Настоящее изобретение относится к новым соединениям, оказывающим местное анестезирующее и аналгезирующее действие, способу их получения и их применению в производстве фармацевтических препаратов.
Часто используемым аналгетиком является петидин, но его местный анестезирующий эффект слаб. Анестеризующее и аналгезирующее действие петидина после спинномозгового введения часто бывает недостаточным в обоих отношениях. Вместо него используют сочетания бупивакоина и фентанила или морфина. Опиоидные аналгетики имеют некоторые серьезные недостатки, например развитие толерантности, привыкание, опасность угнетения дыхания. Существует таким образом потребность в средствах, обеспечивающих местную анестезию и остаточный аналгетический эффект с меньшими побочными эффектами, чем используемые в настоящее время комбинации. Такие средства должны быть использованы во время операции как местные анестетики после спинномозговых и эпидуральных инъекций. В результате соединения хорошо обеспечивали бы ослабление послеоперационных болей.
Известна (J. Med. Chem. 8, стр. 847 - 851 (1965) Hardy D.G. et al) зависимость активности от структуры некоторых аналогов петидина, проявляющих аналгетическую активность. В патенте Швеции N 96 980 описаны 1-метил-4-фенилпиперидин-4-карбоновая кислота и два ее амида, но, кроме того, что эти соединения могут быть использованы в производстве новых лекарственных средств, не раскрыт никакой специфический фармацевтический эффект.
В WO SE 90/00 818 описаны некоторые замещенные 4-фенилпиперидин-4-карбоксамиды, оказывающие как местное анестезирующее, так и аналгезирующее действия.
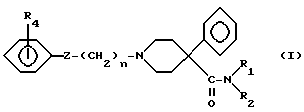
Новые соединения по настоящему изобретению, имеющие следующую формулу (A):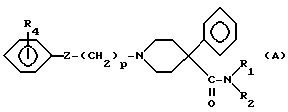
где Z представляет группу
взятую в направлении слева направо в формуле (A), или карбонильную группу; и где:
a) R1 представляет водород или неразветвленную или разветвленную алкильную группу с 1-3 атомами углерода, и
R2 представляет неразветвленную или разветвленную алкильную группу с 1-3 атомами углерода или
b) R1 и R2 вместе образуют цепь -(CH2)n-, где n представляет 3, 4 или 5, или -(CH2)2O(CH2)2-;
m представляет 0 - 1;
p представляет 1 - 2;
R3 представляет водород или -COCH3; и
R4 представляет водород, -CH3, -OH или -OCH3 при условии, что когда Z представляет карбонильную группу, то p = 2, а также фармацевтически приемлемые соли соединений формулы (A).
R4 может находиться в положении 2, 3 или 4.
Предпочтительными соединениями по настоящему изобретению являются те, в который R4 (в бензольном кольце, связанном с группой Z) находится в положении 2 и в которых:
R4 представляет водород, m = 0, R3 представляет водород, p = 2 и R1 и R2 одинаковы и представляют этильную группу; или
R4 представляет водород, m = 0, R3 представляет водород, p = 2, R1 представляет метильную группу и R2 представляет этильную группу; или
R4 представляет водород, m = 0, R3 представляет водород, p = 2 и R1 и R2 вместе образуют цепь -(CH2)4-; или
R4 представляет водород, m = 0, R3 представляет водород, p = 2, R1 представляет метильную группу и R2 представляет изопропильную группу; или
R4 представляет метоксигруппу, m = 0, R3 представляет водород, p = 2, R1 представляет метильную группу и R2 представляет этильную группу; или
R4 представляет метильную группу, m = 0, R3 представляет водород, p = 2, R1 представляет метильную группу и R2 представляет этильную группу; или
R4 представляет водород, m = 0, R3 представляет водород, p = 1 и R1 и R2 одинаковы и представляют этильную группу.
Предпочтительными солями по настоящему изобретению являются фармацевтически приемлемые соли. Особенно предпочтителен гидрохлорид.
Другими примерами солей, которые могут быть использованы, являются цитрат, метансульфонат и малеат.
Соединения по настоящему изобретению более пригодны для устранения боли, потому что они менее токсичны и более эффективны как местные анестезирующие вещества и аналгетики. Соединения формулы A и их фармацевтически приемлемые соли дают не только неожиданно хороший эффект как спинномозговые и эпидуральные анестетики, но и дополнительный аналгетический эффект, продолжающийся долгое время после прекращения анестетического эффекта. Следовательно, нет необходимости в сочетании активных соединений, что позволяет избежать опасностей, связанных с использованием таких сочетаний. Соединения также дают неожиданно более высокий эффект, чем известные соединения указанного комбинированного действия.
Наиболее предпочтительными соединениями по настоящему изобретению, известными в настоящее время, являются соединения в соответствии с примером 13 и примером 6A, т.е. соединения формулы XIII и соединения формулы VI, где:
R4 представляет H, R1 и R2 представляют оба -C2H5;
R4 представляет H, R1 представляет -CH3 и R2 представляет -C2H5;
R4 представляет H и R1+R2 представляет -(CH2)4;
R4 представляет H, R1 представляет -CH3 и R2 представляет изопропил;
R4 представляет 2-OCH3, R1 представляет -CH3 и R2 представляет -C2H5;
R4 представляет 2-CH3, R1 представляет -CH3 и R2 представляет -C2H5.
Получение
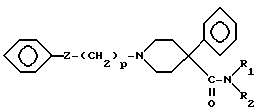
Для получения замещенных пиперидин-4-карбоксамидов формулы (A) по настоящему изобретению соединения можно разделить на шесть групп, которые получали согласно схемам 1 - 5.
Первую группу, ароматически незамещенные 1-(3-ацетокси-3- фенилпропил)-4-фенилпиперидин-4-карбоксамиды (V), и вторую группу, 1-(3-гидрокси-3-фенилпропил)-4-фенилпиперидин-4- карбоксамиды (VI), получали по следующей схеме реакций:
Схема 1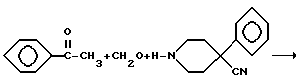

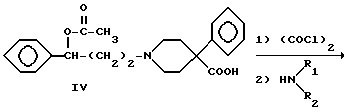
где R1 и R2 - такие, как указаны выше.
