Данное изобретение относится к новым металлоценам с арилзамещенными производными инденила в качестве лигандов, которые можно применять в качестве компонентов катализатора при получении полиолефинов с высокой степенью изотактичности, узким распределением молярной массы и большой молярной массой.
Полиолефины с высокой молярной массой имеют особое значение для получения пленки, пластин или полых изделий большого размера или фасонных деталей, таких как, например, трубы.
Из литературы известно получение полиолефинов с растворимыми металлоценовыми соединениями в комбинации с алюминоксанами или другими сокатализаторами, которые могут благодаря кислотности Льюиса превращать нейтральный металлоцен в катион и стабилизировать его.
Растворимые металлоценовые соединения на основе бис(циклопентадиенил)цирконий-диалкила и -дигалогенида могут в комбинации с олигомерными алюминоксанами способствовать полимеризации этилена с хорошей активностью и пропилена с очень высокой активностью. Получают полиэтилен с узким распределением молярной массы и средней молярной массой. Полученный таким образом полипропилен является атактическим и имеет очень низкую молярную массу. Получение изотактического полипропилена происходит при применении этилен-бис(4,5,6,7-тетрагидро-1-инденил)цирконийдихлорида вместе с алюминоксаном в ходе полимеризации в суспензионной среде (сравни EP 185 918). Полимер имеет узкое распределение молярной массы. Особым недостатком этого способа является то, что при требуемых по технологии температурах полимеризации можно получить только полимеры с очень низкой молярной массой. Был предложен также специальный метод предварительной активации металлоцена с помощью алюминоксана, который ведет к значительному увеличению активности системы катализатора и к явному улучшению морфологии полимерных гранул (сравни DE 37 26 067). Однако предварительная активация не существенно увеличивает молярную массу.
Далее известны катализаторы на основе этиленбисинденилгафнийдихлорида и этиленбис(4,5,6,7-тетрагидро-1-инденил) гафний-дихлорида и метилалюминоксана, с помощью которых путем полимеризации в суспензионной среде можно получить более высокомолекулярные полипропилены (сравни J. Am. Chem. Soc. 1987, 109, 6544). Однако в технически важных условиях полимеризации морфология гранул полимеров, полученных таким образом, является неудовлетворительной и активность используемых систем катализаторов является сравнительно низкой. Из-за больших расходов, связанных с катализаторами, не представляется возможным использование этих систем для относительно дешевой полимеризации. Явного увеличения молярной массы можно было достигнуть путем применения металлоценов, в которых зафиксированные при помощи мостика ароматические п-лиганды имеют заместители в положении 2 (сравни DE 40 35 886) или в положении 2 и 4 (сравни DE 41 28 238). Дальнейшее увеличение молярной массы было достигнуто путем применения ароматических п-лигандов с заместителями в положении 2, 4 и 6 (сравни DE 41 39 596), а также ароматических п-лигандов 4, 5- типа бензоинденила (сравни DE 41 39 595).
Металлоцены, названные последними, с указанными заместителями являются в этом отношении при температуре полимеризации в 70oC уже очень эффективными. Несмотря на это получаемые молярные массы при технически оптимальной температуре полимеризации в 70oC еще слишком низки для многих технических сфер применения, таких как, например, получение полимеров для труб и полых изделий большого формата, а также специальных волокон. Стремясь к более технически дешевому производству, нужно проводить полимеризацию при по возможности высоких температурах реакции, так как при более высоких температурах полимеризации выделяющуюся теплоту реакции можно поглотить при помощи небольшой массы охлаждающей среды. Поэтому можно устанавливать расход охлаждающей воды на низком уровне. Часто встречающийся недостаток растворимых (гомогенных) металлоценовых/метилалюминоксановых систем катализаторов в способах, в которых образованный полимер получают в виде твердого вещества, заключается в интенсивном образовании отложений на стенках ректора и мешалки. Эти отложения образуются из-за агломерации частиц полимеров, когда имеются металлоцен или алюминоксан или оба, растворенные в суспензионной среде. Подобные отложения в системах реакторов необходимо регулярно удалять, так как их толщина быстро растет, они имеют высокую степень твердости и мешают теплообмену с охлаждающей средой.
Поэтому представляется выгодным использовать металлоцены, нанесенными на подложку. Был предложен эффективный и простой способ нанесения металлоценов на подложку, который можно универсально использовать во всех способах полимеризации (сравни EP 92 107331.8).
Еще одним недостатком в случае стереоспецифической полимеризации прохиральных мономеров, например, пропилена, с помощью металлоценовых катализаторов является относительно низкая изотаксичность, которая выражается в случае изотактического полипропилена в низких точках плавления. Особенно металлоцены с заместителями в положении 2 и 4 и ра-диметилсилилбис(2-метил-4-изопропилинденил)цирконийдихлорид в комбинации с метилалюминоксаном дают в случае пропилена полимер с высокой степенью изотактичности и поэтому высокой точкой плавления (ср. E 41 28 238).
Несмотря на это достигнуты точки плавления при наиболее распространенных температурах полимеризации (например, 70oC) являются для некоторых технических сфер применения очень низкими. Разумеется, есть также технические сферы применения, в которых низкие точки плавления являются желательными. Задача заключалась в том, чтобы найти способ полимеризации и/или каталитическую систему, которые позволяют получать полимеры с очень высокой молярной массой и, в случае изоспецифической полимеризации прохиральных мономеров, полимеры с высокой степенью изотаксичности с высоким выходом. При помощи приема нанесения на подложку можно избежать недостатков, известных из уровня техники, возникающих из-за образования отложения и большого количества мелких кристаллов. Путем применения водорода в качестве регулятора молярной массы только с помощью металлоцена можно было бы перекрыть весь диапазон молярных масс, представляющих технический интерес.
Было установлено, что металлоцены со специальными производными инденила в качестве лигандов являются подходящими катализаторами (компонентами катализатора) при получении полиолефинов с высокой молярной массой, особенно при применении прохиральных мономеров изотактических полиолефинов с очень высокой молярной массой и очень высокой степенью изотактичности.
Путем реакции этих растворимых металлоценов с алюминийорганическим компонентом катализатора на носителе, получают систему катализатора, которой не требуется для активации никакого дополнительного сокатализатора, и которая полностью предотвращает образование отложений в реакторе.
Данное изобретение относится поэтому к соединениям формулы I
где
M1 = металл группы IVb, Vb или VIb Периодической системы,
R1 и R2 имеют одинаковое или различное значение и обозначают атом водорода, C1-C10-алкил, C1-C10-алкокси-, C6-C10-арил-, C6-C10-арилокси-, C2-C10-алкенил-, C7-C40-арилалкил, C7-C40-алкиларил-, C8-C40-арилалкенил-, OH-, группу или атом галогена. Остатки R3 имеют одинаковое или различное значение и обозначают атом водорода, атом галогена, C1-C10-алкил-, группу, которая может быть галогенирована, C6-C10-арилгруппу, NR
R4 - R12 имеют одинаковое или различное значение и имеют значения, указанные для R3, или соседние остатки R4 - R12 образуют с соединяющими их атомами одно или несколько ароматических или алифатических колец, или остатки R5 и R6 или R12 образуют с соединяющими их атомами одно ароматическое или алифатическое кольцо,
R13
= BR14, = AlR14, -Ge-, -O-, -S-, =SO, =SO2, =NR14, =CO-, =PR14 или = P(O)R14, при этом R14 и R15 имеют одинаковое или различное значение и обозначают атом водорода, атом галогена, C1-C10-алкил-, C1-C10-фторалкил-, C1-C10-арилокси-, C2-C10-алкенил-, C7-C40-арилалкил-, C7-C40-алкиларил, C8-C40-арилалкенилгруппу, или
R14 и R15 могут образовывать с соединяющими их атомами одно или несколько колец;
M2 является кремнием, германием или оловом.
Данное изобретение относится также к способу получения олефинового полимера путем полимеризации или сополимеризации олефина формулы Ra-CH=CH-Rb, где Ra и Rb имеют одинаковое или различное значение и обозначают атом водорода или остаток углеводорода с 1 - 14 C-атомами или Ra и Rb могут образовывать с соединяющими их атомами одно или несколько колец, при температуре от -60 до 200oC, при давлении от 0,5 до 100 бар, в растворе, в суспензионной среде или в газовой фазе, в присутствии катализатора, который состоит из металлоцена в качестве соединения переходного металла и сокатализатора, отличающийся тем, что металлоцен является соединением формулы I.
Соединения согласно изобретению являются металлоценами формулы I
где
M1 является металлом группы IVb, Vb или VIb Периодической системы, например, титан, цирконий, гафний, ванадий, ниобий, тантал, хром, молибден или вольфрам, преимущественно цирконий, гафний и титан;
R1 и R2 имеют одинаковое или различное значение и обозначают атом водорода, C1-C10-преимущественно C1-C3-алкилгруппу, C1-C10- преимущественно C1-C3-алкоксигруппу, C6-C10- преимущественно, C6-C8-арилгруппу, C6-C10-, преимущественно C6-C8-арилоксигруппу, C2-C10- преимущественно C2-C4-алкенилогруппу, C7-C40- преимущественно C7-C10-арилалкилгруппу, C7-C40-, преимущественно C7-C12-алкиларилгруппу, C8-C40- преимущественно C8-C12; арилалкенилгруппу или атом галогена, преимущественно хлор;
остатки R3 - R12 имеют одинаковое или различное значение и означают атом водорода, атом галогена, преимущественно фтор, хлор или бром, C1-C10-, преимущественно C1-C4-алкилгруппу, которая может быть галогенирована, C6-C10-, преимущественно C6-C8-арилгруппу, -NR
соседние остатки R4 - R12 могут образовывать с соединяющими их атомами ароматическое кольцо, преимущественно ароматическое кольцо с 6 членами, или алифатическое, преимущественно алифатическое кольцо с 4 - 8 членами;
R13 является
=BR14, =AlR14, -Ga, -O-, -S-, =SO-, =SO2, =NR14, =CO, =PR14, или P(O)R14
преимущественно
= BR14, = AlR14, -Ge-, -O-, -S-, = SO-, =SO2, =NR14, =CO, =PR14 или P(O)R14, при этом R14 и R15 имеют одинаковое или различное значение и обозначают атом водорода, атом галогена, C1-C10-преимущественно, C1-C4-алкигруппу, в основном метилгруппу, C1-C10-фторалкил-, преимущественно, CF3-группу, C6-C10-, преимущественно C6-C8-арил-, C6C10-фторарил-, преимущественно пентафторфенилгруппу, C1-C10-, преимущественно C1-C4-алкоксигруппу, в основном метоксигруппу, C2-C10-, преимущественно C2-C4-алкенилгруппу, C7-C40-, преимущественно C7-C10-арилалкилгруппу, C8-C40-, преимущественно C8-C12-арилалкенилгруппу, C7-C40-, преимущественно C7-C12-алкиларигруппу или R14 и R15 могут образовывать с соединяющими их атомами кольцо;
M2 является кремнием, германием или оловом, преимущественно кремнием или германием.
В соединениях формулы I M1 преимущественно является цирконием или гафнием, R1 и R2 имеют одинаковое значение и обозначают C1-C3-алкилгруппу или атом галогена, остатки R3 имеют одинаковое значение и обозначают C1-C4-алкилгруппу,
R4-R12 имеют одинаковое или различное значение и означают водород или C1-C4-алкилгруппу,
R13 обозначает
при этом M2 является силицием или германием и R14 и R15 имеют одинаковое или различное значение и означают C1-C4-алкилгруппу или C6-C10-арилгруппу.
Далее предпочтительными являются соединения формулы I, в которых остатки R4 и R7 обозначают водород и R5, R6 и R8 -R12 обозначают C1-C4-алкилгруппу или водород.
Наиболее предпочтительными являются соединения формулы I, в которых M2 является цирконием, R1 и R2 имеют одинаковое значение и обозначают хлор, остатки R3 имеют одинаковое значение и обозначают C1-C4-алкилгруппу, R4 и R7 означает водород, R5, R6 и R18-R12 имеют одинаковое или различное значение и обозначают C1-C4-алкилгруппу или водород и R13 обозначает
где
M2 означает кремний, и R14 и R15 имеют одинаковое или различное значение и обозначают C1-C4-алкилгруппу или C6-C10-арилгруппу.
Получение металлоценов I происходит методами, известными из литературы, и отражено в нижеследующей схеме реакции



X-нуклеофильная исходная группа, например, галоген или тозил

Производные 2-фенил-бензилгалогенида формулы A имеются в продаже или их можно получить методами, известными из литературы.
