Прототипы изобретения
Многие эндогенные пептиды млекопитающих и амфибий связаны со специфическими опиоидными рецепторами и вызывают аналгезирующий отклик, сходный с эффектом классических наркотических опиатов. Показано, что у высших животных сосуществуют много различных типов опиоидных рецепторов. Например, см. W. Martin и др., J. Pharmacol. Exp. Ther., 197, p. 517 (1975); и J. Lord и др., Nature (London), 257, p. 495 (1977). Идентифицируют три различных типа опиоидных рецепторов. Первый δ показывает видоизменяющееся средство к пептидам, подобным энкефалинам. Второй μ демонстрирует повышенную селективность к морфину и другим полициклическим алкалоидам. Третий k демонстрирует одинаковое сродство к любой группе указанных выше лигандов и преимущественное сродство к динорфину (dynorphin). По-видимому, в основном μ -рецепторы более других включены в аналгезирующие эффекты, δ -рецепторы, по-видимому, имеют дело с поведенческими эффектами, хотя обезболивание также может быть опосредовано δ - и k-рецепторами.
Каждый опиоидный рецептор при взаимодействии с опиатом вызывает специфический биологический отклик, единственный в своем роде для этого типа рецепторов. Когда опиат активирует более одного рецептора, на биологический отклик каждого рецептора при этом влияют произведенные побочные эффекты. При введении опиата менее специфичный и селективный опиат может дать большую вероятность увеличения побочных эффектов.
В прототипах опиаты, пептиды опиоидного типа и их аналоги либо не проявляют специфичности и селективности к тому типу рецептора или рецепторов, с которыми они связаны. Основным местом действия аналгезирующих опиоидов является центральная нервная система (ЦНС). Общеизвестные наркотические аналгетики обычно являются гидрофобными и, следовательно, легко проникают сквозь липидные мембраны, такие как гематоэнцефалический барьер. Благодаря этой физической способности аналгетики имеют тенденцию взаимодействовать с опиоидными рецепторами в пределах центральной нервной системы в головном мозге. Однако они не обязательно связаны с подтипом гомогенного рецептора. Это связывание вызывает нежелательные с медицинской точки зрения побочные эффекты.
Опиаты могут вызывать серьезные и потенциально фатальные побочные эффекты. Неспецифические взаимодействия с рецепторами центральной нервной системы вызывают такие побочные эффекты как угнетение дыхания, выносливость, способность физической зависимости и внезапный абстинентный синдром. См. K. Budd, In International Encyclopedia of Pharmacology and Therapeutics; N.E. Williams and H. Wilkinson, Eds., Pergammon: (Oxford), 112, p. 51 (1983). Следовательно, полагают, что опиоидные аналгетики, действуя преимущественно через опиоидные рецепторы в периферической нервной системе, не вызывают подобных нежелательных побочных эффектов, как эффекты, вызванные опиоидными аналгетиками, действующими на центральную нервную систему. Опиоидные пептиды настоящего изобретения в основном действуют на периферическую нервную систему и поэтому преодолевают некоторые неблагоприятные последствия, характерные для обычных опиатов, в основном посредством предупреждения нежелательных побочных эффектов.
К настоящему времени одним из известных классов агентов, оказывающих периферическое аналгезирующее действие, являются нестероидные противовоспалительные агенты, такие как аспирин, ибупрофен и кеторолак. Эти агенты не взаимодействуют с опиоидными рецепторами, но известно, что они ингибируют циклооксигеназу и ослабляют синтез простагландина. Эти слабые аналгетики не оказывают центрально опосредованных воздействий, но могут вызывать другие побочные эффекты, такие как образование язв желудочно-кишечного тракта. Это является предметом настоящего изобретения - обеспечить пептиды, подобные опиоидам, которые оказывают периферическое воздействие, но по существу не имеют нежелательных побочных эффектов, вызываемых обычными периферически действующими аналгетиками.
Недавно в прототипах было показано, что лекарства типа опиатов обладают значительной периферической аналгезирующей активностью. См. A. Barber и R. Gottschlich, Med. Res. Rev., 12, p. 525 (1992) и C. Stein, Anasth. Analg., 76, p. 182 (1993). Для отличия периферических и центральных аналгезирующих ответных реакций в качестве фармакологических зондов используют четвертичные соли известных центрально действующих опиоидных алкалоидов. Четвертичные соли патентуемых опиатов имеют постоянный положительный заряд и демонстрируют ограниченное прохождение гематоэнцефалического барьера. См. T.W. Smith и др. , Life Sci, 31, p. 1205 (1982); T.W. Smith и др., Int. J. Tiss. Reac., 7, p. 61 (1985); B.B. Lorenzetti и S.H. Ferreira, Braz. J. Med. Biol. Res., 15, p. 285 (1982); D.R. Brown и L.I. Goldberg, Neuropharmacol., 24, p. 181 (1985); G. Bianchi и др. Life Sci, 30, p. 1875 (1982); и J. Russel и др., Eur. J. Pharmacol. , 78, p. 255 (1982). Получены сильно полярные аналоги энкефалинов и дерморфинов, которые сохраняют противоболевую активность, но показывают ограниченное проникновение в центральную нервную систему. См. R. L. Follenfant и др., Br. J. Pharmacol., 93, p. 85 (1988); G. W. Hardy и др., J. Med. Chem. , 32, p. 1108 (1989). Наоборот, в прототипах считают, что липофильные опиоидные пептиды легче проходят гематоэнцефалический барьер. К удивлению, опиоидные пептиды данного изобретения являются сильно липофильными, но незначительно проходят гематоэнцефалический барьер.
В отличие от обычных опиатов опиоидные пептиды являются гидрофобными. Их гидрофобность имеет тенденцию увеличивать скорость их выведения из организма млекопитающего. Гидрофобность увеличивает способность пептидов пересекать эпителиальный барьер. Несмотря на это показано, что введение опиоидных пептидов в организм млекопитающего влияет на центральную нервную систему. Следовательно, максимум усилий было направлено на улучшение абсорбционных свойств этих соединений. Ученые пытались уменьшить проникание пептидов в центральную нервную систему, особенно если пролонгированное воздействие на организм этими препаратами может вызвать вредные побочные эффекты или даже оказаться токсическим.
Полагают, что неполярные пептиды легче проходят в центральную нервную систему, пересекая гематоэнцефалический барьер, чем полярные пептиды. Опубликовано, что TAPP (H-Tyr-D-Ala-Phe-Phe-NH2) демонстрирует противоболевые свойства, периферические и центральные (P. Schiller и др., Proceedings of 20th European Peptide Symposium, 1988). В опровержение заявителями данного изобретения обнаружено, что этот тетрапептид TAPP (H-Tyr-D-Ala-Phe-Phe-NH2) не действует центрально. Этот результат показан отсутствием аналгезирующего действия даже при дозах в 100 мг/кг в тесте с горячей пластиной на мышах (mouse hot plate test). Этот тест является стандартным и известен специалистам. Тест проверяет химические препараты, которые вызывают центрально опосредованную аналгезирующую реакцию.
