Изобретение относится к новым замещенным 1-нафтилпиразол-3-карбоксамидам, обладающим большим сродством к человеческому рецептору нейротензина, к способу их получения и содержащим их в качестве действующих начал фармацевтическим композициям.
Первые потенциальные синтезированные непептидные соединения, способные связываться рецепторами нейротензина описаны в европейском патенте 0477049. Речь идет об амидах пиразол-3-карбоновой кислоты, замещенных аминокислотами, которые вытесняют иодированный нейротензин из его рецептора, в дозах менее микромоля, в случае мембран головного мозга морской свинки. Известно соединение, 2-[1-/7-хлор-4-хинолил/- 5-/2,6-диметоксифенил/-пиразол-3-илкарбониламино] -адамантан-2- карбоновой кислоты, SR 48692, обладающей сильной и селективной антагонистической активностью по отношению к нейротензину [D. Gully и др., Proc. Natl. Acad. Sci USA, 1993, 90, 65 - 69].
Ряд продуктов, описанных в европейском патенте 0477049, характеризуется наличием в положении 1 пиразольного цикла фенильной, нафтильной и 4-хинолильной группы, замещенной или незамещенной. Более конкретно, соединение SR 48692 содержит в положении 1 пиразола 7-хлор-4-хинолильную группу. Продукты, описанные в этом документе и содержащие 1-нафтильную или 4-хлор-1-нафтильную группу в положении 1 пиразольного цикла, обладают чрезвычайно повышенным сродством к рецептору нейротензина морской свинки, так как их IC50 порядка 1 - 1-0 наномоль, тогда как их сродство к человеческому рецептору меньше, ибо их IC50 составляет 10 - 100 наномоль.
В настоящее время найдено, что, замещая положение 4 нафтильной группы 1-нафтилпиразол-3-карбоксамидных соединений особыми группами, повышается сродство к рецептору нейротензина и, преимущественно, увеличивается сродство к человеческому рецептору нейротензина.
Таким образом, настоящее изобретение относится, согласно одному из его аспектов, к новым замещенным 1-нафтилпиразол-3-карбоксамидам формулы /I/: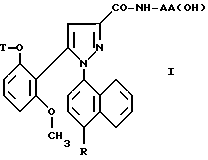
в которой R обозначает группу, выбираемую среди
-CN; -C(NH2)=N-OH, -C(NR4R5)=NR6,
-CONR1R2, -CON(R7)(CH2)pNR1R2,
-CON(R7)(CH2)qCN, -CON(R7)(CH2)qC(NR14R15) = N-R16,
-CH2CN, -CH2CONR1R2, -CH2CON(R7)(CH2)pNR1R2,
-CH2CON(R7)(CH2)qCN, -CH2COOR7,
-O(CH2)nNR1R2, -O(CH2)nCONR1R2,
-O(CH2)nCOOR7, -O(CH2)nSO2NR1R2,
-N(R7)COR3, -N(R7)CO(CH2)nNR1R2,
-N(R7)CO(CH2)nNHCOR3, -N(R7)SO2R8,
-N(R7)CONR9R10, -CH2N(R7)COR3,
-CH2N(R7)SO2R8, -CH2CH2NR11R12,
-CH2CH2N(R7)COR3, -CH2CH2N(R7)SO2R8,
-SO2NR1R2, -SO2N(R7)(CH2)nNR1R2,
-CH2CON(R7)(CH2)qC(NR14 R15) = NR16,
-N(R7)CO(CH2)qCN; -N(R7)CO(CH2)qC(NR14R15) = NR16,
-SO2N(R7)(CH2)qCN; -SO2N(R7)(CH2)qC(NR14R15) = NR16,
или R, связанный с атомом углерода в положении 5 нафтильного радикала, образует группу - CON(R13)CO-;
причем p = 2 - 6; n = 1 - 6; q = 1 - 5;
R1 и R2 каждый независимо друг от друга обозначают водород или (C1-C4)-алкил; или R1 и R2 вместе с атомом азота, с которым они связаны, обозначают гетероцикл, выбираемый в группе, включающей: пирролидин, пиперидин, пиперазин, замещенный в положении 4 радикалом R7; морфолин или тиоморфолин;
R3 обозначает водород, /C1-C8/-алкил; /C3-C8/ циклоалкил; фенил; пиперидил;
R4 и R5 каждый независимо друг от друга, обозначают водород или /C1-C4/-алкил;
R6 обозначает /C1-C4/-алкил;
R7 обозначает водород или /C1-C4/-алкил;
R8 обозначает /C1-C4/-алкил;
R9 и R10, каждый, независимо друг от друга, обозначает водород или /C1-C4/-алкил; кроме того, R10 может обозначать группу - /CH2/nNR1R2;
или R9 и R10 вместе с атомом азота, с которым они связаны, обозначают гетероцикл, выбираемый в группе, включающей: пирролидин, пиперидин, замещенный в положении 4 радикалом R7 пиперазин, морфолин или тиоморфолин;
R11 и R12, каждый, независимо друг от друга, обозначают водород или /C1-C4/-алкил; или R11 и R12 вместе с атомом азота, с которым они связаны, обозначают пирролидин или пиперидин;
R13 обозначает водород, группу -/CH2/nNR1R2; группу - - NHCOR3;
R14 и R15, каждый, независимо друг от друга, обозначают водород или /C1-C4/ - алкил;
R16 обозначает водород; кроме того, R16 может обозначать /C1-C4/ - алкил, когда R14 обозначает водород и R15 обозначает /C1-C4/ - алкил;
или R14 и R16 вместе обозначают этиленовую группу или триметиленовую группу и R15 обозначает водород или /C1-C4/ - алкил;
T обозначает водород; /C1-C4/ - алкил; аллил; /C3-C8/ - циклоалкил;
/C3-C8/ - циклоалкилметил, метоксиэтил;
группа - NH - AA [OH] обозначает остаток аминокислоты: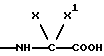
где X обозначает водород и X' обозначает водород, /C1-C5/-алкил или карбоциклический неароматический радикал с 3-15 C-атомами; или X и X' вместе с атомом углерода, с которым они связаны, образуют неароматический карбоцикл с 3-15 C-атомами; и их солям.
Предпочтительно, изобретение относится к соединениями формулы /1/, в которой R обозначает группу, выбираемую среди следующих групп:
-CN, C(NH2) = N-OH, -CONR1R2, -CON(R7)(CH2)pNR1R2,
-O(CH2)nNR1R2, -O(CH2)nCONR1R2, -O(CH2)nCOOR7,
-O(CH2)nSO2NR1R2, -NHCOR3, -NHCO(CH2)nNR1R2,
-CH2CONR1R2, -CH2CON(R7)(CH2)pNR1R2, -CH2COOR7,
-CH2NHCOR3, -SO2NR1R2, -NHSO2R8,
-SO2N(R7)(CH2)nNR1R2,
где p = 2-6; n = 1-6;
R1, R2, каждый, независимо друг от друга, обозначают водород или /C1 - C4/ - алкил; или R1 и R2 вместе с атомом азота, с которым они связаны, обозначают гетероцикл, выбираемый среди пирролидина, пиперидина, пиперазина, морфолина или тиоморфолина;
R3 - обозначает водород; /C1-C8/ - алкил, /C3-C8/ - циклоалкил, фенил;
R7 обозначает водород или /C1-C4/ - алкил;
R8 обозначает /C1-C4/ - алкил;
T обозначает водород, /C1-C4/ - алкил, аллил, /C3-C8/ - циклоалкил,
/C3 - C8/ - циклоалкилметил, метоксиэтил;
группа - NH-AA [OH] обозначает остаток аминокислоты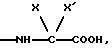
где X обозначает водород и X' обозначает водород, /C1-C5/-алкил или неароматический карбоциклический радикал с 3-15 C-атомами; или еще X и X' вместе с атомом углерода, с которым они связаны, образуют неароматический карбоцикл с 3-15 C-атомами; и их солям.
Согласно настоящему изобретению, под обозначением "/C1-C4/-алкил" или соответственно "/C1-C5/-алкил" или соответственно "/C10C8/-алкил" понимают линейный или разветвленный алкил с 1-4 или соответственно 1-5 или соответственно 1-8 C-атомами.
Карбоциклические неароматические радикалы с 3-15 C-атомами включают моно- или полициклические, конденсированные или связанные мостиками, в случае необходимости, терпеновые, радикалы. Эти радикалы в случае необходимости моно- или полизамещены /C1-C4/-алкилом.
Моноциклические радикалы включают /C3-C15/ - циклоалкильные радикалы, например, как циклопропил, циклопентил, циклогексил, циклогептил, циклооктил, циклододецил.
В вышеуказанном остатке аминокислоты, когда X и X' вместе с атомом углерода, с которым они связаны, образуют неароматический карбоцикл с 3-15 C-атомами, вышеуказанный карбоцикл представляет собой такой, как указанный для соответствующих вышеуказанных радикалов.
Среди неароматических полициклических карбоциклов предпочтителен адамантан, причем соответствующий радикал может представлять собой 1-адамантил, когда X обозначает водород, или 2-адамантилиден, когда X и X' вместе с атомом углерода, с которым они связаны, образуют карбоцикл.
Из неароматических моноциклических карбоциклов особенно предпочтительны циклопентан и циклогексан.
В формуле /I/, R предпочтительно обозначает группу, выбираемую среди: - CONR1R2, -CH2CONR1R2 и -O/CH2/n CONR1R2, причем заместители R1 и R2 предпочтительно обозначают водород и "n" предпочтительно равно 1; -N/R7/ COR3, причем заместитель R3 особенно обозначает /C1-C8/ - алкил, предпочтительно метил, и R7 предпочтительно обозначает водород; -CON/R7/ /CH2/p NR1R2, причем заместитель R7 предпочтительно обозначает водород или метил, R1 и R2 предпочтительно оба обозначают метил и "p" предпочтительно = 2,3 или 4;
-SO2N/R7/ /CH2/nNR1R2, причем заместитель R7 предпочтительно обозначает водород или метил, R1 и R2 предпочтительно оба обозначают метил и n = 2,3 или 4 предпочтительно;
и -CON/R7/ /CH2/q C/NR14R15/ = NR16, причем заместитель R7 предпочтительно обозначает водород или метил, R14 и R16 предпочтительно оба обозначают метил, R15 предпочтительно обозначает водород и q = 2 или 3 предпочтительно. Еще более предпочтительными являются соединения формулы /I/, в которой R имеет вышеуказанное значение и в которой в то же самое время T обозначает водород или метильную или циклопропилметильную группу. Особенно предпочитают соединения формулы /I/, в которой R и T имеют вышеуказанные значения и в которой в то же самое время группа AA/OH/ обозначает 2-карбоксиадамант-2-ильный или α- карбоксициклогексилметильный радикал, причем эти два радикала, вместе с прилегающей группой NH обозначают N-концевые остатки 2-аминоадамантан-2-карбоновой и α- аминоциклогексануксусной /циклогексилглицина/ кислот, соответственно.
Предпочтительными, согласно настоящему изобретению, замещенными 1-нафтилпиразол-3-карбоксамидами являются таковые формулы /I/, в которой
R обозначает аминокарбонильную группу; аминокарбонилметильную группу; ацетамидогруппу; N-/3-N',N'-диметиламинопропил/- аминосульфонильную группу; N-метил-N-/3-N', N'-диметиламинопропил/- аминосульфонильную группу; карбамоилметилокси - группу; N-/3-N', N'- диметиламинопропил/-аминокарбонильную группу; N-/2-N',N'- диметиламиноэтил/-аминокарбонильную группу; N-метил-N-/3-N', N'- диметиламинопропил/-аминокарбонильную группу; N-метил-N-/2-N',N'- диметиламиноэтил/-аминокарбонильную группу; N-метил-N-[2-(N1- метил-N2-метил-амидино)этил]-карбамоильную группу;
T обозначает метильную или циклопропилметильную группу; и группа - NH-AA/OH/ обозначает остаток 2-аминоадамантан-2-карбоновой кислоты или (S) α- аминоциклогексануксусной кислоты;
и их соли
Особенно предпочтительны соединения формулы /Ia/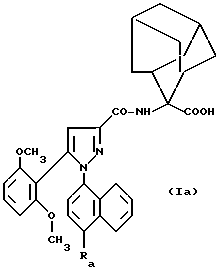
в которой Ra обозначает N-/3-N',N'-диметиламинопропил/ аминокарбонильную или N-/2-N',N'-диметиламиноэтил/ аминокарбонильную группу;
и их соли.
Соли соединений изобретения представляют собой соли со щелочными металлами, предпочтительно с натрием или калием, щелочноземельными металлами, предпочтительно с кальцием и с органическими основаниями, такими, как диэтиламин, трометамин, меглюмин[N-метил-D-глюкамин], лизин, аргинин, гистидин или диэтаноламин.
Соли соединений формулы /I/ согласно настоящему изобретению также включают соли с неорганическими или органическими кислотами, которые позволяют осуществлять надлежащее выделение или кристаллизацию соединений формулы /I/, такими как пикриновая кислота, щавелевая кислота или оптически активная кислота, например, миндальная кислота или камфорсульфокислота, и с такими кислотами, которые образуют фармацевтически приемлемые соли, такие как хлоргидрат, гидросульфат, дигидрофосфат, метансульфонат, малеат, фумарат, 2-нафталин-сульфонат, изэтионат.
Когда соединения формулы /I/ содержат асимметрический атом углерода, энантиомеры составляют часть изобретения.
Когда группа - NH AA [OH] обозначает остаток циклоалифатической аминокислоты, амино- или аминометильные группы могут находиться в эндо- или экзо-положении по отношении к циклической системе; в этих двух случаях соединения формулы /I/ составляют часть изобретения.
Согласно другому из своих аспектов, настоящее изобретение относится к способу получения замещенных 1-нафтилпиразол-3- карбоксамидов формулы /I/ и их солей, отличающемуся тем, что :
1/ Функциональное производное 1 - нафтилпиразол-3-карбоновой кислоты формулы /II/ или /II'/: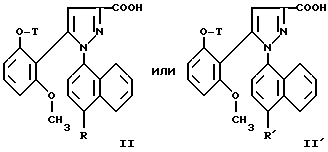
в которой T и R имеют указанные выше для соединения формулы /I/ значения, а R' обозначает предшественник R, выбираемый среди нитро-, амино-, гидроксильной, сульфо-, хлорсульфонильной, карбоксильной группы, обрабатывают аминокислотой, в случае необходимости защищенной обычными в пептидном синтезе защитными группами, формулы:
H - NH - AA /OH/, (III)
в которой -NH-AA/OH имеет указанное выше для соединения формулы /I/ значение;
2/ в случае необходимости, таким образом полученное соединение формулы /I'/:
подвергают последующей соответствующей обработке для превращения заместителя R', предшественника R, в заместитель R;
3/. В случае необходимости, из таким образом полученного в стадии 1/ или стадии 2/ соединения удаляют защитные группы для получения соответствующей свободной кислоты формулы /I/;
4/ в желательном случае, получают соль таким образом полученного соединения формулы /I/.
В качестве функционального производного замещенный 1-нафтилпиразол-3-карбоновой кислоты формулы /II/ или /II'/ можно использовать хлорангидрид, ангидрид, смешанный ангидрид, сложный /C1-C4/-алкиловый эфир, сложный активированный эфир, например, п-нитрофениловый сложный эфир, или свободную кислоту, своевременно активированную, например, с помощью N,N-дициклогексилкарбодиимида или с помощью бензотриазол-1-ил-окси-трис/диметиламино/-фосфоний-гексафторфосфата /BOP/.
Аминокислоты формулы /III/ могут быть использованы такими, какие есть, или после предварительной защиты карбоксильной группы обычными в пептидном синтезе защитными группами, как описано, например, в "Protective Groups in Organic Chemistry" изд. J. F.W. Mc Omie, Plenum Press, 1973, c, 183 или в "Protective Groups in Organic Synthesis" II изд. J.F.W. Greene и P.G.M. Wuts, John Wiley and Sons, 1991, с. 224.
С целью этой защиты, карбоновая группа аминокислоты формулы /III/ может быть просто этерифицирована до сложноэфирной группы, например, в виде сложного метилового, изобутилового или трет.-бутилового эфира, причем этерифицирующую до сложноэфирной группы группу затем удаляют путем омыления или восстановления. Защиту путем этерификации до сложного эфира можно применять только тогда, когда группа R или R' не содержит, также сама, либо сложноэфирной группы, которая должна быть сохранена, как в случае, где, например R обозначает группу O/CH2/nCOOR7 или CH2COOR7 с R7 = алкил, либо, во всяком случае, группы, которая может затрагиваться во время деблокирования сложноэфирной группы. Защита карбоновой группы аминокислоты формулы /III/ также может быть осуществлена путем силилирования, например, с помощью бис-/триметилсилил/-ацетамида, причем вышеуказанная защита может быть осуществлять in situ. Сложный силиловый эфир соединения формулы /I/ затем легко разлагается во время выделения конечного /целевого/ продукта простым подкислением.
Так, на стадии 1/, способа, хлорангидрид 1-нафтилпиразол-3-карбоновой кислоты, полученный путем взаимодействия тионилхлорида или оксалилхлорида с кислотой формулы /II/ или /II'/, можно вводить в реакцию с аминокислотой формулы /III/, в растворителе, таком как ацетонитрил, ТГФ, ДМФ или ДХМ, в инертной атмосфере, при комнатной температуре, в течение времени от нескольких часов до нескольких дней, в присутствии основания, такого как пиридин, гидроксид натрия или триэтиламин.
Один вариант стадии 1/ состоит в получении хлорангидрида или смешанного ангидрида 1 - нафтилпиразол-3-карбоновой кислоты путем взаимодействия изобутилхлорформиата или этилхлорформиата с кислотой формулы /II/ или /II'/, в присутствии основания, такого как триэтиламин, и во введении во взаимодействие с N,O-бис-триметилсилильным производным аминокислоты формулы /III/, полученным путем реакции бис/триметилсилил/-ацетамида или 1,3-бис-/триметилсилил/-мочевины с аминокислотой формулы /III/, в растворителях, таких как ацетонитрил, ДХМ, в инертной атмосфере, при комнатной температуре, в течение времени от одного до нескольких дней.
Другой вариант осуществления стадии 1/ состоит во введении во взаимодействие смешанного ангидрида 1-нафтилпиразол-3-карбоновой кислоты формулы /II/ или /II'/ с аминокислотой формулы /III/, в растворителе, таком как ДХМ, в инертной атмосфере, при комнатной температуре, в течение времени от одного до нескольких дней, в присутствии основания, такого как триэтиламин.
Когда соединение формулы /I/ содержит основную функцию и получено в виде свободного основания, солеобразование осуществляют путем обработки выбранной кислотой в органическом растворителе. Путем обработки свободного основания, растворенного, например, в спирте, таком как метанол, с помощью раствора выбранной кислоты, в том же самом растворителе или в другом растворителе, таком как диэтиловый эфир, получают соответствующую соль, которую выделяют классическими методами. Так, получают, например, хлоргидрат, бромгидрат, сульфат, гидросульфат, дигидрофосфат, метансульфонат, метилсульфат, оксалат, малеат, фумарат, 2-нафталин-сульфонат, изэтионата.
Когда соединение формулы /I/ содержит основную функцию и выделено в форме одной из его солей, например, как хлоргидрат или оксалат, свободное основание может быть получено путем нейтрализации вышеуказанной соли с помощью неорганического или органического основания, такого как гидроксид натрия или триэтиламин, или с помощью карбоната или бикарбоната щелочного металла, такого как карбонат или бикарбонат натрия или калия.
Когда продукт формулы /I/ получен в кислой форме, то его можно превращать в металлическую соль, особенно соль щелочного металла, такую как соль натрия, или щелочноземельного металла, такую как соль кальция, согласно классическим способам.