Превращение в производные формулы B происходит в результате реакции с замещенными сложными эфирами малоновой кислоты в основных условиях, как, например, в этанольных растворах натрийэтанолата.
Соединения формулы B омыляют гидроокисями щелочных металлов, такими как, например, гидроокись калия или гидроокись натрия и декарбоксилируют путем обработки полученной дикарбоновой кислоты с помощью нагрева до получения соединений формулы C.
Замыкание кольца соответствующих фенил-1-инданонов формулы D происходит путем взаимодействия с хлорагентами как, например, SOCl2 до получения соответствующих хлоридов кислоты, и последующей циклизации с помощью катализатора Фриделя-Крафтса в инертном растворителе как, например, AlCl3 или полифосфорная кислота в хлористом метилене или CS2.
Превращение в производные 7-фенил-индена формулы E происходит путем восстановления с помощью реактива, переносящего гидрид-ион, такого как, например, гидрида натрий бора или гидрида литийалюминия или водорода и соответствующего катализатора в инертном растворителе, таком как, например, диэтиловый эфир или тетрагидрофуран (ТГФ) до получения соответствующих спиртов и путем дегидратации спиртов в кислых условиях, как, например, пара- толуолсульфоновой кислоте или содержащей воду минеральной кислоте или путем взаимодействия с обезвоживающими веществами, такими как сульфат магния, сульфат меди, не содержащий воду или молекулярные сита.
Получение системы лигандов формулы G и превращение в хиральные металлоцены формулы H, имеющие мостик, а также выделение требуемой рацемической формы в принципе известно. К тому же производное фенилиндена формулы E с сильным основанием, таким как, например, бутиллитий или гидрид калия, в инертном растворителе депротонируется и вступает во взаимодействие с реактивом формулы F до получения системы лигандов формулы G. Он депротонируется затем с двумя эквивалентами сильного основания, такого как, например, бутиллитий или гидрид калия в инертном растворителе и вступает во взаимодействие с соответствующим металлтетрагалогенидом, таким как, например, цирконийтетрахлорид в подходящем растворителе.
Подходящими растворителями являются алифатические или ароматические растворители, такие как, например, гексан или толуол, эфирные растворители, такие как, например, тетрагидрофуран или диэтиловый эфир или галогенированные углеводороды, такие как, например, хлористый метилен или галогенированные ароматические углеводороды, такие как, например, о-дихлорбензол. Отделение рацемической формы и мезоформы происходит путем выделения (экстракции) или перекристаллизации с подходящими растворителями.
Образование производных металлоценов формулы I может происходить, например, путем реакции с алкилирующими средствами, такими как метиллитий.
Металлоцены I, согласно изобретению, являются высокоактивными компонентами катализатора для полимеризации олефинов. Хиральные металлоцены применяют преимущественно как рацемат. Но применять можно также чистый энантиомер в (+)- или (-) - форме. С помощью чистых энантиомеров получают оптически активный полимер. Однако нужно отделить мезоформу металлоценов, так как полимеризационно активный центр (атом металла) в этих соединениях из-за зеркальной симметрии в центральном атоме металла не является больше хиральным и поэтому нельзя получить никакого высокоизотактического полимера. Если метоформа не отделяется, то наряду с изотактическими полимерами появляется также атактический полимер. Для определения сфер применения, например, мягкие формованные изделия, это может быть очень желательным.
Согласно изобретению, в качестве сокатализатора применяют преимущественно алюминоксан формулы IIa для линейного типа и/или формулы IIb для циклического типа

при этом в формулах 11a и 11b остатки R17 имеют одинаковое или различное значение и обозначают C1-C6-алкилгруппу, C6-C18-арилгруппу, бензил или водород, и p обозначает целое число от 2 до 50, преимущественно от 10 до 35.
Преимущественно остатки R17 имеют одинаковое значение и означают метил, изобутил, фенил или бензил, особое преимущество имеет метил.
Если остатки R17 имеют различное значение, то они являются преимущественно метилом и водородом или альтернативно метилом и изобутилом, при этом они содержат водород или изобутил преимущественно от 0,01 до 40% (число остатков R17)
Алюминоксан можно получить различным образом известными методами. Один из методом заключается, например, в том что алюминийуглеводородное соединение и/или алюмогидридуглеводородное соединение вступает в реакцию обмена с водой (газообразная, твердая, жидкая, или связанная, например, как кристаллизационная вода) в инертном растворителе (как, например, толуол). Для получения алюминоксана с различными остатками R17 два различных алюминийтриалкила вступают, например, во взаимодействие соответственно требуемому составу.
Точная структура алюминоксанов 11a и 11b неизвестна.
Независимо от вида получения общим у всех растворов алюминоксана является меняющееся содержание исходного соединения алюминия, не прореагировавшего, который имеется в свободном виде или в виде аддукта (продукта присоединения).
Представляется возможным провести предварительную активацию металлоцена перед его использованием в реакции полимеризации с помощью алюминоксана формулы 11a и/или 11b. Тем самым заметно увеличивается полимеризационная активность и улучшается морфология полимерных гранул. Предварительную активацию соединения переходного металла проводят в растворе. При этом металлоцен растворяют в растворе алюминоксана в инертном углеводороде. В качестве инертного углеводорода подходит алифатический или ароматический углеводород. Преимущественно используют толуол.
Концентрация алюминоксана в растворе находится в пределах от примерно 1 вес. % до предела насыщения, преимущественно от 5 до 30 вес.%, относительно общего количества раствора. Металлоцен можно использовать в равной (одинаковой) концентрации, однако преимущественно в количестве от 10-4 -1 моль на моль алюминоксана. Предварительная активация составляет от 5 мин до 60 ч преимущественно от 5 до 60 мин. Реакцию проводят при температуре от (-78) до 100oC, преимущественно от 0 до 70oC.
С помощью металлоцена можно провести форполимеризацию. Для форполимеризации преимущественно используют олефин или один из олефинов, используемых в полимеризации.
Металлоцен можно также нанести на подложку. Подходящими материалами подложки являются, например, силикагели, окиси алюминия, твердый алюминоксан или другие неорганические материалы подложки является также мелкодисперсных порошок полиолефина.
Преимущественно сокатализатор, то есть алюминийорганическое соединение, наносят на подложку, как например, силикагели, окиси алюминия, твердый алюминоксан, другие неорганические материалы подложки или также мелкодисперсный порошок полиолефина и затем проводят реакцию взаимодействия с металлоценом.
В качестве неорганических носителей можно использовать окиси, которые были получены пирролитическим путем сжигания галогенидов в пламени гремучего газа, или силикагели с определенными диапазонами размеров частиц и их форм.
Получение сокатализатора с носителем, нанесенным на подложку, может происходить, как описано в EP 93 107 331.8, в реакторе из высококачественной стали, во взрывозащищенных конструкциях с помощью системы перекачивания при давлении в 60 бар, с обработкой инертным газом, с установлением однородного распределения температуры с помощью охлаждающего кожуха и вторичной циркуляцией охлаждения с помощью теплообменника в системе перекачивания. Система перекачивания всасывает содержимое реактора через патрубок в дне реактора с помощью насоса и прогоняет его в мешалку и затем через подающий трубопровод с помощью теплообменника в реактор. Мешалка устроена таким образом, что в подводящем канале имеется суженное поперечное сечение, где возникает скорость потока и в чьей турбулентной зоне аксиально и против направления потока введен тонкий трубопровод, через который периодически можно было добавлять при необходимости определенное количество воды при давлении аргона в 40 бар. Контроль за реакций осуществляют при помощи пробоотборника в цикле перекачивания.
Однако в принципе подходят и другие реакторы.
В описанный выше реактор объемом в 16 дм3 вносят 5 дм3 декана в инертных условиях, 0,5 дм3 (5,2 моля) триметилового алюминия добавляют при 25oC. Затем дополнительно добавляют через трубку с твердым веществом в реактор 250 г силикагеля SD 3216-30 (Grace AG), которые предварительно высушили при 120oC в псевдоожиженном слое аргоне, и с помощью мешалки и системы перекачивания их гомогенно распределили. Общее количество воды, составляющее 76,5 г, добавляют порциями по 0,1 см3 в течение 3,25 ч при необходимости каждые 15 с. Давление, возникающее из-за аргона и выделяемых газов, постоянно поддерживается на уровне 10 бар с помощью вентиля, регулирующего давление. После того как всю воду влили, систему перекачивания отключают и продолжают перемешивание еще в течение 5 ч при 25oC.
Полученный таким образом сокатализатор на носителе используют как 10%-ную суспензию в H-декане. Содержание алюминия составляет 1,06 ммоля A на см3 суспензии. Выделенное твердое вещество содержит 31 вес.% алюминия, суспендирующее средство содержит 0,1 вес.% алюминия.
Другие возможности получения сокатализатора на носителе описаны в EP 92 107 331.8.
Затем металлоцен, согласно изобретению, наносят на сокатализатор на носителе перемешивая растворенный металлоцен с сокатализатором, нанесенным на подложку.
Растворитель удаляют и заменяют углеводородом, в котором не растворяются не только сокатализатор, но и металлоцен. Реакция в системе катализатора на носителе, происходит при температуре от (-20o) до +120oC, преимущественно при 0-100oC, наиболее предпочтительны температуры от 15o до 40oC. Металлоцен вступает во взаимодействие с сокатализатором, нанесенным на подложку, таким образом, что сокатализатор как суспензия с 1-40 вес.%, преимущественно с 5-20 вес.%, вступает в алифатическом, инертном суспендирующем агенте, таком, как, например, H-декан, гексан, гептан, дизельное масло с раствором металлоцена, в инертном растворителе, таком как толуол, гексан, гептан, дихлорметан или с тонко измельченным твердым веществом металлоцена. В обратном порядке можно произвести также взаимодействие металлоцена с твердым веществом сокатализатора.
Реакция обмена происходит при интенсивном смешивании, например, перемешивании в молярном соотношении Al : M1 от 100:1 до 1000:1, преимущественно от 100:1 до 3000:1, а также во время реакции продолжительностью от 5 до 120 мин, преимущественно от 10 до 60 мин, наиболее предпочтительно время от 10 до 30 мин, в инертных условиях. Во время реакции получения системы катализатора на носителе появляются изменения в цвете реакционной смеси, особенно при применении металлоценов, согласно изобретению, максимумами поглощения в видимой области, в процессе чего можно наблюдать за ходом реакции.
После окончания реакции образованный раствор отделяют, например, путем фильтрации или декантирования. Оставшееся твердое вещество промывают от одного до пяти раз с помощью суспендирующего средства, такого как толуол, H-декан, гексан, дизельное масло для удаления растворимых составных частей в образованном катализаторе, в основном для удаления не прореагировавшего и поэтому растворимого металлоцена.
Полученную таким образом каталитическую систему на носителе можно высушить в условиях вакуума в виде порошка или еще с помощью растворителя, который был повторно использован в виде суспензии в одном из вышеназванных инертных суспендирующих средств и добавлен в систему полимеризации. Согласно изобретению, в качестве подходящих сокатализаторов вместо или наряду с алюминоксаном можно применять соединения формул R
В принципе для сокатализатора, согласно изобретению, подходит каждое соединение, которое благодаря кислотности Льюиса может превращать нейтральный металлоцен в катион и стабилизировать его ("лабильная координация"). Кроме этого, сокатализатор или образованный из него анион не вступают в реакции с образованным катионном металлоцена (сравни EP 427 697).
Для удаления каталитических ядов, находящихся в олефине, удачной представляется очистка с помощью алкила алюминия, например, триметил или триэтил алюминия. Эта очистка может происходить как в самой система полимеризации, так и олефин перед добавлением в систему полимеризации вступает в контакт с Al соединением и затем его опять отделяют.
Проводят полимеризацию или сополимеризацию известным способом в растворе, в суспензионной среде или газовой фазе, непрерывно или периодически в одну или несколько стадий, при температуре от - 60o до 200oC, преимущественно от 30 до 80oC, особое преимущество имеют температуры от 50o до 80oC. Проводят полимеризацию или сополимеризацию олефинов формулы Ra - CH = CH - Rb. В этой формуле Ra и Rb имеют одинаковое или различное значение и означают атом водорода или алкильный остаток ч. 1 - 14 C-атомами, однако Ra и Rb могут также образовывать с соединяющими их C- атомами кольцо. В качестве примеров подобных олефинов можно назвать этилен, пропилен, 1-бутен, 1-гексен, 4-метил-1-пентен, 1-октен, норборнен или норборнадиен. В основном проводят полимеризацию пропилена или этилена.