Использованный в данной заявке термин "специфичность" относится к особому или безусловному связыванию опиата или опиоидного пептида с отдельным опиоидным рецептором по сравнению с другим опиоидным рецептором. Специфичность опиоидного пептида указана константой ингибирования связывания Ki. Термин "селективность" относится к способности опиата или опиоидного пептида отличать один среди нескольких опиоидных рецепторов и взаимодействовать только с одним отдельным рецептором. Селективность опиоидного пептида относительно μ -рецепторов указана соотношением констант ингибирования связывания. Например, для определения селективности можно использовать соотношение констант ингибирования связывания K
Для фармакологического применения пептид должен обладать определенными свойствами. Во-первых, пептид должен быть устойчив к протеолетическому разрушению. Во-вторых, пептид должен вызывать усиленную биологическую реакцию. В-третьих, пептид должен быть безопасен для человека. В-четвертых, пептид должен быть таким, чтобы его можно было получить в достаточно большом количестве для клинического исследования на токсичность, и позже для применения на коммерческой основе. В данном случае требуемыми свойствами являются также меньшая липидная растворимость и большая растворимость в воде для того, чтобы предотвратить проникание пептида через гематоэнцефалический барьер и обеспечить быстрое выведение избытка введенного пептида и его метаболитов. Кроме того, для пептидов желательно добиться селективной и специфической активности по связыванию с рецепторами для того, чтобы уменьшить до минимума потенциальные побочные эффекты.
Существует потребность в пептидах, которые действуют на один специфический опиоидный рецептор, особенно μ -рецептор. Желательно найти пептиды с меньшей липидной растворимостью, чем у общеизвестных опиатов, такой, чтобы не разрушать гематоэнцефалический барьер. Кроме того, пептиды с высокой полярностью обычно лучше растворимы в водной среде с физиологическим pH, что повышает их выведение и выведение их метаболитов.
Краткое содержание изобретения