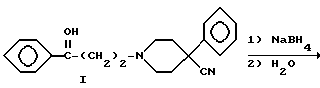
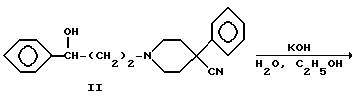
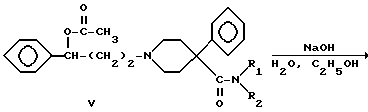
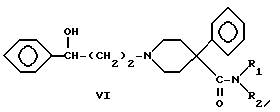
Аминокетон 1 получали путем осуществления реакции Манниха, т.е. взаимодействия 4-фенилпиперидин-4-карбонитрила с формальдегидом и ацетофеноном. Карбонильную группу соединения I восстанавливали боргидридом натрия с получением вторичного спирта II. Гидролиз цианогруппы в щелочной среде дал аминокислоту III. Гидроксильную группу ацетилировали и карбоновую кислоту IV преобразовывали в подходящий амид V посредством хлорангидрида кислоты. Избирательный щелочной гидролиз сложноэфирной группы соединений V дал соответствующие вторичные спирты VI.
Энантиомерные чистые формы соединения VI, где R1 представляет -CH3 и R2 представляет -C2H5 или R1 и R2 одинаковы и представляют -C2H5, получали по следующей схеме реакций:
Схема 2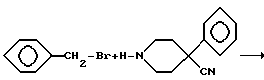
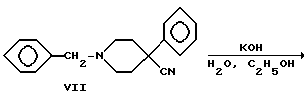

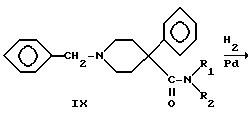
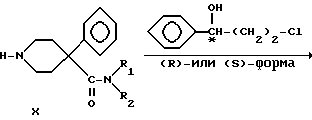
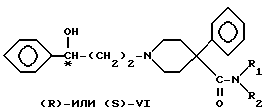
4-Фенилпиперидин-4-карбонитрил бензилировали с получением аминонитрила VII, который гидролизовали в щелочной среде в аминокислоту VIII. Кислоту преобразовывали в подходящий амид IX посредством хлорангидрида кислоты. Гидрогенолиз бензиламинов в присутствии палладиевого катализатора дал вторичные амины X, которые алкилировали либо (R)-, либо (S)-3-хлор-1-фенил-1- пропанолом с получением двух энантиомерных пар VI, где R1 представляет -CH3 и R2 представляет -C2H5 или R1 и R2 одинаковы и представляют -C2H5.
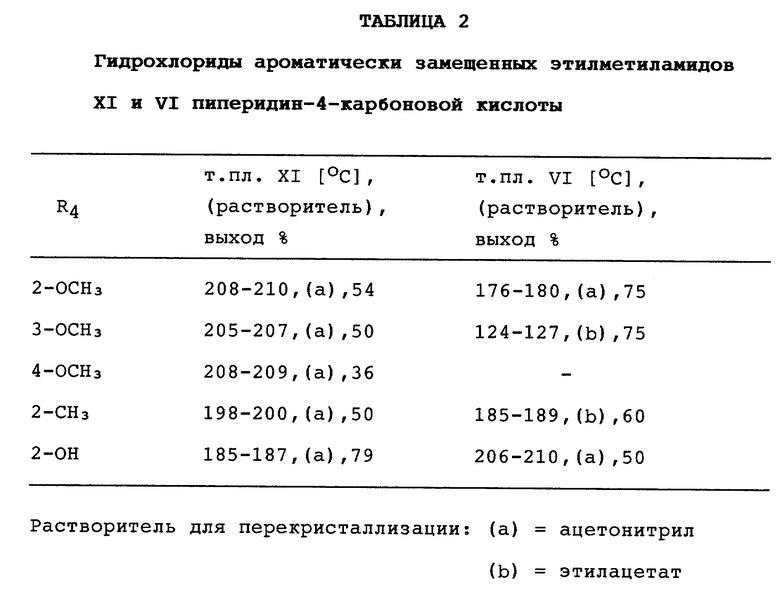
Ароматически замещенные пиперидин-4-карбоксамиды, в которых R1 представляет -CH3, R2 представляет -C2H5 и R4 представляет - CH3, -OH или -OCH3, составляют (вместе с соединением XI, в котором R1 и R2 одинаковы и представляют -C2H5 и R4 представляет водород) третью (кетоны XI) и четвертую (спирты VI) группы новых соединений. Их получали по следующей схеме реакций:
Схема 3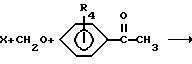
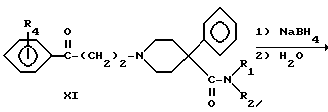
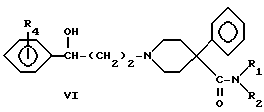
Реакция Манниха между вторичным амином X, где R1 представляет -CH3 и R2 представляет -C2H5 или R1 и R2 одинаковы и представляют -C2H5, формальдегидом и подходящим бензофеноном (R4 представляет водород, -CH3 или -OCH3) дала кетоны XI. Карбонильную группу ароматически замещенных кетонов XI (R4 представляет -CH3, -OCH3 или -OH) восстанавливали боргидридом натрия с получением спиртом VI. Соединение XI, где R4 представляет 2-гидрокси, получали из соответствующего метоксисоединения путем деметилирования трибромидом бора.
Диэтиламид (XIII) 1-(2-гидрокси-2-фенилэтил)-4-фенилпиперидин-4-карбоновой кислоты (пятый тип новых соединений) получали по следующей схеме реакций:
Схема 4
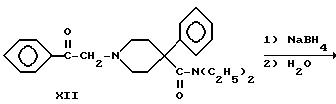
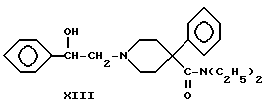
Вторичный амин X, где R1 и R2 одинаковы и представляют -C2H5, алкилировали фенацилбромидом с получением кетона XII, который восстанавливали боргидридом натрия до вторичного спирта XIII.