В качестве регулятора молярной массы и/или для повышения активности добавляют, если это необходимо, водород. Общее давление в системе полимеризации составляет от 0,5 до 100 бар. Преимущественно полимеризация проводится при давлении, представляющем интерес с технологической точки зрения, от 5 до 64 бар.
При этом металлоцен применяют в концентрации в расчете на переходный металл, от 10-3 до 10-8, преимущественно от 10-4 до 10-7 моль переходного металла на дм3 растворителя соответственно дм3 объема реактора. Другие указанные сокатализаторы применяют в эквимолярном количестве по отношению к металлоцену. В принципе и более высокие концентрации также являются возможными.
Если полимеризацию проводят в суспензионной среде или в растворителе, то используют инертный растворитель, применяемый для способа низкого давления, по Циглеру. Реакцию проводят, например, в алифатическом или циклоалифатическом углеводороде, в качестве подобных углеводородов следует назвать пропан, бутан, гексан, гептан, изооктан, циклогексан, метилциклогексан. Далее можно использовать бензиновую или гидрированную фракцию. Применяют также толуол. Преимущественно полимеризацию проводят в жидком мономере.
Если применяют инертные растворители, то дополнительно добавляют газообразные или жидкие мономеры.
Продолжительность полимеризации может быть любой, так как применяемая, согласно изобретению, каталитическая система дает лишь незначительный, не зависящий от времени спад активности полимеризации. Перед добавлением катализатора, особенно системы катализатора на носителе (из металлоцена, согласно изобретению и сокатализатора на носителе, соответственно из металлоцена, согласно изобретению, и алюминийорганического соединения на мелкодисперсном порошке полиолефина), можно дополнительно добавить в реактор другой алкилалюминий, например, триметилалюминий, триэтилалюминий, триизобутилалюминий, триоктилалюминий или изопропенилалюминий для инертизации системы полимеризации (например, для отделения имеющихся в олефине каталитических ядов). Его добавляют в систему полимеризации в концентрации от 100 до 0,01 ммоля Al на 1 кг содержимого реактора. Предпочтительными являются триизобутил алюминий и триэтил алюминий в концентрации от 10 до 0,1 ммоля Al на 1 кг содержимого реактора. Тем самым при синтезе системы катализатора на носителе можно выбрать соотношение Al:M1 на минимальном уровне.
Однако, в принципе, использование других веществ для катализа реакции полимеризации не является необходимым, это значит, что системы, согласно изобретению, можно применять как единственные катализаторы для полимеризации олефинов.
Способ, согласно изобретению, отличается прежде всего тем, что описанные металлоцены при температурах от 50o до 80oC, представляющих интерес с технической точки зрения, при высокой активности катализатора дают очень высокомолекулярные полимеры, в случае прохиральных мономеров с очень высокой молярной массой и очень высокой стереотактичностью.
Особенно цирконоцены, согласно изобретению, отличаются прежде всего тем, что в случае стереоспецифической полимеризации прохиральных олефинов, например пропилена, получают полимеры с высокой изотаксичностью.
Особенно в случае изоспецифической полимеризации пропилена получают изотактический полипропилен с изотактическими единицами большой длины и высокой точкой плавления.
Кроме этого, отложения на стенках реактора можно избежать с помощью систем катализаора на носителе. Нижеследующие примеры должны более подробно осветить изобретение.
Все стеклянные приборы выдерживали при высокой температуре и промывали аргоном. Все операции проводили при отсутствии влажности и кислорода в специальных сосудах. Применяемые растворители были свежеперегнанными под аргоном, через Na/K - сплав и их сохраняли в специальных сосудах. Определение соотношения Al: CH3 в алюминоксане происходило путем разложения с помощью H2SO4 и определения объема полученных в ходе гидролиза газов в нормальных условиях, а также путем комплексометрического титрования алюминия в растворенной затем пробе по Шварценбаху.
В примерах с 3 по 5 с помощью соединения алюминия на носителе метилалюминоксан на силикагеле (ниже это соединение будет называться "MAO на SiO2") была получена примерно 10 вес.%-ная суспензия в Н-декане, которая согласно результатам определения алюминия содержала 60 мг на Al/см3.
В примерах с 26 по 30 при помощи соединения алюминия, нанесенного на подложку (метилалюминоксан на силикагеле SD3216-30/Grace), в дальнейшем "F MAO на SiO2", применяли порошок, не содержащий растворитель, который содержал 20 вес. % алюминия в твердом веществе.
Метилалюминоксан, растворимый в толуоле, использовали в примерах на полимеризацию в суспензионной среде и на полимеризацию в массе с металлоценом, не нанесенным на подложку, как 10 вес.%-ный раствор в толуоле, и он содержал согласно определению алюминия 36 мг Al/см3. Средняя степень олигомеризации согласно понижению точки замерзания в бензоле составила n = 20. Для металлалюминоксана, растворимого в толуоле, было определено соотношение Al : CH3 = 1 : 1,55.
В данном тексте приняты следующие сокращения:
КВ - коэффициент вязкости в см3/г,
Mw - средневесовая молярная масса в г/моль (определенная с помощью гельпроникающей хроматографии),
Mw/Mn = дисперсность молярной массы,
Tпл = точка плавления в oC /определенная по ДСК, 20oC/мин скорость подъема температуры/ скорость охлаждения,
UU= индекс изотактичности (II = mm + 1/2 mr, определенный с помощью 13C-NMR - спектроскопии),
UP 230/5 = индекс расплава, измеренный по DIN/промышленный стандарт ФРГ/53735, в гр/мин,
НВ = насыпной вес полимера в г/дм3.
Синтез метеллоценов I, применяемых в примерах полимеризации, исходные вещества имеются в продаже:
А.рац-диметилсилил-бис(2-метил-4-фенил-инденил)цирконийдихлорид (5)
1) (±)-2-(2-фенил-бензил)-пропионовая кислота (1) К 6,5 г /0,285 молей/ /натрия в 160 см3 EtOH, не содержащего воду/, при комнатной температуре прибавили по каплям 48,6 г /0,279 молей/ диэтилметилмалоната. Затем прибавили по каплям 70,4 г /0,285 молей/ 2-фенил-бензилбромида в 20 см3 EtOH, не содержащего воду, и полученную смесь нагревали в течение 3 часов при флегме. Растворитель перегнали и остаток смешали с 200 см3 H2O. Органическую фазу отделяют, водную фазу насыщают NaCl и дважды экстрагируют, каждый раз с 200 см3 Et2O. Объединенную с экстрактами органическую фазу высушивают /MgSO4/. Остаток, оставшийся после перегонки растворителя, внесли в 500 см3 EtOH и 50 см3 H2O и смешали с 56 г /1 моль/ КОН. Реакционную смесь нагревали в течение 4 часов при флегме. Раствор перегнали в условиях вакуума, остаток внесли в 500 см3 H2O и подкислили с помощью концентрированной, содержащей воду соляной кислоты /HCl/ до величины pH = 1. Выпавший осадок вакуумировали и нагревали в трубке с шаровым расширением в течение 30 минут при сильном кипении до 250oC. Получили 58,3 г /85%/ соединения 1 в виде густого масла.
1H - NMR /100 Мгц, CDCl3/ : 11,7/ с, 1H, COOH/, 7,1 - 7,5/м, 9H, аром. H/, 2,3 - 3,2 /м, 3H, CH и CH2/, 0,9 /d, 3H, CH3/.
/±/-2 метил-4-фенил-индан-OH/2/
Раствор 58,6 /0,242 моля/ соединения 1 перемешивали в 60 см3/ /0,83 моля/ тионилхлорида в течение 18 часов при комнатной температуре. Избыточный тионилхлорид удалили при 10 мбар, и маслянистый осадок путем многократного растворения, каждый раз в 100 см3 толуола, и путем отгонки в условиях вакуума очистили от остатков тионилхлорида.
Хлорангидрид кислоты внесли в 150 см3 толуола и при 10oC добавили по каплям к суспензии 48 г /0,363 моля/ Al Cl3 в 400 см3 толуола. После окончания добавления смесь нагревали еще в течение 3 часов на флегме. Реакционную смесь поместили в 500 г льда и подкислили с помощью концентрированной соляной кислоты до величины pH 1. Органическую фазу отделили и водную фазу дополнительно трижды экстрагировали, каждый раз со 100 см3 Et2O. Объединенные органические фазы промывали насыщенным водным раствором NaHCO3/ и насыщенным водным раствором NaCl и затем высушили /MgSO4/. Получили 50,4 г/93% соединения 2, которое без дальнейшей очистки вводили в дальнейшее взаимодействие.
1H-NMR /100 Мгц, CDCl3/:7,2 - 7,8 /м, 8H, аром. H/ 3,3 /дд, 1H, B - H/ 2,5 - 2,9 /м, 2H, α- и β- -H/,1,3/d, 3H, CH/3/2-метил-7-фенил-инден(3)
50 г /0,226 моля/ соединения 2 растворили в смеси 450 см3 ТГФ с MeOH /2: 1/ и смешали при 0oC при перемешивании порциями с 12,8 г /0,34 моля/ гидрида натрийбора и перемешивали еще в течение 18 часов. Реакционную смесь поместили на лед, смешали с концентрированной HCl до величины pH 1 и несколько раз экстрагировали Et2O. Объединенные органические фазы промыли с помощью насыщенного водного раствора NaHCO3 и NaCl и затем высушили /MgSO4/. Растворитель удалили в условиях вакуума и сырой продукт без дальнейшей очистки перенесли в 1 дм3 толуола, смешали с 2 г пара-толуолсульфоновой кислоты и нагревали в течение 2 часов при флегме.
Реакционную смесь промывали с помощью 200 см3 насыщенного водного раствора NaHCO3 и растворитель удалили в условиях вакуума. Сырой продукт очистили путем фильтрации в 500 г силикагеля /смесь гексана и CH2Cl2/ Получили 42 г /90%/ соединения 3 в виде бесцветного масла.
1H-NMR/100 Мгц, CDCl3 /:7,0 - 7,6/ м, 8H, аром. H/ 6,5/м, 1H, H-C/3/, 3,4/с; 2H, CH2/,2,1/с, 3H, CH3/
4/ Диметилбис(2-метил-4-фенил-инденил)силан(4)
Раствор 15 г /72,7 ммоля/ соединения 3 в 200 см3 толуола, не содержащего H2O и O2 и 10 см3 ТГФ, не содержащего H2O и O2, при комнатной температуре смешали под аргоном с 29 см3/73 ммоля/, 2,5 М раствора бутил лития в гексане и нагреватели в течение часа до 80oC. Затем смесь охладили до 0oC и смешали с 4,7 г/36,4 ммоля/ диметилдихлорсилана. Смесь нагревали в течение 1 часа до 80oC и затем перенесли в 100 см3 H2O. Несколько раз экстрагировали Et2O и объединенные органические фазы высушили /MgSO4/. Сырой продукт, оставшийся после отгонки растворителя в условиях вакуума, подвергли хроматографии в 300 г силикагеля/ смесь гексана и CH2Cl2/ Получили 12,0 /70%/ соединения 4.
1H- -NMR /100 Мгц, CDCl3/: 7,10 - 7,70 /м, 16H, аром. H/, 6,80 /м, 2H, H- C//3/ 3,80/с, 2H, H-C/1/, 2,20/м, 6H, CH3/-0,20/м, 6H, CH3Si /5/ рац-диметилсилилбис(2- метил-4-фенил-инденил)цирконийдихлорид(5)
Раствор 6,0 г /12,9 ммоля/ соединения 4 смешали в 100 см3 толуола, не содержащего H2O и O2 под аргоном при комнатной температуре с 10,6 см3 /26 ммоля/ 2,5 М раствора бутиллития в гексане и нагревали в течение 3 часов при флегме. Затем суспензию дилитиевой соли охладили до -25oC и смешали с 3,2 г/13,6 ммоля/ цирконийтетрахлорида. Смесь нагревали в течение часа до комнатной температуры, перемешивали еще в течение одного часа и отфильтровали затем через специальный фильтр Y 3. остаток экстрагировали с помощью 50 см3 толуола и объединенные фильтраты в условиях вакуума, создаваемого масляным насосом, освободили от растворителя. Получили 9,0 г металлоцена в виде рацемической формы и мезоформы в соотношении 1:1 в виде желтого порошка. Чистый рацемат /5/ был получен путем многократного перемешивания полученной смеси, каждый раз с 20 см3 хлористого метилена, при этом рацемат был получен в виде кристаллического порошка, а мезоформу промыли. Получили 2,74 г /33%/ чистого рацемата.
