ОБЛАСТЬ ТЕХНИКИ
Изобретение относится к 2,6-замещенным-4-монозамещенным соединениям аминопиримидина, их получению, содержащим эти соединения фармацевтическим композициям и их фармацевтическому применению в лечении патологических состояний, которые можно контролировать ингибированием рецептора простагландина D2.
УРОВЕНЬ ТЕХНИКИ
Показано, что локальная стимуляция аллергеном пациентов с аллергическим ринитом, бронхиальной астмой, аллергическим конъюнктивитом и атопическим дерматитом приводит к быстрому росту уровня простагландина D2 «(PGD2)» в назальной и бронхиальной смывной жидкости, слезах и жидкости, собранной из кожного пузыря по методу «кожной камеры». PGD2 может оказывать ряд воспалительных воздействий, например, повышать проницаемость сосудов в конъюнктиве и коже, повышать сопротивление дыхательных путей носа, вызывать сужение дыхательных путей и проникновение эозинофилов в конъюнктиву и трахею.
PGD2 - основной циклооксигеназный продукт арахидоновой кислоты, продуцируемый мастоцитами при иммунологическом стимулировании [Lewis, RA, Soter NA, Diamond PT, Austen KF, Oates JA, Roberts LJ II, prostaglandin D2 generation after activation of rat and human mast cells with anti-IgE, J. Immunol. 129, 1627-1631, 1982]. Активированные мастоциты, один из главных источников PGD2, играют одну из ключевых ролей в возникновении аллергической реакции при таких состояниях, как астма, аллергический ринит, аллергический конъюнктивит, аллергический дерматит и другие заболевания [Brightling CE, Bradding P, Pavord ID, Wardlaw AJ, New Insights into the role of the mast cell in asthma, Clin Exp Allergy 33, 550-556, 2003].
Многие воздействия PGD2 опосредуются его действием на рецептор простагландина D («DP») - рецептор, связанный с G-белком, экспрессируемый на эпителии и в гладких мышцах.
При астме респираторный эпителий уже давно считается главным источником воспалительных цитокинов и хемокинов, которые определяют развитие заболевания [Holgate S, Lackie P, Wilson S, Roche W, Davies D, Bronchial Epithelium as a Key Regulator of Airway Allergen Sensitzation and Remodeling in Asthma, Am J Respir Crit Care Med. 162, 113-117, 2000]. В экспериментальной модели астмы мышей при стимулировании антигеном происходит резкая активация рецептора DP на эпителии дыхательных путей [Matsuoka T, Hirata M, Tanaka H, Takahashi Y, Murata T, Kabashima K, Sugimoto Y, Kobayashi T, Ushikubi F, Aze Y, Eguchi N, Urade Y, Yoshida N, Kimura K, Mizoguchi A, Honda Y, Nagai H, Narumiya S, prostaglandin D2 as a mediator of allergic asthma, Science 287, 2013-2017, 2000]. У нокаутных мышей с отсутствующим рецептором DP заметно снижается гиперактивность дыхательных путей и хроническое воспаление [Matsuoka T, Hirata M, Tanaka H, Takahashi Y, Murata T, Kabashima K, Sugimoto Y, Kobayashi T, Ushikubi F, Aze Y, Eguchi N, Urade Y, Yoshida N, Kimura K, Mizoguchi A, Honda Y, Nagai H, Narumiya S, Prostaglandin D2 as a mediator of allergic asthma, Science 287, 2013-2017, 2000] - два важнейших признака астмы человека.
Считается также, что рецептор DP задействован в аллергическом рините человека, распространенном аллергическом заболевании, характеризующемся такими симптомами, как чихание, зуд, ринорея и заложенность носа. Местное применение PGD2 в носовой полости вызывает зависимое от дозы увеличение заложенности носа [Doyle WJ, Boehm S, Skoner DP, Physiologic responses to intranasal dose-response challenges with histamine, methacholine, bradykinin, and prostaglandin in adult volunteers with and without nasal allergy, J Allergy Clin Immunol. 86(6 Pt 1), 924-35, 1990].
Было показано, что антагонисты рецептора DP снижают воспаление дыхательных путей в экспериментальной модели астмы морских свинок [Arimura A, Yasui K, Kishino J, Asanuma F, Hasegawa H, Kakudo S, Ohtani M, Arita H (2001), Prevention of allergic inflammation by a novel prostaglandin receptor antagonist, S-5751, J. Pharmacol. Exp. Ther. 298(2), 411-9, 2001]. Таким образом, PGD2, по-видимому, действует на рецептор DP и играет важную роль в выявлении некоторых основных особенностей аллергической астмы.
Было показано, что антагонисты DP могут эффективно ослаблять симптомы аллергического ринита у многих видов, в частности было показано, что они могут подавлять индуцированную антигенами заложенность носа, наиболее явный симптом аллергического ринита [Jones, T.R., Savoie, C., Robichaud, A., Sturino, C., Scheigetz, J., Lachance, N., Roy, B., Boyd, M., Abraham, W., Studies with a DP receptor antagonist in sheep and guinea pig models of allergic rhinitis, Am. J. Resp. Crit. Care Med. 167, A218, 2003; and Arimura A., Yasui K., Kishino J., Asanuma F., Hasegawa H., Kakudo S., Ohtani M., Arita H., Prevention of allergic inflammation by a novel prostaglandin receptor antagonist, S-5751. J. Pharmacol. Exp. Ther. 298(2), 411-9, 2001].
Антагонисты DP также эффективны в экспериментальных моделях аллергического конъюнктивита и аллергического дерматита [Arimura A., Yasui K., Kishino J., Asanuma F., Hasegawa H., Kakudo S., Ohtani M., Arita H., Prevention of allergic inflammation by a novel prostaglandin receptor antagonist, S-5751, J. Pharmacol. Exp. Ther. 298(2), 411-9, 2001; and Torisu K., Kobayashi K., Iwahashi M., Nakai Y., Onoda T., Nagase T., Sugimoto I., Okada Y., Matsumoto R., Nanbu F., Ohuchida S., Nakai H., Toda M., Discovery of a new class of potent, selective, and orally active prostaglandin D2 receptor antagonists, Bioorg. & Med. Chem. 12, 5361-5378, 2004].
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение относится к соединению формулы (I):
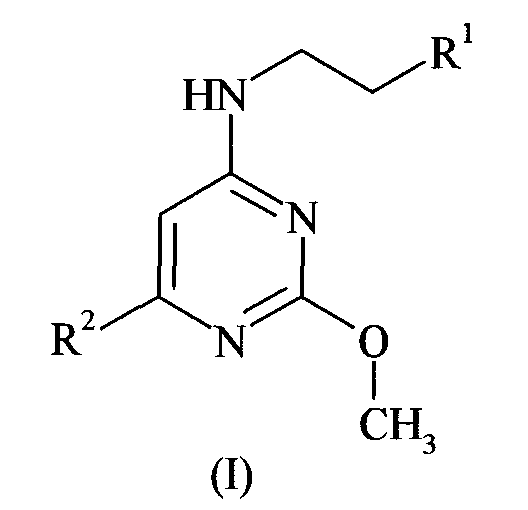
где R1 представляет собой 2,4-дихлорфенил или 4-трифторметоксифенил, и
когда R1 представляет собой 2,4-дихлорфенил, тогда R2 представляет собой 3-карбоксипирролидинил, 3,5-ди-(1-гидрокси-1-метилэтил)фенил, 3-аминопиперидин-1-ил, 4-аминопиперидин-1-ил, 4-ацетамидпиперидин-1-ил, 1-метил-2-карбокси-2,3-дигидро-1H-индол-5-ил, 3-(1-трет-бутилсульфониламинокарбонил-1-метилэтил)фенил, 3-(1-диметиламиносульфониламинокарбонил-1-метилэтил)фенил, 3-(1-тиоморфолин-4-илкарбонил-1-метилэтил)фенил, 3-(1-аминокарбонил-1-метилэтил)фенил, 3-(1-диметиламинокарбонил-1-метилэтил)фенил, 3-карбоксиметилпиперидин-1-ил, 3-метилсульфониламинокарбонилпиперидин-1-ил, 3-этилсульфониламинокарбонилпиперидин-1-ил, 3-трет-бутилсульфониламинокарбонилпиперидин-1-ил, 3-трифторметилсульфониламинокарбонилпиперидин-1-ил, 3-[(1H-тетразол-5-ил)аминокарбонил]пиперидин-1-ил, 3-аминокарбонилпиперидин-1-ил, 3-диметиламинокарбонилпиперидин-1-ил, 3-диметиламиносульфониламинокарбонилпиперидин-1-ил, или 2-карбокси-2,3-дигидробензофуран-5-ил, и
когда R1 представляет собой 4-трифторметоксифенил, тогда R2 представляет собой 3-(1-метил-1-карбоксиэтил)пиперидинил, 3-карбоксипепиридинил, 3-метилсульфониламинокарбонилпиперидин-1-ил, 5-карбокситиофен-2-ил,
или относится к его фармацевтически приемлемой соли, гидрату или сольвату, его фармацевтически приемлемому пролекарству или фармацевтически приемлемой соли, гидрату или сольвату пролекарства.
Другим аспектом настоящего изобретения является фармацевтическая композиция, включающая фармацевтически эффективное количество одного или нескольких соединений настоящего изобретения, или его фармацевтически приемлемой соли, гидрата или сольвата, его фармацевтически приемлемого пролекарства или фармацевтически приемлемой соли, гидрата или сольвата пролекарства в смеси с фармацевтически приемлемым носителем.
Еще один аспект настоящего изобретения представляет собой способ лечения пациента, страдающего заболеванием, опосредованным PGD2, в том числе, не ограничиваясь указанным, аллергическим заболеванием (например, аллергический ринит, аллергический конъюнктивит, атопический дерматит, бронхиальная астма и пищевая аллергия), системным мастоцитозом, нарушениями, сопровождающимися системной активацией мастоцитов, анафилактическим шоком, бронхоконстрикцией, бронхитом, крапивницей, экземой, заболеваниями, сопровождающимися зудом (например, атопический дерматит и крапивница), заболеваниями (например, катаракта, отслоение сетчатки, воспаление, инфекция и нарушения сна), которые возникают в качестве вторичных заболеваний в результате поведения, сопровождающегося зудом (например, расчесывания и растирания), воспалением, хроническим обструктивным заболеванием легких, ишемическим реперфузионным повреждением, расстройством мозгового кровообращения, хроническим ревматоидным артритом, плевритом, неспецифическим язвенным колитом и подобными заболеваниями, включающий введение указанному пациенту фармацевтически эффективного количества соединения настоящего изобретения или его фармацевтически приемлемой соли, гидрата или сольвата, его фармацевтически приемлемого пролекарства или фармацевтически приемлемой соли, гидрата или сольвата пролекарства.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определение терминов
Используемые выше и во всем тексте описания изобретения следующие термины, если не указано иначе, имеют следующие принятые значения:
«Соединения, являющиеся объектом настоящего изобретения» и аналогичные выражения включают описанные в настоящем документе соединения формулы (I), а также фармацевтически приемлемые соли, сольваты, например, гидраты, пролекарства и фармацевтически приемлемые соли, сольваты и гидраты пролекарств, где это допускается контекстом. Точно так же ссылка на промежуточные соединения, независимо от того, включены ли они сами в формулу изобретения или нет, призвана включить их соли и сольваты в случаях, где это допускается контекстом.
Термин «пациент» означает человека и других млекопитающих.
Термин «фармацевтически приемлемые соли» означает нетоксичные, неорганические и органические кислотно-аддитивные и основно-аддитивные соли соединений, являющихся объектом настоящего изобретения. Эти соли могут быть получены in situ на конечной стадии выделения и очистки соединений.
Термин «фармацевтически эффективное количество» означает количество соединения или соединений, которое согласно настоящему изобретению эффективно для создания желательного терапевтического эффекта, описанного в настоящем документе, например, снятия аллергической реакции или воспаления.
Используемый в настоящем документе термин «фармацевтически приемлемые пролекарства» означает пролекарства соединений, являющихся объектом настоящего изобретения, которые в рамках здравого медицинского суждения являются подходящими для использования в контакте с тканями пациента, при этом нежелательная токсичность, раздражение или аллергическая реакция находятся в пределах разумного соотношения пользы и риска, а также являются эффективными для применения по назначению соединений, являющихся объектом данного изобретения. Термин «пролекарство» означает соединения, которые «in vivo» трансформируются с образованием исходного соединения настоящего изобретения, например, посредством гидролиза в крови. Функциональные группы, которые могут подвергаться быстрым превращениям посредством метаболического распада in vivo, образуют класс групп, способных вступать в реакцию с карбоксильной группой соединений, являющихся объектом данного изобретения. К ним относятся, в частности, такие группы, как алканоил (например, ацетил, пропаноил, бутаноил и им подобные), незамещенный или замещенный ароил (например, бензоил или замещенный бензоил), алкоксикарбонил (например, этоксикарбонил), триалкилсилил (например, триметил- или триэтилсилил) и моноэфиры, образуемые дикарбоновыми кислотами (например, сукцинил). Вследствие легкости, с которой подверженные метаболическому распаду группы соединений, являющихся объектом данного изобретения, расщепляются in vivo, соединения с такими группами действуют как пролекарства. Соединения с подверженными метаболическому распаду группами обладают тем преимуществом, что они могут проявлять повышенную биодоступность в результате более высокой растворимости и/или скорости всасывания исходного соединения благодаря наличию подверженной метаболическому распаду группы. Подробное обсуждение приводится в работах Design of Prodrugs, H. Bundgaard, ed., Elsevier (1985); Methods in Enzymology; K. Widder et al., Ed., Academic Press, 42, 309-396 (1985); A Textbook of Drug Design and Development, Krogsgaard-Larsen and H. Bandaged, ed., Chapter 5; "Design and Applications of Prodrugs" 113-191 (1991); Advanced Drug Delivery Reviews, H. Bundgard, 8, 1-38, (1992); J. Pharm. Sci., 77, 285 (1988); Chem. Pharm. Bull., N. Nakeya et al., 32, 692 (1984); Pro-drugs as Novel Delivery Systems, T. Higuchi and V. Stella, 14 A.C.S. Symposium Series, and Bioreversible Carriers in Drug Design, E.B. Roche, ed., American Pharmaceutical Association and Pergamon Press, 1987, которые в силу ссылки на них включаются в настоящий документ.
«Сложноэфирное пролекарство» означает соединение, которое может превращаться «in vivo» в ходе метаболических процессов (например, посредством гидролиза) в соединение настоящего изобретения. Например, сложный эфир соединения настоящего изобретения, содержащего гидроксигруппу, может превращаться посредством гидролиза «in vivo» в исходную молекулу. В качестве альтернативы, сложный эфир соединения настоящего изобретения, содержащего карбоксигруппу, может превращаться посредством гидролиза «in vivo» в исходную молекулу. Примерами эфирных пролекарств являются:

этиловый эфир (1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-ил)уксусной кислоты; и
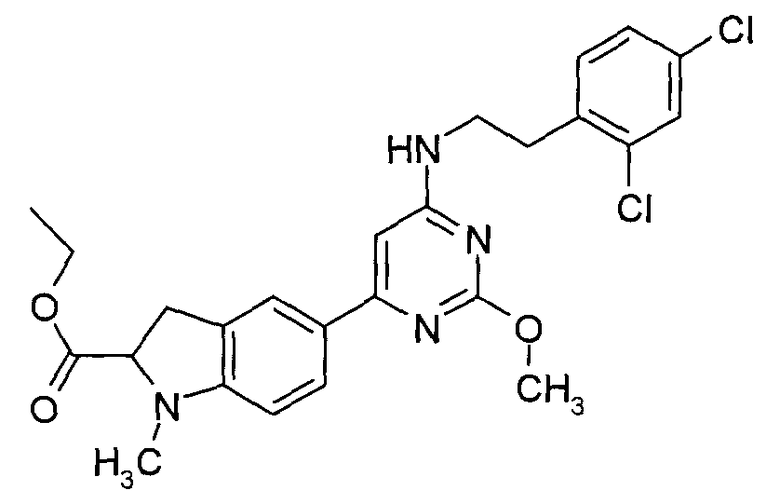
этиловый эфир 5-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}-1-метил-2,3-дигидро-1H-индол-2-карбоновой кислоты.
Подходящими сложными эфирами соединений настоящего изобретения, содержащих гидроксильную группу, являются, например, ацетаты, цитраты, лактаты, тартраты, малонаты, оксалаты, салицилаты, пропионаты, сукцинаты, фумараты, малеаты, метилен-бис-b-гидроксинафтоаты, гентизаты, изетионаты, ди-пара-толуолтартраты, метансульфонаты, этансульфонаты, бензолсульфонаты, пара-толуолсульфонаты, циклогексилсульфаматы и хинаты.
Подходящими сложными эфирами соединений настоящего изобретения, содержащих карбоксильную группу, являются, например, эфиры, описанные в F.J. Leinweber, Drug Metab. Res., 1987, 18, page 379.
Особенно полезный класс сложных эфиров соединений настоящего изобретения, содержащих гидроксильную группу, может быть образован из кислотных групп из числа описанных в работе Bundgaard et al., J. Med. Chem., 1989, 32, page 2503-2507, и включать в себя замещенные (аминометил)бензоаты, например, диалкиламинометилбензоаты, в которых две алкильные группы могут быть соединены и/или разделены атомом кислорода либо атомом азота с возможным заместителем, например, алкилированным атомом азота, а особенно (морфолинометил)бензоаты, например, 3- или 4-(морфолинометил)бензоаты, и (4-алкилпиперазин-1-ил)бензоаты, например, 3- или 4-(4-алкилпиперазин-1-ил)бензоаты.
«Сольват» означает соединение, являющееся объектом данного изобретения, физически связанное с одной или несколькими молекулами растворителя. К физическому связыванию относится, в частности, образование водородной связи. В определенных случаях сольват можно будет выделить, например, когда одна или несколько молекул растворителя включены в кристаллическую решетку твердого кристаллического вещества. Термин «сольват» распространяется как на фазу раствора, так и на выделяемые сольваты. Типичными сольватами являются гидраты, этаноляты и метаноляты.