Настоящее изобретение обеспечивает новые соединения, которые являются селективными и специфическими относительно одного опиоидного рецептора. Настоящее изобретение обеспечивает пептиды, которые демонстрируют предпочтительную селективность и специфичность к μ -рецепторам. Настоящее изобретение также обеспечивает пептиды, которые главным образом взаимодействуют с опиоидными рецепторами на периферических нервных окончаниях и в незначительной степени проходят гематоэнцефалический барьер. Следовательно, настоящее изобретение понижает тяжесть и количество побочных эффектов по сравнению с обычными опиатами и опиоидными пептидами, описанными к настоящему времени.
Следующая формула (1) представляет соединения настоящего изобретения: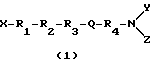
и их производные и аналоги, где X выбирают из группы, состоящей из H и C1-6-алкила;
Y и Z независимо выбирают из группы, состоящей из H, циклического аралкила и C1-6-алкила;
R1 является тирозильным остатком, 2',6'-диметилтирозильным остатком или их аналогом или производным;
R3 является ароматической аминокислотой;
R4 является остатком ароматической аминокислоты;
R2 является аминокислотой, имеющей R-конфигурацию при условии, что когда R1 является тирозильным остатком, R2 является D-аланином, X, Y и Z являются H, и R3 является фенилаланином, то R4 не является незамещенным фенилаланином или фенилаланином, замещенным 4NO2 или 4N3;
когда R1 является тирозильным остатком, R2 является D-аланином, X, Y и Z являются H, и R4 является фенилаланином, то R3 не является незамещенным фенилаланином или фенилаланином, замещенным 4NO2;
когда R1 является тирозильным остатком, R2 является D-аланином, X, Y и Z являются H, и R4 является 1'-нафтилаланином, то R3 не является 1'-нафтилаланином или 2'-нафтилаланином; и
когда R1 является тирозильным остатком, R2 является D-аланином, X, Y и Z являются H, то R3 и R4 каждый не является триптофаном;
и Q является амидной связью или подобием вставленной амидной связи.
Изобретение обеспечивает также фармацевтически приемлемые композиции, включающие эти пептиды, для применения при обезболивании.
Изобретение обеспечивает также применение этих пептидов с целью производства периферических аналгетиков для обезболивания.
Изобретение кроме того обеспечивает применение пептидов формулы H-Tyr-D-Ala-Phe-Phe-NH2 с целью производства периферических аналгетиков для обезболивания.
Таблицы и фигуры
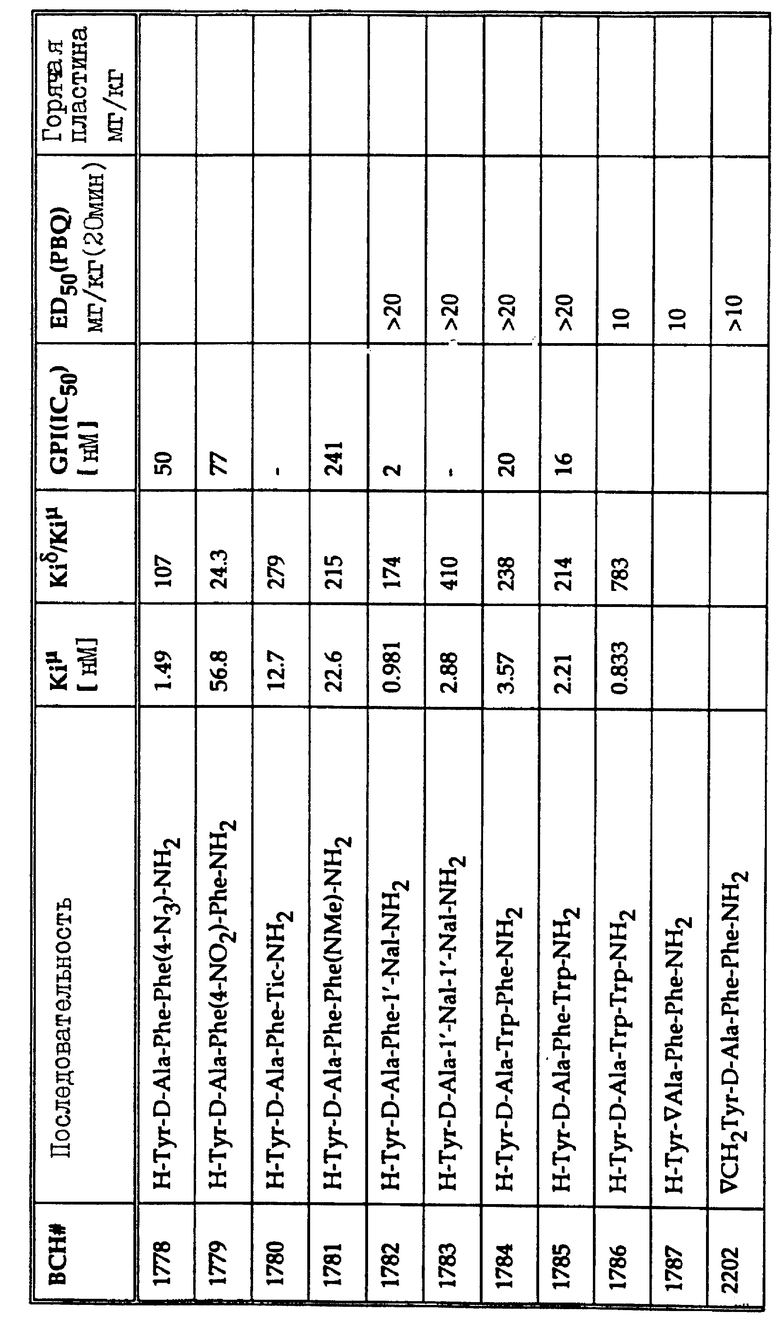
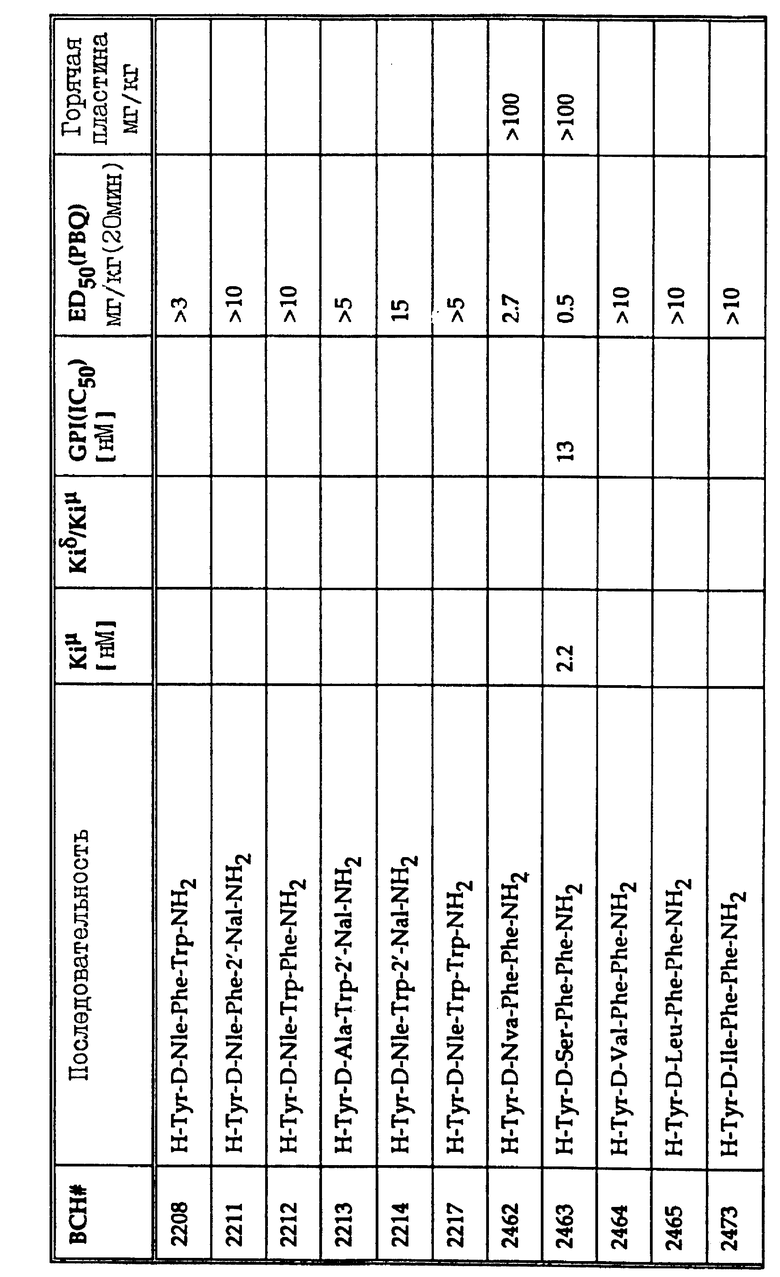
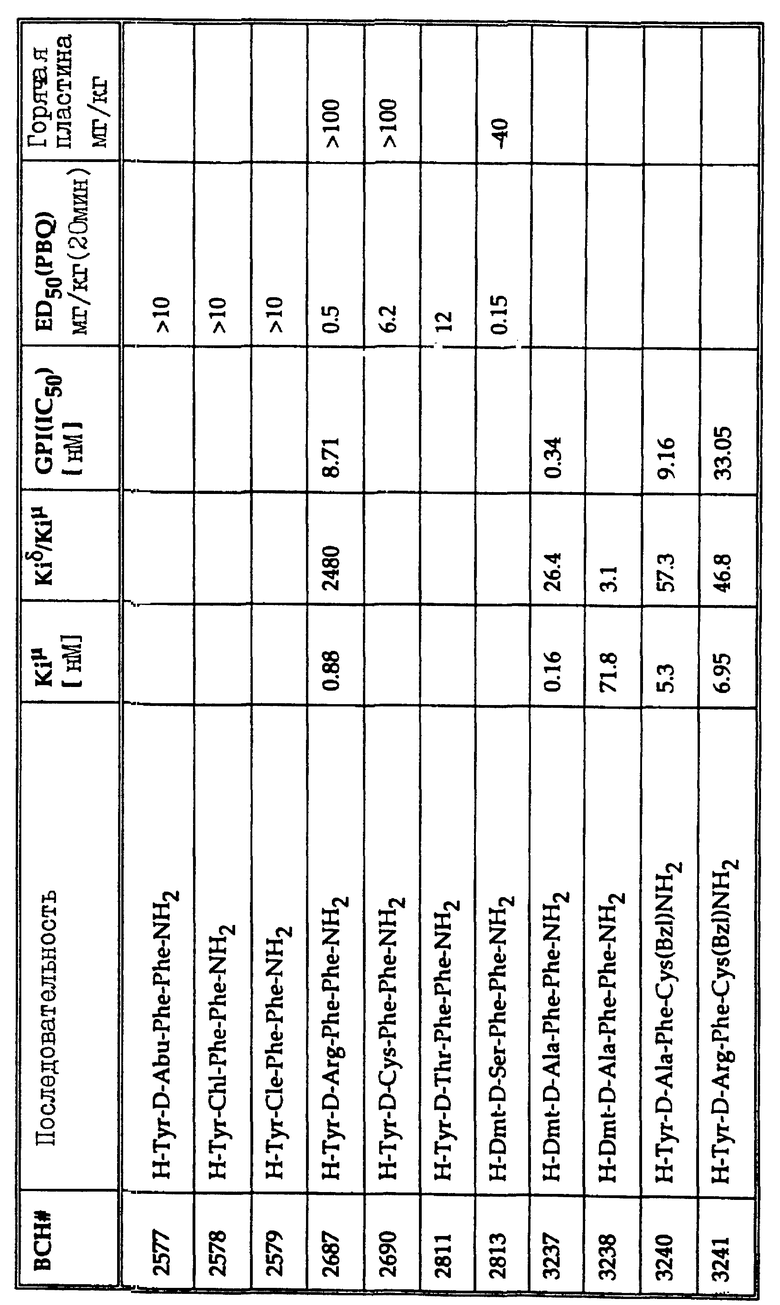
В таблице 1 перечислены in vivo и in vitro активности гидрофобных тетрапептидов, родственных дерморфину.
