольват с этанолом).
Суспендируют 10 г соединения, полученного в примере 1, в 60 мл абсолютного этанола и кипятят с обратным холодильником до полного растворения. Дают охладиться до 20oC ± 3oC и продолжают перемешивание 2 ч. Отфильтровывают образовавшиеся белые кристаллы, промывают этанолом и сушат в вакууме (масса полученного продукта 9,60 г).
DSC : эндотермический пик, центрованный при 102,7oC.
Вычислено, %: C 56,5; H 5,29; N 10,98
Найдено, %: C 56,43; H 5,41; N 11,05
Пример 3.
Хлоргидрат N-пиперидино-5-(4-хлорфенил)-1-(2,4-дихлорфенил)-4-метилпиразол-3-карбоксамида.
К раствору 5,9 г соединения, полученного в примере 1, в 50 мл эфира прибавляют по каплям насыщенный раствор газообразной HCl в эфире до pH 1. Отфильтровывают образовавшийся осадок, промывают эфиром и сушат в вакууме. Получают 6,0 г целевого хлоргидрата, F = 224oC (разложение).
Пример 4.
Хлоргидрат N-пиперидино-5-(4-хлорфенил)-1-/2,4-дихлорфенил)-4-метилпиразол-3-карбоксамид (сольват с этанолом).
Суспендируют 40 г соединения, полученного в примере 3, в 400 мл абсолютного этанола. Нагревают до кипения до полного растворения и перемешивают 20 ч при постепенном охлаждении до 20oC ± 3oC. Отфильтровывают образовавшиеся белые кристаллы, промывают этанолом, сушат в вакууме (масса полученного продукта 29,6 г).
DSC : очень растянутый эндогермический массив (175-230oC). Термогравиметрия : потеря массы (примерно 8,2%), начиная со 100oC.
Вычислено, %: C 53,03; H 5,16; N 10,31
Найдено, %: C 52,68; H 5,23; N 10,34
Пример 5.
Метансульфонат N-пиперидино-5-(4-дихлорфенил)-4-метилпиразол-3-карбоксамида (гемисольват с ацетоном).
К раствору 18,55 г соединения, полученного в примере 1, в 185 мл ацетона прибавляют при 20oC ± 3oC 3,84 г метансульфоновой кислоты и перемешивают 20 ч при этой же температуре. Отфильтровывают образовавшиеся белые кристаллы, промывают ацетоном и сушат в вакууме (масса полученного продукта 21,65 г).
DSC : плавление, рекристаллизация к 175oC, потом плавление при 191,5oC.
Термогравиметрия : потеря массы (5,2% примерно), начиная с 90oC.
Вычислено, %: C 49,90; H 4,75; N 9,5
Найдено, %: C 49,70; H 4,76; N 9,44
Пример 6.
Гемифумарат N-пиперидино-5-(4-хлорфенил)-1-(2,4-дихлорфенил)-4-метилпиразол-3-карбоксамида.
К раствору 0,30 г соединения, полученного в примере 1, в 3 мл ацетона прибавляют по каплям раствор 0,038 г фумаровой кислоты в 6 мл ацетона. Отфильтровывают образовавшиеся белые кристаллы при охлаждении до 0oC, промывают ацетоном и сушат в вакууме. Получают 0,23 г целевой соли, F = 168oC.
Пример 7.
Кислый сульфат N-пиперидино-5-(4-хлорфенил)-1-(2,4-дихлорфенил)-4-метилпиразол-3-карбоксамида
К раствору 0,30 г соединения, полученного в примере 1, в 3 мл ацетона прибавляют 0,018 мл концентрированной серной кислоты, Отфильтровывают образовавшиеся белые кристаллы, промывают ацетоном и эфиром и сушат в вакууме. Получают 0,31 г целевой соли, F = 240oC.
Пример 8.
Паратолуолсульфонат N-пиперидино-5-(4-хлорфенил)-1-(2,4-дихлорфенил)-4-метилпиразол-3-карбоксамида.
К раствору 18,55 г соединения, полученного в примере 1, в 185 мл ацетона прибавляют при 20oC ± 3oC 7,61 г паратолуолсульфоновой кислоты и перемешивают 20 ч при этой температуре. Отфильтровывают образовавшиеся белые кристаллы, промывают ацетоном и сушат в вакууме (масса полученного продукта 24,25 г).
DSC : эндотермический пик, центральный при 236,8oC.
Вычислено, %: C 54,76; H 4,60; N 8,72
Найдено, %: C 54,11; H 4,71; N 8,69
Пример 9.
Дигидрофосфат N-пиперидино-5-(4-хлорфенил)-1-(2,4-дихлорфенил)-4-метилпиразол-3-карбоксамида.
К раствору 18,55 г соединения, полученного в примере 1, в 185 мл метилэтилкетона прибавляют при 20oC ± 3oC 4,61 г 85%-ной фосфорной кислоты. Удаляют воду отгонкой при атмосферном давлении азеотропа метилэтилкетон-вода. Постепенно охлаждают до 20oC ± 3oC, продолжая перемешивание в течение 20 ч. Отфильтровывают образовавшиеся белые кристаллы, промывают метилэтилкетоном и сушат в вакууме (масса полученного продукта 21,0 г).
