Данное изобретение относится к производным арилсульфониламиногидроксамовой кислоты, которые являются ингибиторами матричных металлопротеиназ или продуцирования фактора некроза опухолей (далее обозначаемый как ФНО) и поэтому полезны для лечения заболевания, выбранного из группы, состоящей из артрита, рака, язв тканей, повторного стеноза, периодонталь-ного заболевания, врожденного буллезного эпидермолиза, склерита и других заболеваний, характеризующихся активностью матричной металлопротеиназы, СПИДа, сепсиса, септического шока и других заболеваний, включающих продуцирование ФНО.
Это изобретение также относится к способу применения таких соединений для лечения вышеуказанных заболеваний у млекопитающих, особенно у людей, и к фармацевтическим композициям, применяемым для этого.
Существует ряд ферментов, которые действуют разрушающе на структурные белки и которые являются структурно родственными металлопротеиназами. Матричноразрушающие металлопротеиназы, такие как желатиназа, стромолизин и коллагеназа, участвуют в разрушении структуры тканей (например, коллагеновая недостаточность) и являются причиной многих патологических состояний, включающих аномальный метаболизм соединительной ткани и основной структуры мембран, таких как артрит (например, остеоартрит и ревматоидный артрит), язвы тканей (например, тканей роговицы, язвы кожи и желудка), аномальное заживление ран, периодонтальное заболевание, костные заболевания (например, болезнь Педжета и остеопороз), метастазы или инвазия опухолей, а также ВИЧ-инфекция (J.Leuk.Biol., 52(20): 244-248, 1992).
Известно, что фактор некроза опухолей участвует в патогенезе многих инфекционных и аутоиммунных заболеваний (W.Friers, FEBS Letters, 1991, 285, 199). Кроме того, было показано, что ФНО является прямым медиатором воспалительной реакции наблюдаемой при сепсисе и септическом шоке (С.Е. Spooner et al. Clinical Immunology and Immunopathology, 1992, 62 Sll).
Краткое описание изобретения
Данное изобретение относится к соединениям формулы
или его фармацевтически приемлемым солям,
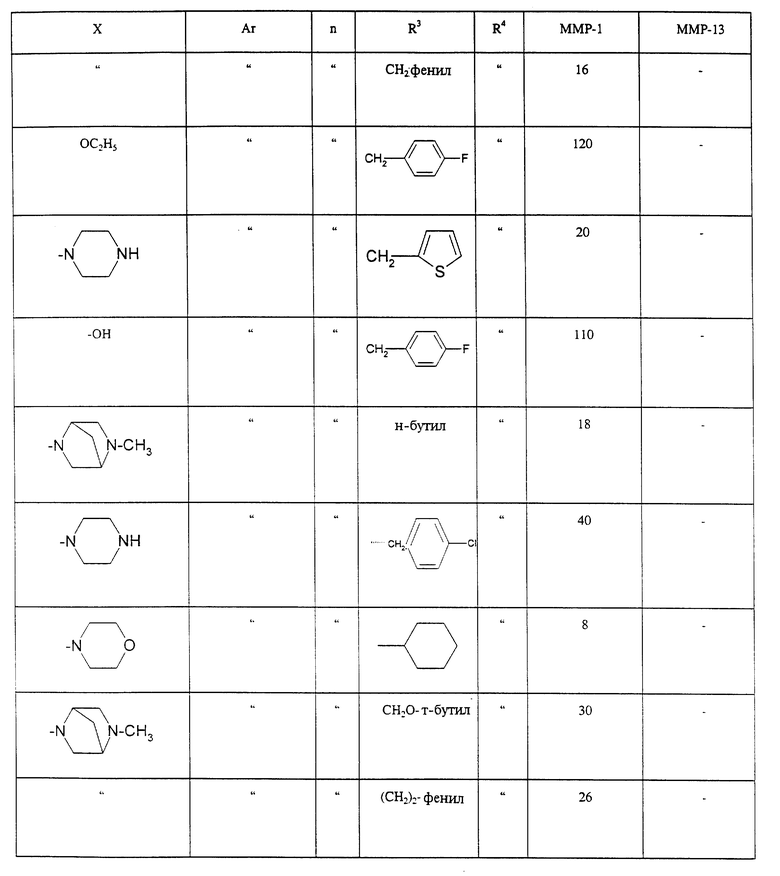
где n равно от 1 до 6;
X является гидроксилом, (C1-C6)алкоксилом или NR1R2, R1 и R2, каждый независимо выбирают из группы, состоящей из (C1-C6)алкила, пиперидила, (C1-C6)алкилпиперидила, (C6-C10)арила, (C5-C7)гетероарила, содержащего в качестве гетероатома азот, (C6-C10)арил(C1-C6)алкила, (C5-C7)гетероарил(C1-C6)алкила, содержащего в качестве гетероатома азот или R1 и R2 вместе образуют пирролидинил, морфолинил, пиперидил, (C1-C6)алкилпиперидил, пиперазил, N-(С1-С6) алкилпиперазил, N-(C6-C10)арилпиперазил,
R3 и R4 каждый независимо выбран из группы, включающей водород, (C1-C6)алкил,(C6-C10)арил, (C5-C9)гетероарил, (C6-C10)арил-(C1-C6) алкил, (C5-C9) гетероарил(C1-C6) алкил, (C6-C10)арил(C6-C10)арил, (C3-C6)циклоалкил и (C3-C6)циклоалкил (C1-C6)алкил, или R3 и R4 вместе могут образовывать (C3-C6)циклоалкил, оксациклогексил, тиоциклогексил, инданил или кольцо тетралинила или пиперидиновое кольцо, замещенное R15, где R15 означает водород, (С1-С6)ацил, (С1-С6)алкил, (С6-С10)арил(С1-С6)алкил,
(С5-С9)гетероарил (С1-С6)алкил или (C1-С6)алкилсульфонил; Ar означает (С6-С10)арил, (С5-С9)гетероарил, (С1-С6)алкил (С6-С10)арил, (С1-С6)алкокси (С6-С10)арил, ((C1-С6) алкокси)2 (С6-С10) арил, (С6-С10) арилокси (С6-С10) арил или (С5-С9)гетероарилокси (С6-С10) арил.
Термин "алкил", как он использован здесь, если не указано иначе, обозначает насыщенные моновалентные углеводородные радикалы, имеющие заместители с прямой, разветвленной цепью или циклические или их комбинации.
Термин "алкокси", как он использован здесь, обозначает О- алкильные группы, где "алкил" имеет указанные выше значения.
Термин "арил", как он использован здесь, если не указано иначе, обозначает органический радикал, полученный от ароматического углеводорода путем удаления одного водорода, такой как фенил или нафтил, необязательно замещенный 1-3 заместителями, выбираемыми из группы, состоящей из фтора, хлора, трифторметила, (С1-C6)алкоксила, (C6-C10)apилоксила, трифторметоксила, дифторметоксила и (С1-С6)алкила.
Термин "гетероарил", как он использован здесь, если не указано иначе, обозначает органические радикалы, полученные от ароматического гетероциклического соединения, содержащего в качестве гетероатома азот.
Термин "ацил", как он использован здесь, если не указано иначе, обозначает радикалы общей формулы RCO, где R является алкилом, алкоксилом, арилом, арилалкилом или ариалкилоксилом, а термины "алкил" или "арил" определены выше.
Термин "ацилоксил", как он использован здесь, включает О-ацильные группы, где "ацил" определен выше.
Соединения формулы I могут иметь хиральные центры и поэтому существуют в разных энантиомерных формах. Данное изобретение относится ко всем оптическим изомерам и стереомерам соединений формулы I и их смесям.
Предпочтительные соединения формулы I включают те, в которых n равно 2.
Другие предпочтительные соединения формулы I включают те, в которых Ar является 4-метоксифенилом или 4-феноксифенилом.
Другие предпочтительные соединения формулы I включают те, в которых или R3 или R4 не является водородом.
Другие предпочтительные соединения формулы I включают те, в которых n равно I.
Другие предпочтительные соединения формулы I включают те, в которых X является гидроксилом, Ar является 4-метоксифенилом или 4-феноксифенилом, а либо R3 или R4 не является водородом.
Другие предпочтительные соединения формулы I включают те, в которых X является алкоксилом, Ar является 4-метоксифенилом или 4-феноксифенилом, а либо R3 или R4 не является водородом.
Другие предпочтительные соединения формулы I включают те, в которых Ar является 4-метоксифенилом или 4-феноксифенилом, a R3 и R4, взятые вместе, образуют (C3-C6)циклоалканил, оксациклогексанил, тиоциклогексанил, инданил или группу формулы
где R15 является (С1-С6)ацилом, (С1-С6)алкилом, (С6-C10)арил- (С1-С6)алкилом, (С5-С9)гетероарил(С1-С6)алкилом или (C1-C6)- алкилсульфонилом.
Более предпочтительными соединениями формулы I являются, те, в которых n равно 2, Ar является 4- метоксифенилом или 4- феноксифенилом, R1 и R2, взятые вместе, образуют пиперазинил, (С1-С6)алкилпиперазинилом, (С6- С10)арилпиперазинилом или (С5-C9)гетероарил(С1-C6)алкилпиперазинилом, а либо R3 или R4 не является водородом, или оба, R3 и R4, не являются водородом.
Более предпочтительными соединениями формулы I являются те, у которых n равно 2, Ar является 4-метоксифенилом или 4- феноксифенилом, R1 является водородом или (С1-С6)алкилом, R2 является 2-пиридилметилом, 3-пиридилметилом или 4-пиридилметилом, а либо R3 или R4 не является водородом, или оба R3 и R4 не являются водородом.
Более предпочтительными соединениями формулы I являются те, в которых n равно 1, Ar является 4-метоксифенилом или 4-феноксифенилом, R2 является 2-пиридилметилом, 3-пиридилметилом или 4-пиридилметилом, а либо R3 или R4 не является водородом, или оба, R3 и R4, не являются водородом.
Конкретные предпочтительные соединения формулы I включают следующие:
2-(R)-N-гидрокси-2- [(4-метоксибензолсульфонил) (3- морфолин-4-ил-3-оксопропил)амино]-3-метилбутирамид;
2-(R) -2-[(2-бензил карбамоилэтил)(4-метоксибензолсульфонил)амино] -N- гидрокси-3-метилбутирамид;
2-(R)-N-гидрокси-2- ((4-метоксибензолсульфонил)(2-[(пиридин-3-илметил) -карбамоил]этил)амино)-3-метилбутирамид;
2-(R)-N-гидрокси-2-([4-метоксибензолсульфонил] [2-(метилпиридин- З-илметилкарбамоил)этил]амино)-3-метилбутирамид;
4-(3-[1-(R)-1-гидроксикарбамоил-2-метилпропил) (4- метоксибензолсульфонил) амино] пропионил) пиперазин-1-карбоновой кислоты трет-бутиловый эфир;
2-(R)-N-гидрокси-2-[(4- метоксибензолсульфонил) (3-оксо-3-пиперазин-1-илпропил)амино)-3- метилбутирамида гидрохлорид;
2-(R)-2-[(бензилкарбамоилметил) (4- метоксибензолсульфонил) амино] N-гидрокси-3-метилбутирамид;
2-(R)-N-гидрокси-2-([4-метоксибензолсульфонил] -[(2-морфолин-4- илэтилкарбамоил) метил] амино) -3-метилбутирамид и
2-(R)-N-гидрокси-2-((4-метоксибензолсульфонил) ([(пиридин-3-илметил) карбамоил)метил)амино)-3-метилбутирамид.
Другие конкретные соединения формулы I включают следующие:
2-(R)-3, 3, 3-трифтор-N-гидрокси-2- [(метоксибензолсульфонил) (3-морфолин-4-ил-3- оксопропил) амино] пропионамид;
2-(R)-N-гидрокси-2-((4-феноксибензолсульфонил) [2- (метил-пиридин-4-илметилкарбамоил)этил)амино)-3-метилбутирамид;
4-[4-метоксибензолсульфонил)(3-морфолин-4-ил-3-оксопропил) амино] -1-метилпипериден-4-карбоновой кислоты гидроксамид;
2-(R)-N-гидрокси-2-((4-метоксибензолсульфонил) - [3-(4-метилпиперазин-1-ил)-3-оксопропил]амино)-3-метилбутирамид;
2-(R)-2-[(2-карбоксиэтил) (4-метоксибензолсульфонил)амино] -N-гидрокси-3-метилбутирамид;
[(2-карбоксиэтил) (3,4-диметоксибензолсульфонил)амино] -N-гидроксиацетамид;
2-(R)-2-[(2-карбамоилэтил) (4-метоксибензолсульфонил) амино]-N-гидрокси-3-метилбутирамид;
2-(R), 3-(R)-3,N-дигидрокси-2-[(4-метоксибензолсульфонил)-(3-оксо- З-пиперидин-1-илпропил)амино]-бутирамид;
2-(R)-N-гидрокси-2- ((4-метоксибензолсульфонил) [3-(метилпиридин-3- илметилкарбамоил)пропил]амино)-3-метилбутирамид;
2-(R)-N-гидрокси-2-((4-метоксибензолсульфонил) [2- (метилкарбоксиметилкарбамоил) этил] амино)-3-метилбутирамид;
2-(R)-N-гидрокси-2- ((4метоксибензолсульфонил) - [(1-метил-пиперидин-4-илкарбамоил) метил] амино)-3-метилбутирамид;
2-(R)-2-циклогексил-N-гидрокси- 2-((4-метоксибензолсульфонил) - [3- (4-метилпиперазин-1-ил) -3- оксопропил]амино)-ацетамид;
2-(R)-N-гидрокси-2-[(метоксибензолсульфонил) (3-морфолин-4-ил-3- оксопропил) амино]-4-(морфолин-4-ил)бутирамид.