Некоторые из соединений, являющихся объектом настоящего изобретения, являются основными, и такие соединения применимы в форме свободных оснований или в форме их фармацевтически приемлемых кислотно-аддитивных солей.
Кислотно-аддитивные соли могут быть более удобной формой для использования, и на практике использование солевой формы по существу означает применение формы свободного основания. К кислотам, которые могут использоваться для приготовления кислотно-аддитивных солей, относятся предпочтительно такие, которые при смешении со свободным основанием приводят к фармацевтически приемлемым солям, то есть солям, анионы которых являются нетоксичными для пациента в фармакологических дозах солей, так что полезные эффекты ингибирования, присущие свободному основанию, не искажаются побочными эффектами, приписываемыми анионам. Хотя фармацевтически приемлемые соли указанных основных соединений являются предпочтительными, все кислотно-аддитивные соли, представляют собой полезные источники формы свободного основания, даже если конкретная соль, сама по себе, нужна только лишь как промежуточный продукт, например, если соль получается исключительно в целях очистки и идентификации или если она используется как промежуточное соединение при получении фармацевтически приемлемой соли с помощью ионообменных процессов. В частности, кислотно-аддитивные соли, могут быть получены независимой реакцией очищенного соединения в форме свободного основания с подходящей органической или неорганической кислотой и выделением полученной таким образом соли. Фармацевтически приемлемые соли, которые относятся к сфере настоящего изобретения, включают соли, полученные из минеральных и органических кислот. Примеры кислотно-аддитивных солей включают гидробромиды, гидрохлориды, сульфаты, бисульфаты, фосфаты, нитраты, ацетаты, оксалаты, валераты, олеаты, пальмитаты, хинаты, стеараты, лаураты, бораты, бензоаты, лактаты, фосфаты, тозилаты, цитраты, малеаты, фумараты, сукцинаты, тартраты, нафтилаты, мезилаты, глюкогептонаты, лактиобионаты, сульфаматы, малонаты, салицилаты, пропионаты, метилен-бис-β-гидроксинафтоаты, гентизаты, изетионаты, ди-пара-толуоилтартраты, этансульфонаты, бензолсульфонаты, циклогексилсульфаматы и лаурилсульфонаты. См., например, работу S.M. Berge, et al., "Pharmaceutical Salts," J. Pharm. Sci., 66, 1-19 (1977), которая включена в настоящий документ в виде ссылки.
Если соединение, являющееся объектом данного изобретения, замещено кислотной группой, могут получаться основно-аддитивные соли, которые являются просто более удобной формой применения, и на практике использование солевой формы по сути равносильно его использованию в форме свободной кислоты. К основаниям, которые могут использоваться для приготовления основно-аддитивных солей, относятся, предпочтительно, такие, которые при смешении со свободной кислотой приводят к фармацевтически приемлемым солям, то есть солям, катионы которых являются нетоксичными для пациента в фармакологических дозах солей, так что полезные эффекты ингибирования, присущие свободному основанию, не искажаются побочными эффектами, приписываемыми катионам. Основно-аддитивные соли можно также получить независимым образом в результате реакции очищенного соединения в его кислой форме с подходящим органическим или неорганическим основанием, образующимся из солей щелочных или щелочноземельных металлов, и выделением полученной таким образом соли. К основно-аддитивным солям относятся фармацевтически приемлемые соли металлов и аминов. К подходящим солям металлов относятся соли натрия, калия, кальция, бария, цинка, магния и алюминия. В конкретном примере солями являются натриевые и калиевые соли. Подходящие неорганические основно-аддитивные соли получают из оснований металлов, к которым относятся гидрид натрия, гидроксид натрия, гидроксид калия, гидроксид кальция, гидроксид алюминия, гидроксид лития, гидроксид магния, гидроксид цинка и им подобные. Подходящие аминные основно-аддитивные соли получают из аминов, основность которых достаточна для образования стабильной соли, и предпочтительно из аминов, часто используемых в медицинской химии по причине их низкой токсичности и пригодности для использования в медицинских целях, например, аммиак, этилендиамин, N-метилглюкамин, лизин, аргинин, орнитин, холин, N,N'-дибензилэтилендиамин, хлорпрокаин, диэтаноламин, прокаин, N-бензилфенетиламин, диэтиламин, пиперазин, трис(гидроксиметил)аминометан, гидроксид тетраметиламмония, триэтиламин, дибензиламин, эфенамин, дегидроабиэтиламин, N-этилпиперидин, бензиламин, тетраметиламмоний, тетраэтиламмоний, метиламин, диметиламин, триметиламин, этиламин, основные аминокислоты, например, лизин и аргинин, и дициклогексиламин.
Являясь полезными сами по себе в качестве активных соединений, соли соединений, являющихся объектом данного изобретения, полезны для целей очистки соединений, например, за счет использования разницы в растворимости солей и исходных соединений, побочных продуктов и/или исходных материалов при помощи известных специалистам способов.
Очевидно, что соединения, являющиеся объектом настоящего изобретения, могут содержать асимметричные центры. Эти асимметричные центры могут независимо друг от друга быть в R- или в S-конфигурации. Для специалиста в данной области очевидно, что определенные соединения, являющиеся объектом данного изобретения, могут также проявлять геометрическую изомерию. Следует понимать, что настоящее изобретение распространяется на отдельные геометрические изомеры и стереоизомеры и их смеси, в том числе рацемические смеси приведенных выше соединений настоящего изобретения. Такие изомеры можно выделить из их смесей при помощи известных методов или их модификаций, например, хроматографических методов или методов перекристаллизации либо получить отдельно из соответствующих изомеров промежуточных соединений. Кроме того, в ситуациях, где возможны таутомеры соединений настоящего изобретения, настоящее изобретение включает все таутомерные формы таких соединений.
Конкретные осуществления настоящего изобретения
Одним из конкретных осуществлений настоящего изобретения являются следующие соединения формулы (I):
1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метилпиримидин-4-ил}пирролидин-3-карбоновая кислота,
2-(1-{2-метокси-6-[2-(4-трифторметоксифенил)этиламино]пиримидин-4-ил}пиперидин-3-ил)-2-метилпропионовая кислота,
2-[3-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}-5-(1-гидрокси-1-метилэтил)фенил]пропан-2-ол,
[6-(3-аминопиперидин-1-ил)-2-метоксипиримидин-4-ил]-[2-(2,4-дихлорфенил)этил]амин,
[6-(4-аминопиперидин-1-ил)-2-метоксипиримидин-4-ил]-[2-(2,4-дихлорфенил)этил]амин,
N-(1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-4-ил)ацетамид,
5-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}-1-метил-2,3-дигидро-1H-индол-2-карбоновая кислота,
[2-(3-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}фенил)-2-метилпропионил]амид 2-метилпропан-2-сульфоновой кислоты,
[2-(3-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}фенил)-2-метилпропионил]амид N,N-диметиламид-2-сульфоновой кислоты,
2-(3-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}фенил)-2-метил-1-тиоморфолин-4-илпропан-1-он,
2-(3-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}фенил)изобутирамид,
2-(3-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}фенил)-N,N-диметилизобутирамид,
(1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-ил)уксусная кислота,
1-{2-метокси-6-[2-(4-трифторметоксифенил)этиламино]пиримидин-4-ил}пиперидин-3-карбоновая кислота,
N-(1-{2-метокси-6-[2-(4-трифторметоксифенил)этиламино]пиримидин-4-ил}пиперидин-3-карбонил)метансульфонамид,
N-(1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбонил)метансульфонамид,
(1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбонил)амид этансульфоновой кислоты,
(1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбонил)амид 2-метилпропан-2-сульфоновой кислоты,
N-(1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбонил)-C,C,C-трифторметансульфонамид,
(1H-тетразол-5-ил)амид 1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбоновой кислоты,
амид 1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбоновой кислоты,
диметиламид 1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбоновой кислоты,
1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбоксамид N,N-диметиламид-2-сульфоновой кислоты,
5-{2-метокси-6-[2-(4-трифторметоксифенил)этиламино]пиримидин-4-ил}-тиофен-2-карбоновая кислота или
5-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}-2,3-дигидробензофуран-2-карбоновая кислота,
или его фармацевтически приемлемая соль, гидрат или сольват, его фармацевтически приемлемое пролекарство или фармацевтически приемлемая соль, гидрат или сольват пролекарства.
Еще одним из конкретных осуществлений настоящего изобретения являются следующие соединения формулы (I) или его сложноэфирные пролекарства:
1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метилпиримидин-4-ил}пирролидин-3-карбоновая кислота,
2-(1-{2-метокси-6-[2-(4-трифторметоксифенил)этиламино]пиримидин-4-ил}пиперидин-3-ил)-2-метилпропионовая кислота,
2-[3-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}-5-(1-гидрокси-1-метилэтил)фенил]пропан-2-ол,
[6-(3-аминопиперидин-1-ил)-2-метоксипиримидин-4-ил]-[2-(2,4-дихлорфенил)этил]амин,
[6-(4-аминопиперидин-1-ил)-2-метоксипиримидин-4-ил]-[2-(2,4-дихлорфенил)этил]амин,
N-(1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-4-ил)ацетамид,
5-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}-1-метил-2,3-дигидро-1H-индол-2-карбоновая кислота,
[2-(3-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}фенил)-2-метилпропионил]амид 2-метилпропан-2-сульфоновой кислоты,
[2-(3-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}фенил)-2-метилпропионил]амид N,N-диметиламид-2-сульфоновой кислоты,
2-(3-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}фенил)-2-метил-1-тиоморфолин-4-илпропан-1-он,
2-(3-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}фенил)изобутирамид,
2-(3-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}фенил)-N,N-диметилизобутирамид,
(1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-ил)уксусная кислота,
1-{2-метокси-6-[2-(4-трифторметоксифенил)этиламино]пиримидин-4-ил}пиперидин-3-карбоновая кислота,
N-(1-{2-метокси-6-[2-(4-трифторметоксифенил)этиламино]пиримидин-4-ил}пиперидин-3-карбонил)метансульфонамид,
этиловый эфир 5-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}-1-метил-2,3-дигидро-1H-индол-2-карбоновой кислоты,
этиловый эфир (1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-ил)уксусной кислоты,
N-(1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбонил)метансульфонамид,
(1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбонил)амид этансульфоновой кислоты,
(1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбонил)амид 2-метилпропан-2-сульфоновой кислоты,
N-(1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбонил)-C,C,C-трифторметансульфонамид,
(1H-тетразол-5-ил)амид 1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбоновой кислоты,
амид 1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбоновой кислоты,
диметиламид 1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбоновой кислоты,
1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбоксамид N,N-диметиламид-2-сульфоновой кислоты,
5-{2-метокси-6-[2-(4-трифторметоксифенил)этиламино]пиримидин-4-ил}-тиофен-2-карбоновая кислота или
5-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}-2,3-дигидрбензофуран-2-карбоновая кислота,
или их фармацевтически приемлемая соль, гидрат или сольват.
Соединения, являющиеся объектом данного изобретения, а также используемые для их получения промежуточные и исходные вещества именуются в соответствии с номенклатурой ИЮПАК, в которой характеристические группы при наименовании имеют следующий приоритет по убыванию важности: кислоты, эфиры, амиды и пр. Тем не менее, считается, что если для какого-либо из соединений, представленного как структурной формулой, так и номенклатурным названием, имеется несоответствие между структурной формулой и номенклатурным названием, правильной необходимо считать структурную формулу.
Соединения, являющиеся объектом данного изобретения, проявляют активность в качестве антагонистов рецептора простагландина D2 и могут использоваться как активные фармакологические вещества. Соответственно, они включаются в фармацевтические композиции и используются в лечении пациентов, страдающих определенными медицинскими нарушениями.
Соединения, на которые распространяется настоящее изобретение, являются антагонистами рецептора простагландина D2 согласно тестам, описанным в литературе, а также в приведенном ниже разделе о фармакологических тестах, результаты которых должны коррелировать с фармакологическим действием в организме человека и других млекопитающих. Таким образом, в еще одном варианте осуществления настоящего изобретения представлены соединения, являющиеся объектом данного изобретения и содержащие их композиции, которые могут использоваться в лечении пациентов, страдающих заболеваниями, которые можно облегчить введением антагониста PGD2, или подверженных таким заболеваниям. Например, соединения настоящего изобретения поэтому могут использоваться для лечения различных заболеваний, опосредованных PGD2, в том числе, без ограничения указанными далее, аллергического заболевания (например, аллергический ринит, аллергический конъюнктивит, атопический дерматит, бронхиальная астма и пищевая аллергия), системного мастоцитоза, нарушений, сопровождающихся системной активацией мастоцитов, анафилактического шока, бронхоконстрикции, бронхита, крапивницы, экземы, заболеваний, сопровождающихся зудом (например, атопический дерматит и крапивница), заболеваний (например, катаракта, отслоение сетчатки, воспаление, инфекция и нарушения сна), которые возникают в качестве вторичных заболеваний в результате поведения, сопровождающегося зудом (например, расчесывания и растирания), воспаления, хронических обструктивных заболеваний легких, ишемического реперфузионного повреждения, расстройства мозгового кровообращения, хронического ревматоидного артрита, плеврита, неспецифического язвенного колита и подобных заболеваний.
Кроме того, соединения, являющиеся объектом настоящего изобретения, могут использоваться для лечения в сочетании с
(i) антигистаминами, например, фексофенадином, лоратадином и цитиризином, для лечения аллергического ринита;
(ii) антагонистами лейкотриенов, например, монтелукастом и зафирлукастом, для лечения аллергического ринита, ХОЗЛ, аллергического дерматита, аллергического конъюнктивита и пр. - точную информацию см. в WO 01/78697 A2;
(iii) бета-агонистами, например, альбутеролом, сальбутеролом и тербуталином, для лечения астмы, ХОЗЛ, аллергического дерматита, аллергического конъюнктивита и пр.;
(iv) антигистаминами, например, фексофенадином, лоратадином и цитиризином, для лечения астмы, ХОЗЛ, аллергического дерматита, аллергического конъюнктивита и пр.;
(v) ингибиторами PDE4 (фосфодиэстеразы 4), например, рофлумиластом и циломиластом, для лечения астмы, ХОЗЛ, аллергического дерматита, аллергического конъюнктивита и пр.; или
(vi) с антагонистами TP (рецептора тромбоксана A2) или антагонистами CrTh2 (молекулы, гомологичной рецептору хемоаттрактанта, экспрессируемой на Th2 клетках), например, раматробраном (BAY-u3405), для лечения ХОЗЛ, аллергического дерматита, аллергического конъюнктивита и т.д.
Конкретным вариантом осуществления терапевтических способов, являющихся объектом настоящего изобретения, является лечение аллергического ринита.
Еще одним конкретным вариантом осуществления терапевтических способов, являющихся объектом настоящего изобретения, является лечение бронхиальной астмы.
В соответствии с еще одним аспектом изобретения предлагается способ лечения пациента (человека или животного), страдающего заболеваниями, которые могут быть облегчены введением антагониста рецептора простагландина D2, или подверженного таким заболеваниям, как, например, описанным выше, включающий введение пациенту эффективного количества соединения, являющегося объектом данного изобретения, или содержащей его композиции. Под «эффективным количеством» подразумевается количество соединения, являющегося объектом настоящего изобретения, которое эффективно в качестве антагониста рецептора простагландина D2 и поэтому способно дать желаемый терапевтический эффект.
Включенные здесь ссылки на лечение распространяются как на профилактическую терапию, так и на лечение диагностированных заболеваний.
Настоящее изобретение распространяется также на фармацевтические композиции, включающие по меньшей мере одно соединение, являющееся объектом данного изобретения, в смеси с фармацевтически приемлемым носителем.
На практике соединения, являющиеся объектом настоящего изобретения, могут вводиться в виде фармацевтически приемлемых лекарственных форм человеку и другим животным посредством местного или системного применения, в том числе перорального, ингаляционного, ректального, назального, буккального, сублингвального, вагинального, кишечного, парентерального (в том числе подкожного, внутримышечного, внутривенного, внутрикожного, интратекального и эпидурального), интрацистернального и внутрибрюшинного. Следует принимать во внимание, что предпочтительный способ введения может варьироваться, например, в зависимости от состояния пациента.
«Фармацевтически приемлемыми лекарственными формами» называются лекарственные формы соединения, являющегося объектом данного изобретения, которые включают, например, таблетки, драже, порошки, эликсиры, сиропы, жидкие составы, в том числе суспензии, спреи, ингаляторы, таблетки, лепешки, эмульсии, растворы, гранулы, капсулы и суппозитории, а также жидкие составы для инъекций, в том числе липосомные препараты. Общее описание методов и составов можно найти в последнем издании Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, PA.
Особым аспектом данного изобретения является соединение, являющееся объектом настоящего изобретения, которое должно вводиться в форме фармацевтической композиции. В соответствии с настоящим изобретением фармацевтические композиции состоят из соединений, являющихся объектом настоящего изобретения, и фармацевтически приемлемых носителей.
Фармацевтически приемлемые носители включают по меньшей мере один из компонентов, которыми могут быть фармацевтически приемлемые носители, разбавители, оболочки, адъюванты, формообразующие или среды, такие как консерванты, наполнители, разрыхлители, смачивающие вещества, эмульгаторы, стабилизаторы эмульсий, суспендирующие вещества, изотонические вещества, подсластители, вкусовые добавки, ароматизаторы, красители, бактерицидные средства, противогрибковые средства, другие терапевтические вещества, скользящие вещества, вещества, замедляющие или ускоряющие всасывание, и дозирующие вещества, в зависимости от особенностей способа введения и лекарственной формы.
Примерами суспендирующих веществ являются этоксилированные изостеариловые спирты, полиоксиэтиленсорбит и сложные эфиры сорбитана, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант или смеси этих веществ.
Примерами бактерицидных и противогрибковых веществ, предотвращающих действие микроорганизмов, являются парабены, хлорбутанол, фенол, сорбиновая кислота и подобные им вещества.
Примерами изотонических веществ являются сахара, хлорид натрия и подобные им вещества.
Примерами веществ, замедляющих и продлевающих всасывание, являются моностеарат алюминия и желатин.
Примерами веществ, ускоряющих и стимулирующих абсорбцию, являются диметилсульфоксид и его аналоги.