Шестую группу соединений, 1-(2-гидрокси-3-фенилпропил)- 4-фенилпиперидин-4-карбоксамиды (XVIII), получали по следующей схеме:
Схема 5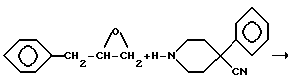

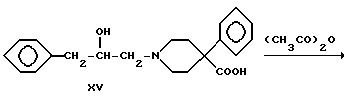
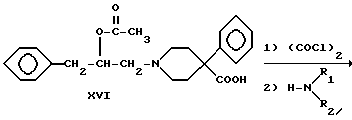
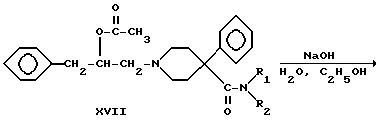
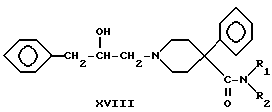
где R1 и R2 - такие, как указаны выше.
4-Фенилпиперидин-4-карбонитрил алкилировали (2,3-эпоксипропил) бензолом с получением вторичного спирта XIV. Соединения XV, XVI, XVII и XVIII получали так, как описано для соединений III, IV, V и VI соответственно.
В следующих далее примерах подробно описано получение соединений (обозначенных аналогичными римскими цифрами) по настоящему изобретению.
ПРИМЕР 1
Получение 1-(3-оксо-3-фенилпропил)-4-фенилпиперидин-4- карбонитрила
(Соединение I)
Смесь 4-фенилпиперидин-4-карбонитрилгидрохлорида (41.6 г, 187 ммоль), формальдегида (8.4 г, 280 ммоль), ацетофенона (33.6 г, 280 ммоль) и 37%-ной хлорводородной кислоты (2 мл) в этаноле (500 мл) нагревали с обратным холодильником в течение 48 ч. После охлаждения собирали путем фильтрования выпавший в осадок материал, в результате чего получили 40.0 г гидрохлорида с т.пл. 207 - 210oC.
ПРИМЕР 2
Получение 1-(3-гидрокси-3-фенилпропил)-4-фенилпиперидин-4- карбонитрила
(Соединение II)
Кетон 1 (48.2 r, 136 ммоль) и боргидрид натрия (20.0 г, 526 ммоль), суспендированные в метаноле (500 мл), нагревали при 50oC в течение 15 ч. Выпаривали растворитель и медленно добавляли 2 н. хлорводородную кислоту (150 мл). Затем раствор подщелачивали 5 н. гидроксидом натрия и экстрагировали тремя порциями хлороформа (3 х 100 мл). Объединенные экстракты сушили над карбонатом калия, фильтровали и выпаривали растворитель. Выход 41.0 г с т. пл. 109 - 112oC.
ПРИМЕР 3
Получение 1-(3-гидрокси-3-фенилпропил)-4-фенилпиперидин-4- карбоновой кислоты
(Соединение III)
Раствор нитрила II (20.3 г, 63 ммоль) и гидроксида калия (17.5 г, 313 ммоль) в этаноле (50 мл) и воде (80 мл) нагревали в автоклаве при 140oC в течение 6 ч. После охлаждения раствор упаривали до одной трети его первоначального объема и затем подкисляли хлорводородной кислотой до pH 2. Отфильтровывали осадок. Получили 22.0 г гидрохлорида с т.пл. 280oC.
ПРИМЕР 4
Получение 1-(3-ацетокси-3-фенилпропил)-4-фенилпиперидин-4- карбоновой кислоты
(Соединение IV)
Смесь соединения III (11.3 г, 30 ммоль), уксусного ангидрида (200 мл) и 4-диметиламинопиридина (0.3 г) нагревали при 50oC в течение 15 ч. Выпаривали растворитель и остаток тщательно высушивали при 80oC в вакуумном шкафу и затем перекристаллизовывали из диоксана. Получили 10.2 г гидрохлорида с т.пл. 189 - 193oC.
ПРИМЕР 5
Получение 1-(3-ацетокси-3-фенилпропил)-4-фенилпиперидин-4- карбоксамидов
(Соединения V)
К суспензии пиперидинкарбоновой кислоты IV (5.0 г, 12 ммоль) в дихлорметане (100 мл) добавляли по каплям с перемешиванием оксалилхлорид (6 мл). Реакционную смесь перемешивали при 50oC в течение 2 ч. Выпаривали растворитель, добавляли несколько миллилитров толуола и опять выпаривали растворитель. Остаток растворяли в дихлорметане (50 мл) и полученный раствор добавляли по каплям при перемешивании к раствору подходящего амина (36 мл) в дихлорметане (20 мл), охлажденному в ледяной воде. Реакционную смесь перемешивали при комнатной температуре 4 ч. Выпаривали растворитель и остаток взбалтывали между разбавленным гидроксидом натрия (20 мл) и дихлорметаном (3 х 20 мл). Органический экстракт сушили над карбонатом калия, фильтровали и выпаривали досуха. Добавлением хлорводорода в диэтиловом эфире к эфирным растворам аминов получали гидрохлориды. Полученные соединения перекристаллизовывали из подходящего растворителя (таблица 1).
ПРИМЕР 6A
Получение 1-(3-гидрокси-3-фенилпропил)-4-фенилпиперидин-4- карбоксамидов
(Соединения VI)
Суспензию сложного эфира V (12 ммоль) в 2 н. гидроксиде натрия (60 мл) и этаноле (40 мл) нагревали при 70oC в течение 3 ч. Раствор упаривали до половины его первоначального объема и затем экстрагировали тремя порциями диэтилового эфира (3 х 50 мл). Эфирный экстракт сушили над карбонатом калия, фильтровали и получали гидрохлорид так, как описано в примере 5. Полученные соединения очищали путем перекристаллизации из указанного растворителя (таблица 1).
ПРИМЕР 7
Получение 1-бензил-4-фенилпиперидин-4-карбонитрила
(Соединение VII)
Смесь 4-фенилпиперидин-4-карбонитрила (41.4 г, 223 ммоль), бензилбромида (41.9 г, 245 ммоль) и карбоната натрия (29.7 г, 281 ммоль) в 1-бутаноле (300 мл) перемешивали при комнатной температуре 12 ч. Выпаривали растворитель и добавляли 1 н. гидроксид натрия (100 мл). Водную фазу экстрагировали три раза дихлорметаном (3 х 150 мл). Объединенные экстракты сушили над карбонатом калия, фильтровали и упаривали досуха. Выход составлял 59.0 г соединения с т.пл. 76 - 79oC.
ПРИМЕР 8
Получение 1-бензил-4-фенилпиперидин-4-карбоновой кислоты
(Соединение VIII)
Нитрил VII (20.0 г, 73 ммоль) гидролизовали в автоклаве так, как описано в примере 3. Реакционную смесь упаривали по одной трети его первоначального объема и нейтрализовали хлорводородной кислотой. Выпавший в осадок материал собирали путем фильтрования, получив в результате 18.6 г аминокислоты с т. пл. 288 - 290oC.
ПРИМЕР 9
Получение 1-бензил-4-фенилпиперидин-4-карбоксамидов
(Соединения IX)
Карбоновую кислоту VIII преобразовывали в подходящий амид так, как описано для соединений V. Амид с R1, представляющим -CH3, и R2, представляющим -C2H5, выделяли в виде свободного основания и перекристаллизовывали из этилацетата, получив выход 62%, т.пл. 108 - 112oC. Амид, в котором R1 и R2 одинаковы и представляют -C2H5, выделяли в виде гидрохлорида и перекристаллизовывали из смеси ацетонитрила и этилацетата. Выход 54%, т.пл. 204 - 206oC.
ПРИМЕР 10
Получение 4-фенилпиперидин-4-карбоксамидов
(Соединение X)
Раствор подходящего третичного амина IX (13 ммоль, получен из гидрохлорида обычным образом) в метаноле (200 мл) гидрировали при атмосферном давлении в присутствии 5%-ного палладия на активированном угле (100 мг), пока не израсходовали расчетное количество водорода. Отфильтровывали катализатор и выпаривали растворитель. Получали так, как описано в примере 5, гидрохлориды и перекристаллизовывали их из ацетонитрила. Получили амид с R1, представляющим -CH3, и R2, представляющим -C2H5, с выходом 94%, т.пл. 214 - 217oC, и амид, в котором R1 и R2 одинаковы и представляют -C2H5, с выходом 91%, т.пл. 238 - 240oC.
ПРИМЕР 6B
Получение оптически активных 1-(3-гидрокси-3-фенилпропил)-4- фенилпиперидин-4-карбоксамидов
(Соединения VI, где R1 представляет -CH3 и R2 представляет -C2H5 или R1 и R2 одинаковы и представляют -C2H5)
Смесь подходящего амина X (4.0 ммоль, получен из гидрохлорида обычным образом), (R)-3-хлор-1-фенил-1-пропанола (0.70 г, 4.12 ммоль; получен по Brown H. C. et al в J. Org. Chem. 53, стр. 2916 - 2920 (1988)) или (S)-энантиомера (промышленного производства), карбоната натрия (0.46 г, 4.3 ммоль) и иодида калия (50 мг) в 1-бутаноле (20 мл) нагревали с обратным холодильником в течение 30 ч. Выпаривали растворитель и добавляли 0.5 н. гидроксид натрия (10 мл). Водную фазу экстрагировали три раза диэтиловым эфиром (3 х 20 мл). Объединенные экстракты сушили над карбонатом калия и фильтровали. Осаждали гидрохлорид так, как описано в примере 5. Соли перекристаллизовывали из указанного растворителя (таблица 1). Ниже приведены значения удельного вращения [ [α]
R1; R2; (R)-VI; (S)-VI;
CH3; C2H5; +22.2 (10.0); -23.4 (10.0);
C2H5; C2H5; +20.4 (2.8); -21.8 (2.8).
ПРИМЕР 11A
Получение 1-(3-оксо-3-фенилпропил)-4-фенилпиперидин-4- карбоксамидов
(Соединения XI)
Реакции Манниха, осуществленные так, как описано в примере 1, между амином X с R1, представляющим -CH3, и R2, представляющим -C2H5, или R1 и R2, одинаковыми и представляющими -C2H5, формальдегидом и подходящим ацетофеноном с R4, представляющим водород, 2-OCH3, 3-OCH3, 4-OCH3 или 2-CH3, дали соответствующие кетоны XI. Соединения XI, где R1 и R2 одинаковы и представляют - C2H5 и R4 представляет водород, перекристаллизовывали из ацетонитрила. Выход 70%, т. пл. 189 - 192oC. Ароматически замещенные кетоны XI перекристаллизовывали из указанного растворителя и имели температуры плавления, приведенные в таблице 2.
ПРИМЕР 11B
Получение этилметиламида 1-[3-(2-гидроксифенил)-3-оксопропил] - 4-фенилпиперидин-4-карбоновой кислоты
(Соединение XI, где R1 представляет -CH3, R2 представляет -C2H5 и R4 представляет 2-OH)
К охлажденному в ледяной воде раствору соединения XI, где R4 представляет 2-OCH3, (1.3 г, 2.9 ммоль) в дихлорметане (25 мл) добавляли 0.5 М раствор трибромида бора (15 мл, 7.5 ммоль) в дихлорметане и смесь перемешивали при комнатной температуре 24 ч. Дихлорметановый раствор взбалтывали с разбавленным аммиаком и органическую фазу отделяли и сушили над сульфатом натрия. Выпаривали растворитель и остаточное основание преобразовывали в гидрохлорид, как описано в примере 5 (таблица 2). Выход составлял 1.0 г.
ПРИМЕР 6C
Получение ароматически замещенных этилметиламидов 1-(3- гидрокси-3-фенилпропил)-4-фенилпиперидин-4-карбоновой кислоты
(Соединения VI)
Кетоны XI (1 ммоль), в которых R4 представляет 2-OCH3, 3-OCH3, 4-OCH3, 2-CH3 или 2-OH, и боргидрид натрия (6 ммоль) суспендировали в тетрагидрофуране и перемешивали при комнатной температуре 48 ч. Продукт выделяли так, как описано в примере 2, и получали гидрохлорид так, как описано в примере 5. Растворители для перекристаллизации и температуры плавления указаны в таблице 2.