Данное изобретение также относится к фармацевтической композиции для (а) лечения заболевания, выбранного из группы, состоящей из артрита, рака, язв тканей, возвратного стеноза, периодонозного заболевания, врожденного буллезного эпидермолиза, склерита и других заболеваний, характеризующихся активностью матричных протеиназ, СПИДа, сепсиса, септического шока и других заболеваний, которые связаны с продуцированием фактора некроза опухолей (ФНО) или (б) подавления матричных металлопротеиназ или продуцирования фактора некроза опухолей (ФНО) у млекопитающих, включая людей, содержащей количество соединения по пункту 1 или его фармацевтически приемлемой соли, эффективного для такого лечения и фармацевтически приемлемый носитель.
Данное изобретение также относится к способу ингибирования (а) матричных металлопротеиназ или (б) продуцирования фактора некроза опухолей (ФНО) у млекопитающих, включая людей, который включает введение указанному млекопитающему эффективного количества соединения по пункту 1 или его фармацевтически приемлемой соли.
Данное изобретение также относится к способу лечения заболевания, выбранного из группы, состоящей из артрита, рака, язв тканей, повторного стеноза, периодонтозного заболевания, врожденного буллезного эпидермолиза, склерита и других заболеваний, характеризующихся активностью матричных протеиназ, СПИДа, сепсиса, септического шока и других заболеваний, которые связаны с продуцированием фактора некроза опухолей (ФНО) у млекопитающих, включая людей, включающий введение указанному млекопитающему количества соединения по пункту 1 или его фармацевтически приемлемой соли, эффективного для лечения такого заболевания.
Детальное описание изобретения
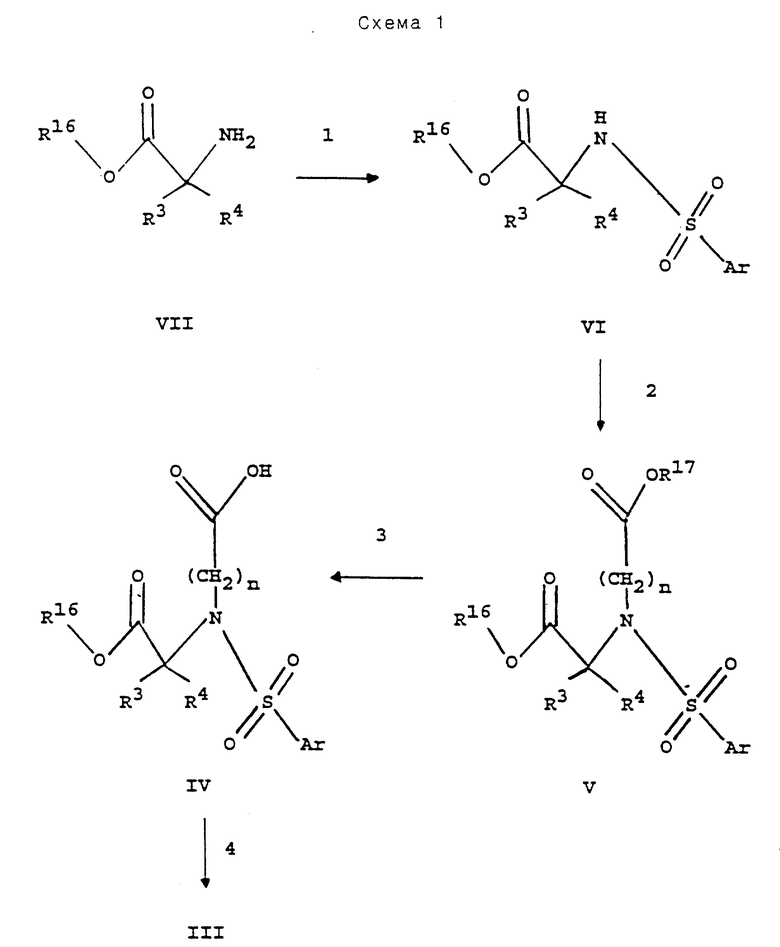
Следующие далее схемы реакций иллюстрируют получение соединений данного изобретения. Если не указано иначе, R1, R2, R3, R4, n и Ar в схемах реакций и в описании, которое следует далее, имеют те же значения, что и указанные выше.
В реакции 1 схемы 1 аминокислотное соединение формулы VII, в котором R16 является (С1-С6алкилом, бензилом, аллилом или трет-бутилом, превращают в соответствующее соединение формулы VI реакцией УП с реактивным функциональным производным арилсульфоновой кислоты, таким как арилсульфонилхлорид, в присутствии основания, такого как триэтиламин, и полярного растворителя, такого как тетрагидрофуран, диоксан, вода или ацетонитрил, предпочтительно, смеси диоксана и воды. Реакционную смесь перемешивают при комнатной температуре в течение периода времени от примерно 10 минут до примерно 24 часов, предпочтительно, примерно 60 минут.
В реакции 2 схемы 1 арилсульфониламино-соединение формулы VI, в котором R16 является (С1-С6алкилом, бензилом, аллилом или трет-бутилом, превращают в соответствующее соединение формулы V, в котором n равно 1,3,4,5 или 6, реакцией VI с реактивным производным спирта формулы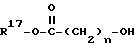
таким как хлорид, бромид или иодид, предпочтительно, бромид, где R17 защитная группа является (С1-С6)алкилом, бензилом, аллилом или трет-бутилом, в присутствии основания, такого как карбонат калия или гидрид натрия, предпочтительно, гидрид натрия, и полярного растворителя, такого как диметилформамид. Реакционную смесь перемешивают при комнатной температуре в течение периода времени от примерно 60 минут до примерно 48 часов, предпочтительно, примерно 18 часов. R17 защитную группу выбирают так, чтобы ее можно было селективно удалить в присутствии и без потери R16 защитной группы, поэтому R17 не может быть такой же, как и R16. Удаление R17 защитной группы из соединения формулы V, с получением соответствующей карбоновой кислоты формулы IV, в реакции 3 схемы 1, проводится в условиях, подходящих для этой конкретной используемой R17 защитной группы, которые не влияют на R16 защитную группу. Такие условия включают: (а) омыление, когда R17 является (С1-С6)алкилом, a R16 является трет-бутилом, (б) гидрогенолиз, когда R17 является бензилом, a R16 является трет-бутилом или (С1-С6)алкилом, (в) обработка сильной кислотой, такой как трифторуксусная кислота или хлористоводородная кислота, когда R17 является трет-бутилом, a R16 является (C1-С6)алкилом, бензилом или аллилом, или (г) обработка трибутил-оловогидридом и уксусной кислотой в присутствии каталитического бис(трифенилфосфин) палладия (П) хлорида, когда R17 является аллилом, a R16 является (С1-С6)алкилом, бензилом или трет-бутилом.
В реакции 4 схемы 1 карбоновую кислоту формулы IV конденсируют с амином, R1R2NH, или их солью с получением соответствующего амидного соединения формулы III. Образование амидов из первичных или вторичных аминов или аммиака и карбоновых кислот достигается путем превращения карбоновой кислоты в активированное функциональное производное, которое в дальнейшем реагирует с первичным или вторичным амином или аммиаком с образованием амида. Активированное функциональное производное может быть выделено перед реакцией с первичным или вторичным амином или аммиаком. Альтернативно карбоновую кислоту можно обработать оксалилхлоридом или тионилхлоридом, чистым или в инертном растворителе, таком как хлороформ, при температуре от примерно 25oC до примерно 80oC, предпочтительно, при 50oC, с получением соответствующего функционального хлоридного производного кислоты. Инертный растворитель и оставшийся оксалилхлорид или тиотилхлорид затем удаляют выпариванием в вакууме. Оставшееся функциональное хлоридное производное кислоты затем реагирует с первичным или вторичным амином или аммиаком в инертном растворителе, таком как метиленхлорид с образованием амида. Предпочтительным методом конденсации карбоновой кислоты формулы IV с амином для получения соответствующего амидного соединения формулы III является обработка IV (бензотриазол-1-илокси)трис(диметиламино)фосфония гексафторфосфатом в присутствии основания, такого как триэтиламин с получением in situ бензотриазол-1-окси-эфира, который в свою очередь реагирует с амином, R1R2N, в инертном растворителе, таком как метиленхлорид, при комнатной температуре с получением амидного соединения формулы III.
Удаление R16 защитной группы у соединения формулы III с получением соответствующей карбоновой кислоты формулы II в реакции 5 схемы 1, проводят в условиях, подходящих для конкретной используемой R16 защитной группы. Такие условия включают: (а) омыление, когда R16 является низшим алкилом, (б) гидрогенолиз, когда R16 является бензилом, (в) обработку сильной кислотой, такой как трифторуксусная кислота или хлористоводородная кислота, когда R16 является трет-бутилом или (г) обработку гидридом трибутилолова и уксусной кислотой в присутствии каталитического бис (трифенилфосфин) палладия (П) хлорида, когда R16 является аллилом.
В реакции 6 схемы 1 соединение карбоновой кислоты формулы II превращают в соединение гидроксамовой кислоты формулы I обработкой П 1-(3-диметиламинопропил)-3-этилкарбодиимидом и 1-гидроксибензтриазолом в полярном растворителе, таком как диметилформамид, с последующим добавлением гидроксиламина к реакционной смеси через от примерно 15 минут до примерно 1 часа, предпочтительно через 30 минут. Предпочтительно, гидроксиламин образуется in situ, из солевой формы, такой как гидрохлорид гидроксиламина, в присутствии основания, такого как N- метилморфолин. Альтернативно, может быть использовано защищенное производное гидроксиламина или его соль, где гидроксильная группа защищена путем получения третбутилового, бензилового или аллилового эфира, в присутствии (бензотриазол-1- илокси)трис(диметиламино)фосфония гексафторфосфата и основания, такого как N-метилморфолин. Удаление защитной группы гидроксиламина производится гидрогенолизом в случае бензильной защитной группы или обработкой сильной кислотой, такой как трифторуксусная кислота, в случае третбутильной защитной группы. Аллильная защитная группа может быть удалена обработкой гидридом трибутилолова и уксусной кислотой в присутствии каталитического бис (трифенилфосфин) палладия (П) хлорида. В качестве защищенного гидроксиламинового производного можно также использовать N,0- бис(4-метоксибензил)гидроксиламин, причем удаление защиты достигается с применением смеси метансульфоновой кислоты и трифторуксусной кислоты.
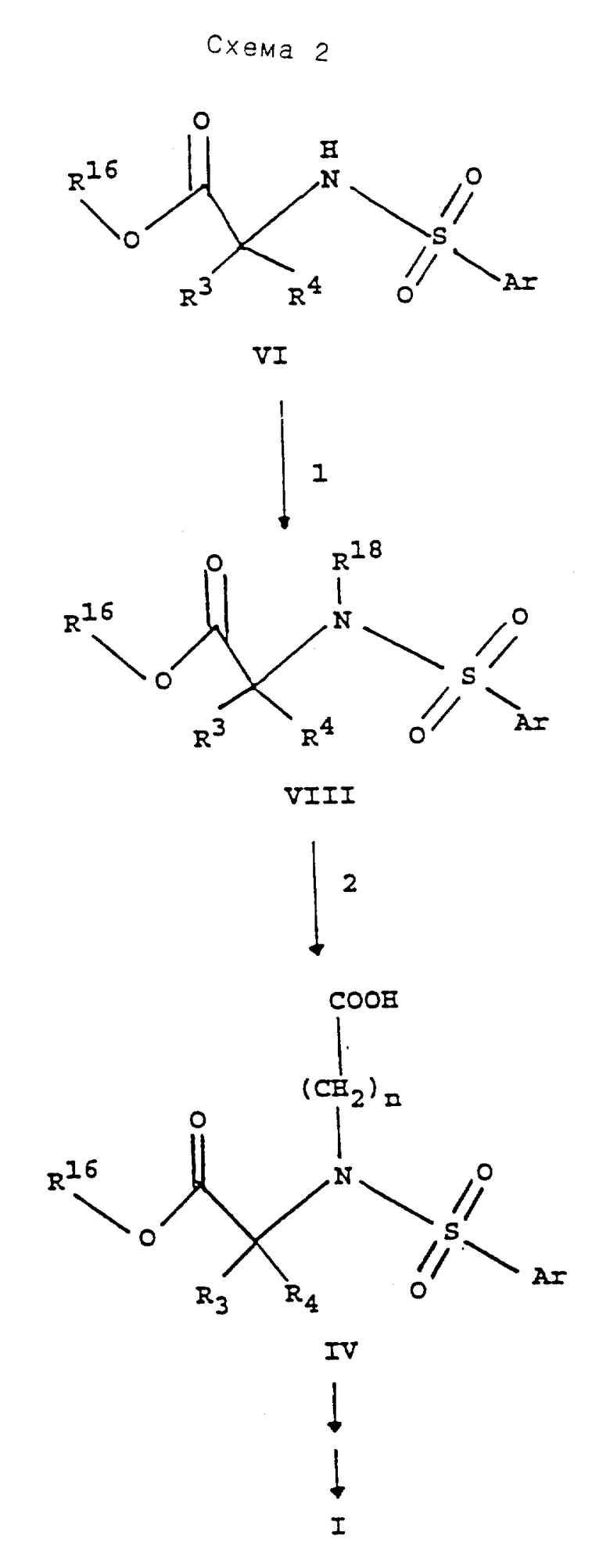
В реакции 1 схемы 2 арилсульфонаминовое соединение формулы VI, где R16 является (С1-С6)алкилом, бензилом или трет-бутилом, превращают в соответствующее соединение формулы VIII, где R18 является 2-пропенилом или З-бутенилом, реакцией IX с реактивным функциональным производным, таким как галогенид, предпочтительно йодным производным, в 2-пропен-1-ол, когда R18 является 2-пропенилом или в З-бутен-1-ол, когда R18 является 3-бутенилом, в присутствии основания, такого как карбонат калия, карбонат цезия или гидрид натрия, предпочтительно, гидрид натрия, когда R18 является 2-пропенилом, или карбонат цезия, когда R18 является 3-бутенилом. Реакционную смесь перемешивают в полярном растворителе, таком как диметилформамид, при комнатной температуре в течение периода времени, равного от примерно 2 часов до примерно 48 часов, предпочтительно, в течение 18 часов.
В реакции 2 схемы 2 соединение по формуле VIII превращают в соединение карбоновой кислоты формулы IV, где n равно 2. Соединение формулы VIII, где R18 является 2-пропенилом, превращают в соединение формулы IV, где n равно 2, реакцией VIII с боран-диметилсульфидным комплексом с последующим немедленным окислением с использованием трехокиси хрома в водной уксусной кислоте. Окислительное отщепление концевых олефинов до карбоновых кислот может быть проведено несколькими известными в этой области технологии методами. Предпочтительный метод окислительного расщепления соединения формулы VIII, где R18 является 3-бутенилом для получения карбоновой кислоты формулы IV состоит в том, что соединение VIII реагирует с перйодатом натрия в присутствии каталитического количества хлорида рутения (III) в смеси четыреххлористого углерода, ацетонитрила и воды.
Соединение формулы IV, где n равно 2, далее реагирует с получением соединения гидроксамовой кислоты формулы I, где n равно 2, по методике, описанной выше для реакций 4, 5 и 6 схемы 1.
Альтернативный способ синтеза соединения гидроксамовой кислоты формулы I, где n равно 1, a R3 и R4 оба являются водородами, показан в реакции 1 схемы 3, начиная с реакции иминоуксусной кислоты или ее соли с металлом или аммонием формулы X с функциональным производным соединения арилсульфоновой кислоты, таким как арилсульфонилхлорид, при комнатной температуре в присутствии подходящего основания, такого как триэтиламин, и полярного растворителя, такого как тетрагидрофуран, диоксан, вода или ацетонитрил, предпочтительно, смеси диоксана и воды, с получением соответствующего соединения дикарбоновой кислоты формулы XI.
В реакции 2 схемы 3 соединение дикарбоновой кислоты формулы XI дегидрируют с получением циклического ангидрида формулы XII. Образование циклических ангидридов дегидрированием дикарбоновых кислот может быть осуществлено различными способами. Предпочтительный способ дегидрирования дикарбоновой кислоты формулы XI с получением циклического ангидрида формулы XII состоит в обработке XI избытком уксусного ангидрида при температуре в интервале от примерно 25oC до примерно 80oC, предпочтительно при примерно 60oC. Избыток уксусного ангидрида и уксусной кислоты, побочный продукт реакции, удаляют выпариванием при пониженном давлении, причем остается циклический ангидрид формулы XII.
В реакции 3 схемы 3 циклический ангидрид формулы XII реагирует при комнатной температуре с амином, NR1R2, или солью этого амина, такой как гидрохлорид, в присутствии основания, такого как триэтиламин, с получением карбоновой кислоты формулы II, где n равно 1, a R3 и R4 оба являются водородами. Подходящими растворителями для реакции являются те, которые не будут реагировать с исходными материалами, и которые включают хлороформ, метиленхлорид и диметилформамид, предпочтительно, метиленхлорид.
Соединение формулы II далее реагирует с получением соединения гидроксамовой кислоты формулы I, где n равно 1, a R3 и R4 оба являются водородом, в соответствии с методикой, описанной выше для реакции 6 схемы 1.
В реакции 1 схемы 4 соединение карбоновой кислоты формулы IV, где n равно 2, превращают в соответствующее соединение формулы V, где R19 является (C1-C6)aлкилом или трет-бутилом, реакцией IV с соединением формулы
(R19O)2CHN(CH3)2,
где R19 является (С1-C6)алкилом или трет-бутилом, в инертном растворителе, таком как толуол, при температуре в интервале от примерно 60oC до примерно 100oC, предпочтительно, примерно 100oC, в течение периода времени от примерно 1 часа до примерно 3 часов, предпочтительно, 2 часов. В реакции 2 схемы 4 арилсульфониламино-соединение формулы VI, где n равно 1, 3,4,5 или 6, a R16 является (С1-С6)алкилом, бензилом, аллилом или трет-бутилом, превращают в соответствующее соединение формулы XIII, где R19 является (С1-C6)алкилом или трет-бутилом, реакцией VI с реактивным производным спирта формулы
таким, как хлоридное, бромидное или йодное производное, предпочтительно бромидное производное, где R19 является (С1-С6)- алкилом или трет-бутилом, в присутствии основания, такого как карбонат калия или гидрид натрия, предпочтительно гидрид натрия, и полярного растворителя, такого как диметилформамид. Реакционную смесь перемешивают при комнатной температуре в течение периода времени от примерно 60 минут до примерно 48 часов, предпочтительно в течение примерно 18 часов. Защитная группа R16 соединений формулы IV и VI выбирается так, чтобы ее можно было селективно удалить в присутствии и без потери R19 (С1-С6)алкильной или трет-бутильной группы, поэтому R16 не может быть такой же, как и R19. Удаление защитной группы R16 у соединения формулы XIII с получением соответствующей карбоновой кислоты формулы XIV, где n равно 1-6, в реакции 3 схемы 4 производят в условиях, подходящих для данной конкретной использованной R16 защитной группы, которые не будут влиять на R19(С1-С6)алкильную или трет-бутильную группу. Такие условия включают: (а) омыление, когда R16 является (С1-С6)алкилом, a R19 является трет-бутилом, (б) гидрогенолиз, когда R16 является бензилом, a R19 является трет-бутилом или (C1-С6)алкилом, (в) обработку сильной кислотой, такой как трифторуксусная кислота или хлористоводородная кислота, когда R19 является трет-бутилом, а R19 является (С1-C6)алкилом, или (г) обработку гидридом трибутилолова и уксусной кислотой в присутствии каталитического бис (трифенилфосфин) палладия (П) хлорида, когда R16 является аллилом, a R19 является (С1-С6)-алкилом или трет-бутилом.
В реакции 4 схемы 4 карбоновую кислоту формулы XIV превращают в соединение гидроксамовой кислоты формулы XV, где n равно 1-6, обработкой XIV 1-(3-диметиламинопропил)-3-этилкарбодиимидом и 1-гидроксибензтриазолом в полярном растворителе, таком как диметилформамид с последующим добавлением гидроксиламина в реакционную смесь через от примерно 15 минут до примерно 1 часа, предпочтительно примерно через 30 минут. Гидроксиламин предпочтительно получают in situ из солевой формы, такой как гидроксиламина гидрохлорид, в присутствии основания, такого как N- метилморфолин. Альтернативно, защищенное производное гидроксиламина или его солевую форму, где гидроксильная группа защищена в виде трет-бутилового, бензилового или аллилового эфира, может использоваться в присутствии (бензотриазол-1-илокси) трис (диметиламино) фосфония гексафторфосфата и основания, такого как N-метилморфолин. Удаление групп, защищающих гидроксиламин, производится гидрированием в случае бензильной защитной группы или обработкой сильной кислотой, такой как трифторуксусная кислота, в случае трет-бутильной защитной группы. Аллильная защитная группа может быть удалена обработкой гидридом трибутилолова и уксусной кислотой в присутствии каталитического бис (трифенилфосфин) палладия (П) хлорида. В качестве защищенного производного гидроксиламина может также использоваться N,0-бис(4-метоксибензил) гидроксиламин, когда R19 является (С1-С6)алкилом, в этом случае удаление защитной группы проводится при использовании смеси метансульфоновой кислоты и трифторуксусной кислоты.
В реакции 5 схемы 4 амидное соединение формулы XV, если требуется, превращается в соответствующее соединение карбоновой кислоты формулы XVI (а) омылением, когда R19 является низшим алкилом или (б) обработкой сильной кислотой, такой как трифторуксусная кислота или хлорводородная кислота, когда R19 является трет-бутилом.
Фармацевтически приемлемыми солями кислотных соединений данного изобретения являются соли, образованные с основаниями, то есть катионные соли, такие как соли щелочных и щелочноземельных металлов, таких как натрий, литий, калий, кальций, магний, а также соли аммония, триметиламмония, диэтиламмония и трис-(гидроксиметил) -метиламмония.
Подобным же образом возможно получение солей присоединения кислот, таких как минеральные кислоты, органические карбоновые и органические сульфоновые кислоты, например, хлористоводородная кислота, метансульфоновая кислота, малеиновая кислота, благодаря тому, что основная группа, такая как пиридильная, составляет часть структуры.
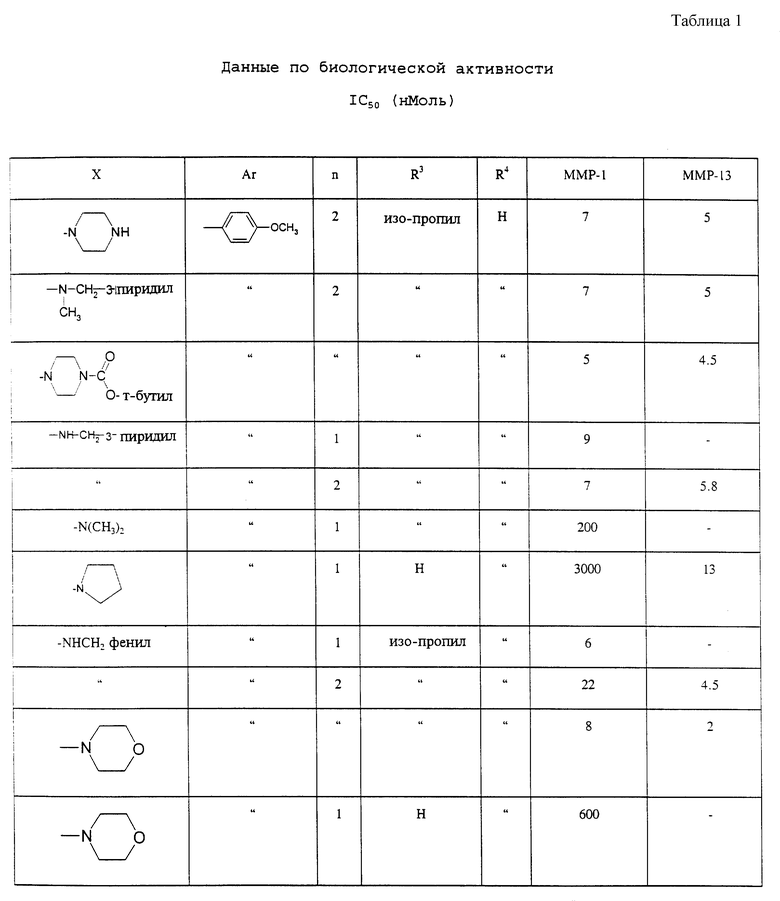
Способность соединений формулы I или их фармацевтически приемлемых солей (здесь далее называемых соединениями данного изобретения) подавлять матричные металлопротеиназы или продуцирование фактора некроза опухолей (ФНО) и, следовательно, демонстрация их эффективности для лечения заболеваний, характеризующихся активностью матричных протеиназ или продуцированием фактора некроза опухолей, показана следующими исследованиями in vitro.
Биологическое испытание на ингибирование человеческой коллагеназы (ММР-1)
Человеческую рекомбинантную коллагеназу активируют трипсином, используя следующее соотношение: 10 мкг трипсина на 100 мкг коллагеназы. Трипсин и коллагеназу инкубируют при комнатной температуре в течение 10 минут, затем добавляют пятикратный избыток (50 мкг/10 мкг трипсина) ингибитора трипсина соевых бобов.
Готовят стандартные 10 мМ растворы ингибиторов в диметилсульфоксиде и затем разбавляют их, используя следующую схему.
10 мМ ---> 120 мкМ ---> 12 мкМ ---> 1,2 мкМ ---> 0,12 мкМ
Двадцать пять микролитров раствора каждой концентрации затем добавляют в три повторные соответствующие ячейки 96-ячеечного микротитровального плата. Конечная концентрация ингибитора будет разведением 1:4 после добавления фермента и субстрата. Положительные контроли (фермент, без ингибитора) помещают в ячейки D1-D6, а растворы сравнения (без фермента, без ингибиторов) помещают в ячейки D7-D12.
Коллагеназу разводят до 400 нг/мл и затем добавляют в соответствующие ячейки микротитровального плата. Конечная концентрация коллагеназы в исследовании составляет 100 нг/мл.