Примерами разбавителей, растворителей, носителей, солюбилизирующих добавок, эмульгаторов и стабилизаторов эмульсии являются вода, хлороформ, сахароза, этанол, изопропиловый спирт, этиловый эфир угольной кислоты, этилацетат, бензиловый спирт, тетрагидрофурфуриловый спирт, бензилбензоат, полиолы, пропиленгликоль, 1,3-бутиленгликоль, глицерин, полиэтиленгликоли, диметилформамид, Tween® 60, Span® 60, цетостеариловый спирт, миристиловый спирт, глицерилмоностеарат и лаурилсульфат натрия, сложные эфиры сорбитана и жирных кислот, растительные масла (такие как хлопковое масло, арахисовое масло, кукурузное масло, оливковое масло, касторовое масло и кунжутное масло) и инъецируемые органические сложные эфиры, такие как этилолеат и ему подобные, или подходящие смеси этих соединений.
Примерами формообразующих наполнителей являются лактоза, молочный сахар, цитрат натрия, карбонат кальция и дикальцийфосфат.
Примерами разрыхлителей являются крахмал, альгиновые кислоты и некоторые сложные силикаты.
Примерами скользящих веществ являются стеарат магния, лаурилсульфат натрия, тальк, а также полиэтиленгликоли с высоким молекулярным весом.
Выбор фармацевтически приемлемого носителя, в целом, определяется в соответствии с химическими свойствами активного соединения, такими как растворимость, метод применения и нормы, которым необходимо следовать в фармацевтической практике.
Фармацевтические композиции, являющиеся объектом настоящего изобретения, пригодные для перорального применения, могут представлять собой отдельные единицы, такие как твердые лекарственные формы, такие как капсулы, облатки или таблетки, каждая из которых содержит определенное количество активного ингредиента, либо такие как порошки или гранулы или жидкие лекарственные формы, такие как растворы или суспензии в водной или неводной жидкой среде или жидкой эмульсии масла в воде или воды в масле. Активный ингредиент также может быть в виде болюса, электуария или пасты.
«Твердая лекарственная форма» означает лекарственную форму соединения, являющегося объектом данного изобретения, в виде твердого вещества, например, капсул, таблеток, пилюль, порошка, драже или гранул. В таких лекарственных формах соединение, являющееся объектом данного изобретения, добавлено в как минимум один традиционно используемый инертный формообразующий наполнитель (или носитель), например, цитрат натрия или дикальцийфосфат или (a) наполнители или добавки, такие как, например, крахмал, лактоза, сахароза, глюкоза, маннит и кремниевая кислота, (b) связующие вещества, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и камедь, (c) увлажняющие вещества, такие как, например, глицерин, (d) разрыхлители, такие как, например, агар-агар, карбонат кальция, картофельный или маниоковый крахмал, альгиновая кислота, определенные сложные силикаты и Na2CO3, (e) замедлители образования растворов, такие как, например, парафин, (f) ускорители абсорбции, такие как, например, четвертичные аммониевые соединения, (g) увлажняющие вещества, такие как, например, цетиловый спирт и глицеринмоностеарат, (h) адсорбенты, такие как, например, каолин или бентонит, (i) скользящие вещества, такие как, например, тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия, (j) замутняющие компоненты, (k) буферные вещества и вещества, отсроченно высвобождающие соединение или соединения, являющиеся объектом данного изобретения, в определенной части кишечного тракта.
Таблетка может быть приготовлена прессованием или формовкой и может иметь один или несколько вспомогательных компонентов. Прессованные таблетки можно получать прессованием в подходящем аппарате активного ингредиента в сыпучей форме, такой как порошок или гранулы, которая может быть смешана со связующим веществом, скользящим веществом, инертным разбавителем, консервантом, поверхностно-активным или диспергирующим веществом. Могут использоваться такие инертные наполнители, как лактоза, цитрат натрия, карбонат кальция, дикальцийфосфат, и разрыхлители, такие как крахмал, альгиновые кислоты и определенные сложные силикаты, смешанные со скользящими веществами, такими как стеарат магния, лаурилсульфат натрия и тальк. Смесь порошкообразных соединений, смоченную инертным жидким разбавителем, можно формовать на подходящем автомате для получения формованных таблеток. Таблетки могут иметь покрытия или насечки, а также могут иметь состав, обеспечивающий медленное или контролируемое высвобождение содержащегося в них активного ингредиента.
Твердые составы также могут использоваться в качестве наполнителей в желатиновых капсулах с мягким или твердым наполнением с использованием таких инертных наполнителей, как лактоза или молочный сахар, а также полиэтиленгликолей с большим молекулярным весом и подобных им веществ.
В случае необходимости, а также для более эффективного распространения, соединения могут быть микроинкапсулированными в системах медленного или направленного высвобождения или присоединены к ним, такими системами являются биосовместимые, биоразлагаемые полимерные матрицы (например, сополимер d,l-лактида с гликолидом), липосомы и микросферы для подкожного и внутримышечного инъецирования методом, называемым подкожной или внутримышечной инъекцией замедленного всасывания, обеспечивающей медленное высвобождение соединения или соединений в течение 2 недель или дольше. Соединения могут быть стерилизованы, например, фильтрацией через задерживающий бактерии фильтр или добавлением стерилизующих веществ в форме стерильных твердых составов, которые могут быть растворены в стерильной воде или другой стерильной инъецируемой среде непосредственно перед применением.
«Жидкая лекарственная форма» означает форму активного соединения, которая вводится пациенту в жидкой форме, например, в виде фармацевтически приемлемых эмульсий, растворов, суспензий, сиропов или эликсиров. Помимо активных соединений жидкие лекарственные формы могут содержать обычно применяемые в данной области инертные разбавители, такие как растворители, солюбилизирующие вещества и эмульгаторы.
Используемые водные суспензии могут содержать эмульгаторы или вещества, которые способствуют образованию суспензий.
Фармацевтические композиции, пригодные для местного применения, - это составы в форме, допускающей местное введение пациенту. Составы могут иметь форму мазей местного применения, бальзамов, порошков, спреев и ингаляторов, гелей (на водной или спиртовой основе), кремов, обычно используемых в данной области, или быть включенными в матричную основу для применения в качестве пластыря, обеспечивающего контролируемое высвобождение соединения через кожный барьер. В форме мази активные ингредиенты могут использоваться или с парафиновыми, или с водорастворимыми основами. В альтернативном варианте активные ингредиенты могут иметь форму крема с масляно-водной основой. Составы, предназначенные для местного применения в глаза, представляют собой глазные капли, в которых активный ингредиент растворен или взвешен в подходящем носителе, как правило водном растворителе. Составы, предназначенные для местного применения через слизистую рта, включают лекарственные леденцы, имеющие в своем составе активный ингредиент во вкусовой добавке, как правило сахарозе и камеди или трагаканте; пастилки, имеющие в своем составе активный ингредиент на инертной основе, такой как желатин и глицерин или сахароза и камедь; и составы для полоскания рта, имеющие в своем составе активный ингредиент в подходящем жидком носителе.
Масляная фаза эмульсионных фармацевтических композиций может быть получена обычным способом из известных ингредиентов. Эта фаза может иметь в своем составе только эмульгатор (также называемый эмульгирующим веществом), однако желательно, чтобы в нее также входил как минимум один эмульгатор, содержащий жир или масло, или эмульгатор, содержащий и жир, и масло. В одном конкретном осуществлении изобретения в состав включен гидрофильный эмульгатор наряду с липофильным эмульгатором, который действует как стабилизатор. Эмульгатор (эмульгаторы) вместе со стабилизатором (стабилизаторами) или без него (них) образуют эмульгирующийся воск, а вместе с маслом или жиром образуют эмульгирующуюся основу мази, которая является масляной диспергированной фазой кремовых составов.
При необходимости водная фаза кремовой основы может включать, например, не менее 30% вес. многоатомного спирта, то есть спирта с двумя или более гидроксильными группами, например, пропиленгликоль, бутан-1,3-диол, маннит, сорбит, глицерин или полиэтиленгликоль (в том числе PEG 400) и их смеси. Составы для местного применения могут, если нужно, иметь в своем составе соединение, стимулирующее всасывание или проникновение активного ингредиента через кожу или другие пораженные зоны.
Выбор подходящих масел или жиров для использования в составе зависит от свойств, которые необходимо получить. Желательно, чтобы крем был нежирным, не оставляющим пятен и смываемым продуктом подходящей консистенции, не допускающей утечки из тюбиков и других емкостей. Могут использоваться линейные и разветвленные одно- и двухосновные алкиловые эфиры, такие как диизопропилмиристат, децилолеат, изопропилпальмитат, бутилстеарат, 2-этилгексилпальмитат или смесь эфиров с разветвленной цепью, известной как Crodamol CAP. Они могут использоваться отдельно или в смесях, в зависимости от желаемых свойств. В качестве альтернативы могут использоваться липиды с высокой температурой плавления, такие как белый мягкий парафин и/или жидкий парафин и другие минеральные масла.
Фармацевтическими композициями для ректального или вагинального применения называются составы, форма которых допускает ректальное или вагинальное введение пациенту и которые содержат как минимум одно соединение, являющееся объектом данного изобретения. Суппозитории представляют собой одну из форм таких составов, которую можно получить смешиванием соединений, являющихся объектом данного изобретения, с подходящими нераздражающими инертными наполнителями или носителями, как, например, масло какао, полиэтиленгликоль или восковая основа суппозитория, которые находятся в твердом состоянии при обычных температурах, но становятся жидкими при температуре тела, а поэтому плавятся при ректальном или вагинальном введении и высвобождают активный ингредиент.
Фармацевтические композиции, вводимые посредством инъекции, могут вводиться внутримышечно, внутривенно, внутрибрюшинно и/или подкожно. Составы, являющиеся объектом настоящего изобретения, готовят в жидких растворах, в частности в физиологически совместимых буферах, таких как раствор Хенка или раствор Рингера. Кроме того, составы могут быть приготовлены в твердой форме и растворены или взвешены непосредственно перед применением. Возможны также лиофилизированные формы. Составы являются стерильными и включают эмульсии, суспензии, водные и неводные растворы для инъекций, которые могут содержать суспендирующие вещества, загустители и антиоксиданты, буферы, бактериостаты и добавки, делающие состав изотоническим, и иметь правильно подобранный уровень pH, соответствующий показателю крови пациента, которому будет вводиться состав.
Фармацевтическими композициями, являющимися объектом настоящего изобретения, пригодными для назального или ингаляционного применения, называются составы, форма которых пригодна для введения пациенту назально или ингаляционно. Состав может содержать носитель в форме порошка с размером частиц, например, в диапазоне от 1 до 500 микрон (в том числе с размерами частиц в диапазоне от 20 до 500 микрон с шагом 5 микрон, то есть с размерами 30 микрон, 35 микрон и т.д.). К подходящим составам с жидким носителем для применения, например, в качестве спрея или капель для носа, относятся водные или масляные растворы активного ингредиента. Составы, пригодные для аэрозольного введения, могут быть получены в соответствии с традиционными методами и вводиться с другими терапевтическими веществами. Для введения составов, являющихся объектом данного изобретения, в качестве ингаляционной терапии могут использоваться дозирующие ингаляторы.
Действительная дозировка активного ингредиента (ингредиентов) в составах, являющихся объектом данного изобретения, может варьироваться с целью получения количества активного ингредиента (ингредиентов), эффективного для получения желаемого терапевтического эффекта для определенного состава и метода его введения пациенту. Поэтому дозировка, выбираемая для каждого пациента, зависит от множества факторов, таких как желаемый терапевтический эффект, способ введения, желаемая длительность лечения, этиология и тяжесть заболевания, состояние пациента, вес, пол, диета и возраст, тип и активность каждого активного ингредиента, скорость абсорбции, метаболизма и/или выделения и других факторов.
Полная дневная доза соединений, являющихся объектом данного изобретения, вводимая пациенту в виде одной или нескольких доз, может составлять, например, примерно от 0,001 до 100 мг/кг массы тела в день, предпочтительно от 0,01 до 10 мг/кг/день. Например, для взрослого дозы, как правило, составляют примерно от 0,01 до 100, предпочтительно примерно от 0,01 до 10 мг/кг массы тела в день при ингаляции, примерно от 0,01 до 100, предпочтительно от 0,1 до 70, желательно от 0,5 до 10 мг/кг массы тела в день при пероральном введении и примерно от 0,01 до 50, предпочтительно от 0,01 до 10 мг/кг массы тела в день при внутривенном введении. Процентное содержание активного ингредиента в композиции может быть разным, но оно должно обеспечивать получение подходящей дозировки. Лекарственные формы могут содержать дольные единицы дозы, позволяющие получить желаемую дневную дозу. Естественно, что формы, содержащие несколько единиц дозы, могут вводиться примерно одновременно. Введение доз может быть настолько частым, насколько необходимо для достижения желаемого терапевтического эффекта. Некоторые пациенты могут проявлять быструю ответную реакцию на более высокую или более низкую дозу, и для них могут оказаться достаточными гораздо более слабые поддерживающие дозы. Для других пациентов могут понадобиться долгосрочные курсы лечения с частотой от 1 до 4 доз в день в соответствии с физиологическими потребностями каждого конкретного пациента. Само собой разумеется, для других пациентов может потребоваться не более одной или двух доз в день.
Стандартные дозы составов могут быть получены любым из традиционно используемых в фармацевтике способов. Такие способы включают стадию связывания активного ингредиента с носителем, состоящим из одного или нескольких вспомогательных ингредиентов. Как правило, составы получают однородным и тесным связыванием активного ингредиента с жидкими носителями или мелкозернистыми твердыми носителями, или и теми и другими, с последующим формованием продукта, если это необходимо.
Составы могут быть в упаковках по одной дозе или по несколько доз, например, в запаянных ампулах и пузырьках с эластичными пробками и могут храниться в лиофилизированном состоянии, требующем только добавления стерильного жидкого носителя, например, воды для инъекций, непосредственно перед применением. Индивидуальные растворы для инъекций и суспензии могут быть приготовлены из стерильных порошков, гранул или таблеток описанных выше типов.
Соединения, являющиеся объектом данного изобретения, могут быть получены посредством применения или адаптации известных способов, под которыми подразумеваются способы, использовавшиеся прежде либо описанные в литературе, например описанные в работе R.C. Larock in Comprehensive Organic Transformations, VCH publishers, 1989.
Согласно еще одному признаку изобретения образованные кислотно-аддитивные соли соединений, являющихся объектом данного изобретения, можно получить в результате реакции свободного основания с соответствующей кислотой при помощи известных способов или их модификаций. Например, образованные кислотно-аддитивные соли соединений, являющихся объектом данного изобретения, можно получить либо растворением свободного основания в воде или водном растворе спирта или других подходящих растворителях, содержащих соответствующую кислоту, и выделением соли выпариванием раствора, либо в результате реакции свободного основания и кислоты в органическом растворителе, где соль выпадает в осадок или выпаривается из раствора.
Соединения, являющиеся объектом данного изобретения, можно регенерировать из их кислотно-аддитивных солей при помощи известных способов или их модификаций. Например, исходные соединения, являющиеся объектом данного изобретения, можно регенерировать из их кислотно-аддитивных солей щелочами, например, водным раствором бикарбоната натрия или водным раствором аммиака.
Соединения, являющиеся объектом данного изобретения, можно регенерировать из их солей присоединения основания при помощи известных способов или их модификаций. Например, исходные соединения, являющиеся объектом данного изобретения, можно регенерировать из их солей присоединения основания обработкой кислотой, например соляной кислотой.
Соединения, являющиеся объектом настоящего изобретения, можно без труда получить или образовать в процессе, являющемся объектом настоящего изобретения, в виде сольватов (например, гидратов). Гидраты соединений, являющихся объектом настоящего изобретения, могут быть без труда получены путем перекристаллизации из смеси водного/органического растворителя с помощью таких органических растворителей, как диоксан, THF или MeOH.
Согласно еще одному признаку изобретения основно-аддитивные соли соединений, являющихся объектом данного изобретения, можно получить в результате реакции свободной кислоты с соответствующим основанием при помощи известных способов или их модификаций. Например, основно-аддитивные соли соединений, являющихся объектом данного изобретения, можно получить либо растворением свободной кислоты в воде, или водном растворе спирта, или других подходящих растворителях, содержащих соответствующее основание, и выделением соли выпариванием раствора, либо в результате реакции свободной кислоты и основания в органическом растворителе, где соль выпадает в осадок или выпаривается из раствора.
Исходные или промежуточные соединения можно получить с помощью способов, описанных в настоящей заявке или адаптацией известных способов.
Соединения изобретения, способы их получения, а также их биологическая активность будут более очевидны из анализа следующих примеров, которые приводятся только в качестве иллюстрации и не должны рассматриваться как ограничивающие объем изобретения. Соединения настоящего изобретения идентифицировали, например, с помощью следующих аналитических методов.
Эксперименты по жидкостной хроматографии высокого давления и масс-спектрометрии (ЖХ-МС) для определения времени удерживания (Rt) и соответствующих масс ионов проводились с использованием одного из следующих методов.
Масс-спектры (МС) записывали на масс-спектрометре Micromass LCT. Метод включает ионизацию положительным электрораспылением и сканирование массы м/z от 100 до 1000. Жидкостную хроматографию осуществляли при помощи бинарного насоса и дегазатора Hewlett Packard 1100 Series; неподвижная фаза: колонка Phenomenex Synergi 2 µ Hydro-RP 20×4,0 мм, подвижная фаза: A=0,1% муравьиная кислота (FA) в воде, B=0,1% FA в MeCN. Объем ввода 5 мкл при помощи системы CTC Analytical PAL. Скорость потока элюента составляет 1 мл/мин. Градиент составляет от 10% B до 90% B за 3 минуты и от 90% B до 100% B за 2 минуты. Вспомогательные детекторы: УФ-детектор Hewlett Packard 1100 Series, длина волны = 220 нм, и испарительный детектор светорассеяния (ELS) Sedere SEDEX 75, температура детектора = 46°C, давление N2=4 бар.
Спектры ядерного магнитного резонанса (ЯМР) 300 МГЦ 1H записывали при комнатной температуре на спектрометре Varian Mercury (300 МГц) с 5 мм датчиком ASW. В ЯМР величины химического сдвига (δ) указаны в миллионных долях (м.д.) относительно тетраметилсилана (TMS) в качестве внутреннего стандарта.