1H-NMR/300 Мгц, CDCl3 /: 7,0 - 7,7/м, 16H, аром. H/6,9/с, 2H, H-C/3/, 2,2/ с, 6H, CH3/,1,3/м, 6H, CH3Si/.
молярная масса: 626 M+, правильное разложение
Пример Б
рац-метилфенилсиландиилбис-(2-метил-4-фенилинденил) цирконийдихлорид(7)
1/ Метилфенилбис-(2-метил-4-фенилинденил)силан (6)
Раствор 10,3 г /50 ммолей/ 3 в 90 мл толуола, не содержащего H2O и O2 и 10 мл ТГФ, не содержащего H2O и O2, смешали при комнатной температуре под аргоном с 21 мл/52 ммоля/ 2,5 М раствора бутиллития в гексане и нагревали в течение 1 часа до 80oC.
Затем охладили до 0oC и смешали с 4,8 г/25 ммолей/ метилфенилдихлорсилана и перемешивали далее в течение ночи при комнатной температуре. Выпавший в осадок LiCl /хлорид лития/ отделили путем фильтрации, и сырой продукт, полученный в результате отгонки растворителя в условиях вакуума, подвергли хроматографии на 300 г силикагеля /смесь гексана и CH2Cl2 в соотношении 9:1/ Получили 4,6 г /35%/ соединения 6.
1H-NMR /100 Мгц, CDCl3 /7,0 - 7,8 /м, 16H, аром. H/6,9/с, 2H, H-C(3) 3,9/м, 2H, H-C (1), 2,3/ м, 6H, CH3/, -0,1 /с, 3H, CH3Si/
2/ рац-метилфенилсиландиилбис(2-метил-4-фенилинденил) цирконийдихлорид(7)
2,3 г /4,4 ммоля/ 6 в 25 мл толуола, не содержащего H2O и O2 перемешивали под аргоном с 3,6 мл/8,9 ммолей/2,5 М раствора бутиллития в гексане и нагревали в течение 3 часов до 80oC. Затем суспензию дилитиевой соли охладили до -30oC и смешали с 1,1 г /4,5 ммоля/ цирконийтетрахлорида. Нагревали в течение часа до комнатной температуры, перемешивали в течение 1 часа /дополнительно/. После фильтрации на специальном фильтре 3
удалили растворитель из фильтрата, и остаток выкристаллизовался из 10 мл хлористого метилена. Получили 0,2 г рацемической формы 7 в виде оранжевых кристаллов.
1H-NMR /100 Мгц, CDCl3/: 7,0 - 8,2/м, 21H, аром. H/, 6,9/м, 2H, -C/3/ 2,4/с, 3H, CH3/, 2,0/с, 3H, CH3/1,3/с, 3H, CH3Si /Масс.-спектр :690 M+, правильное разложение.
Пример B
рац-диметилсиландиилбис(4-фенилинденил)цирконийдихлорид (12)
1/ 3-(2-фенил-фенил)пропионовая кислота (8)
К 14 г/0,61 ммоля/ натрия в 400 см3 EtOH, не содержащего воду, прибавили при комнатной температуре 93 см3/0,61 ммоля/ диэтилового эфира малоновой кислоты, растворенного в 50 см3 EtOH, не содержащего H2O. Затем прибавили по каплям 150 г/0,61 ммоля/ 2-фенилбензилбромида в 200 см3 EtOH, не содержащего H2O, и в течение 3 часов нагревали при флегме. При комнатной температуре прибавили 102 г/ 1,83 ммоля/ KOH, растворенного в 150 см3 H2O, и еще в течение 4 часов нагревали при флегме. Растворители удалили в условиях вакуума, остаток смешивали до получения истинного раствора с H2O и подкислили с помощью концентрированной соляной кислоты до величины pH 1. Выпавший осадок вакуумировали, высушили и в течение 1 часа нагревали до 130oC. Получили 112 г/81 %/ 8 в виде густого масла.
1H-NMR/100 Мгц, CDCl3/:9,1/ с, 1H, COOH/,6,9-7,5/ м, 9H, аром. H/ 2,3 - 3,0 м, 4H, 2 CH2/
2/ 4-фенил-1-инданон (9)
Раствор 102 г/0,45 ммоля/ 8 в 37 см3 /0,5 ммоля/ тионилхлорида перемешивали в течение 18 часов при комнатной температуре. Избыточный тионилхлорид удалили при 10 мбар, и маслянистый осадок путем многократного растворения, каждый раз в 100 см3 толуола и отгонки в условиях вакуума освободили от образовавшихся остатков тионилхлорида.
Хлорид кислоты внесли в 200 см3 толуола и добавили по каплям при 10oC к суспензии 72 г/0,54 ммоля/ AlCl3 в 1000 см3 толуола и нагревали в течение 1 часа до 80oC. Реакционную смесь поместили в лед /1000 г льда/ и подкислили с помощью концентрированной HC до величины pH 1. Органическую фазу отделили, и водную фазу дополнительно трижды экстрагировали, каждый раз с помощью 200 см3 E O. Объединенные органические фазы промыли с помощью насыщенного водного раствора NaHCO3, насыщенного водного раствора NaCl и затем высушили /MgSO4/. Получили 96, /96%/ 9, который подвергают дальнейшему превращению без дополнительной очистки.
1H-NMR-/100 Мгц. CDCl3/: 6,9 - 7,5/м, 8H, аром. H/, 2,5 - 3,4''/м, 4H,2 CH2/
3) 7-фенил-инден (10)
Раствор 86 г /0,41 ммоля/ 9 в 300 см3 смеси ТГФ с метанолом в соотношении 2:1 смешали при 0oC порциями с 23 г/0,62 ммоля/ NaBH и перемешивали в течение 18 часов при комнатной температуре. Реакционную смесь поместили на 300 г льда, смешали с концентрированной, HCl до величины pH 1 и несколько раз экстрагировали с Et2O. Объединенные органические фазы промыли насыщенным водным раствором NaHCO3 - насыщенным водным раствором NaCl, высушили /MgSO4/ и освободили от растворителя в условиях вакуума.
Сырой продукт внесли в 1000 см3 толуола, смешали с 4,5 г пара-толуолсульфоновой кислоты и нагревали в водоотделителе в течение 2 часов до флегмы. Реакционную смесь трижды промыли 250 см3 насыщенного водного раствора NaHCO3 и растворитель удалили в условиях вакуума. После перегонки при 0,1 мбар, получили при 96 - 108oC 33 г /41%/ соединения 10 в виде бесцветного масла.
1H-NMR/100 Мгц, CDCl3/:7,1 - 7,7/м, 8H, аром.H/ , 6,9- и 6,5/ /2м, 2H, CH/ 3,5/м, 2H, CH2/
4/ Диметилбис-(4-фенилинденид)-силан (II)
Раствор 10 г /50 ммолей/ 10 в 100 см3 толуола, не содержащего H2O и O2 и 5 мл ТГФ, не содержащего H2O и O2, смешали при комнатной температуры с 18,7 см3/50 ммолей/ 20%-ного раствора бутиллития в толуоле и нагревали в течение 2 часов до 80oC. Затем желтую суспензию охладили до 0oC и смешали с 3,2 г/25 ммолей/ диметилдихлорсилана. Реакционную смесь нагревали еще в течение 1 часа до 80oC и затем промыли при помощи 50 см3 H2O. Растворитель удалили в условиях вакуума, и остаток перекристаллизовался из гептана при - 20oC. Получили 6,7 г /62%/ соединения 11 в виде бесцветных кристаллов /точка плавления 109-110oC/ 1H-NMR/100 Мгц, CDCl3 / : 7,0-7,7/ м, 18H, аром.H/ и H-C /3/ 6,8 /дд, 2H, H-C /2/, 3,8 /м, 2H, H-C/1/ - 0,2/с, 6H, CH3Si/.
5/ рац-диметилсиландиилбис(4-фенилинденил)цирконийдихлорид (12)
Раствор 6,6 (16 ммолей/ 11 в 70 см3 Et2O, не содержащего H2O и O2, смешали под аргоном при комнатной температуре с 12 см3 /32 ммоля/ 20%-ного раствора бутиллития в толуоле и затем нагревали в течение 3 часов на флегме. Растворитель удалили в условиях вакуума, и осадок отфильтровали с помощью 50 мл гексана, не содержащего H2O и O2, на специальном фильтре Y3, дополнительно промыли с помощью 50 мл гексана, не содержащего H2O и O2, и высушили /0,1 мбар, комнатная температура/
Дилитиевую соль добавили при - 78oC к суспензии 3,6 г /16 ммолей/ цирконийтетрахлорида в 80 см3 хлористого метилена и нагревали в течение 18 часов при перемешивании с помощью магнитной мешалки до комнатной температуры. Смесь отфильтровали на фильтре Y3 и остаток дополнительно экстрагировали порциями с общим объемом хлористого метилена в 200 см3. Объединенные фракции освободили в условиях вакуума от растворителя, и они перекристаллизовались из смеси хлористого метилена с гексаном /в соотношении 1:1/. Получили 5,6 г рацемической и мезоформы в соотношении 1:1. Путем повторной перекристаллизации из хлористого метилена получили рацемческий комплекс в форме желтых кристаллов.
1H-NMR/100 Мгц, CDCl3 : 7,0-7,8/м, 22H, аром. H и H-C (3), 6,1 - (д, 2H, H-C(2), 1,1 (с, 6H, CH3Si). Масс-спектр : 598 M+, правильное разложение.
Пример Г
рац-диметилсиландиилбис/2-этил-4-фенилинденил/цирконийдихлорид (17)
1/(±)-2-(2-фенилбензил)-масляная кислота /13/
К 23 г /1 моль/ натрия в 400 см3 EtOH, не содержащего H2O, прибавили по каплям при комнатной температуре 188 г /1 моль/ диэтилового эфира тилмалоновой кислоты, растворенного в 100 см3EtOH, не содержащего H2O. Затем 247 г /1 моль/ 2-фенилбензил-бромида прибавили по каплям в 300 см3 EtOH, не содержащего H2O, и нагревали в течение 3 часов на флегме. При комнатной температуре добавили 170 г /3 моля/ KOH, растворенного в 300 см3 H2O и нагревали в течение 4 часов на флегме. Растворители удалили в условиях вакуума, остаток смешивали до получения истинного раствора с H2O, затем подкислили с помощью концентрированной, содержащей воду HCl до величины pH 1. Выпавший осадок вакуумировали, высушили и нагревали в течение часа до 130oC. Получили 236 г /93%/ соединения 13 в виде густого масла.
1H-NMR/ 100 Мгц, CDCl3 /10,3/с, 1H, COOH/, 7,0-7,3/ м, 9H, аром. H/, 2,5-3,0/м, 3H, CH и CH2/, 1,5-1,9/м, 2H, CH2/, 0,9/, т, 3H, CH3/
2/ (±)-2-этил-4-фенил-1-инданон (14)
Раствор 236 г /0,93 г моля/ соединения 13 в 81 см3 /1,2 моля/ тионилхлорида перемешивали в течение 18 часов при комнатной температуре. Избыточный тионилхлорид удалили при 10 мбар, и маслянистый осадок путем многократного растворения, каждый раз в 200 см3 толуола и отгонки в условиях вакуума освободили от образовавшихся остатков тионилхлорида.
Хлорид кислоты внесли в 400 см3 толуола и прибавили по каплям при 10oC к суспензии 133 г /1,0 моль/ AlCl в 2000 см3 толуола и нагревали в течение 1 часа до 80oC. Реакционную смесь поместили на 2000 г льда и подкислили с помощью концентрированной HCl до величины pH 1. Органическую фазу отделили, и водную фазу трижды дополнительно экстрагировали, каждый раз с 200 см3 Et2OH. Объединенные органические фазы промыли при помощи насыщенного водного раствора NaHCO3, насыщенного водного раствора NaCl и затем высушили /MgSO4/
Получили 187 г /85%/ соединения 14, который подвергался дальнейшему превращению без дополнительной очистки
1H-NMR -/100 Мгц, CDCl3/:7,0-7,8 (м, 8H, аром. H), 3,1-3,4 (м, 1H, H-C (3), 2,5-2,9 (м, 2H, H-C(2) и H-C(3), 1,3-2,0 (м, 2H, CH3), 0,9 (т, 3H, CH3)
3/ 2-этил-7-фенилинден (15)
Раствор 50 г/0,21 моля) соединения 14 в 600 см3 смеси ТГФ с метанолом в соотношении 2: 1 смешали при 0oC порциями с 8 г /0,21 моля) NaBH4 и перемешивали в течение 18 часов при комнатной температуре. Реакционную смесь поместили на 600 г льда, смешали с концентрированной HC до величины pH 1 и несколько раз экстрагировали Et2OH. Объединенные органические фазы промыли с помощью насыщенного водного раствора NaHCO3, насыщенного водного раствора NaXl и затем высушили /MgSO4/
Сырой продукт внесли в 1000 см3 толуола, смешали в 4,5 p-толуолсульфоновой кислоты и нагревали в течение 2 часов в водоотделителе при флегме. Реакционную смесь трижды промывали с 250 см3 насыщенного водного раствора NaHCO3 и растворитель удалили в условиях вакуума. После перегонки при 0,1 мбар получили при 135oC 33 г /72%/ соединения 15 в виде бесцветного масла.