Замещенные 1-нафтилпиразол-3-карбоновые кислоты формулы /II/ или /II'/: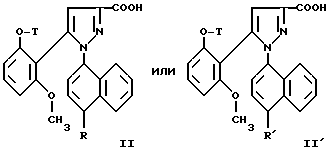
в которой T и R имеют указанные выше для соединений формулы /I/ значения, а R' обозначает предшественник R, выбираемый среди нитро-, амино-, гидроксильной, сульфо-, хлорсусльфонильной, карбоксильной групп; так же, как их функциональные производные кислотной функции, представляют собой главные промежуточные соединения при получении соединений формулы /I/. Когда R1 другой, чем карбоксил, соединения формул /II/ и /II'/ являются новыми и составляют следующий предмет настоящего изобретения.
Кислоты формул /II/ и /II'/, хлорангидриды кислот формул /II/ и /II'/, сложные /C1-C4/-алкиловые эфиры кислот формулы /II и /II'/, которые также могут быть предшественниками вышеуказанных кислот [особенно метиловый, этиловый и трет-бутиловый сложные эфиры] и смешанный ангидрид кислот формул /II/ и /II'/ с изобутил- или этил-хлорформиатом представляет собой особенно предпочтительные промежуточные продукты.
Способ получения соединений формул /II/ или /II'/ через сложные эфиры формул /IIa/ или /II'a/ представлен схемой 1.
На первой стадии а/ сильное основание, такое как метилат натрия, вводят во взаимодействие с кетоном формулы 1, в которой T имеет вышеуказанное значение: затем [стадия b/] вводят во взаимодействие с эквимолярным количеством этилоксалата в спирте, как, например, метанол, согласно L.Claisen, Ber. 1909. 42, 59. После осаждения в эфире, таком как диэтиловый эфир или диизопропиловый эфир, еноляты натрия формулы 2 отделяют путем фильтрации. Можно также получать енолят лития, согласно W.V.Myrray и сотр., J. Heterocyclic Chem. 1989, 26, 1389.
Енолят металла формулы 2, полученный таким образом, и избыток производного нафтилгидразина формулы 3, или его соли затем нагревают при температуре кипения с обратным холодильником в уксусной кислоте [стадия c/] для получения сложных эфиров формул /IIa/ или /II'a/.
Путем омыления сложных эфиров формул /IIa/ или /II'a/ за счет воздействия щелочного агента, как, например, гидроксид калия, гидроксид натрия или гидроксид лития, затем подкисления, получают кислоты формул /II/ или /II'/ [стадия d/].
Особенно соединение формулы /II/, в которой R, связанный с атомом углерода в положении 5 нафтильного радикала, образует группу -CO-N/R13/ CO - с R13 = -NHCOR3, получают при использовании способа, проиллюстрированного в схеме 2.
На первой стадии a'/., калиевую соль 4-сульфо-1,8-нафталинового ангидрида вводят во взаимодействие с гидразин-моногидратом, нагревая при температуре 120oC в течение 36 часов. После осаждения в воде соединение формулы 5 отфильтровывают. Реакция гидразина формулы 5 с енолятом металла формулы 2 [стадия b'/] , согласно способу, описанному в стадии c/. Схемы 1, позволяет получать сложные эфиры формулы /II''a/. Путем омыления сложных эфиров формулы /II''a/ [стадия c'/] согласно способу, описанному в стадии d/. Схемы 1, получают кислоты формулы /II''/. Путем взаимодействия кислот формулы /II''/ с соответствующими хлорангидридами или ангидридами кислот получают целевые соединения формулы /II/.
Из соединения формулы 3 некоторые являются новыми и составляют дальнейший предмет настоящего изобретения.
Таким образом, соединения формулы 3'
в которой Ry обозначает циано- или карбоксиметильную группу; и их соли являются новыми и составляют часть изобретения.
Производные нафтилгидразина формулы /3/, содержащие заместитель R или заместитель R', могут быть получены путем диазотирования соответствующего нафтиламина в присутствии нитрита натрия, затем восстановления соли диазония, например, путем воздействия хлорида олова- /II/. Замещенные нафтиламины известны или получаются известными методами.
Производные нафтилгидразина, в которых R, связанный с атомом кислорода в положении 5 нафтильного радикала, образует группу - CON/R13/CO- с R13 -=H или группу - /CH2/n NR1 R2, получают путем применения способа, проиллюстрированного в схеме 3.
Реакция соединения формулы 4 с амином формулы H2NP13, в которой R13 обозначает водород или группу - /CH2/nNR1 R2, позволяет получать соединения формулы 6 [стадия a''/]. Путем нагревания соединений формулы 6 с гидразин-моногидратом в водном растворе получают целевые гидразины формулы 3 [стадия b''/.].
Превращение соединения формулы /1'/ или, соответственно, формулы /II'/ или формулы /II'a/, в котором нафтильнпя группа замещена радикалом R', в соединении формулы /I/ или, соответственно, формулы /II/ или формулы /IIa/, в котором нафтильная группа замещена радикалом R, осуществляют хорошо известными специалисту классическими методами.
Например, когда R'=SO3H, получают соединение формулы /II'a/, в котором R'=-SO2Cl, затем его превращают в другое соединение формулы /IIa/, в котором R обозначает аминосульфонильную группу, в случае необходимости замещенную, путем воздействия соответствующего амина формулы NHR1 R2, HN[R7] /CH2/n NR1 R2 или HN /R7/ /CH2/q CN, в которой R1, R2, R7, "n" и "q" имеют указанные выше для соединения формулы /I/ значения.
Соединения формулы /IIa/, в которых R обозначает карбоксиметильную группу, позволяют получать соединения формулы /IIa/, в которых R обозначает группу - CH2CONR1 R2 или, соответственно, группу - CH2CON/R7/ /CH2/p NR1 R2 или группу - CH2CON/R7/ /CH2 q CN, путем взаимодействия промежуточно полученного хлорангидрида кислоты с амином HNR1 R2 или, соответственно, с диамином HN /R7//CH2/pNR1 R2 или с аминосоединением HN/R7//CH2/qCN.
Путем реакции соединений формулы /I/, в которых R обозначает группу -CH2CONH2, с пероксидом натрия получают соединения формулы /I/, в которых R обозначает карбоксиметильную группу. Во время получения соединений формулы /I/, в которых R обозначает группу - CH2CONH2, из соединений формулы /II/, в которых R обозначает группу - CH2CONH2, также получают соединения формулы /I/, в которых R обозначает группу - CH2CN, которые разделяют путем хроматографии. Путем восстановления соединений формулы /I/, в которых R обозначает группу - CH2CN, например, путем гидрирования в присутствии катализатора, такого как никель Ренея®, получают соединения формулы /I/, в которых R обозначает группу - CH2CH2NH2. Эти последние соединения позволяют получать соединения формулы /I/, в которых R обозначает группу - CH2CH2NR1 R2, группу - CH2CH2N/R7/COR3 или группу - CH2CH2N/R7/SO2R8, известными специалисту методами.
Соединения формулы /II'a/, или, соответственно, соединения формулы /II'/ или соединения формулы /I'/, в которых R' обозначает нитрогруппу, можно превращать в соединения формулы /II'a/ или, соответственно, формулы /II'/ или формулы /I'/, в которых R' обозначает аминогруппу, затем, известными методами, получают соединения формулы /IIa/ или, соответственно, формулы /II/ или формулы /I/ в которых R обозначает группу - N/R7/COR3 группу - N[R7] CO[CH2] n NR1 R2, группу N/R7/CO - /CH2 n - NHCOR3, группу - N/R7/SO2R8 группу - N/R7/CONR9 R10 или группу - N/R7/CO/CH2 q CN.
Соединения формулы /II'a/ или, соответственно, формулы /II'/ или формулы /I'/, в которых R' обозначает аминогруппу, также позволяет получать соединения формулы /II'a/ или, соответственно, формулы /I'/, в которых R' обозначает гидроксильную группу, затем, известными методами, получают соединения формулы /IIa/ или, соответственно, формулы /II/ или формулы /I/, в которых R обозначает группу - O/CH2/n NR1 R2, группу - O/CH2/nCONR1 R2, группу - O/CH2/nCOOR7 или группу - O/CH2/nSO2NR1R2.
Соединения формулы /I/, в которых R обозначает цианогруппу, позволяют получать путем реакции с пероксидом водорода в присутствии основания, такого как гидроксид натрия, соединения формулы /I/, в которых R обозначает карбамоильную группу. Таким же образом получают соединения формулы /II/, в которых R обозначает карбамоильную группу, исходя из соединений формулы /IIa/, в которых R обозначает цианогруппу.
Соединения формулы /I/, в которых R обозначает цианогруппу, позволяет также получать, путем реакции с гидроксиламином в присутствии основания, такого как карбонат калия, соединения формулы /I/, в которых R обозначает группу - C/NH2/=NOH.
Путем восстановления соединений формулы /IIa/ или, соответственно, формулы /II/ или формулы /I/, в которых R обозначает цианогруппу, например, путем гидрирования в присутствии катализатора, такого как оксид платины, затем взаимодействия с соответствующим хлорангидридом, или ангидридом кислоты, или, соответственно, с сульфонилхлоридом, получают соединения формулы /IIa/ или, соответственно, формулы /II/ или формулы /I/, в которых R обозначает группу - CH2NHCOR3 или, соответственно, группу - CH2NHSO2R8. Точно также получают соединения формулы /IIa/ или, соответственно, формулы /II/ или формулы /I/, в которых R обозначает группу - CH2N/R7/ COR3 или группу - CH2N/R7/SO2R8 с R7 другим, чем водород, осуществляя реакцию алкилирования промежуточного полученного амина.
Соединения формулы /II'a/, в которых R' обозначает карбоксильную группу, позволяют получать соединения формулы /IIa/, в которых R обозначает группу - CONR1 R2 или, соответственно, группу - CON/R7//CH2/p - NR1 R2 или группу - CON/R7//CH2/q CN, путем взаимодействия промежуточно полученного хлорангидрида кислоты с амином HNR1 R2 или, соответственно, с амином HN/R7/ /CH2/p NR1 R2 или с амином HN/R7/ /CH2/q CN.
Реакция соединений формулы /I/, в которых R обозначает группу - CON/R7/ /CH2/q CN или группу - CH2 CON/R7/ /CH2/q CN или группу - N/R7/CO/CH2/q CN или группу - SO2N/R7/ /CH2/q CN, с HCl в спиртовом растворе позволяет промежуточно получать соответствующий имидат. Если имидат вводят во взаимодействие с эквимолярным количеством амина HNR14 R15, то получают соединения формулы /I/, в которых R обозначает группу - CON/R7/ /CH2/q C[NR14 R15]=NR16 или группу - CH2CON/R7/ /CH2/q C[NR14 R15]=NR16 или группу - N/R7/CO/CH2 q C[NR14 R15] = NR16 или группу - SO2N/R7/ /CH2/q - C/NR14 R15/=NR16 с R16=H. Если имидат вводят во взаимодействие с избытком амина HNR14 R15, то получают соединения формулы /I/, в которых R обозначает группу - CON/R7/ /CH2/q C/NR14 R15/=N-R16 или группу - CH2CON/R7//CH2/q C/NR14 R15/=NR16 или группу - N/R7/CO/CH2/q C/NR14 R15/=NR16 или группу - SO2N/R7 /CH2 q - C/NR14 R15/= NR16, c R16 другим, чем водород. Если имидат вводят во взаимодействие с незамещенным или N-замещенным /C1-C4/ - алкилом этилендиамином или с незамещенным или N-замещенным /C1-C4/ - алкилом 1,3 - диаминопропаном, то получают соединения формулы /I/, в которых R обозначает группу - CON/R7/ /CH2/q C/NR14 R15/=NR16 или группу - CH2CON/R7/ /CH2/q C/NR14 R15=NR16 или группу
-N/R7/CO/CH2/q C/NR14 R15/ =NR16 или группу
- SO2N/R7/ /CH2/q C[NR14 R15]=NR16, в которой R14 и R16 вместе образуют этиленовую или триметиленовую группу и R15 обозначает водород или /C1-C4/ - алкил.
Соединения формулы /I/, в которых R обозначает карбамоильную группу, монозамещенную на азоте, позволяют получать, путем реакции с пентахлоридом фосфора, затем взаимодействия с амином HNR4 R5, соединения формулы /I/, в которых R обозначает группу - C/NR4 R5/=N-R6.
Аминокислоты формулы /III/ включают, например, глицин, аланин, лейцин, норлейцин, изолейцин, валин, 1 - адамантиоглицин, 2 - адамантилглицин, циклопропилглицин, циклопентилглицин, циклогексилглицин, циклогептилглицин, 1 - аминоциклопропанкарбоновую кислоту, 1 - аминоциклобутанкарбоновую кислоту, 1 - аминоциклопентанкарбоновую кислоту, 1 - аминоциклогексанкарбоновую кислоту, 1-аминоциклогептанкарбоновую кислоту, 1-амино-4-метилцилкогексанкарбоновую кислоту, 2-аминоадамантан-2-карбоновую кислоту, 2-аминобицикло/3.2.1/октан-2-карбоновую кислоту, 9-аминобицикло/3.3.1/нонан-9-карбоновую кислоту, 2-аминобицикло/2.2.1/-гептан-2-карбоновую кислоту {или 2-аминонорборнан-2-карбоновую кислоту}.
Аминокислоты формулы /III/ представляют собой торговые продукты или могут быть очень легко получены классическими способами. Особенно не имеющиеся в продаже аминокислоты формулы /III/ получают согласно синтезу Strecker, Ann. 1850, 75, 27, или согласно синтезу H.T.Bucherer и др., J.Pract. Chem. 1934, 141, 5; с последующим гидролизом для получения аминокислот; например, 2-амионадамантан-2-карбоновую кислоту получают согласно H.T.Nagasawa и др. J. Med. Chem. 1973, 16 /7/, 823.
α- Амино-1-адамантилуксусную кислоту и α- амино-2-адамантилуксусную кислоту получают согласно B.Gaspert и др. Croatica Chemica Acta, 1976, 48 /28/, 169 - 178.
2-Аминонорборнан-2-карбонову кислоту получают согласно H.S.Tager и др., J. Am.Chem. Soc., 1972, 94, 968.
α- Аминоциклоалкилкрабоновые кислоты получают согласно J.W. Tsang и др., J.Med. Chem., 1984, 27, 1663.
Циклопентилглицины R и S конфигурации получают согласно европейской заявке на патент 477049.
R и S Циклогекилглицины получают согласно Rudman и др., J. Am. Chem. Soc., 1952, 741, 551.
R и S Циклогексилглицины также можно получать путем каталитического гидрирования R и S фенилглицинов.
α- Аминоциклоалкилкарбоновые кислоты /R/ - или /S/ - конфигурации также моно получать путем ферментативного стереоспецифического гидролиза соответствующих рацемических N-ацетилированных производных согласно J.Hill и др., J. Org. Chem. 1965, 1321.
Согласно формулы /I/ и их соли обладают очень большим сродством к человеческим рецепторам нейротензина, в тестах, описанных в публикации D. Gully и др. , цитированной выше. В особенности, по отношению к 1-нафтильным и 4-хлор-1-нафтлиьным производным, описанным в европейском патенте 0477049, которые имеет IC50 равную или выше 100 нмоль, соединения изобретения имеют показатель IC50 значительно меньший, доходящий от 1 нмоль до 50 нмоль. Особенный интерес представляют продукты формулы /I/, в которой T обозначает метил и R обозначает аминокарбонил, аминокарбонилметил, ацетамидогруппу, N-метил-N-/3-N,N-диметиламинопропил/-аминосульфонил, карбамоилметоксигруппу, N-/3-N', N'-диметиламинопропил/-аминокарбонил, N-/2-N', N'-диаметиламиноэтил/-аминокарбонил. Эти соединения, следовательно, даже более активны, чем SR 48692, что неожиданно, если обращаются к активности описанных в европейском патенте 0477049 1-нафтилпиразол-3-карбоксамидов.
Соединения формулы /I/ и их соли исследовали ин виво. Поступая согласно способу, описанному M.Poncelet и др., в Naunyn Schmiedberg's Arch. Pharmacol. , 1994, 60, 349 - 357, наблюдают, что соединения согласно изобретению, вводимые орально, антагонизируют контралатеральные ротации, вызываемые интрастриатальной односторонней инъекцией нейротензина в случае мыши.
Кроме того, работая согласно способу, описанному D.Nisato и др., Life Aciences 1994, 54, 7, 95-100, констатируют, что соединения согласно изобретению, введенные внутривенно, ингибируют повышение артериального давления, вызываемое внутривенной инъекцией нейротензина в случае анестезированной морской свинки.
Соединения, описанные в европейском патенте 0477049, в этих тестах проявляют активность ниже таковой соединений согласно настоящему изобретению.
Соединения формулы /I/ и их фармацевтически приемлемые соли малотоксичны, в частности, их острая токсичность совместима с их использованием в качестве лекарственного средства. Млекопитающим вводят эффективное количество соединения формул /I/ или одной из фармацевтически приемлемых солей, для лечения зависящих от нейротенрзина патологий. Так, вышеуказанные соединения могут быть использованы для лечения нейропсихиатрических расстройств, в особенности таких, которые связаны с дисфункционированием допаминергических систем, например, психозов, особенно шизофрении, и заболеваний, связанных с нарушением координации движений, например болезни Паркинсона [D.R.Handrich и др. , Brain Research, 1982, 231, 216-221, и C.B. Nemeroff, Biological Psychiatzy, 1980, 15 /28, 283-302].
Они могут быть пригодны для диагносцирования и/или лечения раковых заболеваний, например, хирургически недоступных человеческих менингитом [B.Mailleux, Peptides, 1990, 11, 1245-1253]; некоторых видов рака простаты [I.Sehgal и др. Proc. Nat. Acad. Sci. 1994, 91, 4673-4677]; рака легких в малых клетках [T. Sethi и др., Cancer Res. 1991, 51, 3621 - 3623]. Их можно применять для лечения моторных, секректорных, язвенных и/или опухолевых нарушений желудочно-кишечного тракта [Обзор A.Shulkes в "Gut Peptides: Biochemistry and Physiology. изд. J. Waish и G.J. Dockray, 1994"]. Таким образом, соединения формулы /I/ согласно изобретению могут быть пригодны для лечения таких заболеваний, как: раздражительный синдром ободочной кишки, диарея, колиты, язвы, опухоли желудочно-кишечного тракта, диспепсия, панкреатит, эзофагит. Соединения согласно изобретению могут быть показаны в случае сердечно-сосудистых нарушений, и также в случае патологий, связанных с выделением гистамина, таких как воспалительные процессы {D.E. Cochrane и др. Faseb. J. , 1994, 8, 7, 1195} . Соединения настоящего изобретения также могут представлять интерес в области анальгезии, действую наподобие эффекта морфина [M.O. Urban, J. Pharm. Exp. Ther. 1993, 265, 2, 580-586].
Таким образом, предметом настоящего изобретения, являются фармацевтически композиции, содержащие в качестве действующих начал соединения формул /I/ или их фармацевтически приемлемые возможные соли.
В фармацевтических композициях настоящего изобретения для орального, подъязычного, подкожного, внутримышечного, внутривенного, чрескожного или ректального введения действующие начала можно вводить животным и людям в единичных дозах приема, в виде смеси или вместе с классическими фармацевтическими носителями. Соответствующие единичные формы введения включают пероральные формы, такие как таблетки, желатиновые капсулы с лекарством, порошки, гранулы и оральные растворы или суспензии, подъязычные и через рот, подкожные, внутримышечные или внутривенные формы введения и ректальные формы введения.
Для достижения желательного эффекта, доза действующего начала может изменяться в пределах 0,5-1000 мг в день, предпочтительно 2-500 мг.
Каждая единичная доза может содержать 0,5-250 мг действующего начла, предпочтительно 1-125 мг, в комбинации с фармацевтическим носителем. Эта унитарная доза может вводиться 1-4 раза в день.
