Данное изобретение относится к производным арилсульфонил гидроксамовой кислоты, которые являются ингибиторами матричных металлопротеиназ или образования фактора некроза опухоли (здесь и далее обозначаемый как TNF) и как таковые применяются при лечении болезненных состояний, таких как артрит, карцинома, образование язвенной ткани, рестеноз, периодонтальная болезнь, врожденный буллезный эпидермолиз, склерит и других заболеваний, характеризуемых активностью матричной металлопротеиназы, а также СПИДа, сепсиса, септического шока и других заболеваний, включающих образование TNF.
Настоящее изобретение, кроме того, относится к способу использования таких соединений при лечении вышеупомянутых заболеваний у млекопитающих, особенно людей, и к фармацевтическим композициям, используемым для этого.
Существует ряд ферментов, которые расщепляют структурные белки и которые по структуре относятся к металлопротеиназам. Матрично-деградирующие металлопротеиназы, такие как желатиназа, стромелизин и коллагеназа, вовлекаются в матричную деградацию ткани (например, коллагеновый коллапс) и являются причиной многих патологических состояний, включающих абномальный матричный метаболизм соединительной ткани и базальной мембраны, таких как артрит (например, остеоартрит и ревматоидный артрит), изъязвление ткани (например, корнеальная, эпидермальная язва и язва желудка), абномальное течение заживления раны, периодонтальная болезнь, заболевания кости (например, болезнь Педжета и остеопороз), опухолевые метастазы или инвазия, а также ВИЧ-инфекция (J. Leuk Biol., 52(2): 244-248, 1992).
Известно, что фактор некроза опухоли участвует во многих инфекционных и аутоиммунных заболеваниях (W. Fries, FEBS Letters, 1991, 285, 199). Кроме того, показано, что TNF является первичным медиатором воспалительного ответа, наблюдаемого при сепсисе и септическом шоке (C.E. Spooner et al., Clinical Immunology and Immunopathology, 1992, 62 Sll).
Данное изобретение относится к соединению формулы
или фармацевтически приемлемой его соли, в котором пунктирная линия обозначает необязательную двойную связь;
X - углерод, кислород или сера;
Y - углерод, кислород, сера, сульфоксид, сульфон или азот;
R1, R2, R3, R4, R5, R6, R7, R8 и R9 выбирают из группы, состоящей из водорода, (C1-C6)алкила, произвольно замешенного (C1-C6)алкиламино, (C1-C6)алкилтио, (C1-C6)алкокси, трифторметилом, (C6-C10)арилом, (C5-C9)гетероарилом, (C6-C10)- ариламино, (C6-C10)арилтио, (C6-C10)арилокси,
(C5-C9)гетероариламино, (C5-C9)гетероарилтио, (C5-C9)гетероарилокси, (C6-C10)арил(C6-C10)арилом, (C3-C6)циклоалкилом, гидрокси(C1-C6)- алкилом,
(C1-C6)алкил(гидроксиметиленом), пиперазинилом, (C6-C10)арил(C1-C6)алкокси,
(C5-C9)гетероарил(C1-C6)алкокси, (C1-C6)ациламино, (C1-C6)ацилтио,
(C1-C6)ацилокси, (C1-C6)алкилсульфинилом, (C6-C10)арилсульфинилом, (C1-C6)алкилсульфонилом, (C6-C10)арилсульфонилом, амино, (C1-C6)алкиламино или ((C1-C6) алкиламино)2;
(C2-C6)алкенила, (C6-C10)арил(C2-C6)алкенила, (C5-C9)гетероарил(C2-C6)алкенила, (C2-C6)алкинила, (C6-C10)арил(C2-C6)алкинила, (C5-C9)гетероарил(C2-C6)алкинила, (C1-C6)алкиламино, (C1-C6)алкилтио, (C1-C6)алкокси, трифторметила, (C1-C6)алкил(дифторметилена), (C1-C3)алкил(дифторметилен) (C1-C3)алкила,