В приведенных далее примерах и описаниях синтеза, а также в остальной части заявки используемые термины имеют следующие значения: «кг» - килограммы, «г» - граммы, «мг» - миллиграммы, «мкг» - микрограммы, «моль» - моли, «ммоль» - миллимоли, «М» - моли на литр, «мМ» - миллимоли на литр, «мкМ» - микромоли на литр, «нМ» - наномоли на литр, «л» - литры, «мл» - миллилитры, «мкл» - микролитры, «°C» - градусы Цельсия, «т.пл.» - точка плавления, «т.к.» - точка кипения, «мм рт.ст.» - давление в миллиметрах ртутного столба, «см» - сантиметры, «нм» - нанометры, «абс.» - абсолютный, «конц.» - концентрированный, «c» - концентрация в г/мл, «к.т.» - комнатная температура, «ТСХ» - тонкослойная хроматография, «ВЭЖХ» - высокоэффективная жидкостная хроматография, «в/б» - внутрибрюшинно, «в/в» - внутривенно, «с» - синглет, «д» - дублет; «т» - триплет; «кв» - квартет; «м» - мультиплет, «дд» - дублет дублетов; «уш.» - уширенный, «ЖХ» - жидкостной хроматограф, «МС» - масс-спектрограф, «ESI/MS» - ионизация электрораспылением/масс-спектрограф, «Rt» - время удерживания, «М» - молекулярный ион, «фунтов на кв. дюйм» - фунтов на квадратный дюйм, «ДМСО» - диметилсульфоксид, «DMF» - N,N-диметилформамид, «CDI» - 1,1'-карбонилдиимидазол, «DCM» или «CH2Cl2» - дихлорметан, «HCl» - соляная кислота, «SPA» - SPA-анализ, «ATTC» - Американская коллекция типовых культур, «FBS» - эмбриональная бычья сыворотка, «МПС» - минимальная поддерживающая среда, «имп./мин.» - количество импульсов в минуту, «EtOAc» - этилацетат, «PBS» - фосфатный буферный физиологический раствор, «ТМД» - трансмембранный домен, «IBMX» - 3-изобутил-1-метилксантин, «cAMP» - циклический аденозинмонофосфат, «ИЮПАК» - Международный союз теоретической и прикладной химии, «МГц» - мегагерц, «ПЭГ» - полиэтиленгликоль, «MeOH» - метанол, «N» - нормальность, «THF» - тетрагидрофуран, «ч» - часы, «мин» - минута(ы), «MeNH2» - метиламин, «N2» - газообразный азот, «O.D.» - внешний диаметр, «MeCN» или «CH3CN» - ацетонитрил, «Et2O» - этиловый эфир, «преп ЖХ» - препаративная жидкостная флэш-хроматография, «ТФЭ» - твердофазная экстракция, «K2CO3» = карбонат калия, «Na2CO3» = карбонат натрия, «пмол» - пикомолярный, «гептан» - н-гептан, смола «HMBA-AM» - аминометильная смола 4-гидроксиметилбензойной кислоты, «PdCl2(dppf)2» - комплекс дихлорида 1,1'-бис(дифенилфосфино)ферроценпалладия(II) в DCM, «~» - приблизительно и «IC50» = концентрация соединения, дающая 50% ингибирование в SPA-анализе цАМФ в T-клетках LS174 человека.
ПРИМЕРЫ
Пример 1
1-{6-[2-(2,4-Дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пирролидин-3-карбоновая кислота

Стадия 1: Раствор 4,6-дихлор-2-метоксипиримидина (0,7 г), 2-(2,4-дихлорфенил)этиламина [0,74 г) и Na2CO3 (0,88 г) в EtOH (25 мл) нагревают при 80°С в течение трех часов и выливают в воду (400 мл). Полученное твердое вещество фильтруют и сушат воздухом, получая (6-хлор-2-метоксипиримидин-4-ил)-[2-(2,4-дихлорфенил)этил]амин.
Стадия 2: В пробирке смешивают (6-хлор-2-метоксипиримидин-4-ил)-[2-(2,4-дихлорфенил)этил]амин (300 мг), гидрохлорид 3-пирролидинкарбоновой кислоты (341 мг), K2CO3 (373 мг) и 1-метил-2-пирролидинон (5 мл). Пробирку запаивают, нагревают до 140°С и перемешивают в течение 16 часов. Смесь охлаждают до комнатной температуры, разбавляют водой (60 мл), подкисляют с помощью 3М HCl и трижды экстрагируют этилацетатом (60 мл). Органические экстракты объединяют, сушат над сульфатом магния, концентрируют и очищают хроматографией на силикагеле (40 г) с элюированием смесью от 0 до 20% МеОН в дихлорметане, получая 1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метилпиримидин-4-ил}пирролидин-3-карбоновую кислоту (190 мг) в виде твердого вещества. ЖХ-МС Rt =2,22 мин, МС: 411 (M+H). 1H ЯМР [300 МГц, (CD3)2SO]: δ 7,57 (1H, с); 7,36 (2H, с); 6,77 (1H, с); 5,01 (1H, с); 3,72 (3H, с); 3,5 (6H, м); 3,12 (1H, м); 2,91 (2H, т); 2,09 (2H, м). IC50=9 нМ.
Пример 2
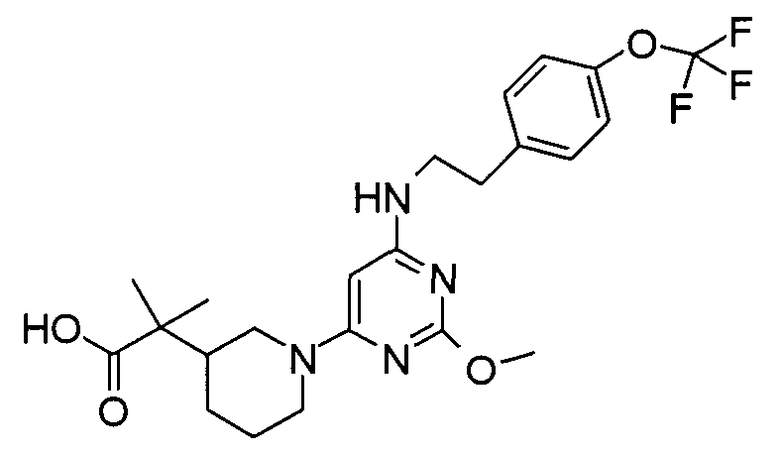
2-(1-{2-Метокси-6-[2-(4-трифторметоксифенил)этиламино]пиримидин-4-ил}пиперидин-3-ил)-2-метилпропионовая кислота
Стадия 1: Смесь этилового эфира пирид-3-илуксусной кислоты (12,6 г) и родия на окиси алюминия (12,6 г) в этаноле (200 мл) помещают в аппарат Парра при 60°C и давлении 60 фунтов на кв. дюйм на 16 часов. Суспензию фильтруют через целит. Фильтр промывают этанолом и фильтрат концентрируют до объема приблизительно 50 мл и добавляют воду (600 мл). Раствор экстрагируют EtOAc (3×100 мл). Объединенные органические слои промывают солевым раствором, сушат (Na2SO4), фильтруют и выпаривают в вакууме. Остаток растворяют в THF (150 мл) с добавлением триэтиламина (10,7 мл). Раствор охлаждают до 0°C и добавляют по каплям бензилхлорформиат (11 мл). Раствор перемешивают при 0°C в течение двух часов. Раствор концентрируют до объема приблизительно 50 мл и добавляют воду (600 мл). Раствор экстрагируют EtOAc (2×150 мл). Объединенный органический слой промывают солевым раствором, сушат (Na2SO4), фильтруют и выпаривают в вакууме, получая бензиловый эфир 3-этоксикарбонилметилпиперидин-1-карбоновой кислоты (21,5 г), который используют на следующей стадии без дополнительной очистки. МС: 306 (M+H); 1H ЯМР (300 МГц, ДМСО-d6) δ 7,3 (м, 5H); 5,05 (с, 2H); 3,8-4,1 (м, 4H); 2,5-2,6 (м, 1H); 1,5-1,7 (м, 4H); 1-1,4 (м, 4H).
Стадия 2: К 1M суспензии трет-бутоксида калия в THF (200 мл) при -78°C добавляют по каплям раствор бензилового эфира 3-этоксикарбонилметилпиперидин-1-карбоновой кислоты (21,5 г) в THF (25 мл) в течение десяти минут. Одной порцией добавляют метилиодид (6,85 мл). Суспензию перемешивают при -78°C в течение одного часа, при -40°C в течение одного часа и доводят до комнатной температуры в течение ночи. Суспензию выливают в воду (800 мл) и экстрагируют EtOAc (2×150 мл). Объединенные органические слои промывают солевым раствором, сушат (Na2SO4), фильтруют и выпаривают в вакууме. Остаток очищают хроматографией на силикагеле с элюированием смесью 100% гептана до 30% EtOAc в гептане, получая бензиловый эфир 3-(1-этоксикарбонил-1-метилэтил)пиперидин-1-карбоновой кислоты (151,1 г). МС: 334 (M+H); 1H ЯМР (300 МГц, ДМСО-d6) δ 7,3 (м, 5H); 5,05 (с, 2H); 3,8-4,1 (кв, 2H); 2,5-2,6 (м, 1H); 1,5-1,7 (м, 4H); 1-1,4 (м, 4H); 1 (с, 6H).
Стадия 3: Суспензию бензилового эфира 3-(1-этоксикарбонил-1-метилэтил)пиперидин-1-карбоновой кислоты (3,3 г) и 10% палладия на углероде (500 мг) в ледяной уксусной кислоте (2 мл)/метаноле (200 мл) помещают в аппарат Парра при давлении 50 фунтов на кв. дюйм на 90 минут при комнатной температуре. Суспензию фильтруют через целит. Фильтр промывают метанолом, и фильтрат концентрируют до объема приблизительно 50 мл. Раствор метанола разбавляют THF (50 мл) и водным раствором 2 н. гидроксида калия (50 мл). Раствор перемешивают при комнатной температуре в течение 16 часов и концентрируют до объема 70-80 мл в вакууме. Раствор охлаждают до 5°C и медленно добавляют концентрированную водную HCl (8,5 мл). Раствор экстрагируют EtOAc (3×100 мл). Объединенный органический слой промывают солевым раствором, сушат (Na2SO4), фильтруют и выпаривают в вакууме, получая 2-метил-2-пиперидин-3-илпропионовую кислоту (1,1 г), которую используют на следующей стадии без дополнительной очистки. МС: 172 (M+H); 1H ЯМР (300 МГц, ДМСО-d6) δ 2,5 (м, 1H); 1,5-1,7 (м, 4H); 1-1,4 (м, 5H); 1 (с, 6H).
Стадия 4:
Метод A. Раствор (4-трифторметоксифенил)ацетонитрила (5,05 г) в MeOH (75 мл) насыщают газообразным аммиаком и обрабатывают никелем Ренея в воде (2 мл, 50%). Суспензию помещают в аппарат Парра при давлении 50 фунтов на кв. дюйм и 50°C на 3 часа и фильтруют через целит. Фильтрат выпаривают и остаточное маслянистое вещество распределяют между водой и этилацетатом. Органическую фазу сушат над сульфатом натрия, фильтруют и выпаривают. Остаток растворяют в MeOH и добавляют раствор, обработанный концентрированной соляной кислотой (1 мл). Раствор выпаривают в вакууме до получения твердого вещества, которое растирают в порошок в эфире и сушат воздухом, получая гидрохлорид 2-(4-трифторметоксифенил)этиламина (5,15 г). МС: 206 (M+H), 1H ЯМР (CDCl3): δ 8,2 (2H, м); 7,4 (2H, д, J=5 Гц); 7,3 (2H, д, J=5 Гц); 3-3,1 (2H, м); 2,9-3 (2H, м).
Метод B. Раствор 4-трифторметоксибензальдегида (1 г) и нитрометана (0,96 г) в уксусной кислоте (10,6 мл) обрабатывают ацетатом аммония (1,01 г) и нагревают в микроволновой печи до 150°C в течение 15 минут. Реакционную смесь разбавляют водой и трижды экстрагируют DCM (50 мл). Объединенные экстракты последовательно промывают 2 н. гидроксидом натрия, водой и солевым раствором, сушат над сульфатом натрия и концентрируют. Остаток хроматографируют на силикагеле и получают 4-трифторметокси-(2-нитровинил)бензол (1,23 г) в виде твердого вещества. Часть 4-трифторметокси-(2-нитровинил)бензола (0,504 г) гидрируют водородом из баллона, 10% Pd/C (115 мг) в MeOH (22 мл), с содержанием концентрированной соляной кислоты (0,27 мл), при комнатной температуре в течение 15 часов. Смесь фильтруют и фильтрат концентрируют до твердого вещества, которое промывают Et2O, получая гидрохлорид 2-(4-трифторметоксифенил)этиламина (0,3 г) в виде твердого вещества. ЖХ-МС: МС: 206 (M+H).
Стадия 5: Методом, аналогичным описанному в примере 1, стадия 1, но с использованием 4,6-дихлор-2-метоксипиримидина (0,39 г), гидрохлорида 2-(4-трифторметоксифенил)этиламина (0,38) и бикарбоната натрия (0,74 г) получают (6-хлор-2-метоксипиримидин-4-ил)-[2-(4-трифторметоксифенил)этил]амин (0,61 г). МС: 360 (M+H), 1H ЯМР (CDCl3): δ 7,4 (2H, д, J=7 Гц); 7,3 (2H, д, J=7 Гц); 6,2 (1H, с); 3,8 (3H, с); 3,5-3,6 (2H, м); 2,8 (2H, т).
Стадия 6: Раствор 2-метил-2-пиперидин-3-илпропионовой кислоты (0,6 г), (6-хлор-2-метоксипиримидин-4-ил)-[2-(4-трифторметоксифенил)этил]амина (0,46 г) и K2CO3 (0,46 г) в 1-метилпирролидин-2-оне (10 мл) нагревают при 140°C в течение 16 часов. Раствор охлаждают и выливают в воду (200 мл). Водный раствор подкисляют до pH~6 ледяной уксусной кислотой и экстрагируют EtOAc (3×100 мл). Объединенные органические слои промывают солевым раствором, сушат (Na2SO4), фильтруют и выпаривают в вакууме. Остаток очищают хроматографией на силикагеле с элюированием 5% MeOH в EtOAc, получая 2-(1-{2-метокси-6-[2-(4-трифторметоксифенил)этиламино]пиримидин-4-ил}пиперидин-3-ил)-2-метилпропионовую кислоту (105 мг). МС: 483 (M+H); 1H ЯМР (300 МГц, ДМСО-d6) δ 7,45 (д, J=3, 2H); 7,3 (д, J=3, 2H); 5,5 (с, 1H); 3,95 (с, 3H); 3,6 (м, 2H); 2,9 (т, 2H); 2,7 (м, 1H); 1,7-1,9 (м, 4H); 1,3-1,4 (м, 3H); 1,1 (д, J=3, 6H). IC50=2 нМ.
Пример 3
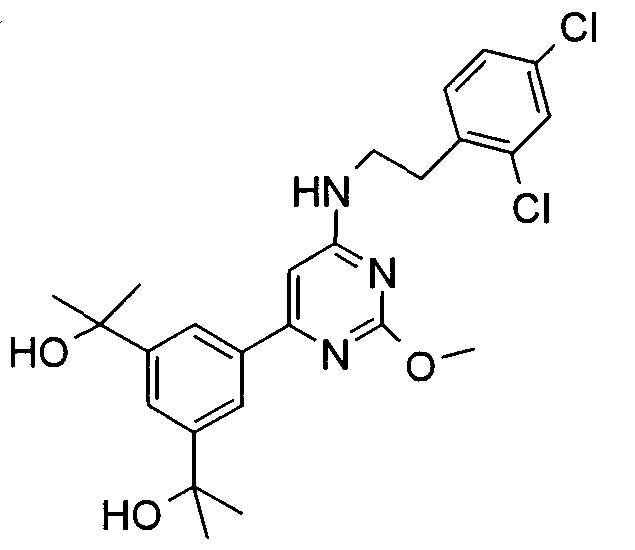
2-[3-{6-[2-(2,4-Дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}-5-(1-гидрокси-1-метилэтил)фенил]пропан-2-ол
Стадия 1: Раствор диметил-5-бромизофталата (5 г) в THF (250 мл) охлаждают до
-78°C и добавляют по каплям раствор 3M метилмагнийбромида в эфире (36,6 мл), поддерживая температуру ниже -70°C. Раствор перемешивают при -78°C в течение 2 часов и доводят до комнатной температуры в течение ночи. Раствор разбавляют эфиром (300 мл) и охлаждают до 0°C. 1 н. водную HCl (100 мл) добавляют по каплям. Объединенные органические слои промывают солевым раствором, сушат (Na2SO4), фильтруют и выпаривают в вакууме. Остаток очищают хроматографией на силикагеле с элюированием 60% EtOAc в гептане, получая 2-[3-бром-5-(1-гидрокси-1-метилэтил)фенил]пропан-2-ол (4,1 г). МС: 272 (M+H); 1H ЯМР (300 МГц, ДМСО-d6) δ 7,5 (с, 1H); 7,4 (с, 2H); 5,15 (с, 2H); 1,4 (с, 12H).
Стадия 2: 2-[3-Бром-5-(1-гидрокси-1-метилэтил)фенил]пропан-2-ол (1,08 г), 4,4,5,5,4',4',5',5'-октаметил-[2,2']би[[1,3,2]-диоксабороланил] (1,12 г), ацетат калия (0,78 г) и PdCl2(dppf)2 (42 мг) суспедируют в ДМСО (20 мл) и дегазируют в течение 20 минут. Суспензию нагревают при 90°C в течение 16 часов. Раствор выливают в воду (300 мл) и экстрагируют EtOAc (2×150 мл). Объединенные органические слои промывают солевым раствором, сушат (Na2SO4), фильтруют и выпаривают в вакууме. Остаток очищают хроматографией на силикагеле с элюированием 50% EtOAc в гептане, получая 2-[3-(1-гидрокси-1-метилэтил)-5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)фенил]пропан-2-ол (0,9 г). МС: 285 (M+H); 1H ЯМР (300 МГц, ДМСО-d6) δ 7,5 (с, 1H); 7,2 (с, 2H); 5,15 (с, 2H); 1,6 (с, 12H); 1,4 (с, 12H).