Субстрат (DNP-Pro-Cha-Gly-Cys (Me) -His-Ala-Lys (NMA) - NH2) готовят в виде 5 мМ стандартного раствора в диметилсульфоксиде и затем разводят до 20 мкМ буфером для исследований. Исследование начинают добавлением 50 мкл субстрата в ячейку микротитровального плата с получением конечной концентрации 10 мкМ.
Показатели флуоресценции (возбуждение 360 нм, излучение 460 нм) определяли в момент 0 и затем через 20-минутные интервалы. Исследование проводят при комнатной температуре, причем обычное время исследования равно 3 часам.
Затем строили график зависимости флюоресценции от времени как для растворов сравнения, так и для образцов, содержащих коллагеназу (берут средние значения, определенные из трех повторений). Момент времени, в который получен хороший сигнал (раствор сравнения) и который находится на линейной части кривой (обычно около 120 минут) выбран для определения значений IC50. Нулевой момент времени использован в качестве сравнения для каждого соединения при каждой концентрации, и эти значения вычитали из показаний для 120 минут. Строили график зависимости концентрации ингибитора от % контроля (флюоресценция раствора с ингибитором деленная на флюоресценцию раствора с одной коллагеназой х 100). Значения IC50 определены по концентрации ингибитора, которая дает сигнал, равный 50% контрольного.
Если показано, что значения IC50 < 0,03 мкМ, то ингибиторы оценивают при концентрациях 0,3 мкМ, 0,03 мкМ и 0,003 мкМ.
Ингибирование желатиназы (ММР-2)
Ингибирование активности желатиназы исследовали, используя субстрат Dnp-Pro-Cha-Gly-Cys(Me)-His-Ala-Lys(NMA)-NH2 (10 мкМ) в тех же самых условиях, как и при ингибировании человеческой коллагеназы (ММР-1).
Желатиназу в 72 кД активируют 1 мМ АФРА (n-аминофенил-ртути ацетата) в течение 15 часов при 4oC и разводят с получением конечной концентрации для исследования, равной 100 мг/мл. Ингибиторы разводят таким же образом, как и для определения ингибирования человеческой коллагеназы (ММР-1) с получением конечных концентраций исследования, равных 30 мкМ, 3 мкМ, 0,3 мкМ и 0,03 мкМ. Каждую концентрацию исследуют трижды.
Показатели флюоресценции (возбуждение 360 нм, излучение 460 нм) определяли в нулевой момент времени и затем через 20-минутные интервалы в течение 4 часов.
Значения IC50 определены как и при ингибировании человеческой коллагеназы (ММР-1). Если сообщено, что значения IC50 менее 0,03 мкМ, то ингибиторы исследовали при конечных концентрациях, равных 0,3 мкМ, 0,03 мкМ, 0,003 мкМ и 0,003 мкМ (см. табл. 1).
Ингибирование активности стромелизина (ММР-З)
Изучение ингибирования активности стромелизина основано на модифицированном спектрофотометрическом исследовании, описанном Weingarten Н. and Feder J. (Weingarten H. and Feder J., Spectrophotometric Assay for Vertebrate Collagenase, Anal. Boochem. 147, 437-440 (1985)). Гидролиз тиопептолидного субстрата [Ac-Pro-Leu-Gly-SCH[CH2CH(CH3)2] CO-Leu-Gly-OC2H5] дает меркаптановый фрагмент, который может быть зафиксирован в присутствии реагента Эллмана.
Человеческий рекомбинантный простромелизин активируют трипсином, используя соотношения: 1 мкл стандартного раствора трипсина с концентрацией 10 мг/мл на 26 мкг стромелизина. Трипсин и стромелизин инкубируют при 37oC в течение 15 минут с последующей инкубацией с 10 мкл раствора ингибитора соевого трипсина с концентрацией 10 мг/мл в течение 10 минут при 37oC для погашения активности трипсина.
Исследования проводили в общем объеме 250 мкл буфера для исследований (200 мМ хлорида натрия, 50 мМ MES и 10 мМ хлорида кальция, рН 6,0) в 96-ячеечных платах. Активированный стромелизин разводят буфером для исследований до 25 мкг/мл. Реактив Эллмана (3-карбокси-4-нитрофенилдисульфид) готовят в виде 1М стандартного раствора в диметилформамиде, разводят до 5 мМ буфером для исследований, получая 50 мкл на ячейку с конечной концентрацией 1 мМ.
Готовят 10 мМ стандартные растворы ингибиторов в диметилсульфоксиде и разводят серийно буфером для исследований так, что добавление 50 мкл в соответствующие ячейки дает конечные концентрации, равные 3 мкМ, 0,3 мкМ. 0,003 мкМ и 0.0003 мкМ. Все испытания проводились в трех повторных ячейках.
300 мМ стандартного раствора пептидного субстрата в диметилсульфоксиде разводят до 15 мМ буфером для исследования, и исследование начинают добавлением 50 мкл этого раствора в каждую ячейку с получением конечной концентрации 3 мМ субстрата. Растворы сравнения состоят из пептидного субстрата и реагента Эллмана без фермента. Образование продукта фиксировали при 405 нм с помощью считывающего устройства для плат Molecular Devices Uvmax.
Значения IC50 определяли таким же образом, как и для коллагеназы.
Ингибирование ММР-13
Человеческую рекомбинантную ММР-13 активируют с помощью 2 мМ АФРА ацетата n-аминофенилртути в течение 1,5 часов при 37oC и разводят до 400 мг/мл буфером для исследований (50 мМ Трис, рН 7,5, 200 мМ хлорида натрия, 5 мМ хлорида кальция, 20 мкМ хлорида цинка, 0,02% brij). Добавляют двадцать пять микролитров разведенного фермента на ячейку 96- ячеечного плата. Фермент затем при исследовании разводят в соотношении 1:4 добавлением ингибитора и субстрата с получением конечной концентрации исследования, равной 100 мг/мл.
10 мМ стандартные растворы ингибиторов готовят в диметилсульфоксиде и затем разводят буфером для исследований, также по схеме разведения ингибитора для ингибирования человеческой коллагеназы (ММР-1). Двадцать пять микролитров раствора каждой концентрации добавляют в трехкратном повторении в ячейки микротитровального плата (для определения флюоресценции). Конечные концентрации при исследовании составляют 30 мкМ, 3 мкМ, 0,3 мкМ и 0,03 мкМ.
Субстрат (Dnp-Pro-Cha-Gly-Cys (Me) -His-Ala-Lys (NMA)-NH2) готовят так же, как и для ингибирования человеческой коллагеназы (ММР-1) и добавляют в каждую ячейку 50 мкл с получением конечной концентрации исследования, равной 10 мкМ.
Показатели флюоресценции (возбуждение - 360 нм, излучение - 450 нм) снимали в момент времени 0 и через каждые 5 минут в течение 1 часа.
Положительные контроли включали фермент и субстрат без ингибитора, а растворы сравнения включали только субстрат.
IC50 определяют так же, как и при подавлении человеческой коллагеназы (ММР-1). Если показано, что IC50 менее 0,03 мкМ, ингибиторы затем оценивали при конечных концентрациях, равных 0,3 мкМ, 0,03 мкМ, 0,003 мкМ и 0,0003 мкМ.
Ингибирование продуцирования ФНО
Способность соединений или их фармацевтически приемлемых солей ингибировать продуцирование ФНО, и, следовательно, демонстрация их эффективности для лечения заболеваний, включающих продуцирование ФНО, показана следующими исследованиями in vitro.
Человеческие моноядерные клетки были выделены из человеческой антикоагулированной крови с использованием одностадийной методики выделения Фиколл-гипак (Ficoll-hypaque). Моноядерные клетки промывали три раза сбалансированным солевым раствором Хенкса (CCPX) с двухвалентными катионами и повторно суспендировали до плотности 2 • 106 мл в CCPX, содержащем 1% БСА. Дифференциальные числа, определенные с использованием анализатора клеток Abbot Dyn 3500, показали, что моноциты в этих препаратах составляли от 17 до 24% от всего числа клеток.
Образцы по 180 мкл клеточной суспензии помещали в плоскодонные 96-ячеечные платы (Costar). Добавление соединений и ЛПС (конечная концентрация 100 нг/мл) давало конечный объем 200 мкл. Все испытания повторялись трижды. После четырехчасовой инкубации при 37oC в увлажняемом инкубаторе при повышенной концентрации CO2 платы вынимали и центрифугировали (10 минут при примерно 250 об/мин), и супернатант удаляли и исследовали на присутствие DHO α , используя набор ELlSA R&D).
Для введения людям для ингибирования матричных металлопротеиназ или продуцирования фактора некроза опухолей (ФНО), можно использовать ряд обычных способов введения, включая пероральный, парентеральный и местный. Как правило, активное соединение может быть введено перорально или парентерально в дозах от примерно 0,1 до 25 мг/кг веса тела субъекта в сутки, который нуждается в лечении, предпочтительно от примерно 0,3 до 5 мг/кг. Однако, при необходимости могут быть сделаны некоторые изменения дозы в зависимости от состояния субъекта, которого нужно лечить. Лицо, ответственное за назначение препарата в любом случае будет определять дозу, подходящую для конкретного субъекта.
Соединения данного изобретения можно вводить в виде ряда различных дозированных форм, в основном терапевтически эффективные соединения этого изобретения присутствуют в таких дозированных формах в концентрации, в интервале от примерно 5% до примерно 70% по весу.
Для перорального введения могут использоваться таблетки, содержащие различные наполнители, такие как микрокристаллическая целлюлоза, цитрат натрия, карбонат кальция, двузамещенный кальция фосфат и глицин вместе с разными разрыхлителями, такими как крахмал (и предпочтительно крахмал кукурузы, картофеля или тапиоки), альгиновая кислота и некоторые сложные силикаты вместе со связывающими веществами для грануляции, такими как поливинилпирролидон, сахароза, желатин и камедь. Дополнительно, для таблетирования предпочтительно использовать смазывающие вещества, такие как стеарат магния, лаурилсульфат натрия и тальк. Твердые композиции подобного типа могут также использоваться в качестве наполнителей для желатиновых капсул предпочтительные ингредиенты в данном случае включают лактозу или молочный сахар, а также высокомолекулярные полиэтиленгликоли. Когда для перорального введения желательны водные суспензии и/или эликсиры, активный ингредиент может сочетаться с различными подслащивающими или улучшающими вкус веществами, подкрашивающими веществами или красителями и, если желательно, с эмульгирующими веществами и/или суспендирующими веществами, а также вместе с такими разбавителями (растворителями) как вода, этанол, пропиленгликоль, глицерин и разные их комбинации.
Для парентерального введения (внутримышечного, внутриперитонального, подкожного и внутривенного) готовят стерильный раствор активного ингредиента для инъекций. Могут использоваться растворы терапевтического соединения данного изобретения или в кунжутном или в арахисовом масле или в водном пропиленгликоле. Водные растворы должны быть соответствующим образом доведены и забуферены, предпочтительно до рН более 8, если необходимо, жидкий растворитель делают сначала изотонически. Эти водные растворы пригодны для внутривенных инъекций. Масляные растворы пригодны для внутриартериальных, внутримышечных и подкожных инъекций. Получение этих растворов в стерильных условиях легко выполняется с помощью стандартных фармацевтических методов, хорошо известных специалистам.
Данное изобретение иллюстрируется следующими примерами, но не ограничивается их деталями.
Пример 1
2-(R)-N-Гидрокси-2-[(метоксибензолсульфонил) (2- морфолин-4-ил-2-оксоэтил)амино]-3-метилбутирамид
К раствору гидрохлорида бензилового эфира D-валина (2,4 грамма, 10 ммолей) и триэтиламина (2,5 грамма, 3,5 мл, 25 ммолей)в воде (50 мл) и 1,4-диоксана (50 мл) добавляли 4-метоксибензолсульфонилхлорид (2,3 грамма, 11 ммолей). Смесь перемешивали при комнатной температуре в течение 1 часа и затем большую часть растворителя удаляли выпариванием в вакууме.
Смесь разводили этилацетатом и последовательно промывали разбавленным раствором хлористоводородной кислоты, водой и раствором соли. Органический раствор сушили над сульфатом магния и концентрировали, получая и-(4-метоксибензолсульфонил)-D- валинбензиловый эфир в виде белого твердого вещества, 3,6 грамма (97%); т.пл. 92-94oC.
N-(4-метоксибензолсульфонил)-D- валинбензиловый эфир (1,50 грамм, 4,0 ммоля) добавляли к суспензии гидрида натрия (0,1 грамма, 4,2 ммоля) в сухом диметилформамиде (20 мл) и через 30 минут добавляли трет-бутилбромацетат (0,8 мл, 4,2 ммоля). Полученную смесь перемешивали в течение ночи при комнатной температуре и затем реакцию останавливали добавлением насыщенного раствора хлорида аммония (3 мл). Диметилформамид удаляли выпариванием в вакууме. Остаток растворяли в этилацетате и промывали водой и раствором соли. После высушивания над сульфатом магния этилацетат выпаривали, получая масло, из которого выделяли бензиловый эфир 2-(R)-2-[трет-бутоксикарбонилметил (4- метоксибензолсульфонил) амино] -3-метилмасляной кислоты с использованием тонкослойной хроматографии на силикагеле и элюирования 15% этилацетатом в гексане в виде прозрачного масла (1,92 г, 98%).
К холодному (0oC раствору бензилового эфира 2-(R)-2-
[трет-бутоксикарбонилметил (4-метоксибензолсульфонил)амино] -3-метилмасляной кислоты (1,92 грамма, 3,9 ммоля) в метиленхлориде (28 мл) добавляли трифторуксусную кислоту (7 мл). Полученному раствору давали нагреться до комнатной температуры и перемешивали в течение ночи. Метиленхлорид и трифторуксусную кислоту выпаривали в вакууме, получая бензиловый эфир 2-(R)-2- [карбоксиметил(4-метоксибензолсульфонил)амино)] - 3-метилмасляной кислоты в виде прозрачного масла, 1,70 грамм (100%).
К раствору бензилового эфира 2-(R)-2- [карбоксиметил(4-метоксибензолсульфонил)амино)] -3-метилмасляной кислоты (573 мг, 1,32 ммоля) в метиленхлориде (12 мл) добавляли последовательно триэтиламин (0,46 мл, 3,28 ммоля), морфолин (0,127 мл, 1,46 ммоля) и гексафторфосфат (бензотриазол-1-илокси)трис (диметиламино)фосфония (646 мг, 1,46 ммоля). Смесь перемешивали при комнатной температуре в течение ночи и затем разводили этилацетатом. Раствор промывали 0,5 N раствором хлористоводородной кислоты и солевым раствором, сушили над сульфатом магния и концентрировали в вакууме. Остаток хроматографировали на силикагеле с использованием 40% этилацетата в гексане, получая бензиловый эфир 2-(R)-2-[(4-метоксибензолсульфонил) (2-морфолин-4- илоксоэтил)амино]-3-метилмасляной кислоты в виде прозрачного масла, 590 мг (89%).
К раствору бензилового эфира 2-(R)-2-[(4- метоксибензолсульфонил) (2-морфолин-4-ил-2-оксиэтил)амино] -3- метилмасляной кислоты (590 мг, 1,17 ммоля) в этаноле (50 мл) добавляли 10% палладия на активированном угле (200 мг). Смесь взбалтывали при давлении 3 атмосферы водорода в шейкере Парра в течение 2 часов. Катализатор удаляли фильтрацией через найлон (размер пор 0,45 мкм) и растворитель выпаривали, получая 2-(R)-2-[(4- метоксибензолсульфонил) (2-морфолин-4-ил-2-оксоэтил) амино] -3- метилмасляную кислоту в виде белой пены, 485 мг (100%).
К раствору 2-(R)-2-[(4-метоксибензолсульфонил) (2-морфолин-4-ил-2-оксоэтил) амино]-3-метилмасляной кислоты (485 мг, 1,17 ммоля) в метиленхлориде (12 мл) добавляли последовательно триэтиламин (0,52 мл, 3,71 ммоля), гидрохлорид о-бензилгидроксиламина (205 мг, 1,28 ммоля) и гексафторфосфат (бензотриазол-1-илокси)трис (диметиламино)фосфония (570 мг, 1,29 ммоля). Смесь перемешивали при комнатной температуре в течение ночи и затем разводили этилацетатом. Раствор промывали последовательно 0,5 N раствором хлористоводородной кислоты, водой, насыщенным раствором гидрокарбоната натрия и солевым раствором, сушили над сульфатом магния и концентрировали в вакууме. Остаток хроматографировали на силикагеле, используя 20% гексан в этилацетате с получением 2-(R)-N-бензилокси-2-[(4- метоксибензолсульфонил) (2-морфолин-4-ил-2-оксоэтил)амино] - метилбутирамида в виде белой пены, 510 мг (84%).
К раствору 2-(R)-N-бензилокси-2-[(4-метоксибензолсульфонил) (2-морфолин-4-ил-2-оксоэтил) амино]-3-метилбутирамида (510 мг, 0,98 ммоля) в метаноле (50 мл) добавляли 5% палладия на активированном угле (120 мг). Смесь взбалтывали при 2 атмосферах водорода в шейкере Парра в течение 2 часов. Катализатор удаляли фильтрованием через найлон (размер пор 0,45 мкм) и растворитель выпаривали, получая 2-(R)-N-гидрокси-2-[(метоксибензолсульфонил) (2-морфолин-4-ил-2- оксоэтил)амино] -3-метилбутирамид в виде белого твердого вещества, 418 мг
(99%); 1H-ЯМР (CDCI3): δ 10,3 (шир. с.,1H), 7,90 (шир. с, 1H, перекрытый), 7,86 (д, J =8,8 Гц, 2H, перекрытый), 6,94 (д, J = 8,8 Гц, 2H), 4,39 (д, J= 17,1 Гц, 1H), 4,09(д, J= 17,1 Гц, 1H), 3,84(с, 3H), 3,80-3,48(м, 9H), 2,20-1,95(м, 1H), 0,82(д, J=6,5 Гц, 3H), 0,45(д, J=6,5Гц, 3H), MC(LSIMS): м/з 430 (М+Н).
Пример 2
2-(R)-N-гидрокси-2-[(4-метоксибензолсульфонил) (3-морфолин-4-ил- З-оксопропил) амино]-3-метилбутирамид
К раствору N-(4-метоксибензолсульфонил)-D-валинбензилового эфира (2,2 грамма, 5,83 ммоля) в сухом диметилформамиде (40 мл) добавляли карбонат цезия (2,3 грамма, 7,1 ммоля) и 1-иод-3-бутен (1,3 г, 7,1 ммоля). Смесь перемешивали при комнатной температуре в течение ночи и затем выливали в воду. Смесь экстрагировали дважды эфиром и объединенные эфирные экстракты промывали солевым раствором, сушили над сульфатом магния и концентрировали при пониженном давлении. Остаток собирали в 20% этилацетат/гексан; исходный материал N-(4-метоксибензолсульфонил)-D-валинбензиловый эфир (1,5 г) кристаллизовали из смеси и отделяли фильтрованием. Фильтрат концентрировали в вакууме и остаток хроматографировали на силикагеле, используя 20% этилацетат/гексан в качестве элюента с получением бензилового эфира 2-(R)-2-[бут-3-енил (4- метоксибензолсульфонил) амино] -3-метилмасляной кислоты в виде прозрачного масла 404 мг (16%).
К смеси бензилового эфира 2-(R)-2- [бут-3-енил (4-метоксибензолсульфонил) амино] -3-метилмасляной кислоты (780 мг, 1,81 ммоля) и гидрата рутения (III) хлорида (10 мг, 0,048 ммоля) в ацетонитриле (6 мл), четыреххлористого углерода (6 мл) и воды (8 мл) добавляли периодат натрия (1,7 грамма, 7,9 ммоля). После перемешивания при комнатной температуре в течение 2 часов смесь разводили метиленхлоридом и фильтровали через диатомову землю. Органический слой отделяли, промывали разведенным раствором хлористоводородной кислоты и солевым раствором, сушили над сульфатом магния и концентрировали, получая бензиловый эфир 2-(R)-2-[2-карбоксиэтил(4-метоксибензолсульфонил)амино] -3-метилмасляной кислоты в виде масла, 710 мг (87%).
Альтернативно промежуточное соединение бензиловый эфир 2-(R)-2- [2-карбоксиэтил (4-метоксибензолсульфонил) амино] -3- метилмасляной кислоты получали по следующей методике с более высоким выходом.
N-(4-метоксибензолсульфонил)-D-валинбензиловый эфир (18,8 г, 49,8 ммоля) добавляли к суспензии гидрида натрия (1,3 г, 54 мМ) в сухом диметилформамиде (200 мл) и через 1,5 часа добавляли раствор аллилбромида (4,7 мл, 54 ммоля). Полученную смесь перемешивали в течение ночи при комнатной температуре и затем реакцию останавливали добавлением насыщенного раствора хлорида аммония. Диметилформамид удаляли выпариванием в вакууме. Остаток растворяли в эфире и промывали водой и солевым раствором. После высушивания над сульфатом магния эфир выпаривали, получая бензиловый эфир 2-(R)-2-[(4-метоксибен золсульфонил) проп-2- ениламино] -3-метилмасляной кислоты, прозрачное чистое масло (18,1 г, 87%) было выделено с помощью тонкослойной хроматографии на силикагеле с элюированием 10% этилацетатом в гексане и затем 20% этилацетатом в гексане.
К 1 М раствору комплекса боран/дисульфид в метиленхлориде (1,45 мл, 2,9 моля) добавляли раствор бензилового эфира 2-(R)-2-[(4-метоксибензолсульфонил) проп-2- ениламино] -3-метилмасляной кислоты (3,6 г, 8,6 ммоля) в метиленхлориде (8 мл). Раствор перемешивали при комнатной температуре в течение 4 часов, причем в течение этого времени дополнительно добавляли 1 М раствор комплекса боран/дисульфид в метиленхлориде (2,0 мл, 4,0 ммоля). Смесь перемешивали при комнатной температуре в течение ночи и затем по каплям добавляли к механически перемешиваемому раствору трехокиси хрома (5,1 г, 51,6 моля) в уксусной кислоте (31 мл) и воде (3,5 мл) при поддержании внутренней температуры от -5oC до 10oC. После перемешивания при комнатной температуре в течение ночи смесь разводили водой и экстрагировали метиленхлоридом. Экстракт промывали солевым раствором, сушили (сульфатом магния) и концентрировали. Остаток хроматографировали на силикагеле, элюируя последовательно хлороформом и 2% метанолом в хлороформе с получением бензилового эфира 2-(R)-2-[2-карбоксиэтил(4-метоксибензолсульфонил)амино]-3- метилмасляной кислоты в виде масла (2,42 грамма, 63%).
К раствору бензилового эфира 2-(R)-2-[2-карбоксиэтил(4- метоксибензолсульфонил)амино]-3-метилмасляной кислоты (710 мл, 1,58 ммоля) в метиленхлориде (15 мл) последовательно добавляли триэтиламин (0,47 мл, 3,35 ммоля) морфолин (0,15 мл, 1,72 ммоля) и (бензотриазол-1-илокси)трис(диметиламино)фосфония гексафторфосфат (769 мг, 1,74 ммоля). Смесь перемешивали при комнатной температуре в течение ночи и затем разводили метиленхлоридом. Раствор промывали 0,5 N раствором хлористоводородной кислоты и солевым раствором, сушили над сульфатом магния и концентрировали в вакууме. Твердый остаток подвергали хроматографированию на силикагеле, используя 20% гексан в этилацетате с получением бензилового эфира 2-(R)-2-[(4-метоксибензолсульфонил) (3- морфолин-4-ил-3-оксопропил) амино] -3-метилмасляной кислоты в виде прозрачного масла, 725 мг (88%).
К раствору бензилового эфира 2-(R)-2-[(4-метоксибензолсульфонил) -(3-морфолин-4-ил-3-оксопропил) амино] -3- метилмасляной кислоты (725 мг, 1,40 ммоля) в этаноле (35 мл) добавляли 10% палладия на активированном угле (50 мг). Смесь взбалтывали при 3 атмосферах водорода в шейкере Парра в течение 3 часов. Катализатор удаляли фильтрованием через найлон (размер пор 0,45 мкм) и растворитель выпаривали, получая 2-(R)-(2)-[(4- метоксибензолсульфонил)-(3-морфолин-4-ил-3-оксопропил) амино]-3- метилмасляную кислоту в виде белого твердого вещества, 540 мг (90%).
К раствору 2-(R)-2-[(4-метоксибензолсульфонил)-(3-морфолин- 4-ил-3-оксопропил) амино]-3-метилмасляной кислоты (540 мг, 1,26 ммоля) и 1-гидроксибензтриазолгидрата (205 мг, 1,33 ммоля) в сухом диметилформамиде (12 мл) добавляли 1-(3-диметиламинопропил)-3- этилкарбодиимидгидрохлорид (289 мг, 1,51 ммоля). После перемешивания в течение 30 минут добавляли гидроксиламина гидрохлорид (350 мг, 5,04 ммоля) и триэтиламин (1,0 мл, 7,17 ммоля). Смесь перемешивали при комнатной температуре в течение ночи и затем разводили этилацетатом. Раствор последовательно промывали водой, 0,5 N раствором хлористоводородной кислоты и солевым раствором. Раствор затем сушили над сульфатом магния и концентрировали в вакууме, получая белое пенообразное вещество. Вещество растворяли в толуоле, фильтровали и концентрировали. Остаток растирали с эфиром с получением 2-(R)-N-гидрокси-2-[(4- метоксибензолсульфонил)-(3-морфолин-4-ил-3-оксопропил) амино]-3- метилбутирамида в виде твердого вещества, 200 мг (36%);
1H-ЯМР (CDCl3) δ 9,35 (шир. с, 1H), 7,73 (д, J=8,9 Гц, 2H), 6,95 (д, J = 8,9 Гц, 2H), 3,86 (с, 3H), 3,83-3,73 м,1H),
3,70-3,52 (м, 7H), 3,46-3,43 (м, 2H), 3,41-3,29 (м,1H), 2,92-2,69 (м, 2H), 2,30-2,17 (м, 1H), 0,84 (д, J=6,5 Гц, 3H), 0,41 (д, J=6,5 Гц, 3H); МС (пучок частиц); м/з 444 (М+ Н), 428, 383, 329; HR Масс-спектр вычислено для C19H30N3O7S (М+Н): 444.1804, найдено: 464.1818.
Соединения, названные в заголовках примеров 3-6 были получены методом, аналогичным описанному в примере 2, с использованием бензилового эфира 2-(R)-2-[2- карбоксиэтил(4-метокси-бензолсульфонил)амино] -3-метилмасляной кислоты в качестве исходного материала, который реагировал с указанным амином.
Пример 3
2-(R)-2-[(2-Бензилкарбамоилэтил) (4- метоксибензолсульфонил)амино]-N-гидрокси-3-метилбутирамид
Реакция с бензиламином: 1H ЯМР (ДМСО-d6): δ 10,72 (с,1H) 8,89 (с,1H), 8,39 (м, 1H), 7,74 (д, J=9,Q Гц, 2H), 7,32-7,21 (м, 5H), 7,05 (д, J=9,0 Гц, 2H), 4,21 (д, J=5,9 Гц, 2H), 4,02 - 3,87 (м, 1H), 3,82 (с, 3H), 3,63 (д, J = 10,8 Гц, 1H), 3,29-3,17 (м, 1H), 2,71-2,57 (м, 1H), 2,52-2,40 (м, 1H), 2,06-1,94 (м, 1H), 0,77 (д, J= 6,6 Гц, 3H), 0,74 (д, J=6,5 Гц, 3H), МС (LSIMS): м/з 464 (М+Н); HR Macc-спектр рассчитано для С22H30N3O6S (М+Н): 464,1855. Найдено: 464,1832.
Пример 4
2-(R)-N-гидрокси-2-((4-метоксибензолсульфонил) (2-[(пиридин-3-илметил)карбамоил]этил)амино)-3-метилбутирамид
Реакция с 3-пиридилметиламином: 1H ЯМР (ДМСО-d6): δ 10,72 (c,1H), 8,89 (с, 1H), 8,49-8,42 (м, 3H), 7,73(д, J=8,9 Гц, 2H), 7,63-7,60 (м, 1H), 7,32 (дд, J =4,8; 7,8 Гц, 1H), 7,05 (д, J=8,9 Гц, 2H), 4,23 (д, J =5,8 Гц, 2H), 4,00-3,88 (и. , 1H), 3,81 (с, 3H), 3,62 (д, J =10,8 Гц, 1H), 3,27-3,17 (м, 1H), 2,69- 2,58 (м, 1H), 2,52-2,41 (м, 1H), 2,07-1,94 (м, 1H), 0,76 (д, J = 6,5 Гц, 3H), 0,72 (д, J=6,4 Гц, 3H); МС (LSIMS): м/з 465 (М+Н).
Пример 5
2-(R)-N-гидрокси-2-([4-метоксибензолсульфонил] [2-(метилпиридин-3-илметилкарбамоил)этил]амино)-3- метилбутирамид
Реакция с 3-(N-метиламинометил) пиридином 1H ЯМР (ДМСО- d6): δ 10,75 (шир. с. , 1H), 8,92 (с,1H), 8,52-8,29 (м,2H), 7,75 (д, J =8,8 Гц, 1,4 Н), 7,67 (д, J =8,8 Гц, 0,6Н), 7,62-7,58 (м, 1H), 7,42-7,32 (м, 1H), 7,06 (д, J = 8,8 Гц, 1,4 Н), 7,01 (д, J =8,8 Гц, 0,6Н), 4,55-4,41 (м,2H), 3,94-3,82 (м, 1H), 3,81 (с, 2,1H), 3,80(c, 09H), 3,68-3,60 (м,1H), 3,33-3,19(м,1H), 2,90-2,50(м, 2H), 2,88(c, 2,1H, перекрытый), 2,79 (с, 0,9H), 2,05-1,80 (м, 1H), 0,79-0,69(м,6H); МС (термораспыление): м/з 479(М+Н), 364.
Пример 6
4-(3-[(1-(R)-1-Гидроксикарбамоил-2-метилпропил) (4-метоксибензолсульфонил) амино] пропионил) пиперазин-1-карбоновой кислоты трет-бутиловый эфир
Реакция с трет-бутил-1-пиперазинкарбоксилатом: 1H ЯМР (ДМСО-d6): δ 10,77 (шир, с, 1H), 8,93 (с,IH), 7,74 (д, J =8,9 Гц, 2H); 7,06 (д, J=8,9 Гц, 2H), 3,90-3,80(м, 1H), 3,82(с, 3H, перекрытый), 3,64 (д, J =10,8 Гц, 1H), 3,60-3,16(м, 9H), 2,80- 2,71 (м,1H), 2,59-2,47 (м, 1H), 2,03-1,91(м,1H), 1,39 (c, 9H), 0,77 (д, J =6,5 Гц, 3H), 0,71 (д, J =6,5, 3H); МС (термораспыление): м/з 543 (М+Н), 443, 382, 328.
Пример 7
2-(R)-N-гидрокси-2-[(4-метоксибензолсульфонил) (3-оксо-3-пиперазин-1-илпропил) амино] -3-метилбутирамида гидрохлорид
Раствор 4-(3-[(1-(R)-1-гидроксикарбамоил-2-метилпропил) - (4-метоксибензолсульфонил) амино] пропионил) пиперазин-1-карбоновой кислоты трет-бутилового эфира (пример 6) (430 мг, 0,79 ммоля) в метиленхлориде (11 мл) охлаждали до 0oC. Затем через раствор пропускали газообразный хлористый водород в течение примерно 0,5 минуты. Раствору давали нагреться до комнатной температуры при перемешивании в течение 1 часа. Летучие вещества удаляли выпариванием и остаток отфильтровывали, промывая метиленхлоридом, получая твердый гидрохлорид 2-(R)-N-гидрокси-2- [(4-метоксибензолсульфонил) (3-оксо-3-пиперазин-1-илпропил) амино]-3-метилбутирамида, 375 мг (99%).
1H ЯМР (ДМСО-d6): δ 10,78 (шир. с, 1H), 9,16 (шир. с, 1H), 7,74 (д, J = 8,8 Гц, 2H), 7,07 (д, J =8,9 Гц, 2H), 3,82 (с, 3H), 3,62 (шир. с, 4H), 3,38-3,18 (м, 1H), 3,16-3,07 (шир. с, 2H), 3,07-2,98 (шир. с, 2H), 2,83-2,73 (м, 1H), 2,65- 2,53 (м, 1H), 2,06-1,90 (м, 1H), 0,76 (д, J=6,5 Гц, 3H), 0,72 (д, J =6,5 Гц, 3H). Широкий пик воды между δ 4,0 и 3,5 делает неясными некоторые сигналы данного соединения; МС (термораспыление): м/з 443 (М+Н), 382, 328.
Пример 8
2-(R)-2-[(Бензилкарбамоилметил) (4- метоксибензолсульфонил)амино]-N-гидрокси-3-метилбутирамид
К раствору бензилового эфира 2-(R)-2-[карбоксиметил(4- метоксибензолсульфонил) амино]-3-метилмасляной кислоты (пример 1) (905 мг, 2,08 ммоля) в метиленхлориде (18 мл) добавляли последовательно триэтиламин (0,72 мл, 5,14 ммоля), бензиламин (0,25 мл, 2,29 ммоля) и (бензотриазол-1-илокси)трис(диметиламино) фосфония гексафторфосфат (1,01 грамм, 2,28 ммоля). Смесь перемешивали при комнатной температуре в течение ночи и затем разводили этилацетатом. Раствор промывали 0,5 N раствором хлористоводородной кислоты и солевым раствором, сушили над сульфатом магния и концентрировали в вакууме. Остаток хроматографировали на силикагеле, используя метиленхлорид/этилацетат/гексан в соотношении 2:5:16 с получением бензилового эфира 2-(R)-2-[(бензилкарбамоилметил) (4-метоксибензолсульфонил) амино]-3-метилмасляной кислоты в виде прозрачного масла, 933 мл (86%).
К раствору бензилового эфира 2-(R)-2- [(бензилкарбамоилметил) (4-метоксибензолсульфонил) амино]-3- метилмасляной кислоты (933 мг, 1,17 ммоля) в этаноле (50 мл) добавляли 10% палладия на активированном угле (85 мг). Смесь перемешивали при 3 атмосферах водорода в шейкере Парра в течение 4 часов. Катализатор удаляли фильтрованием через найлон (размер пор 0,45 мкм) и растворитель выпаривали, получая 2-(R)-2- [(бензилкарбамоилметил) (4-метоксибензолсульфонил) амино]-3- метилмасляную) кислоту в виде белой пены, 755 мг (98%).
К раствору 2-(R)-2-[(бензилкарбамоилметил)(4-метоксибензолсульфонил) -амино-3-метилмасляной кислоты (655 мг, 1,51 ммоля) и 1-гидроксибензтриазолгидрата (224 мг, 1,46 ммоля) в сухом диметилформамиде (15 мл) добавляли гидрохлорид 1-(3-диметиламинопропил)- 3-этилкарбодиимида (316 мг, 1,65 ммоля). После перемешивания в течение 30 минут добавляли гидрохлорид гидроксиламина (416 мг, 6,0 ммоля) и затем N-метилморфолин (0,99 мл, 9,0 ммоля). Смесь перемешивали при комнатной температуре в течение ночи и затем разводили этилацетатом. Раствор промывали последовательно водой, 0,5 N раствором хлористоводородной кислоты и солевым раствором. Раствор затем сушили над сульфатом магния и концентрировали в вакууме, получая белое пенообразное вещество, которое подвергали хроматографии на силикагеле, элюируя этилацетатом с получением 2-(R)-2-[(бензилкарбамоилметил) - (4-метоксибензолсульфонил)-амино] -N-гидрокси-3-метилбутирамида в виде белой пены, 570 мг (84%); 1H ЯМР (ДМСО-d6): δ 10,75 (шир. с, 1H), 8,90 (с, 1H), 8,47 м, 1H), 7,85 (д, J = 8,9 Гц, 2H), 7,83-7,19 (м, 5H), 7,04 (д, J= 8,9 Гц, 2H), 4,37 (д, J = 11,4 Гц, 1H), 4,28 (д, J =5,9 Гц, 2H), 3,89(д, J =11,4 Гц, 1H), 3,82(с, 3H), 3,45(д, J = 10,3 Гц, 1H), 1,90-1,79 (м, 1H), 0,73 (д, J =6,3 Гц, 6H); МС (LSIMS): м/з 450 (М+Н).
Пример 9
2-(R)-2-[(Бензилметилкарбамоилметил) (4- метоксибензолсульфонил) амино] -N-гидрокси-3-метилбутирамил
К раствору бензилового эфира 2-(К)-2-[карбоксиметил(4- метоксибензолсульфонил) амино]-3-метилмасляной кислоты (пример 1) (1,05 г, 2,41 ммоля) в метиленхлориде (20 мл) добавляли последовательно триэтиламин (0,84 мл, 6 ммолей), N-бензилметиламин (0,34 мл, 2,63 ммоля) и (бензотриазол-1-илокси)трис- (диметиламино)фосфония гексафторфосфат (1,17 г, 2,69 моля). Смесь перемешивали при комнатной температуре в течение ночи и затем разводили этилацетатом. Раствор промывали 0,5 N раствором хлористоводородной кислоты и солевым раствором, сушили над сульфатом магния и концентрировали в вакууме. Остаток подвергали хроматографии на силикагеле, используя 35% этилацетат в гексане (плюс небольшое количество метиленхлорида, чтобы загрузить образец в колонку), с получением бензилового эфира 2-(R)-2- [бензилметилкарбамоилметил) (4-метоксибензолсульфонил)амино] -3- метилмасляной кислоты в виде прозрачного масла, 1,14 г (88%).
К раствору бензилового эфира 2-(R)-2-[(бензилметилкарбамоилметил) (4-метоксибензолсульфонил) амино]-3-метилмасляной кислоты (1,14 г, 2,12 ммоля) в этаноле (100 мл) добавляли 10% палладия на активированном угле (400 мг). Смесь взбалтывали при 3 атмосферах водорода в шейкере Парра в течение 3 часов. Катализатор удаляли фильтрованием через найлон (размер пор 0,45 мкм) и растворитель выпаривали, получая 2-(R)-2-[(бензилметилкарбамоилметил) (4- метоксибензолсульфонил) аминов-3-метилмасляную кислоту в виде белой пены, 902 мг (95%).
К раствору 2-(R)-2- [(бензилметилкарбамоилметил) (4-метоксибензолсульфонил) амино]-3- метилмасляной кислоты (902 мг, 2,01 ммоля) в метиленхлориде (20 мл) добавляли последовательно триэтиламин (0,90 мл, 6,42 ммоля), гидрохлорид о-аллилгидроксиламина (242 мг, 2,21 ммоля) и гексафторфосфат(бензотриазол-1-илокси)трис(диметиламино)фосфония (978 мг, 2,21 ммоля).
Смесь перемешивали при комнатной температуре в течение ночи и затем разводили этилацетатом. Раствор промывали 0,5 N раствором хлористоводородной кислоты и солевым раствором, сушили над сульфатом магния и концентрировали в вакууме. Остаток хроматографировали на силикагеле, используя 40% гексан в этилацетате с получением 2-(R)-N-аллилокси-2-[(бензилметилкарбамоилметил) (4-метоксибензолсульфонил) амино] - 3 -метилбутирамида в виде масла, 1,008 грамма (100%).