DSC : эндотермический пик, центрованный при 185,5oC.
Вычислено, %: C 47,04; H 4,31; N 9,97
Найдено, %: C 46,96; H 4,62; N 9,98
Заявленные соединения являются сильными и селективными антагонистами рецепторов центральных каннабиноидов (CB).
Испытания на сродство к указанным рецепторам было осуществлено согласно методике, описанной Devane и сотр. в журнале Molecular Pharmacology, 1988, 34, 605-613.
Так, соединение по примеру 1 показало активность, выраженную показателем Ki, равным около 2 нМ. Кроме того, соединения настоящего изобретения проходят гематоэнцефалический барьер, тест на такое испытание приведен ниже.
Обычно соединение проходит гематоэнцефалический барьер, если оно вызывает смещение специфического связывания значительно меньшее 100%. В случае соединений согласно изобретению это смещение составляет ниже 10%, что означает, что они очень легко переходят гематоэнцефалический барьер.
Острая токсичность заявленных соединений на мышах превышает значение 500 мг/кг.
Протокол испытаний на связывание ex vivo.
Для проведения испытаний на связывание EX VIVO соединение примера 1 растворяют в двух каплях Tween 80, разводят в дистиллированной воде и вводят орально в объеме 20 мл/кг самцам мышей перед тем, как их забивают путем обезглавливания. Мозг (без мозжечка) удаляют и гомогенизируют в 20 мл буфера A. Затем выполняют количественные определения связывания in vivo как описано ниже.
Для количественных оценок связывания in vivo аликвотные части по 0,8 мл гомогенатов мозга инкубируют при 30oC с 0,2 нМ [3H]-CP 55,940 в 1 мл буфера A (Трис-HCl 50 нМ, pH 7,7) в течение 1 ч. Для сбора и промывания мембран (трижды 5 мл холодного буфера A, содержащего 0,25% BSA) используют методику быстрой фильтрации с использованием фильтров с использованием фильтров Whatman GF/C (предварительно обработанных 0,5%-ным полиэтиленимином (мас./об.) и 48-ячеечного фильтрационного устройства. Радиоактивность, связанную с фильтрами, рассчитывают с помощью 4 мл жидкого сцинтиллирующего биофтора. Неспецифическое связывание определяют в присутствии 1 мкМ CP 55,940. Данные выражаются в виде процентной доли смещения специфического связывания в тканях необработанных мышей (100%).
Соединение примера 1 показало значение смещения 9,3%. Испытания с соединениями других примеров дают значения аналогичного порядка.
Далее заявитель направляет ряд примеров на типы галеновых формулировок:
Желатиновая капсула, мг:
Соединение примера 1 - 3,00
Кукурузный крахмал - 51,00
Моногидрат лактозы - 101,33
Поливидон - 4,3
Лаурилсульфат натрия - 0,17
Сшитая натрийкарбоксиметилцеллюлоза - 8,50
Очищенная вода - Достаточное количество
Стеарат магния - 1,70 - 170,00
Желатиновая капсула, мг:
Соединение примера 1 - 10,00
Кукурузный крахмал - 51,00
Моногидрат лактозы - 94,33
Поливидон - 4,30
Лаурилсульфат натрия - 0,17
Сшитая натрийкарбоксиметилцеллюлоза - 8,50
Очищенная вода - Достаточное количество
Стеарат магния - 1,70 - 170,00
Желатиновая капсула, мг:
Соединение примера 4 - 30,00
Кукурузный крахмал - 51,00
Моногидрат лактозы - 74,33
Поливидон - 4,30
Лаурилсульфат натрия - 0,17
Сшитая натрийкарбоксиметилцеллюлоза - 8,50
Очищенная вода - Достаточное количество
Стеарат магния - 1,70 - 170,00р

N-Пиперидино-5-(4-хлорфенил)-1-(2,4-дихлорфенил)-4-метилпиразол-3-карбоксамид, обладающий антагонистической активностью к рецепторам центральных каннабиноидов, получают обработкой соединения формулы I 1-аминопиперидином в органическом растворителе в присутствии основания. 3 с. и 7 з.п. ф-лы.
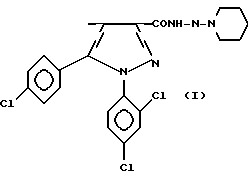
или его соли присоединения фармацевтически приемлемых кислот, или их сольваты.
1-аминопиперидином в органическом растворителе и в присутствии основания и возможно превращают полученное таким образом соединение в одну из его солей или в один из его солей или в один из их сольватов.
Приоритет по пунктам
02.12.93 по пп.1 - 10.
| Способ получения 6-(ациламиноарил)-4,5-дигидро-3(2Н)-пиридазинопроизводных или их фармацевтически приемлемых солей | 1984 |
|
SU1648250A3 |
| EP 0483851 A1, 1992 | |||
| Трамбовка для уплотнения грунта | 1973 |
|
SU480794A1 |
| Теплоизоляционный клееный арболит | 2016 |
|
RU2642757C1 |