1H-NMR/100 Мгц, CDCl3/ : 7,0-7,5 (м, 8H, аром, H), 6,5 (м, 1H, CH), 3,2 (м, 2H, CH2), 2,5 (дкв, 2H, CH2), 1,1 (т, 3H, CH3)
4/ диметилбис(2-этил-4-фенилинденил)силан(16)
Раствор 17 г (77 молей) соединения 15 в 160 см3 толуола, не содержащего H2O и O2 и 8 мл ТГФ, не содержащего H2O и O2, смешали при комнатной температуре с 29 см3 /77 молей/ 20%-ного раствора бутиллития в толуоле и нагревали в течение 2 часов до 80oC. Затем желтую суспензию охладили до 0oC и смешали с 5 г (38 ммолей) диметилдихлорсилана. Реакционную смесь нагревали еще в течение 1 часа до 80oC и затем промыли со 100 см2 H2O. Растворитель удалили в условиях вакуума и остаток очистили путем хроматографии в 200 г силикагеля (смесь гексана с хлористым метиленом в соотношении 9:1) Получили 9 г /47% соединения 16 в виде густого масла.
1H-NMR (100 Мгц, CDCl3): 6,9-7,4 (м, 16H, аром.H), 6,5 (м, 2H, H-C (3), 3,7 (м, 2H, H-C(1), 2,4 (м, 4H, CH2), 1,1 (т, 6H, CH3),-0,1 (s, 6H, CH3Si)
5/ рац-диметилсиландилбис(2-этил-4-фенилинденил)цирконийдихлорид (17)
Раствор 5,6 г/11 ммолей) соединения 16 в 50 см3 Et2OH, не содержащего H2O и O2 смешали под аргоном при комнатной температуре с 8,4 см 20%-ного раствора бутиллития в толуоле и нагревали затем в течение 3 часов при флегме. Растворитель освободили в условиях вакуума, и осадок отфильтровали с 50 мл гексана, не содержащего H2O и O2, через специальный фильтр Y3, дополнительно промыли с 50 мл гексана, не содержащего H2O и O2, и высушили /0,1 мбар, комнатная температура/ Дилитиевую соль добавили при - 78oC к суспензии 2,5 г /11 ммолей/ цирконийтетрахлорида в 50 см3 хлористого метилена и в течение 18 часов нагревали до комнатной температуры с помощью магнитной мешалки. Смесь отфильтровали через специальный фильтр Y3, и остаток дополнительно экстрагировали порциями с помощью хлористого метилена /общий объем 100 см3/. Объединенные фильтраты освободили в условиях вакуума от растворителя, и они перекристаллизовались из смеси толуола с гексаном (в соотношении 1:1/. Получили 2 г /27%/ рацемической и мезоформы в соотношении 1:1. Путем повторной перекристаллизации из толуола получили рацемический комплекс 17 в форме желтых кристаллов.
1H-NMR/100 Мгц, CDCl3/ : 6,8 - 7,7 (м, 16H, аром. H), 6,6 (м, 2H, H-C (3) 2,3 - 3,9 (м, 4H, CH2), 1,0 - 1,4 (м, 12H, CH3 и CH3Si)
Масс-спектр: 654 M+
правильное разложение.
Пример Д.
рац-диметилсиландиилбис (2-метил-4-(1-нафтил)инденил) цирконийдихлорид(24)
1/ 2-(1-нафтил)-толуол (18)
13,9 г /0,57 ммоля/ магниевых опилок обработали 150 мл Et2O, не содержащего H2O, и обеспечили проведение реакции Гриньяра с 5 г 2-бромтолуола и несколькими кристалликами йода. Затем 93 г /0,57 моля/ 1-бромтолуола в 450 мл Et2O, не содержащего H2O, прибавили по каплям таким образом, что реакционная смесь во время находилась в стадии кипения. После окончания добавления нагревали до кипения до тех пор, пока магний полностью не прореагировал.
Раствор Гриньяра затем добавили по каплям к раствору 118 г /0,57 моля/ 1-бромнафталина и 3,5 г бис(трифенилфосфин) никельдихлорида в 800 см3 толуола, так что температура внутри не превышала 50oC. Затем нагревали еще в течение 3 часов на флегме, смешали с 500 мл 10%-ной HCl, фазы отделили и органическую фазу освободили в условиях вакуума от растворителя. После фильтрации на силикагеле /гексан/ получили 115 г /92%/ соединения 18 в виде бесцветного масла.
1H-NMR /100 Мгц, CDCl3/: 7,2 - 8,0 (м, 11H, аром. H) 2,0 (с, 3H, CH3)
2/ 2-(1-нафтил)-бензилбромид (19)
114 г /0,52 моля/ 18 и 103 г /0,58 моля/ N-бромсукцинимида растворили при комнатной температуре в 2000 см3 тетрахлоруглероде, смешали с 3 г азобисизобутиронитрила и в течение 4 часов нагревали на флегме. Образовавшийся сукцинимид отфильтровали, растворитель удалили в условиях вакуума и осадок очистили путем фильтрации в 1000 г силикагеля (смесь гексана с хлористым метиленом в соотношении 9:1). Получили 141 г /82% соединения 19 в виде бесцветного масла со слезоточивыми свойствами.
1H-NMR /100 Мгц, CDCl3/: 7,1 - 8,0 (м, 11H, аром. H), 4,2 (кв, 2H, CH2Br)
3/ (±) -2-(2-(1-нафтил)бензил)-пропионовая кислота (20)
К 10 г /0,43 ммоля/ натрия в 100 см3 EtOH, не содержащего H2O, прибавили по каплям при комнатной температуре 75 г (0,43 моля) диэтилового эфира метилмалоновой кислоты, растворенного в 50 см3 EtOH, не содержащего H2O. Затем прибавили по каплям 140 г /0,43 ммоля/ 2-фенилбензилбромида в 200 см3 EtOH, не содержащего H2O, и нагревали в течение 3 часов на флегме. При комнатной температуре добавили 85 г /1,3 ммоля/ KOH, растворенного в 100 см3 H2O, и нагревали в течение еще 4 часов на флегме. Растворители удалили в условиях вакуума, остаток смешали до получения истинного раствора с H2O и подкислили с помощью концентрированной HCl до величины pH 1. Выпавший осадок вакуумировали, высушили и в течение 1 часа нагревали до 120oC. Получили 96 г /77%/ соединения 20 в виде густого масла.
1H-NMR (100 Мгц, CDCl3): 10,1 (с, 1H, COOH), 6,9 - 8,0 (м, 11H, аром. H), 2,3 - 3,0 (м, 3H, CH2 и CH), 0,8 (д, 3H, CH3)
4/ (±)-2-метил(1-нафтил)-1-инданон (21)
Раствор 96 г /0,33 моля/ 20 в 37 см3 /0,5 моля/ тионилхлорида перемешивали в течение 18 часов при комнатной температуре. Избыточный тионилхлорид удалили при 10 мбар, и маслянистый осадок путем многократного растворения, каждый раз в 100 см3 толуола и отгонки в условиях вакуума освободили от образовавшихся остатков тионилхлорида.
Хлорид кислоты внесли в 200 см3 толуола и добавили по каплям при 10oC к суспензии 44 г /0,33 моля/ Al Cl3 в 1000 см3 толуола и нагревали в течение 3 часов до 80oC. Реакционную смесь поместили на 1000 г льда и подкислили с помощью концентрированной HCl до величины pH 1. Органическую фазу отделили и водную фазу дополнительно трижды экстрагировали, каждый раз с 200 см3 хлористого метилена. Объединенные органические фазы промыли с помощью насыщенного водного раствора NaHCO3 - насыщенного водного раствора NaCl и затем высушили /MgSO4/. После хроматографии в 1000 г силикагеля (смесь гексана и хлористого метилена) получили 12 г /13%/ соединения 21
1H-NMR / 100 Мгц, CDCl3/: 7,3 - 8,0 (м, 10H, аром. H) 2,2 - 3,2 (м, 3H, CH2 и CH) 1,2 (д, 3H, CH3)
5/ 2-метил-7-(1-нафтил)инден (22)
К раствору 12 г (44 моля) 21 в 100 см3 смеси ТГФ с метанолом в соотношении 2: 1 добавили при 0oC 1,3 г - 33 моля/ NaBH4 и перемешивали в течение 18 часов при комнатной температуре. Реакционную смесь поместили на 100 г льда, подкислили с помощью концентрированной содержащей воду HCl до величины pH 1 и несколько раз экстрагировали Et2O. Объединенные органические фазы промыли с помощью насыщенного водного раствора NaHCO3 насыщенного водного раствора NaCl и затем высушили /MgSO4/.
Сырой продукт внесли в 200 см3 толуола, смешали с 0,5 г пара-толуолсульфоновой кислоты и в течение 2 часов нагревали в водоотделителе до флегмы. Реакционную смесь трижды промывали с помощью насыщенного водного раствора NaHCO3, состоящего из 50 см3, и растворитель удалили в условиях вакуума. После фильтрации через 200 г силикагеля (смесь гексана и хлористого метилена) получили 10 г (86%) соединения 22 в виде бесцветного масла.
1H-NMR (100 Мгц, CDCl3): 7,0 - 8,0 (м, 10H, аром. H), 6,6 (м, 1H, CH) 3,0 (м, 2H, CH2), 2,0 (м, 3H, CH3)
6/ диметилбис(2-метил-4-(1-нафтил)-инденил)силан (23)
Раствор 10 г (38 ммоля) 22 в 100 см3 толуола, не содержащего H2O и O2, и 5 мо ТГФ, не содержащего H2O и O2, смешали при комнатной температуре с 14,4 см3 /50 ммолей/ 20%-ного раствора бутиллития в толуоле и нагревали в течение 2 часов до 80oC. Затем желтую суспензию охладили до 0oC и смешали с 2,5 г /19 ммолей/ диметилдихлорсилана. Реакционную смесь нагревали еще в течение 1 часа до 80oC и затем промыли с 50 см3 H2O. Раствор удалили в условиях вакуума, и остаток перекристаллизовался из гептана при - 20oC. Получили 8,2 г /75%/ 23 в виде бесцветных кристаллов.
1H-NMR (100 Мгц, CDCl3): 7,2 - 8,1 (м, 20H, аром. H), 6,4 (м, 2H, H-C(3) 4,0 (м, 2H, H-C(1)-0,1 (с, 6H, CH3Si)
7/ рац-диметилсиландиилбис)2-метил-4(1-нафтил)инденил) цирконийдихлорид(24)
Раствор 8,0 г /14 ммолей/ 23 в 70 см3 Et2O, не содержащего H2O и O2, смешали при аргоне при комнатной температуре с 10,5 см, 20%-ного раствора бутиллития в толуоле и затем нагревали в течение 3 часов до флегмы. Растворитель удалили в условиях вакуума, и осадок отфильтровали с 50 мл гексана, не содержащего H2O и O2, через специальный фильтр Y3, дополнительно промыли с помощью гексана, не содержащего H2O и O2, и высушили /0,1 мбар, комнатная температура/
Дилитиевую соль добавили при - 78oC к суспензии, 3,2 г /14 ммолей/ цирконийтетрахлорида в 80 см3 хлористого метилена и в течение 18 часов нагревали при перемешивании с помощью магнитной мешалки до комнатной температуры. Смесь отфильтровали через специальный фильтр Y3, и остаток дополнительно экстрагировали порциями с общим объемом хлористого метилена 400 см3. Объединенные фильтраты освободили в условиях вакуума от растворителя, и они перекристаллизовались из хлористого метилена. Получили 1,5 г/15%/ рацемической и мезоформы в соотношении 1:1. Путем повторной перекристаллизации из хлористого метилена получили рацемический комплекс в форме желтых кристаллов.
1H-NMR/100 Мгц, CDCl3) : 7,0-8,0(м, 22 PH, аром.H), 6,5 (с, 2H, H-C (3)
2,2 (с, 6H, CH3), 1,3 (с, 6H, CH3Si). Масс-спектр: 729 M+, правильное разложение.
