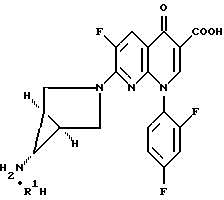
Изобретение относится к новым способам и промежуточным продуктам для получения фармацевтически приемлемых солей кислот формулы нафтиридонового антибиотика 7- (1α,5α,6α)- (6-амино-3-азабицикло[3,1,0]гекс- 3-ил)-1-(2,4-дифторфенил)-6-фтор-1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновой кислоты.
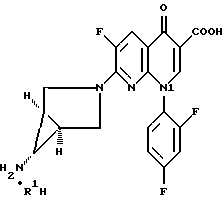
где R1H обозначает фармацевтически приемлемую кислоту, выбранную из группы, включающей R4SO3H, R4PO3H и YH, где R4 выбран из (C1-C6)алкила и необязательно замещенного фенила или нафтила, где заместителем является (C1-C6)алкил; и Y выбран из Cl, SO4, HSO4, NO3, HPO3H или PO4.
Антибактериальная активность вышеуказанного нафтиридонового антибиотика описана в пат. США 5164402 и 5229396, выданных 17.11.92 и 20.07.93 соответственно, причем изложенное в них полностью входит в данное описание в качестве ссылки. Настоящая заявка является продолжением вышеуказанных патентов.
Описание изобретения
В первом осуществлении настоящее изобретение относится к способу получения соединения формулы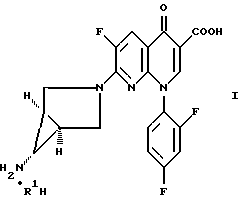
где R1H обозначает фармацевтически приемлемую кислоту, выбранную из группы, включающей R4SO3H и R4PO3H и YH, где R4 выбран из (C1-C6)алкила и необязательно замещенного фенила или нафтила, где заместителем является (C1-C6)алкил; и Y выбран из Cl, SO4, HSO4, NO3, HPO3H или PO4, который включает обработку соединения формулы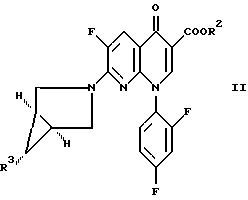
где R2 обозначает (C1-C6)алкил, арил (C1-C6)алкил или водород и R3 обозначает NO2 или NH2;
а) когда R3 является NH2, то соединением формулы R1H, указанным выше;
б) когда R3 является NO2, то восстановителем в присутствии соединения формулы R1H, где R1H определен выше.
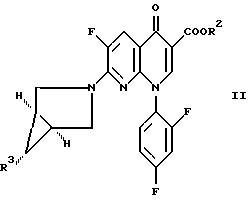
Изобретение также относится к способу получения соединения формулы II, где R3 обозначает NH2 и R2 определен выше, путем обработки соединения формулы II, где R3 обозначает NO2, восстановителем в присутствии соединения формулы R1H, где R1H определен выше.
В другом исполнении настоящее изобретение относится к способу получения соединения формулы II, где R3 обозначает NO2, включающему взаимодействие соединения формулы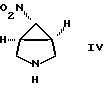
с соединением формулы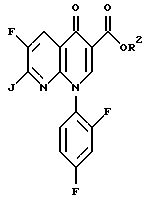
где R2 определен выше и J представляет собой подходящую уходящую группу.
В соответствии с другим осуществлением изобретения соединение формулы IV получают обработкой N-деалкилирующим агентом соединения формулы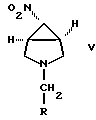
где R выбран из (C1-C6)алкила или (C6-C10)арила, где указанная арильная группа может быть замещена, необязательно, одним или более заместителями, независимо выбранными из галогена, нитро, (C1-C6)алкила, (C1-C6)алкокси, амино или трифторметила. Предпочтительно, R представляет собой фенил или водород. Соединение формулы V может быть получено обработкой восстановителем соединения формулы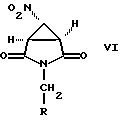
где R определен выше. Соединение формулы VI получают взаимодействием соединения формулы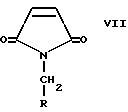
где R определен выше, с соединением формулы X-XH2-NO2, где X является подходящей уходящей группой, в присутствии основания. Предпочтительным основанием является 1,2-диметил-1,4,5,6-тетрагидропиримидин.