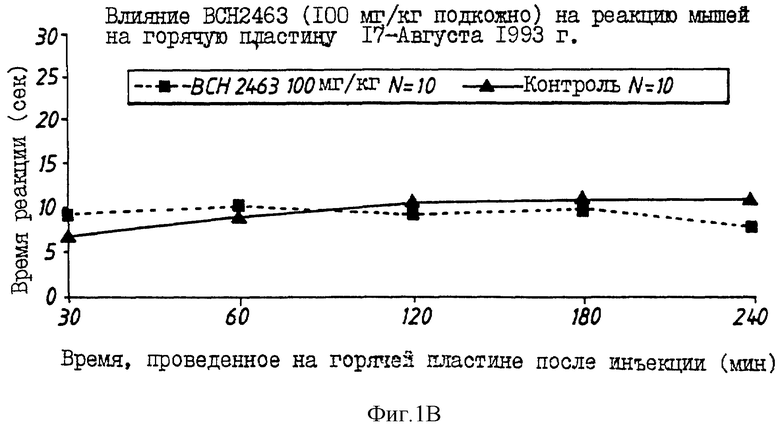
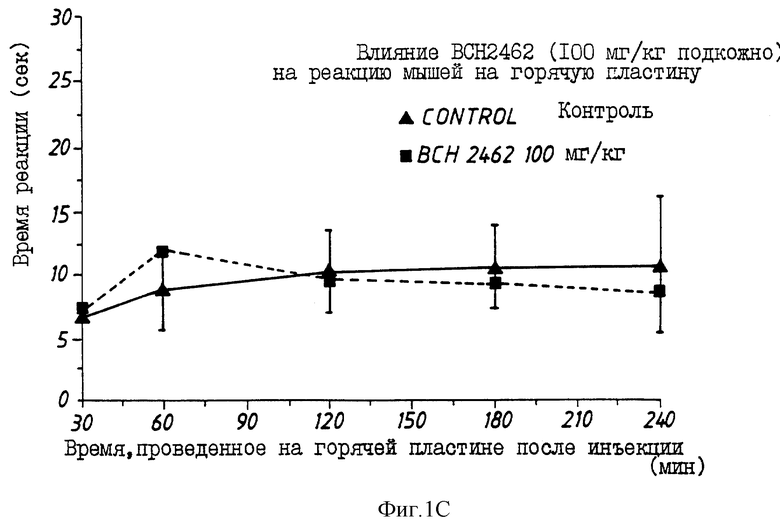
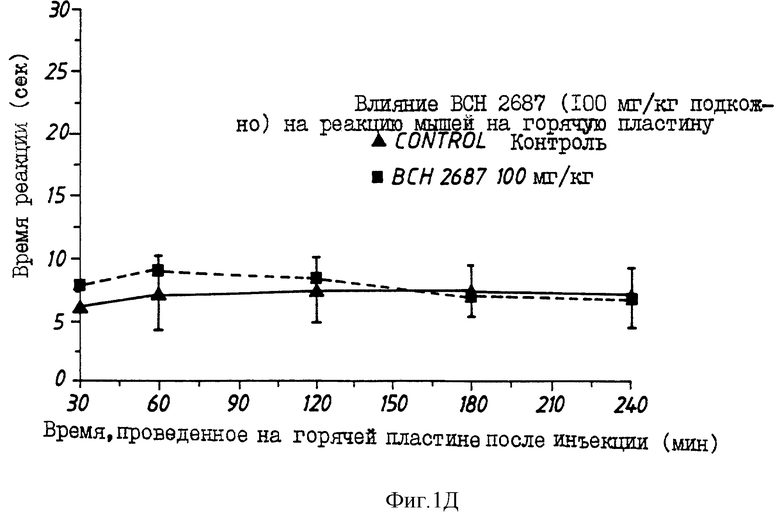
На фиг.1 показано изменение во времени аналгезирующего действия морфина (10 мг/кг) (фиг. 1A) и типичных проверяемых соединений (фиг. B: BCH2463; C: BCH2462; D: BCH2687).
На фиг. 2 показана диаграмма зависимости реакции от дозы для BCH2463 в опытах по исследованию конвульсий, вызванных фенилхиноном, который вводят мышам подкожно, ЭД50 = 0.5 мг/кг через 20 мин после введения.
На фиг. 3 приведено сравнительное изменение во времени аналгезирующего действия BCH1774 и BCH2463 в опытах по исследованию конвульсий, вызванных фенилхиноном, который вводят мышам подкожно.
Описание изобретения
В описании и формуле изобретения использованы следующие общие обозначения:
Abu - аминомасляная кислота; Ala - аланин; Arg - аргинин; Cle - циклолейцин; Gln - глутамин; Gly - глицин; His - гистидин; Hph - гомофенилаланин; Aib - аминоизомаляная кислота; Chl - циклогомолейцин; Cis(Bzl) - цистеин (бензил); Dmt - 2',6'-диметилтирозил; Glu - глутаминовая кислота; GPI - подвздошная кишка морской свинки; Ile - изолейцин; Leu - лейцин; Nle - норлейцин; Phe - фенилаланин; Phg - фенилглицин; Ser - серин; Trp - триптофан; Met - метионин; MVD - семенной проток мыши; Nva - норвалин; Pro - пролин; Thr - треонин; Tyr - тирозин; Nal - 1'-, или 2'-нафтилаланин; PBQ - фенил-пара-бензохинолин; Tic - тетрагидроизохинолин-3-карбоновая кислота; TAPP - H-Tyr-D-Ala-Phe-Phe-NH2; TSPP - H-Tyr-D-Ser-Phe-Phe-NH2.
Использованные здесь термины "аминокислота" и "ароматическая аминокислота" включают встречающиеся в природе аминокислоты, а также не встречающиеся в природе аминокислоты, их производные и аналоги, обычно используемые специалистами в области химического синтеза и химии пептидов. Включены также аналоги TAPP, где фенилаланин является пара-замещенным по положению 4 нитро- или азидо-остатком. Перечень не встречающихся в природе аминокислот можно найти в "The Peptide", т. 5, 1983, Academic Press, Глава 6, D.C. Roberts и F. Vellaccio, эта монография включения здесь в виде ссылки. Примеры ароматических аминокислот включают тирозин, триптофан, фенилглицин, гистидин, нафтилаланин, тетрагидроизохинилин-3-карбоновую кислоту и бензилцистеин. Другие примеры ароматических аминокислот включают фенилаланин, замещенный по ароматическому кольцу, например, группами CH3, C2H5, F, Cl, Br, NO2, OH, SH, CF3, CN, COOH и CH2COOH или замещенный по β -углероду низшими алкильными радикалами, OH, SH или бензольной группой. Ароматическое кольцо может иметь несколько заместителей. Ароматические аминокислоты могут также включать ароматические карбоциклы типа фенилглицина, где ароматическое кольцо фенилглицина замещено группами CH3, C2H5, F, Cl, Br, NO2, OH, SH, CF3, CN, COOH и CH2COOH. Эти примеры приведены только для того, чтобы проиллюстрировать изобретение, но ни коим образом его не ограничивают.
Термин "ЭД50" (ED50), как показано в таблице 1 для исследования конвульсий, вызванных PBQ, определяют как дозу лекарственного препарата, которая включает 50% снижение конвульсий, наблюдаемое по сравнению с контролем. Термин "ЭД50", использованный в опытах с горячей пластиной, определяют как дозу лекарства, необходимую для двукратного увеличения времени скрытой реакции по сравнению с контрольными образцами и определяют путем параллельного анализа эффективности.
Термин "подобие вставленной амидной связи" обозначает связь, в которой карбонильная группа и NH-группа амидной связи меняются местами.
Термин "Ki" обозначает константу ингибирования связывания.
Термин "K
Термин "R-конфигурация" относится к трехмерному расположению заместителей вокруг хирального элемента. Основная система для обозначения абсолютной конфигурации основана на системе приоритетов, которая хорошо известна специалистам и кратко описана далее. Каждую группу, присоединенную к хиральному центру, обозначают номером согласно приоритетам. Конфигурацию определяют как "R", если при переходе от наиболее приоритетной группы к менее приоритетным группам глаз следует по направлению движения часовой стрелки.
Термин "остаток", примененный к аминокислоте, обозначает радикал, производный от соответствующей аминокислоты при удалении гидроксила карбоксильной группы и одного водорода аминогруппы.
Соединениями настоящего изобретения являются соединения, представленные формулой (1):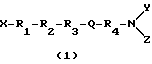
и их производные и аналоги, где X выбирают из группы, состоящей из H и C1-6-алкила;
Y и Z независимо выбирают из группы, состоящей из H, циклического аралкила и C1-6-алкила;
R1 является тирозильным остатком, 2',6'-диметилтирозильным остатком или его аналогом или производным;
R3 является аминокислотным остатком, выбранным из группы, состоящей из ароматических аминокислот;
R4 является остатком ароматической аминокислоты;
R2 является аминокислотой, имеющей R-конфигурацию при условии, что когда R1 является тирозильным остатком, R2 является D-аланином, X, Y и Z являются H, и R3 является фенилаланином, то R4 не является незамещенным фенилаланином или фенилаланином, замещенным 4NO2 или 4N3;
когда R1 является тирозильным остатком, R2 является D-аланином, X, Y и Z являются H, и R4 является фенилаланином, то R3 не является незамещенным фенилаланином или фенилаланином, замещенным 4NO2;
когда R1 является тирозильным остатком, R2 является D-аланином, X, Y и Z являются H, и R4 является 1'-нафтилаланином, то R3 не является 1'-нафтилаланином или 2'-нафтилаланином; и
когда R2 является D-аланином, R1 является тирозильным остатком и X, Y и Z являются H, то R3 и R4 каждый не является триптофаном; и
Q является амидной связью или подобием вставленной амидной связи.