Пример E
рац-диметилсиландиилбис(2-метил-4-(-2-нафтил)инденил)цирконийдихлорид (31)
1/2-(2-нафтил)-толуол (25)
14 г /0,57 моля/ магниевых опилок соединили мол 150 мл Et2O, не содержащего H2O и обеспечили проведение реакции Гриньяра с 5 г бромтолуола и несколькими кристалликами йода. Затем прибавляли по каплям 95 г /0,58 ммоля/ бромтолуола в 450 мл Et2O, не содержащего H2O, таким образом, что реакционная смесь все время находилась в стадии кипения. После окончания добавления еще нагревали до тех пор, пока магний полностью не прореагировал.
Раствор Гриньяра затем добавляли по каплям к раствору 120 г /0,57 моля/ 2-бромнафталина и 3,5 г бис/трифенилфосфин/ никельдихлорида в 800 см3 толуола в 800 см3 толуола, так что температура внутри не превысила 50oC. Затем нагревали еще в течение 3 часов на флегме, смешали с 500 мл 10%-ной содержащей воду HCl, фазы отделили и органическую фазу освободили в условиях вакуума от растворителя. После фильтрации через силикагель /гексан/ получили 107 г /87%/ соединения 25 в виде бесцветного масла.
1H-NMR/100 Мгц, CDCl3/:7,0-7,9 (м, 11H, аром.H) 1,9(с, 3H, CH3)
2/2-(2-нафтил)-бензилбромид (26)
105 г /0,48 моля/ соединения 25 и 90 г /0,5 моля/ N-бромсукцинимида растворили при комнатной температуре в 2000 см3 тетрахлоруглерода, смешали с 3 г азобисизобутиронитрила и нагревали в течение 4 часов при флегме. Образованный сукцинимид очистили путем фильтрации в 1000 г силикагеля (смесь гексана и хлористого метилена в соотношении 9:1) Получили 112 г/79%/ 26 в виде бесцветного масла со слезоточивыми свойствами.
1H-NMR(100 Мгц, CDCl3):6,9-8,0 (м, 11H, аром.H), 4,1 (с, 2H, CH2Br)
3/(±)-2-(2-(2-нафтил)-бензил)-пропионовая кислота (27)
К 8,5 г/0,37 моля/ натрия в 100 см3EtOH, не содержащего H2O, прибавляли по каплям при комнатной температуре 70 г (0,37 ммоля) диэтилового эфира метилмалоновой кислоты, растворенного в 50 см3EtOH, не содержащего H2O. Затем добавили 110 г/0,37 моля/ соединения 26 в 200 см3 и нагревали в течение 3 часов при флегме. При комнатной температуре добавили 62 г/1,1 моля/ KOH, растворенного в 100 см3H2O, и еще в течение 4 часов нагревали до флегмы. Растворители удалили в условиях вакуума, остаток смешали до получения истинного раствора с H2O и подкисляли с помощью концентрированной содержащей воду HCl до величины pH 1. Выпавший осадок вакуумировали, высушили и нагревали в течение 1 часа до 130oC. Получили 90 г /84%/ соединения 27 в виде густого масла.
1H-NMR/100 Мгц, CDCl3): 10,9 (с, 1H, COOH), 7,0-8,1 (м, 11H, аром.H) 2,3-3,0(м, 3H, CH2, CH), 1,0 (д, 3H, CH3)
4/(±) 2-метил-42-(2-нафтил)-1-инданон (28)
Раствор 89 г /0,31 моля/ 27 в 37 см3 /0,5 моля/ тионилхлорида перемешивали в течение 18 часов при комнатной температуре. Избыточный тионилхлорид удаляли при 10 мбар, и маслянистый осадок путем многократного растворения, каждый раз в 100 см3 толуола и отгонки в условиях вакуума освободили от образовавшихся остатков тионилхлорида.
Хлорид кислоты внесли в 200 см3 толуола и добавили по капля\м при 10o к суспензии 44 г /0,33 моля/ AlCl3 1000 см3 толуола и нагревали в течение 3 часов до 80oC. Реакционную смесь поместили на 1000 г льда и подкислили с помощью концентрированной HCl до величины pH 1. Органическую фазу отделили в водную фазу дополнительно трижды экстрагировали, каждый раз с помощью хлористого метилена общим объемом в 200 см3. Объединенные органические фазы промыли при помощи насыщенного водного раствора NaHCO3, насыщенного водного раствора NaCl и затем высушили /MgCO4/. После хроматографии в 1000 г силикагеля /смесь гекасана и AeOEt получили 27 г/ 33%/ соединения 28.
1H-NMR/100 Мгц, CDCl3/:7,1-8,0 (м, 10H, аром.H), 2,2-3,3 (м, 3H, CH2 и CH) 1,1 (д, 3H, CH3)
5/2-метил-7-(2-нафтил)инден (29)
К раствору 27 г /100 молей/ соединения 28 в 200 см3 смеси ТГФ с метанолом в соотношении 2:1 добавили при 0oC 3,8 г /100 молей/ NaBH4 и перемешивали в течение 18 часов при комнатной температуре. Реакционную смесь поместили в 100 г льда, смешали с концентрированной HCl до величины pH 1 и несколько раз экстрагировали с Et2O. Объемные органические фазы промыли с помощью насыщенного водного раствора NaHCO3 и насыщенного водного раствора NaCl и затем высушили /MgSO4/.
Сырой продукт внесли в 500 см3 толуола, смешали с 1,5 г паратолуольсульфоновой кислоты и нагревали в течение 2 часов в водоотделителе до флегмы. Реакционную смесь трижды промыли с 50 см3 насыщенного водного раствора NaHCO3 и растворитель удалили в условиях вакуума. После фильтрации через 200 г силикагеля (смесь гексана с хлористым метиленом) получили 18,4 /72%/ соединения 29 в виде бесцветного масла.
1H-NMR (100 Мгц, CDCl3):7,0-8,0 (м, 10H, аром.H), 6,6 (м, 1H, CH), 3,0 (м, 2H, CH2), 2,0 (м, 3H, CH3)
6/ диметилбис(2-метил-4-(2-нафтил)инденил)силан (30)
Раствор 18 г (70 ммолей) соединения 29 в 70 см3 толуола, не содержащего H2O и O2 и 4 мл ТГФ, не содержащего H2O и O2, смешали при комнатной температуре с 26 см3 /70 ммолей/ 20%-ного раствора бутиллития в толуоле и нагревали в течение 2 часов до 80oC. Затем желтую суспензию охладили до 0oC и смешали с 4,5 г /35 ммолей/ диметилдихлорсилана. Реакционную смесь нагревали еще в течение 1 часа до 80oC и затем промыли с помощью 50 см3 H2O. Растворитель удаляли в условиях вакуума и остаток перекристаллизовывался из гептана при -20oC. Получили 10,8 г /54%/ соединения 30 в виде бесцветных кристаллов.
1H-NMR/100 Мгц, CDCl3):7,0-8,1 (м, 20H, аром.H), 6,4 (м, 2H, H-C (3) 4,0 (м, 2H, H-C (1), -0,1(с, 6H, CH3Si)
7/рац-диметилсиландиилбис(2-метил-4-(2-нафтил)инденил) цирконийдихлорид(31)
Раствор 10,5 г /18 ммолей/ соединения 30 в 70 см3 Et2O, не содержащего H2O и O2, смешали при аргоне при комнатной температуре с 13,6 см3 20%-ного раствора бутиллития в толуоле и затем нагревали в течение 3 часов при флегме. Растворитель удалили в условиях вакуума и осадок отфильтровали с помощью 50 мл гексана, не содержащего H2O и O2, в специальном фильтре Y3, промыли с помощью 50 мл гексана, не содержащего H2O и O2 и высушили /0,1 мбар, комнатная температура/. Дилитиевую соль добавили при - 78oC к суспензии, 4,2 г /18 ммолей/ цирконийтетрахлорида в 80 см3 хлористого метилена и нагревали в течение 18 часов при перемешивании с помощью магнитной мешалки до комнатной температуры. Смесь отфильтровали через фильтр Y 3 и остаток дополнительно экстрагировали порциями с общим объемом хлористого метилена 400 см3. Объединенные фильтраты освободили в условиях вакуума от растворителя, и они перекристаллизовались из хлористого метилена. Получили 3,1 г /23%/ рацемической и мезоформы в соотношении 1:1. Путем повторной перекристаллизации из хлористого метилена получили рацемический комплекс в форме желтых кристаллов.
1H-NMR/100 Мгц, CDCl3:7,0-8,0 (м, 22H, аром. H), 6,9 (с, 2H-C (3), 2,2 (с, 6H, CH3), 1,3 (с, 6H, CH3Si). Масс/спектр: 729 М+ правильное разложение.
Пример Ж.
рац-этандиилбис(2 метил-4-фенилинденил)цирконийдихлорид(33)1/1,2- бис-(2-метил-4-фенилинденил)этан (32)
Раствор 50 г /0,24 ммоля/ соединения 3 в 500 мл ТГФ смешали под аргоном при комнатной температуре с 90 см3 /0,24 ммоля/ 20%-ного раствора бутилового лития в толуоле и дополнительно перемешивали в течение 2 часов при 60oC. Смесь охладили до - 78oC добавили 22,5 г /0,12 моля/ 1,2-дибромэтана и нагревали в течение 18 часов до комнатной температуры. Реакционную смесь промыли с помощью 50 см3 H2O. Растворитель удалили в условиях вакуума и остаток подвергли хроматографии 500 г силикагеля /смесь гексана с хлористым метиленом в соотношении 9:1/. Получили 2,5 г /5% соединения 32 в виде желтого масла, которое медленно загустело при - 20oC/.
1H-NMR/100 Мгц, CDCl3): 7,0-8,1 (м, 20H, аром. H), 6,4 (м, 2H,H-C(3), 4,0 (м, 2H, H-C (1) - 0,1 (с, 6H, CH3Si).
2/рац-этандиилбис/2-метил-4-фенилинденил/цирконийдихлорид(33)
Раствор 2,3 г /5 ммолей/ соединения 32 в 20 мл Et2O, не содержащего H2O и O2, смешали под аргоном при комнатной температуре с 4 см3 /10 ммолей/ 20%-ного раствора бутилового лития в толуоле и нагревали в течение 3 часов при флегме. Растворитель удалили в условиях вакуума и осадок отфильтровали с помощью 30 мл гексана, не содержащего H2O и O2, через специальный фильтр Y3, дополнительно промыли с помощью 30 мл гексана, не содержащего H2O, и O2, и высушили /0,1 мбар, комнатная температура/.
Дилитиевую соль добавили при - 78oC к суспензии 1,2 г /5 ммолей/ цирконийтетрахлорида в 30 см3 хлористого метилена и нагревали в течение 18 часов при перемешивании магнитной мешалки до комнатной температуры. Осадок отфильтровали с помощью фильтра Y 3 и остаток дополнительно экстрагировали порциями с общим объемом хлористого метилена 100 см3. Объединенные фильтраты освободили в условиях вакуума от растворителя, и они перекристаллизовались из смеси хлористого метилена с гексаном. Получили 0,5 г /18%/ рацемической и мезоформы в соотношении 1:1. Путем повторной перекристаллизации из толуола получили рацемический комплекс в форме желтых кристаллов.
1H-NMR/100 Мгц, CDCl3): 7,0-7,7 м, 16H, аром. H), 6,6 (м, 2H,H-C(3), 3,4-4,1 (м, 4H, H2C-CH2), 2,1 (с, 6H, CH3) Масс-спектр: 598 М+ правильное разложение.
Пример 3
Me2Si(2-Me-4-Ph-инденил)2ZrMe (BPh4)(35)
1/Рац-диметилсиландиилбис(2-метил-4-фенил-инденил/ цирконийдиметил(34)
0,5 г /0,8 ммолей/ рац- соединения 5 смешали в 10 см3 Et2O, не содержащего H2O и O2, при - 30oC с 1 см3 1,6 М /1,6 ммолей/ раствора метилового лития в Et2O и перемешивали в течение 1 часа при 0oC. Затем растворитель удалили в условиях вакуума, остаток внесли в 20 см3 гексана, не содержащего H2O и O2, и отфильтровали через фильтр Y 3. Получили 0,34 г /72%/ соединения 34. Масс-спектр: 588 М+, правильное разложение
2/Me2Si(2-Me-4-Ph-инденил)2ZrMe(BPh4)/35/
0,2 г /0,3 ммоля/ 34 добавляли при 0oC к 0,25 г /молль/ трибутиламонийтетрафенилбората в 30 см3 толуола. При перемешивании нагрели до 50oC и смесь перемешивали в течение 15 минут при этой температуре. Для полимеризации применили аликвотную часть растворителя.