Когда готовят твердую композицию в форме таблеток, то действующее начало смешивают с фармацевтическим эксципиентом, таким как желатина, крахмал, лактоза, стеарат магния, тальк, гуммиарабик, или аналогичные вещества. Таблетки можно покрывать сахарозой или другими соответствующими материалами или можно их обрабатывать таким образом, чтобы они обладали пролонгированной или замедленной активностью и высвобождали, непрерывно, заданной количество действующего начала.
Препарат в виде желатиновых капсул с лекарством получают путем смешения действующего начала с разбавителем и путем введения полученной смеси в мягкие или твердые желатиновые капсулы.
Препарат в форме сиропа или эликсира может содержать действующее начало вместе с подслащивающим, предпочтительно некалорийным, веществом, метилпарабеном и пропилпарабеном в качестве антисептиков, также как с агентом, придающим соответствующий вкус и цвет.
Диспергируемые в воде гранулы или порошки могут содержать действующее начало в смеси в диспергаторами или смачивателями или суспендирующими агентами, такими как поливинилпирролидон и подобные вещества, точно так же, как с подслащивающими веществами или улучшающими вкус веществами.
Для ректального введения прибегают к свечам, которые получают со связующими, плавящимися при ректальной температуре, например, с маслом какао или полиэтиленгликолем.
Для парентерального ведения используют водные суспензии, изотонические солевые растворы или стерильные растворы для инъекций, которые содержат диспергаторы и/или смачиватели, которые фармакологически совместимы, например, как полиэтиленгликоль или бутиленгликоль.
Действующее начало также может быть сформулировано в форме микрокапсул, в случае необходимости с одним или несколькими носителями или добавками.
Для улучшения растворимости продуктов изобретения, соединения формулы /I/ или их фармацевтически приемлемые соли также могут находиться в виде комплексов с циклодекстринами.
В описании и в примерах используются следующие сокращения: MeOH = метанол, EtOH = этанол, эфир = диэтиловый эфир; изо-эфир = диизопропиловый эфир, AcOEt = этилацетат; MeCN = ацетонитрил; ДХМ = дихлорметан; ДМФ = диметилфомамид; ДМСО = диметилсульфоксид; ТГФ = тетрагидрофуран; HCl = соляная кислота; AcOH = уксусная кислота; ТФК = трифторукскусная кислота; H2SO4 = серная кислота; NaOH = гидроксид натрия; KOH = Гидроксид калия; NH4OH = аммиак; Na2SO4 = сульфат натрия; P2O5 - фосфорный ангидрид, Me, MeO = метил, метокси; Et = этил, т.пл. = температура плавления, ТК = комнатная температура, силикагель = силикагель 50 H, выпускаемый в продажу фирмой MER CK /Дармштадт/;
ЯМР = ядерный магнитный резонанс;
с. = синглет, с.ш. = уширенный синглет, д. = дублет, т. = триплет;
кд. = квадруплет, кт. = квинтуплет, м. = массив, мт. - мультиплет;
Приготовление 1
Натриевая соль (метил)-4-/2,6-диметоксифенил/-4-оксидо-2- оксобут-3-еноата
Раствор 100 г 2,6-диметоксиацетофенона и 7,5 мл этилоксалата в 520 мл безводного метанола медленно добавляют к раствору метилата натрия, приготовленному из 12,7 г натрия и 285 мл безводного метанола. В течение 7 часов кипятят с обратным холодильником и оставляют на ночь при комнатной температуре. Реакционную смесь выливают в 2 л изо-эфира и перемешивают в течение 15 минут. Получают целевой продукт путем отфильтовывания, промывки изо-эфиром и высушивания в вакууме. Масса = 120 г.
Т.пл. = 178oC.
Приготовление гидразинов формулы 3:
Приготовление 2.1.: 1-Гидразино-4-нитронафталин-хлоргидрат
К охлажденной до -5oC суспензии 5,2 г 4-нитро-1-нафтиламина в 150 мл концентрированной HCl, 100 мл 1н. HCl и 150 мл
уксусной кислоты, добавляют раствор 1,9 г нитрита натрия в 10 мл воды. Оставляют на 1 ч 15 минут при перемешивании, при температуре от -5oC до 0oC. Охлаждают до -15oC и очень медленно добавляют раствор 12,5 г дигидрата хлорида олова - /II/ в 30 мл концентрированной HCl. Оставляют температуру подниматься до комнатной и выдерживают 2,5 часа при перемешивании. Реакционную среду фильтруют, затем твердое вещество осаждают водой и снова отфильтровывают. Получают 5,9 г целевого продукта.
Приготовление 2.2: 4-Циано-1-гидразинонафталин
К охлажденному до 0oC раствору 8,35 г 4-циано-1-нафтиламина в 180 мл 1 н. HCl добавляют раствор 4,09 г нитрита натрия в 30 мл воды. Оставляют стоять при перемешивании в течение 1 часа 15 минут при 0oC. Охлаждают до -10oC и медленно добавляют раствор 41,75 г дигидрата хлорида олова - /II/ в 42 мл концентрированной HCl. Перемешивают в течение 1 часа, причем температура повышается до комнатной. Реакционную смесь фильтруют. Остаток суспендируют в воде и добавляют 20 мл концентрированного раствора NaOH. Получают целевой продукт [после отфильтровывания, промывки водой, затем высушивают в вакууме] . Масса = 8,6 г.
Приготовление 2.3: Хлоргидрат 4-гидразинонафталин-1-сульфокислоты
К суспензии 22,33 г 4-аминонафталин-1-сульфокислоты в 125 мл воды добавляют 4 г NaOH. Смесь охлаждают до 0oC и добавляют 2 мл концентрированного раствора NaOH, затем 7,5 г нитрита натрия и 125 г льда для поддерживания температуры 0oC. Таким образом полученную суспензию выливают в 75 мл концентрированной HCl, предварительно охлажденной до 0oC, и оставляют на 2 часа 15 минут при перемешивании при этой температуре. Реакционную смесь медленно добавляют к раствору 55 г дигидрата хлорида олова -/II/ в 50 мл концентрированной HCl и 25 воды, предварительно охлажденному до -10oC. Спустя ночь получают целевой продукт путем отфильтровывания, промывки 1 н. раствором HCl и водой.
Масса = 23,96 г.
Приготовление 2.4. Хлоргидрат 4-гидразинонафталин-1-уксусной кислоты.
А/. Хлоргидрат 4-аминонафталин-1-уксусной кислоты
Это соединение получают по способу, описанному Y. Ogata и сотр., J. Org. Chem. 1951, 16, 1588.
Б/. Хлоргидрат 4-гидразинонафталин-1-уксусной кислоты
При -5oC, 3,18 г полученного в предыдущей стадии соединения смешивают с 30 мл концентрированной HCl и 30 мл уксусной кислоты. Быстро добавляют раствор 1 г нитрата натрия в 15 мл воды и оставляют на 2 часа при перемешивании при температуре от -5oC до +4oC. Затем добавляют раствор 14,2 г дигидрата хлорида олова -/II/ в 19 мл концентрированной HCl и оставляют на 30 минут при перемешивании при 0oC. Оставляют температуру повышаться до комнатной и перемешивают в течение 1 часа. Реакционную смесь отфильтровывают и собранные кристаллы перемешивают в течение 1 часа в MeCN. Получают целевой продукт [после отфильтровывания]. Масса = 20,4 г. Т.пл. = 180oC.
Приготовление 2.5. Хлоргидрат 4-гидразинонафталин-1-карбоновой кислоты
А/. Метансульфонат 4-аминонафталин-1-карбоновой кислоты
К охлажденному до -15oC раствору 5 г 4-бром-1-нафтиламина в 90 мл эфира, в атмосфере аргона, добавляют 2,8 мл 1,6 М раствора н-бутиллития в гексане и перемешивают в течение 1 часа при -15oC. Затем добавляют раствор 5 г 1,2-бис-/хлордиметилсилил/-этана в 50 мл эфира, перемешивают в течение 30 минут при -10oC и оставляют температуру повышаться до комнатной. Охлаждают до -5oC, добавляют 15 мл 1,6 М раствора н-бутиллития в гексане и перемешивают в течение 1 часа при 0oC. Затем в реакционную смесь пропускают путем барботирования ток диоксида углерода, в течение 2-х часов и при температуре 0-5oC. После этого добавляют 2,86 мл хлортриметилсилана и оставляют на 30 минут при перемешивании. Затем добавляют воду, экстрагируют этилацетатом, органическую фазу сушат над сульфатом натрия и растворитель выпаривают в вакууме.
Остаток растворяют в ацетоне, добавляют 1,46 мг метансульфокислоты, образовавшийся осадок отсасывают и промывают его эфиром. Получают 4 г целевого продукта. Т.пл. = 180oC /разложение/.
Это соединение также получают, следуя нижеописанной методике.
А'/. 4-Аминонафталин-1-карбоновая кислота
Смесь 32 г 4-циано-1-нафтиламина в 400 мл 50%-ного водного раствора KOH кипятят с обратным холодильником в течение ночи. После охлаждения в реакционную смесь добавляют 800 мл воды и отфильтровывают нерастворимую часть. Фильтрат охлаждают до +5oC и подкисляют до pH 5 за счет добавления концентрированной HCl. Образовавшийся осадок отсасывают и после высушивания получают 35 г целевого продукта.
Это соединение также получают, следуя двум стадиям нижеописанного способа:
А''/. 4-Нитронафталин-1-карбоновая кислота
Это соединение получают согласно J. Am. Chem. Soc., 1929, 51, 1831 - 1836, исходя из ангидрида 4-нитро-1,8-нафталиновой кислоты.
Б''/. Хлоргидрат 4-аминонафталин-1-карбоновой кислоты
В аппарате Парра, под давлением 8 бар, гидрируют смесь 35 г полученного в стадии А''/. соединения, 1 л метанола, 200 мл ДМФ и в присутствии никеля Ренея®. Спустя 4 часа, катализатор отфильтровывают и фильтрат выпаривают в вакууме. Остаток обрабатывают водой, оставляют на ночь при перемешивании и осадок отсасывают. Осадок растворяют в насыщенном растворе хлороводорода в метаноле и добавляют эфир вплоть до осаждения. После отсасывания, затем высушивания, получают 21,4 г целевого продукта.
Это соединение также получают следуя трем стадиям нижеописанного способа:
А''. N-трет-Бутоксикарбонил-4-бром-1-нафтиламин
Смесь 5 г 4-бром-1-нафтиламина, 6,4 г ди-трет-бутилдикарбоната в 120 мл трет. -бутанола в течение 20 часов кипятят с обратным холодильником. Реакционную смесь выливают в 250 мл воды, отсасывают образовавшийся осадок и промывают водой. Осадок растворяют в ДХМ, органическую фазу сушат над сульфатом натрия и растворитель выпаривают в вакууме. Получают 6,9 г целевого продукта. Т.пл. = 134oC
Б''/. 4-трет-Бутоксикарбониламинонафтил-1-карбоновая кислота
К охлажденному до -10oC раствору 5 г полученного в предыдущей стадии соединения в 100 мл эфира, в атмосфере азота, добавляют 21,3 мл 1,6 М раствора н-бутиллития в гексане, и перемешивают в течение 1,5 часов при 0oC. Охлаждают до -10oC, и в реакционную смесь пропускают путем барбонирования ток диоксида углерода, в течение 15 минут. Затем добавляют 250 мл воды, и, после декантации, водную фазу промывают эфиром. Водную фазу подкисляют до pH 6 путем добавления уксусной кислоты, экстрагируют с помощью ДХМ, органическую фазу сушат над сульфатом натрия и растворитель выпаривают в вакууме. Получают 2,7 г целевого продукта. Т.пл. = 214 oC.
В''/. Метансульфонат 4-аминонафталин-1-карбоновой кислоты
К суспензии 7 г полученного в предыдущей стадии соединения в 70 мл ДХМ, при комнатной температуре, прикапывают раствор 15,8 мл метансульфокимслоты в 50 мл ДХМ. После перемешивания в течение 45 минут при комнатной температуре, к реакционной смеси добавляют 150 мл эфира и образовавшийся осадок отсасывают. После промывки осадка эфиром и высушивания получают 6,5 г целевого продукта. Т.пл. = 196oC /разложение/.
Б/. Хлоргидрат 4-гидразинонафталин-1-карбоновой кислоты
При 0oC, 2,4 г хлоргидрата 4-аминонафталин-1-карбоновой кислоты смешивают с 80 мл концентрированной HCl. Добавляют раствор 0,89 г нитрита натрия в 19 мл воды и перемешивают 2 часа при 2-3oC. Охлаждают до -10oC и медленно добавляют раствор 9,7 г дигидрата хлорида олова -/II/ в 90 мл концентрированной HCl. Оставляют температуру повышаться до комнатной и выдерживают 30 минут при перемешивании. Добавляют 200 мл воды, оставляют на 30 минут при перемешивании и отсасывают образовавшееся твердое вещество. Твердое вещество обрабатывают с помощью MeCN, отсасывают, промывают эфиром и высушивают. Получают 2,2 г целевого продукта, Т.пл. = 190oC.
Приготовление 2.6: Хлоргидрат 4-гидразино-1,8-нафталимида
А/. Калиевая соль 4-сульфо-1,8-нафталимида
В течение 2-х часов при 60-70oC перемешивают смесь 25 г калиевой соли ангидрида 4-сульфо-1,8-нафталиновой кислоты и 300 мл водного 30%-ного раствора аммиака. Спустя ночь стояния при комнатной температуре, отсасывают выпавший осадок, промывают его водой, затем этанолом. Получают 21 г целевого продукта. Т.пл. > 300oC.
Б/. Хлоргидрат 4-гидразин-1,8-нафталимида
В течение 4-х дней при 80oC нагревают смесь 7 г полученного в предыдущей стадии соединения, 3,5 мл гидразин-моногидрата и 100 мл воды. После охлаждения подкисляют до pH 1 за счет добавления 1 н. раствора HCl, отсасывают выпавший осадок. Получают, после порошкования в смеси этанола с эфиром, затем отсасывают, целевой продукт в количестве 3,5 г. Т.пл. = 278oC.
Приготовление 2.7: N-Амино-4-гидразино-1,8-нафталимид /соединение 5/
В течение 1,5 дней при 120oC нагревают смесь 25 г калиевой соли 4-сульфо-1,8-нафталинового ангидрида и 50 мл гидразин-моногидрата. После охлаждения к реакционной смеси добавляют воду, отсасывают выпавший осадок и высушивают его. Получают 15,7 г целевого продукта.
Приготовление сложных эфиров формул /IIa/, /II'a/ и /II''a/.
Приготовление 3.1:
Сложный метиловый эфир 5-/2,6-диметоксифенил/-1-/4-нитронафт-1- ил/-пиразол-3-карбоновой кислоты
[Формула /II'a': R' = NO2; T = метил].
В течение 5,5 часов кипятят с обратным холодильником смесь 10 г полученного в Приготовлении 2.1 соединения, 13,5 г полученного в Приготовлении 1 соединения и 200 мл уксусной кислоты. После отфильтровывания нерастворимой части фильтрат выливают в 2 л смеси воды со льдом. Отфильтровывают образовавшийся осадок и перемешивают этот последний в 300 мл изоэфира. Получают, после отфильтровывания, 14,8 г целевого продукта. Т. пл. = 180oC.
Приготовление 3.2.
Сложный метиловый эфир 1-/4-цианонафт-1-ил/-5- /2,6-диметоксифенил/-пиразол-3-карбоновой кислоты
[формула /IIa/, R = CN, T = метил]
В течение 5 часов 15 минут кипятят с обратным холодильником смесь 8,6 г полученного в Приготовлении 2.2 соединения, 13,5 г полученного в Приготовлении 1 соединения и 85 мл уксусной кислоты. Спустя ночь выдерживания при комнатной температуре, реакционную смесь выливают в смесь воды со льдом. Отфильтровывают образовавшийся осадок и промывают его водой. Хроматографируют на диоксиде кремния, элюируя ДХМ, затем смесью ДХМ [98: 2 по объему]. Получают 5,38 г целевого продукта. Т. пл. = 165oC.
Приготовление 3.3.
Сложный метиловый эфир 5-/2,6-диметоксифенил/-1-/4-сульфонафт- 1-ил/-пиразол-3-карбоновой кислоты
[формула /II'a/, R' = SO3H, T = метил]
В течение 2-х часов кипятят с обратным холодильником смесь 22,9 г полученного в Приготовлении 2.3 соединения, 21,65 г полученного в Приготовлении 1 соединения и 150 мл уксусной кислоты. После охлаждения реакционную смесь выливают в смесь воды со льдом, и отфильтровывают образовавшийся осадок. Фильтрат охлаждают и доводят до pH 5 - 6 путем добавки концентрированного раствора NaOH. Образовавшийся осадок отфильтровывают и промывают водой. Получают 28,66 г целевого продукта. Т. пл. = 236oC.
Приготовление 3.4:
Сложный метиловый эфир 1-[4-/карбоксиметил/-нафт-1-ил] -5- (2,6-диметоксифенил)-пиразол-3-карбоновой кислоты
[формула /IIa/: R = CH2COOH, T = метил]
В течение 2-х часов 30 минут при 60 - 70oC нагревают смесь 2,4 г полученного в Приготовлении 2.4 соединения, 2,8 г полученного в Приготовлении 1 соединения и 50 мл уксусной кислоты. После охлаждения добавляют в реакционную смесь воды и отделяют образовавшееся вязкое масло. Это масло растворяют в этаноле и медленно добавляют его к водному раствору. Перемешивают в течение 1 часа и отфильтровывают образовавшийся осадой. Получают 2,8 г целевого продукта. Т. пл. = 200oC.
Приготовление 3.5.
Метиловый эфир 5-/2,6-диметоксифенил/-1-[4- /этанкарбоксамидо/-нафт-1-ил]-пиразол-3-карбоновой кислоты
[формула (IIa): R = NHCOEt; T = метил]
При 80oC, в течение 33-х часов и при атмосферном давлении, гидрируют смесь 0,87 г полученного в Приготовлении 3.1 соединения 0,3 г 10%-ного палладия - на-угле и 5 мл пропионового ангидрида в 5 мл ДМФ. Затем добавляют 0,5 мл пиридина и оставляют на ночь при перемешивании и при комнатной температуре. Фильтруют через Целит®, промывают метанолом и выпаривают в вакууме. Хроматографируют на силикагеле, элюируя смесью толуола с этилацетатом /70 : 30 по объему/. Получают 0,45 г целевого продукта, Т. пл. = 110oC.
Приготовление 3.6
Метиловый эфир 1-[4-/ацетиламинометил/-нафт-1-ил]-5- [2,6-диметоксифенил]-пиразол-3-карбоновой кислоты
[формула /IIa/: R = -CH2NHCOMe, T = метил]
В течение 12 часов при 60oC, затем в течение 8 часов при 100oC, и при атмосферном давлении, гидрируют смесь 2 г полученного в Приготовлении 3.2 соединения, 30 мл уксусного ангидрида и 0,2 г оксида платины. После отфильтровывания на Целите®, промывки этилацетатом, добавляют к фильтрату воду. Экстрагируют этилацетатом, сушат над сульфатом натрия и выпаривают растворитель в вакууме. После хроматографирования на силикагеле, элюируя градиентом смеси ДХМ /этилацетат от [98/2 по объему] до [60/40 по объему], получают целевое соединение. Получают 0,9 г целевого продукта.
ЯМР - спектр при 200 мГц в ДМСО: δ, м.д. = 1,95 /с., 3H/; 3,5 /с.ш., 6H/; 3,9 /с., 3H/; 4,75 /д., 2H/; 6,55 /д., 2H/; 6,95 /с., 1H/; 7,15 - 8,25 /м., 7H/; 8,5 /т., 1H/;
Приготовление 3.7.
Сложный метиловый эфир 5-/2,6-диметоксифенил/-1-{4- /диметиламиносульфонил/-нафт-1-ил} -пиразол-3-карбоновой кислоты [формула /IIa/:
R = SO2N /Me/2; T = метил].