(C6-C10)арила, (C5-C9)гетероарила, (C6-C10)ариламино, (C6-C10)арилтио, (C6-C10)арилокси, (C5-C9)гетероариламино, (C5-C9)гетероарилтио, (C5-C9)гетероарилокси, (C3-C6)циклоалкила, (C1-C6)алкил(гидроксиметилена), пиперидила, (C1-C6)-алкилпиперидила, (C1-C6)ациламино, (C1-C6)ацилтио, (C1-C6)-ацилокси, R13 (C1-C6)алкила, где R13 есть (C1-C6)ацилпиперазино, (C6-C10)арилпиперазино, (C5-C9)гетероарилпиперазино, (C1-C6)алкилпиперазино, (C6-C10) арил (C1-C6)алкилпиперазино, (C5-C9)гетероарил(C1-C6)алкилпиперазино, морфолино, тиоморфолино, пиперидино, пирролидино, пиперидил, (C1-C6)алкилпиперидил, (C6-C10)арилпиперидил, (C5-C9)гетероарилпиперидил, (C1-C6) алкилпиперидил(C1-C6)алкил, (C6-C10)арилпиперидил(C1-C6)алкил, (C5-C9)гетероарилпиперидил(C1-C6)алкил или (C1-C6)ацилпиперидил;
или группы формулы
в которой n - целое число от 0 до 6;
Z - гидрокси, (C1-C6)алкокси
или NR14R15, в которой R14 и R15 каждый независимо выбирают из группы, состоящей из водорода, (C1-C6)алкила, произвольно замещенного (C1-C6)-алкилпиперидилом, (C6-C10)арилпиперидилом, (C5-C9)гетероарилпиперидилом, (C6-C10)арилом, (C5-C9)гетероарилом, (C6-C10)-арил(C6-C10)арилом или (C3-C6)циклоалкилом; пиперидила, (C1-C6)алкилпиперидила, (C6-C10)арилпиперидила, (C5-C9) гетероарилпиперидила, (C1-C6)ацилпиперидила, (C6-C10)арила, (C5-C9)гетероарила, (C6-C10)арил(C6-C10)арила, (C3-C6)циклоалкила, R16 (C2-C6)алкила, (C1-C5)алкил(CHR16)(C1-C6)алкила, где R16 есть гидрокси, (C1-C6)ацилокси,
(C1-C6)алкокси, пиперазино, (C1-C6)ациламино, (C1-C6)алкилтио, (C6-C10)арилтио, (C1-C6)алкилсульфинил, (C6-C10)арилсульфинил, (C1-C6)алкилсульфоксил, (C6-C10)арилсульфоксил, амино, (C1-C6)алкиламино,
((C1-C6)алкил)2амино, (C1-C6)ацилпиперазино, (C1-C6)алкилпиперазино,
(C6-C10)арил(C1-C6)алкилпиперазино, (C5-C9)гетероарил(C1-C6)алкилпиперазино, морфолино, тиоморфолино, пиперидино или пирролилино; R17 (C1-C6)алкила, (C1-C5)алкил- (CHR17) (C1-C6)алкила, где R17 есть пиперидил или (C1-C6)алкилпиперидил; и CH(R18)COR19, где R18 есть водород, (C1-C6)-алкил, (C6-C10)арил(C1-C6)алкил, (C5-C9)гетероарил(C1-C6)алкил, (C1-C6)алкилтио(C1-C6)алкил, (C6-C10)арилтио(C1-C6)алкил, (C1-C6)алкилсульфинил(C1-C6)алкил, (C6-C10)арилсульфинил(C1-C6)алкил, (C1-C6)алкилсульфинил(C1-C6)алкил,
(C6-C10)арилсульфонил(C1-C6)алкил, гидрокси(C1-C6)алкил, амино(C1-C6)алкил,
(C1-C6)алкиламино(C1-C6)алкил, ((C1-C6)алкиламино)2 (C1-C6)алкил,
R20R21 NCO(C1-C6)алкил или R20OCO(C1-C6)алкил, где R20 или R21 каждый независимо выбирают из группы, состоящей из водорода, (C1-C6)алкила, (C6-C10)арил(C1-C6)алкила и (C5-C9)гетероарил(C1-C6)алкила; и R19 есть R22O или R22R23N, где R22 и R23 каждый независимо выбирают из группы, состоящей из водорода, (C1-C6)алкила, (C6-C10)арил(C1-C6)алкила и (C5-C9)гетероарил(C1-C6)алкила;
или R14 и R15, или R20 и R21, или R22 и R23 могут соединяться с образованием азетидинила, пирролидинила, морфолинила, тиоморфолинила, индолинила, изоиндолинила, тетрагидрохинолинила, тетрагидроизохинолинила, (C1-C6)ацилпиперазинила, (C1-C6)алкилпиперазинила, (C6-C10)арилпиперазинила,
(C5-C9)гетероарилпиперазинила или диазабициклоалкильного кольца с мостиковой связью, выбранного из группы, состоящей из
где r - 1, 2 или 3;
m - 1 или 2;
p - 0 или 1; и