Еще одно осуществление изобретения относится к способу получения соединения формулы I, включающему стадии:
а) взаимодействия соединения формулы VII с соединением формулы X-CH2-NO2, где X является уходящей группой, в присутствии основания с образованием соединения формулы VI, которое затем обрабатывают восстановителем с образованием соединения формулы V;
б) обработки соединения формулы V деалкилирующим агентом с образованием соединения формулы IV;
в) взаимодействия соединения формулы IV соединением формулы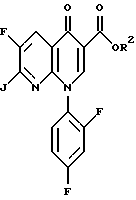
где R2 определен выше и J представляет собой подходящую уходящую группу, с образованием соединения формулы II, где R - NO2; и
г) обработки соединения формулы II, где R представляет NO2, восстанавливающим агентом, содержащим водород, в присутствии катализатора или металлом и кислотой формулы R1H, указанной выше, с образованием:
I) соединения формулы I, когда гидрирование осуществляют водородом в присутствии кислоты R1H, указанной выше, или когда R1H представляет собой соединение формулы YH или R4SO3H, где Y и R4 указаны выше; или
II) соединения формулы II, где R3 представляет NH2, с последующей обработкой указанного соединения формулы R1H, которое может быть таким же или отличаться от R1H со стадии восстановления, или соединением формулы R4CO2H, где R4 определен выше, с образованием соединения формулы I.
В другом осуществлении изобретение относится к соединению формулы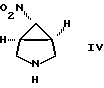
Еще одно осуществление изобретения относится к соединению формулы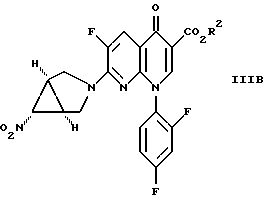
где R2 определен выше.
Другое осуществление изобретения относится к соединению формулы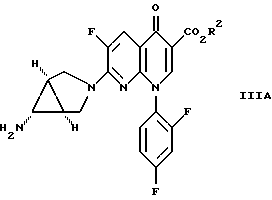
где R2 определен выше.
Используемый здесь термин "галоген" относится к фтору, хлору, брому или иоду, как приемлемо.
Используемый здесь термин "алкил" включает прямые, и, когда содержит более двух атомов углерода, разветвленные углеводородные цепи и углеводородные циклы, и сочетание прямых или разветвленных углеводородных цепей и углеводородных циклов.
Подробное описание изобретения
Способы по настоящему изобретению и получение соединений по настоящему изобретению проиллюстрированы нижеследующей схемой реакций. В схеме реакции и последующем обсуждении заместители R, R1H, R2, R3 и X определены ранее, если иное не указано особо.
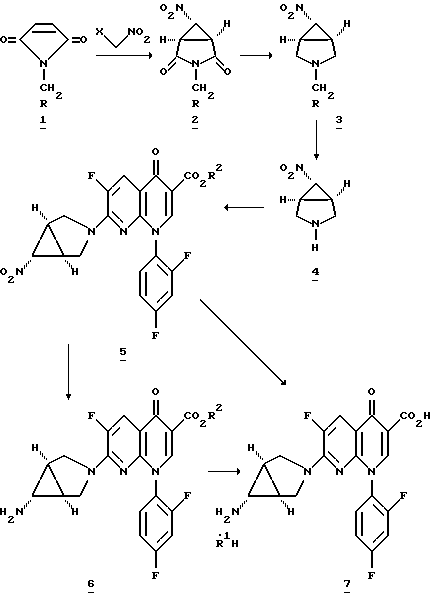
Вышеприведенная схема реакций иллюстрирует получение солей нафтиридонового антибиотика формулы I, новых промежуточных продуктов, используемых в указанных синтезах, и способы получения указанных промежуточных продуктов.
Согласно приведенной схеме, взаимодействие соединения 1 с соединением формулы X-CH2-NO2, где X обозначает подходящую уходящую группу, такую как хлор и бром, в присутствии основания, приводит к соответствующему соединению 2. Обычно эту реакцию проводят в инертном полярном апротонном растворителе, таком как диметилформамид (ДМФ), диметилсульфоксид (ДМСО) или диметилацетамид (ДМАА), в эфирном растворителе, таком как диэтиловый эфир, глим, диглим, диоксан или тетрагидрофуран (ТГФ), или в ароматическом растворителе, таком как необязательно хлорированном бензоле или толуоле. Предпочтительнее использовать толуол. Подходящая температура реакции находится в области от около -78oC до около 80oC, предпочтительно от около 0oC до около -20oC. Предпочтительно добавлять основание последним. Примеры подходящих оснований включают карбонатные основания, такие как карбонат натрия или калия, фосфоринамидные основания, такие как 2-трет-бутилимино-2-диэтиламино-1,3-диметилпергидро-1,3,2-диазафосфорин, и аминовые основания, такие как триэтиламин, гуанидин, диизопропилэтиламин, тетраметилгуанидин, 1,8-диазабицикло[5,4,0]ундец-7-ен (ДБУ), 1,5-диазацикло-[4,3,0]нон-5-ен (ДБН) и 1,2-диметил-1,4,5,6-тетрагидропиримидин. Преимущественно используются аминовые основания. Предпочтительным является 1,2-диметил-1,4,5,6-тетрагидропиримидин.
Восстановление соединения формулы 2 в инертном эфирном растворителе дает соответствующее соединение 3. Подходящие восстанавливающие агенты включают боран, боргидрид натрия и трифторид бора, эфиратные комплексы. Используемые при восстановлении инертные эфирные растворители включают глим, диглим, диизопропиловый эфир, диметилсульфид, ДМСО, диэтиловый эфир и ТГФ. Предпочтительным восстановителем является боран, а предпочтительными растворителями - ТГФ или диэтиловый эфир. Обычно восстановление проводят при температуре в области от около 25oC до около 90oC. Предпочтительно проводить восстановление при температуре в области от около 25o до около 65oC, и наиболее предпочтительно в области от около 25o до около 45oC в ТГФ. Этот способ описан в пат. США 5256791, включенном в данное изобретение в качестве ссылки.
Соединение 3, где R обозначает (C1-C5)алкил или (C6-C10)арил, преобразуют в соединение 4 путем обработки соединения 3:
а) когда R является (C6-C10)арилом, водородом или (α-хлорэтил)хлорформиатом; или
б) когда R является (C1-C6)алкилом, (α-хлорэтил)хлормиатом.
Когда R представляет собой (C6-C10)арил, то удаление гидрированием RCH2-группы соединения 3 обычно проводят путем взаимодействия указанного соединения с газообразным водородом при давлении от около 10 до около 2000 psi, предпочтительно, от около 14 до около 60 psi в присутствии катализатора благородного металла, такого как палладий, платина или родий, или их солей. Предпочтительным является палладий, или гидроксид палладия, на угле. Температура может изменяться от около 20oC до около 80oC, предпочтительной является температура около 25oC. Растворителем обычно являются (C1-C6)алкиловый спирт, предпочтительно метанол.
Соединение 4 преобразуют в соединение 5 путем взаимодействия с соединением формулы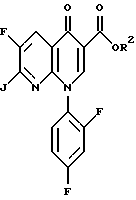
где R2 определен выше, а J является подходящей уходящей группой, такой как хлор и бром. Предпочтительной уходящей группой является хлор и бром, наиболее предпочтительной уходящей группой является хлор.
Реакцию можно проводить как в растворителе, так и без него. Если применяется растворитель, то он должен быть инертен в условиях реакции. Подходящими растворителями являются ацетонитрил, тетрагидрофуран, этанол, хлороформ, диметилсульфоксид, диметилформамид, пиридин, вода или их смеси.
Температура реакции обычно изменяется от около 20oC до около 150oC.
Удобным может быть проведение реакции в присутствии связывающего кислоту агента, такого как неорганическое или органическое основание, например, карбонат или бикарбонат щелочного или щелочноземельного металла, третичный амин, например, триэтиламин, пиридин или пиколин.
Соединение 5 преобразуют в соединение 6 обработкой металлом и кислотой формулы R1H, где R1H определена выше в присутствии водного апротонного растворителя, такого как ацетонитрил или ДМФ. Предпочтительным металлом является цинк. Подходящие кислоты включают неорганические кислоты, такие как хлористоводородная и серная кислоты, и органические кислоты, такие как сульфоновые кислоты, например, метан-, трифторметан- и п-толуолсульфоновые кислоты. Метансульфокислота или хлористоводородная кислота являются предпочтительными. Реакцию обычно проводят в водных (C1-C6)алкиловых спиртах, таких как этанол, метанол, 1-пропанол и 2-пропанол, предпочтительно, этанол, при температуре от около 0oC до около 80o, предпочтительно, при температуре около 25oC.
Альтернативно, соединение 5 может быть преобразовано в 6 путем взаимодействия с водородом в присутствии никеля Ренея или катализатора благородного металла. Никель Ренея является предпочтительным катализатором.
Гидрирование обычно проводится в смешанном водном растворителе. Подходящие растворители включают (C1-C6)алкиловые спирты, такие как этанол, метанол, 1-пропанол и 2-пропанол, и смешивающиеся с водой апротонные растворители, такие как ДМФ, ТГФ, диметилацетамид, диоксан и (C1-C6)алкиловые эфиры. Используемое давление водорода находится в области от около 14 до около 100 psi, предпочтительно, в области от около 40 до около 60 psi, и температура находится в области от около 15oC до около 80oC, предпочтительно, от около 20oC до около 30oC.
Соединение 6 преобразуют в соединение 7 путем взаимодействия с соединением формулы R1H, определенной выше, в водной среде.
Альтернативно, соединение 5 может быть прямо преобразовано в соединение 7 обработкой металлом и кислотой формулы R1H, такой, как определено выше, в водной среде. Предпочтительным металлом является цинк, а предпочтительной кислотой является метансульфокислота.
Фармацевтически приемлемые соли прибавления кислот, в которых кислотой является соединение формулы R4CO2H или R1H, где R4 и R1H определены выше, получают обычным способом обработкой раствора или суспензии соединения 1 в форме свободного основания примерно одним химическим эквивалентом фармацевтически приемлемой кислоты. Для выделения солей применяются обычные способы концентрирования и перекристаллизации. Примерами подходящих кислот являются уксусная, молочная, янтарная, малеиновая, винная, лимонная, глюконовая, аскорбиновая, бензойная, метансульфоновая, п-толуолсульфоновая, коричная, фумаровая, фосфоновая, хлористоводородная, бромистоводородная, иодистоводородная, сульфаминовая и сульфоновая кислоты.
Антибактериальные соединения формулы I и родственные антибиотики - производные азабициклонафтиридонкарбоновой кислоты, которые могут быть синтезированы с использованием способов и промежуточных продуктов по данному изобретению, применяются при лечении бактериальных инфекций у животных и человека. Они применяются при обработке широкого спектра бактериальных инфекций, в частности, грамположительных штаммов бактерий.
Соединения формулы I могут вводиться самостоятельно, но обычно вводятся в смеси с фармацевтическим носителем, выбранным с учетом предполагаемого пути введения и обычной фармацевтической практики. Например, они могут вводиться перорально или в виде таблеток, содержащих такие добавки, как крахмал или лактоза, или в капсулах как самостоятельно, так и в смеси с добавками, или в виде эликсиров или суспензий, содержащих отдушки и красители. В случае животных, удобно включать их в корм животных или в питьевую воду в концентрациях от около 5 до около 5000 миллионных частей, предпочтительно от около 25 до около 500 миллионных частей. Они могут вводиться парентально, например, внутримышечно, внутривенно или подкожно. Для парентального введения лучше всего использовать соединения в виде стерильного водного раствора, который может содержать другие растворители, например достаточные для придания изотоничности количества растворов соли и глюкозы. В случае животных, соединения формулы I могут вводиться внутримышечно или подкожно на уровне дозировки от около 0,1 до около 50 мг/кг/день, преимущественно от около 0,2 до около 10 мг/кг/день в виде разовой дневной дозы или до 3 раздельных доз.
Изобретение также относится к фармацевтическим композициям, содержащим антибактериально эффективное количество соединения формулы I вместе с фармацевтически приемлемым растворителем или носителем.
Соединения по изобретению могут вводиться для лечения бактериальных инфекций у людей пероральным либо парентальным путями, и могут вводиться перорально на уровне дозировки от около 0,1 до около 500 мг/кг/день, удобным образом, 0,5-50 мг/кг/день, в виде разовой дозы или до 3 раздельных доз. Для внутримышечного или внутривенного введения уровни дозировки составляют от около 0,1 - 200 мг/кг/день, удобным образом, 0,5-50 мг/кг/день. Если внутримышечное введение может осуществляться как однократной инъекцией, так и в виде вплоть до 3 раздельных доз, то внутривенное введение может включать использование капельницы. В зависимости от веса и состояния пациента, подвергаемого лечению, и выбранного конкретного пути введения, обязательно будут проведены изменения, как понятно специалисту в данной области.
Антибактериальная активность соединений по изобретению показана исследованиями в соответствии с репликаторной методикой Стирса, которая является стандартным бактериальным исследованием in vitro, описанным E. Steers et al. , Antibiotocs and Chemotherapy, 9, 307 (1959).
Следующие примеры иллюстрируют способы и соединения по настоящему изобретению. Следует учесть, однако, что изобретение не ограничивается частными деталями этих примеров.
Пример 1
(1a,5a,6a)-3-Бензил-6-нитро-2,4-диоксо-3-азабицикло-[3,1,0]-гексан
22-литровый сосуд, снабженный верхней мешалкой, термометром, капельной воронкой, охлаждающей баней, прямым холодильником, барботером и вводом азота, продувают азотом. В продутый азотом сосуд загружают N-бензилмалеимид (500 г, 2,67 моль), толуол (12 л), бромнитрометан (751, 90%-ный, 4,83 моль) и порошок молекулярных сит (2020 г) и перемешивают при от около 10 до около 15oC. Взвесь в течение 3 часов обрабатывают, прибавляя по каплям 1,2-диметил-1,4,5,6-тетрагидропиримидин (ДТГП) (616 г., 5,49 моль). Прибавление ДТГП приводит к образованию большого количества смолы, собранной молекулярными ситами. Реакционную смесь нагревают до около 25oC и перемешивают в течение 60-90 минут. Молекулярные сита собирают на большой воронке Бюхнера и дважды промывают 2 л толуола. Фильтрат трижды промывают 750 мл 2 М HCl. В 22-литровый сосуд, снабженный обратным холодильником, загружают фильтрат и Darco (торговая марка) КВВ (50 г). Смесь нагревают при 60-70oC и перемешивают 1 час. Затем смесь охлаждают до около 25oC, фильтруют через покрытую Celite (торговая марка) фильтром воронку Бюхнера, и осадок промывают толуолом дважды по 500 мл. Обработанный углем фильтрат упаривают в вакууме в 12-литровой круглодонной колбе, снабженной верхней мешалкой, термометром, вакуумным штуцером, перегонной насадкой, прямым холодильником и 22-литровым приемником. Упаривание в вакууме считают законченным, когда остается от около 2 до около 3 литров концентрата. Концентрированный раствор медленно обрабатывают 4 литрами 2-пропанола. Азеотропную перегонку (25oC) продолжают до тех пор, пока не удалят весь толуол (что выражается в подъеме температуры кипения на 10oC). Желто-оранжевое твердое вещество собирают на фильтровальной воронке, промывают дважды 500 мл 2-пропанола и высушивают в вакууме при 40oC. Выход 175,38 г (26,7%), т. пл. 108-112oC. Чистоту определяют с помощью ВДЖХ по сравнению с чистым образцом (89-96%). 1H ЯМР (CDCl3) δ, 7,3 (с, 5H), 4,55 (с, 2H), 4,45 (с, 1H), 3,36 (с, 2H).
Пример 2
Гидрохлорид (1a,5a,6a)-6-нитро-3-азабицикло[3,1,0]гексана
В 250-миллилитровую 3-горлую круглодонную колбу, снабженную прямым холодильником, верхней мешалкой и капельной воронкой, загружают 1,2-дихлорэтан (115 мл), (1a,5a,6a)-3-бензил-6-нитро-3-азабицикло-[3,1,0]-гексан (полученный из соединения, указанного в заголовке примера 1 способом по примеру 2 патента США 5256791 (включен здесь в качестве ссылки в данное описание) (25,1 г, 115 ммоль). Раствор охлаждают до от около 0 до около 5oC и в течение 20 минут прибавляют по каплям α- хлорэтилхлорформиат (АХЭХФ) (25,3 г. , 172 ммоль). Реакционную смесь нагревают до от около 50 до около 55oC и выдерживают около от 2 до 3 часов (о завершении реакции судят по данным ТСХ). Растворитель и избыток АХЭХФ отгоняют на роторном испарителе. Полученный черный остаток растворяют в метаноле (100 мл) и нагревают до от около 55 до около 60oC в течение 3 часов. Полученную взвесь охлаждают до комнатной температуры и гранулируют в течение 18 часов. Взвесь затем обрабатывают концентрированной хлористоводородной кислотой (10 мл, 115 ммоль) и перемешивают 1,5 часа. Продукт выделяют на фильтре с отсосом. Лепешку на фильтре промывают хлороформом (25 мл) и высушивают в вакууме. Выход: 9,99 г, 60 ммоль (53%), т.пл. 170-180oC (разл.). 1H ЯМР (ДМСО-d6) 9,8 (ушир. с, 2H), 4,9 (с, 1H), 3,5 (м, 4H), 2,9 (с, 2H).
Пример 3
Этиловый эфир 7-([1a, 5a, 6a]-6-нитро-3-азабицикло[3,1,0]гекс-3-ил)-6- фтор-1-(2,4-дифторфенил)-1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновой кислоты
В 500-миллилитровую 3-горлую круглодонную колбу, снабженную верхней мешалкой, прямым холодильником и термометром, загружают ацетонитрил (190 мл), этиловый эфир 7-хлор-6-фтор-1-(2,4-дифторфенил)-1,4-дигидро- 4-оксо-1,8-нафтиридинкарбоновой кислоты (19,07 г, 50 ммоль) соединения, указанного в заголовке примера 2 (9,88 г, 60 ммоль), триэтиламин (15,3 г, 151 ммоль). Смесь нагревают до кипения (82oC), перемешивают 6,5 часов и проверяют полноту протекания реакции с помощью ТСХ (этилацетат: гексан 3:2, УФ). Полученную взвесь охлаждают до комнатной температуры и обрабатывают водой (115 мл). Взвесь затем гранулируют при от около 0 до около 5oC в течение 1 часа. Продукт собирают на воронке для фильтрования в виде белого твердого вещества и промывают CH3CN:водой 1:1 (50 мл). Продукт высушивают в вакууме при 40oC. Выход: 21,17 г, 44,6 ммоль (89,2%), т.пл. 245-250oC. 1H ЯМР (CDCl3) δ 8,4 (с, 1H), 8,1 (д, 1H), 7,4 (м, 2H), 7,05 (м, 1H), 4,35 (кв, 2H), 4,1 (с, 1H), 3,95 (м, 2H), 3,65 (м, 2H), 2,75 (м, 2H), 1,35 (т, 3H).
Пример 4
Этиловый эфир 7-([1a, 5a, 6a]-6-амино-3-азабицикло[3,1,0]гекс-3-ил)-6- фтор-1-(2,4-дифторфенил)-1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновой кислоты
А. В 250-миллитровую 3-горлую круглодонную колбу, снабженную прямым холодильником, термометром и верхней мешалкой, загружают соединение, указанное в заголовке примера 3 (10,0 г, 21,1 ммоль), ацетонитрил (50 мл), воду (50 мл) и цинковую пыль (6,9 г, 105,6 ммоль). Серую взвесь обрабатывают метансульфоновой кислотой (70%, 25,5 мл, 241 ммоль), в результате чего температура поднимается до 40oC. Оранжево-желтую реакционную смесь нагревают до 50-55oC и выдерживают 3 часа (окончание реакции по ВДЖХ). Смесь охлаждают до комнатной температуры, обрабатывают водой (100 мл) и Celite (торговая марка) (1 г) и перемешивают 15 минут. Взвесь фильтруют через покрытую Celite воронку, получая темно-янтарный раствор. Раствор подщелачивают 50%-ным водным NaOH до pH 10,1. Оранжево-янтарную взвесь обрабатывают дихлорметаном (250 мл) и фильтруют для удаления нерастворимых частиц. Органический слой упаривают досуха, получают сырой продукт (2,57 г, 27,4%). Образец сырого продукта (0,55 г) хроматографируют на колонке с силикагелем. Элюируют семь раз 50 мл этилацетата и тринадцать раз 50 мл метанола. Последние пять фракций объединяют и концентрируют с получением чистого указанного в заголовке соединения (0,14 г, 27,2% степень извлечения с колонки). Общий выход 5,73%. Продукт характеризуют сравнительной ВДЖХ (в сравнении с чистым образцом) и FAB МС [M+H] + = 445. 1H ЯМР (CDCl3) δ 8,35 (с, 1H), 7,8 (д, 1H), 7,35 (м, 1H), 7,05 (м, 2H), 4,35 (кв, 2H), 3,6 (ушир. с, 2H), 3,5 (ушир. с, 2H), 2,05 (с, 1H), 1,57 (с, 2H), 1,51 (с, 2H), 1,39 (т, 3H).
Б. В 600-миллилитровый аппарат Parr (торговая марка), снабженный газовым расходомером Preteric Ltd. Pressflow (торговая марка) Gas Controller (Model 1502), загружают соединение, указанное в заголовке примера 3 (2,04 г, 4,3 ммоль), никель Ренея [A-4000, Activated Metals and Chemicals Inc., Seviorille, TN] (1,44 г, вес влажного) N,N-диметилформамид (70 мл) и воду (20 мл). Аппарат запечатывают, дважды продувают азотом (35 psi), наполняют водородом (50 psi) и нагревают в течение 45 минут до от около 40 до около 45oC. Давление затем поднимают примерно до около 57 psi и выдерживают в течение 24 часов. Реакционную смесь охлаждают до комнатной температуры, продувают азотом и проверяют на полноту завершения реакции по ТСХ (89 CHCl3 : 10 метанол : 1 NH4OH). Катализатор собирают на воронку для фильтрования, покрытую Celite, и промывают водой (25 мл). К фильтрату прибавляют воду (40 мл) и трижды экстрагируют 100 мл этилацетата. Этилацетатный слой затем концентрируют до 100 мл и экстрагируют водой (100 мл) для удаления остаточного ДМФ. Этилацетатный слой упаривают досуха на роторном испарителе. Выход сырого продукта: 1,36 г (71,1%). Анализ ВДЖХ на чистоту (76,5%). Выход чистого вещества (54%). Продукт был охарактеризован ВДЖХ (в сравнении с чистым образцом). Данные 1H ЯМР аналогичны полученным для продукта в разделе А выше.
В. Способ раздела Б повторяют с загрузкой, включающей соединение, указанное в заголовке примера 3 (10,0 г, 21,1 ммоль), 4,3 г никель Ренея (4,3 г влажного), тетрагидрофуран (ТГФ) (180 мл) и воду (40 мл). Аппарат запечатывают и дважды продувают азотом (35 psi). Аппарат затем наполняют водородом (50 psi) и перемешивают при 25-29oC 2,5 часа (до поглощения водорода). Реактор продувают азотом и проверяют полноту завершения реакции по ТСХ (89 CHCl3 : 10 метанол : 1 NH4OH.
Катализатор отфильтровывают через покрытую Celite воронку. Лепешку на фильтре дважды промывают ТГФ (20 мл). ТГФ отгоняют на роторном испарителе, получая светло-желтую взвесь. К взвеси добавляют этанол (25 мл), затем взвесь гранулируют при от около 20 до около 25oC в течение 30 минут. Продукт собирают на воронке Бюхнера. Выход: 7,56 г (80,6%). Анализ ВДЖХ на чистоту (97,1%). Выход чистого (78,3%). Продукт охарактеризован ВДЖХ (в сравнении с чистым образцом). Данные 1H ЯМР соответствуют полученным для продукта в разделе А. Была собрана вторая порция (выход по массе 7,5%), однако чистота его ниже, как показано 1H ЯМР.
Пример 5
Метансульфонат 7-([1a, 5a, 6a]-6-амино-3-азабицикло[3,1,0]гекс-3-ил)-6- фтор-1-(2,4-дифторфенил)-1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновой кислоты
А. В 200-миллилитровую 3-горлую круглодонную колбу, снабженную прямым холодильником, термометром и верхней мешалкой, загружают соединение, указанное в заголовке примера 4 (1,54 г, 3,46 ммоль), и воду (25 мл). Белую взвесь обрабатывают 70%-ой водной метансульфоновой кислотой (5,25 г, 38,4 ммоль) и нагревают до от около 45 до около 50oC. Исходный продукт медленно переходит в раствор. Смесь перемешивают в течение 18 часов (завершение реакции по данным ТСХ). Смесь охлаждают до комнатной температуры и продукт выделяют путем фильтрации с отсасыванием. Выход: 1,48 г (83,5%). ВДЖХ % чистота (в сравнении с чистым образцом) 96,1%. 1H ЯМР (ДМСО-d6) δ 8,85 (с, 1H), 8,17 (ушир. м, 2H), 8,11 (д, 1H), 7,83 (м, 1H), 7,62 (м, 1H), 7,37 (м, 1H), 3,67 (ушир. с, 3H), 2,45 (с, 1H), 2,37 (с, 4H), 2,08 (с, 2H).
Б. В 100-миллилитровую 3-горлую круглодонную колбу, снабженную прямым холодильником, верхней мешалкой и капельной воронкой, загружают соединение, указанное в заголовке примера 3, 1,01 г (2,13 ммоль), цинк (0,70 г, 10,7 ммоль), ацетонитрил (20 мл) и воду (20 мл). Серую взвесь нагревают до около 50oC и обрабатывают 5 мл раствора 70%-ной водной метансульфоновой кислоты (3,3 г, 24 ммоль). Ход реакции периодически контролируют по ВДЖХ для завершения (23 часа). Реакционную смесь нагревают до от около 80 до около 85oC, затем обрабатывают дополнительным количеством 70%-ной водной метансульфоновой кислотой (2,6 г, 19 ммоль) для полноты гидролиза эфира (ВДЖХ). Реакционную смесь охлаждают до комнатной температуры и обрабатывают водой (250 мл) с получением желто-коричневой взвеси. Взвесь отфильтровывают, к фильтрату прибавляют 500 мл воды. Полученный раствор упаривают на роторном испарителе для удаления ацетонитрила. К концентрату добавляют 2-пропанол (50 мл), после чего упаривают досуха. Остаток обрабатывают водой (50 мл) и ацетоном (50 мл) с получением коричневой взвеси. Взвесь фильтруют для отделения нерастворимых частиц. Фильтрат охлаждают до 0-5oC для кристаллизации продукта. Получают указанное в заголовке соединение в виде желтых кристаллов (105 мг, выход 10,5%). ВДЖХ (20% CH3CN : 80% pH2, 50 мМ H3PO4; 270 нм, 1,00 мл/мин; Zorbax (торговая марка) RX C18 5 мк 4,6 мм х 15 см) в сравнении со стандартным образцом указанного в заголовке соединения подтверждает структуру. Аналитическая ВЭЖХ с чистым образцом указанного в заголовке соединения показывает, что продукт в данном примере является соединением, указанным в заголовке. Данные 1H ЯМР продукта те же, что и для продукта из раздела А выше.
Описывается способ получения солей нафтиридонкарбоновой кислоты формулы I, где R1Н обозначает фармацевтически приемлемую кислоту, выбранную из группы, включающей R4SO3H, R4PO3H и YH, где R4 выбран из (С1 - C6)алкила и необязательно замещенного фенила или нафтила, где заместителем является (С1 - C6)алкил, и Y выбран из Cl, NO3, отличающийся тем, что соединение формулы II, где R2 является (С1 - C6)алкилом, арил (С1 - C6)алкилом или водородом и R3 обозначает NO2, подвергают взаимодействию с восстанавливающим агентом, необязательно, в присутствии соединения формулы R1H, где R1 определен выше, с последующей, если необходимо, обработкой полученного соединения соединением формулы R1H, которое может быть таким же или отличным от R1H на стадии восстановления. Описываются также промежуточные продукты соединения, проявляющие антибактериальную активность. Технический результат - упрощение процесса. 4 с. и 30 з.п. ф-лы.