A/ Сложный метиловый эфир 1-[4-/хлорсульфонил/-нафт-1-ил]- 5-[2,6-диметоксифенил]пиразол-3-карбоновой кислоты [формула /II'a/:
R = -SO2Cl; T = метил].
В течение 5 часов при комнатной температуре перемешивают смесь 5 г полученного в Приготовлении 3.3 соединения, 3,21 г пентахлорида фосфора в 100 мл ДХМ. Выпаривают в вакууме досуха и остаток нагревают при 110oC в течение 1 часа. Оставляют стоять в течение ночи при комнатной температуре. Остаток обрабатывают несколько раз толуолом, ДХМ, 1,2-дихлорэтаном, с последующим выпариванием в вакууме после каждой обработки. Получают 5,9 г целевого продукта, который используют таким, какой есть, в следующей стадии.
Б/. Сложный метиловый эфир 5-[2,6-диметоксифенил]-1- {4-/диметиламино-сульфонил/-нафт-1-ил}-пиразол-3-карбоновой кислоты
[формула /IIa/: R = -SO2N /Me/2; T = метил]
В течение 45 минут газообразный диметиламин пропускают в раствор 5,9 г полученного в предыдущей стадии соединения в 100 мл ДХМ, к которому добавляют 1,2-дихлорэтан и ДМФ до растворения соединения. Затем выдерживают при перемешивании и при комнатной температуре в течение 3,5 часов. После отфильтровывания нерастворимой части, фильтрат выпаривают в вакууме. Остаток экстрагируют этилацетатом, промывают водой, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Получают 2,67 г целевого продукта.
ЯМР - спектр при 200 мГц в ДМСО: δ, м.д. = 2,65 /с., 6H/; 3,35 /с.ш., 6H/; 6,45 /д., 2H/; 6,95 /с., 1H/; 7,15 /т., 1H/; 7,3 - 8,8 /м., 6H/.
Приготовление 3.8
Сложный метиловый эфир 5-/2,6-диметоксифенил/-1-{4- [N-метил-N-/3-N', N'-диметиламинопропил/-аминосульфонил] -нафт-1-ил}- пиразол-3-карбоновой кислоты
[формула /IIa/: R = -SO2N/Me/ /CH2/3 N/Me/2; T = метил].
При температуре кипения с обратным холодильником, 2,34 г полученного в стадии А/. Приготовление 3.7. соединения растворяют в 50 мл толуола, охлаждают до комнатной температуры и добавляют 1,4 мл N,N,N1-триметил-1,3-пропандиамина. Затем в течение 2-х часов 45 минут перемешивают при комнатной температуре и добавляют 0,7 мл N,N,N'-триметил-1,3-пропандиамина. После перемешивания в течение 45 минут при комнатной температуре нерастворимую часть отфильтровывают и фильтрат выпаривают в вакууме. Остаток экстрагируют этилацетатом, экстракт промывают водой, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Хроматографируют на диоксиде кремния, элюируя смесью ДХМ /метанол/ 100 : 6 по объему/. Получают 0,82 г целевого продукта. Т. пл. = 87oC.
Приготовление 3.9
Сложный метиловый эфир 1-[4-/карбамоилметил/нафт-1-ил] - 5-/2,6-диметоксифенил/пиразол-3-карбоновой кислоты
[формула /IIa/: R = -CH2CONH2; T = метил].
При 40oC в течение 3-х часов нагревают смесь 1,4 г полученного в Приготовлении 3.4 соединения, 3 мл тионилхлорида в 30 мл ДХМ; затем выпаривают в вакууме досуха. Добавляют раствор хлорангидрида кислоты, полученного выше, в ТГФ к 20 мл 0,4 н. раствора аммиака в ТГФ. Перемешивают в течение ночи при комнатной температуре, и реакционную смесь выпаривают в вакууме. Остаток обрабатывают водой и отфильтровывают образовавшийся осадок. Получают целевой продукт, который используют таким какой есть, в Приготовлении 4.8.
Приготовление 3.10
Сложный метиловый эфир 1-/4-аминонафт-1-ил/-5-/2,6- диметоксифенил/-пиразол-3-карбоновой кислоты
[формула /II'a/: R' = NH2; T = метил].
В течение 6 дней, при комнатной температуре и атмосферном давлении, гидрируют смесь 7,5 г полученного в Приготовлении 3.1 соединения, 0,75 г никеля Ренея® в 200 мл метанола. Реакционную смесь фильтруют через Целит®, промывают 100 мл ДМФ и растворители выпаривают досуха. После кристаллизации из изо-эфира, получают 0,7 г целевого продукта.
Т.пл. = 246oC.
Приготовление 3.11
Сложный метиловый эфир 5-/2,6-диметоксифенил/-1- /4-гидроксинафт-1-ил/пиразол-3-карбоновой кислоты
[формула /II'a/: R' = OH; T = метил]
К охлажденной до 0oC смеси 2,5г полученного в Приготовлении 3.10 соединения в 50 мл 35%-ной серной кислоты добавляют раствор 0,475 г нитрата натрия в 25 мл воды. Перемешивают в течение 1,5 часов при 3oC и, после отфильтровывания образовавшихся кристаллов, затем высушивания, получают 1,3 г соли диазония. Таким образом полученную соль диазония добавляют к раствору 130 г нитрита меди в 800 мл воды и перемешивают в течение 30 минут. Затем добавляют 1 г сульфата железа и перемешивают 2 часа. После отфильтровывания реакционной среды, остаток обрабатывают метанолом и перемешивают в присутствии животного угля. Отфильтровывают через Целит® и фильтрат выпаривают в вакууме. После кристаллизации из этанола получают 0,69 г целевого продукта. Т. пл. = 240oC.
Приготовление 3.12
Сложный метиловый эфир 5-/2,6-диметоксифенил/-1-{4- /3-N,N-диметиламино-пропокси/нафт-1-мл}-пиразол-3-карбоновой кислоты
[формула /IIa/: R = -O/CH2/3N /Me/2; T = метил].
Смешивают 0,44 г полученного в Приготовлении 3.11 соединения 0,19 мл 50%-ного водного раствора гидроксида цезия и 1 мл метанола, затем выпаривают досуха. Остаток обрабатывают с помощью 5 мл ДМФ и добавляют 0,48 г, хлоргидрата 3-хлор-N,N-диметилпропиламина, затем 1,44 г карбоната калия. Нагревают 3 часа при 60oC. После охлаждения добавляют воду, экстрагируют этилацетатом, экстракт сушат над сульфатом натрия и растворитель выпаривают в вакууме. Хроматографируют на диоксиде кремния, элюируя смесью ДXM /метанол/ NH4OH [100 : 5 : 0,5 по объему]. Получают 0,25 г целевого продукта. ЯМР - спектр при 200 мГц в ДМСО; δ, м.д. = 1,9 /кт., 2H/; 2,6 /с., 6H/; 2,4 /мт., 2H/, 3,35 /c. , 6H/; 3,7 / c., 3H/; 4,1 /т., 2H/; 6,35 /д., 2H/; 6,8 /с., 1H/; 6,95 - 8,1 /м., 7H/.
Приготовление 3.13
Сложный метиловый эфир 1 -{4-/карбамоилметокси/-нафт-1-ил}-5-/2,6-диметоксифенил/-пиразол-3- карбоновой кислоты
[формула /IIа/:
R=OCH2CONH2; T = метил].
Смешивают 0,39 г полученного в Приготовлении 3,11, соединения, 0,17 мл 50%-ного водного раствора гидроксида цезия и 1 мл метанола, затем выпаривают досуха. Остаток обрабатывают с помощью 5 мл ДМФ и добавляют 0,193 г 2-бромацетамида. Нагревают 2 часа при 60oC. После охлаждения добавляют воду, экстрагируют этилацетатом, сушат экстракт над сульфатом натрия и растворитель выпаривают в вакууме. Получают 0,28 г целевого продукта, который используют таким, какой есть, в Приготовлении 4,10
Приготовление 3.14
Сложный метиловый эфир 1-/4-карбоксинафт-1-ил/-5-/2,6- диметоксифенил/-пиразол-3-карбоновой кислоты
[формула /II'a/: R' = -COOH; T=метил].
В течение 3-х часов кипятят с обратным холодильником смесью 2,2 г полученного в Приготовлении 2.5 соединения, 2,65 г полученного в Приготовлении 1 соединения и 200 мл уксусной кислоты. После охлаждения добавляют 700 мл воды и отсасывают образовавшийся осадок. Осадок обрабатывают 1,4 - диоксаном и выпаривают растворитель в вакууме. После высушивания получают 2,75 г целевого продукта. Т.пл. = 240oC.
Приготовление 3.15
Сложный метиловый эфир 5-/2,6-диметоксифенил/-1-{4-/N-[3- (N',N'-диметиламино)-пропил]-карбамоил/нафт-1-ил}-пиразол-3-карбоновой кислоты
[формула /IIа/: R = -CONH/CH2/3N/Me/2; T = метил].
Выдерживают в течение 2-х часов при перемешивании смесь 0,5 г полученного в Приготовлении 3.14 соединения и 5 мл тионилхлорида, затем концентрируют в вакууме. Таким образом полученный хлорангидрид кислоты растворяют в 5 мл ДХМ, затем этот раствор прикапывают к раствору 0,165 мл N,N-диметил-1,3-пропандиамина, 0,172 мл триэтиламина в 10 мл ДХМ, и перемешивают в течение ночи при комнатной температуре. К реакционной смеси добавляют воду, органическую фазу, после декантации, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Получают 0,52 г целевого продукта.
ЯМР - спектр при 200 мГц в ДМСО: δ, м.д. = 1,8 - 2,0 /мт, 2H/; 2,8/ с., 6H/, 3,05 - 3,40/ 2т., 4H/; 3,4/ с., 6H/; 6,5 /д., 2H/; 7,0/ с., 1H/ 7,1 - 7,2/ т., 1H/ 7,4 /д.,1H/: 7,45 - 7,70 /м., 4H/; 8,20 /мт., 1H/.
Приготовление 3.16
Сложный метиловый эфир 5-/2,6-диметоксифенил/1-{4-[N-[2-(N'-N'-диметиламино)-этил]-карбамоил]- нафт-1-ил]-пиразол-3-карбоновой кислоты
[формула /IIа/ : R = -CONH/CH2/2N/Me/2; T = метил].
Это соединение получают согласно методике, описанной в Приготовлении 3.15, исходя из 0,5 г полученного в Приготовлении 3,14, соединения, 5 мл тионилхлорида, затем 0,155 мл N,N-диметилэтилендиамина и 0,196 мл триэтиламина. Получают 0,6 г целевого продукта.
ЯМР - спектр при 200 мГц в ДМСО: δ, м.д. = 2,8 /с., 6H/; 3,2 - 3,8 /м., 10H/ ; 3,9 /с., 3H/ с., 3H/; 6,8 /д., 2H/; 7,0 /с.,1H/; 7,2/ т., 1H/; 7,4 /д., 1H/; 7,45 - 7,70 /м., 3H/; 7,8 /д., 1H/; 8,3 /м., 1H/; 10,2 /с.ш.,1H/.
Приготовление 3.17
Сложный метиловый эфир 1-[4-/N-(цианометил)-карбамоил/-нафт-1-ил]-5-/2,6-диметоксифенил/ пиразол-3-карбоновой кислоты
[формула /IIа/ :R = -CONHCH2CN;T = метил].
Это соединение получают согласно методике, описанной в Приготовлении 3,15, исходя из 1,22 г, полученного в Приготовлении 3.14, соединения, 12 мл тионилхлорида, затем 0,269 г хлоргидрата аминоацетонитрила и 0,8 мл триэтиламина. Получают, после кристаллизации их этанола, 0,77 г целевого продукта. Т.пл. = 138 -140oC.
Приготовление 3.18
Сложный метиловый эфир 1-[4-[N-(2-цианоэтил)-N-метилкабамоил-нафт-1-ил] -5-/2,6-диметоксифенил/- пиразол-3-карбоновой кислоты
[формула /IIа/ : R = -CON/Me/ CH2CH2CN; T = метил].
Это соединение получают согласно методике, писанной в Приготовлении 3,15, исходя из 10 г полученного в Приготовлении 3,14, соединения, 15 мл тионилхлорида, затем 2,4 мл 3-метиламинопропионитрила и 3,3 мл триэтиламина. Получают 12 г целевого продукта.
ЯМР - спектр при 200 мГц в ДМСО : δ, м.д. = 2,6 /с., 3H/; 2,8 /т., 2H/; 3,3 /с., 6H/; 3,9 /м, 5H/; 6,4 /д., 2H/; 6,9/c., 1H/; 7,1 /т., 1H/; 7,2 -7,8 /м., 6H/
Приготовление 3.19
Сложный метиловый эфир 5-/2,6-диметоксифенил/-1-[4-/N-метилкарбамоил/нафт-1-ил]-пиразол-3- карбоновой кислоты
[формула /IIа/ :R = -CONHME; T = метил]
При 40oC в течение 2-х часов нагревают смесь 4 г полученного в Приготовлении 3,14 соединения 20 мл тионилхлорида в 20 мл ДХМ, затем концентрируют в вакууме. Таким образом полученный хлорангидрид кислоты обрабатывают с помощью 40 мл ДХМ и полученный раствор прикапывают к раствору 4 мл водного 40%-ного раствора метиламина в 80 мл метанола, предварительно охлажденному до 5oC. Перемешивают в течение ночи при комнатной температуре, остаток обрабатывают водой и выпавший осадок отсасывают. После высушивания получают целевой продукт, который используют таким, какой есть, в Приготовлении 4.15,
Приготовление 3.20
Сложный метиловый эфир 5-/2,6-диметоксифенил/-1-[4-/6-ацетамидогексаноиламино/нафт-1-ил]- пиразол-3-карбоновой кислоты.
[формула /IIа/:R = -NHCO/CH2/5NHCOMe, T= метил].
A/. Хлорангидрид - 6- ацетамидогексановой кислоты
В течение 24-х часов при комнатной температуре перемешивают смесь 0,37 г 6-ацетамидлогексановой кислоты и 2,5 мл тионилхлорида. Концентрируют в вакууме и используют таким образом полученный хлорангидрид кислоты таким, какой есть, в следующей стадии.
Б. Сложный метиловый эфир 5-/2,6-диметоксифенил/-1-[4-/6- ацетамидогексаноиламино/нафт-1-ил]-пиразол-3-карбоновой кислоты
В течение ночи и при комнатной температуре, перемешивают смесь 5 г полученного в Приготовлении 3.10 соединения, 0,5 мл бис/триметилсилил/ ацетамида в 5 мл МECN. Затем добавляют раствор соединения, полученного в предыдущей стадии, в 5 мл MeCN, после этого добавляют 2 мл триэтиламина. Перемешивают в течение 2-х дней при комнатной температуре, добавляют 5 мл метанола и 5 мл воды, перемешивают 15 минут и концентрируют в вакууме. Остаток экстрагируют с помощью 50 мл ДХМ, органическую фазу промывают водой, 1 н. раствором HCl, сушат над сульфатом натрия и растворитель удаляют в вакууме. Получают 0,7 г целевого продукта, который используют таким, какой есть, в Приготовлении 4.16.
Приготовление 3.21
Сложный метиловый эфир 5-/2,6-диметоксифенил/1-{4-[N-метил-N-[3- -(N', N'-диметиламино)пропил]-карбамоил]нафт-1-ил}-пиразол-3- карбоновой кислоты
[формула /IIa/ R = - CON/Me//CH2/3N/Me/2; T = метил]
Это соединение получают согласно методике, описанной в Приготовлении 3,15, исходя из 0,5 г полученного в Приготовлении 3,14, соединения, 5 мл тионилхлорида, затем 0,120 мл N,N,N'-триметил-1,3-пропандиамина и 0,170 мл триэтиламина. Получают 0,43 г целевого продукта.
Приготовление 3.22
Сложны метиловый эфир 5-/2,6-диметоксифенил/-1-{ 4-[N-метил-N-[2-(N', N'-диметиламино)этил] карбамоил]нафт-1-ил]пиразол-3-карбоновой кислоты
[формула /IIa/ : R= -CON/Me/ /CH2/2N/Me/2; T= метил]
Это соединение получают согласно методике, описанной в Приготовлении 3,15, исходя из 0,5 г полученного в Приготовлении 3,14, соединения, 5 мл тионилхлорида, затем 0,179 мл N,N,N'-триметилэтилендиамина и 0,2 мл триэтиламина. Получают 0,4 г целевого продукта.
Приготовление 3.23
Сложный метиловый эфир 5-/2,6-диметоксифенил/-1-[1,3-/2H/-диоксо-1H-бенз/de/изохинол-6-ил]- пиразол-3-карбоновой кислоты
[формула /IIa/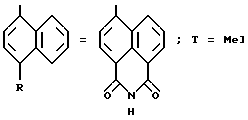
В течение 3-х часов кипятят с обратным холодильником смесью 2 г полученного в Приготовление 2.6 соединения, 2,4 г полученного в Приготовлении 1 соединения и 50 мл уксусной кислоты. После охлаждения добавляют воду и отсасывают образовавшийся осадок. Хроматографируют осадок на диоксиде кремния, элюируя смесью ДХМ /метанол [100:2 по объему].
Получают 2,4 г целевого продукта. Т.пл. = 138oC /разложение/.
Приготовление 3.24
Сложный метиловый эфир 5-/2,6-диметоксифенил/-1-[2-амино-1,3/2H/-диоксо-1H-бенз/de/изохинол-6-ил] пиразол-3-карбоновой кислоты
[формула /II''a/: T=метил].
В течение 2-х часов при 70oC нагревают смесь 3 г полученного в Приготовлении 2.7 соединения, 3,6 г полученного в Приготовлении 1 соединения и 70 мл уксусной кислоты. Концентрируют в вакууме, остаток обрабатывают водой, образовавшийся осадок отсасывают и высушивают. Осадок хроматографируют на диоксиде кремния, элюируя смесью ДХМ /уксусная кислота [100:5 по объему]. Получают 3,3 г целевого продукта, который используют таким, какой есть, в Приготовлении 4.20.
Приготовление кислот формул /II/, /II'/ и /II''/.
Приготовление 4.1
5-/2,6-Диметоксифенил/-1-/4-нитронафт-1-/пиразил-3-карбоновая кислота
[формула /II'/: R'=NO2; T=метил].
В течение 1,5 часов при 40oC нагревают смесь 14,8 г полученного в Приготовлении 3.1 соединения, 5,8 г KOH, 12 мл воды и 50 мл метанола. Растворители выпаривают в вакууме и остаток обрабатывают с помощью 250 мл воды. Подкисляют при 10oC до pH 1 за счет добавления 1 н. HCl. Образовавшийся осадок отфильтровывают и промывают два раза водой. Получают 14,64 г целевого продукта, после перекристаллизации из изо-эфира. Т.пл. = 255oC.
Приготовление 4.2
5-/2,6-Диметоксифенил/-1-[4-/этанкарбоксамидо/нафт-1-ил] - пиразол-3-карбоновая кислота
[формула /II/: R = -NHCOEt; T = метил].
В течение 4,5 часов при комнатной температуре перемешивают смесь 0,35 г полученного в Приготовлении 3.5 соединения, 0,07 г моногидрата гидроксида лития в 2 мл этанола. Выпаривают в вакууме, остаток обрабатывают 5 мл 1 н. HCl и оставляют на 1 час при перемешивании. После отфильтровывания и промывок водой, получают 0,27 г целевого продукта. Т.пл. = 168oC.