Пример 1.
Высушенный реактор емкостью 16 дм3 промыли вначале азотом, затем пропиленом и наполнили 10 дм3 жидкого пропилена. Затем ввели 30 см3 толуольного раствора метилалюминоксана и смесь перемешивали при 30oC в течение 15 минут.
Параллельно с этим 1,1 мг рац-соединения 5 растворили в 20 см3 толуольного раствора метилалюминоксана /27 ммолей A1/ и обеспечили проведение реакции путем 15 минутного отстаивания. Раствор затем влили в реактор, путем подвода тепла обеспечили температуру полимеризации в 50oC/4oC/мин/ и поддерживали процесс полимеризации путем охлаждения в течение 1 часа при 50oC. Полимеризацию прекратили, добавив 20 см3 изопропанола. Избыточный мономер откачали, полимер высушили в условиях вакуума. Получили 0,9 кг полипропилена. В реакторе образовался тонкий налет на внутренней стенке и в мешалке. Активность катализатора составила 818 кг полипропилена /г металлоцена в час. Коэффициент вязкости = 905 см3/ г, точка плавления = 159,4oC, индекс изотактичности = 98,8% mmmm = 95,4%, Mw = 1100000 г/моль, Mw/Mw = 2,5.
Пример 2
Повторили полимеризацию из примера 1, отличие заключается в том, что в качестве катализатора применяли 0,9 мг рац-соединения 5 и температура полимеризации была 70oC. Получили 1,4 кг полипропилена. В реакторе образовали сильные отложения на внутренней стенке и на мешалке.
Активность катализатора составила 1555 кг полипропилена /г металлоцена в час, коэффициент вязкости = 719 см3/ г, точка плавления = 157,7oC
Пример 3
22 см3 суспензии "МАО в SiO" /49 ммолей A1/ внесли под аргоном в специальный фильтр Y 3 и смешали с раствором 4,5 мг рац-соединения 5 в 10 см3 толуола /7,2 μmol Zr/
Реакционную смесь перемешивали в течение 30 минут при комнатной температуре, при этом спонтанное покраснение постепенно бледнело. Затем смесь отфильтровали и твердое вещество трижды промыли с 10 см3 гексана. Оставшийся на фильтре осадок со следами гексана вновь суспендировали в 20 см3 гексана для полимеризации. Параллельно с этим высушенный реактор емкостью 16 дм3 вначале промыли азотом, а затем пропиленом и наполнили 10 дм3 жидкого пропилена. Затем 3 см3 триизобутилового алюминия (чистого, без примесей 12 ммолей) разбавили с помощью 30 см3 гексана, подали в реактор и смесь перемешивали при 30oC в течение 15 минут. Затем суспензию катализатора подали в реактор, путем подвода тепла обеспечили температуру полимеризации в 50oC/4oC/мин/ и поддерживали процесс полимеризации путем охлаждения в течение 1 часа при 50oC. Полимеризацию прекратили, добавив 20 см3 изопропанола. Избыточный мономер откачали, полимер высушили в условиях вакуума. Получили 300 г полипропилена. В реакторе не было отложений на внутренней стенке и на мешалке. Активность катализатора составила 67 кг полипропилена /г металлоцена в час. Коэффициент вязкости = 1380 см3/ г, точка плавления = 156oC
Пример 4
Синтез системы катализатора на носителе из примера 3 повторили, отличие заключается в том, что использовали 13 см3 /29 ммолей A1/ суспензии "МАО в SiO2" и 1,8 мг рац-5 /2,9 μmol Zr/
Полимеризация происходила аналогично примеру 3 при 70oC. Получили 420 г полипропиленового порошка. В реакторе не было отложений на внутренней стенке и на мешалке. Активность катализатора составила 233 кг полипропилена /г металлоцена в час. Коэффициент вязкости = 787 см3/ г, точка плавления = 149,5oC.
Пример 5
Синтез системы катализатора на носителе из примера 3 повторили, отличие заключается в том, что применяли 150 см3 /335 ммолей A1/ суспензии "МАО в SiO2" и 44,2 мг рац-5 /70,3 μmol Zr / и реакционную смесь перемешивали в течение 60 минут при комнатной температуре. Затем твердое вещество отфильтровали и трижды промыли с 50 см3 гексана. Оставшийся на фильтре осадок со следами гексана высушили в условиях вакуума и получили бледно-розовый порошок с хорошей сыпучестью. Получили 33,3 г высушенного катализатора с носителем, нанесенным на подложку.
Для полимеризации вновь суспендировали 2,98 г /4 мг = 6,3/ этого высушенного катализатора в 20 см3 гексана.
Полимеризация происходила аналогично примеру 3 при 70oC. Получили 1,05 кг полипропиленового порошка. В реакторе не было отложений на внутренней стенке и на мешалке. Активность катализатора составила 263 кг полипропилена /г металлоцена в час. Коэффициент вязкости = 944 см3/г, точка плавления = 156oC
Пример 6
Высушенный реактор емкостью 1,5 дм3 промыли с помощью N2 и наполнили при 20oC 750 см3 деароматизированного бензина с пределом границ кипения 100 - 120oC / ''RExxsol 100 /120"/ Затем газовое пространство реактора промыли от азота путем пятикратного заполнения пропиленом под давлением 8 бар и сброса давления. Затем добавили 3,75 см3 толуольного раствора метилалюминоксана /10 вес. % МАО/ При перемешивании содержимое реактора нагревали в течение 15 минут до 30oC и после добавки пропилена при скорости перемешивания 500 оборотов в минуту было установлено общее давление в 8 бар. Параллельно с этим 0,1 мг рац-соединения 5 растворили в 1,25 см3 толуольного раствора метилалюминоксана и путем 15- минутного отстаивания обеспечили проведение реакции. Затем раствор влили в реактор, процесс полимеризации проводили при температуре в 50oC и путем соответствующего охлаждения поддерживали в течение 1 часа при этой температуре. Путем соответствующего введения пропилена давление в течение этого времени поддерживалось на уровне 8 бар, затем реакцию прекратили, добавив 2 см3 изопропанола, полимер отфильтровали и высушили в условиях вакуума.
Получили 16 г полипропилена. В реакторе был налет на внутренней стенке и в мешалке. Активность катализатора / KZAred / составила 20 кг полипропилена/г металлоцена • час • бар. Коэффициент вязкости = 833 см3/г, точка плавления = 159oC
Пример 7
полимеризацию из примера 6 повторили, отличие заключается в том, что температура полимеризации составила 60oC. Получили 35 г полипропилена. В реакторе был налет на внутренней стенке и в мешалке. Активность катализатора / KZAred /составила 44 кг полипропилена/г металлоцена • час • бар. Коэффициент вязкости = 484 см3/г, точка плавления = 159oC
Пример 8
Полимеризацию из примера 6 повторили, отличие заключается в том, что температура полимеризации составила 70oC. Получили 88 г полипропилена. В реакторе был налет на внутренней стенке и в мешалке. Активность катализатора / KZAred /составила 110 кг полипропилена/г металлоцена • час • бар. Коэффициент вязкости = 414 см3/г, точка плавления = 159oC.
Примеры 9 - 12.
Реакцию проводили как в примере 2. Однако, перед тем как реактор заполнили жидким пропиленом, добавили водород.
Примеры 9 - 12 (см. таблицу) показывают хорошую чувствительность к водороду металлоцена, согласно изобретению. Можно регулировать молярную массу вплоть до образования воска /смотри пример 12/.
Пример 13.
Реакцию проводили как в примере 3. Однако перед добавлением катализатора в реактор подали водород при давлении 0,2 бар, температура полимеризации составила 60oC. Во время полимеризации однако дополнительно равномерно добавили этилен. В целом в реактор подали 12 г этилена. Получили 0,4 кг этилен-пропиленового сополимера. Активность металлоцена была 88 кг сополимера /г металлоцена в час. Содержание этилена в полимере составило 2,4 вес.%, при этом этилен находился преимущественно в виде изолированных фрагментов. Коэффициент вязкости = 200 см3/г. точка плавления = 143oC.
Пример 14
Реакцию проводили как в Примере 13. Однако во время полимеризации в целом добавили 34 г этилена. Получили 0,38 кг этиленпропиленового сополимера с 7 вес.% этилена. Коэффициент вязкости = 120 см3/г, точка плавления = 121oC.
Пример 15
Реакцию проводили как в примере 4. Во время полимеризации однако дополнительно добавили 4 г этилена и перед полимеризацией заполнили водородом при давлении 0,1 бар. Получили 0,52 кг этилен-пропиленового сополимера. Активность металлоцена составила 286 кг сополимера /г металлоцена в час. Содержание этилена в полимере составило 6,1 вес.%, при этом этилен находился преимущественно в виде изолированных фрагментов. Коэффициент вязкости = 150 см3/г, точка плавления = 116oC.
Пример 16
Высушенный реактор емкостью 150 дм3 промыли с помощью азота и заполнили при 20oC 80 дм3 деароматизированного бензина с пределом границ кипения 100 - 120oC. Затем газовое пространство промыли от азота путем пятикратного заполнения пропиленом под давлением 2 бар и сброса давления. После того как добавили 50 л жидкого пропилена, добавили 64 см3 толуольного раствора метилалюминоксана/ соответственно 100 ммолей Al, молярная масса согласно криоскопическому определению 1080 г/моль/ и содержимое реактора нагрели до 50oC. Путем дополнительной дозировки водорода содержание водорода в газовом пространстве реактора было установлено 2,0% и путем добавочной дозировки во время первой стадии полимеризации постоянно поддерживалось.
9,8 мг рац-соединения 7 растворили в 32 мл толуольного раствора метилалюминоксана /соответственно 50 ммолей A1/ и через 15 минут подали в реактор. Полимеризацию проводили теперь на первой стадии полимеризации при 50oC в течение 5 часов. Затем давление в реакторе было снижено до 3 бар и добавили 2000 г газообразного этилена. Давление в реакторе увеличивалось при этом до 8 бар и полимеризацию проводили еще 14 часов при 40oC, пока реакцию не прекратили, введя углекислый газ /CO2/.
Получили 18,6 кг блоксополимера, соответственно активности металлоцена 99,9 кг сополимера/г металлоцена в час. Коэффициент вязкости = 230 см3/г, ИР (230/5) = 11 гр/ мин, ИР (230/2.16) = 6,7 гр/мин, точка плавления полимера первой стадии полимеризации: 159oC, температура стеклования полимера второй стадии полимеризации: - 38oC. Блоксополимер содержал 5% этилена. В результате фракционирования продукта получили следующий состав: 69 вес.% гомополимера, 31 вес.% сополимера, при этом сополимер имел содержание этилена, составляющее 15 вес.%, средняя длина блока C2 была 2,2.
Пример 16-а.
Проводили реакцию как в примере 16.
3 мг рац-соединения 24 растворили в 32 мл толуольного раствора метилалюминоксана /соответственно 50 ммолей Al/ и через 15 минут подали в реактор. Полимеризацию проводили теперь на первой стадии полимеризации при 50oC в течение 2,5 часов. Затем давление в реакторе было снижено до 3 бар и добавили 3000 г газообразного этилена. Давление в реакторе увеличилось при этом до 8 бар и полимеризацию проводили еще в течение 8 часов при 40oC, до тех пор, пока ее не прекратили, добавив углекислый газ /CO2/. Получили 16,5 кг блоксополимера, соответственно активности металлоцена 524 кг сополимера/г металлоцена в час. Коэффициент вязкости = 480 см3/г, ИР (230/5) = 2 гр/мин, точка плавления полимера первой стадии полимеризации : 162oC, температура стеклования полимера второй стадии полимеризации : - 54oC. Блок сополимера содержал 15% этилена.
Пример 17.
Процесс проводили как в примере 1. Использовали однако 12,5 мг металлоцена рац-7. Получили 1,5 кг полипропилена, активность металлоцена была 120 кг полипропилена/г металлоцена в час. Коэффициент вязкости = 1050 см3/г, точка плавления : 159oC.
Пример 18
Проводили реакцию как в примере 2. Применяли однако 4,1 металлоцена рац-7. Получили 1,3 кг полипропилена, активность металлоцена была 317 кг полипропилена /г металлоцена в час. Коэффициент вязкости = 555 см3/г, точка плавления: 157oC.