Стадия 3: Раствор 2-[3-(1-гидрокси-1-метилэтил)-5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)фенил]пропан-2-ола (0,35 г), (6-хлор-2-метоксипиримидин-4-ил)-[2-(2,4-дихлорфенил)этил]амина (0,2 г), карбоната цезия (0,58 г) и тетракис(трифенилфосфин)палладия(0) (41 мг) в 20 мл воды/80 мл диметоксиэтана дегазируют в течение 20 минут и нагревают при 90°C в течение 16 часов. Раствор выпаривают в вакууме. Остаток очищают хроматографией с элюированием 70% EtOAc в гептане, получая 2-[3-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}-5-(1-гидрокси-1-метилэтил)фенил]пропан-2-ол (0,44 г). МС: 491 (M+H); 1H ЯМР (300 МГц, ДМСО-d6) δ 7,9 (с, 2H), 7,8 (с, 1H); 7,45 (с, 1H); 7,2-7,3 (м, 2H); 6,5 (с, 1H); 3,95 (с, 3H); 3,85 (м, 2H); 3,1 (т, 2H); 1,6 (с, 12H). IC50=730 нМ.
Пример 4
[6-(3-Аминопиперидин-1-ил)-2-метоксипиримидин-4-ил]-[2-(2,4-дихлорфенил)этил]амин
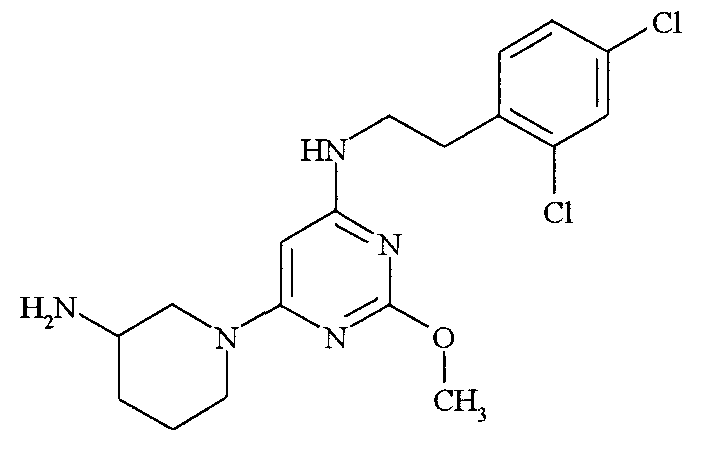
Стадия 1: Методом, аналогичным описанному в примере 1, стадия 2, но используя 3-N-Boc-аминопиперидин (450 мг) вместо гидрохлорида 3-пирролидинкарбоновой кислоты и очищая продукт реакции флэш-хроматографией на колонке с силикагелем (40 г) с элюированием смесью от 20 до 50% EtOAc в гептане, получают трет-бутиловый эфир (1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-ил)карбаминовой кислоты (281 мг). 1H ЯМР [300 МГц, (CD3)2SO]: δ 7,57 (1H, с); 7,36 (2H, с); 6,9 (2H, м); 5,29 (1H, с); 4 (2H, м); 3,71 (3H, с); 3,41 (5H, м); 2,91 (2H, т); 2,65 (2H, м); 1,82 (1H, с); 1,63 (1H, с); 1,39 (9H, с).
Стадия 2: Раствор трет-бутилового эфира (1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-ил)карбаминовой кислоты (234 мг) в дихлорметане (4 мл) обрабатывают трифторуксусной кислотой (4 мл). Смесь перемешивают при комнатной температуре в течение 3 часов и концентрируют в вакууме. Остаток растворяют в насыщенном растворе бикарбоната натрия (25 мл) и дважды экстрагируют этилацетатом (25 мл). Органические экстракты объединяют, промывают солевым раствором (20 мл), сушат над сульфатом магния, концентрируют и очищают хроматографией на силикагеле (12 г) с элюированием смесью от 0 до 10% MeOH в дихлорметане, получая [6-(3-аминопиперидин-1-ил)-2-метоксипиримидин-4-ил]-[2-(2,4-дихлорфенил)этил]амин (157 мг) в виде твердого вещества. ЖХ-МС Rt = 1,77 мин, МС: 396 (M+H). 1H ЯМР [300 МГц, (CD3)2SO]: δ 7,59 (1H, с); 7,36 (2H, с); 6,86 (2H, м); 5,93 (1H, уш.); 5,29 (1H, с); 4,16 (2H, д); 3,82 (2H, д); 3,73 (3H, с); 3,41 (4H, м); 2,91 (4H, м); 1,91 (1H, м); 1,69 (1H, м); 1,41 (2H, м); 1,23 (1H, с). IC50=985 нМ.
Пример 5
(a) [6-(4-Аминопиперидин-1-ил)-2-метоксипиримидин-4-ил]-[2-(2,4-дихлорфенил)этил]амин
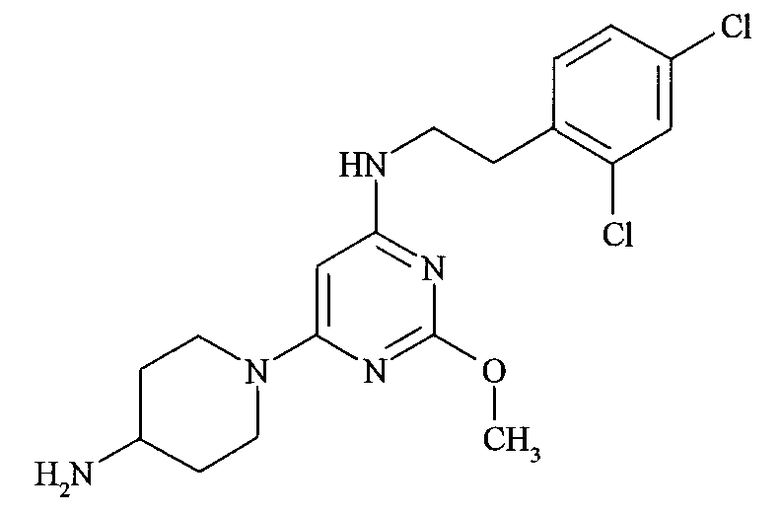
Стадия 1: Методом, аналогичным описанному в примере 1, стадия 2, но используя 4-N-Boc-аминопиперидин (450 мг) вместо гидрохлорида 3-пирролидинкарбоновой кислоты и очищая продукт реакции флэш-хроматографией на колонке с силикагелем (40 г) с элюированием смесью от 0 до 40% EtOAc в гептане, получают трет-бутиловый эфир (1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-4-ил)карбаминовой кислоты (320 мг).
Стадия 2: Раствор трет-бутилового эфира ((1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-4-ил)карбаминовой кислоты (300 мг) в DCM (5 мл) обрабатывают триэтилсиланом (194 мкл) с последующим добавлением трифторуксусной кислоты (106 мкл). Смесь перемешивают при комнатной температуре в течение 20 часов и концентрируют в вакууме. Остаток растворяют в насыщенном растворе бикарбоната натрия (30 мл) и дважды экстрагируют этилацетатом (30 мл). Органические экстракты объединяют, промывают солевым раствором (20 мл), сушат над сульфатом магния и концентрируют, получая [6-(3-аминопиперидин-1-ил)-2-метоксипиримидин-4-ил]-[2-(2,4-дихлорфенил)этил]амин (230 мг) в виде твердого вещества.
(b) N-(1-{6-[2-(2,4-Дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-4-ил)ацетамид

К смеси [6-(3-аминопиперидин-1-ил)-2-метоксипиримидин-4-ил]-[2-(2,4-дихлорфенил)этил]амина (190 мг), триэтиламина (134 мкл, 0,96 ммоль) и N,N-диметиламинопиридина (6 мг) в тетрагидрофуране (6 мл) добавляют ацетилхлорид (41 мкл, 0,58 ммоль). Реакционную смесь перемешивают в течение 17 часов, гасят добавлением воды (20 мл) и дважды экстрагируют этилацетатом (25 мл). Органические экстракты объединяют, сушат над сульфатом магния, концентрируют и очищают хроматографией на силикагеле (12 г) с элюированием смесью от 0% до 12% MeOH в CH2Cl2, получая N-(1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-4-ил)ацетамид (48 мг) в виде твердого вещества. ЖХ-МС Rt =1,9 мин, МС: 438 (M+H). 1H ЯМР [300 МГц, (CD3)2SO]: δ 7,81 (1H, д); 7,59 (1H, с); 7,36 (2H, с); 6,79 (2H, м); 5,31 (1H, с); 4,07 (2H, м); 3,78 (1H, д); 3,71 (3H, с); 3,41 (2H, м); 2,91 (4H, м); 1,78 (3H, с); 1,73 (1H, м); 1,25 (4H м). IC50=26 нМ.
Пример 6
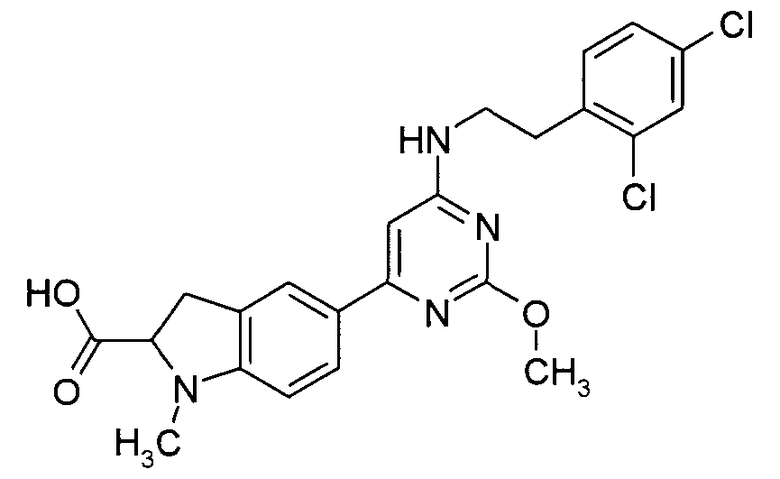
5-{6-[2-(2,4-Дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}-1-метил-2,3-дигидро-1H-индол-2-карбоновая кислота
Стадия 1. К смеси этил-5-броминдол-2-карбоксилата (2,5 г) в DMF (20 мл) добавляют раствор 60% NaH (485 мг) в DMF (10 мл). Полученную смесь перемешивают в течение 15 минут и добавляют через шприц иодметан (0,638 мл). Реакционную смесь перемешивают при комнатной температуре в течение 20 минут. Добавляют воду (200 мл) и смесь дважды экстрагируют этилацетатом (100 мл). Органические экстракты объединяют, промывают водой (3×50 мл) и один раз солевым раствором (50 мл), сушат над сульфатом магния и концентрируют, получая этиловый эфир 5-бром-1-метил-1H-индол-2-карбоновой кислоты (1,28 г) в виде твердого вещества. 1H ЯМР [300 МГц, CDCl3]: δ 7,79 (1H, д); 7,41 (1H, дд); 7,27 (1H, т); 7,2 (1H, с); 4,39 (2H, кв); 4,05 (3H, с); 1,41 (3H, т).
Стадия 2. К раствору этилового эфира 5-бром-1-метил-1H-индол-2-карбоновой кислоты (1,28 г) в трифторуксусной кислоте (10 мл) добавляют цианоборгидрид натрия (680 мг) при 0°C. Реакционную смесь доводят до комнатной температуры, перемешивают в течение 20 часов и гасят водой (100 мл). Подщелачивают NaOH и экстрагируют Et2O (3×50 мл). Органические экстракты объединяют, промывают солевым раствором (30 мл), сушат над сульфатом магния, концентрируют и очищают хроматографией на силикагеле (34 г) с элюированием смесью от 0% до 25% этилацетата в гептане, получая этиловый эфир 5-бром-1-метил-2,3-дигидро-1H-индол-2-карбоновой кислоты (800 мг) в виде твердого вещества. 1H ЯМР [300 МГц, CDCl3]: δ 7,19 (1H, д); 7,21 (1H, с); 6,34 (1H, д); 4,25 (2H, кв.д); 4,06 (1H, т); 3,21 (2H, м); 2,82 (3H, с); 1,30 (3H, т).
Стадия 3. Смесь этилового эфира 5-бром-1-метил-2,3-дигидро-1H-индол-2-карбоновой кислоты (800 мг), бис(пинаколато)дибора (1,5 г), ацетата калия (1,47 г) и PdCl2(dppf)2 (139 мг) в диметилсульфоксиде (10 мл) дегазируют продуванием азота в течение 5 минут. Смесь нагревают до 90°C в течение 4 часов. Реакционную смесь охлаждают, разбавляют водой (75 мл) и этилацетатом (100 мл) и перемешивают с обесцвечивающим углем. Двухфазную смесь фильтруют через целит и фильтрат дважды экстрагируют EtOAc (50 мл). Органические экстракты объединяют, промывают трижды водой (50 мл), один раз солевым раствором (30 мл), сушат над сульфатом магния, концентрируют и очищают хроматографией на силикагеле (34 г) с элюированием смесью от 0% до 20% этилацетата в гептане, получая этиловый эфир 1-метил-5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-2,3-дигидро-1H-индол-2-карбоновой кислоты (903 мг) в виде твердого вещества. 1H ЯМР [300 МГц, (CD3)2SO]: δ 7,39 (1H, д); 7,28 (1H, с); 6,46 (1H, д); 4,18 (3H, м); 3,3 (1H, д); 2,97 (1H, м); 2,79 (3H, с); 1,24 (12H, с); 1,22 (3H, т).
Стадия 4: Смесь (6-хлор-2-метоксипиримидин-4-ил)-[2-(2,4-дихлорфенил)этил]амина (200 мг), этилового эфира 1-метил-5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-2,3-дигидро-1H-индол-2-карбоновой кислоты (300 мг), Cs2CO3 (390 мг) и тетракис(трифенилфосфин)палладия (35 мг) в воде (0,4 мл) и диметилового эфира этиленгликоля (1,6 мл) дегазируют продуванием азота в течение 5 минут и нагревают при 90°C в течение 19 часов. Реакционную смесь охлаждают, разбавляют водой (50 мл) и дважды экстрагируют этилацетатом (50 мл). Органические экстракты объединяют, сушат над сульфатом магния, концентрируют и очищают хроматографией на силикагеле (40 г) с элюированием смесью от 0% до 40% этилацетата в гептане, получая этиловый эфир 5-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}-1-метил-2,3-дигидро-1H-индол-2-карбоновой кислоты (110 мг) в виде твердого вещества. ЖХ-МС Rt =5,57 мин, МС: 501 (M+H). 1H ЯМР [300 МГц, (CD3)2SO]: δ 7,72 (2H, м); 7,59 (1H, с); 7,37 (3H, с); 6,54 (1H, д); 6,42 (1H, с); 4,30 (1H, м); 4,17 (2H, кв.д); 3,84 (3H, с); 3,54 (2H, уш.); 3,41 (1H, м); 3,06 (1H, м); 2,97 (2H, т); 2,83 (3H, с); 1,23 (3H, т).
Стадия 5: Моногидрат гидроксида лития (1,28 ммоль) добавляют к перемешиваемой смеси этилового эфира 5-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}-1-метил-2,3-дигидро-1H-индол-2-карбоновой кислоты (0,43 ммоль) в MeOH/H2O (10 мл, 9:1). Реакционную смесь перемешивают в течение ночи при комнатной температуре. Реакционную смесь разбавляют водой и летучие вещества удаляют в вакууме. Водную фазу однократно экстрагируют Et2O, подкисляют до pH 4 (1н, HCl) и дважды экстрагируют этилацетатом. Объединенный органический слой сушат (MgSO4) и концентрируют в вакууме, получая 5-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}-1-метил-2,3-дигидро-1H-индол-2-карбоновую кислоту.
Пример 7
(a) [2-(3-{6-[2-(2,4-Дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}фенил)-2-метилпропионил]амид 2-метилпропан-2-сульфоновой кислоты

Стадия 1: К раствору диизопропиламида лития в смеси THF/н-гептан/этилбензол (1,8M, 17 мл) при 0°C добавляют раствор 2-(3-бромфенил)пропионовой кислоты (3 г) в THF (5 мл) по каплям в течение 15 минут. Раствор перемешивают в течение 1 часа с последующим добавлением по каплям метилиодида (4,93 г) в THF (5 мл) в течение 10 минут. Реакционную смесь перемешивают в течение 15 часов, гасят 2 н. соляной кислотой, концентрируют в вакууме и растворяют в эфире (150 мл). Слой эфира промывают 2 н. соляной кислотой и трижды экстрагируют 2 н. гидроксидом натрия (50 мл). Объединенные слои гидроксида натрия подкисляют 6 н. соляной кислотой до pH~1 и трижды экстрагируют эфиром (75 мл). Объединенные органические слои промывают солевым раствором, сушат над сульфатом натрия и концентрируют, получая 2-(3-бромфенил)-2-метилпропионовую кислоту в виде твердого вещества (3,08 г), которую используют далее без дополнительной очистки. ЖХ-МС: 243 (M+H)
Стадия 2: В раствор 2-(3-бромфенил)-2-метилпропионовой кислоты (2,18 ммоль) в безводном эфире (20 мл) добавляют трет-бутиллитий (1,7M в пентане, 5,4 мл, 9,16 ммоль) по каплям при -78°C и эту смесь перемешивают в течение 30 минут с трибутилборатом (2,34 мл, 8,72 ммоль). Реакционную смесь доводят до комнатной температуры, перемешивают в течение 15 часов, разбавляют эфиром и гасят 1M H3PO4. После перемешивания в течение 30 минут эфирный слой отделяют и экстрагируют 2 н. водным гидроксидом натрия (3×20 мл). Объединенные экстракты гидроксида натрия подкисляют 6 н. соляной кислотой до pH~1 и трижды экстрагируют эфиром (50 мл). Объединенные органические экстракты промывают солевым раствором, сушат над сульфатом натрия и концентрируют, получая 3-(1-карбокси-1-метилэтил)фенилбороновую кислоту, которую далее используют без дополнительной очистки.