Приготовление 4.3
1-/4-Цианонафт-1-ил/-5-/2,6-диметоксифенил/-пиразол-3-карбоновая кислота
[формула /II/: R = -CN; T =метил]
В течение ночи при комнатной температуре перемешивают смесь 4,88 г полученного в приготовлении 3.2, сложного эфира, 1,2 мл концентрированного раствора NaOH в 500 мл метанола. Нагревают 3 часа при 40oC и реакционную смесь концентрируют в вакууме наполовину. Добавляют 1,2 мл концентрированного раствора NaOH и нагревают 6 часов при 40oC. Спустя 48 часов выдерживания при комнатной температуре, частично концентрируют реакционную среду и выливают в смесь воды со льдом. Экстрагируют эфиром, водную фазу подкисляют до pH 4 за счет добавления 1,2 н. раствора HCl, экстрагируют этилацетатом, экстракт сушат и над сульфатом натрия и растворитель выпаривают в вакууме.
Получают 4,6 г целевого продукта. Т.пл. = 216oC.
Приготовление 4.4.
1-/4-Карбамоил-нафт-1-ил/-5-/2,6-диметоксифенил/-пиразол-3- карбоновая кислота
[формула /II/: R = -CONH2; T = метил]
В течение 1 часа 20 минут кипятят с обратным холодильником смесь 1,48 г полученного в Приготовлении 3.2 соединения, 3 мл 6 н. раствора NaOH и 3 мл пероксида водорода /водный 3%-ный раствор/ в 15 мл 95o этанола. Затем перемешивают при комнатной температуре в течение ночи. Образовавшийся осадок отфильтровывают и промывают 95o этанолом. К водной суспензии полученного твердого вещества добавляют концентрированную HCl вплоть до pH 1. Получают 0,92 г целевого продукта [после фильтрации, промывок водой и высушивания]. Т.пл. = 305oC.
Приготовление 4.5
1-[4-/Ацетиламинометил/-нафт-1-ил] -5-/2,6-диметоксифенил/-пиразол-3-карбоновая кислота
[формула /II/: R = -CH2NHCOMe; T = метил]
В течение 4-х дней при комнатной температуре перемешивают смесь 0,8 г полученного в приготовлении 3,6 соединения и 0,1 г моногидрата гидроксида лития в 12 мл метанола и 1,5 мл воды. Реакционную смесь выливают в смесь воды со льдом и промывают эфиром. Водную фазу подкисляют до pH 2 за счет добавления 1,2 н. раствора HCl. После отфильтровывания образовавшегося осадка, промывки его водой и высушивания в вакууме, над P2O5, получают 0,58 целевого продукта. Т.пл. = 252oC.
Приготовление 4.6
5-/2,6-Диметоксифенил/-1-[4-/диметиламиносульфонил/нафт-1-ил]-пиразол-3- карбоновая кислота
[формула /II/: R = -SO2N/Me/2; T = метил]
При комнатной температуре раствор 0,6 г KOH в 0,5 мл воды добавляют к раствору 2,15 г полученного в Приготовлении 3.7 соединения в 15 мл диоксана. Перемешивают при комнатной температуре в течение 1 часа, затем реакционную смесь оставляют стоять в течение ночи. Вышеуказанную среду частично концентрируют в вакууме и перемешивают в течение 4-х часов при комнатной температуре. Выливают в смесь воды со льдом и промывают эфиром. Водную фазу подкисляют до pH 4-5 путем добавления 1,2 н. раствора HCl. После отфильтровывания образовавшегося осадка, промывки его водой и высушивания в вакууме над P2O5, получают 2 г целевого продукта, Т. пл. = 209oC.
Приготовление 4,7.
5-/2,6-Диметоксифенил/-1-[4-/N-метил-N-(3-N', N'-диметиламинопропил) -аминосульфонил/-нафт-1-ил]-пиразол-3-карбоновая кислота
[формула /II/: R=SO2 N/Me/ /CH2/3 N/Me2; T = метил].
При комнатной температуре раствор 0,2 г KOH в 0,5 мл воды добавляют к раствору 0,82 г полученного в Приготовлении 3.8 соединения в 16 мл диоксана. Перемешивают при комнатной температуре в течение 1 дня, затем выливают в смесь воды со льдом. Промывают эфиром и водную фазу подкисляют до pH 1 за счет добавления 1 н. раствора HCl. Затем экстрагируют с помощью ДХМ, экстракт сушат над сульфатом натрия и растворитель выпаривают в вакууме. После кристаллизации из эфира получают 0,68 г целевого продукта. Т.пл. = 176oC.
Приготовление 4.8
1-[4-/Карбамоилметил/-нафт-1-ил] -5-(2,6-диметоксифенил)пиразол-3- карбоновая кислота
[формула /II/: R = -CH2CONH2; T = метил]
В течение 2-х часов кипятят с обратным холодильником смесь полученного в Приготовлении 3.9 соединения, 0,35 г KOH в 10 мл 95o этанола.
Реакционную смесь выпаривают в вакууме и остаток обрабатывают 1 н. раствором HCl. После отфильтровывания образовавшегося осадка, затем высушивания его получают 1,3 г целевого продукта. Т.пл. = 262oC.
Приготовление 4.9
5-/2,6-Диметоксифенил/-1-[4-/3-N,N-диметиламинопропокси/нафт-1-ил]- пиразол-3-карбоновая кислота
[формула /II/: R = -O/CH2/3N/Me2/; T =метил]
В течение 2-х часов при 60oC нагревают смесь 0,25 г полученного в Приготовлении 3.12 соединения, 0,025 г моногидрата гидроксида лития в 3 мл метанола и 0,4 мл воды. Выпаривают в вакууме и нейтрализуют водную среду до pH 7 добавлением 1 н. раствора HCl. Получают после отфильтровывания образовавшегося осадка, затем высушивания 0,19 г целевого продукта, который используют таким, какой есть, в примере 16.
Приготовление 4.10
1-[4-/Карбамоилметокси/нафт-1-ил]-5-/2,6-диметоксифенил/пиразол-3- карбоновая кислота
[формула /II/: R = OCH2CONH2; T =метил]
В течение 4-х часов при 60oC нагревают смесь 0,28 г полученного в Приготовлении 3.13 соединения, 0,029 г моногидрата гидроксида лития в 3 мл метанола и 3 мл воды. Выпаривают в вакууме, добавляют воду и подкисляют до pH 2 с помощью 1 н. раствора HCl. После отфильтровывания образовавшегося осадка, затем его высушивания, получают 0,31 г целевого продукта. Т.пл. = 224oC.
Приготовление 4.11
5-/2,6-Диметоксифенил/-1-{ 4-[N-[3-(N', N'-диметиламино)- пропил]-карбамоил]-нафт-1-ил}-пиразол-3-карбоновая кислота
[формула /II/ : R = - СОNH /CH2/3 N/Me/2; T = метил]
В течение 2-х часов кипятят с обратным холодильником смесью 0,5 г полученного в Приготовлении 3.15. соединения, 0,058 г моногидрата гидроксида лития в 15 мл метанола и 5 мл воды. Добавляют 1 н. раствора HCl до pH 6 и концентрируют в вакууме. Остаток обрабатывают насыщенным раствором NaCl, добавляют ДХМ и отсасывают образовавшееся твердое вещество. Это твердое вещество обрабатывают этанолом, отфильтровывают нерастворимую часть и фильтрат выпаривают в вакууме. Получают 0,41 г целевого продукта.
ЯМР-спектр при 200 мГц в ДМСО; δ, м.д. = 1,5 - 1,7 /кт., 2H/; 2,2 /с., 6H/; 2,4 /м., 2H/; 3,2 /м., 2H/; 3,3 /с., 6H/; 6,4 /д., 2H/; 6,7 /с., 1H/; 7,1 /т., 1H/; 7,2 /д., 1H/; 7,3 - 7,5 /м., 4H/; 8,0 /м., 1H/; 8,6 /т., 1H/.
Приготовление 4.12
5-/2,6-Диметоксифенил/-1-{4-[N-/2-(N',N'- диметиламино)этил]- карбамоил] -нафт-1-ил}-пиразол-3-карбоновая кислота
[формула /II/ : R = -CONH/CH2/2N/Me/2; T = метил]
В течение 2-х часов при 70oC нагревают смесь 0,6 г полученного в Приготовлении 3.16. соединения, 0,088 г моногидрата гидроксида лития в 10 мл метанола и 10 мл воды. Концентрируют в вакууме, остаток обрабатывают насыщенным раствором NaCl, добавляют 1 н. раствора HCl до pH 6,5, экстрагируют с помощью ДХМ, органическую фазу сушат над сульфатом натрия и растворитель выпаривают в вакууме. Получают 0,44 г целевого продукта, который используют таким, какой есть в Примере 20.
Приготовление 4.13
1-[4-[N-(Цианометил)-карбамоил] -нафт-1-ил] -5-/2,6- диметоксифенил/-пиразол-3-карбоновая кислота
[формула /II/ : R = -CONHCH2CN, T = метил]
В течение 1,5 часов кипятят с обратным холодильником смесь 0,77 г полученного в Приготовлении 3.17 соединения, 0,118 г моногидрата гидроксида лития в 15 мл метанола и 10 мл воды, затем концентрируют в вакууме. Остаток обрабатывают насыщенным раствором NaCl, экстрагируют с помощью ДХМ, органическую фазу сушат над сульфатом натрия и растворитель выпаривают в вакууме. Получают 0,55 г целевого продукта.
ЯМР - спектр при 200 мГц в ДМСО; δ, м.д. = 3,4 /с.,6H/; 3,8 /с., 2H/; 6,4 /д., 2H/; 6,8 /с., 1H/; 7,1 /т., 1H/; 7,3 /д., 1H/; 7,4 - 7,6 /м., 4H/; 8,2 /м., 1H/; 8,8 /м., 1H/.
Приготовление 4.14
1-{ 4-[N-/2-Цианоэтил/-N-метилкарбамоил] нафт-1-ил}-5-/2,6- диметоксифенил/-пиразол-3-карбоновая кислота
[формула /II/ : R = -CON/Me/CH2CH2CN; T = метил]
В течение ночи перемешивают при комнатной температуре смесь 8 г полученного в Приготовлении 3.18. соединения, 0,77 г моногидрата гидроксида лития в 100 мл метанола и 100 мл воды. Реакционную смесь разбавляют путем добавления воды, затем добавляют 1 н. раствор HCl вплоть до pH 2 и отсасывают образовавшийся осадок. После высушивания, получают 7,36 г целевого продукта. Т.пл. = 145oC.
Приготовление 4.15
5-/2,6-Диметоксифенил/-1-[4-/N-метилкарбамоил/-нафт-1-ил]- пиразол-3-карбоновая кислота
[формула /II/ : R = -CONHMe -; T = метил]
В течение 3-х часов при 50oC нагревают смесь полученного в Приготовлении 3.19 соединения, 0,69 г моногидрата гидроксида лития в 50 метанола и 50 мл воды. Добавляют 10%-ный раствор HCl до pH 2 и отсасывают образовавшийся осадок. После высушивания, соединение перекристаллизуют из метанола и получают 3,3 г целевого продукта. Т.пл. > 260oC.
ЯМР - спектр при 200 мГц в ДМСО; δ, м.д. = 2,8 /с., 3H/; 3,4 /c., 6H/; 6,4 /д. , 2H/; 6,8 /с., 1H/; 7,1 /т., 1H/; 7,35 - 8,20 /м., 6H/; 8,45 /кд., 1H/.
Приготовление 4.16
5-/2,6-Диметоксифенил/-1-[4-/6-ацетамидогексаноиламино/- нафт-1-ил]-пиразол-3-карбоновая кислота
[формула /II/ : R = -NHCO/CH2/5 NHCOMe; T = метил]
В течение ночи при комнатной температуре перемешивают смесь 0,7 г полученного в Приготовлении 3.20 соединения, 0,18 г моногидрата гидроксида лития в 5 мл 1,4-диоксана и 1 мл воды. Концентрируют в вакууме, остаток обрабатывают с помощью 10 мл метанола и 5 мл воды и в течение 2-х часов нагревают при 45oC в ультразвуковой бане. Концентрируют в вакууме, остаток обрабатывают 20 мл воды, промывают 30 мл эфира, подкисляют водную фазу до pH 1 за счет добавления концентрированной серной кислоты, экстрагируют 2 раза по 500 мл этилацетатом, сушат экстракт над сульфатом натрия и выпаривают его в вакууме. Получают 0,4 г целевого продукта. Т.пл. = 140oC.
Приготовление 4.17
5-/2,6-Диметоксифенил/1-{ 4-[N-метил-N-[3-(N'-диметиламино)- пропил]карбамоил]-нафт-1-ил}-пиразол-3-карбоновая кислота
[формула /II/ : R = -CON/Me/ /CH2/3 N/Me2; T = метил].
В течение 2-х часов кипятят с обратным холодильником смесь 0,41 г полученного в Приготовлении 3.21 соединения, 0,43 г моногидрата гидроксида лития в 15 мл метанола и 5 мл воды. Концентрируют в вакууме остаток обрабатывают насыщенным раствором NaCl, добавляют 1н раствор HCl до pH 6, экстрагируют с помощью ДХМ, органическую фазу сушат над сульфатом натрия и растворитель выпаривают в вакууме.
Получают 0,33 г целевого продукта.
Приготовление 4.18
5-/2,6-Диметоксифенил/-1-[4-[N-метил-N-[2-(N', N'-диметиламино)- этил]-карбамоил]-нафт-1-ил]-пиразол-3-карбоновая кислота.
[формула /II/ : R = -CON/Me/ /CH2/2N/Me/2; T - метил.]
В течение 2-х часов кипятят с обратным холодильником смесь 0,38 г полученного в Приготовлении 3.22 соединения, 0,052 г моногидрата гидроксида лития в 5 мл метанола и 5 мл воды. Концентрируют в вакууме, остаток обрабатывают насыщенным раствором NaCl, добавляют 1н раствор HCl до pH 6,5, экстрагируют с помощью ДХМ, органическую фазу сушат над сульфатом натрия и растворитель выпаривают в вакууме. Получают 0,32 г целевого продукта.
Приготовление 4.19
5-/2,6-Диметоксифенил/-1-[1,3/2H/-диоксо-1H-бенз/d e/изохинол-6-пиразол-3-карбоновая кислота
[формула /II/: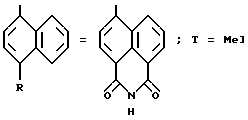
В течение ночи при комнатной температуре перемешивают смесь 2,4 г полученного в Приготовлении 3.23 соединения, 0,375 г моногидрата гидроксида лития, 10 мл метанола и 10 мл воды, затем нагревают 2 часа при 60oC. После охлаждения подкисляют до pH 2 путем добавления 1 н. раствора HCl и образовавшийся осадок отсасывают. Получают 2,3 г целевого продукта. Т.пл. = 190oC /разложение/.
Приготовление 4.20
Хлоргидрат 5-/2,6-диметоксифенил/-1-[2-амино-1,3-/2H/-диоксо-1H- бенз-/de/-ихохинол-6-ил]-пиразол-3-карбоновой кислоты
[формула /II''/ : T = метил].
В течение ночи при комнатной температуре перемешивают смесь 3,3 г полученного в Приготовлении 3.24 соединения, 0,7 г моногидрата гидроксида лития, 15 мл метанола и 15 мл воды. Реакционную смесь подкисляют до pH 2 путем добавления 1 н. раствора HCl, образовавшийся осадок отсасывают и высушивают. Получают 2,76 г целевого продукта.
Т.пл. = 180oC.
Приготовление 4.21
5-/2,6-Диметоксифенил/-1-[2-ацетамидо-1,3/3H/-диоксо-1H- бенз/de/-изохинол-6-ил]-пиразол-3-карбоновая кислота
Формула /II/: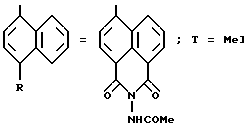
В течение ночи при комнатной температуре перемешивают смесь 2,26 г полученного в Приготовлении 4.20 соединения, 1,13 мл уксусного ангидрида, 0,407 г бикарбоната натрия и 75 мл уксусной кислоты. К реакционной смеси добавляют воду, образовавшийся осадок отсасывают и высушивают. Получают 1,9 г целевого продукта. Т. пл. = 250oC.
Приготовление соединения формулы /III/
Хлоргидрат сложного метилового эфира /S/-циклогексилглицина
A/. /S/-N-трет-Бутоксикарбонилциклогексилглицин
В течение 4-х дней при комнатной температуре и давлении 70 бар гидрируют смесь 10,7 г /S/-N-трет-бутоксикарбонилглицина, 2г. 5%-ного родия-на-оксиде алюминия, в 100 мл метанола. Катализатор отфильтровывают на Целите® и фильтрат выпаривают в вакууме. Получают 11,1 г целевого продукта. 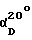 = 3,5o /с = 1, ДМФ/.
= 3,5o /с = 1, ДМФ/.
Б/. /S/-Циклогексилглицин-трифторацетат
50 мл ТФК быстро добавляют к охлажденному до 0oC раствору 11 г полученного в предыдущей стадии соединения в 50 мл ДХМ. Перемешивают в течение 1 часа при комнатной температуре и выпаривают в вакууме. Остаток обрабатывают изо-эфиром и отфильтровывают образовавшийся осадок. Получают 7,6 г целевого продукта. Т.пл.>260oC.
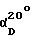 = +19,2o / с=2; 5H раствор HCl/.
= +19,2o / с=2; 5H раствор HCl/.
B// Хлоргидрат сложного метилового эфира /S/-циклогексилглицина
К охлажденному до -10oC раствору 1,2 г полученного в предыдущей стадии соединения в 200 мл метанола прикапывают 10 мл тионилхлорида. Оставляют температуру возвращаться к комнатной и в течение 2-х часов кипятят с обратным холодильником. Выпаривают досуха и остаток обрабатывают толуолом, затем выпаривают в вакууме; эту операцию повторяют 2 раза.
Получают 1 г целевого продукта. Т.пл. > 260oC.
ЯМР - спектр при 200 мГц в ДМСО; δ, м.д. = 0,8 - 2,1 /м., 11H/; 2,6 /ДМСО/; 3,4 /DHO/; 3,8 /с., 3H/; 3,95 /д., 1H/; 8,6 /с.ш. 3H/;
Пример 1
2-[5-/2,6-Диметоксифенил/-1-/4-формамидонафт-1-ил/пиразол-3-ил- карбониламино]-адамантан-2- -карбоновая кислота
[формула /I/: R=-NHCHO; T=метил; AA/OH/=2-карбоксиадамант-2-ил].
А/ Хлорангидрид 5-/2,6-диметоксифенил/-1-/4-нитронафт-1-ил/-пиразол-3-карбоновой кислоты.
В течение 4-х часов кипятят с обратным холодильником раствор 14,6 г полученной в Приготовлении 4,1 кислоты в 100 мл ДХМ и 30 мл тионилхлорида. Выпаривают в вакууме, остаток обрабатывают с помощью ДХМ, затем снова выпаривают. Таким образом полученный хлорангидрид кислоты используют таким, какой есть, в следующей стадии.
Б/. 2-[5-/2,6-Диметоксифенил/-1-/4-нитронафт-1-ил/- пиразол-3-ил-карбониламино]адамантан-2-карбоновая кислота
[формула /I'/:R'=-NO2; T=метил, AA/OH/=2-карбоксиадамат-2-ил].
При комнатной температуре, раствор полученного в предыдущей стадии хлорангидрида кислоты в 20 мл ДХМ добавляют в суспензию 6,8 г 2-аминоадамантан-2-карбоновой кислоты в 100 мл ДМФ и 20 мл пиридина. Перемешивают в течение ночи при комнатной температуре и растворители выпаривают в вакууме. Остаток обрабатывают с помощью 200 мл 1 н. раствора HCl и образовавшийся осадок отфильтровывают. Получают 10 г целевого продукта /после двух последовательных перекристаллизацией из MeCN/. Т.пл. = 268oC.
В/. 2-[1-/4-Аминонафт-1-ил/-5-/2,6-диметоксифенил/пиразол-3- ил-карбониламино]адамантан-2-карбоновая кислота
[формула/I'/: R'=-NH2; T=метил, AA/OH/=2-карбоксиадамант-2-ил].