Пример для сравнения A
Реакцию проводили как в примере 1. Однако использовали 12,5 мг рац-фенил(метил) силандиилбис(2-метил-1-инденил) цирконийдохлорида. Получили 1,35 кг полипропилена, активность металлоцена была 108 кг/ г металлоцена в час. Коэффициент вязкости = 1050 см3/г, точка плавления : 149oC.
Пример для сравнения B.
Реакцию проводили как в примере 1. Однако применяли 12,5 мг рац-фенил(метил) силандиилбис(1-инденил)цирконийдихлорида. Получили 0,28 кг полипропилена, активность металлоцена была 22,4 кг полипропилена /г металлоцена в час. Коэффициент вязкости - 74 см3/г, точка плавления = 141oC.
Пример 19
Реакцию проводили как в примере 1. Применяли однако 3,3 мг соединения 24. Получили 0,78 кг полипропилена, активность металлоцена была 237 кг полипропилена /г металлоцена в час. Коэффициент вязкости = 1700 см3/г, точка плавления: 163oC, Mw = 2,1* • 106 г/моль, ИР 230 /21,6 = 1 гр/мин, Mw/Mw = 2,1.
Пример 19-а
Реакцию проводили как в примере 2. Однако применяли 1,0 мг рац-соединения 24. Получили 1,2 кг полипропилена. Активность металлоцена была 1200 кг полипропилена/г металлоцена в час. Коэффициент вязкости = 1100 см3/г, точка плавления = 161oC.
Пример 20
Реакцию проводили как в примере 1, однако температура полимеризации была 40oC. Применяли 6,0 мг 17. Получили 1,95 кг полипропилена, активность металлоцена была 325 кг полипропилена/г металлоцена в час. Коэффициент вязкости = 1320 см3/г, точка плавления 162oC, Mw = 1,79 • 106 г/моль, Mw/Mw = 2,3
Пример для сравнения C
Реакцию проводили как в примере 20. Однако применяли металлоцен рац-диметилсиландиилбис/2-этил-1-инденил/цирконийдихлорид, не согласно изобретению. Получили 0,374 кг полипропилена, активность металлоцена была 62,3 кг полипропилена/г металлоцена, в час. Коэффициент вязкости = 398 см3/г, точка плавления 147oC, Mw = 450.000 г/моль, Mw/Mw = 2,5.
Пример 21
Реакцию проводили как в примере 1. Однако применяли 5,2 мг 31. Получили 1,67 кг полипропилена, активность металлоцена была 321 кг полипропилена/г металлоцена в час. Коэффициент вязкости = 980 см3/г, точка плавления 158oC.
Пример 22
Реакцию проводили как в примере 1. Однако полимеризацию проводили при 30oC. Однако применяли 3,7 мг соединения 33. Получили 0,35 кг полипропилена, активность металлоцена была 94 кг полипропилена/г металлоцена в час. Коэффициент вязкости = 440 см3/г, точка плавления 153oC.
Пример 23
Высушенный реактор емкостью 16 дм3 промыли с помощью пропилена и наполнили 10 дм3 жидкого пропилена. Затем 1,1 см3 продукта реакции из примера H2. (соответственно 7,5 мг соединения 34) растворили в 20 см3 толуола и подали при 30oC в реактор. Реактор нагрели до 50oC (10oC/мин) и систему полимеризации поддерживали в течение часа путем охлаждения при этой температуре. Реакцию прекратили, добавив углекислый газ /CO2/. Избыточный мономер откачали и полимер высушили в условиях вакуума при 80oC. Получили 2,45 кг полипропилена. Коэффициент вязкости = 875 см3/г, точка плавления 160oC.
Пример 24
Высушенный реактор емкостью 16 дм3 промыли с помощью азота и наполнили при 20oC 10 дм3 деароматизированного бензина с пределом границ кипения 100 - 120oC. Затем газовое пространство реактора промыли от азота путем пятикратного заполнения этилена под давлением 2 бар и сброса давления. Затем добавили 30 см3 толуольного раствора метилалюминоксана /соответственно 45 ммолей A1, согласно криоскопическому определению 700 г/моль. При перемешивании содержимое реактора нагревали в течение 15 минут до 30oC и после добавки этилена при скорости перемешивания 250 оборотов в минуту было установлено общее давление в 5 бар. Параллельно с этим 3,2 г соединения 12 растворили в 20 см3 толуольного раствора метилалюминоксана и провели предварительную активацию путем 15-минутного отстаивания. Затем раствор влили в реактор, процесс полимеризации проводили при температуре в 50oC и путем соответствующего охлаждения поддерживали в течение 4 часов при этой температуре. Общее давление поддерживалось в это время путем соответствующего добавления /соответствующей подачи/ этилена на уровне 5 бар. Полимеризацию прекратили, добавив 20 мл изопропанола, полимер отфильтровали и высушили в условиях вакуума. Получили 0,7 кг полиэтилена. Коэффициент вязкости = 690 см3/г.
Пример 25
Соблюдали инструкцию примера 24. В отличие от примера 23 использовали 1,8 кг рац-соединения 7, полимеризацию проводили при 70oC и в течение 1 часа поддерживали при этой температуре. Получили 0,9 кг полиэтилена. Коэффициент вязкости = 730 см3/г.
Пример 26
15 г "F-МАО" в SiO2 /111 ммолей Al/ суспендировали при перемешивании в 100 см3 толуола и охладили до - 20oC. Одновременно 155 мг /0,246 ммолей/ соединения рац-5 растворили в 75 см3 толуола и в течение 30 минут прибавляли по каплям к суспензии. Медленно нагрели до комнатной температуры при перемешивании, при этом суспензия приобрела красную окраску. Затем перемешивали в течение часа при 80oC и затем после охлаждения до комнатной температуры смесь отфильтровали и твердое вещество трижды промыли, каждый раз со 100 см3 толуола и один раз с 100 см3 гексана. Фильтрат был красного цвета. Оставшийся на фильтре осадок со следами гексана высушили в условиях вакуума. Получили 13,2 г светло-красного катализатора с хорошей сыпучестью с носителем, нанесенным на подложку. Анализ показал содержание 3,2 мг цирконоцена на 1 г катализатора.
Полимеризация: для полимеризации суспендировали 2,08 г катализатора в 50 см3 деароматизированного бензина с пределом границ кипения 100 - 120oC. Полимеризацию проводили аналогично примеру 3 при 60oC. Получили 1100 г полипропиленового порошка. В реакторе не было налета на внутренней стенке и в мешалке. Активность = 165 кг полипропилена /г металлоцена в час/. Коэффициент вязкости = 1100 см3/г, точка плавления = 153oC, Mw = 1.485.000, Mw/Mw = 3,2.
ИР 230/5 = 0,1 гр/мин, HB = 440 г/дм3
Пример 27
1,31 г катализатора из примера 26 суспендировали в 50 см3 деароматизированного бензина с пределом границ кипения 100 - 120oC. Полимеризация происходила аналогично примеру 3 при 70oC. Получили 1300 г полипропиленового порошка. В реакторе не было налета на внутренней стенке и в мешалке. Активность = 310 кг полипропилена /г металлоцена в час/. Коэффициент вязкости = 892 см3/г, точка плавления = 150oC, Mw = 1.290.000, Mw/Mw = 3,0, HB = 410 г/дм3
Пример 28
Способ нанесения из примера 26 повторили, отличие заключается в том, что 0,845 г соединения рац-5, растворили в 500 см3 толуола, с 90 г "F МАО в SiO2", суспендированными в 500 см3 толуола. Получили 84 г красного, порошкового катализатора. Анализ показал содержание 9 мг металлоцена на грамм твердого вещества, красный фильтрат содержал 13 мг циркония.
Полимеризация: 1,1 г катализатора на носителе, суспендировали в 50 мл деароматизированного бензина с пределом границ кипения 100 - 120oC. Полимеризация происходила аналогично примеру 3 при 70oC. Получили 2850 г полипропиленового порошка. В реакторе не было налета на внутренней стенке и на мешалке. Активность = 288 кг полипропилена г металлоцена в час/. Коэффициент вязкости = 638 см3/г, точка плавления = 150oC, ИР 230/5 = 0,5 гр/мин, HB = 410 г/дм3
Пример 29
Микропористый полипропиленовый порошок /фирма AKZO/ с размером частиц меньше 100 мкм очистили от примесей экстракцией толуолом в сосуде Сокслета в инертных условиях и затем промыли с помощью 20 вес.%-ного раствора триметилового алюминия в толуоле и высушили в условиях вакуума. Параллельно с этим 51,1 мг соединения рац-5 растворили в 40 см3 толуольного раствора метилалюминоксана и обеспечили проведение реакции путем 15-минутной выдержки. Дополнительно добавили 16,5 г полипропиленового порошка и путем кратковременного создания вакуума удалили газ, находящийся в порах, и часть растворителя, и раствор катализатора был полностью поглощен. Путем интенсивного встряхивания реактора получили 46 г гомогенного, мелкодисперсного порошка с хорошей сыпучестью, порошка красного цвета. 10 г порошка катализатора с носителем подвергали форполимеризации в инертных условиях в ротационном испарителе с этиленом в течение 30 минут. Избыточное давление этилена постоянно поддерживалось на уровне в 0,1 бар при помощи вентиля, регулирующего давление, перемешивание порошка катализатора происходило путем непрерывного вращения реактора при охлаждении при 0oC. Получили 12 г катализатора, полученного в результате форполимеризации
Полимеризация: 4,6 г катализатора на носителе, полученного в результате форполимеризации, суспендировали в 50 см3 деароматизированного бензина с пределом границ кипения 100 - 120oC. Полимеризация происходила аналогично примеру 3 при 70oC. Получили 250 г полипропиленового порошка. В реакторе не было налета на внутренней стенке и в мешалке, средний размер частиц был 1000 мкм. Активность = 59 кг полипропилена / г металлоцена в час/, коэффициент вязкости = 734 см3/г, точка плавления = 152oC, насыпной вес = 390 г/дм3.
Пример 30
1 г катализатора на носителе, не подвергшегося форполимеризации, из примера 29 суспендировали в 50 см3 п-декана, для полимеризации. Полимеризация происходила аналогично примеру 3 при 70oC. Получили 600 г полипропилена. В реакторе был тонкий налет на внутренней стенке и на мешалке, средний размер частиц > 2000 мкм. Активность = 540 кг полипропилена /г металлоцена в час/, коэффициент вязкости = 1400 см3/г, точка плавления = 157,7oC, насыпной вес = 280 г/дм3.

Предложены металлоцены с арилзамещенными производными инденила в качестве лигандов, способ их получения и их применение в качестве катализаторов. Очень эффективная каталитическая система для полимеризации или сополимеризации олефинов состоит из сокатализатора, преимущественно алюминоксана или алюминоксана, нанесенного на подложку, и металлоцена формулы 1
где М1 преимущественно обозначает Zr или Hf, R1 и R2 обозначают галоген или алкил, R3 обозначает алкил, R4 - R12алкил или водород и R13 обозначает (замещенный) алкиленовый мостик или мостик гетероатома. Металлоцены, особенно цирконоцелы, дают при наиболее интересных в техническом отношении температурах между 50o и 80oC при высокой активности катализатора полимеры с очень высокой молярной массой, высокой стереотактичностью и высокой точкой плавления. Кроме этого, с помощью систем катализаторов с носителем, нанесенными на подложку, удается избежать образования отложений в реакторе. 4 с. и 9 з.п. ф-лы.

где M1 - титан, цирконий или гафний;
R1 и R2 - одинаковые или различные, означают атом галогена или C1 - C10-алкил,
R3 - одинаковые или различные, означают водород, C1 - C10-алкил, C6 - C10 арил или радикал SiR
R4 - R12 - галоген или галогенированный алкил, а также значения, указанные для R3, или смежные радикалы R11 и R12 вместе образуют с соединяющими их атомами одно или несколько ароматических колец;
R13 - остаток формулы M2R14R15 или CR14R15-CR14R15, где M2 - кремний, германий или олово, R14 и R15 - одинаковые или различные и означают C1 - C10-алкил или C6 - C10-арил.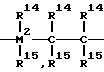
при этом M2 является кремнием или германием и R14 и R15 имеют одинаковое или различное значение и означают C1 - C4-алкилгруппу или C6 - C10-арилгруппу.
при этом M2 - кремний, R14 и R15 имеют одинаковое или различное значение и означают C1 - C4-алкилгруппу или C6 - C10-арилгруппу.
при этом M2 обозначает кремний, и R14 и R15 имеют одинаковое или различное значение и обозначают метил, этил, н-пропил, изопропил или фенил.
| EP, 3808268, кл | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| EP, 3726067, кл | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |

