Изобретение относится к новым бензоконденсированным соединениям, к содержащим их фармацевтическим композициям, к способу их получения, а также к способу получения промежуточного продукта. Соединения по изобретению подавляют эффекты лейкотриена B4 (ЛТВ4) и поэтому могут быть использованы при лечении болезней, вызванных ЛТВ4, например, воспалительных заболеваний, включая ревматоидный артрит, остеоартрит, воспалительные заболевания пищеварительного тракта, псориаз и другие кожные болезни, например, экзему, эритему, зуд, угри, внезапные приступы (припадки) и другие формы реперфузионных нарушений, отторжение трансплантата, аутоиммунные заболевания, астму и другие состояния, когда наблюдается заметная нейрофильная инфильтрация.
Антагонисты ЛТВ4 описаны в европейских патентных публикациях N 276064 и 292977 и относятся к дифениловым эфирам, бензофенонам и другим соединениям, содержащим две фенильные группы, и производным 7-(3-алкокси-4-алканоилфеноксил)алкоксибензопирана, соответственно.
В соответствии с изобретением было обнаружено, что соединения формулы I обладают антагонистическими свойствами в отношении ЛТВ4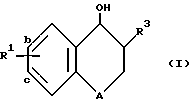 ,
,
где
A обозначает атом кислорода или группу -CH2-;
R1 обозначает заместитель в положении "b" или "c" и имеет формулу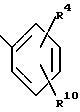 ,
,
где
R4 представляет карбоксигруппу, тетразолил или оксазолил;
R10 обозначает атом водорода или один или любые два заместителя, выбранные из атома фтора или хлора, C1-C6-алкила, C1-C6-алкоксигруппы или C1-C4-перфторалкила;
R3 является группой -(CH2)q-CHR11R12 или -(CH2)q-R12,
где
q равно 0, 1, 2 или 3;
R11 представляет атом водорода, C1-C6-алкил или R8 - замещенный фенил, где R8 обозначает атом водорода или один, или любые два заместителя, выбранные из атома фтора или хлора, C1-C6-алкила, C1C6-алкоксигруппы или C1-C4-перфторалкила;
R12 представляет атом водорода, фенил или нафтил, где указанный фенил может быть замещен фенилом, R9 или R9 - замещенным фенилом, где R9 имеет значения, указанные для R8 выше,
или их соли или сложные эфиры таких соединений общей формулы I, которые содержат карбоксигруппу, при этом эфирные группы выбраны из групп, содержащих C1-C6-алкил, фенил-C1-C6-алкил, C3-C7-циклоалкил, либо фенил или бензил, замещенные атомом фтора или хлора, C1-C6-алкилом или C1-C6-алкоксигруппой.
Ингибирование рецепторного связывания ЛТВ4 можно осуществлять путем введения субъекту в случае необходимости такого ингибирования соединения формулы I, которое определено выше.
Изобретение, кроме того, включает способ получения полупродуктов формулы II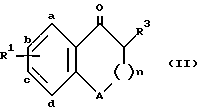 ,
,
где
A обозначает атом кислорода или группу -CH2-;
R1 обозначает заместитель в положении "b" или "c" и имеет формулу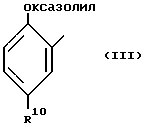 ,
,
где
R10 обозначает атом водорода или один, или любые два заместителя, выбранные из атома фтора или хлора, C1-C6-алкила, C1-C6-алкоксигруппы или C1-C4-перфторалкила;
R3 является группой -(CH2)q-CHR11R12 или -(CH2)q-R12,
где
q равно 0, 1, 2 или 3;
R11 представляет атом водорода, C1-C6-алкил или R8 - замещенный фенил, где R8 обозначает атом водорода или один, или любые два заместителя, выбранные из атома фтора или хлора, C1-C6-алкила, C1-C6-алкоксигруппы или C1-C4-перфторалкила;
R12 представляет фенил или нафтил, где указанный фенил может быть замещен фенилом, R9 или R9-замещенным фенилом, где R9 имеет значения, указанные для R8 выше,
или их солей,
путем взаимодействия соединения формулы IV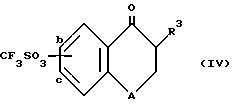 ,
,
где
R3 и A указаны выше, а группа CF3SO3 находится в положении "b" или "c", с соединением формулы V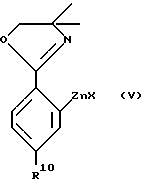 ,
,
где
X является атомом хлора, брома или йода, а R10 указан выше,
которое получают путем взаимодействия соединения формулы VI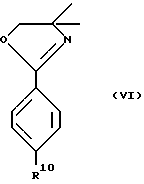 ,
,
с н-бутиллитием и затем с ZnX2, где X указан выше.
Термин "C1-C6-алкил" всякий раз, когда он используется здесь в описании, например при определениях от R1 до R14, означает насыщенные одновалентные прямые или разветвленные алифатические углеводородные радикалы, имеющие от одного до шести углеродных атомов, например метил, этил, пропил, трет-бутил, гексил и т.д.
Подобно термины C3-C7 - циклоалкил и C3-C8 - циклоалкил означают циклоалкильную группу, имеющую от 3 до 7 или 8 атомов углерода, например, соответственно циклопропил, циклооктил и т.д.
Когда в соединении формулы I A является кислородом, соединение может быть описано или как 3,4-дигидробензопиран, или как хроман.
Соединения изобретения имеют два асимметричных углеродных атома, обозначенных звездочками в следующей формуле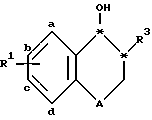 .
.
Стереоизомеры можно обозначить ссылаясь на R и S в соответствии со стандартной номенклатурой. Когда здесь ссылаются на S, R или R, S, это означает одно энантиомерно чистое соединение, тогда как S*, R* и R*, S* означают рацемические смеси и оптические изомеры формулы I.
В соответствии с конкретным способом изобретения полупродукты вышеприведенной формулы II, где R1 является заместителем формулы III, получают путем взаимодействия соединения формулы IV, которое определено выше, с соединением формулы V, которое определено выше. Реакция обычно происходит в растворителе, таком как эфирный растворитель, например в тетрагидрофуране, диэтиловом эфире, этиленгликольдиметиловом эфире, 1,4-диоксане и предпочтительно тетрагидрофуране. Реакцию проводят в присутствии катализатора, в особенности палладиевого катализатора, который является источником палладия и обеспечивает в реакционных условиях палладий (Pdo), например, тетракистрифенилфосфинпалладия. Реакцию обычно проводят при температуре перегонки используемого растворителя, предпочтительно при 78oC. Время реакции обычно составляет от 1 до 24 ч, например около 3 ч.
Соединения формулы V получают in situ из соединения вышеприведенной формулы VI путем взаимодействия с н-бутиллитием или втор-бутиллитием в гексане при температурах ниже -78oC и затем с ZnX2, где X - йод, бром или хлор, обычно при температуре от 0 до 78oC в течение от 1 до 4 ч.
Кетоны формулы II, в которой A, R4 и R3 являются такими, как это определено при ссылке на формулу I, могут быть восстановлены до соответствующих гидроксильных соединений формулы I путем взаимодействия с боргидридом натрия. Обычно восстановление проводят в растворителе. Подходящими растворителями являются низшие спирты, имеющие от одного до шести углеродных атомов, смеси низших спиртов с органическими растворителями, например тетрагидрофураном или диоксаном, и смеси водосмешивающихся низших спиртов или других водосмешивающихся органических растворителей с водой. Растворителем предпочтительно является низший спирт, например метанол или этанол. Температура реакции обычно составляет от -78 до 100oC и обычно 0 до 25oC. В результате стадии восстановления получают стереоизомерную смесь соединений формулы I, имеющих следующие структуры: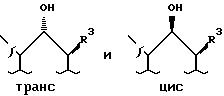 .
.
Эти цис- и транс-изомеры можно разделить обычной колоночной хроматографией.
Повторное растворение энантиомерной смеси, следующее после разделения цис- и трансизомеров, можно выполнить, способами, известными в данной области. По одному из способов соединение формулы 1, где R1 содержит карбоксильную группу (COOH), взаимодействует с хиральным основанием, например метилбензиламином в полярном растворителе, например эфире, с образованием диастереомерных солей, которые разделяют и затем превращают в оптически чистые кислоты путем обработки кислотой, например водным или метанольным хлористым водородом. По другому способу соединение формулы 1, где R1 содержит эфирную группу, взаимодействует с оптически активной кислотой, например, R-миндальной кислотой или N-трет-бутоксикарбонил-D-триптофаном с образованием диастереомерных эфиров, которые разделяют на оптически чистые эфиры, например хроматографией.
Удаление разделяющей эфирной группы и гидролиз эфирной группы карбоновой кислоты в R1 обычно проводят водным основанием, например гидроксидом щелочного металла, например гидроксидом натрия, при температурах от комнатной до температуры перегонки или кипения растворителя или используемой смеси растворителей. Реакцию можно проводить в присутствии сорастворителя, например метанола, этанола или тетрагидрофурана.
Соединения формулы 1, где R4 является оксазолилом, превращают в соответствующие соединения формулы 1, где R4 является карбокси, сначала путем взаимодействия с метилиодидом при возможном присутствии диметилсульфоксида и затем с основанием, например водным раствором гидроксида натрия.
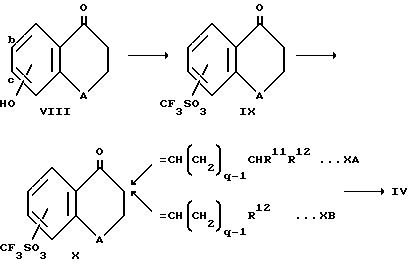
Соединения формулы IV, где R3 является (CH2)q CHR11R12 или (CH2)q R12, можно получить в соответствии с реакционной схемой 1 из соединения формулы VIII, где А является таким, как это определено при ссылке на формулу 1.
Соединение формулы VIII взаимодействует с трифторметансульфоновым ангидридом в подходящем растворителе, например метиленхлориде, в присутствии триэтиламина с образованием соединения формулы IX.
Группу R3, когда она определена как -(CH2)q CHR11R12 или -(CH2)q R12, можно ввести в соединение формулы IX двухстадийным способом, включающим взаимодействие с альдегидом формулы R11R12CH (CH2)q, CHO или R12 (CH2)q-1 CHO с образованием соединения соответственно формулы XA или XB и затем гидрирование. Реакцию с альдегидом проводят в присутствии пирролидинового катализатора или хлористоводородной кислоты в уксусной кислоте. Гидрирование проводят водородом в присутствии палладиевого катализатора обычным образом (схема 1).
 .
.
Соединения формулы VIII являются обычно коммерчески доступными. Если нет, то их можно получить способами, известными в предшествующей области. Например, соединения формулы VIII, где А является кислородом, можно получить из замещенного 2', 4' -дигидрокси-3-хлорпропиофенона (далее как соединение 1) путем циклизации с гидроксидом натрия. Соединение 1 можно получить из замещенного резорцина и 3-хлорпропионовой кислоты в присутствии кислоты, предпочтительно трифторметансульфоновой кислоты. Соединения формулы VIII, в которых А является серой, можно аналогично получить из замещенного 4'-гидрокси-2'-сульфгидрид-3-хлорпропиофенона, который можно получить из замещенного 3-гидрокситиофенола.
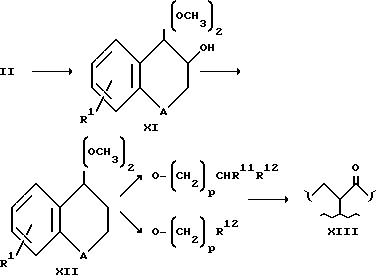
Группу 3, когда она определена как -O(CH2)p CHR11R12 или -O(CH2)R12, можно ввести в соединения формулы VIII по методике, обрисованной в общих чертах в схеме 11.
Соединения формулы XI можно получить из соединения формулы II, в которой R3 является водородом, путем их смешивания с 20% раствором гидроксида калия и добавления фенилдиацетоксииодида.
 .
.
Соединения формулы XI при смешивании с Br(CH2)pR12 или Br(CH2)pR12 образуют соединения формулы XII, которые превращают в соединения формулы XIII путем гидролиза с кислотой, такой как соляная кислота. Соединения формулы XIII при восстановлении образуют соединения формулы 1. Восстановление осуществляют обычным образом боргидридом натрия в спиртовом растворителе при окружающей температуре. Вышеприведенные соединения формулы IV можно превратить в соединения формулы 1, в которой R1 является таким, как это определено при ссылке на формулу 1, а R4 является карбокси в соответствии с реакционной схемой III.
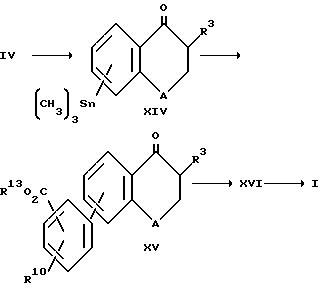 .
.
Соединение формулы XIV образуется путем взаимодействия соединения формулы IV с (CH3)3Sn Sn(CH3)3 и палладиевым катализатором, например тетракистрифенилфосфинпалладием (Pd(PPd3)4) или бисбензонитрилпалладийхлоридом, в присутствии фосфинового лиганда, например трифенилфосфина, в количестве от 0,5 до 5 молярных эквивалентов на моль используемой основы. Соединение формулы XIV превращают в соединение формулы XV путем взаимодействия с эфирозащищенным соединением формулы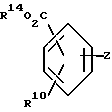 ,
,
в которой
R10 является таким, как это определено при ссылке на формулу 1, R14 является C1-C6 алкилом, фенилом или бензилом, а Z - йод, бром, или CF3SO3. Реакция сочетания происходит в присутствии палладиевого катализатора, например тетракистрифенилфосфинпалладия или бистрифенилфосфинхлорида палладия.
Эфиры кетона формулы XV сначала восстанавливают до соответствующих гидроксильных соединений XVI (формула не показана) и затем гидролизуют до соответствующей кислоты формулы 1. В реакции принимает участие боргидрид натрия, который описан ранее при ссылке на восстановление кетонов формулы II. Гидролиз до образования кислоты можно проводить водным раствором основания, таким как гидроксид щелочного металла, например гидроксидом натрия, при необязательном присутствии сорастворителя, например, метанола или этанола при температурах от комнатной до температуры перегонки или кипения используемого растворителя.
Соединения формулы 1, в которой R1 является: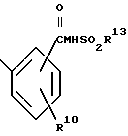 ,
,
где
R10 и R13 являются такими, как это определено выше при ссылке на формулу 1, могут быть получены путем взаимодействия соединений формулы 1, в которой R1 является: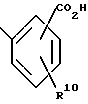 ,
,
с сульфонамидом формулы R13SO2NH2 в присутствии связующего вещества, например 1,3-дициклогексилкабодиимида или 1-/3-(диметиламино)пропил/-3-этилкарбодиимида, и в присутствии органического основания, например пиридина, диметиламинопиридина, триэтиламина, диизопропилэтиламино или диазобицикло (5,4,0) ундена-7. Реакцию проводят в растворителе, например тетрагидрофуране, диэтиловом эфире, толуоле и хлорбензоле при температуре от комнатной до температуры кипения используемого в реакции растворителя.
Соединения формулы 1, в которой R4 является тетразолилом, могут быть получены из соответствующих эфирных соединений формулы 1, в которой R4 является карбоксил-C1-C4 алкилэфирной группой (-CO2/C1-C4/алкил). Эфирное соединение сначала взаимодействует с третбутилдиметилсилилхлоридом в присутствии органического основания, например триэтиламина или пиридина, или предпочтительно имидазола в полярном апротонном растворителе, предпочтительно диметилформамиде, для защиты гидроксильной группы, которая является известной в данной области. Защищенное эфирное соединение взаимодействует с аммиаком и три (C1-C8)алкилалюминием в ксилоле для замещения карбоксилэфирной группы цианогруппой. Цианогруппа взаимодействует с триметилстаннилазидом в толуоле при 110oC. Превращения в тетразолил и удаления силильной защитной группы достигают посредством реакции с тетрабутиламмонийфторидом в тетрагидрофуране для получения соединений формулы 1, в которой R4 является тетразолилом.
Соли соединений формулы 1, содержащие карбоксигруппу, могут быть получены обычной реакцией взаимодействия с основанием, таким как гидроксид щелочного металла, например гидроксидом натрия, или гидроксид щелочноземельного металла, например гидроксидом магния. Сложные эфиры соединений 1, содержащие карбоксигруппу, можно получить обычным образом путем взаимодействия кислотной группы с C1-C6 спиртом, например этанолом, фенил (C1-C6) спиртом, C3-C7 -циклоалканолом, фенолом, замещенным одной-тремя группами, выбранными из фтора, хлора, C1-C6-алкила или C1-C6 -алкила или C1-C6 - алкокси.
Соединения изобретения можно назначать человеку для лечения заболеваний, вызванных ЛTB4, различными путями: перорально, парентерально и локально, а также с использованием суппозиториев и клизм.