Стадия 3: Раствор (6-хлор-2-метоксипиримидин-4-ил)-[2-(2,4-дихлорфенил)этил]амина (0,51 ммоль) и 3-(1-карбокси-1-метилэтил)фенилбороновой кислоты (0,61 ммоль) в MeCN (2,5 мл) и водном растворе Na2CO3 (0,4M, 2,5 мл) дегазируют азотом в течение 5 минут, а затем добавляют тетракис(трифенилфосфин)палладий(0) (29,5 мг). Реакционную емкость герметизируют и нагревают в микроволновой печи до 130°C в течение 30 минут. К реакционной смеси добавляют 2 мл воды, pH доводят до ~7 при помощи 2 н. водной соляной кислоты и эту смесь трижды экстрагируют EtOAc (30 мл). Объединенные экстракты промывают солевым раствором, сушат над сульфатом натрия и концентрируют. Полученное маслянистое вещество хроматографируют на силикагеле с элюированием смесью от 0% до 7% MeOH в DCM, получая 2-(3-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}фенил)-2-метилпропионовую кислоту (205 мг) в виде твердого вещества. ЖХ-МС: Rt =2,39 мин, МС: 460,2 (M+H). 1H ЯМР [300 МГц, (CD3)2SO]: δ 12,38 (1H, с), 7,36-8 (7H, м), 6,58 (1H, с), 3,84 (3H, с), 3,58 (2H, м), 2,98 (2H, м), 1,54 (6H, с).
Стадия 4: Гидрохлорид N-(3-диметиламинопропил)-N-этилкарбодиимида (0,23 ммоль) добавляют в перемешиваемый ледяной раствор 2-(3-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}фенил)-2-метилпропионовой кислоты (0,22 ммоль), трет-бутилсульфонамида (0,23 ммоль) и 4-диметиламинопиридина (0,22 ммоль) в сухом DCM в атмосфере азота. Ледяную баню убирают, и реакционную смесь перемешивают в течение ночи при 60°C. Летучие вещества удаляют при пониженном давлении, остаток растворяют в этилацетате, промывают 0,1 н. HCl, солевым раствором и водой, сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Сырой остаток очищают хроматографией (колонка с SiO2) с элюированием EtOAc/DCM, получая [2-(3-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}фенил)-2-метилпропионил]амид 2-метилпропан-2-сульфоновой кислоты (25 мг). ЖХ-МС: Rt =2,67 мин, МС: 579, 581 (M+H). IC50=2 нМ.
(b) [2-(3-{6-[2-(2,4-Дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}фенил)-2-метилпропионил]амид N,N-диметиламид-2-сульфоновой кислоты
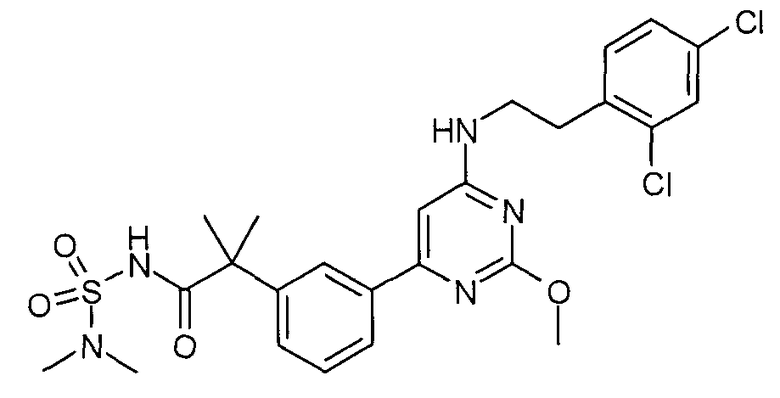
Методом, аналогичным описанному в примере 7(a), стадия 4, но используя N,N-диметилсульфамид вместо трет-бутилсульфонамида, получают [2-(3-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}фенил)-2-метилпропионил]амид N,N-диметиламид-2-сульфоновой кислоты (185 мг). ЖХ-МС: Rt =2,26 мин, МС: 566, 568 (M+H).
(c) 2-(3-{6-[2-(2,4-Дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}фенил)-2-метил-1-тиоморфолин-4-илпропан-1-он
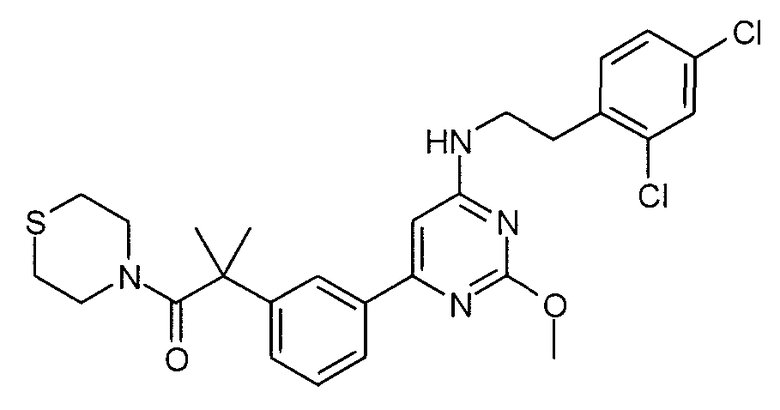
Методом, аналогичным описанному в примере 7(a), стадия 4, но используя тиоморфолин вместо трет-бутилсульфонамида, получают 2-(3-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}фенил)-2-метил-1-тиоморфолин-4-илпропан-1-он (120 мг). ЖХ-МС: Rt=2,68 мин, МС: 545, 547 (M+H). IC50=383 нМ
(d) 2-(3-{6-[2-(2,4-Дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}фенил)изобутирамид
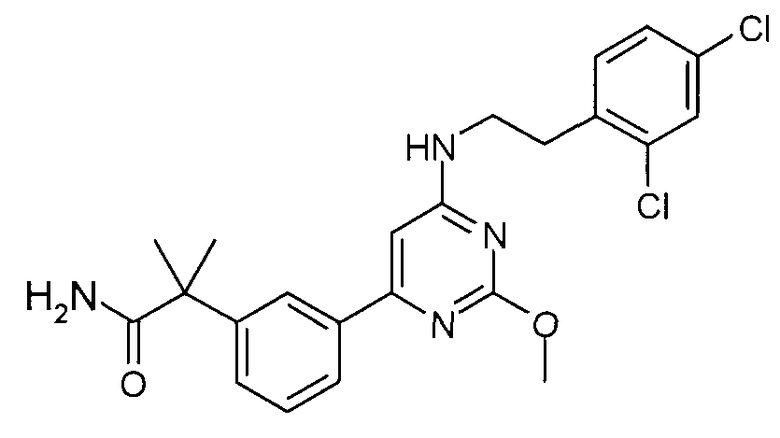
Методом, аналогичным описанному в примере 7(a), стадия 4, но используя бикарбонат аммония вместо трет-бутилсульфонамида, получают 2-(3-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}фенил)изобутирамид (120 мг). ЖХ-МС: Rt=2,01 мин, МС: 459, 461 (M+H).
(e) 2-(3-{6-[2-(2,4-Дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}фенил)-N,N-диметилизобутирамид
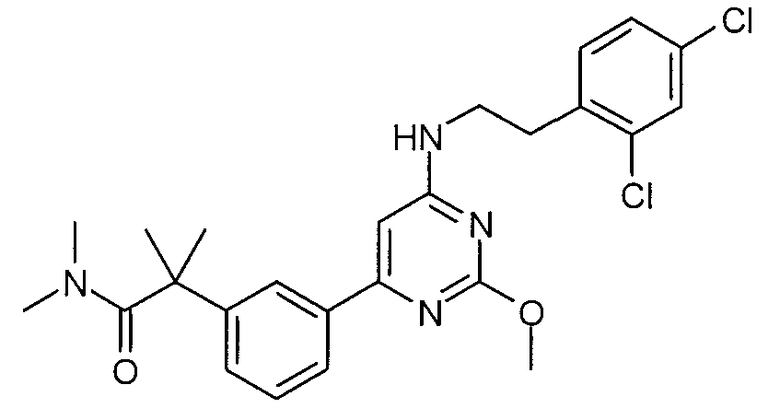
Методом, аналогичным описанному в примере 7(a), стадия 4, но используя диметиламин вместо трет-бутилсульфонамида, получают 2-(3-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}фенил)-N,N-диметилизобутирамид (186 мг). ЖХ-МС: Rt=2,44 мин, МС: 487, 489 (M+H).
Пример 8
(1-{6-[2-(2,4-Дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-ил)уксусная кислота
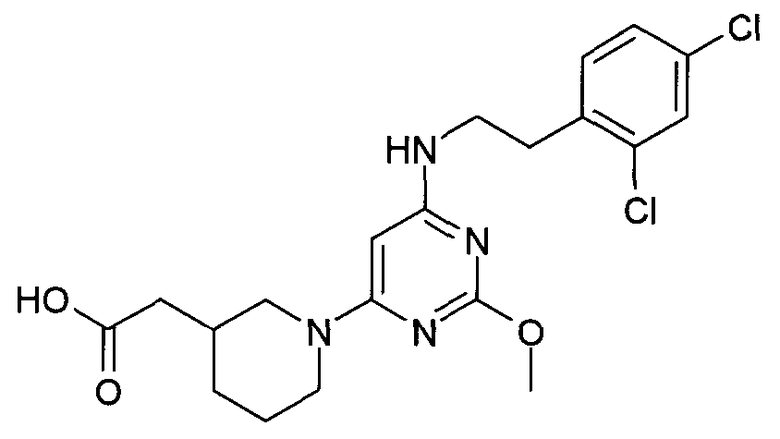
Стадия 1: Раствор (6-хлор-2-метоксипиримидин-4-ил)-[2-(2,4-дихлорфенил)этил]амина (3 ммоль), этилового эфира пиперидина уксусной кислоты (7,5 ммоль) и K2CO3 (9 ммоль) в 1-метил-2-пирролидиноне (10 мл) перемешивают в течение ночи при 145°C. Реакционную смесь охлаждают до комнатной температуры, разбавляют водой (60 мл) и дважды экстрагируют DCM. Водный слой медленно подкисляют 1 н. соляной кислотой до pH 4, энергично перемешивая, и перемешивание продолжается в течение 1,5 часа. Образовавшийся осадок фильтруют отсасыванием и сушат на воздухе, получая этиловый эфир (1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-ил)уксусной кислоты (1,42 г). ЖХ-МС: Rt=2,35 мин, МС: 467, 469 (M+H).
Стадия 2: Моногидрат гидроксида лития (54 мг) добавляют к перемешиваемому раствору этилового эфира (1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-ил)уксусной кислоты (0,2 г) в MeOH/H2O (10 мл, 9:1). Реакционную смесь перемешивают в течение ночи при комнатной температуре. Реакционную смесь разбавляют водой и летучие вещества удаляют в вакууме. Водную фазу однократно экстрагируют Et2O, подкисляют до pH 4 (1н, HCl) и дважды экстрагируют этилацетатом. Объединенный органический слой сушат (MgSO4) и концентрируют в вакууме, получая (1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-ил)уксусную кислоту (180 мг). ЖХ-МС: Rt=2,08 мин, МС: 439, 441 (M+H). IC50=0,5 нМ.
Пример 9
1-{2-Метокси-6-[2-(4-трифторметоксифенил)этиламино]пиримидин-4-ил}пиперидин-3-карбоновая кислота
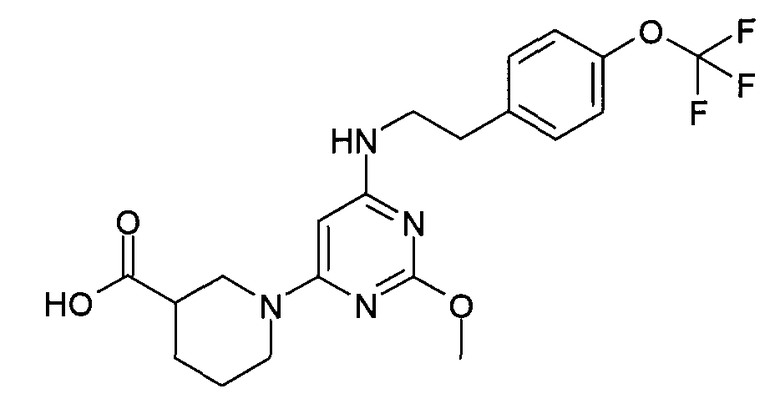
Раствор (6-хлор-2-метоксипиримидин-4-ил)-[2-(4-трифторметоксифенил)этил]амина (1 г), нипекотиновой кислоты (0,93 г) и K2CO3 (1,19 г) в 1-метил-2-пирролидиноне (10 мл) перемешивают в течение ночи при 145°C. Реакционную смесь охлаждают до комнатной температуры, разбавляют водой (60 мл) и дважды экстрагируют дихлорметаном. Водный слой медленно подкисляют 1 н. соляной кислотой до pH 4, энергично перемешивая, и перемешивание продолжается в течение 1,5 часа. Образовавшийся осадок фильтруют отсасыванием и сушат на воздухе, получая 1-{2-метокси-6-[2-(4-трифторметоксифенил)этиламино]пиримидин-4-ил}пиперидин-3-карбоновую кислоту в виде порошка (0,99 г). ЖХ-МС: Rt=2,07 мин, МС: 441 (M+H). IC50=9 нМ.
Пример 10
N-(1-{2-Метокси-6-[2-(4-трифторметоксифенил)этиламино]пиримидин-4-ил}пиперидин-3-карбонил)метансульфонамид
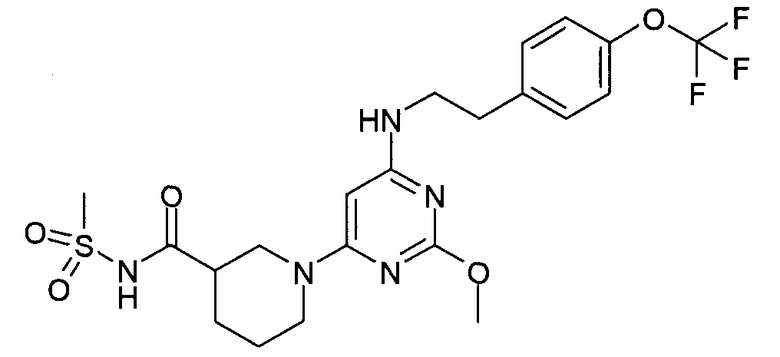
Гидрохлорид N-(3-диметиламинопропил)-N-этилкарбодиимида (68 мг) добавляют в перемешиваемый ледяной раствор 1-{2-метокси-6-[2-(4-трифторметоксифенил)этиламино]пиримидин-4-ил}пиперидин-3-карбоновой кислоты (150 мг), метансульфонамида (48,6 мг) и 4-диметиламинопиридина (50 мг) в сухом DCM в атмосфере N2. Ледяную баню убирают, и реакционную смесь перемешивают в течение ночи, доводя до комнатной температуры. Смесь концентрируют в вакууме. Остаток растворяют в этилацетате, промывают 0,1 н. HCl, солевым раствором и водой, сушат (Na2SO4), фильтруют и концентрируют. Остаток очищают хроматографией (колонка с SiO2) с элюированием EtOAc/DCM, получая N-(1-{2-метокси-6-[2-(4-трифторметоксифенил)этиламино]пиримидин-4-ил}пиперидин-3-карбонил)метансульфонамид (65 мг). ЖХ-МС: Rt=2,09 мин, МС: 518 (M+H). IC50=22 нМ.
Пример 11
(a) N-(1-{6-[2-(2,4-Дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбонил)метансульфонамид
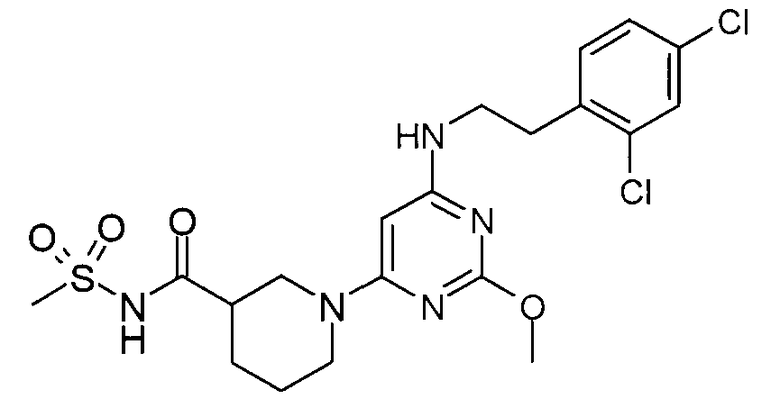
Стадия 1: В пробирке смешивают (6-хлор-2-метоксипиримидин-4-ил)-[2-(2,4-дихлорфенил)этил]амин (200 мг), нипекотиновую кислоту (194 мг), K2CO3 (249 мг) и 1-метил-2-пирролидинон (2,5 мл). Пробирку запаивают, нагревают до 140°C и перемешивают в течение 5 часов. Смесь охлаждают до комнатной температуры, оставляют на 12 часов, разбавляют водой (20 мл) и подкисляют с помощью 3M водной HCl. Образующийся осадок собирают фильтрацией и сушат в высоком вакууме, получая 1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбоновую кислоту (121 мг) в виде твердого вещества. ЖХ-МС Rt=2,15 мин, МС: 425 (M+H).
Стадия 2: N-(3-Диметиламинопропил)-N-этилкарбодиимидгидрохлорид (71 мг) добавляют в перемешиваемый ледяной раствор 1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбоновой кислоты (150 мг), метансульфонамида (43,6 мг) и 4-диметиламинопиридина (52 мг) в сухом дихлорметане в присутствии N2. Ледяную баню убирают, и реакционную смесь перемешивают в течение ночи при комнатной температуре. Смесь концентрируют в вакууме. Остаток растворяют в этилацетате, промывают 0,1 н. HCl, солевым раствором и водой, сушат (Na2SO4), фильтруют и концентрируют. Остаток очищают хроматографией (колонка с SiO2) с элюированием EtOAc/DCM, получая N-(1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбонил)метансульфонамид (145 мг). ЖХ-МС: Rt=1,88 мин, МС: 502, 504 (M+H). IC50=1 нМ.