Предпочтительными являются соединения, представленные формулой (1), и их производные и аналоги, где X является H.
Другими предпочтительными являются соединения, представленный формулой (1), и их производные и аналоги, где
R2 является аминокислотным остатком, имеющим R-конфигурацию при условии, что, когда R1 является тирозильным остатком, R2 является D-аланином, X, Y и Z являются H, то R3 и R4 разные и выбраны из группы, состоящей из фенилаланина и триптофана.
Другими предпочтительными являются соединения, представленные формулой (1), и их производные и аналоги, где
Q является амидной связью или подобием вставленной амидной связи формулы Q1-Q2, где Q1 выбирают из группы, состоящей из CH2, CHOH, C=O, C=S и CH=, и Q2 выбирают из группы, состоящей из CH2, NH, S, SO, SO2, O и CH=, при условии, что когда Q1 является CH=, то Q2 является CH=.
Кроме того, предпочтительными являются соединения, представленные формулой (1), и их производные и аналоги, где
Y и Z являются H;
R3 и R4 независимо являются ароматическими аминокислотами, и
R2 является аминокислотой, имеющей R-конфигурацию, при условии, что когда R1 является тирозильным остатком, и R2 является D-аланином, то R3 и R4 разные и выбраны из группы, состоящей из фенилаланина и триптофана.
Кроме того, предпочтительными являются соединения, представленные формулой (1), и их производные и аналоги, где
R2 является аминокислотой, имеющей R-конфигурацию, при условии, что R2 не является D-аланином, а R3 и R4 являются фенилаланиновыми остатками.
Еще предпочтительными являются соединения, представленные формулой (1), и их производные и аналоги, где R1 является тирозильным остатком, R2 выбирают из группы, состоящей из D-норвалина, D-серина и D-аргинина;
R3 и R4 являются фенилаланиновыми остатками; и
Q является пептидной связью.
Более предпочтительными являются соединения, представленные формулой (1), и их производные и аналоги, где
X является H,
Y и Z независимо выбирают из группы, состоящей из H, аралкила и C1-6-алкила;
R1 является тирозильным остатком, 2',6'-диметилтирозильным остатком или его аналогом или производным;
R3 является ароматической кислотой,
R4 независимо выбирают из группы, состоящей из ароматических и алифатических аминокислот; и
R2 является аминокислотным остатком, имеющим R-конфигурацию, при условии, что когда R2 является D-аланином, R1 является тирозильным остатком, и Y и Z являются H, то R3 и R4 независимо выбирают из группы, состоящей из фенилаланина и триптофана, но они не являются одинаковыми, Q являются амидной связью или подобием вставленной амидной связи формулы Q1-Q2, где Q1 выбирают из группы, состоящей из CH2, CHOH, C=O, C=S и CH=, и Q2 выбирают из группы, состоящей из CH2, NH, S, SO, SO2, O и CH=, при условии, что когда Q1 является CH=, то Q2 является CH=.
Более предпочтительными являются соединения, представленные формулой (1), и их производные и аналоги, где X является H, Y и Z являются H;
R1 является тирозильным остатком, 2',6'-диметилтирозильным остатком или его аналогом или производным;
R3 и R4 независимо являются ароматическими аминокислотами;
R2 является аминокислотой, имеющей R-конфигурацию при условии, что когда R2 является D-аланином и R1 является тирозильным остатком, то R3 и R4 независимо выбирают из группы, состоящей из фенилаланина и триптофана, но они не являются одинаковыми, Q является амидной связью или подобием вставленной амидной связи формулы Q1-Q2, где Q1 выбирают из группы, состоящей из CH2, CHOH, C=O, C=S и CH=, и Q2 выбирают из группы, состоящей из CH2, NH, S, SO, SO2, O и CH=, при условии, что когда Q1 является CH=, то Q2 является CH=.
Более предпочтительными являются соединения, представленные формулой (1), и их производные и аналоги, где X является H, Y и Z являются H,
R1 является тирозильным остатком, 2',6'-диметилтирозильным остатком или его аналогом или производным;
R2 является аминокислотой, имеющей R-конфигурацию при условии, что R2 не является аланином,
R3 и R4 являются фенилаланиновыми остатками;
Q является амидной связью или подобием вставленной амидной связи формулы Q1-Q2, где Q1 выбирают из группы, состоящей из CH2, CHOH, C=O, C=S и CH=, и Q2 выбирают из группы, состоящей из CH2, NH, S, SO, SO2, O и CH=, при условии, что когда Q1 является CH=, то Q2 является CH=.
Наиболее предпочтительными являются соединения, представленные формулой (1), и их производные и аналоги, где X является H; Y и Z являются H;
R1 является тирозильным остатком, R2 выбирают из группы, состоящей из D-норвалина, D-серина и D-аргинина; R3 и R4 являются фенилаланиновыми остатками; и
Q является пептидной связью.
Предпочтительные соединения I и более предпочтительные соединения II настоящего изобретения даны в конце описания.
Наилучшим известным в настоящее время способом осуществления данного изобретения является применение соединения H-Tyr-D-Arg-Phe-Phe-NH2.
Изобретение также включает применение в качестве периферического аналгетика соединения TAPP H-Tyr-D-Ala-Phe-Phe-NH2.
В качестве лигандов опиоидных рецепторов и аналгезирующих агентов общего действия получено и исследовано большое количество тетрапептидов на основе общей формулы (1). Эти соединения перечислены в таблице 1 вместе с соответствующими константами ингибирования связывания и отношениями селективности к рецепторам.
2', 6'-диметилтирозин (Dmt) можно заменить в опиоидных пептидных соединениях тирозином. Эксперименты показывают, что замена Dmt на тирозин по положению R1 в общей формуле 1 (первый аминокислотный остаток) повышает потенциал опиоидного пептида относительно μ -рецептора на два порядка величины. Селективность к μ -рецепторам увеличивается, когда соединение включает Dmt в положении R1. Это замещение вызывает соответствующий сдвиг в соотношении констант ингибирования связывания, что отражает увеличенную селективность к μ -рецепторам.
Многие соединения, перечисленные в таблице 1, демонстрируют хорошее связывание с μ -рецепторами, но проявляют слабое аналгезирующее действие в исследовании конвульсий на мышах (mouse writhing assay). Эта аномалия может наблюдаться вследствие быстрого протеолиза, быстрого очищения или обеих причин. Например, когда липофильный пептид деморфина TAPP (BCH1774) прототипа подвергают воздействию на границе почечных мембран, наблюдают быструю деградацию за 15-30 мин. Среди пептидов, перечисленных в таблице 1, три предпочтительных соединения, помимо самого TAPP, демонстрируют повышенный аналгезирующий эффект in vivo. Эти три соединения H-Tyr-Nva-Phe-Phe-NH2 (BCH2462), H-Tyr-D-Ser-Phe-Phe-NH2 (BCH2463) и H-Tyr-D-Arg-Phe-Phe-NH2 (BCH2687). Показано, что BCH2462, BCH2463 и BCH2687 демонстрируют периферическое аналгезирующее действие. При использовании этих пептидов центрального аналгезирующего действия не наблюдают, даже при дозах 100 мг/кг в тесте на мышах с горячей пластиной.
Как показано в таблице 1, величина ЕД50 для TAPP (BCH1774) составляет 1.4. Соответствующие величины для H-Tyr-D-Nva-Phe-Phe-NH2 (BCH2462), H-Tyr-D-Ser-Phe-Phe-NH2 (BCH2463) и H-Tyr-D-Arg-Phe-Phe-NH2 (BCH2687) равны 2.7, 0.5 и 0.5 соответственно. Величины ЕД50 для остальных соединений в таблице 1 больше этих значений. Хотя значение ЕД50 для BCH2813 составляет только 0.15, обнаружено, что это соединение действует центрально при дозах около 40 мг/кг в тесте с горячей пластиной.
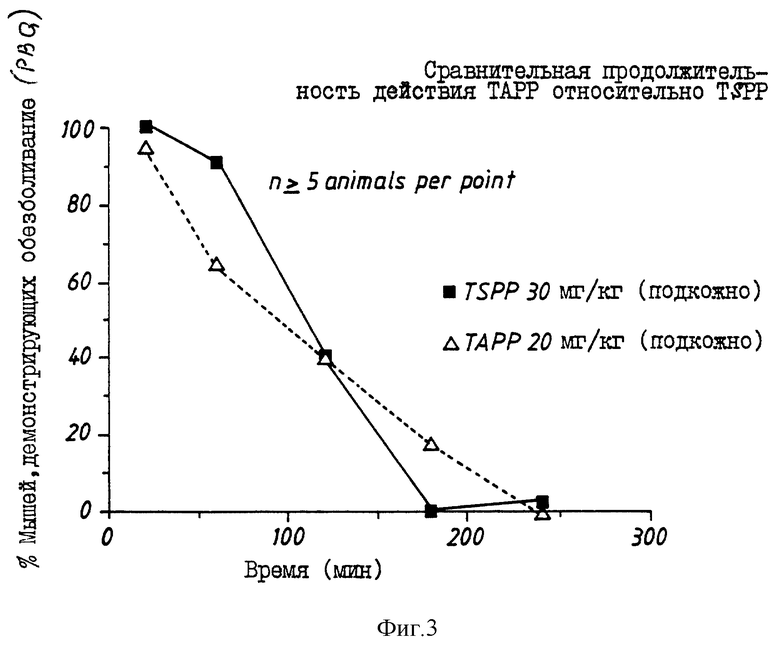
Результаты показывают, что соединения BCH1774, BCH2462 и BCH2463 еще подвергаются протеолизу, но имеют больший период полураспада и, следовательно, являются более эффективными аналгетиками. На фиг. 3 сравнивают продолжительности обезболивания in vivo, вызванного BCH1774 (TAPP) и BCH2463 (TSPP). При использовании 30 мг/кг BCH2463 (подкожно) и 20 мг/кг BCH1774 (подкожно) на фиг. 3 показано, что аналгезирующее действие BCH1774 продолжается дольше, чем BCH2463, указывая, вероятно, несколько ускоренный протеолиз BCH2463 по сравнению с BCH1774 in vivo.
На фиг. 1A-D показаны влияния морфина, BCH2463 (TSPP), BCH2462 (TNPP) и BCH2687 на мышей при оценке реакции мышей в тесте с горячей пластиной. Как показано на фиг. 1A, время реакции мыши, обработанной 10 мг/кг морфина, составляет примерно 17 с. Время реакции мыши, обработанной 100 мг/кг BCH2463 (фиг. 1B) составляет примерно 9 с по сравнению с контрольным значением приблизительно 7 с. Эти результаты указывают, что в то время, как морфин ингибирует болевой термический раздражитель, BCH2463 - нет; но BCH2463 является потенциальным аналгезирующим агентом, как показано посредством ингибирования химически-индуцированных конвульсий (фиг. 2). Время реакции мыши, обработанной BCH2462 и BCH2687 (фиг.1C, 1D) составляет приблизительно 8 с, что указывает аналогичный результат, что и для BCH2463.
Исследованы эффекты ингибирования протеолитического метаболизма BCH2463 ингибитором DL-тиофаном, а также метаболический распад BCH2463, опосредованный граничными почечными мембранами (brush border kindney membranes). Полученные данные показывают, что почки могут быть основным местом очищения и метаболизма для соединения BCH2463. На фиг.2 ясно, что фермент эндопептидаза EC24-11, который ингибируется DL-тиофаном, является предварительным медиатором протеолиза BCH2463 посредством экстракта почек (brush border kidney extract).
И BCH1774 (TAPP) и BCH2462 (TNPP) производят летальное действие на мышей при введении дозы лекарства 1-5 мг/кг внутривенно. В отличие от этого BCH2463 (TSPP), к удивлению, не оказывает летального действия при дозах до 20 мг/кг внутривенно. К тому же пептиды безопасны при введении подкожно (s. c. ) при дозах более 100 мг/кг. Следовательно, желательным способом введения этих соединений является подкожное введение. Таким образом, структурный пример, представленный BCH1774, можно модифицировать, сохраняя исключение из центральной нервной системы даже при дозах выше 100 мг/кг подкожно, и можно свести к минимуму вредную токсичность при внутривенном введении. Таким образом, BCH2463 не является летальным для мышей при дозах по крайней мере выше 20 мг/кг (внутривенно).
Фармацевтически пригодные соли пептидов настоящего изобретения можно получить обычным образом, добавляя такие кислоты как соляная, бромистоводородная, фосфорная, уксусная, фумаровая, салициловая, лимонная, молочная, миндальная, винная, оксалиновая, метансульфоновая и другие подходящие кислоты, известные специалистам.
Настоящее изобретение также обеспечивает фармацевтические композиции. Подходящие композиции содержат фармацевтически эффективное количество пептида настоящего изобретения или его фармацевтически пригодные соли и фармацевтически пригодный носитель или разбавитель.
Настоящее изобретение также обеспечивает способ обезболивания у таких животных как млекопитающие, включая человека. Способ включает стадии введения пациенту фармацевтически эффективного количества пептида формулы 1 или его фармацевтически пригодной соли. Можно также использовать описанные выше фармацевтические композиции.
Следующие далее примеры приведены для лучшего описания изобретения. Эти примеры даны только с целью иллюстрации и ни коим образом не ограничивают изобретение.
Примеры
Опиоидную активность пептидов оценивают in vitro, используя препарат продольной мышцы подвздошной кишки морских свинок (GPI), а их противоболевую активность определяют на грызунах in vivo на модель конвульсий, вызванных PBQ (периферическая активность), и в тесте с горячей пластиной (центральная активность). Антагонизм обезболиванию периферическим опиоидным антагонистом N-метилналорфином и сравнение активности при конвульсиях и в тестах с горячей пластиной показывают, что аналгезирующее действие преимущественно опосредовано на периферии. На периферическое обезболивание указывает высокий потенциал при наблюдении конвульсий и при этом низкий потенциал в тесте с горячей пластиной.
Вызванные PBQ (фенил-пара-бензохиноном) конвульсии мышей служат оценкой центрального и периферического обезболивания. Условия проведения экспериментов смотри в работе Sigmund и др., Proc. Soc. Exp. Biol. Med., 95, p. 729 (1957), которая включена здесь в качестве ссылки. Центральное обезболивание определяют посредством ингибирования реакции мышей на горячую пластину. Условия проведения экспериментов смотри в работе G. Woolf и A. Macdonald, J. Pharmacol. Exp. Ther., 80, p. 300 (1944), которая включена здесь в качестве ссылки. Исследования, определяющие связывание опиоидных рецепторов (для и рецепторов), так же как и GPI и MVD оценки были сделаны в экспериментальных условиях, описанных в работе Schiller и др., Biophis. Res. Commun., 85, p. 1322 (1975), которая включена здесь в качестве ссылки.
Соединения настоящего изобретения получают, применяя твердофазный синтез, описанный в общих чертах ниже и, как правило, известный специалистам.
Пример 1. Твердофазный синтез опиоидных пептидов
Синтетические пептиды получают, используя смолу Rink*1 [4-(2',4'-диметоксифенил-Fmoc-аминометил)-фенокси смолу] (Novabiochem of Advanced Chemtech) и соответствующий C-терминальный N-Fmoc-L-аминокислотный остаток каждого синтезируемого пептида (* - торговая марка).
Все L- и D-аминокислоты (Novabiochem of Advanced Chemtech) имеют свои Fmoc-защищенные (9-флуоренил-метилоксикарбонил) альфа-группы и указанные далее боковые защитные группы: простой трет-бутиловый эфир (tBu) для серина, треонина и тирозина; сложный трет-бутиловый эфир (OtBu) для аспарагиновой кислоты и глутаминовой кислоты; трет-бутилоксикарбонил (tBoc) для лизина, 2,2,5,7,8-пентаметилхроман-6-сульфонил (pmc) для аргинина и тритил (trt) для цистеина.
Не содержащий диметиламина диметилформамид (DMF) (Anachemia) обрабатывают активированными молекулярными ситами 4A. Пиперидин (Advanced Chemtech) используют без дополнительной очистки. DCC (дициклогексилкарбодиимид) и HOBt (гидроксибензотриазол) получают от Fluka и Advanced Chemtech соответственно.
Твердофазный синтез пептидов осуществляют вручную на смоле Rink*2(* - торговая марка). Загрузка составляет примерно 0.6 ммоля/г. Конденсацию пептида проводят, применяя:
1) взаимодействие 2 эквивалентов каждой Fmoc-аминокислоты, HOBt и DCC в DMF в течение 1-4 ч при комнатной температуре;
2) повторное взаимодействие 1 эквивалента каждой Fmoc-аминокислоты, HOBt и DCC;
3) ацетилирование 20% (объем/объем) (CH2CO)2O/DMC в течение 1 ч при комнатной температуре;
4) снятие N-Fmoc-защиты: 20% (объем/объем) пиперидин в DMF в течение 25 мин.
Удаление боковых защитных групп (tBu, Boc, Trt, Pmc) и отщепление пептида от смолы осуществляют при воздействии коктейля, содержащего TFA:
TFA/Анизол/DCM = 55/5/40 (объем/объем) в течение 90 мин при комнатной температуре под N2. Пептид осаждают из диэтилового эфира, фильтруют и сушат. Сырой пептид чистят и анализируют способом высокоэффективной жидкостной хроматографии (HPLC) на колонке с обращенной фазой при градиентном элюировании, используя 0.06% TFA/H2O и 0.06% TFA/ацетонитрил.
Пример 2. Исследование с горячей пластиной
Определение аналгезирующей активности
Для теста используют самцов мыши CD#1 весом от 20 до 25 г. Мышей взвешивают, маркируют и делят на группы по 10 животных.
Обычно мышам делают подкожную инъекцию соединения (или стандарта, или среды)), вводимый объем составляет 0.1 мл на 10 г веса мыши (10 мл/кг). Если используют антагонист, такой как Nalaxone или N-метил-леваллорфан (N-methyl-Levallorphan), то его вводят внутрибрюшинно за 20 мин до введения соединения (или стандарта, или среды). Вводимый объем также составляет 0.1 мл на 10 г веса мыши. Доза антагониста равна 10 мл/кг.
Для каждой мыши оценивают время реакции на горячей пластине. Температуру горячей пластины (Sorel, модель DS37) устанавливают 55oC. За мышью наблюдают, отмечая признаки дискомфорта, такие как облизывание лап или встряхивание, попытки побега (спрыгивает с пластины) или дрожь. Время реакции (в с) засекают, когда появляется один из этих признаков. Каждую мышь наблюдают максимум в течение 30 с, так чтобы предотвратить повреждение ткани лап. За мышами можно наблюдать, спустя различные временные интервалы после введения исследуемого соединения (или среды, или стандарта). Временные интервалы могут составлять 30, 60 или 120 мин (или другие).
Для каждого временного показателя среднее время реакции контрольной группы умножают на 1.5. Время реакции каждой обработанной мыши сравнивают со "средним контрольным • 1.5". Если время реакции меньше "среднего контрольного • 1.5", считают, что мышь не получила обезболивания. Если время реакции выше "среднего контрольного • 1.5", то считают, что на мышь оказано обезболивающее действие. Количество мышей, испытавших обезболивание, в группе определяют процент обезболивания данным соединением для данного показателя. Если процент обезболивания менее 30%, соединение считают неактивным.
Пример 3. Исследование конвульсий
Определение количества вывихов
Для теста используют самцов мыши CD#1 весом от 18 до 22 г. Мышей взвешивают, маркируют. Внутрибрюшинно вводят мышам 0.02% раствор фенилхинона, 0.3 мл/20 г веса. Считают количество вывихов за период 15 мин после инъекции. Фенилхинон вводят подкожно с интервалами 5, 20 или 60 мин после введения исследуемого соединения (или среды, или стандарта). Фенилхинон вводят орально с интервалом 60 мин после введения исследуемого соединения (или среды, или стандарта).
0.02% раствор фенилхинона - (2-фенил-1,4-бензохинон (Sigma) - получают следующим способом. 20 мг фенилхинона растворяют в 5 мл 90% этанола (Sigma, реагент, спирт). Растворенный фенилхинон, непрерывно встряхивая и предварительно подогревая (не до кипения), медленно добавляют к 95 мл дистиллированной воды. Раствор фенилхинона постоянно защищают от света и каждый день для тестирования готовят новый раствор. Перед использованием раствора фенилхинона рекомендуют подождать 2 ч.
Тестирование можно проводить на 5 мышах одновременно. Каждая группа обычно включает 10 мышей. Если применяют антагонист, такой как налоксон (naloxone), то его вводят внутрибрюшинно за 20 мин до исследуемого соединения (или среды, или стандарта).