ПРИМЕР 12
Получение диэтиламида 1-(2-оксо-2-фенилэтил)-4-фенилпиперидин- 4-карбоновой кислоты
(Соединение XII)
Смесь вторичного амина X (1.0 г, 3.8 ммоль), в котором R1 и R2 одинаковы и представляют -C2H5, фенацилбромида (0.81 г, 4.1 ммоль) и карбоната натрия (1.0 г, 9.4 ммоль) в 1-бутаноле перемешивали при комнатной температуре 5 дней. Продукт выделяли так, как описано в примере 7. Получали гидрохлорид так, как описано в примере 5. Перекристаллизация из смеси ацетонитрила и диоксана дала 0.8 г продукта с т.пл. 209 - 212oC.
ПРИМЕР 13
Получение диэтиламида 1-(2-гидрокси-2-фенилэтил)-4-фенилпиперидин- 4-карбоновой кислоты
(Соединение XIII)
Смесь кетона XII (0.5 г, 1.2 ммоль, преобразован в свободный амин обычным образом) и боргидрида натрия (0.3 г, 7.9 ммоль) в тетрагидрофуране (20 мл) перемешивали при комнатной температуре 4 дня. Продукт выделяли так, как в примере 2. Гидрохлорид осаждали так, как описано в примере 5, и продукт перекристаллизовывали дважды из смеси ацетонитрила и этилацетата. Выход 0.2 г, т.пл. 203 - 205oC.
ПРИМЕР 14
Получение 1-(2-гидрокси-3-фенилпропил)-4-фенилпиперидин-4- карбонитрила
(Соединение XIV)
Смесь 4-фенилпиперидин-4-карбонитрила (6.3 г, 34 ммоль) и (2,3-эпоксипропил) бензола (5.0 г, 37 ммоль) в диоксане нагревали с обратным холодильником в течение семи дней. Выпаривали растворитель и остаток взбалтывали между разбавленным гидроксидом натрия и диэтиловым эфиром. Эфирный экстракт сушили над карбонатом калия, фильтровали и продукт преобразовывали в гидрохлорид так, как описано в примере 5. Перекристаллизация из смеси этанола и ацетонитрила дала 10.0 г продукта с т.пл. 246 - 249oC.