При пероральном приеме уровень дозировки составляет от 0,5 до 1000 мг/день, преимущественно около 5-500 мг/день, которая может быть дана как однократная доза или как три небольших дозы, повторяемых через определенные интервалы времени. При внутривенном введении уровень дозировки составляет около 1-500 мг/день, преимущественно около 1,0-100 мг/день. Внутривенное введение может включать непрерывное капельное вливание. В зависимости от возраста, веса и состояния пациента неизбежны изменения в выбранном способе введения лекарственного средства, которые известны специалистам в данной области.
Соединения изобретения можно вводить отдельно, но обычно их вводят в смеси с фармацевтическим носителем, выбранным в зависимости от способа приема лекарственного средства и стандартной фармацевтической практики. Например, их можно принять перорально в виде таблеток, содержащих такие наполнители, как крахмал или лактозу, или в виде отдельных капсул или в смеси с наполнителями, или в виде эликсиров или суспензий, содержащих средства для придания вкуса, или окрашивающие средства. Их можно вводить парентерально, например, внутримышечно, внутривенно или подкожно. При парентеральном введении их лучше всего использовать в виде стерильного водного раствора, который может содержать другие растворенные вещества, например, достаточное количество соли или глюкозы, чтобы раствор стал изотоническим.
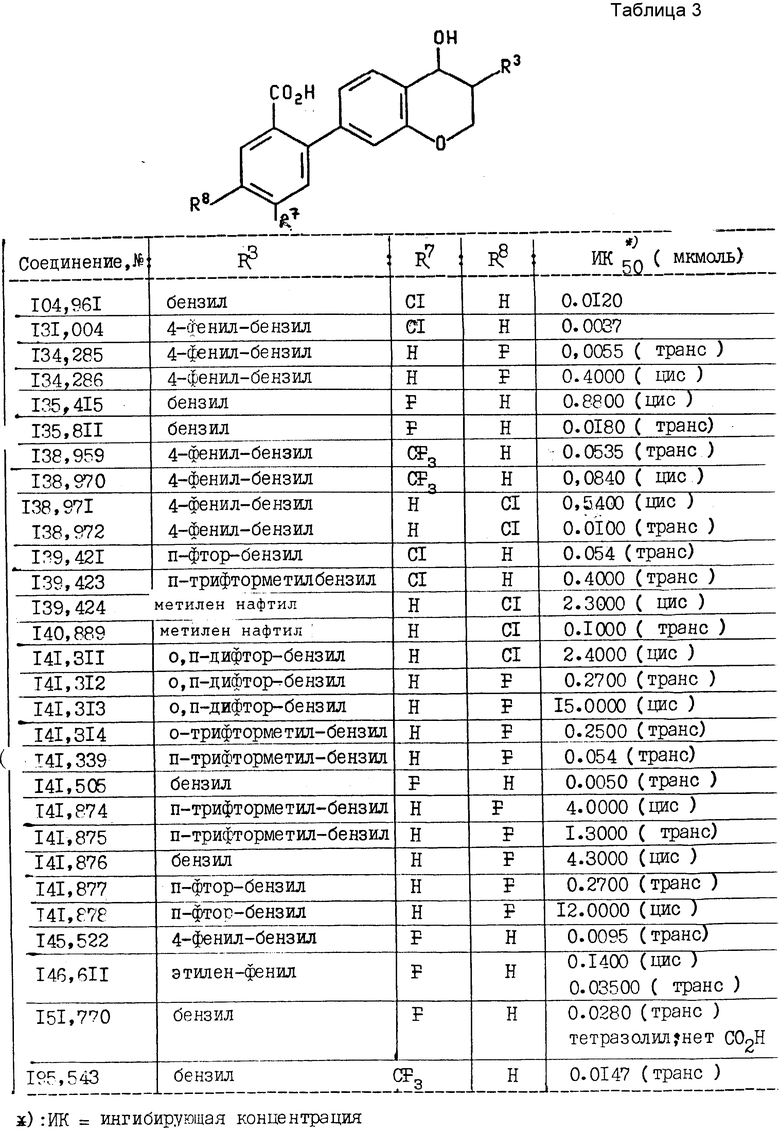
Активность соединений изобретения в отношении ЛТВ4 может быть определена путем сравнения способности соединений изобретения конкурировать с меченым радиоактивным изотопом ЛТВ4 для конкурентных клеточных рецепторов ЛТВ4 на оболочках селезенки морской свинки. Оболочки селезенки морской свинки получали как описано Cheng et. al. (J. Pharmacology and Experimental Therapeutics, 232:80, 1985). Анализ связывания 3H- ЛТВ4 проводили в 150 мкл, содержащих 50 мМ трис pH 7,3, 10 мМ MgCl2, 9% метанола, 0,7 нМ3H- ЛТВ4 (NEN, приблизительно 200 Ci (ммоль) и 0,33 мг/мл оболочки селезенки морской свинки. Добавляли немеченый радиоактивным изотопом ЛТВ4 при концентрации 5 мкМ для того, чтобы определить неспецифическое связывание. Пробные соединения добавляли при различных концентрациях, чтобы оценить их воздействие на связывание 3H ЛТВ4. Реакции проводили в термостате при 4oC в течение 30 мин. 3H-ЛТВ4, связанный оболочкой, собирали фильтрацией через стекловолоконный фильтр, и связанное количество определяли путем подсчета сцинтилляций. Значением 1C50 для пробного соединения является концентрация, при которой подавляют 50% специфического связывания 3H- ЛТВ4. Данные испытаний даны в табл. 3. Последующие примеры иллюстрируют получение соединений изобретения.
Пример 1. A. 2',4-дигидрокси-3-хлорпропиофенон.
К перемешиваемой смеси резорцина (200 г, 1,82 ммоль) и 3-хлорпропионовой кислоты (200 г, 1,84 моль) добавили трифторметансульфоновую кислоту ( 1 кг) одной порцией. Раствор медленно нагревали в течение 45 мин до 80oC, затем охлаждали до комнатной температуры в течение 15 мин и влили в хлороформ (4 л). Органическую часть медленно влили в воду (4,0 л) и слои разделили. Водный слой экстрагировали хлороформом (2х2,0 л). Объединенные органические слои промыли рассолом, сушили над сульфатом натрия и фильтровали. При концентрировании в вакууме получили оранжевое полутвердое вещество (244, 1 г), которое использовали сырым на следующей стадии.
1H-ЯМР (300 MHz, CDCl3): 12,56 (1H, s), 7,63 (1H, d, J = 7,6 Hz), 6,37 - 6,46 (2H, m), 3,92 (2H, t), J = 6,3 Hz), 3,41 (2H, t, J = 6,3 Hz).
В. 7 - Гидроксибензопиранон-4.
К охлажденному раствору (5oC/2N гидроксида натрия (10 л) добавили соединение стадии A (244, 1 г) одной порцией. Раствор нагревали до комнатной температуры в течение 2 ч, используя баню с теплой водой, затем повторно охлаждали до 5oC и устанавливали pH, равный 2,6 М серной кислотой (1,2 л). Смесь экстрагировали 3х3,0 л этилацетата, промывали рассолом (1х2,0 л), сушили над сульфатом магния и фильтровали. При концентрировании в вакууме получили рыжевато-коричневое твердое вещество. После растирания в порошок в гексане и фильтрации получили 173,7 г (выход 58%) основного продукта. Температура плавления - 136 - 137oC.
C. 7-//Трифторметилсульфонил/окси/бензопиранон-4.
К перемешиваемому раствору соединения стадии B (173,7 г, 1,05 моль) в метиленхлориде (3,0 л) при -78oC добавили триэтиламин (320 г, 3,16 моль) и диметиламинопиридин (2,5 г). После полного растворения добавляли по каплям трифторметансульфоновый ангидрид (327 г, 1,16 моль) в течение 20 мин, смесь перемешивали в течение 30 мин при -78oC, и затем нагревали до комнатной температуры в течение 2 ч. Реакционную смесь влили в насыщенный раствор хлорида аммония (2,5 л) и слои разделили. Водный слой экстрагировали 2х2,0 л метиленхлорида. Объединенные органические фракции промыли водой (1х1,0 л), сушили над сульфатом магния и фильтровали. При концентрировании в вакууме получили красное масло. Хроматография на силикагеле ( 1 кг) при элюировании смесью гексана и этилацетата (8: 1) обеспечила при удалении растворителя 211,1 г (выход 69%) основного продукта. Температура плавления 43 - 44oC.