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ОПИОИДОПОДОБНЫЕ ПЕПТИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2165432C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ НА ИХ ОСНОВЕ | 1995 |
|
RU2145599C1 |
| ПЕПТИД ИЛИ ЕГО ОРГАНИЧЕСКИЕ ИЛИ НЕОРГАНИЧЕСКИЕ СОЛИ, СПОСОБ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБЫ СТИМУЛИРОВАНИЯ ВЫСВОБОЖДЕНИЯ ГОРМОНА РОСТА И УВЕЛИЧЕНИЯ ЕГО СОДЕРЖАНИЯ В КРОВИ | 1992 |
|
RU2126014C1 |
| ОСНОВАННЫЕ НА АМИДАХ ПРОЛЕКАРСТВА ПЕПТИДОВ ГЛЮКАГОНОВОГО НАДСЕМЕЙСТВА | 2009 |
|
RU2550696C2 |
| АНТАГОНИСТЫ РЕЦЕПТОРОВ СОМАТОСТАТИНА | 1997 |
|
RU2179172C2 |
| Амиды гептапептида для лечения HMGB1-зависимых заболеваний | 2021 |
|
RU2760133C1 |
| СОСТАВЫ ДЛЯ ПЕРОРАЛЬНОГО ПРИМЕНЕНИЯ АГОНИСТОВ КАППА-ОПИОИДНОГО РЕЦЕПТОРА | 2019 |
|
RU2835627C2 |
| АГОНИСТЫ И АНТАГОНИСТЫ УРОТЕНЗИНА-II | 2001 |
|
RU2263679C2 |
| БИОТИНОВЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1996 |
|
RU2171807C2 |
| СИНТЕТИЧЕСКИЕ ПЕПТИДНЫЕ АМИДЫ И ИХ ДИМЕРЫ | 2007 |
|
RU2510399C2 |





Опиоидные пептиды формулы (I): X-R1-R2-R3-Q-R4-N(Y)Z и его аналоги с периферическим аналгезирующим действием, где Х - Н или C1-6-алкил; Y, Z - Н, циклический аралкил и C1-6-алкил; R1 - тирозильный остаток, 2',6'-диметил тирозильный остаток или их аналог; R3, R4 остаток ароматической аминокислоты (другие обозначения см. в п.1 формулы изобретения). 2 с. и 19 з.п. ф-лы, 3 ил., 1 табл.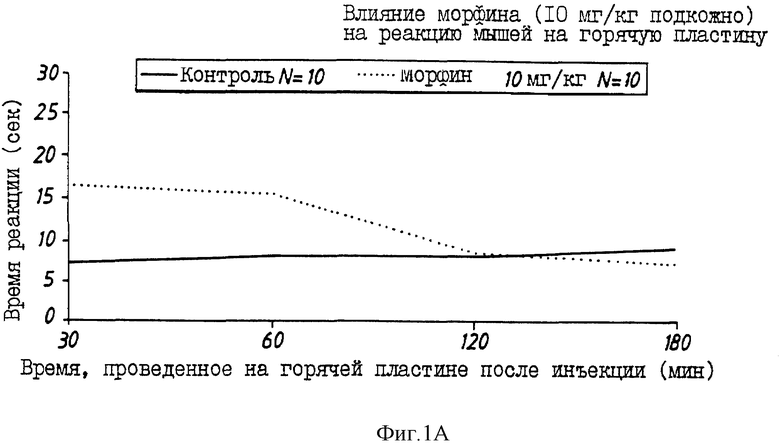
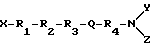
и их производные и аналоги с периферическим анальгезирующим эффектом, где X = водород; Y и Z = водород; R1 является тирозильным остатком, 2',6'-диметилтирозильным остатком или их аналогом или их производным; R3 - остаток ароматической аминокислоты; R4 - остаток ароматической аминокислоты; R2 - остаток аминокислоты, имеющей R - конфигурацию, или выбран из группы, состоящей из аминоизомасляной кислоты, циклопропилаланина, циклогомолейцина, циклолейцина при условии, что когда R1 является тирозильным остатком, R2 является - аланином, X, Y и Z являются водородом, и R3 является фенилаланином, то R4 не является незамещенным фенилаланином или фенилаланином, замещенным 4NO2 или 4N3; когда R1 является тирозильным остатком, R2 является D-аланином, X, Y и Z являются водородом и R4 является фенилаланином, то R3 не является незамещенным фенилаланином или фенилаланином, замещенным 4NO2; когда R1 является тирозильным остатком, R2 является D-аланином, X, Y и Z являются водородом и R4 является 1'-нафтилаланином, то R3 не является 1'-нафтилаланином или 2'-нафтилаланином; и когда R1 является тирозильным остатком, R2 является D-аланином, X, Y и Z являются H, то R3 и R4 не являются триптофаном; и Q является амидной связью, и при этом исключены
H-Tyr-D-Phe-Phe-Phe-NH2
H-Tyr-D-NMePhe-Phe-Phe-NH2
H-Tyr-D-Tic-Phe-Phe-NH2
2. Соединение формулы I по п.1, отличающееся тем, что R2 является аминокислотным остатком, имеющим R-конфигурацию при условии, что, когда R1 является тирозильным остатком, R2 является D-аланином, Y и Z являются H, то R3 и R4 разные и выбраны из группы, состоящей из фенилаланина и триптофана.
H-Tyr-Aib-Phe-Phe-NH2
H-Tyr-D-Nle-Phe-Phe-NH2
H-Tyr-D-Ala-Phe-2'-Nal-NH2
H-Tyr-D-Ala-D-Phe-Phe-NH2
H-Tyr-D-Ala-Phe(4NO2)-Phe(4NO2)-NH2
H-Tyr-D-Ala-Phe-Tic-NH2
H-Tyr-D-Ala-Phe-1'-Nal-NH2
H-Tyr-D-Ala-Trp-Phe-NH2
H-Tyr-D-Ala-Phe-Trp-NH2
H-Tyr-D-Nle-Phe-Trp-NH2
H-Tyr-D-Nle-Phe-2'-Nal-NH2
H-Tyr-D-Nle-Trp-Phe-NH2
H-Tyr-D-Ala-Trp-2'-Nal-NH2
H-Tyr-D-Nle-Trp-2'-Nal-NH2
H-Tyr-D-Nle-Trp-Trp-NH2
H-Tyr-D-Nva-Phe-Phe-Phe-NH2
H-Tyr-D-Ser-Phe-Phe-NH2
H-Tyr-D-Val-Phe-Phe-NH2
H-Tyr-D-Leu-Phe-Phe-NH2
H-Tyr-D-Ile-Phe-Phe-NH2
H-Tyr-D-Abu-Phe-Phe-NH2
H-Tyr-Chl-Phe-Phe-NH2
H-Tyr-Cle-Phe-Phe-NH2
H-Tyr-D-Arg-Phe-Phe-NH2
H-Tyr-D-Cys-Phe-Phe-NH2
H-Tyr-D-Thr-Phe-Phe-NH2
H-DMT-D-Ser-Phe-Phe-NH2
H-Tyr-D-Ala-Phe-Phg-NH2 - соль трифторуксусной кислоты
H-Tyr-D-Arg-Phe-Hgh-NH2 - соль бис-трифторуксусной кислоты
H-DMT-D-Ala-Phe-Phe-NH2 - трифторуксусная кислота
H-D-DMT-D-Ala-Phe-Phe-NH2 - соль трифторуксусной кислоты
H-Tyr-D-Ala-Phe-Hph-NH2 - соль трифторуксусной кислоты
H-Tyr-D-Ala-Phe-Cys(Bzl)-NH2 - соль трифторуксусной кислоты
H-Tyr-D-Arg-Phg-Phe-NH2 - соль бис-трифторуксусной кислоты
H-Tyr-D-Arg-Hph-Phe-NH2 - соль бис-трифторуксусной кислоты
H-Tyr-D-Ala-Phe-Phe(pf)-NH2 - соль трифторуксусной кислоты
H-Tyr-D-Ala-Phe-D-Phe(pf)-NH2 - соль трифторуксусной кислоты
H-Tyr-D-Ala-Hph-Phe-NH2 - соль трифторуксусной кислоты
H-Tyr-D-Met-Phe-Phe-NH2 - соль трифторуксусной кислоты
H-Tyr-D-Arg-Phe-D-Phe-NH2 - соль бис-трифторуксусной кислоты
H-Tyr-D-Ala-Phg-Phe-NH2 - соль трифторуксусной кислоты
H-Tyr-D-Ala-D-Phg-Phe-NH2 - соль трифторуксусной кислоты
H-Tyr-D-Arg-Phe-Phe(pf)-NH2 - соль бис-трифторуксусной кислоты
H-Tyr-D-Arg-Phe-D-Phe(pf)-NH2 - соль дитрифторуксусной кислоты
9. Соединение по п.1, отличающееся тем, что указанное соединение является H-Tyr-D-Nva-Phe-Phe-NH2.
| Способ получения тетрапептидов или их кислотно-аддитивных солей | 1981 |
|
SU1082319A3 |
| Способ получения производных тетрапептидов или их солей | 1978 |
|
SU908246A3 |
| US 4350627 A, 1982 | |||
| Приспособление для подачи в топку мелкого сыпучего топлива | 1928 |
|
SU14719A1 |
| Способ получения ацетиленовых спиртов | 1961 |
|
SU141483A1 |