(b) (1-{6-[2-(2,4-Дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбонил)амид этансульфоновой кислоты
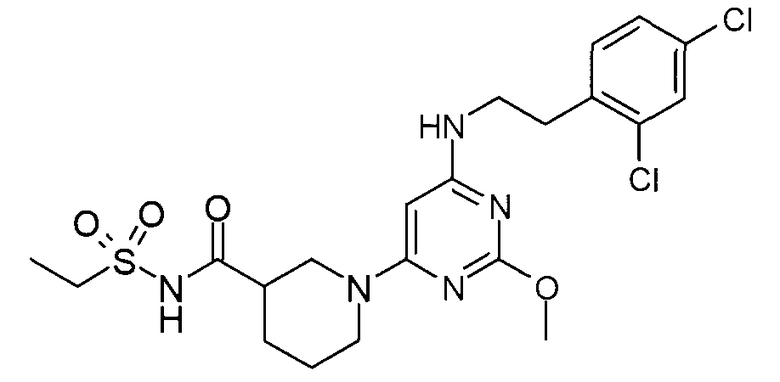
Методом, аналогичным описанному в примере 11(a), стадия 2, но используя этансульфонамид вместо метансульфонамида, получают (1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбонил)амид этансульфоновой кислоты (125 мг). ЖХ-МС: Rt=2,12 мин, МС: 516, 518 (M+H).
(c) (1-{6-[2-(2,4-Дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбонил)амид 2-метилпропан-2-сульфоновой кислоты
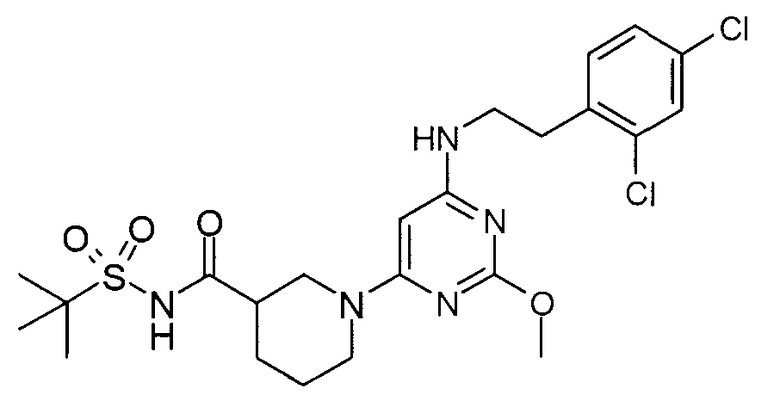
Методом, аналогичным описанному в примере 11(a), стадия 2, но используя трет-бутилсульфонамид вместо метансульфонамида, получают (1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбонил)амид 2-метилпропан-2-сульфоновой кислоты (132 мг). ЖХ-МС: Rt=2,2 мин, МС: 544, 546 (M+H).
(d) N-(1-{6-[2-(2,4-Дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбонил)-C,C,C-трифторметансульфонамид
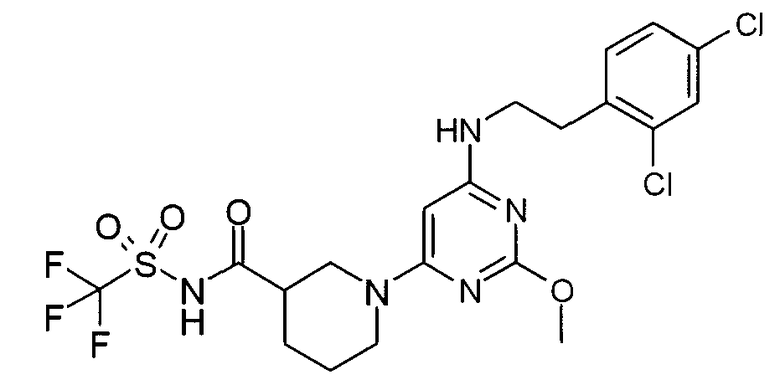
Методом, аналогичным описанному в примере 11(a), стадия 2, но используя трифторметилсульфонамид вместо метансульфонамида, получают N-(1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбонил)-C,C,C-трифторметансульфонамид (257 мг). ЖХ-МС: Rt=2,3 мин, МС: 556, 558 (M+H). IC50=18 нМ.
(e) (1H-Тетразол-5-ил)амид 1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбоновой кислоты
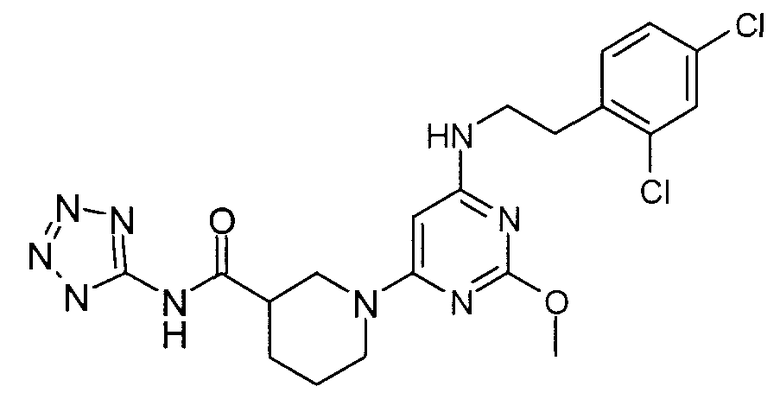
Методом, аналогичным описанному в примере 11(a), стадия 2, но используя 1H-тетразол-5-иламин вместо метансульфонамида, получают (1H-тетразол-5-ил)амид 1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбоновой кислоты (15 мг). ЖХ-МС: Rt=1,81 мин, МС: 492, 494 (M+H). IC50=4,4 нМ.
(f) Амид 1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбоновой кислоты
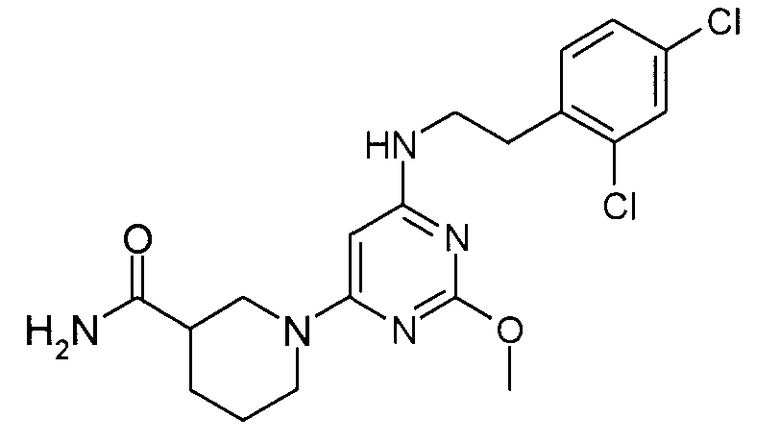
Гидрохлорид N-(3-диметиламинопропил)-N-этилкарбодиимида (0,23 ммоль) добавляют в перемешиваемый ледяной раствор 1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбоновой кислоты (0,22 ммоль), этансульфонамида (0,23 ммоль) и 4-диметиламинопиридина (0,22 ммоль) в сухом дихлорметане в атмосфере N2. Ледяную баню убирают, и реакционную смесь перемешивают в течение ночи при 60°C. Смесь концентрируют в вакууме. Остаток растворяют в этилацетате, промывают 0,1н HCl, солевым раствором и водой, сушат (Na2SO4), фильтруют и концентрируют при пониженном давлении. Остаток очищают хроматографией (колонка с SiO2) с элюированием EtOAc/DCM, получая амид 1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбоновой кислоты (75 мг). ЖХ-МС: Rt=1,77 мин, МС: 424, 426 (M+H).
(g) Диметиламид 1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбоновой кислоты
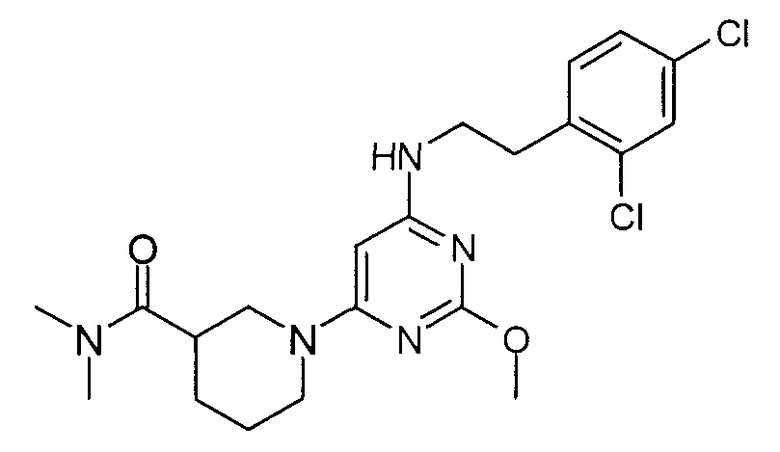
Методом, аналогичным описанному в примере 11(a), стадия 2, но используя диметиламин вместо метансульфонамида, получают диметиламид 1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбоновой кислоты (65 мг). ЖХ-МС: Rt=1,88 мин, МС: 452, 454 (M+H).
(h) 1-{6-[2-(2,4-Дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбоксамид N,N-диметиламид-2-сульфоновой кислоты
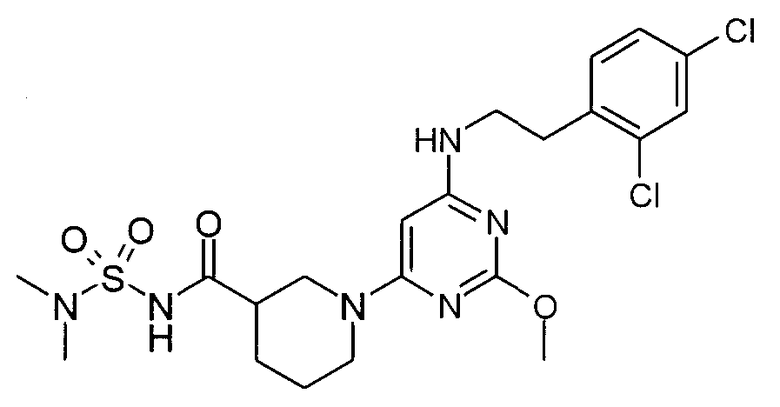
Методом, аналогичным описанному в примере 11(a), стадия 2, но используя N,N-диметилсульфамид вместо метансульфонамида, получают 1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбоксамид N,N-диметиламид-2-сульфоновой кислоты (241 мг). ЖХ-МС: Rt=2,5 мин, МС: 531, 533 (M+H). IC50=14 нМ.
Пример 12
5-{2-Метокси-6-[2-(4-трифторметоксифенил)этиламино]пиримидин-4-ил}-тиофен-2-карбоновая кислота
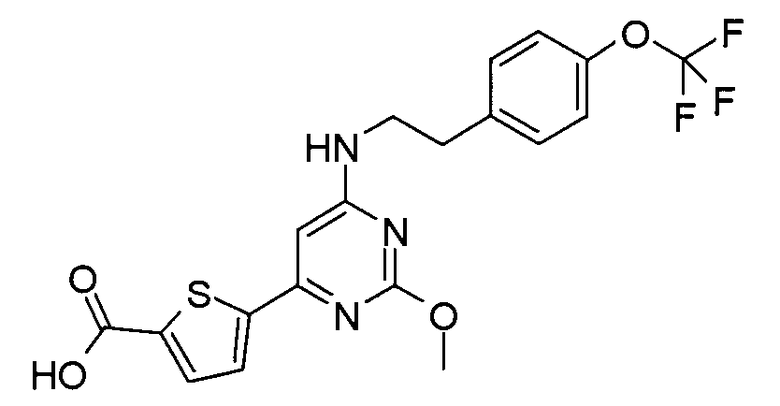
Стадия 1: 5-(Дигидроксиборил)-2-тиофенкарбоновую кислоту (527 мг) и 2,2-диметилпропан-1,3-диол (361 мг) перемешивают при комнатной температуре в THF (10 мл) в течение 19 часов и концентрируют в вакууме, получая 5-(5,5-диметил-[1,3,2]диоксаборинан-2-ил)-тиофен-2-карбоновую кислоту (748 мг) в виде твердого вещества. ЖХ-МС: Rt=1,15 мин; 1H ЯМР [300 МГц, (CD3)2SO]: δ 13,15 (1H, с); 7,7 (1H, м); 7,45 (1H, м); 3,75 (4H, с); 0,95 (6H, с).
Стадия 2: Смесь (6-хлор-2-метоксипиримидин-4-ил)-[2-(4-трифторметоксифенил)этил]амина (267 мг), 5-(5,5-диметил-[1,3,2]диоксаборинан-2-ил)-тиофен-2-карбоновой кислоты (277 мг), фторида цезия (351 мг) и тетракис(трифенилфосфин)палладия (71 мг) в воде (1,6 мл) и диметиловом эфире этиленгликоля (6,4 мл) дегазируют продуванием азота в течение 5 минут и нагревают до 85°C в течение 16 часов. Реакционную смесь охлаждают, разбавляют водой (150 мл) и солевым раствором (25 мл), дважды экстрагируют EtOAc (100 мл), и экстракты концентрируют в вакууме. Остаток очищают флэш-хроматографией на колонке с силикагелем (10 г) с элюированием смесью от 0 до 5% MeOH в EtOAc. Полученное твердое кристаллическое вещество растирают в порошок в DCM (5 мл) и эфире (5 мл) и сушат, получая 5-{2-метокси-6-[2-(4-трифторметоксифенил)этиламино]пиримидин-4-ил}-тиофен-2-карбоновую кислоту (42 мг) в виде твердого вещества. МС: 440; ЖХ-МС: Rt=3,48 мин; 1H ЯМР [300 МГц, (CD3)2SO]: δ 7,7 (3H, м); 7,35 (2H, м); 7,25 (2H, м), 6,6 (1H, с); 3,85 (3H, с); 2,55 (2H; м); 1,9 (2H, т, J=7 Гц). IC50=2 нМ.
Пример 13
Гидрохлорид 5-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}-2,3-дигидробензофуран-2-карбоновой кислоты

Стадия 1: К раствору 2,3-дигидробензофуран-2-карбоновой кислоты (510 мг) в ледяной уксусной кислоте (4 мл) добавляют по каплям бромин (497 мг). Через 16 часов реакцию гасят водой (100 мл) и бисульфитом натрия (1 г) и дважды экстрагируют EtOAc (100 мл). Экстракты концентрируют в вакууме и сушат в высоком вакууме, получая 5-бром-2,3-дигидробензофуран-2-карбоновую кислоту (811 мг) в виде твердого вещества. МС: 241 (M+H), 1H ЯМР [300 МГц, (CD3)2SO]: δ 13,05 (1H, с); 7,4 (1H, с); 7,25 (1H, д); 6,8 (1H, м); 5,25 (1H, кв), 3,55 (1H, дд); 3,25 (1H, м).
Стадия 2: Смесь 5-бром-2,3-дигидробензофуран-2-карбоновой кислоты (0,74 г), бис(пинаколато)дибора (1,51 г), ацетата калия (1,47 г, 15 ммоль) и PdCl2(dppf)2 (115 мг, 0,14 ммоль) в диметилсульфоксиде (10 мл) дегазируют продуванием азота в течение 5 минут. Смесь нагревают до 90°C в течение 16 часов. Реакционную смесь охлаждают, разбавляют водой (200 мл) и солевым раствором (25 мл) и фильтруют через целит, а затем добавляют воду (200 мл) и EtOAc (200 мл). Фильтрат дважды экстрагируют EtOAc (200 мл), и экстракты концентрируют в вакууме. Остаток очищают флэш-хроматографией на колонке с силикагелем (4 г) с элюированием смесью от 80 до 100% EtOAc в гептане, получая 5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-2,3-дигидробензофуран-2-карбоновую кислоту (715 мг) в виде маслянистого вещества. МС: 289 (M-H), 1H ЯМР [300 МГц, (CD3)2SO]: δ 13,05 (1H, с); 7,5 (2H, м); 6,8 (1H, м); 5,2 (1H, м); 3,6 (1H, м); 3,3 (1H, м); 1,05 (12H, с).
Стадия 3: Смесь (6-хлор-2-метоксипиримидин-4-ил)-[2-(2,4-дихлорфенил)этил]амина (212 мг), 5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-2,3-дигидробензофуран-2-карбоновой кислоты (124 мг), карбоната цезия (414 мг) и тетракис(трифенилфосфин)палладия (49 мг) в воде (1,2 мл) и диметиловом эфире этиленгликоля (4,8 мл) дегазируют продуванием азота в течение 5 минут и нагревают до 70°C в течение 64 часов. Реакционную смесь охлаждают, разбавляют водой (150 мл) и солевым раствором (25 мл) и дважды экстрагируют EtOAc (150 мл), и экстракты концентрируют в вакууме. Остаток очищают флэш-хроматографией на колонке с силикагелем (4 г) с элюированием смесью от 0 до 25% MeOH в EtOAc, получая 5-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}-2,3-дигидробензофуран-2-карбоновую кислоту (80 мг) в виде маслянистого вещества. МС: 460; ЖХ-МС: Rt=2,81 мин.
Стадия 4: Часть 5-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}-2,3-дигидробензофуран-2-карбоновой кислоты обрабатывают флэш-хроматографией на колонке с силикагелем (5 г) с элюированием смесью от 0% до 25% MeOH в EtOAc. Продукт растворяют в MeOH и обрабатывают 0,5M соляной кислотой в MeOH и концентрируют в вакууме. Продукт растворяют в THF (3 мл) и эфире (10 мл). Осадок удаляют и сушат, получая гидрохлорид 5-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}-2,3-дигидробензофуран-2-карбоновой кислоты (20 мг) в виде твердого вещества. ЖХ-МС: Rt=2,79 мин; МС: 460. IC50=2 нМ.