В течение 4-х часов при комнатной температуре и при атмосферном давлении гидрируют смесь 0,2 г полученного в предыдущей стадии соединения, 0,05 г 10%-ного палладия -на- угле, в 200 мл этанола. Реакционную смесь отфильтровывают и растворитель выпаривают в вакууме. Получают 0,15 г целевого продукта /после кристаллизации из изо-эфира и перекристаллизации из метанола/ Т.пл. = 222oC.
Г/. 2-[5-/2,6-Диметоксифенил/ 1-/4-формамидонафт-1-ил/-пиразол-3-ил-карбониламино]адамантан-2-карбоновая кислота.
В течение 1 часа при комнатной температуре перемешивают смесь 0,2 г полученного в предыдущей стадии соединения с 1 мл муравьиной кислоты /99-100%-ная, d= 1,22/ и в 2 мл уксусного ангидрида. Реакционную смесь отфильтровывают и промывают полученное твердое вещество изо-эфиром.
После высушивания в вакууме при 100oC, получают 0,17 г целевого продукта. Т. пл. = 270oC.
Пример 2
2-{ 5-/2,6-Диметоксифенил/-1-[4-/метилсульфонамидо/ нафт-1-ил]-пиразол-3-ил-карбониламино}-адамантан-2-карбоновая кислота.
[формула /I/: R=-NHSO2Me; T=метил; AA/OH/=2-карбоксиадамант-2-ил]
В течение 1 часа при комнатной температуре перемешивают смесь 0,3 г полученного в стадии В/. Примера 1 соединения, 0,3 г триметилсилилхлорида и 1,6 мл триэтиламина в 30 мл ТГФ. Добавляют 0,09 мл метансульфонилхлорида и кипятят с обратным холодильником в течение 23-х часов. Выпаривают в вакууме, экстрагируют с помощью ДХМ, промывают 1н раствором HCl, водой, сушат над сульфатом натрия и выпаривают в вакууме. Хроматографируют на диоксиде кремния, элюируя с помощью ДХМ, затем с помощью смеси ДХМ с метанолом /97:3 по объему вплоть до /88 : 12 по объему/. Получают 0,15 г целевого соединения, Т.пл. = 200oC. /разложение/.
Пример 3
2-[1-/4-Ацетамидонафт-1-ил/-5-/2,6-диметоксифенил/пиразол-3-ил- карбониламино]-адамантан-2-карбюоновая кислота
[формула /I/: R=-NHCOMe; T=метил; AA/OH/=карбоксиадамант-2-ил].
А/. 2'-[1-/4-Ацетамидонафт-1-ил/-5-/2,6-диметоксифенил/ пиразол-3-ил]-спиро[адамантан-2,4'-/2'-оксазолин-5'-он/].
В течение 50 минут при комнатной температуре перемешивают смесь 0,2 г полученного в стадии В/. Примера 1 соединения с 6 мл уксусного ангидрида. Выпаривают в вакууме и остаток обрабатывают водным 5%-ным раствором карбоната натрия. Осадок отфильтровывают, промывают водой и высушивают в эксикаторе. Хроматографируют на силикагеле, элюируя смесью ДХМ / этилацетат [95 : 5 по объему]. После кристаллизации из изо-эфира получают 0,093 г целевого продукта. Т.пл. = 213oC /разложение/.
Б/. 2-[1-/4-Ацетамидонафт-1-ил/5-/2,6-диметоксифенил/ пиразол-3-ил-карбониламино]-адамантан-2-карбоновая кислота
В течение 15 дней при комнатной температуре выдерживают раствор 0,09 г полученного в предыдущей стадии соединения в 2 мл ТФК и 2 мл ДХМ. Растворители выпаривают в вакууме и остаток обрабатывают эфиром. После отфильтрования и промывки эфиром получают 0,065 г целевого продукта. Т.пл. = 213oC /разложение/.
Пример 4
2-{ 5-/2,6-Диметоксифенил/-1-[4-/этанкарбоксамидо/- нафт-1-ил]-пиразол-3-ил-карбониламино}-адамантан-2-карбоновая кислота
[формула /I/: R=-NHCOEt; T=метил, AA/OH/=2-карбоксиадамант-2-ил].
В течение 2,5 часов в атмосфере азота кипятят с обратным холодильником раствор 0,14 г 2-аминоадамантан-2-карбоновой кислоты, 0,3 мл бис/триметилсилил/ ацетамида в 7 мл MeCN. Выдерживают при комнатной температуре. С другой стороны, 0,1 мл изобутилхлорфтормиата добавляют к охлажденному до +5oC раствору 0,31 г полученного в Приготовлении 4,2 соединения, 0,1 мл триэтиламина в 5 мл ДХМ. перемешивают 3 часа при комнатной температуре. Этот раствор выливают в вышеполученный раствор силилированного производного и выдерживают 4 дня при комнатной температуре в атмосфере азота. Подкисляют до pH 1 путем добавления 1 н. раствора HCl и экстрагируют с помощью ДХМ, экстракт сушат над сульфатом натрия и выпаривают в вакууме. Остаток обрабатывают циклогексаном при температуре кипения с обратным холодильником и после декантации продукт экстрагируют изо-эфиром при температуре кипения с обратным холодильником. После кристаллизации при охлаждении до комнатной температуры, получают 0,15 г целевого продукта. Т. пл. = 192oC.
Пример 5
2-[1-/4-Цианонафт-1-/ил/-5-/2,6-диметоксифенил/пиразол-3-ил- карбонил-амино]-адамантан-2-карбоновая кислота
[формула /I/; R=-CN; N=метил, AA/OH/=2-карбоксиадамант-2-ил]
А/. Хлорангидрид 1-/4-цианонафт-1-ил/-5-/2,6-диметоксифенил/ пиразол-3-карбоноваой кислоты
Это соединение получают согласно методике, описанной в стадии А/. Примера 1, исходя из 0,5 г полученного в Приготовлении 4.3 соединения и 0,32 мл тионилхлорида. Получают 0,47 г целевого продукта и используют его таким, какой есть, в следующей стадии.
Б/. 2-[2-/4-Цианонафт-1-ил/-5-/2,6-диметоксифенил/пиразол-3-ил-карбонил- амино]-адамантан-2-карбоновая кислота
Это соединение получают согласно методике, описанной в стадии Б/ Примера 1, исходя из 0,47 г хлорангидрида кислоты предыдущей стадии и 0,25 г 2-аминоадамантан-2-карбоновой кислоты.
После выпаривания в вакууме остаток обрабатывают с помощью ДХМ и образовавшийся осадок отфильтровывают. Хроматографируют на силикагеле, элюируя смесью ДХМ /этилацетат/уксусная кислота [95:4,5:0,5 по объему]. После кристаллизации из эфира, получают 0,152 г целевого продукта. Т.пл. = 290oC.
Пример 6
2-{ 1-[4-/Гидроксииминокарбоксамидо/-нафт-1-ил] -5-/2,6-диметоксифенил/- пиразол-3-ил-карбониламино}-адамантан-2-карбоновая кислота
[формула /I/: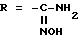
T = метил; AA/OH/=2-карбоксиадамант-2-ил].
Раствор 0,2 г гидроксиламин-хлоргидрата в 10 мл метанола добавляют к раствору 0,2 г полученного в Примере 5 соединения в 20 мл этанола; затем вводят раствор 0,2 г карбоната калия в 4 мл воды. В течение 2-х часов кипятят с обратным холодильником, затем частично концентрируют в вакууме реакционную смесь. После добавления воды и отфильтровывания образовавшегося осадка получают 0,15 г целевого продукта. Т.пл. = 230oC.
Пример 7
2-[1-/4-Карбамоилнафт-1-ил/-5-/2,6-диметоксифенил/пиразол-3-ил- карбониламино]-адамантан-2-карбоновая кислота
[формула /I/: R=-CONH2; T = метил; AA/OH/=2-карбоксиадамант-2-ил].
В течение 2-х дней кипятят с обратным холодильником смесь 0,2 г полученного в Примере 5 соединения, 0,4 мл пероксида водорода [водный 33%-ный раствор], 0,4 мл 6 н. раствора NaOH в 25 мл 95o этанола. Затем в реакционную среду добавляют 0,4 мл пероксида водорода, 0,4 мл 6 н. раствора NaOH и продолжают кипятить с обратным холодильником в течение 1 дня. После отфильтровывания фильтрат разбавляют водой и экстрагируют с помощью ДХМ. Водную фазу подкисляют до pH=2 путем добавления концентрирования HCl. Образовавшийся осадок отфильтровывают и промывают водой. Получают, после кристаллизации из эфира, 0,128 г целевого продукта. Т.пл. = 287oC.
Соединение Примера 7 также может быть получено, следуя нижеописанному способу:
2-[1-/4-Карбамоилнафт-1-ил/-5-/2,6-диметоксифенил/пиразол-3-ил-карбониламино] адамантан-2-карбоновая кислота
Это соединение получают согласно методике, описанной в Примере 4, исходя из 0,45 г полученного в Приготовлении 4.4 соединения и 0,22 г 2-аминоадамантан-2-карбоновой кислоты. Соединение очищают путем хроматографии на диоксиде кремния, элюируя смесью ДХМ /метанола/ уксусная кислота [100:4:0,5 по объему] , затем кристаллизации из эфира. Получают 0,3 г целевого продукта. Т. пл. = 292oC.
Пример 8
2-[1-/4-Карбамоилнафт-1-ил/-5-/2,6-диметоксифенил/пиразол-3-ил- карбониламино]адамантан-2-карбоксилат натрия
[формула /I/: R = -CONH2; T = метил; AA/OH/=2-карбоксиадамант-2-ил; соль натрия].
0,05 г полученного в Примере 7 соединения добавляют к раствору 0,005 г карбоната натрия в 0,3 мл воды и 3 мл метанола. Выдерживают 48 часов при -18oC. После выпаривания в вакууме досуха остаток порошкуют в 3 мл пропан-2-ола. После отфильтровывания и высушивания в вакууме над P2O5, получают 0,04 г целевого продукта. Т.пл. = 335oC /разложение/.
Пример 9
(N-Метил-D-глюкамин)-2-[1-/4-карбамоилнафт-1-ил/-5-/2,6-диметоксифенил/ пиразол-3-ил-карбониламино]адамантан-2-карбоксилат
[формула /I/: R =-CONH2; T = метил; AA/OH/=-2-карбоксиадамант-2-ил; соль N-метил-D-глюкамина].
0,05 г полученного в Примере 7 соединения добавляют к раствору 0,017 г N-метил-D-глюкамина в 4 мл метанола; затем добавляют 8 мл эфира. Выдерживают 3 дня при -18oC. После отфильтровывания образовавшихся кристаллов и высушивания в вакууме над P2O5, получают 0,04 г целевого продукта. Т.пл. = 170-172oC.
Пример 10
[2S] -2-[1-/4-Карбамоил-нафт-1-ил/-5-/2,6-диметоксифенил/пиразол-3-ил- карбониламино]-2-циклогексилуксусная кислота
[формула /I/: R=-CONH2; T = метил; AA/OH/= α -карбоксициклогексилметил].
A/ Сложный метиловый эфир [2S]-2-[1-/4-карбамоил-нафт-1-ил/-5-/2,6-диметоксифенил/пиразол-3-ил- карбониламино]-2-циклогексилуксусной кислоты
[формула /I/: R= -CONH2; T = метил; AA/OH/=сложный метиловый эфир α -карбоксициклогексилметильной группы]
В течение 1,5 часов, при комнатной температуре и в атмосфере азота, перемешивают смесь 0,45 г полученного в Приготовлении 4.4 соединения, 0,15 мл триэтиламина и 0,15 мл изобутилхлорформиата в 10 мл ДХМ. Затем медленно добавляют раствор 0,224 г хлоргидрата сложного метилового эфира /S/-циклогексилглицина, 0,15 мл триэтиламина в 5 мл ДХМ. Перемешивают в течение 5 дней при комнатной температуре. После отфильтровывания нерастворимой части, фильтрат промывают 1н раствором HCl, сушат над сульфатом натрия и выпаривают в вакууме. После перекристаллизации из MeCN, получают 0,37 г целевого продукта. Т.пл. = 198oC.
Б/. [2S}-2-[1-/4-Карбамоилнафт-1-ил/-5-/2,6-диметоксифенил/пиразол-3-ил- карбониламино]-2-циклогексилуксусная кислота
При комнатной температуре, раствор 0,081 г KOH в 1 мл воды добавляют к раствору 0,33 г полученного в предыдущей стадии соединения в 5 мл диоксана и перемешивают 5 часов при комнатной температуре. Затем реакционную смесь выпаривают в вакууме и остаток обрабатывают водой. Подкисляют до pH 1 путем добавления 1 н. раствора HCl. После отфильтровывания промывают водой и после высушивания получают 0,28 г целевого продукта. Т.пл. = 186oC. αD = +4o[с = 0,5; этанол].
Пример 11
2-{ 1-[4-(Ацетиламинометил)-нафт-1-ил] -5-(2,6-диметоксифенил) пиразол-3-ил-карбониламино}адамантан-2-карбоновая кислота
[формула /I/: R = -CH2NHCOMe; T=Me; AA/OH/=2-карбоксиадамант-2-ил]
А/. Хлорангидрид 1-[4-(ацетиламинометил)нафт-1-ил]-5-/2,6-диметоксифенил/-пиразол-3- карбоновой кислоты
Это соединение получают согласно методике, описанной в стадии А/. Примера 1, исходя из 0,55 г полученного в Приготовлении 4.5. соединения и 0,31 мл тионилхлорида. Полученный продукт используют таким, какой есть, в следующей стадии.
Б/. 2-{1-[4-/Ацетиламинометил/нафт-1-ил]-5-(2,6-диметоксифенил/пиразол-3- карбониламино}адамантан-2-карбоновая кислота
При комнатной температуре и в атмосфере азота, раствор хлорангидрида кислоты, полученного в предыдущей стадии, в 7 мл ДХМ добавляют к смеси 0,29 г 2-аминоадамантан-2-карбоновой кислоты и 15 мл пиридина. Перемешивают 72 часа при комнатной температуре. Затем отфильтровывают нерастворимую часть и фильтрат выпаривают в вакууме. Остаток экстрагируют с помощью ДХМ, экстракт промывают буфером с pH 2, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Затем хроматографируют на двуокиси кремния H, элюируя смесью ДХМ /метанол (100/4 по объему). После кристаллизации из эфира получают 0,15 г целевого продукта.
Т.пл. = 211oC.
Пример 12
2-{ 5-(2,6-Диметоксифенил)-1-[4-/диметиламиносульфонил/нафт-1-ил] -пиразол- 3-ил-карбониламино}адаментан-2-карбоновая кислота
[формула /I/: R=-SO2N (Me)2; T = метил; AA/OH/=2-карбоксиадамант-2-ил]
А/. Хлорангидрид-5-(2,6-диметоксифенил/-1-[4-/диметиламиносульфонил/-нафт-1-ил] пиразол-3-карбоновой кислоты
Это соединение получают согласно методике, описанной в стадии А/. Примера 1, исходя из 1,46 г соединения, полученного в Приготовлении 4.6, и 0,77 мл тионилхлорида. Полученный продукт используют непосредственно в следующей стадии.
Е/. 2-{5-(2,6-Диметоксифенил/-1-[4-/диметиламиносульфонил/нафт-1-ил]-пиразол-3- ил-карбониламино}-адамантан-2-карбоновая кислота
Это соединение получают согласно методике, описанной в стадии Б/. Примера 11, исходя из полученного в предыдущей стадии соединения и 0,7 г 2-аминадамантан-2-карбоновой кислоты. Продукт очищают путем хроматографии на двуокиси кремния H, элюируя смесью ДХМ /этилацетат/ уксусная кислота (90:10: 0,5 по объему). После кристаллизации из гексана получают 0,6 г целевого продукта. Т.пл. = 269oC.
Пример 13
Хлоргидрат 2-{ 5-/2,6-диметоксифенил/-1-[4-/N-метил-N-(3-N', N'-диметиламинопропил) аминосульфонил/нафт-1-ил]-пиразол-3-ил-карбониламино}-адамантан-2- карбоновой кислоты.
[формула /I/: R = -SO2N(Me) (CH2)3-N(Me)2; T = метил; AA/OH/=2-карбоксиадамант-2-ил].
Это соединение получают согласно методике, описанной в Примере 4, исходя из 0,37 г полученного в Приготовлении 4.7 соединения и 0,13 г 2-аминоадамантан-2-карбоновой кислоты. Продукт очищают путем хроматографии на двуокиси кремния H, элюируя смесью ДХМ /метанол/ уксусная кислота (100:8:1 по объему). Получают, после кристаллизации из эфира, 0,11 г целевого продукта. Т. пл. = 246oC (разложение).
Соединение Примера 13 также может быть получено, следуя двум стадиям нижеописанного способа.
А/. Хлорангидрид-5-/2,6-диметоксифенил/-1-[4-/N-метил-N-(3-N',N'- диметиламинопропил)аминосульфонил/нафт-1-ил]-пиразол- 3-карбоновой кислоты.
В течение 3-х часов, в атмосфере азота и при комнатной температуре, перемешивают смесь 0,2 г полученного в Приготовлении 4.7 соединения и 2 мл тионилхлорида. Затем добавляют еще 2 мл тионилхлорида и продолжают перемешивание при комнатной температуре в течение 1,5 часов.
Реакционную смесь выпаривают в вакууме, остаток обрабатывают с помощью ДХМ и снова выпаривают в вакууме. Таким образом полученный хлорангидрид кислоты используют таким, какой есть, в следующей стадии.
Б'/. 2-{5-(2,6-Диметоксифенил)-1-[4-/N-метил-N-(3-N',N'-диметиламинопропил) аминосульфонил/ нафт-1-ил] пирозол-3-ил-карбониламино}адамантан-2-карбоновая кислота
В течение 15 минут в атмосфере азота кипятят с обратным холодильником смесь 0,071 г 2-аминоаламантан-2-карбоновой кислоты, 0,36 мл бис (триметилсилил)ацетамида в 10 мл MeCN. После охлаждения добавляют раствор полученного в предыдущей стадии хлорангидрида кислоты в 10 мл ДХМ и оставляют на 48 часов при перемешивании при комнатной температуре. Добавляют воду, подкисляют до pH 2 путем добавления 1,2 н. раствора HCl, экстрагируют с помощью ДХМ, экстракт сушат над сульфатом натрия и выпаривают в вакууме. Получают 0,23 г целевого продукта.
Пример 14.
2-{ 1-/4-(Карбамоилметил)нафт-1-ил/5-(2,6-диметоксифенил)пиразол-3 -ил-карбониламино}адамантан-2-карбоновая кислота
(формула /I/:R= - CH2CONH2; T = метил; AA(OH) = 2-карбоксиадамант-2-ил)
Это соединение получают согласно методике, описанной в Примере 4, исходя из 0,8 г полученного в Приготовлении 4.8 соединения и 0,4 г 2-аминоаламантан-2-карбоновой кислоты. После выпаривания реакционной смеси, к остатку добавляют 1н раствор HCl и этанол. После перемешивания, обращавшийся осадок отфильтровывают и высушивают этот последний в вакууме. Затем хроматографируют на силикагеле, элюируя смесью ДХМ /метанол/ уксусная кислота (100:4:0,5 по объему). Полученный продукт растворяют 1н растворе NaOH и этаноле, подкисляют до pH 1 путем добавления 1 н. раствора HCl, экстрагируют с помощью ДХМ, экстракт сушат над сульфатом натрия и растворитель выпаривают в вакууме. После кристаллизации из этанола получают 0,53 г целевого продукта. Т.пл. = 224oC.