К раствору 2-(R)-N- аллилокси-2-[(бензилметилкарбамоилметил) (4-метоксибензолсульфонил) амино] -3-метилбутирамида (500 мг, 0,99 ммоля) в метиленхлориде (40 мл) добавляли бис-(трифенилфосфин) палладия (П) хлорид (280 мг, 0,4 ммоля) и затем, по каплям, гидрид трибутилолова (0,43 мл, 2,2 ммоля). Раствор перемешивали при комнатной температуре в течение 1 часа, разводили метиленхлоридом, промывали 1 N раствором хлористоводородной кислоты, сушили над сульфатом магния и концентрировали. Остаток растворяли, в этилацетате и фильтровали для удаления твердых веществ. После концентрирования фильтрат хроматографировали на силикагеле, элюируя хлороформом и затем 2% метанолом в хлороформе с получением 2-(R)-2-[(бензилметилкарбамоилметил) (4-метоксибензолсульфонил) амино] -N-гидрокси-3-метилбутирамида в виде белого твердого вещества (340 мг, 74%). 1H ЯМР (ДМСО-d6): δ 10,66 (шир.с, 1H), 8,87 (шир. с, 0,6Н), 8,84 (с, 0,4Н), 7,91 (д, J = 8,9 Гц, 1,2Н), 7,78 (д, J= 8,9 Гц, 0,8Н), 7,43-7,21 (м, 5H), 7,05 (д, J =8,9 Гц, 1,2 Н), 7,00 (д, J=8,9 Гц, 0,8Н), 4,72 (д, J =17,7 Гц, 0,4Н), 4,70 (д, J =17,7 Гц, 0,6Н), 4,59-4,42 (м,1H), 4,25 (д, J =17,8 Гц, 0,6Н), 4,07 (д, J =17,7 Гц, 0,4Н), 3,82 (с,3H), 3,46-3,40 (м, 1H), 2,91 (с, 1,8Н), 2,83 (с, 1,2Н), 1,92-1,70 (м,1H), 0,75-0,69 (м, 6H); МС (термораспыление) м/з 464 (М+Н), 307, 239.
Соединения из заголовка примеров 10-11 получали методом, аналогичным описанному в примере 9, используя бензиловый эфир 2- (R)-2-[карбоксиметил (4-метоксибензолсульфонил) амино] -3-метилмасляной кислоты (пример 1) в качестве исходного материала, который реагировал с указанным амином.
Пример 10
2-(R)-N-Гидрокси-2-([4-метоксибензолсульфонил] - [(2-морфолин-4-илэтилкарбамоил) метил] амино) -3-метилбутирамид
Реакция с 4-(2-аминоэтил)морфолином: 1H ЯМР (ДМСО-d6): δ 10,74 (шир.с, 1H), 8,90 (шир.с, 1H), 7,84 (шир.с, 1H, перекрытый), 7,84 (д, J=8,8 Гц, 2H), 7,06 (д, J =8,8 Гц, 2H), 4,33 (д, J=17,5 Гц, 1H), 3,83 (с,3H), 3,78 (д, J = 17,5 Гц, 1H), 3,57-3,54 (м, 4H), 3,49(д, J =10,2 Гц, 1H), 3,28-3,06 (м, 2H), 2,34-2,30 (м, 6H), 1,93-1,77 (м, 1H), 0,77-0,74 (м, 6H).
Пример 11
2-(R)-N-Гидрокси-2-[(4-метоксибензолсульфонил) (2-оксо-2-пирролидин-1-илэтил) амино]-3-метилбутирамид
Реакция с пирролидином: 1H ЯМР (CD3OD): δ 7,90 (д, J = 8,9 Гц, 2H), 7,04 (д, J =8,9 Гц, 2H), 4,50 (д, J =17,6 Гц, 1H), 4,15 (д, J =17,6 Гц, 1H), 3,87 (с, 3H), 3,56-3,39(м,5H), 2,07-1,82 (м,5H), 0,83 (д, J=6,6 Гц, 3H), 0,73 (д, J= 6,6 Гц, 3H) МС (термораспыление): м/з 414 (M+1); HRMC вычислено для C18H28N3O6S (М+Н): 414,1699. Найдено 414,1703.
Пример 12
2-[Диметилкарбамоилметил (4-метоксибензолсульфонил) амино] -N- гидрокси-3-метилбутирамид
Раствор бензилового эфира 2- (R) -2- [карбоксиметил (4- метоксибензолсульфонил) амино] -3-метилмасляной кислоты (пример 1) (1,89 г, 4,34 ммоля) в тионилхлориде (25 мл) нагревали с обратным холодильником в течение 1 часа. После охлаждения избыток тионилхлорида выпаривали. Остаток растворяли в метиленхлориде (50 мл) и раствор охлаждали на ледяной бане. Через раствор медленно пропускали газ диметиламина в течение 1 часа. После выпаривания растворителя остаток растворяли в этилацетате, промывали 1 N раствором хлористоводородной кислоты, водой и солевым раствором, сушили над сульфатом магния и концентрировали, получая бензиловый эфир диметилкарбамоилметил-(4- метоксибензолсульфонил)амино-3-метилмасляной кислоты в виде масла, 1,77 г (88%).
К раствору бензилового эфира диметилкарбамоилметил (4-метоксибензолсульфонил) амино-3-метилмасляной кислоты (1,77 г, 3,83 ммоля) в этаноле (100 мл) добавляли 10% палладия на активированном угле (644 мг). Смесь взбалтывали при 3 атмосферах водорода в шейкере Парра в течение 1,5 часов. Катализатор удаляли фильтрованием через найлон (размер пор 0,45 мкм) и растворитель выпаривали, получая диметилкарбамоилметил (4- метоксибензолсульфониламино-3-метилмасляную кислоту в виде белой пены, 1,42 г (100%).
K раствору диметилкарбамоилметил(4-метоксибензолсульфонил)амино- 3-метилмасляной кислоты (1,42 г, 3,81 ммоля) и 1- гидроксибензтриазолгидрата (687 мг, 4,48 ммоля) в сухом диметилформамиде (7 мл) добавляли 1-(3-диметиламинопропил)-3- этилкарбодиимида гидрохлорид (974 мг, 5,08 ммоля). После перемешивания в течение 30 минут добавляли гидроксиламина гидрохлорид (1,17 г, 16,8 ммоля) и затем N-метилморфолин (2,8 мл, 25,5 ммоля). Смесь перемешивали при комнатной температуре в течение ночи и затем концентрировали в вакууме. Остаток растворяли в этилацетате и полученный раствор промывали последовательно водой, 0,5 N раствором хлористоводородной кислоты и солевым раствором. Раствор затем сушили над сульфатом магния и концентрировали в вакууме, получая масло, которое хроматографировали на силикагеле, элюируя последовательно этилацетатом, 5% метанолом в хлороформе и 10% метанолом в хлороформе с получением 2-[диметилкарбамоилметил(4-метоксибензолсульфонил)амино] -N-гидрокси-3- метилбутирамида в виде белого твердого вещества. 390 мг (26%). 1H ЯМР (ДМСО-d6): δ 10,70 (шир.с,1H), 8,89 (с, 1H), 7,80 (д, J =8,9 Гц, 2H), 7,10 (д, J=8,9 Гц, 2H), 4,62 (д, J =17,7 Гц, 1H), 4,14 (д, J = 17,7 Гц, 1H), 3,84 (с,3H), 3,40 (д, J =10,4 Гц, 1H), 2,97 (с,3H), 2,82 (с, 3H), 1,88-1,72 (м, 1H), 0,72 (д, J = 6,5 Гц, 6H); МС (термораспыление): м/з (М+Г); HRMC вычислено для C16H26N3O6S (М+Н): 388, 1542. Найдено: 388, 1592.
Пример 13
2-R)-2-N-Гидрокси-2- ((4- метоксибензолсульфонил) ([пиридин-3-илметил) -карбамоил] метил) амино) -3-метилбутирамид
2-(R)-N-Гидокси-2-((4-метоксибензолсульфонил) ([пиридин- З-илметил) -карбамоил] метил) амино) -3-метилбутирамид получали по методике, подобной методике примера 12, исходя из бензилового эфира 2-(R)-2-[карбоксиметил (4-метоксибензолсульфонил)-амино] -3- метилмасляной кислоты (пример 1), которая реагирует с 3-пиридилметиламином через промежуточный хлорангидрид кислоты. 1H ЯМР (CD3OD): δ 8,55-8,53 (м, 1H), 8,43-8,40 (м, 1H), 7,90-7,82 (м, 1H, перекрытый), 7,86 (д, J = 8,9 Гц, 2H), 7,40 (дд, J = 4,8, 7,8 Гц, 1H), 7,04 (д, J = 8,9 Гц, 2H), 4,50 (д, J=17,5 Гц, 1H), 4,39 (д, J =17,5 Гц, 1H), 4,32 (д, J =17,7 Гц, 1H), 4,02 (д, J =17,7 Гц, 1H), 3,87 (с,1H), 3,60 (д, J =10,3 Гц, 1H), 2,08-1,93 (м, 1H), 0,85 (д, J =6,5 Гц, 3H), 0,70 (д, J =6,5 Гц, 2H); МС (термораспыление): м/з 451 (М+Н), 336, 320.
Пример 14
N-Гидрокси-[(4-метоксибензолсульфонил) (2- морфолин-4-ил-2-оксоэтил) амино] ацетамид
К раствору моногидрата динатриевой соли иминоуксусной кислоты (5,0 грамм, 25,6 ммоля) в диоксане (50 мл) и воде (50 мл) добавляли триэтиламин (5,3 мл, 38 ммолей) с последующим добавлением 4-метоксибензолсульфонилхлорида (5,8 г, 28,0 ммоля). Смесь перемешивали в течение ночи при комнатной температуре и разводили метиленхлоридом. Раствор промывали 1N раствором хлористоводородной кислоты, водой и солевым раствором, сушили над сульфатом магния и концентрировали в вакууме, получая [карбоксиметил (4-метоксибензолсульфонил)амино] уксусную кислоту в виде белого твердого вещества, 3,83 г (49%).
[Карбоксиметил (4-метоксибензолсульфонил) аминоуксусную кислоту (0,5 г, 1,65 ммоля) растворяли в уксусном ангидриде (15 мл) при легком нагревании. Полученный раствор перемешивали при комнатной температуре в течение ночи. Уксусный ангидрид удаляли выпариванием в вакууме, остаток растворяли в метиленхлориде и добавляли морфолин (0,16 мл, 1,82 ммоля).
Смесь перемешивали в течение ночи при комнатной температуре и концентрировали в вакууме. Остаток растворяли в этилацета-е, промывали I N раствором хлористоводородной кислоты, водой и солевым раствором, сушили над сульфатом магния и концентрировали с получением [(4- метоксибензолсульфонил) (2-морфолин-4-ил-2-оксоэтил)амино] уксусной кислоты в виде масла, 0,33 грамма (54%).
К раствору [(4- метоксибензолсульфонил) (2-морфолин-4-ил-2-оксоэтил)-амино] уксусной кислоты (0,33 г, 0,89 ммоля) в метиленхлориде (10 мл) последовательно добавляли триэтиламин (0,43 мл, 3,1 ммоля) гидрохлорид о-бензилгидроксиламина (0,15 г, 0,94 ммоля) и гексафторфосфат (бензотриазол-1-илокси)трис-(диметиламино)фосфония (0,43 г, 0,97 ммоля). Смесь перемешивали при комнатной температуре в течение ночи и затем разбавляли этилацетатом. Раствор последовательно промывали 0,5 N раствором хлористоводородной кислоты, водой и солевым раствором, сушили над сульфатом магния и концентрировали в вакууме. Остаток хроматографировали на силикагеле, используя этилацетат с получением N-бензилокси-[(4- метоксибензолсульфонил) (2-морфолин-4-ил-2- оксоэтил)амино]ацетамида в виде белого твердого вещества, 0,33 г (78%).
К раствору N-бензилокси-[(4-метоксибензолсульфонил) (2- морфолин-4-ил-2-оксоэтил)амино]ацетамида (0,33 г, 0,69 ммоля) в метаноле (35 мл) добавляли 5% палладия на активированном угле (85 мг). Смесь взбалтывали при 2 атмосферах водорода в шейкере Парра в течение 1,5 часов. Катализатор удаляли фильтрованием через найлон (размер пор 0,45 мкм) и растворитель выпаривали. Остаток хроматографировали на силикагеле, элюируя 5% метанолом в метиленхлориде с получением N-метокси[(4-метоксибензолсульфонил) (2-морфолин-4-ил-2-оксоэтил) амино] ацетамида в виде белого твердого вещества, 65 мг (24%); 1H ЯМР CD3OD): δ 7,82 (д, J = 9,0 Гц, 2H), 7,08 (д, J =9,0 Гц, 2H), 4,24 (с, 2H), 3,88 (с, 3H), 3,84 (с, 2H), 3,68-3,64 (м,4H), 3,58-3,50 (м,4H); МС (термораспыление): м/з 388 (М+1), 387 (М); HRMC вычислено для C16H22N3O7S (М+Н): 388,1178. Найдено: 338,1180.
Вещества, названные в заголовках примеров 15-16, были получены методом, аналогичным методу, описанному в примере 14 при использовании [карбоксиметил(4-метоксибензолсульфонил)-амино] уксусной кислоты в качестве исходного вещества, которая после обработки уксусным ангидридом реагировала с указанным амином.
Пример 15
N-Гидрокси [(4-метоксибензолсульфонил) (2-оксо-2- пирролидин-1-илэтил)амино]ацетамид
Реакция с пирролидином: 1H ЯМР (ДМСО-d6): δ 11,26 (шир. с, 1H), 8,89 (с, 1H), 7,81 (д, J =8,9 Гц, 2H), 7,10 (д, J = 8,9 Гц, 2H), 4,09 (с,2H), 3,85 (с,3H), 3,74 (с,2H), 3,45-3,25 (м,4H), 1,93-1,72 (м, 4H); МС (термораспыление): м/з 372 (M+1): Анализ, рассчитанный для С15H21N3O6S: С, 48,51; H, 5,70; N 11,31. Обнаруженное: С, 48,51; H 5,82; N 11,24.