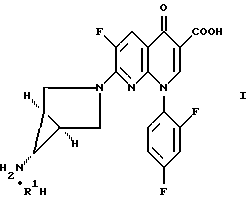
где R1H обозначает фармацевтически приемлемую кислоту, выбранную из группы, включающей R4SO3H, R4PO3H и YH, где R4 выбран из (C1-C6)алкила и необязательно замещенного фенила или нафтила, где заместителем является (C1-C6)алкил, и Y выбран из Cl, NO3,
отличающийся тем, что соединение формулы II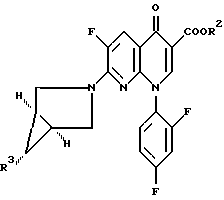
где R2 является (C1-C6)алкилом, арил(C1-C6)алкилом или водородом;
R3 обозначает NO2,
подвергают взаимодействию с восстанавливающим агентом, необязательно, в присутствии соединения формулы R1H, где R1 определен выше, с последующей, если необходимо, обработкой полученного соединения соединением формулы R1H, которое может быть таким же или отличным от R1H на стадии восстановления.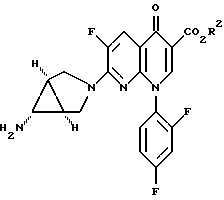
где R2 имеет значения, определенные в п.1,
и затем осуществляют обработку указанного соединения соединением формулы R1H, которое может быть таким же или отличным от R1H на стадии восстановления.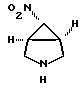
с соединением формулы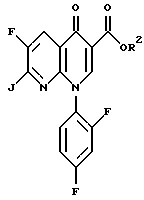
где R2 определен выше и J представляет собой уходящую группу.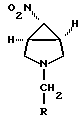
где R выбран из (C1-C6)алкила или (C6-C10)арила, где указанный арил может быть замещен необязательно одним или несколькими заместителями, независимо выбранными из галогена, нитро, (C1-C6)алкила, (C1-C6)алкокси, амино и трифторметила.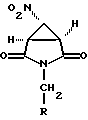
где R имеет вышеуказанные значения.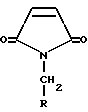
где R имеет вышеуказанные значения,
с соединением формулы
X-CH2-NO2,
где X обозначает уходящую группу,
в присутствии 2-диметил-1,4,5,6-тетрагидропиримидина.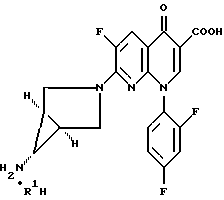
где R1H обозначает фармацевтически приемлемую кислоту, выбранную из сульфоновой кислоты R4SO3H, R4PO3H и YH, где R4 выбран из (C1-C6)алкила и необязательно замещенного фенила или нафтила, где заместителем является (C1-C6)алкил;
Y выбран из Cl, NO3, отличающийся тем, что он включает стадии: a) взаимодействия соединения формулы VII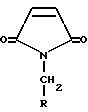
где R выбран из (C1-C6)алкила или (C6-C10)арила, где указанный арил может быть необязательно замещен одним или несколькими заместителями, независимо выбранными из галогена, нитро, (C1-C6)алкила, (C1-C6)алкокси, амино и трифторметила,
с соединением формулы X-CH2-NO2, где X является уходящей группой, предпочтительно хлором или бромом, в присутствии основания с образованием соединения формулы VI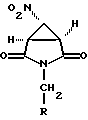
в которой R имеет вышеуказанные значения, предпочтительно бензил или метил,
и обработки полученного соединения формулы VII восстанавливающим агентом с получением соединения формулы V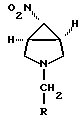
где R имеет вышеуказанные значения, предпочтительно бензил или метил;
b) обработки соединения формулы V деалкилирующим агентом с образованием [1а,5а,6а]-6-нитро-3-азабицикло[3, 1, 0]-гексана формулы IV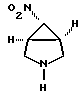
c) взаимодействия соединения формулы IV с соединением формулы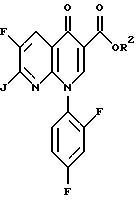
где R2 является (C1-C6)алкилом, арил(C1-C6)алкилом или водородом и V является подходящей уходящей группой, такой, как хлор или бром,
в присутствии основания с получением соединения формулы IIIB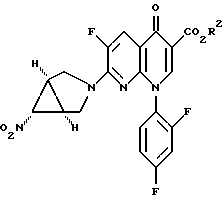
в которой R2 имеет вышеуказанные значения,
и d) I) обработки полученного соединения формулы IIIB восстанавливающим агентом - металлом и кислотой формулы R1H, определенной выше, в присутствии водного апротонного растворителя или водородом в присутствии никеля Ренея или катализатора - благородного металла с получением соединения формулы IIIA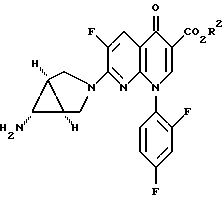
которое затем преобразуют в соединение формулы I взаимодействием с кислотой формулы R1H, определенной выше, в водной среде,
II) или, альтернативно, соединение формулы IIIB прямо преобразуют в соединение формулы I обработкой металлом и кислотой формулы R1H, определенной выше.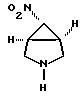
34. Сложный эфир 7-([1а,5а,6а]-6-нитро-3-азабицикло-[3,1,0]гекс-3-ил)-6-фтор-1-(2,4-дифторфенил)-1,4-дигидро-4-оксо-1,8-нафтиридин-3-карбоновой кислоты формулы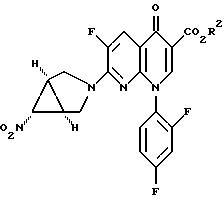
III B
где R2 является (C1-C6)алкилом, арил(C1-C6)алкилом или водородом.
| US 5164402 A, 17.11.92 | |||
| US 5229396 A, 20.07.93. |