Пример 15
2-{ 1-/4-(Карбоксиметил)нафт-1-ил/5-(2,6-диметоксифенил)пиразол-3-ил -карбониламино}адамантан-2-карбоновая кислота
(формула /I/: R = -CH2 CO2H; T=метил; AA(OH)=2-карбоксиадамант-2-ил)
В течение 1 дня при 60oC нагревают смесь 0,17 г полученного в Примере 14 соединения, 0,135 г пероксида натрия в 5 мл воды и при добавке нескольких капель метанола. Подкисляют до pH 1 путем добавления 1 н. раствора HCl и оставляют стоять при перемешивании. После отфильтровывания, путем кристаллизации из MeCN, получают целевой продукт.
Масса = 0,1 г Т.пл. = 267oC.
Пример 16
2-{ 5-(2,6-Диметоксифенил)-1-[4-(3-N,N-диметиламинопропокси)нафт-1- ил-пиразол-3-ил-карбониламино}-аламантан-2-карбоновая кислота
(формула /I/: R = -O(CH2)3N(Me)2; T=метил;
AA(OH)= 2 - карбоксиадамант-2-ил)
Это соединение получают согласно методике, описанной в Примере 4, исходя из 0,19 г полученного в Приготовлении 4.9 соединения и 0,14 г 2-аминоадамантан-2-карбоновой кислоты. Соединение очищают путем хроматографии на диоксиде кремния, элюируя смесью ДХМ /метанол/ NH4OH (100:15:1 по объему). Получают 0,04 г целевого продукта. Т.пл. = 200oC.
Пример 17
2-{1-[4-(Карбамоилметокси)нафт-1-ил]-5-(2,6-диметоксифенил)пиразол-3-ил- карбониламино}адамантан-2-карбоновая кислота
(формула /I/: R=-OCH2CONH2; T = метил; AA(OH) = 2 - карбоксиадамант-2-ил)
Это соединение получают согласно методике, описанной в Примере 1, исходя из 0,31 г соединения, полученного в Приготовлении 4.10 и 0,27 г 2-аминоадамантан-2-карбоновой кислоты.
Соединение очищают путем хроматографии на силикагеле, элюируя смесью ДХМ /метанол/уксусная кислота (100: 1:0,5 по объему). Получают 0,14 г целевого продукта.
Т.пл. = 200oC.
Пример 18
2-{ 5-(2,6-Диметоксифенил)-1-[4-[N-[3-(N,N'-диметиламино)пропил}карбамоил] нафт-1-ил]пиразол-3-ил-карбониламино}адамантан-2-карбоновая кислота
(формула /I/: R = -CONH(CH2)3N(Me)2; T = метил; AA(OH) = 2-карбоксиаламант-2-ил)
В течение 1 часа при 35oC нагревают смесь 2 г полученного в Приготовлении 4.11 соединения, 10 мл тионилхлорида в 30 мл ДХМ. Концентрируют в вакууме и таким образом полученный хлорангидрид кислоты используют таким, какой есть. С другой стороны, в течение 1 часа кипятят с обратным холодильником смесь 1,2 г 2-аминоадамантан-2-карбоновой кислоты, 3 мл бис(триметилсилил)ацетамида, в 70 мл MeCN. После охлаждения до комнатной температуры, добавляют раствор вышеполученного хлорангидрида кислоты в 50 мл ДХМ, затем 0,57 мл триэтиламина и перемешивают в течение 48 часов при комнатной температуре. Реакционную смесь концентрируют в вакууме, остаток обрабатывают водой, подкисляют до pH 2 путем добавления 1 н. раствора HCl, перемешивают в течение 1 часа и отсасывают образовавшийся осадок. Осадок растворяют в подогретой воде, добавляют 5%-ный раствор NaOH до pH 6, экстрагируют с помощью ДХМ, органическую фазу сушат над сульфатом натрия и растворитель выпаривают в вакууме. После кристаллизации из пропан-2-ола и перекристаллизации из метанола, получают 0,48 г целевого продукта.
Т.пл. = 244oC.
ЯМР -спектр при 200 мГц в ДМСО; δ, м.д. = 1,4 - 2,1 (м., 14H); 2,2 (с., 6H); 2,4 (т., 2Н); 2,55 (м., 2H); 3,4 (кд., 2H); 3,6 (с., 6Н); 6,5 (д., 2H); 6,9 (с., 1H); 7,2 (т., 1H); 7,4 - 7,8 (м., 6H); 8,2 (м., 1H); 8,7 (т., 1H).
Пример 19
п-Толуолсульфонат 2-{5-(2,6-диметоксифенил)-1-[4-N-[3-(N',N-диметиламино)пропил] карбамоил] нафт-1 -ил]пиразол-3-ил-карбониламино}адамантан-2-карбоновой кислоты
К охлажденному до +5oC раствору 0,4 г полученного в Приготовлении 4.11 соединения, 0,127 мл триэтиламина в 10 мл ДХМ, добавляют 0,130 мл изобутилхлорформиата и перемешивают в течение 2-х дней при комнатной температуре. С другой стороны, в течение 1,5 часов кипятят с обратным холодильником смесь 0,231 г 2-аминоадамантан-2-карбоновой кислоты, 0,87 мл бис (триметилсилил) ацетамида в 15 мл MeCN. После охлаждения до комнатной температуры, добавляют раствор вышеполученного смешанного ангидрида и перемешивают в течение 2-х дней при комнатной температуре. К реакционной смеси добавляют воду, подкисляют до pH 2 путем добавления 1н раствора HCl, перемешивают и концентрируют в вакууме. Остаток обрабатывают водой, добавляют 5%-ный раствор NaOH вплоть до pH 6,5, экстрагируют с помощью ДХМ, отфильтровывают нерастворимую часть, фильтрат сушат над сульфатом натрия и растворитель выпаривают в вакууме. Получают 0,23 г сырого продукта; 0,11 г полученного продукта растворяют в минимальном количестве MeCN, добавляют 0,032 г моногидрата п-толуолсульфокислоты и добавляют эфир до осаждения. После отсасывания и высушивания получают 0,050 г целевого продукта.
ЯМР - спектр при 200 мГц в ДМСО; δ, м.д. = 1,6 - 2,2 (м., 14H); 2,4 (с., 3H); 2,5 (м., 2H); 2,9 (с., 6H); 3,2 (т., 2H); 3,3 - 3,8 (м., 8H); 6,5 (д., 2H); 6,9 (с., 1H); 7,2 (д., 3H); 7,4 - 7,8 (м., 7H); 8,2 (м., 1H); 8,7 (т., 1H).
Пример 20
2-{ 5-(2,6-Диметоксифенил)-1-[4-[N-[2-(N', N'-диметиламино)этил] -карбамоил] нафт-1-ил]пиразол-3-ил-карбониламино}адамантан-2-карбоновая кислота
(формула /I/: R=-CONH(CH2)2N(Me)2; T=метил; AA(OH)=2-карбоксиадамант-2-ил).
В течение 1 часа нагревают при 35oC смесь 2 г полученного в Приготовлении 4,12 соединения, 10 мл тионилхлорида, в 30 мл ДХМ. Концентрируют в вакууме и таким образом полученный хлорангидрид кислоты используют таким, какой есть. С другой стороны, в течение 1 часа кипятят с обратным холодильником смесь 1,2 г 2-аминоадамантан-2-карбоновой кислоты, 3 мл бис (триметилсилил) ацетамида в 70 мл MeCN. После охлаждения до комнатной температуры, добавляют раствор вышеполученного хлорангидрида кислоты в 50 мл ДХМ, затем 0,58 мл триэтиламина и перемешивают в течение ночи при комнатной температуре. Реакционную смесь концентрируют в вакууме, остаток обрабатывают водой (кристаллизация продукта), подкисляют до pH 2 путем добавления 1 н. раствора HCl и перемешивают в течение 1 часа. Выкристаллизовавшийся продукт отсасывают и, после высушивания, этот последний хроматографируют на силикагеле, элюируя градиентом смеси ДХМ /метанол/ H2O от (100:5:0,2 по объему) до (100/10:0,75 до объему). Получают 0,62 г целевого продукта (после кристаллизации из ацетона).
ЯМР - спектр при 200 мГц в ДМСО; δ, м.д. = 1,4 - 2,2 (м., 12H); 2,6 (м., 2H); 2,8 (с., 6H); 3,3 (т., 2H); 3,45 (с., 6H); 3,7 (т., 2H); 6,4 (д., 2H); 6,9 (с., 1H) 7,2 (т., 1H); 7,4 (д., 1H); 7,6 (м., 3H); 7,7 (д., 1H); 8,2 (м. , 1H); 8,4 (т., 1H).
Это соединение также получают согласно нижеописанной методике.
К охлажденному до +5oC раствору 0,44 г полученного в Приготовлении 4.12 соединения, 0,140 мл триэтиламина в 10 мл ДХМ, добавляют 0,139 мл изобутилхлорформиата и перемешивают в течение 3-х дней при комнатной температуре.
С другой стороны, в течение 1,5 часов кипятят с обратным холодильником смесь 0,321 г 2-аминоаламантан-2-карбоновой кислоты, 0,49 мл бис (триметилсилил) ацетамида в 20 мл MeCN. После охлаждения до комнатной температуры, добавляют вышеполученный раствор смешанного ангидрида и перемешивают в течение 3-х дней при комнатной температуре. Добавляют 1 н. раствор HCl до pH 2, затем метанол и концентрируют в вакууме. Остаток обрабатывают насыщенным раствором NaCl, экстрагируют с помощью ДХМ, органическую фазу сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток обрабатывают водой, добавляют раствор аммиака до pH 7 - 8, образовавшийся осадок отсасывают и высушивают. Осадок хроматографируют на силикагеле, элюируя смесью ДХМ /метанол/ H2O (80:10:0,75 по объему), затем смесью ДХМ /метанол/H2O/ 80:15:2 по объему). Получают 0,04 г целевого продукта.
Пример 21
2-{1-[4-[N-(Цианометил)карбамоил]нафт-1-ил]-5-(2,6-диметоксифенил)- пиразол-3-илкарбониламино}адамантан-2-карбоновая кислота
(формула /I/: R = -CONHCH2CN; T = Me; AA(OH)=2-карбоксиадамант-2-ил)
К раствору 0,55 г полученного в Приготовлении 4.13 соединения, 0,176 мл триэтиламина в 10 мл 1,4 - диоксана добавляют 0,181 мл изобутилхлорформиата и перемешивают в течение 6 дней при комнатной температуре. С другой стороны, в течение 1,5 часов кипятят с обратным холодильником смесь 0,351 г 2-аминоадамантан-2-карбоновой кислоты, 0,106 мл бис (триметилсилил) ацетамида в 20 мл MeCN. После охлаждения до комнатной температуры добавляют вышеполученный раствор смешанного ангидрида и перемешивают в течение 3-х дней при комнатной температуре. После этого добавляют 1н раствор HCl до pH 2 и перемешивают 2 часа при комнатной температуре. Концентрируют в вакууме, остаток обрабатывают насыщенным раствором NaCl, экстрагируют с помощью ДХМ и отфильтровывают нерастворимую часть. После декантации, органическую фазу сушат над сульфатом натрия и растворитель выпаривают в вакууме.
Остаток хроматографируют на силикагеле, элюируя смесью ДХМ /метанол/ H2O (80:5:0,3 по объему). Получают 0,080 г целевого продукта.
ЯМР-спектр при 200 мГц в ДМСО; δ, м.д.= 1,4 - 2,3 (м., 12H); 2,6 (м., 2H); 3,5 (с., 6H); 4,0 (с., 2H); 6,4 (д., 2H); 6,9 (с., 1H); 7,2 (т., 1H); 7,4 - 7,8 (м., 5H); 8,4 (м., 1H); 8,8 (т., 1H).
Пример 22
2-{ 1-[4-[N-(2-Цианоэтил)-N-метилкарбамоил] нафт-1-ил] -5-(2,6- диметокси-фенил)пиразол-3-ил-карбониламино}адамантан-2-карбоновая кислота
(формула /I/; R=-CON(Me) CH2CH2CN; T=метил; AA(OH)=2-карбоксиадамант-2-ил)
В течение 2-х часов нагревают при 35oC смесь 4 г полученного в Приготовлении 4.14 соединения, 40 мл тионилхлорида в 40 мл ДХМ. Концентрируют в вакууме и таким образом полученный хлорангидрид кислоты используют таким, какой есть. С другой стороны, в течение 1 часа кипятя с обратным холодильником смесь 2,34 г 2-аминоадамантан-2-карбоновой кислоты, 6 мл бис (триметилсилил) ацетамида в 150 мл MeCN. После охлаждения до комнатной температуры добавляют раствор вышеполученного хлорангидрида кислоты в 100 мл ДХМ, затем 1,12 мл триэтиламина и перемешивают в течение ночи при комнатной температуре. Концентрируют в вакууме, остаток обрабатывают водой, добавляют 1н. раствор HCl до pH= 2, перемешивают и отсасывают образовавшееся твердое вещество. После высушивания твердое вещество хроматографируют на силикагеле, элюируя смесью ДХМ /метанол/ H2O (100:10:1) по объему).
После кристаллизации из ацетона получают целевой продукт.
Т.пл.=264oC.
Пример 23
2-{5-(2,6-Диметоксифенил)-1-[4-[N-метил-N-[2-(N1-метил -N2-метиламидино)этил] карбамоил]нафт-1-ил]пиразол-3-ил- карбониламино}адамантан-2-карбоновая кислота
(формула /I/:R=CON(Me) CH2CH2C(=NMe) NHME; T=метил; AA(OH)=2-карбоксиадамант-2-ил)
1 г Соединения, полученного в Примере 22, растворяют в 30 мл этанола и 10 мл эфира, затем в течение 2-х часов пропускают путем барботирования газообразный HCl. Оставляют стоять в течение ночи при +5oC и концентрируют в вакууме. Остаток обрабатывают 50 мл этанола и в течение 2-х часов при комнатной температуре пропускают путем барбортирования газообразный метиламин. Концентрируют в вакууме, остаток обрабатывают с помощью смеси этанола с эфиром, отфильтровывают выкристаллизовавшийся хлоргидрат метиламина и фильтрат выпаривают в вакууме. Остаток обрабатывают водой, образовавшиеся кристаллы отсасывают и высушивают. После перекристаллизации из этанола получают 0,7 г целевого продукта. Т.пл.=235oC.
ЯМР-спектр при 200 мГц в ДМСО; δ, м.д.=1,4 - 2,4 (м., 12H); 2,5 - 4,0 (м., 21H); 6,5 (д., 2H); 6,9 (с., 1H); 7,15 (т., 1H); 7,3 - 7,8 (м, 6H);
Пример 24
2-{ 5-(2,6-Диметоксифенил)-1-/4-(N1-метил-N2- метиламино)нафт-1-ил/-пиразол-3-ил-карбониламино}адамантан-2- карбоновая кислота
(формула/I/:R=-C(=NME) NHME; T=метил; AA(OH)=2-карбоксиадамант-2-ил).
A/. 2-{ 5-(2,6-Диметоксифенил)-1-[4-(N-метилкарбамоил)нафт-1-ил] -пиразол-3-ил-карбониламино}адамантан-2-карбоновая кислота
Это соединение получают согласно методике, описанной в Примере 22, исходя из 2 г соединения, полученного в Приготовлении 4,15, 25 мл тионилхлорида, затем 1,39 г 2-аминоадамантан-2-карбоновой кислоты, 3,5 мл бис (триметилсилил) ацетамида и 0,64 мл триэтиламина. Хроматографируют на силикагеле, элюируя смесью ДХМ /метанол/ H2O (100:3:0,2 по объему), затем (100: 5:0,4 по объему). Получают 1,2 г целевого продукта. Т.пл.=170oC.
Б/. 2-{5-(2,6-Диметоксифенил)-1-[4-(N1-метил-N2) метиламидино)нафт-1-ил] -пиразол-3-ил-карбониламино}адамантан-2 -карбоновая кислота
В течение 20 минут кипятят с обратным холодильником смесь 0,5 г полученного в предыдущей стадии соединения, 0,34 г пентахлорида фосфора в 15 мл толуола. После охлаждения отсасывают выкристаллизовавшееся твердое вещество, кристаллы обрабатывают 30 мл этанола и через раствор пропускают путем барботирования газообразный метиламин.
Концентрируют в вакууме и остаток хроматографируют на силикагеле, элюируя смесью ДХМ/ метанол/ H2O (100:2:0,2 по объему). Получают 0,18 г целевого продукта. Т.пл.=180oC.
Пример 25
2-{ 1-[4-(2,6-Ацетамидогексаноиламино)нафт-1-ил] -5-(2,6- диметокси-фенил)пиразол-3-ил-карбониламино}адамантан-2-карбоновая кислота
(формула /I/:R=NHCO(CH2)5 NHCOMe; T=метил; AA(OH)=2-карбоксиадамант-2-ил)
К охлажденному до 5oC раствору 0,4 г соединения, полученного в Приготовлении 416, 0,2 мл триэтиламина в 5 мл MeCN добавляют 0,12 мл изобутилхлорформиата и перемешивают 2 часа при комнатной температуре. С другой стороны, в течение 2-х часов перемешивают, при комнатной температуре, смесь 0,21 г 2-аминоадамантан-2-карбоновой кислоты, 2 мл бис (триметилсилил) ацетамида в 3 мл MeCN. Затем этот раствор добавляют к вышеполученному раствору смешанного ангидрида и перемешивают в течение 16 дней при комнатной температуре. Затем к реакционной смеси добавляют 5 мл метанола и 1 мл воды, в течение 30 минут перемешивают и концентрируют в вакууме. Остаток обрабатывают с помощью 100 мл ДХМ, отфильтровывают нерастворимую часть, фильтрат промывают водой, 1 н. раствором HCl, органическую фазу сушат над сульфатом натрия и выпаривают в вакууме. Остаток хроматографируют на силикагеле, элюируя смесью ДХМ /метанол/ уксусная кислота (100:8:1 по объему). После кристаллизации из эфира, получают 0,07 г целевого продукта. Т.пл.=190oC.
Пример 26
2-{ 1-[4-(2-Аминоэтил)нафт-1-ил] -5-(2,6-диметоксифенил)пиразол- 3-ил-карбоновая кислота
(формула /I/:R=-CH2CH2NH2; T=Me; AA(OH)=2-карбоксиадамант-2-ил)
А/. 2-{ 1-[4-(Цианометид)нафт-1-ил] -5-(2,6-диметоксифенил) пиразол-3-ил-карбониламино}адамантан-2-карбоновая кислота
В течение 1 часа нагревают при 40oC смесь 5 г соединения, полученного в Приготовлении 4.8, 50 мл тионилхлорида в 135 мл ДХМ, затем концентрируют в вакууме. С другой стороны, в течение 1 часа кипятят с обратным холодильником смесь 3,38 г 2-аминоадамантан-2-карбоновой кислоты, 8,75 мл бис(триметилсилил) ацетамида в 150 мл MeCN. После охлаждения до комнатной температуры добавляют раствор вышеполученного хлорангидрида кислоты в 100 мл ДХМ, затем 1,6 мл триэтиламина и перемешивают в течение ночи при комнатной температуре. Концентрируют в вакууме, остаток растворяют в этаноле и добавляют эфир вплоть до кристаллизации. После отсасывания кристаллов (соединение Примера 14), фильтрат концентрируют в вакууме. Остаток обрабатывают этанолом, добавляют 3 н. раствор HCl до pH 1, отсасывают образовавшийся осадок и высушивают его. Осадок хроматографируют на силикагеле, элюируя смесью ДХМ /метанол/ уксусная кислота (100:3:0,5 по объему). Отделяют соединение Примера 14 и затем получают 1,2 г целевого продукта.
Б/. 2-{ 1-[4-(2-Аминоэтил)нафт-1-ил]-5-(2,6-диметоксифенил) пиразол-3-ил-карбониламино}адамантан-2-карбоновая кислота
При комнатной температуре и атмосферном давлении гидрируют смесь 1,2 г полученного в предыдущей стадии соединения, 0,12 г никеля Ренея®, 60 мл метанола и 10 мл концентрированного аммиака. Катализатор отфильтровывают и фильтрат частично концентрируют в вакууме. Образовавшиеся кристаллы отсасывают и высушивают. Получают 0,89 г целевого продукта. Т.пл.=250oC.