Пример 16
2 [Диметилкарбамоилметил (4-метоксибензолсульфонил) амино]-N- гидроксиацетамид
Реакция с диметиламином: т.пл.: 170oC (разл.): 1H ЯМР (ДМСО-d6): δ 10,69 (шир. с, 1H), 8,88 (с, 1H), 7,91 (д, J = 8,9 Гц, 2H), 7,06 (д, J= 8,9 Гц, 2H), 4,19 (с, 2H), 3,85 (с, 3H), 3,73 (с, 2H), 2,94 (с, 3H), 2,84 (с, 3H); МС (термораспыление): м/з 346 (М+1); Анализ, рассчитанный для C13H19N3O6S С, 45,21; H 5,55; N, 12,17. Обнаруженное: С, 44,93; H, 5,61; N 12,03.
Пример 17
2-(R)-2-[(2-Карбамоилэтил) (4-метоксибензолсульфонил)-амино] -N-гидрокси-3-метилбутирамид
К раствору бензилового эфира 2-(R)-2-[(2-карбоксиэтил (4- метоксибензолсульфонил)амино] -3-метилмасляной кислоты (пример 2) (900 мг, 2,0 ммоля) в метиленхлориде (10 мл) добавляли тионилхлорид (0,16 мл, 2,2 ммоля). Реакционную смесь перемешивали в течение 1,5 часов при комнатной температуре и затем концентрировали в вакууме. После растворения остатка в метиленхлориде (10 мл), через раствор пропускали газообразный аммиак в течение 0,5 минуты. Раствор перемешивали при комнатной температуре в течение ночи и концентрировали в вакууме. Тонкослойная хроматография остатка на силикагеле с элюированием 2% метанолом в хлороформе давала бензиловый эфир 2-(R)- 2- [(2-карбамоилэтил) (4-метоксибензолсульфонил) амино] -N-гидрокси- З-метилмасляной кислоты в виде прозрачного масла (275 мг, 31%).
К раствору бензилового эфира 2-(R)-2-[(2-карбамоилэтил)-(4- метоксибензолсульфонил) амино] -N-гидрокси-3-метилмасляной кислоты (275 мг, 0,61 ммоля) в этаноле (15 мл) добавляли 10% палладия на активированном угле (30 мг). Смесь взбалтывали при 3 атмосферах водорода в шейкере Парра в течение 5 часов. Катализатор удаляли фильтрованием через диатомову зумлю и растворитель выпаривали, получая 2-(R)-2-[(2-карбамоилэтил)-(4-метоксибензолсульфонил) амино] -N-гидрокси-3-метилмасляную кислоту в виде белой пены, 211 мг (96%).
К раствору 2-(R)-2-[(2-карбамоилэтил) (4- метоксибензол-сульфонил) амино]-N-гидрокси-3-метилмасляной кислоты (205 мг, 0,57 ммоля) и 1-гидроксибензотриазолгидрата (85 мг, 0,55 ммоля) в сухом диметилформамиде (5 мл) добавляли 1-(3- диметиламинопропил)-3-этилкарбодиимида гидрохлорид (120 мг, 0,63 ммоля). После перемешивания в течение 30 минут добавляли гидроксиламина гидрохлорид (158 мг, 2,3 ммоля) и затем N-метилморфолин (0,37 мл, 3,4 ммоля). Смесь перемешивали в течение ночи и затем разводили этилацетатом. Раствор промывали водой и солевым раствором. Затем раствор сушили над сульфатом магния и концентрировали в вакууме, получая масло, которое хроматографировали на силикагеле, элюируя 2% метанолом в хлороформе с получением 2-(R)-2-[(2-карбамоилэтил) (4-метоксибензолсульфонил) амино]-N-гидрокси-3-метилбутирамида в виде белого твердого вещества, 45 мг (21%); 1H ЯМР (ДМСО-d6) δ 10,74 (шир. с, 1H), 8,91 (шир.с, 1H), 7,74 (д, J =8,8 Гц, 2H), 7,33 (шир.с, 1H), 7,07 (д, J =8,8 Гц, 2H), 6,79 (шир.с, 1H), 3,93-3,82 (м 1H, перекрытый), 3,83 (с, 3H), 3,64 (д, J =10,7 Гц, 1H), 3,25- 3,12 (М, 1H), 2,62-2,48 (м. 1H), 2,42-2,30 (М. , 1H), 2,06-1,94 (м, 1H), 0,79 (д, J =6,6 Гц, 3H), 0,76 (д, J =6,6 Гц, 3H); МС (термораспыление): м/з 374 (М+Н).
Пример 18
2-(R)-2-[(2-трет-бутоксикарбонилэтил) (4- метоксибензолсульфонил) амино] -N-гидрокси-3-метилбутирамид
Раствор N, N-диметилформамид-ди-трет-бутилацеталя (1,9 мл, 7,9 ммоля) в толуоле (15 мл) по каплям добавляли к раствору бензилового эфира 2-(R)-2-[(2-карбоксиэтил(4-метоксибензолсульфонил) амино] -3-метилмасляной кислоты (пример 2) (900 мг, 2,0 ммоля) в толуоле при 80oC. После нагревания в течение 2 часов при 80oC смесь охлаждали и концентрировали, получая янтарное масло, которое хроматографировали на силикагеле, элюируя хлороформом с получением бензилового эфира 2-(R)-2-[(2-трет-бутоксикарбонилэтил) (4-метоксибензолсульфонил) амино] -3-метилмасляной кислоты в виде масла, 3,75 мг (37%).
К раствору бензилового эфира 2-(R)-2-[(2-трет-бутоксикарбонилэтил) (4-метоксибензолсульфонил) амино]-3-метилмасляной кислоты (370 мг, 0,73 ммоля) в этаноле (20 мл) добавляли 10% палладия на активированном угле (40 мг). Смесь взбалтывали при 3 атмосферах водорода в шейкере Парра в течение 5 часов. Катализатор удаляли фильтрованием через диатомову землю и растворитель выпаривали, получая 2-(R)-2-[(2-трет-бутоксикарбонилэтил) (4- метоксибензолсульфонил) амино] -3-метилмасляную) кислоту в виде белой пены, 30 мг (100%).
К раствору 2-(R)-2-[(2-трет-бутоксикарбонилэтил) (4-метоксибензолсульфонил)амино] -3- метилмасляной кислоты (303 мг, 0,73 ммоля) и 1- гидроксибензтриазолгидрата (108 мг, 0,70 ммоля) в сухом диметилформамиде (10 мл) добавляли гидрохлорид 1-(3- диметиламинопропил)-3-этилкарбодиимида (153 мг, 0,80 ммоля). После перемешивания в течение 45 минут добавляли гидрохлорид гидроксиламина (203 мг, 2,9 ммоля) и затем N-метилморфолин (0,48 мл, 4,4 ммоля). Смесь перемешивали при комнатной температуре в течение ночи и затем концентрировали в вакууме. Остаток хроматографировали на силикагеле, элюируя 2% метанолом в хлороформе и снова 10% этилацетатом в гексане с получением 2-(R)- 2-[(2-трет-бутоксикарбонилэтил) (4-метоксибензолсульфонил)-амино]-N-гидрокси-3-метилбутирамида в виде белой пены, 135 мг, (43%); 1 ЯМР (ДМСО-d6): δ 10,77 (шир.с, 1H), 7,74 (д, J = 8,9 Гц, 2H), 7,08 (д, J =8,9 Гц, 2H), 3,93-3,82 (м, 1H, перекрытый), 3,83 (с, 3H), 3,64 (д, J =10,8 Гц, 1H), 3,26-3,14 (м, 1H), 2,70-2,60 (м, 1H), 2,50-2,38 (м, 1H), 2,04- 1,91 (м, 1H), 1,38 (с, 9H), 0,78 (д, J =6,5 Гц, 3H), 0,72 (д, J =6,5 Гц, 3H); МС (термораспыление): м/з 431 (М+Н), 375, 314.
Пример 19
2-(R)-2-[(2-Карбоксиэтил) (4-метоксибензолсульфонил)амино] -N-гидрокси- З-метилбутирамид
К раствору 2-(R)-2-[(2-тpeт-бутoкcикapбoнилэтил)(4-метoксибензолсульфонил) -амино]-N-гидрокси-3-метилбутирамида (пример 18) (100 мг, 0,23 ммоля) в метиленхлориде (1 мл) при 0oC
добавляли трифторуксусную кислоту (1 мл). Смеси давали нагреться до комнатной температуры при перемешивании в течение ночи. После выпаривания трифторуксусной кислоты и метиленхлорида остаток хроматографировали на силикагеле, элюируя 5% метанолом в хлороформе. Концентрирование соответствующих фракций давало 2-(R)-2-[2-карбоксиэтил (4-метоксибензолсульфонил)-амино]- N-гидрокси-3-метилбутирамид в виде белого твердого вещества, 35 мг (41%). 1H ЯМР (ДМСО-d6AMCO-d6): δ 10,79 (шир.с, 1H), 8,97 (шир.с, 1H), 7,76 (д, J =8,9 Гц, 2H), 7,09 (д, J =8,9 Гц, 2H), 3,95-3,82 (м, 1H, перекрытый), 3,84 (с, 3H), 3,66 (д, J =10,8 Гц, 1H), 3,30-3,20 (м,1H), 2,73-2,62 (м,1H), 2,50- 2,40 (м, 1H), 2,07-1,94 (м, 1H), 0,80 (д, J = 6,5 Гц, 3H), 0,74 (д, J = 6,5 Гц, 3H); МС (термораспыление): м/з 375 (М+Н), 314.











Описываются производные арилсульфониламиногидроксамовой кислоты общей формулы I, где значение Ar, X, n, R3, R4 указаны в п.1 формулы изобретения, которые являются ингибиторами матричных металлопротеиназ или продуцирования фактора некроза опухолей. Могут быть использованы для лечения артрита, рака, язв тканей, септического шока. Описывается способ их получения, способ ингибирования, способ лечения и фармкомпозиции на основе соединений формулы I. 5 с. и 13 з.п. ф-лы, 1 табл.


или их фармацевтически приемлемые соли,
где n равно от 1 до 6;
X означает гидрокси, (C1 - C6)алкокси или NR1R2, где R1 и R2 каждый независимо выбирают из группы, состоящей из (C1 - C6)алкила, пиперидинила, (C1 - C6)алкилпиперидила, (C6 - C10)арила, (C5 - C7)гетероарила, содержащего в качестве гетероатома азот, (C6 - C10)арил(C1 - C6)алкила, (C5 - C7)гетероарил(C1 - C6)алкила, содержащего в качестве гетероатома азот, или R1 и R2 вместе образуют пирролидинил, морфолинил, пиперидил, (C1 - C6)алкилпиперидил, пиперазил, N-(C1 - C6)алкилпиперазил, N-(C6 - C10)арилпиперазинил;
R3 и R4 каждый независимо выбран из группы, включающей водород, (C1 - C6)алкил, (C6 - C10)арил, (C5 - C9)гетероарил, (C6 - C10)арил(C1 - C6)алкил, (C5 - C9)гетероарил(C1 - C6)алкил, (C6 - C10)арил(C6 - C10)арил, (C3 - C6)циклоалкил и (C3 - C6)циклоалкил(C1 - C6)алкил, или R3 и R4 вместе могут образовывать (C3 - C6)циклоалкил, оксациклогексил, тиоциклогексил, инданил или кольцо тетралинила или пиперидиновое кольцо, замещенное R15, где R15 означает водород, (C1 - C6)ацил, (C1 - C6)алкил, (C6 - C10)арил(C1 - C6)алкил, (C5 - C9)гетероарил(C1 - C6)алкил или (C1 - C6)алкилсульфонил;
Ar означает (C6 - C10)арил, (C5 - C9)гетероарил, (C1 - C6)алкил(C6 - C10)арил, (C1 - C6)алкокси(C6 - C10)арил, ((C1 - C6)алкокси)2(C6 - C10)арилокси(C6 - C10)арил или (C5 - C9)гетероарилокси(C6 - C10)арил.
где R15 является (C1 - C6)ацилом, (C1 - C6)алкилом, (C6 - C10)арил, (C1 - C6)алкилом, (C5 - C9)гетероарил(C1 - C6)алкилом или (C1 - C6)алкилсульфонилом.
где R3, R4, Ar, X, n принимают значения, указанные в п.1,
отличающийся тем, что соединение формулы
где R3, R4, Ar, X, n принимают значения, указанные выше,
подвергают взаимодействию с 1-(3-диметиламинопропил)-3-этил-карбодиимидом, 1-гидроксибензтриазолом и гидроксиламином.
| Турбохолодильник для системы кондиционирования воздуха | 1976 |
|
SU606046A1 |
| WO 9005719 A, 31.05.90 | |||
| Способ получения 4-фениловых эфиров 3-амино-5 сульфамоилбензойных кислот | 1973 |
|
SU523635A3 |
| US 4373106 A, 08.02.83 | |||
| Машковский М.Д | |||
| Лекарственные средства | |||
| - Вильнюс, 1994, ч.2, с.383. | |||