ФАРМАКОЛОГИЧЕСКИЙ ТЕСТ
Ингибиторное действие соединений, являющихся объектом данного изобретения, оценивают при помощи функционального анализа на простагландин D человека. Применяется анализ цАМФ с использованием линии клеток человека LS174T, экспрессирующих эндогенный рецептор простагландина D. Протокол аналогичен описанному ранее (Wright DH, Ford-Hutchinson AW, Chadee K, Metters KM, The human prostanoid DP receptor stimulates mucin secretion in LS174T cells, Br J Pharmacol. 131(8):1537-45 (2000)).
Протокол SPA-анализа цАМФ в клетках LS174 T человека
Материалы
- PGD2 (Cayman Chemical, кат. №12010)
- IBMX (Sigma кат. №5879)
- Система прямого скринингового SPA-анализа цАМФ (код Amersham RPA 559)
- 96-луночные планшеты для клеточных культур (Wallac кат. №1450-516)
- Сцинтилляционный счетчик Wallac 1450 Microplate Trilux (PerkinElmer)
- Приспособления для заклеивания планшетов
- Центрифужные пробирки Eppendorf
- Физиологический раствор в фосфатном буфере (PBS) Дульбекко (Invitrogen кат. №14040-133)
- Дистиллированная вода
- Встряхиватель
- Магнитная мешалка и набор якорей
Подготовка реагентов
Перед разбавлением все реагенты должны быть доведены до комнатной температуры.
Аналитический буфер 1X
Переносят содержимое бутылки в 500 мл мензурку многократным промыванием дистиллированной водой. Доводят полный объем до 500 мл, добавив дистиллированную воду, и тщательно перемешивают.
Лизирующий реагент 1 и 2
Растворяют каждый из лизирующих реагентов 1 и 2 в 200 мл аналитического буфера, соответственно. Оставляют на 20 минут при комнатной температуре для растворения.
Гранулы антител кролика для SPA-анализа
Добавляют в бутылку 30 мл лизирующего буфера 2. Осторожно встряхивают бутылку в течение 5 минут.
Иммунная сыворотка
Добавляют 15 мл лизирующего буфера 2 в каждый сосуд и осторожно перемешивают содержимое до полного растворения.
Маркер (I 125 -цАМФ)
Добавляют 14 мл лизирующего буфера 2 в каждый сосуд и осторожно перемешивают содержимое до полного растворения.
Подготовка иммунореагента
1) Добавляют равные объемы маркера, иммунной сыворотки и SPA-реагента антител кролика в сосуд, проследив за тем, чтобы был приготовлен достаточный объем для нужного количества лунок (150 мкл на лунку).
2) Тщательно перемешивают.
3) Необходимо готовить свежий раствор иммунореагента перед каждым анализом и не использовать его повторно.
Стандарт
1) Добавляют 1 мл лизирующего буфера 1 и осторожно перемешивают содержимое до полного растворения.
2) Конечный раствор содержит цАМФ в концентрации 512 пмоль/мл.
3) Помечают 7 полипропиленовых или полистирольных пробирок 0,2 пмоль, 0,4 пмоль, 0,8 пмоль, 1,6 пмоль, 3,2 пмоль, 6,4 пмоль и 12,8 пмоль.
4) Пипеткой добавляют 500 мкл лизирующего буфера 1 во все пробирки.
5) В пробирку «12,8 пмоль» добавляют пипеткой 500 мкл стандартной смеси (512 пмоль/мл) и тщательно перемешивают. Переносят 500 мкл из пробирки «12,8 пмоль» в пробирку «6,4 пмоль» и тщательно перемешивают. Повторяют процедуру разбавления вдвое с остальными пробирками.
6) Порции по 50 мкл в двух экземплярах из каждого последовательного разбавления и стандарт исходного раствора позволяют получить 8 стандартных уровней цАМФ в диапазоне от 0,2 до 25,6 пмоль.
Буфер разбавления соединения
Добавляют 50 мкл 1 мМ IBMX в 100 мл PBS, чтобы получить конечную концентрацию 100 мкМ, и обрабатывают ультразвуком при 30°C в течение 20 минут.
Подготовка PGD2
Растворяют 1 мг PGD2 (FW, 352,5) в 284 мкл ДМСО, чтобы получить исходный раствор 10 мМ, и хранят его при 20°C. Перед каждым анализом необходимо готовить свежий раствор. Добавляют 3 мкл 10 мМ исходного раствора в 20 мл ДМСО, тщательно перемешивают и переносят 10 мл в 40 мл PBS.
Разбавление соединения
Разбавление соединения проводят в Biomex 2000 (Beckman) при помощи метода 11 точек 1_цАМФ DP.
5 мкл каждого соединения из 10 мМ планшетов стандартных растворов соединений переносят в лунки 96-луночного планшета, как указано ниже.
Наполняют планшет 45 мкл ДМСО, кроме колонки 7, куда добавляют 28 мкл ДМСО. Тщательно отмеряют пипеткой содержимое колонки 1 и переносят 12 мкл параллельно в колонку 7. Выполняют последовательное разбавление 1:10 от колонки 1 к колонке 6 и от колонок 7 к колонке 11, перенося от 5 мкл до 45 мкл ДМСО, чтобы получить следующие концентрации:
Добавляют в новый 96-луночный планшет 247,5 мкл буфера разбавления соединения. Переносят 2,5 мкл последовательно разбавленных соединений из приготовленного выше планшета в новый (разбавление 1:100) следующим образом:
Рост клеток
1. LS174 T всегда выращивают в MEM (ATCC кат. №30-2003), 10% FBS (ATCC кат. №30-2020) и дополнительных 2 мМ L-глутамина, при 37°C и 5% CO2.
2. Нагревают 0,05% трипсина и версина (Invitrogen кат. №25300-054) при 37°C на водяной бане.
3. Удаляют среду для роста из клеток. Клетки в колбе T165 дважды промывают 4 мл трипсина, а затем инкубируют при 37°C и 5% CO2 в течение 3 минут.
4. Добавляют 10 мл среды и пипеткой тщательно отделяют и пересчитывают клетки.
5. Доводят плотность клеток до 2,25×105 клеток на мл и высевают 200 мкл клеток на лунку (45000 клеток на лунку) в 96-луночном планшете за 1 день до анализа.
Порядок проведения анализа
День 1
Высевают 45000 клеток на лунку в 200 мкл среды в 96-луночных планшетах. Инкубируют планшеты с клетками при 37°C, 5% CO2 и 95% влажности в течение ночи.
День 2
1. Выполняют разбавление соединений.
2. Готовят аналитический буфер, лизирующие буферы 1 и 2, PGD2 и стандарт.
3. Удаляют среду из клеток отсасыванием и добавляют 100 мкл раствора соединения, следуя протоколу цАМФ DP Zymark Sciclone-ALH/FD.
4. Инкубируют клетки при 37°C, 5% CO2 и 95% влажности в течение 15 минут.
5. Добавляют 5 мкл 300 нМ PGD2 (20×15 нМ конечной концентрации) в каждую лунку при помощи протокола Zymark цАМФ DP PGD2 и инкубируют клетки при 37°C, 5% CO2 и 95% влажности еще 15 минут.
6. Удаляют среду из клеток отсасыванием и добавляют 50 мкл лизирующего буфера 1 при помощи протокола Zymark цАМФ DP лизиса и инкубируют при комнатной температуре с встряхиванием в течение 30 минут.
7. Добавляют во все лунки 150 мкл иммунореагента (полный объем 200 мкл на лунку).
8. Герметизируют планшет, встряхивают в течение 2 минут и помещают в камеру сцинтилляционного счетчика для микротитровальных планшетов Wallac на 16 часов.
День 3
Проводят счет количества [125I] цАМФ в течение 2 минут в сцинтилляционном счетчике 1450 Trilux.
Обработка данных
Откладывают стандартную зависимость цАМФ от количества импульсов в минуту.
Концентрации цАМФ (пмоль/мл) неизвестных образцов вычисляются по стандартной зависимости цАМФ от количества импульсов в минуту. Процент ингибирования вычисляется по следующей формуле:
Осуществления настоящего изобретения могут принимать другие конкретные формы, которые не будут затрагивать сущности изобретения или его основных положений.
| название | год | авторы | номер документа |
|---|---|---|---|
| 2,6-ЗАМЕЩЕННЫЕ-4-МОНОЗАМЕЩЕННЫЙ АМИНО-ПИРИМИДИНЫ КАК АНТАГОНИСТЫ РЕЦЕПТОРА ПРОСТАГЛАНДИНА D2 | 2005 |
|
RU2417990C2 |
| ЗАМЕЩЕННЫЕ N-ФЕНИЛ-БИПИРРОЛИДИНМОЧЕВИНЫ И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2008 |
|
RU2478094C2 |
| ПЕРВИЧНАЯ КИСЛАЯ ФОСФАТНАЯ СОЛЬ АНТАГОНИСТА РЕЦЕПТОРА ПРОСТАГЛАНДИНА D2 | 2006 |
|
RU2419614C2 |
| АМИДНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ АКТИВАТОРОВ GK | 2002 |
|
RU2374236C2 |
| ПРОИЗВОДНЫЕ 3-ЗАМЕЩЕННОГО 1,5-ДИФЕНИЛПИРАЗОЛА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ СВ1 МОДУЛЯТОРОВ | 2005 |
|
RU2375349C2 |
| НОВЫЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ В КАЧЕСТВЕ ИНГИБИТОРОВ КАТЕПСИНА | 2004 |
|
RU2346943C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНМОЧЕВИНЫ | 2014 |
|
RU2666894C2 |
| АМИНОТЕТРАГИДРОПИРАНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ДИПЕПТИДИЛПЕПТИДАЗЫ-IV ДЛЯ ЛЕЧЕНИЯ ИЛИ ПРЕДУПРЕЖДЕНИЯ ДИАБЕТА | 2010 |
|
RU2550508C2 |
| ТИЕНОПИРАЗОЛЫ | 2004 |
|
RU2358978C2 |
| СОЛИ ГЕТЕРОЦИКЛИЛАМИДЗАМЕЩЕННЫХ ИМИДАЗОЛОВ С СУЛЬФОНОВОЙ КИСЛОТОЙ | 2012 |
|
RU2606639C2 |
Изобретение относится к новым производным 2,6-замещенных-4-монозамещенных аминопиримидинов формулы (I) или их фармацевтически приемлемым солям, которые обладают свойствами антагониста рецептора простагландина D2. В формуле (I):
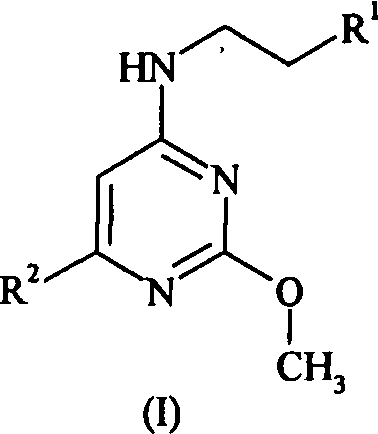
R1 представляет собой 2,4-дихлорфенил или 4-трифторметоксифенил, и когда R1 представляет собой 2,4-дихлорфенил, тогда R2 представляет собой 3-карбоксипирролидинил, 3,5-ди-(1-гидрокси-1-метилэтил)фенил, 3-аминопиперидин-1-ил, 4-аминопиперидин-1-ил, 4-ацетамидпиперидин-1-ил, 1-метил-2-карбокси-2,3-дигидро-1Н-индол-5-ил, 3-(1-трет-бутилсульфониламинокарбонил-1-метилэтил)фенил, 3-(1-диметиламиносульфониламинокарбонил-1-метилэтил)фенил, 3-(1-тиоморфолин-4-илкарбонил-1-метилэтил)фенил, 3-(1-аминокарбонил-1-метилэтил)фенил, 3-(1-диметиламинокарбонил-1-метилэтил)фенил, 3-карбоксиметилпиперидин-1-ил, 3-метилсульфониламинокарбонилпиперидин-1-ил, 3-этилсульфониламинокарбонилпиперидин-1-ил, 3-трет-бутилсульфониламинокарбонилпиперидин-1-ил, 3-трифторметилсульфониламинокарбонилпиперидин-1-ил, 3-[(1Н-тетразол-5-ил)аминокарбонил]пиперидин-1-ил, 3-аминокарбонилпиперидин-1-ил, 3-диметиламинокарбонилпиперидин-1-ил, 3-диметиламиносульфониламинокарбонилпиперидин-1-ил или 2-карбокси-2,3-дигидробензофуран-5-ил, и когда R1 представляет собой 4-трифторметоксифенил, тогда R2 представляет собой 3-(1-метил-1-карбоксиэтил)пиперидинил, 3-карбоксипиперидинил, 3-метилсульфониламинокарбонилпиперидин-1-ил, 5-карбокситиофен-2-ил. Изобретение также относится к фармацевтической композиции, содержащей указанные соединения. 2 н. и 1 з.п. ф-лы, 1 табл.
1. Соединение формулы (I)
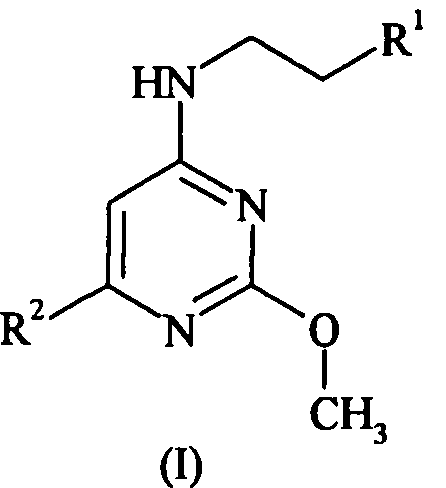
где R1 представляет собой 2,4-дихлорфенил или 4-трифторметоксифенил, и
когда R1 представляет собой 2,4-дихлорфенил, тогда R2 представляет собой 3-карбоксипирролидинил, 3,5-ди-(1-гидрокси-1-метилэтил)фенил, 3-аминопиперидин-1-ил, 4-аминопиперидин-1-ил, 4-ацетамидпиперидин-1-ил, 1-метил-2-карбокси-2,3-дигидро-1Н-индол-5-ил, 3-(1-трет-бутилсульфониламинокарбонил-1-метилэтил)фенил, 3-(1-диметиламиносульфониламинокарбонил-1-метилэтил)фенил, 3-(1-тиоморфолин-4-илкарбонил-1-метилэтил)фенил, 3-(1-аминокарбонил-1-метилэтил)фенил, 3-(1-диметиламинокарбонил-1-метилэтил)фенил, 3-карбоксиметилпиперидин-1-ил,
3-метилсульфониламинокарбонилпиперидин-1-ил,
3-этилсульфониламинокарбонилпиперидин-1-ил,
3-трет-бутилсульфониламинокарбонилпиперидин-1-ил,
3-трифторметилсульфониламинокарбонилпиперидин-1-ил,
3-[(1Н-тетразол-5-ил)аминокарбонил]пиперидин-1-ил,
3-аминокарбонилпиперидин-1-ил, 3-диметиламинокарбонилпиперидин-1-ил,
3-диметиламиносульфониламинокарбонилпиперидин-1-ил или
2-карбокси-2,3-дигидробензофуран-5-ил,
и когда R1 представляет собой 4-трифторметоксифенил, тогда R2 представляет собой 3-(1-метил-1-карбоксиэтил)пиперидинил, 3-карбоксипиперидинил,
3-метилсульфониламинокарбонилпиперидин-1-ил, 5-карбокситиофен-2-ил,
или его фармацевтически приемлемая соль.
2. Соединение по п.1, которое представляет собой
1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пирролидин-3-карбоновую кислоту,
2-(1-{2-метокси-6-[2-(4-трифторметоксифенил)этиламино]пиримидин-4-ил}пиперидин-3-ил)-2-метилпропионовую кислоту,
2-[3-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}-5-(1-гидрокси-1-метилэтил)фенил]пропан-2-ол,
[6-(3-аминопиперидин-1-ил)-2-метоксипиримидин-4-ил]-[2-(2,4-дихлорфенил)этил]амин,
[6-(4-аминопиперидин-1-ил)-2-метоксипиримидин-4-ил]-[2-(2,4-дихлорфенил)этил]амин,
N-(1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-4-ил)ацетамид,
5-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}-1-метил-2,3-дигидро-1Н-индол-2-карбоновую кислоту,
[2-(3-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}фенил)-2-метилпропионил]амид 2-метилпропан-2-сульфоновой кислоты,
[2-(3-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}фенил)-2-метилпропионил]амид N,N-диметиламид-2-сульфоновой кислоты,
2-(3-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}фенил)-2-метил-1-тиоморфолин-4-илпропан-1-он,
2-(3-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}фенил)изобутирамид,
2-(3-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}фенил)-N,N-диметилизобутирамид,
(1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-ил)уксусную кислоту,
1-{2-метокси-6-[2-(4-трифторметоксифенил)этиламино]пиримидин-4-ил}пиперидин-3-карбоновую кислоту,
N-(1-{2-метокси-6-[2-(4-трифторметоксифенил)этиламино]пиримидин-4-ил}пиперидин-3-карбонил)метансульфонамид,
N-(1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбонил)метансульфонамид,
(1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбонил)амид этансульфоновой кислоты,
(1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбонил)амид 2-метилпропан-2-сульфоновой кислоты,
N-(1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбонил)-С,С,С-трифторметансульфонамид,
(1Н-тетразол-5-ил)амид 1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбоновой кислоты,
амид 1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбоновой кислоты,
диметиламид 1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбоновой кислоты,
1-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}пиперидин-3-карбоксамид N,N-диметиламид-2-сульфоновой кислоты,
5-{2-метокси-6-[2-(4-трифторметоксифенил)этиламино]пиримидин-4-ил}-тиофен-2-карбоновую кислоту, или
5-{6-[2-(2,4-дихлорфенил)этиламино]-2-метоксипиримидин-4-ил}-2,3-дигидробензофуран-2-карбоновую кислоту,
или его фармацевтически приемлемая соль.
3. Фармацевтическая композиция, обладающая свойствами антагониста рецептора простагландина D2, включающая фармацевтически эффективное количество соединения по п.1 или его фармацевтически приемлемой соли в смеси с фармацевтически приемлемым носителем.
| WO 03066047 А, 14.08.2003 | |||
| TORISU K et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| СПОСОБ ПОЛУЧЕНИЯ 3-ЗАМЕЩЕННЫХ 2-ОКСОИНДОЛОВ | 1991 |
|
RU2039042C1 |