ЯМР-спектр при 200 мгц в ДМСО: δ, м.д.= 1,4 - 2,2 (м., 12H); 2,5 (м., 2H); 3,0 - 3,6 (м., 10H); 6,4 (д., 2H); 6,9 (с., 1H); 7,1 (т., 1H); 7,2 - 7,6 (м., 5H); 8,1 (м., 1H);
Пример 27
Хлоргидрат 2-{5-(2,6-диметоксифенил)-1-[4-[N-метил-N-[3- (N',N'-диметиламино)пропил] карбамоил] нафт-1-ил]пиразол-3-ил- карбомиламино}-адамантан-2-карбоновая кислота.
(формула /I/: R=-CON(Me) (CH2)3 N(Me); T=метил; AA(OH)=2-карбокси-адамант-2-ил).
Это соединение получают согласно методике, описанной в Примере 18, исходя из 0,32 г соединения, полученного в Приготовлении 4.17, 2 мл тионилхлорида, 5 мл ДХМ, затем 0,185 г 2-аминоадамантан-2-карбоновой кислоты, 0,465 мл бис (триметилсилил) ацетамида, 10 мл MeCN и 0,09 мл триэтиламина. Сырой продукт растворяют в минимальном количестве этанола, добавляют насыщенный раствор хлороводорода в эфире и концентрируют в вакууме. После кристаллизации из эфира получают 0,1 г целевого продукта.
Пример 28
Хлоргидрат 2-{5-(2,6-диметоксифенил)-1-[4-[N-метил-N-[2- (N',N'-диметиламино)этил] карбамоил] нафт-1-ил]пиразол-3-ил- карбониламино}-адамантан-2-карбоновой кислоты
(формула /I/: R = -CON(Me) (CH2)2N(Me)2;
T = метил; AA (OH) = 2-карбоксиадамант-2-ил)
Это соединение получают согласно методике, описанной в Примере 18, исходя из 0,31 г соединения, полученного в Приготовлении 4.18, 2 мл тионилхлорида, 5 мл ДХМ, затем 0,185 г 2-аминоадамантан-2-карбоновой кислоты, 0,465 мл бис(триметилсилил)ацетамида, 10 мл MeCN и 0,09 мл триэтиламина. Сырой продукт растворяют в минимальном количестве этанола, добавляют насыщенный раствор хлороводорода в эфире и концентрируют в вакууме. После кристаллизации из эфира получают 0,08 г целевого продукта.
Пример 29
2-{ 5-(2,6-Диметоксифенил)-1-[1,3(2H)-диоксо-1H-бенз /de/изохинол-6-ил] -пиразол-3-ил-карбониламино}адамантан-2-карбоновая кислота
(формула /I/: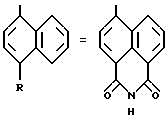
T = метил; AA(OH) = 2-карбоксиадамант-2-ил).
К охлажденному до 5oC раствору 1,4 г соединения, полученного в Приготовлении 4.19, 0,28 мл пиридина в 20 мл MeCN, добавляют 0,43 мл изобутилхлорформиата и перемешивают в течение 1 часа при комнатной температуре. С другой стороны, в течение 1 часа кипятят с обратным холодильником смесь 0,64 г 2-аминоадамантан-2-карбоновой кислоты, 1,54 мл бис(триметилсилил)ацетамида в 320 мл MeCN. После охлаждения до комнатной температуры добавляют вышеполученный раствор смешанного ангидрида и перемешивают 3 дня при комнатной температуре и в атмосфере азота. Реакционную смесь подкисляют до pH 2 путем добавления 1 н. раствора HCl и концентрируют в вакууме. Остаток обрабатывают водой, экстрагируют этилацетатом, органическую фазу сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток хроматографируют на силикагеле, элюируя смесью ДХМ /метанол/ уксусная кислота (100:1:0,5 по объему). Полученный продукт обрабатывают метанолом, подщелачивают путем добавления концентрированного раствора NaOH и концентрируют в вакууме. Продукт в форме натриевой соли обрабатывают метанолом, подкисляют до pH 1 путем добавления 1 н. раствора HCl и отсасывают образовавшийся осадок. Получают 0,17 г целевого продукта. Т.пл. = 210oC.
Пример 30
2-{ 5-(2,6-Диметоксифенил)-1-[2-ацетамидо-1,3(2H)-диоксо-1H- бенз/de/-изохинол-6-ил]пиразол-3-ил-карбониламино}адамантан-2- карбоновая кислота
[формула /I/: T = метил; AA(OH) = 2-карбоксиадамант-2-ил;]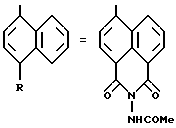
Это соединение получают согласно методике, описанной в Примере 29, исходя из 0,9 г соединения, полученного в Приготовлении 4.21, 0,25 мл триэтиламина, 0,25 мл изобутилхлороформиата и 15 мл ДХМ, затем 0,36 г 2-аминоадамантан-2-карбоновой кислоты, 0,89 мл бис(триметилсилил)ацетамида и 20 мл MeCN. Сырой продукт хроматографируют на силикагеле, элюируя смесью ДХМ /метанол/ уксусная кислота (100:3:0,5 по объему), до pH 3 путем добавления 1 н. раствора HCl, экстрагируют этилацетатом, экстракт сушат над сульфатом натрия и растворитель выпаривают в вакууме. После кристаллизации из эфира, получают 0,45 г целевого продукта.
Т.пл. = 292oC.
Поступая согласно методике, описанной в Примере 7 (второй способ) и исходя из соединения, полученного в Приготовлении 4.4, получают соединения согласно изобретению, представленные в таблице 1.
Поступая согласно методике, описанной в Примере 10, и исходя из соединения, полученного в Приготовлении 4.4, получают соединения согласно изобретению, представленные в таблице 2.
Пример 40
Хлоргидрат 2-{5-(2,6-диметоксифенил)-1-[4-[N-[3-(N',N'- диметиламино)-пропил]карбамоил]нафт-1-ил]пиразол-3-ил- карбониламино}адамантан-2-карбновой кислоты
Это соединение получают согласно методике, описанной в Примере 18, исходя из 5,6 г соединения, полученного в Приготовлении 4.11, 20 мл оксалилхлорида и 20 мл ДХМ, затем 3,4 г 2-аминоадамантан-2-карбоновой кислоты, 8,4 мл MeCN и 1,6 мл триэтиламина. После кристаллизации из пропан-2-ола и перекристаллизации из метанола, получают 1,3 г целевого продукта в форме основания. (Т. пл. = 242oC). 20 мг Полученного в форме основания продукта растворяют в минимальном количестве метанола, подкисляют до pH 1 путем добавления насыщенного раствора хлороводорода в эфире и концентрируют в вакууме. После кристаллизации из смеси ацетона с пентаном получают целевой хлоргидрат. Т.пл. = 200oC (разложение).
ЯМР-спектр при 200 мгц в ДМСО; δ, м.д. = 0,4 - 2,3 (м., 14H); 2,6 (с.ш, 2H); 2,8 (с.ш, 6H); 3,15 (с.ш, 2H); 3,4 (с.ш, 2H); 3,55 (с., 6H); 6,5 (д., 2H); 6,9 (с., 1H); 7,2 (т., 1H); 7,3 - 8,3 (м., 7H); 8,8 (с.ш, 1H); 10 - 13 (2 с.ш., 2H).
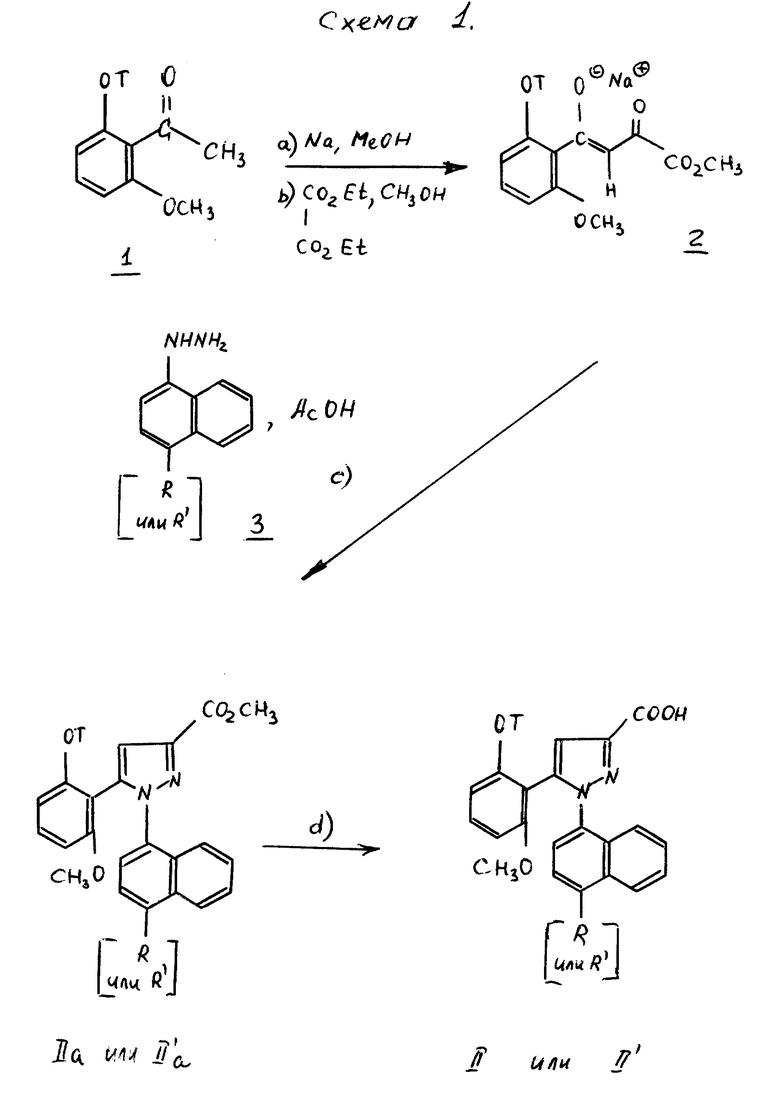
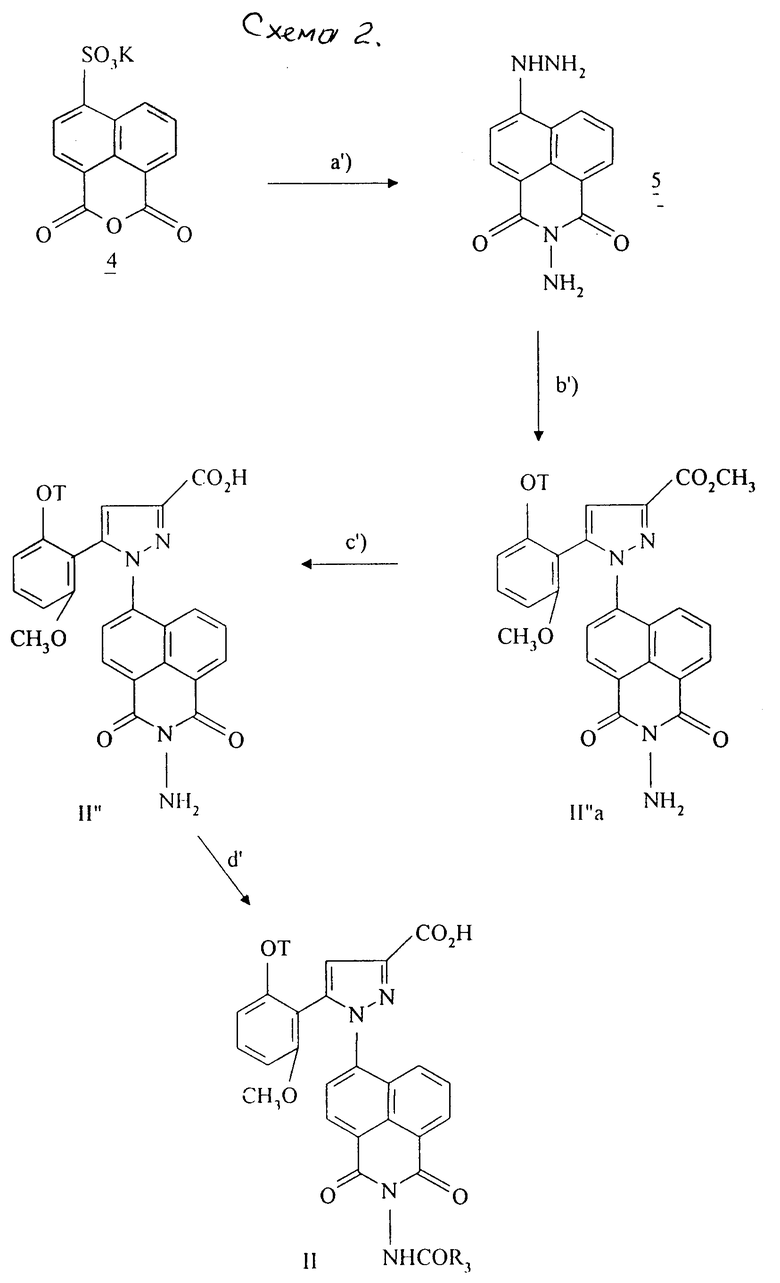
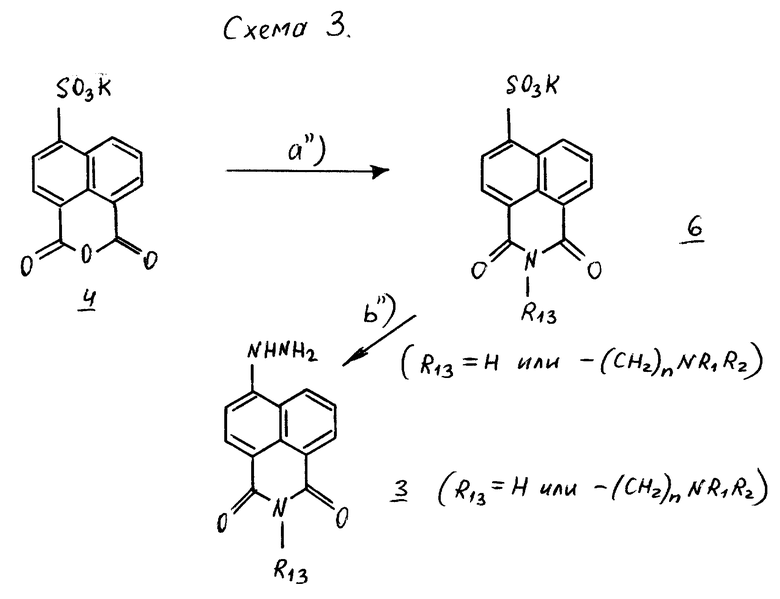
Описываются новые замещенные 1-нафтилпиразол-3-карбоксамиды общей формулы (I), где значения R, p, n, q, R1-R8, R11-R17, T указаны в п.1 формулы изобретения, обладающие большим сродством к человеческому рецептору нейтротензина. Описывается их способ получения, а также содержащая их фармацевтическая композиция и промежуточные соединения синтеза. 5 с. и 4 з.п. ф-лы, 3 ил., 2 табл.
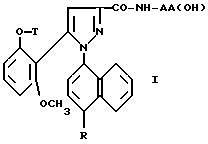
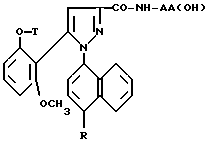
в которой R обозначает группу, выбираемую среди -CN, -C(NH2)=N-OH, -C(NR4R5)=NR6, -CONR1R2, -CON(R7)(CH2)pNR1R2, -CON(R7)(CH2)qCN,
-CON(R7)(CH2)qC(NR14R15)= N-R16, -CH2CONR1R2, -CH2COOR7, -O(CH2)nNR1R2,
-O(CH2)nCONR1R2, -N(R7)COR3, -N(R7)CO(CH2)nNHCOR3, -N(R7)SO2R8, -CH2N(R7)COR3, -CH2CH2NR11R12, -SO2NR1R2, -SO2N(R7)(CH2)nNR1R2, или R, связанный с атомом углерода в положении 5 нафтильного радикала, образует группу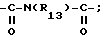
p = 2 - 6;
n = 1 - 6;
q = 1 - 5;
R1 и R2 каждый независимо друг от друга - водород или (C1-C4)-алкил;
R3 - водород или (C1-C8)-алкил;
R4 и R5 каждый независимо друг от друга - водород или (C1-C4)-алкил;
R6 - (C1-C4)-алкил;
R7 - водород или (C1-C4)-алкил;
R8 - (C1-C4)-алкил;
R11 и R12 каждый независимо друг от друга - водород или (C1-C4)-алкил;
R13 - водород или группа NHCOR3;
R14 и R15 каждый независимо друг от друга - водород или (C1-C4)-алкил;
R16 - водород, кроме того, R16 может обозначать (C1-C4)-алкил, когда R14 - водород и R15 - (C1-C4)-алкил;
T - (C1-C4)-алкил;
группа -NH-AA(OH) - остаток аминокислоты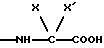
где X - водород и X' - водород, (C1-C5)-алкил или карбоциклический неароматический радикал с 3 - 15 атомами углерода или X и X' вместе с атомами углерода, с которым они связаны, образуют неароматический карбоцикл с 3 - 15 атомами углерода,
или их соли.
T обозначает метильную группу и группа -NH-AA-(OH) обозначает остаток 2-аминоадамантан-2-карбоновой или (S)-α-аминоциклогексануксусной кислоты, и их соли.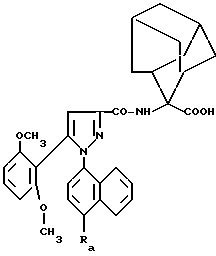
в которой Ra обозначает N-(3-N',N'-диметиламинопропил)аминокарбонильную или N-(2-N',N'-диметиламиноэтил)аминокарбонильную группу и их соли.
а) функциональное производное 1-нафтилпиразол-3-карбоновой кислоты формулы (II)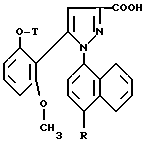
или формулы (II')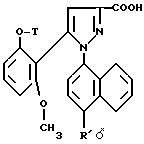
в которой T и R имеют указанные для соединения формулы (I) значения;
R' - предшественник R, выбираемый среди нитро-, амино-, гидроксильной, сульфо-, хлорсульфонильной, карбоксильной группы,
обрабатывают аминокислотой, в случае необходимости защищенной обычными в пептидном синтезе защитными группами, формулы (III):
H-HN-AA(OH),
в которой -NH-AA(OH) имеет значение, указанное для соединения формулы (I);
б) в случае необходимости таким образом полученное соединение формулы (I')
подвергают последующей соответствующей обработке для превращения заместителя R', предшественника R, в заместитель R;
в) в случае необходимости из таким образом полученного на стадии а) или на стадии б) соединения удаляют защитные группы для получения соответствующей свободной кислоты формулы (I);
г) в случае необходимости, получают соль полученного соединения формулы (I).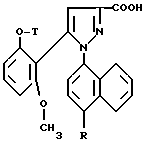
или формулы (II')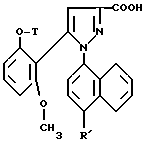
в которой T и R имеют значения, указанные в п.1 для соединения формулы (I);
R' - предшественник R, выбираемый среди нитро-, амино-, гидроксильной, сульфо-, хлорсульфонильной групп,
или их функциональные производные по кислотной функции.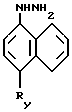
в которой Rу обозначает циано- или карбоксиметильную группу,
и их соли.
| Спасательный плот | 1972 |
|
SU477049A1 |
| 1972 |
|
SU418845A1 | |
| RU 2066317 A1, 10.06.96. | |||