Q - водород, (C1-C3)алкил, (C1-C6)ацил или (C1-C6)алкокси карбамоил;
или R1 и R2, или R3 и R4, или R5 и R6 могут соединяться вместе с образованием карбонила;
или R1 и R2, или R3 и R4, или R5 и R6, или R7 и R8 могут соединяться вместе с образованием (C3-C6)циклоалкила, оксациклогексила, тиоциклогексила, инданилового или тетралинилового кольца или группы формулы
где R24 - водород, (C1-C6)ацил, (C1-C6)алкил, (C6-C10)арил- (C1-C6)алкил, (C5-C9)гетероарил(C1-C6)алкил или (C1-C6)алкилсульфонил; и
Ar - (C6-C10)арил или (C5-C9)гетероарил, каждый из которых может быть необязательно замещен (C1-C6)алкилом, одним или двумя (C1-C6)алкокси, (C6-C10)арилокси или (C5-C9)гетероарилокси;
при условии, что R7 отличен от водорода только в случае, когда R8 отличен от водорода;
при условии, что R6 отличен от водорода только в случае, когда R5 отличен от водорода;
при условии, что R3 отличен от водорода только в случае, когда R4 отличен от водорода;
при условии, что R2 отличен от водорода только в случае когда R1 отличен от водорода;
при условии, что, когда R1, R2 и R9 являются заместителем, включающим гетероатом, то этот гетероатом не может быть непосредственно присоединен по 2- или 6-положениям;
при условии, что, когда X - азот, R4 не присутствует;
при условии, что, когда X - кислород, сера, сульфоксид, сульфон или азот и когда один или более из заместителей группы, состоящей из R1, R2, R5 и R6, представляет собой заместитель, включающий гетероатом, то этот гетероатом не может быть непосредственно присоединен по 4- или 6-положениям;
при условии, что, когда Y - кислород, сера, сульфоксид, сульфон или азот и когда один или более заместителей из группы, состоящей из R3, R4, R7 и R8, являются независимо заместителем, включающем гетероатом, то этот гетероатом не может быть непосредственно присоединен по 3- или 5-положениям;
при условии, что, когда X - кислород, сера, сульфоксид или сульфон, R3 и R4 не присутствуют;
при условии, что, когда Y - азот, R4 не присутствует;
при условии, что, когда Y - кислород, сера, сульфоксид или сульфон, R5 и R6 не присутствуют;
при условии, что, когда Y - азот, R6 не присутствует;
при условии, что, когда пунктирная линия является двойной связью, R4 и R6 не присутствуют;
при условии, что, когда R3, R5 независимо являются заместителем, включающем гетероатом, когда пунктирная линия является двойной связью, то гетероатом не может быть непосредственно присоединен по положениям X и Y.
при условии, что, когда либо X, либо Y положение представляет кислород, серу, сульфоксид, сульфон или азот, то другое положение X или Y представляет углерод;
при условии, что, когда X или Y определен как гетероатом, пунктирная линия не является двойной связью;
при условии, что, когда R1, R2, R3, R4, R5, R6, R7, R8 и R9 все определены как водород или (C1-C6)алкил, то либо X или Y представляют собой кислород, серу, сульфоксид, сульфон или азот, либо пунктирная линия представляет двойную связь.
Используемый здесь термин "алкил", если не оговорено особо, включает насыщенные одновалентные углеводородные радикалы, имеющие неразветвленные, разветвленные или циклические части или комбинации их.
Используемый здесь термин "алкокси" включает O-алкильные группы, в которых "алкил" такой, как определено выше.
Используемый здесь термин "арил", если не оговорено особо, включает органический радикал, получаемый из ароматического углеводорода путем удаления одного водорода, такой как фенил или нафтил, необязательно замещенный 1-3 заместителями, независимо выбранными из группы, состоящей из фтора, хлора, циано, нитро, трифторметила, (C1-C6) алкокси, (C6-C10)-арилокси, трифторметокси, дифторметокси и (C1-C6)алкила.
Используемый здесь термин "гетероарил", если не оговорено особо, включает органический радикал, полученный из ароматического гетероциклического соединения путем удаления одного водорода, такой как пиридил, фурил, пироил, тиенил, изотиазолил, имидазолил, бензимидазолил, тетразолил, пиразинил, пиримидил, хинолил, изохинолил, бензофурил, изобензофурил, бензотиенил, пиразолил, индолил, изоиндолил, пуринил, карбазолил, изоксазолил, тиазолил, оксазолил, бензтиазолил или бензоксазолил, необязательно замещенный 1-2 заместителями, независимо выбранными из группы, состоящей из фтора, хлора, трифторметила, (C1-C6) алкокси, (C6-C10)арилокси, трифторметокси, дифторметокси и (C1-C6)алкила.
Используемый здесь термин "ацил", если не оговорено особо, включает радикал общей формулы RCO, в котором R - алкил, алкокси, арил, арилалкил или арилалкилокси, а термины "алкил" и "арил" такие, как определено выше.
Используемый здесь термин "ацилокси" включает O-ацильные группы, в которых "ацил" такой, как определено выше.
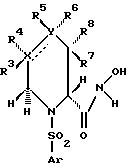
Положения в кольце формулы I, как используется здесь, определены следующим образом:
Предпочтительная структура соединения формулы I включает гидроксамовую кислоту, аксиально расположенную в 2-положении.
Соединение формулы I может иметь хиральные центры и поэтому существует в различных энантиомерных формах. Изобретение относится ко всем оптическим изомерам и стереоизомерам соединений формулы I и их смесям.
К предпочтительным соединениям формулы I относятся соединения, в которых Y представляет кислород, азот или серу.
К другим предпочтительным соединениям формулы I относятся соединения, в которых Ar представляет 4-метоксифенил или 4-феноксифенил.
К другим предпочтительным соединениям формулы I относятся соединения, в которых R8 представляет (C6-C10)арил, (C5-C9)гетероарил, (C6-C10)арил(C1-C6)алкил, (C5-C9)гетероарил(C1-C6)алкил, карбоновую кислоту или карбоновую кислоту (C1-C6)алкил.
К другим предпочтительным соединениям формулы I относятся соединения, в которых R2, R3, R6, R7 и R9 являются водородом.
Более предпочтительными соединениями формулы I являются соединения, в которых Y является углеродом, Ar представляет 4-метоксифенил или 4-феноксифенил и R8 представляет (C6-C10)арилалкинил или (C5-C9)гетероарилалкинил. Более предпочтительными соединениями формулы I являются соединения, в которых Y является кислородом, Ar представляет 4-метоксифенил или 4-феноксифенил и R8 представляет (C6-C10)арилалкинил или (C5-C9)гетероарилалкинил.
Более предпочтительными соединениями формулы I являются соединения, в которых Y является углеродом, Ar представляет 4-метоксифенил или 4-феноксифенил и R8 представляет карбоновую кислоту или карбоновую кислоту (C1-C6)алкил.
Более предпочтительными соединениями формулы I являются соединения, в которых Y является кислородом, Ar представляет 4-метоксифенил или 4-феноксифенил и R8 представляет карбоновую кислоту или карбоновую кислоту (C1-C6) алкил.
Более предпочтительными соединениями формулы I являются соединения, в которых Y является углеродом, Ar представляет 4- метоксифенил или 4-феноксифенил и R5 представляет (C6-C10)арилалкинил или (C5-C9)гетероарилалкинил. Более предпочтительными соединениями формулы I являются соединения, в которых Y является кислородом, Ar представляет 4- метоксифенил или 4-феноксифенил и R5 представляет (C6-C10)арилалкинил или (C5-C9)гетероарилалкинил.