D. 7-//Трифторметилсульфонил/окси/-3-фенилметилбензопиранон-4.
К перемешиваемому раствору продукта стадии C (27 г, 91,2 ммоль) в 183 мл метанола добавили бензальдегид (11,1 мл, 109 ммоль), а затем пирролидин (9,1 мл, 109 ммоль).
Смесь перемешивали при комнатной температуре всю ночь, охлаждали до 0oC и фильтровали. Твердый продукт один раз промывали 50 мл метанола, охлажденного льдом, и сушили в вакууме; выделили 35,2 г (выход 75%) основного продукта. Температура плавления 133 - 135oC.
1H ЯМР (300 MHz, CDCl3):8,11 (1H, d, J = 8,7Hz), 7,91/1H,bs/, 7,40 - 7,51 (2H, м) 7,24 - 7,38 (3H, м), 6,97 (1H, dd, J = 8,7 Hz, 2,4 Hz), 6,91 (1H, d, J = 2,4 Hz), 5,40 (1H, bs).
E. 7//Трифторметилсульфонил/окси/-3-фенилметилбензопиран-4-он.
К раствору соединения стадии D (26,6 г, 69,2 ммоль) в 250 мл этилацетата, находящемуся в 500-мл качающейся колбе Парра, добавили 10%-ный палладий на углеродном катализаторе (1,3 г). Смесь гидрировали под давлением 40 psi (фунтов/дюйм2) до прекращения поглощения водорода в течение 3 ч. Смесь фильтровали через броунмиллерит (торговое название диатомовой земли) для удаления палладиевого катализатора и подвергали хроматографии на силикагеле (гексан-эфир); получили 25,1 г (выход 94%) основного продукта. Температура плавления 56 - 58oC.
1H ЯМР (300 MHz, CDCl3): 8,01 (1H, d, J = 8,5 Hz), 7,20 - 7,35 (5H, м), 6,981 - 6,96 (2H, м), 4,42 (1H, dd, J=11,6, 4,4 Hz), 4,22 (1H, dd, J=11,6 Hz, 8,7 Hz), 3,26 (1H, dd, J=14,0, 4,4 Hz), 2,90 - 3,05 (1H, dd, J=14,0 8,7 Hz.
F. 7-(Триметилстанил)-3-фенилметилбензипиран-4-он.
К перемешиваемому раствору соединения стадии E (9,20 г, 25,0 ммоль) в 200 мл диоксана добавили хлорид лития (3,20, 75,0 ммоль), Pd(PPd3)4 (1,15 г, 1,0 ммоль), 3 кристаллика бутилированного гидрокситолуола и гексаметилдитин (9,0 г, 27,5 ммоль). Смесь нагревали с обратным холодильником в течение 1,5 ч, охлаждали до комнатной температуры и влили в 150 мл насыщенного водного раствора хлорида аммония. Смесь экстрагировали 3х150 мл диэтилового эфира, объединенные органические фракции промыли солевым раствором, сушили над сульфатом натрия и фильтровали. Выпаривание в вакууме привело к получению желтого полутвердого вещества, которое подвергли хроматографии на силикагеле (при отношении гексана к эфиру 5:1) для того, чтобы получить 8,90 г (выход 89%) основного продукта. Температура плавления 84-86oC.
1H ЯРМ (300 МНz, CDCl3): 7,85 (1H, d, J=8,7 Hz), 7,18-7,37 (5H, м), 7,14 (1H, d, J= 8,7 Hz), 7,11 (1H, S), 4,38 (1H, dd, J=11,6, 4,5 Hz), 4,17 (1H, dd, J= 11,6 Hz, 8,4 Hz), 3,28 (1H, dd, J=14,0, 4,4 Hz), 2,84-2,95 (1H, м), 2,71 (1H, dd, J=14 Hz, J=11,0 Hz), 0,31 (9H, δ).
G. 7-(3-карбометоксифенил)-3-фенилметилбензопиран-4-он.
К перемешиваемому раствору соединения стадии F (7,0 г, 17,5 ммоль) в диметилформамиде (ДМФ) (35 мл) добавили Pd(PPh3)2 Cl2 (490 мл, 0,7 ммоль), 3 кристалла ВНТ и метил-3-иодобензоат (5,0 г, 19,1 ммоль). Смесь перемешивали при дефлегмации в течение 1,5 ч, охладили до комнатной температуры и влили в 150 мл насыщенного водного раствора хлорида аммония. Смесь экстрагировали 3х150 мл диэтилового эфира, объединенный экстракт промыли 2х100 мл воды, а затем солевым раствором. Раствор сушили над сульфатом натрия, фильтровали и выпаривали в вакууме до получения желтого масла. Хроматографией на силикагеле (элюирование смесью гексана и эфира при соотношении компонентов 4:1) получили 6,51 г основного продукта в виде вязкого масла).
1H ЯРМ (300 MHz), CDCl3): 8,29 (1H, t, J=1,6 Hz), 8,06 (1H, dd, J=7,6 1,6 Hz), 8,00 (1H, d, J=8,2 Hz), 7,79 (1H, dd, J=7,6 Hz, 1,6 Hz), 7,53 (1H, t, J= 7,6 Hz), 7,22-7,36 (7H, м), 4,41 (1H, dd, J=11,6, 4,5 Hz), 4,21 (1H, dd, J=11,6, 8,5 Hz), 3,94 (3H, S), 3,31 (1H, dd, J=14,0, 4,4 Hz), 2,91-2,99 (1H, м), 2,73 (1H, м), 2,73 (1H, dd, J=14,0, 11,1 Hz).
H. 7-(3-карбометоксифенил)-4-гидрокси-3-фенилметил бензопиран.
К перемешиваемому раствору соединения стадии G (6,50 г, 17,5 ммоль) в 35 мл метанола при комнатной температуре добавили боргидрид натрия (940 мг, 26,0 ммоль) одной порцией. Смесь темного цвета перемешивали при комнатной температуре в течение 2 ч, затем влили в насыщенной водный раствор хлорида аммония (75 мл) и экстрагировали 3х75 мл диэтилового эфира. Объединенные экстракты промыли рассолом, сушили над сульфатом натрия и концентрировали в вакууме до получения низкосортного желтого масла. Хроматографией на силикагеле при элюировании смесью гексана и эфира с соотношением компонентов в смеси 4:1 получили сначала 3,26 г цис-циклического изомера основного соединения, а затем 1,98 г, транс-изомера основного соединения в виде вязкого масла, общий выход составил 81%.
Цис-циклический изомер:
1H ЯРМ (300 MHz, CDCl3): 8,26 (1H, t, J=1,7, Hz), 8,02 (1H, dt, J=7,8, 1,7 Hz), 7,76 (1H, dt, J=7,8, 1,7 Hz), 7,50 (H,t, D=7,8 Hz), 7,41 (1H, d, J= 7,9 Hz), 7,31 (1H, d, 7,3 Hz); 7,14-7,25 (6H, м), 4,58 (1H, t, J=7,2 Hz), 4,28 (1H, dd, J=9,1, 2,5 Hz), 4,03 (1H, dd, J=9,1, 5,4 Hz), 3,93 (3H, S), 2,78 (1H), 2,77 (1H, dd, J=13,7, 6,2 Hz), 2,58 (1H, dd, J=13,7, 9,1 Hz), 2,20-2,29 (1H, м), 1,83 (1H, d, J=7,2 Hz).
Транс-циклический изомер:
1H ЯМР (300 MHz, CDCl3): 8,23 (1H, t, J=1,7 Hz), 7,98 (1H, dt, J=7,8 Hz), 7,74 (1H, t, J=7,8 Hz, 1,7 Hz), 7,48 (1H, t, J=7,8 Hz), 7,20-7,36 (6H, м), 7,15 (1H, dd, J=8,0, 1,8 Hz) 7,09 (1H, d, J=1,8 Hz), 4,56 (1H, dt, J= 4,7, 3,8 Hz), 4,12-4,19 (2H, м), 3,92 (3H, S), 2,90 (1H, dd, J=13,6, 8,4 Hz), 2,70 (1H, dd, J=13,6, 7,2 Hz), 2,36-2,39 (1H, м), 1,75 (1H, d, J=4,7 Hz).
J. N- α -трет-бутоксикарбонил-L-триптофан-7-[(3-карбометоксифенил)-3-фенилметил]хроманил-4 эфир.
К перемешиваемому раствору соединения стадии H (2,5 г, 6,7 ммоль) в 70 мл CH2Cl2 добавили DMAP (897 мг, 7,34 ммоль, 1,1 эквивалент) и N-трет-бок- α -триптофан (2,4 г, 8,01 г ммоль, 1,2 эквивалента). Смесь перемешивали при комнатной температуре в течение 12 ч, фильтровали и промывали 1M HCl и солевого раствора. Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме. Хроматографией (на силикагеле при соотношении циклогексана к эфиру 3:1) получили 860 мг менее полярного диастереомера (Rf=0,3) и 700 мг более полярного диастереоизомера (Rf=0,2).
Менее полярный продукт (3, S, 4R):
1H-ЯРМ (300 MHz, CDCl3); 8,29 (1H, S), 8,03 (2H, d, J=7,8 Hz), 7,77-7,83 (2H, м), 7,52 (2H, t, J=7,6 Hz), 7,02-7,33 (5H, м), 6,64 (H,S), 5,65 (1H, S), 5,06 (1H, d J=8,4 Hz), 4,58-4,62 (1H, м), 3,95 (3H, S), 3,73-3,85 (2H, м), 3,18-3,28 (2H, м), 2,45-2,61 (2H, м), 2,09-2,15 (1H, brds), 1,39 (H, S).
Более полярный продукт (3R, 4S):
1H-ЯМР (300 MHz, CDCl3): 8,25 (1H, S), 8,01 (1H, d, J = 7,8 Hz), 7,94 (1H, brds), 7,74 (1H, d, J = 8,2 Hz), 7,54 (1H, d, J = 11,9 Hz), 7,48 (1H, t, J = 7,8 Hz), 7,09-7,38 (H, м), 6,95 (1H, S), 5,61 (1H, S), 5,08 (1H, d, J = 8,2 H), 4,55-4,60 (1H, м), 3,94 (3H, S), 3,73-3,76 (2H, м), 3,22-3,35 (2H, м), 2,42-2,60 (2H, м), 1,90-1,96 (1H, м), 1,39 (9H, S).
K. 3S,4R-7-(3-карбоксифенил)-4-гидрокси-3-фенилметил-2H-1-бензопиран.
К перемешиваемому раствору менее полярного 4R, 3S-триптофанового эфира стадии J (840 мг, 1,08 ммоль) в 10 мл метанола добавили 10 мл 2 M раствора NaOH. Смесь нагревали с обратным холодильником в течение 8 ч, охлаждали и подкисляли до pH 4 IM HCl. Мутную эмульсию экстрагировали 3х20 мл этилацетата, затем объединенные органические фракции промыли рассолом и сушили над MgSO4. После фильтрации и удаления растворителя в вакууме получили желтую пену. Хроматографией на силикагеле (при соотношении этилацетат:гексан:уксусная кислота 35:75:1) получили 210 мг продукта.
1H ЯМР (300 MHz, CD3CN): 8,22 (1H, t, 1,7 Hz), 7,97 (1H, dt, J = 7,8, 1,7 Hz), 7,87Б(1H, dt, J=7,8, 1,7 Hz), 7,55 (1H, t, J = 7,8 Hz), 7,42 (1H, d, J= 7,9 Hz),7,15-7,36 (6H, м), 7,10 (1H, d, J = 1,8 Hz), 4,44 (1H, d, J = 4,9 Hz), 4,19 (1H, dd, J = 9,1, 2,5 Hz), 3,97 (1H, dd, J = 9,1, 5,4 Hz), 2,72 (1H, dd, J = 13,7, 6,2 Hz, 2,51 (1H, dd, J = 13,7, 9,1 Hz), 2,04-2,20 (3H, м), [α]D = +11,1 при c= 1,00 в метаноле, температура плавления 210-212oC.
При омылении более полярного 3R, 4S триптофанового эфира (700 мг) получили 3R, 4S энантиомер.
1H-ЯМР (300 MHz, CD3CN): 8,22 (1H, t, 1,7 Hz), 7,97 (1H, dt, J = 7,8, 1,7 Hz), 7,87 (1H, dt, J = 7,8, 1,7 Hz), 7,55 (1H, t, J = 7,8 Hz), 7,42 (1H, d, J = 7,9 Hz), 7,15-7,36 (6H, м), 7,10 (1H, d, J = 1,8 Hz), 4,44 (1H, d, J= 4,9 Hz), 4,19 (1H, dd, J=9,1, 2,5 Hz), 3,97 (1H, dd, J = 9,1, 5,4 Hz), 2,72 (1H, dd, J = 13,7, 6,2 Hz), 2,51 (1H, dd, J = 13,7, 9,1 Hz), 2,04-2,20 (3H, м), [α]D = 11,0 при c=1,01 в метаноле; температура плавления 209-211oC.
L. Транс 3-фенилметил-4-гидрокси-7-(3-карбоксифенил)-2H-I-бензопиран.
Омылением, как на стадии K, транс-циклического изомера стадии H, получили соответствующую кислоту.
1H-ЯМР (300 MHz, CD3CN): 8,22 (1H, t, 1,7 Hz), 7,97 (1H, dt, J= 7,8, 1,7 Hz), 7,87 (1H, dt, J = 7,8, 1,7 Hz), 7,55 (1H, t, J= 7,8 Hz), 7,42 (1H, d, J = 7,9 Hz), 7,15-7,36 (6H, м), 7,10 (1H, d, J = 1,8 Hz), 4,44 (1H, d, J = 4,9 Hz, 4,19 (1H, dd, J= 9,1, 2,5 Hz), 3,97 (1H, dd, J= 9,1; 5,4 Hz), 2,72 (1H, dd, J= 13,7, 6,2 Hz), 2,51 (1H, dd, J = 13,7, 9,1 Hz), 2,04-2,20 (3H, м). Температура плавления 210-212oC.
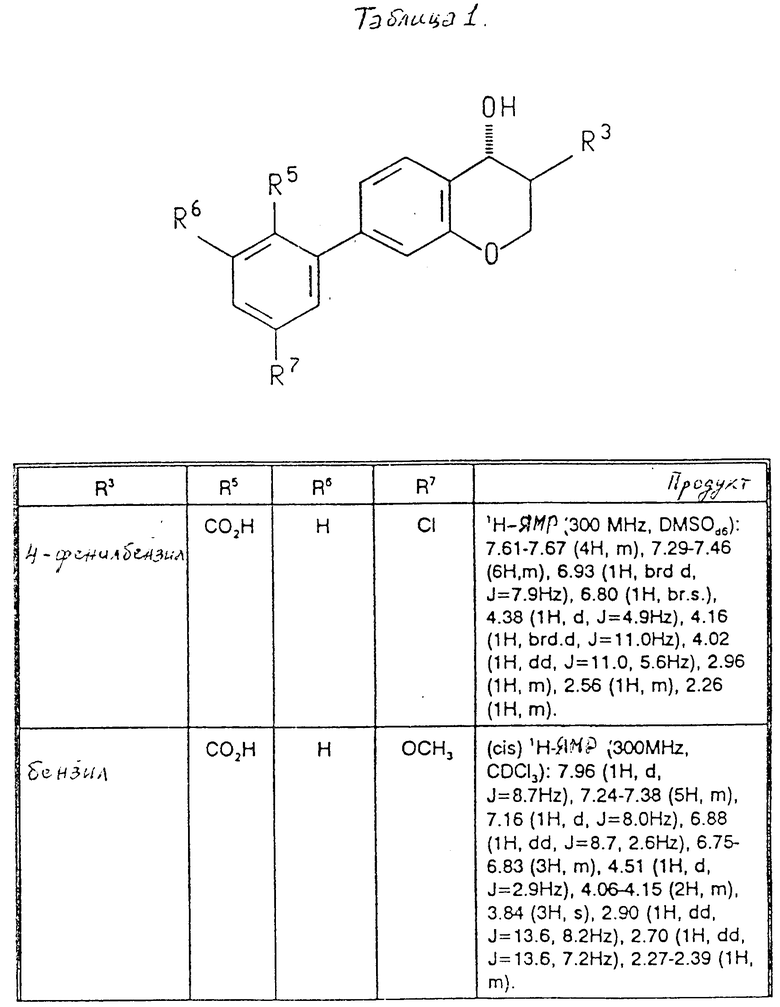
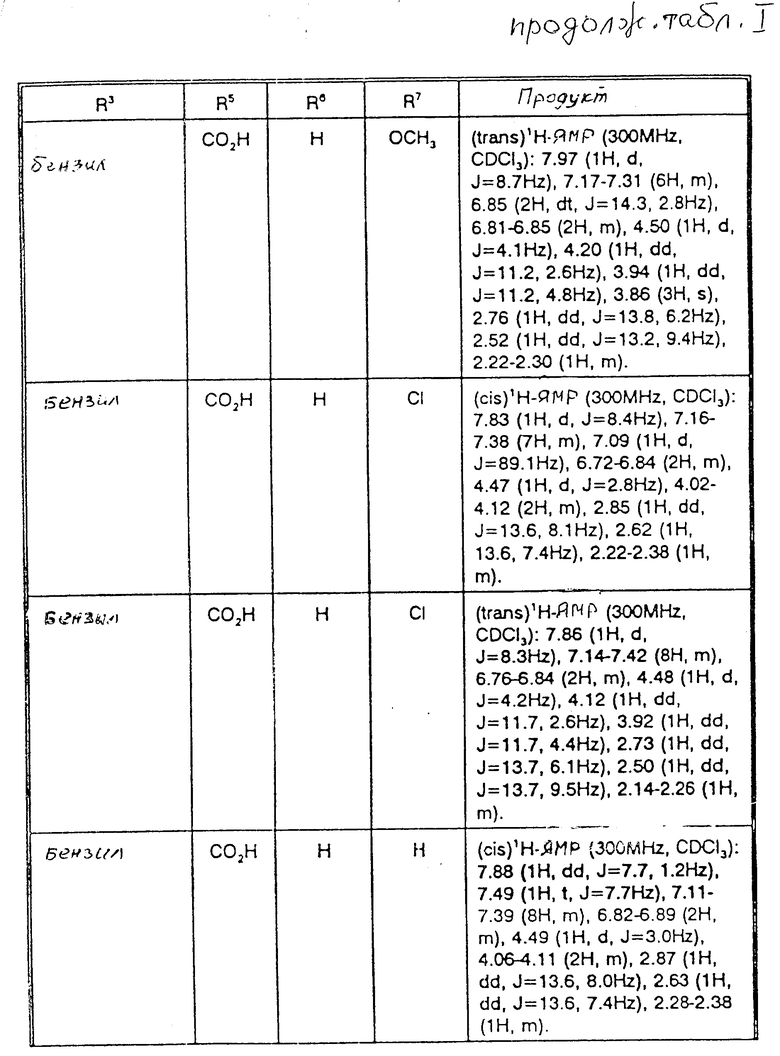
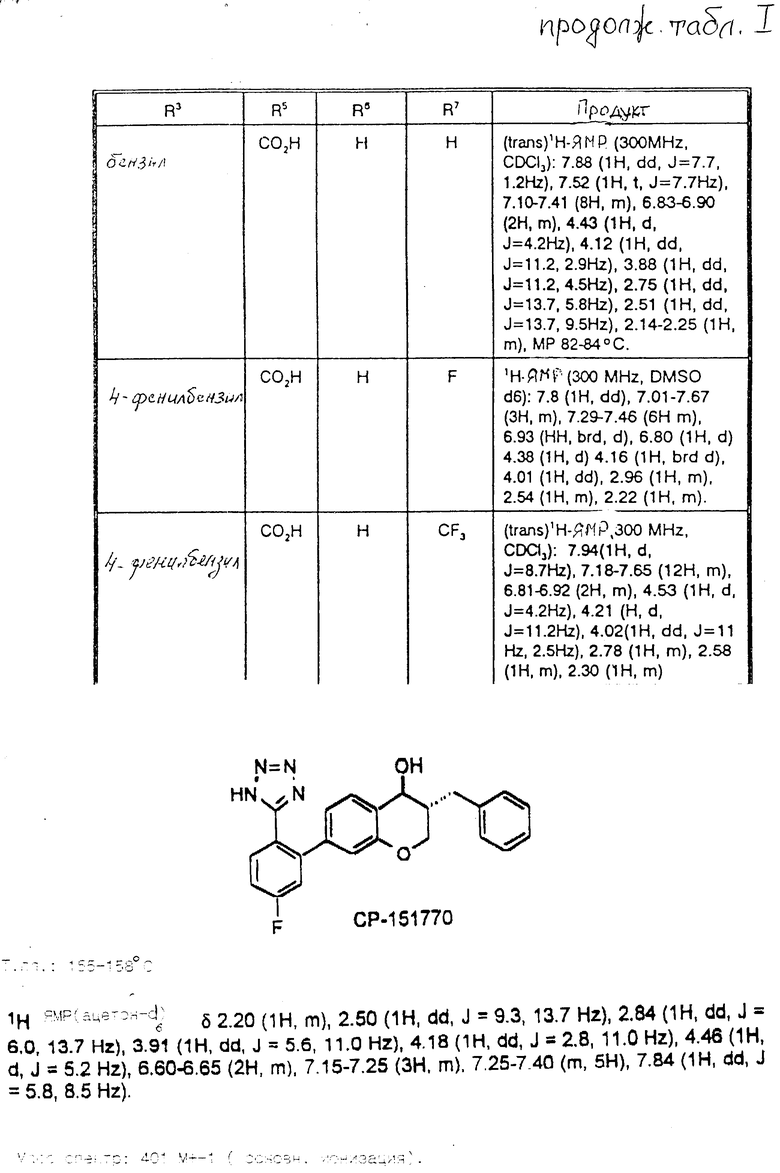
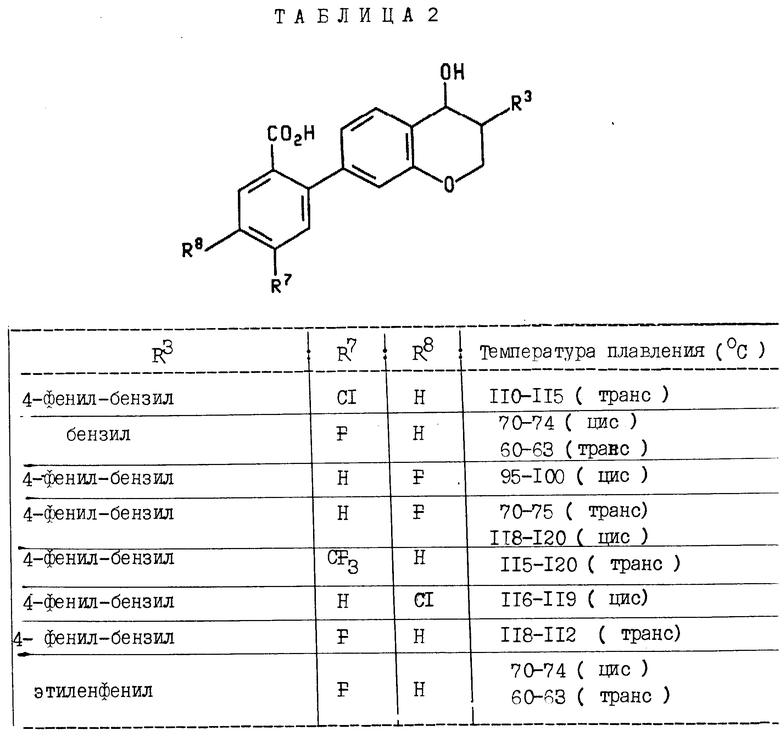
Пример 2. Следующие соединения в табл. 1 были получены омылением в соответствии с примером 1J. Температура плавления приведена в градусах Цельсия в табл.2.
Пример 3. При омылении соответствующего эфира в соответствии с примером 1J, образовался 7-(4-гидрокси-3-карбоксифенил)-4-гидрокси-3-фенилметил-2H-1-бензопиран, имеющий температуру плавления 158-160oC (цис) и 173-175oC (транс).
Пример 4. A. 7-[5-фтор-2-(4,4-диметил-2-оксазолинил)фенил]-3-фенилметилен-1-бензопиранон-4.
К перемешиваемому раствору 2-(4-фторфенил)-4,4-диметил-2-оксазолина (1,0 эквивалент в тетрагидрофуране, концентрация 0,5 м) при -78oC в атмосфере азота добавили н-бутиллитий в гексане (1,1 эквивалент, 2,2 M раствор). Смесь перемешивали при -78oC в течение 1 ч, затем добавили ZnCl2 (IM раствор в эфире, 1,1 эквивалент). Смесь нагревали до 10oC в течение 1 ч до получения 2-(4-фторфенил-2-хлорцинк)-4,4-диэтил-2-оксазолина (не отделен).
К этому раствору добавили 7-[((трифторметил)сульфонил-окси] 3-фенилметилен-1-бензопиранон-4-(1,0 эквивалент) и Pd(PPh3)4 (0,2 эквивалента). Смесь нагревали с обратным холодильником (68oC) в течение 3 ч, охлаждали до комнатной температуры и влили в раствор NH4OH. Раствор экстрагировали 3 раза диэтиловым эфиром и объединенную органическую фракцию сушили над сульфатом магния. Затем осуществляли фильтрацию, удаление растворителя в вакууме и колоночную хроматографию на силикагеле (при соотношении гексан:эфир 2:1, при этом получили основное соединение в виде желтого твердого, выход 65%, температура 110-112oC.
1H-ЯМР (300 MHz, CDCl3):8,04 (1H, d), 7,91 (1H, S), 7,78 (1H, dd), 7,41 - 7,52 (3H, м), 7,31 (2H, d), 7,06 - 7,18 (3H, м), 7,02 (1H, S), 5,40 (2H, S), 3,86 (2H, S) 1,31 (6H, S).
B. (3S*, 4R*)7-[5-фтор-2-(4,4-диметил-2-оксазолинил) фенил]4-гидрокси-3-фенилметил-2H-1-бензопиран.
К перемешиваемому раствору соединения со стадии А в ТГФ (тетрагидрофуране) (0,1М) при 0oC добавляли по каплям LiAlH4 (1М в эфире, 2,2 эквивалента) в течение 10 мин. Смесь нагревали до комнатной температуры и перемешивали в течение 12 ч. Смесь охлаждали до 0oC, гасили сегнетовой солью и фильтровали через инфузорную землю (кизельгур). Водный слой дважды экстрагировали этилацетатом, объединенные органические слои промыли солевым раствором и сушили над MgSO4. После фильтрации и удаления растворителя получили желтое масло. Хроматографией на силикагеле получили белое твердое, выход 60%. Температура плавления 65 - 70oC (при разложении).
Вычислено, %: C 75,15; H 6,07; N 3,25.
C27H26NO3F
Найдено: C 74,75; H 6,02; N 3,09.
1H-ЯМР (300 МНz, CDCl3): 7,70 (1H, dd), 7,02 - 7,37 (8H, м), 6,96 (1H, dd), 7,91 (1H, d), 4,51 (1H, d), 4,23 (1H, dd), 4,39 (1H, dd), 3,87 (2H, dd), 2,74 (1H, dd) 2,55 (1H, dd), 2,18 - 2,28 (1H, m), 1,31 (6H, d).
C. (3S*, 4R*) 7-/2-карбокси-5-фторфенил-4-гидрокси-3-фенилметил-2H-1-бензопиран.
Соединение со стадии B растворяли в метилиодиде (0,5М) при комнатной температуре и перемешивали в течение 24 ч. Метилиодид удалили в вакууме, маслянистое твердое растворили в CH2Cl2, а растворитель удалили в вакууме. Операцию повторяли для удаления следов метилиодида. Твердый продукт растворили в метаноле (0,5М) и добавили 2М NaOH (0,5M). Смесь нагревали с обратным холодильником в течение 5 ч, охлаждали до комнатной температуры и подкисляли до pH 1 1M HCl. Смесь дважды экстрагировали этилацетатом, промывали рассолом и сушили над MgSO4. Осуществляли фильтрацию и удаление растворителя в вакууме, затем хроматографию на силикагеле (при соотношении метилхлорида к метанолу 10:1), в результате чего получили желаемую кислоту, выход 93%.
1H-ЯМР (300 МНz, CD3COCD3): 7,80 (1H,dd), 7,48 (1H, d), 7,18 (7H, м), 7,13 (1H, dd), 6,91 (1H, dd), 6,80 (1H, d), 4,52 (1H, d), 4,23 (1H, dd), 3,96 (1H, dd), 2,89 (1H, dd), 2,54 (1H, dd), 2,19 - 2,30 (1H, м).
D1. (3S, 4R)-7/2карбокси-5-фторфенил/-4-гидрокси-3-фенилметил- 2H-1-бензопиран.
Соединение со стадии C растворяли в диэтиловом эфире (0,1M) и нагревали с обратном холодильником. К раствору добавляли по каплям S(-)метилбензиламин (1 эквивалент) в диэтиловом эфире (0,1 M) в течение 10 мин. Смесь охлаждали до комнатной температуры и перемешивали в течение 48 ч. Осажденную соль фильтровали, затем повторно перемешивали 2 раза при кипячении в диэтиловом эфире (0,1H) в течение 24 ч и фильтровали. Соль (температура плавления 170 - 173oC) помещали в метиленхлорид и промывали 3 раза 1M HCl, затем один раз рассолом, сушили над MgSO4 и фильтровали. После удаления растворителя в вакууме и рекристаллизации (гексан:эфир = 1:1) получили белые мелкие кристаллы при более чем 99,8%-ном энантиомерном избытке ВЭЖХ анализом (высокоэффективной жидкостной хроматографией).
[α]
Вычислено, %:C 73,01; H 5,06.
C23H19O4F
Найдено, %: C 72,88; H 4,76.
D2. (3R, 4S/7-/2-карбокси-5-фторфенил/-4-гидрокси-3-фенил метил-3H-1-бензопиран.
Фильтрат объединенных суспензий соли стадии D1 три раза промыли 1M HCl, один раз солевым раствором и сушили над MgSO4. После фильтрации и удаления растворителя получили твердое желтое. При использовании подобной процедуры, которая описана на стадии D1, и применении R(+) метилбензиламина получили желаемый продукт.
[α]
Вычислено,%: C 73,01; H 5,06.
C23H19O4F
Найдено,%: C 73,03; H 4,84.
Пример 5. 2-(6-Бензил-5-гидрокси-5,6,7,8-тетрагидро-нафталин-2-ил)- 4-фтор-бензойная кислота.
А. 2-Бензилиден-6-метокси-3,4-дигидро-2H-нафталин-1-он.
К перемешиваемому раствору 6-метокси-12-тетралона (227 ммоль, 40 г) и бензальдегида (272 ммоль, 27,5 мл) в 450 мл метанола добавляют пирролидин (272 ммоль, 23,6 мл). Смесь перемешивают при комнатной температуре в течение 2-х дней до тех пор, пока ТСХ не покажет, что в смеси отсутствует исходный тетралон. Смесь концентрируют в вакууме, затем растворяют в этилацетате, промывают четырьмя порциями (частями) 10%-ной HCl, двумя порциями (частями) насыщенного раствора NaHCO3 и одной порцией (частью) рассола. Растворитель удаляют в вакууме и сырое масло порошкуют с помощью диэтилового эфира, получая 38 г целевого соединения этого примера получения 1А; т.пл. 100-102oC.
Анализ для C18H16O2:
рассчитано: 264.1146,
найдено: 264.1149.
B. 2-Бензил-6-метокси-3,4-дигидро-2H-нафталин-1-он.
Склянку для гидрирования Parr® загружают нафталин-1-оном (15 г), этилацетатом (150 мл) и 1 г 10%-ного палладия-на-угле. Смесь гидрируют на аппарате для встряхивания Parr® в течение 15 ч при давлении водорода 1,406 кг/см3. Полученную смесь фильтруют через слой (прокладку) целита (Celite®) и концентрируют в вакууме до получения красного цвета масла, которое очищают с помощью флэш-хроматографии (гексан/диэтиловый эфир = 3:1), получая 14,1 г бензилтетралона с т.пл. 50-51oC.
Анализ для C18H18O2:
рассчитано: 266.1302,
найдено: 266.1308.
C. 2-Бензил-6-гидрокси-3,4-дигидро-2H-нафталин-1-он.
К перемешиваемому раствору бензилтетралона (5 г, 19 ммоль) в метиленхлориде (40 мл) при температуре около -78oC добавляют трибромид бора (1,95 мл, 21 ммоль). Охлаждающую баню удаляют и реакционную смесь перемешивают в течение ночи при комнатной температуре, затем повторно добавляют дополнительные 1,5 мл трибромида бора. Перемешивание продолжают при комнатной температуре в течение следующих примерно 4 ч, после чего выливают в воду со льдом и перемешивают в течение около 0,5 ч. Водную смесь насыщают хлоридом натрия и экстрагируют четырьмя порциями метиленхлорида. Слои разделяют и органическую фазу промывают водой и сушат над безводным сульфатом натрия. Фильтрация и удаление в вакууме приводят к твердому веществу коричневого цвета, которое очищают путем флэш-хроматографии (гексан/эфир = 3:2), получая 3 г фенола; т.пл. 160-162oC.
Анализ для C17H18O2:
рассчитано: 252.1146,
найдено: 252.1144.
D. 6-Бензил-5-оксо-5,6,7,8-тетрагидро-нафталин-2-иловый эфир трифторметансульфокислоты.
К перемешиваемому раствору фенола (2,75 г, 11 ммоль), триэтиламина (4,56 мл, 33 ммоль) и DMAP (0,05 г) в метиленхлориде (100 мл) при температуре примерно -78oC добавляют ангидрид трифторметансульфокислоты (2 мл, 12 ммоль). Охлаждающую баню удаляют и реакционную смесь нагревают до комнатной температуры и перемешивают в течение ночи. Смесь затем выливают в воду со льдом и экстрагируют этилацетатом. Полученный органический слой промывают водой, сушат над безводным сульфатом натрия, отфильтровывают и растворитель удаляют в вакууме. Сырой продукт очищают с помощью флэш-хроматографии, получая 3,9 г трифлата; т.пл. 52-53,7oC.