ПРИМЕР 15
Получение 1-(2-гидрокси-3-фенилпропил)-4-фенилпиперидин-4- карбоновой кислоты
(Соединение XV)
Нитрил XIV (10.0 г, 28.1 ммоль) гидролизовали в автоклаве так, как описано в примере 8. Получили 9.5 г аминокислоты с т.пл. 288 - 290oC.
ПРИМЕР 16
Получение 1-(2-ацетокси-3-фенилпропил)-4-фенилпиперидин-4- карбоновой кислоты
(Соединение XVI)
Вторичный спирт XV (2.0 г, 5.9 ммоль) ацетилировали так, как описано в примере 4. Перекристаллизация из диоксана дала 2.0 г аминокислоты с т.пл. 179 - 182oC.
ПРИМЕР 17
Получение 1-(2-ацетокси-3-фенилпропил)-4-фенилпиперидин-4- карбоксамидов
(Соединения XVII)
Карбоновую кислоту XVI преобразовывали в подходящие амиды так, как описано для соединений V. Гидрохлориды перекристаллизовывали из этилацетата. Получили амид, в котором R1 представляет -CH3 и R2 представляет -C2H5, с выходом 83%, т. пл. 185 - 187oC, и амид, в котором R1 и R2 одинаковы и представляют - C2H5, с выходом 75%, т.пл. 164 - 167oC.
ПРИМЕР 18
Получение 1-(2-гидрокси-3-фенилпропил)-4-фенилпиперидин-4- карбоксамидов
(Соединения XVIII)
Сложные эфиры XVII гидролизовали так, как описано в примере 6A. Гидрохлориды перекристаллизовывали из смеси ацетонитрила и этилацетата. Получили соединение XVIII, в котором R1 представляет -CH3 и R2 представляет -C2H5, с выходом 60%, т.пл. 171 - 175oC, и соединение XVIII, в котором R1 и R2 одинаковы и представляют -C2H5, с выходом 55%, т.пл. 166 - 169oC.
ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ
Для изготовления фармацевтических препаратов одно из новых соединений растворяют в жидком носителе, пригодном для инъекции, например, в физиологическом растворе. Используемые препараты представляют собой водные растворы, содержащие 0.1 - 100 мг/мл, предпочтительно 3 - 20 мг/мл активного соединения в расчете на хлористоводородную соль.
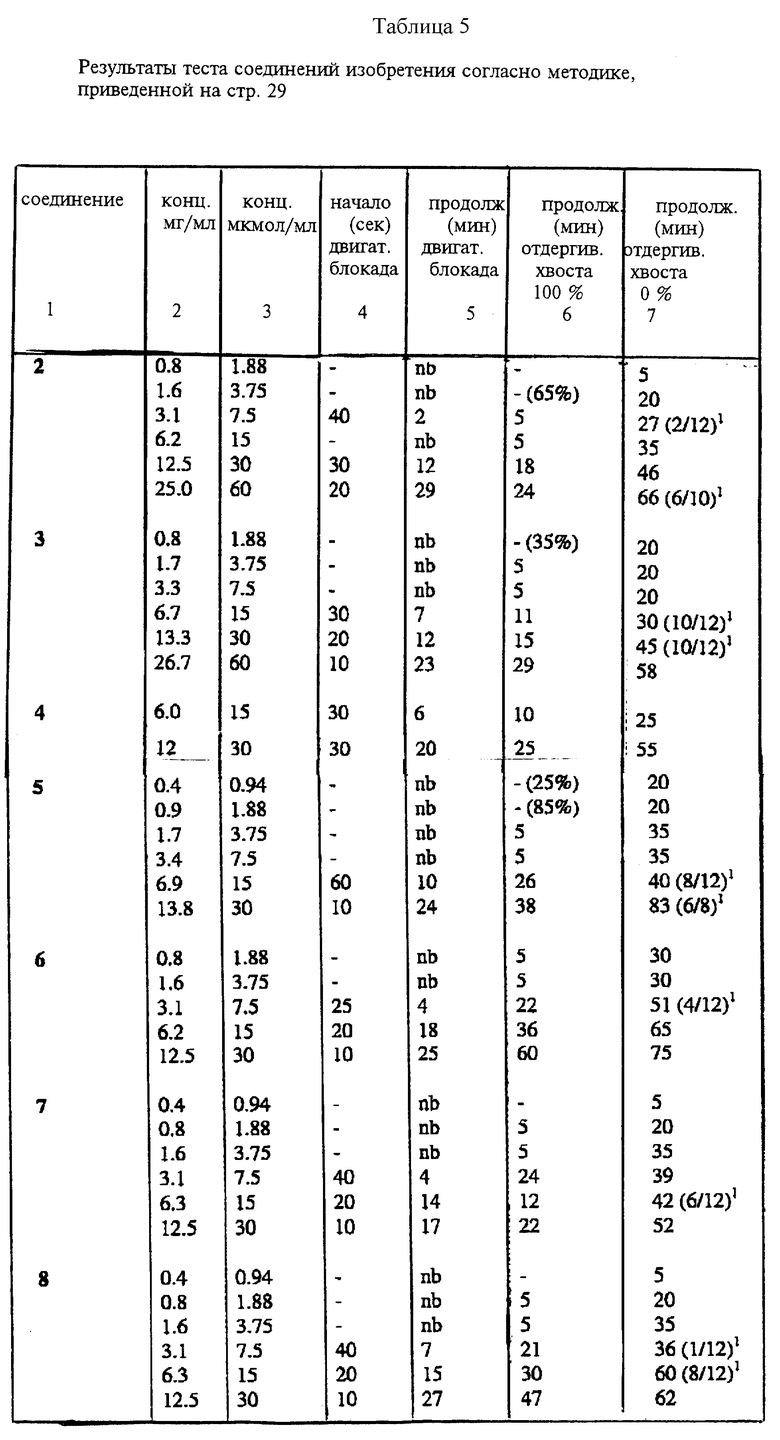
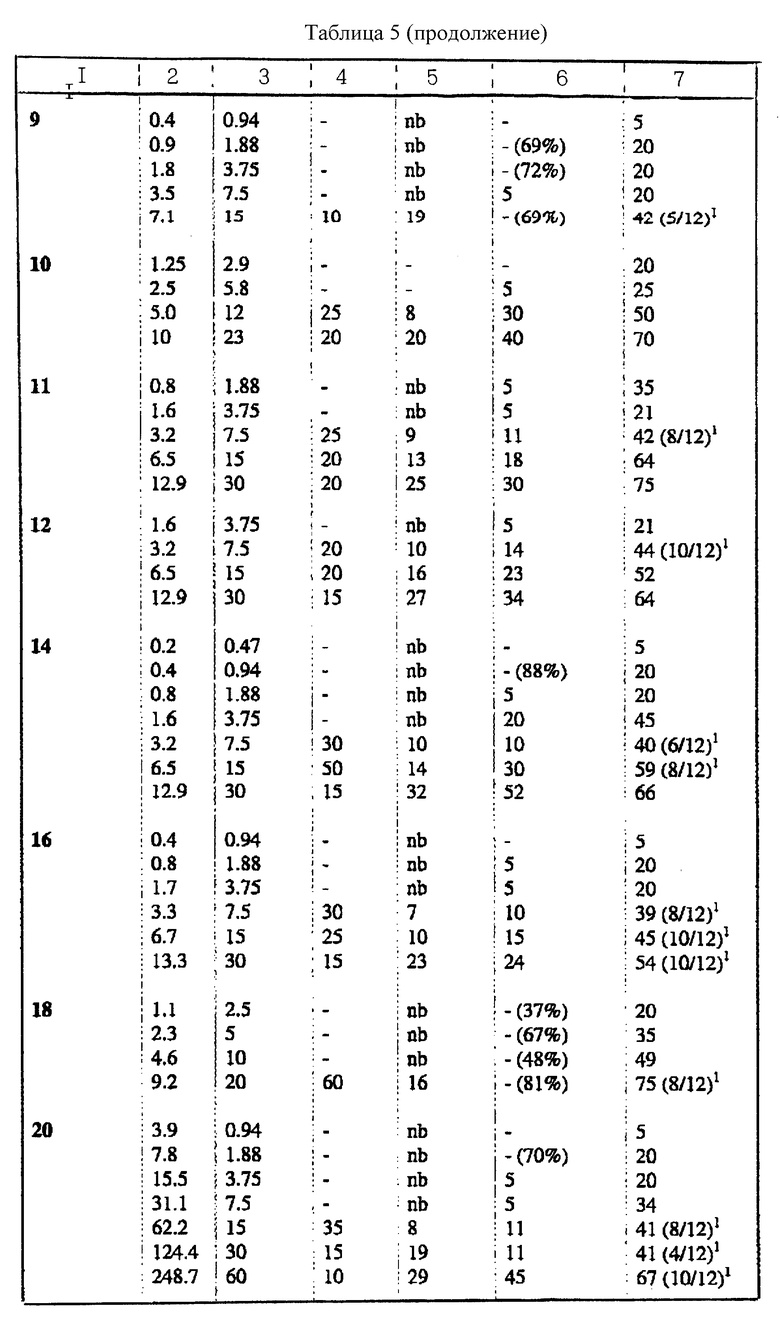
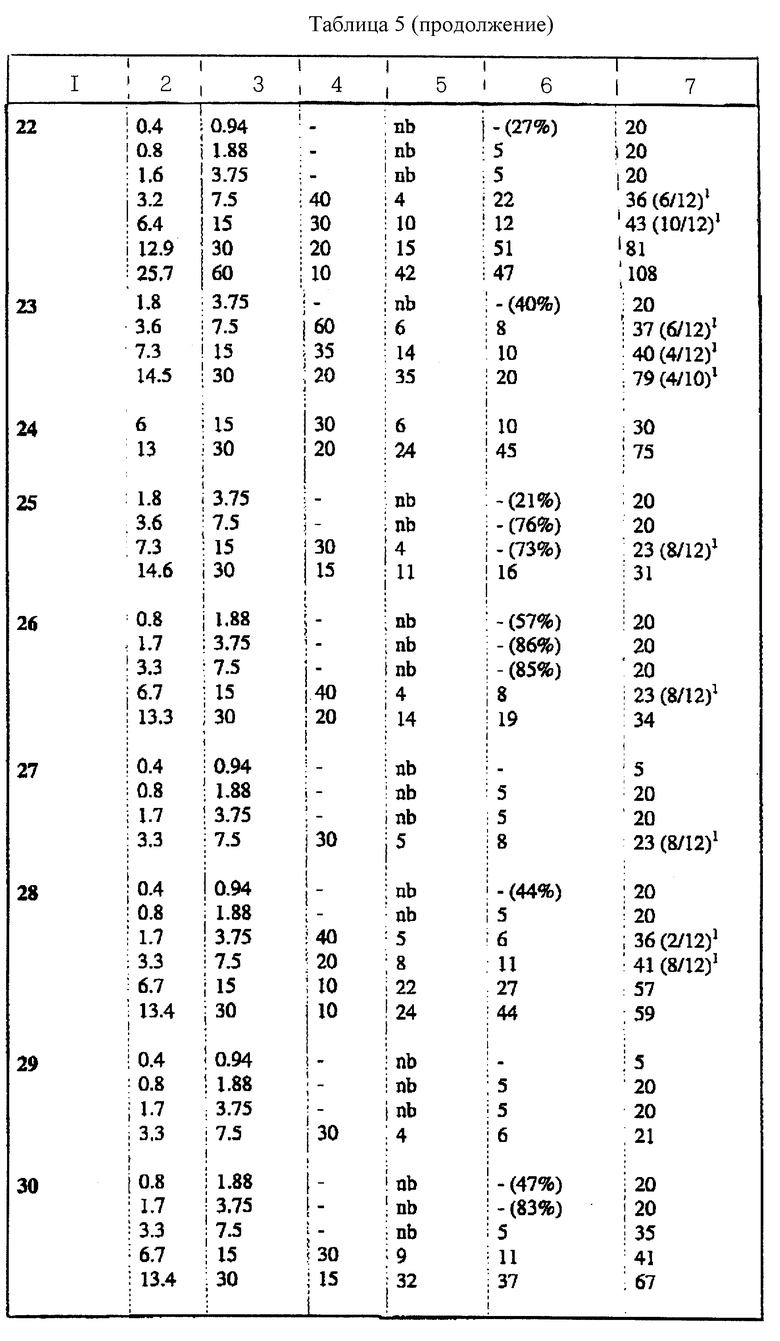
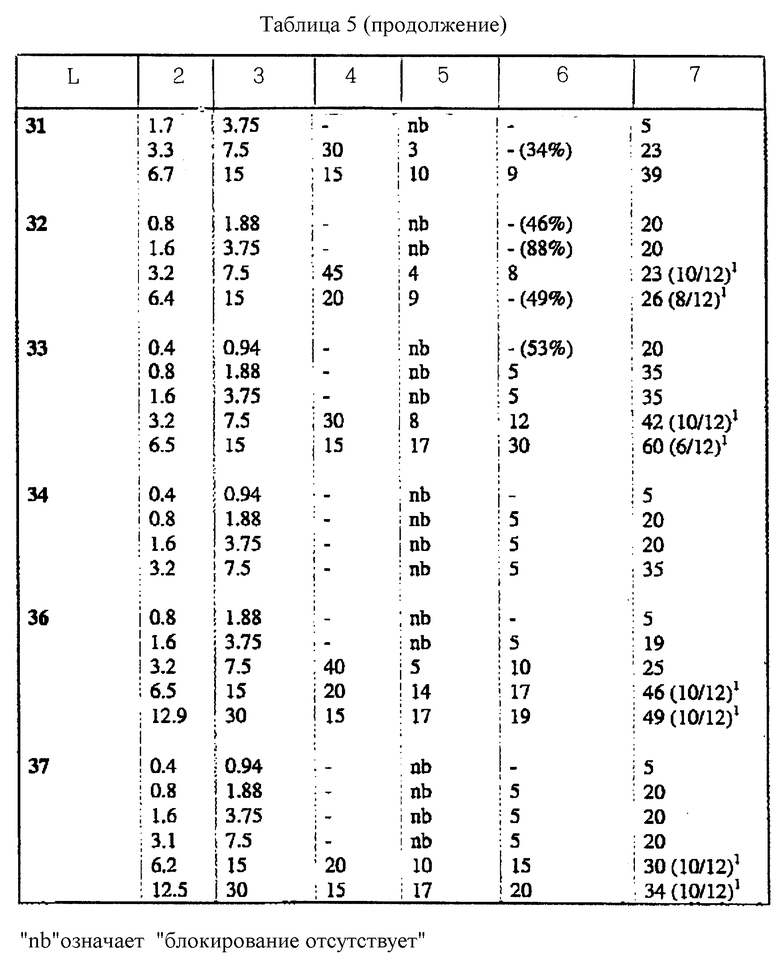
БИОЛОГИЧЕСКИЕ ИССЛЕДОВАНИЯ
Спинномозговая анестезия
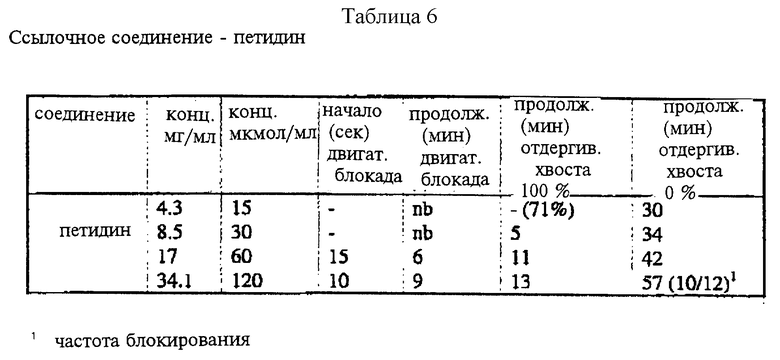
Соединения по настоящему изобретению испытывали на спинномозговую анестезию мыши. В каждой группе было шесть животных. В качестве соединения для сравнения использовали петидин.
Измеряли среднюю продолжительность (минуты) двигательной блокады и полной аналгезии (подергивание хвостом) у мышей после внутриоболочечной (спинномозговой, спинальной) инъекции 5 мкл испытуемого раствора. Продолжительность действия считали от момента инъекции.
Вывод
Поскольку местный анестезирующий эффект сильно проявляется в сочетании с аналгетическим эффектом, соединения по настоящему изобретению могут быть более полезными, чем петидин. Появляется также возможность заменить часто используемые комбинации аналгезирующего и анестезирующего средств. Результаты биологических исследований представлены в таблицах 1 - 6.