Анализ для C18H15O4SF3:
рассчитано: 384.0638,
найдено: 384.0602.
E. 2-Бензил-6-/2-(4,4-диметил-4,5-дигидро-оксазол-2-ил)-5- фторфенил/-3,4 дигидро-2H-нафталин-1-он.
К перемешиваемому раствору н-бутиллития (3,6 мл, 2,5 М раствора в гексане, 9 ммоль) в толуоле (10 мл) при температуре около -40oC добавляют раствор арилоксазолина (1,76 г, 9 ммоль) в толуоле (5 мл) путем прикапывания пипеткой. Смесь перемешивают примерно при -40oC в течение примерно 0,5 ч, затем нагревают примерно до -25oC и перемешивают в течение следующего примерно 1 ч. К этой смеси добавляют хлорид цинка (9 мл 1 М раствора в диэтиловом эфире, 9 ммоль). Охлаждающую баню удаляют и смесь нагревают до комнатной температуры и перемешивают в течение около 1 ч. Полученную смесь с помощью пипетки добавляют к раствору тетралон-трифлата (3,5 г, 9 ммоль) и тетракистрифенилфосфин-палладия (0,5 ммоль, 0,63 г) в тетрагидрофуране (15 мл). Реакционную смесь кипятят с обратным холодильником в течение примерно 22 ч., охлаждают до комнатной температуры и выливают насыщенный водный раствор хлорида аммония. Водную смесь экстрагируют тремя порциями этилацетата. Органическую фазу промывают тремя порциями 0,1 M раствора HCl насыщенным водным раствором бикарбоната натрия и рассолом. Органическую фазу затем сушат над безводным сульфатом натрия, отфильтровывают и растворитель удаляют в вакууме. Сырой продукт очищают путем флэш-хроматографии (диэтиловый эфир/гексан = 2:1), получая 2,07 г продукта реакции сочетания; т. пл. 114-115oC.
Анализ для C28H26NO2F:
рассчитано: 427,1948,
найдено: 427,1956.
F. 2-Бензил-6-/2-(4,4-диметил-4,5-дигидро-оксазол-2-ил)- 5-фторфенил/-1,2,3,4-тетрагидро-нафталин-1-ол.
К перемешиваемому раствору тетралоноксазолина (1,5 г, 3,5 ммоль) в метаноле (35 мл) добавляют боргидрид натрия (0,20 г, 5,25 ммоль). Полученную смесь коричневого цвета перемешивают при комнатной температуре в течение примерно 1 ч., затем выливают в рассол и экстрагируют тремя порциями этилацетата. Органическую фазу сушат над безводным сульфатом натрия и растворитель удаляют в вакууме, получая 1,20 г смеси 1:1 цис- и транс-спиртов; т.пл. 88-89oC.
Анализ для C28H28NO2D:
рассчитано: 429.2087,
найдено: 429.2067.
G. 2-(6-Бензил-5-гидрокси-5,6,7,8-тетрагидро-нафталин-2-ил)- 4-фторбензойная кислота.
Оксазолин (1,0 г, 2,34 ммоль) растворяют в 5 мл метилиодида и перемешивают при комнатной температуре в течение примерно 5 дней, после чего метилиодид удаляют в вакууме. Остаток обрабатывают метиленхлоридом и концентрируют с целью удаления следов остаточного метилиодида. Темно-красного цвета остаток растворяют в метаноле (5 мл) и добавляют 2н раствор NaOH (5 мл). Полученную смесь кипятят с обратным холодильником при перемешивании в течение примерно 5 ч. Смесь затем охлаждают до комнатной температуры и подкисляют с помощью 3н раствора HCl. Полученную суспензию экстрагируют тремя порциями этилацетата и объединенную органическую фазу промывают рассолом. Органическую фазу сушат над безводным сульфатом натрия и растворитель удаляют в вакууме, получая 0,80 г карбоновой спиртокислоты.
1H-ЯМР (250 мГц, метанол, -d4) : 7,83 (dd, 1H, J = 7,0, 7,5); 7,50 (d, 1H, J = 7,0); 7,30-7,00 (м, 9H x 2); 4,50 (d, 1H, J = 2,0); 4,41 (d, 1H, J = 8,0); 3,15 (dd, 1H, J = 5,4, 13,9); 3,00-2,57 (м, 4H); 2,42 (dd, 1H, J = 11,4, 13,5); 2,09-1,35 (м, 5H x 2).
Бензопиран и другие бензоконденсированные антагонисты лейкотриена В4 имеют формулу (I), в которой R1 является фенилом или замещенной фенильной группой, а А, n, и R3 являются такими, как определено в описании. Изобретение включает фармацевтическую композицию на основе соединений формулы I, способ получения соединений формулы I и способ получения промежуточного продукта формулы II. 4 с.и 8 з.п.ф-лы, 3 табл.
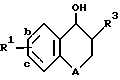
где A - атом кислорода или группа -CH2-;
R1 - заместитель в положении "b" или "c" и имеет формулу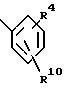
где R4 - карбоксигруппа, тетразолил или оксазолил;
R10 - атом водорода или один или любые два заместителя, выбранные из атома фтора или хлора, C1-C6-алкила, C1-C6-алкоксигруппы или C1-C4-перфторалкила;
R3 - группа -(CH2)q-CHR11R12 или -(CH2)q-R12,
где q равно 0, 1, 2 или 3;
R11 - атом водорода, C1-C6-алкил или R8 - замещенный фенил, где R8 - атом водорода или один, или любые два заместителя, выбранные из атома фтора или хлора, C1-C6-алкила, C1-C6-алкоксигруппы или C1-C4-перфторалкила;
R12 - атом водорода, фенил или нафтил, где указанный фенил может быть замещен фенилом, R9 или R9-замещенным фенилом, где R9 имеет значения, указанные для R8 выше,
или их соли, или сложные эфиры таких соединений общей формулы I, которые содержат карбоксигруппу, при этом эфирные группы выбраны из групп, содержащих C1-C6-алкил, фенил-C1-C6-алкил, C3-C7-циклоалкил, либо фенил или бензил, замещенные атомом фтора или хлора, C1-C6-алкилом или C1-C6-алкоксигруппой.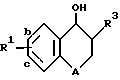
где значения A, R1 и R3 приведены в п. 1,
отличающийся тем, что восстанавливают соединение формулы XIII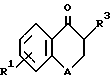
где значения A, R1 и R3 приведены выше.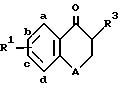
где A - атом кислорода или группа -CH2-;
R1 - заместитель в положении "b" или "c" и имеет формулу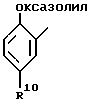
где R10 - атом водорода или один, или любые два заместителя, выбранные из атома фтора или хлора, C1-C6-алкила, C1-C6-алкоксигруппы или C1-C4-перфторалкила;
R3 - группа -(CH2)q-CHR11R12 или -(CH2)q-R12,
где q равно 0, 1, 2 или 3;
R11 - атом водорода, C1-C6-алкил или R8 - замещенный фенил, где R8 - атом водорода или один, или любые два заместителя, выбранные из атома фтора или хлора, C1-C6-алкила, C1-C6-алкоксигруппы или C1-C4-перфторалкила;
R12 - фенил или нафтил, где указанный фенил может быть замещен фенилом, R9 или R9-замещенным фенилом, где R9 имеет значения, указанные для R8 выше,
или их солей, отличающийся тем, что осуществляют взаимодействие соединения формулы IV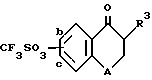
где R3 и A указаны выше, а группа CF3SO3 находится в положении "b" или "c",
с соединением формулы V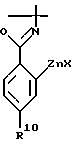
где
X - атом хлора, брома или йода, а R10 указан выше,
которое получают путем взаимодействия соединения формулы VI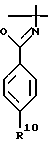
с н-бутиллитием и затем с ZnX2, где X указан выше.
| EP, A, 0276064 (Eli Lilly), 27.07.88, C 07 C 65/40 | |||
| EP, A, 0404440 (Pfize r), 27.12.90, C 07 D 417/12 | |||
| US, A, 5059609 (J.F | |||
| Eggler et al), 22.10.91, C 07 D 401/12. |