Изобретение относится к производным пиперидина общей формулы (I), где Z представляет группу -(CH2)m-CH(OR3)- или карбонильную группу, R1 - водород или (C1 - C3)алкил, R2 - (C1 - C3)алкил или R1 и R2 вместе образуют цепь -(CH2)n, где n число 3 - 5, или -(CH2)2-O-(CH2)2-, m = 0 - 1, n = 1 - 2, R3 - водород или -COCH3 и R4 - водород, -CH3, -OH или -OCH3, при условии, что когда Z представляет карбонильную группу, h = 2, или их фармацевтически приемлемым солям. Соединения формулы (I) обладают аналгетическим и местным анестезирующим действием и могут найти применение в медицине. 8 с. и 27 з.п. ф-лы, 6 табл.
где Z представляет группу
взятую в направлении слева направо в формуле A,
или карбонильную группу,
где а) R1 представляет водород или неразветвленную или разветвленную алкильную группу с 1 - 3 атомами углерода,
R2 представляет неразветвленную или разветвленную алкильную группу с 1 - 3 атомами углерода; или
b) R1 и R2 вместе образуют цепь - (CH2)n-, где n представляет 3, 4 или 5, или -(CH2)2O(CH2)2-;
m представляет 0 - 1;
р представляет 1 - 2;
R3 представляет водород или -СОСН3; и
R4 представляет водород, -СН2, -ОН или -ОСН3 при условии, что когда Z представляет карбонильную группу, то р = 2, а также их фармацевтически приемлемые соли.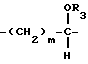
где R3 представляет
R4 представляет водород;
m = 0, p = 2,
R1 и R2 такие, как указаны в п.1.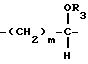
R3 представляет водород,
R4 представляет водород;
m = 0, р = 2;
R1 и R2 такие, как указаны в п.1.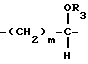
где R3 представляет водород;
m = 0, p = 2;
R1, R2 и R4 такие, как указаны в п.1.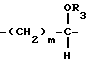
где R3 представляет водород;
R4 представляет водород;
m = 1, p = 1;
R1 и R2 такие, как указаны в п.1.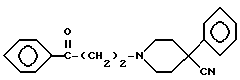
(реакция Манниха), после чего карбонильную группу соединения I восстанавливают боргидридом натрия с получением вторичного спирта II,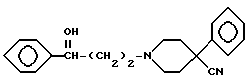
после чего гидролиз цианогруппы в щелочной среде дает аминокислоту III,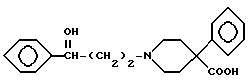
гидроксильную группу соединения III ацетилируют с получением сложного эфира IV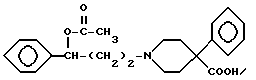
и карбоксильную группу соединения IV преобразуют в подходящий амид V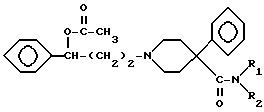
хлорангидридом кислоты, после чего избирательным щелочным гидролизом сложноэфирной группы соединений V получают соединение VI,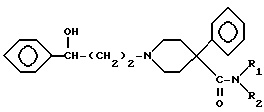
соответствующее соединение формулы (А), где R4 представляет водород, Z представляет  р = 2 и R1 и R2 такие, как указано в n.1, и полученное соединение преобразовывают в его фармацевтически приемлемую соль.
р = 2 и R1 и R2 такие, как указано в n.1, и полученное соединение преобразовывают в его фармацевтически приемлемую соль.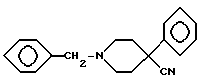
который гидролизуют в щелочной среде в аминокислоту VIII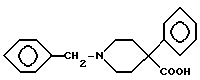
после чего соединение VIII преобразуют в подходящий амид IX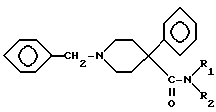
посредством хлорангидрида кислоты и осуществляют гидрогенолиз бензиламинов в присутствии палладиевого катализатора с получением вторичных аминов Х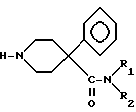
которые алкилируют (R)- или (S)-3-хлор-1-фенил-1-пропанолом с получением двух энантиомерных пар соединения VI, и полученное соединение преобразуют в его фармацевтически приемлемую соль.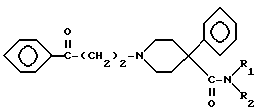
после чего карбонильную группу соединения XI восстанавливают боргидридом натрия с получением соединения формулы VI и полученное соединение преобразовывают в его фармацевтически приемлемую соль.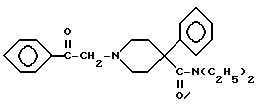
который затем восстанавливают боргидридом натрия с получением вторичного спирта XIII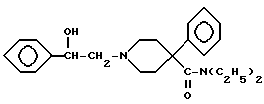 который является соединением, соответствующим формуле (А), где R4 представляет водород, р = 1, Z представляет
который является соединением, соответствующим формуле (А), где R4 представляет водород, р = 1, Z представляет 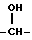 и R1 и R2 представляют каждый С2Н5 и полученное соединение преобразовывают в его фармацевтически приемлемую соль.
и R1 и R2 представляют каждый С2Н5 и полученное соединение преобразовывают в его фармацевтически приемлемую соль.
после чего соединение XIV преобразуют в соединение XVIII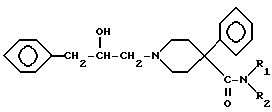
которое является соединением, соответствующим формуле (A), где R4 представляет водород, Z представляет 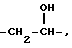 р = 1 и R1 и R2 такие, как указаны в п.1, и полученное соединение преобразовывают в его фармацевтически приемлемую соль.
р = 1 и R1 и R2 такие, как указаны в п.1, и полученное соединение преобразовывают в его фармацевтически приемлемую соль.
| US 3850935 A, 1974 | |||
| US 2951080 A, 1960 | |||
| Сталь | 1980 |
|
SU931789A1 |
| Способ получения производных 1-циклогексил-4-арил-4-пиперидинкарбоновых кислот,или их кислотно-аддитивных солей,или их стереохимических изомерных форм (его варианты) | 1981 |
|
SU1230467A3 |