Более предпочтительными соединениями формулы I являются соединения, в которых Y является углеродом, Ar представляет 4- метоксифенил или 4-феноксифенил и R5 представляет карбоновую кислоту или карбоновую кислоту (C1-C6)алкил.
Более предпочтительными соединениями формулы I являются соединения, в которых Y является кислородом, Ar представляет 4- метоксифенил или 4-феноксифенил и R5 представляет карбоновую кислоту или карбоновую кислоту (C1-C6)алкил.
Более предпочтительными соединениями формулы I являются соединения, в которых Y является углеродом, Ar представляет 4- метоксифенил или 4-феноксифенил и R5 представляет (C1-C6)алкиламино.
Более предпочтительными соединениями формулы I являются соединения, в которых Y является кислородом, Ar представляет 4-метоксифенил или 4-феноксифенил и R8 представляет (C1-C6) алкиламино.
К конкретным предпочтительным соединениям формулы I относятся следующие:
(2R, 3S)-N-гидрокси-3-этинил-1-(4-метоксибензолсульфонил) -пиперидин-2-карбоксамид;
(2R, 3S)-N-гидрокси-1-(4-метоксибензолсульфонил)-3-(5- метокситиофен-2-ил-этинил)-пиперидин-2-карбоксамид;
(2R, 3S)-N-гидрокси-1-(4-метоксибензолсульфонил)-3-(3- пиридин-3-ил-проп-2-инил)-пиперидин-2-карбоксамид;
(2S, 3R)-N-гидрокси-4-(4-метоксибензолсульфонил)-2- пиридин-3-ил-морфолин-3-карбоксамид;
(2S,3R)-N-гидрокси-2-гидроксикарбамоил-4-(4-метоксибензолсульфонил)- морфолин-3-карбоксамид;
(2R,3R)-N-гидрокси-2-гидроксикарбамоил-4-(4-метоксибензолсульфонил)- пиперидин-2-карбоксамид;
(2R, 3S)-N-гидрокси-1-(4-метоксибензолсульфонил)-3-(4- фенилпиридин-2-ил)-пиперидин-2-карбоксамид;
(2S, 3R)-N-гидрокси-1-(4-метоксибензолсульфонил)-2-(4- фенилпиридин-2-ил)-морфолин-2-карбоксамид;
(2R, 3S)-N-гидрокси-3-(2-хлоро-4-фторофенил)-1-(4- метоксибензолсульфонил)-пиперидин-2-карбоксамид; и
(2S, 3R)-N-гидpoкcи-2-(2-хлоро-4-фторофенил)-1-(4- метоксибензолсульфонил)-пиперидин-3-карбоксамид.
Данное изобретение, кроме того, относится к фармацевтической композиции для
(a) лечения таких состояний, как артрит, карцинома, образование язвенной ткани, рестеноз, периодонтальной болезни, врожденного буллезного эпидермолиза, склерит и других заболеваний, характеризуемых матричной металлопротеиназной активностью, СПИДа, сепсиса, септического шока и других заболеваний, включающих образование фактора некроза опухоли (TNF) или
(b) ингибирования матричных металлопротеиназ или образования фактора некроза опухоли (TNF) у млекопитающего, включая человека, содержащая количество соединения по п. 1 или его фармацевтически приемлемой соли, эффективное для такого лечения или ингибирования, и фармацевтически приемлемый носитель.
Данное изобретение также относится к способу ингибирования (a) матричных металлопротеиназ или (b) образования фактора некроза опухоли (TNF) у млекопитающего, включая человека, включающему введение указанному млекопитающему эффективного количества соединения по п.1 или его фармацевтически приемлемой соли.
Кроме того, данное изобретение относится к способу лечения таких состояний, как артрит, карцинома, образование язвенной ткани, рестеноз, периодонтальной болезни, врожденного буллезного эпидермолиза, склерита и других заболеваний, характеризуемых матричной металлопротеиназной активностью, СПИДа, сепсиса, септического шока и других заболеваний, включающих образование фактора некроза опухоли (TNF), у млекопитающего, включая человека, включающему введение указанному млекопитающему количества соединения по п.1 или его фармацевтически приемлемой соли, эффективного для лечения такого состояния.
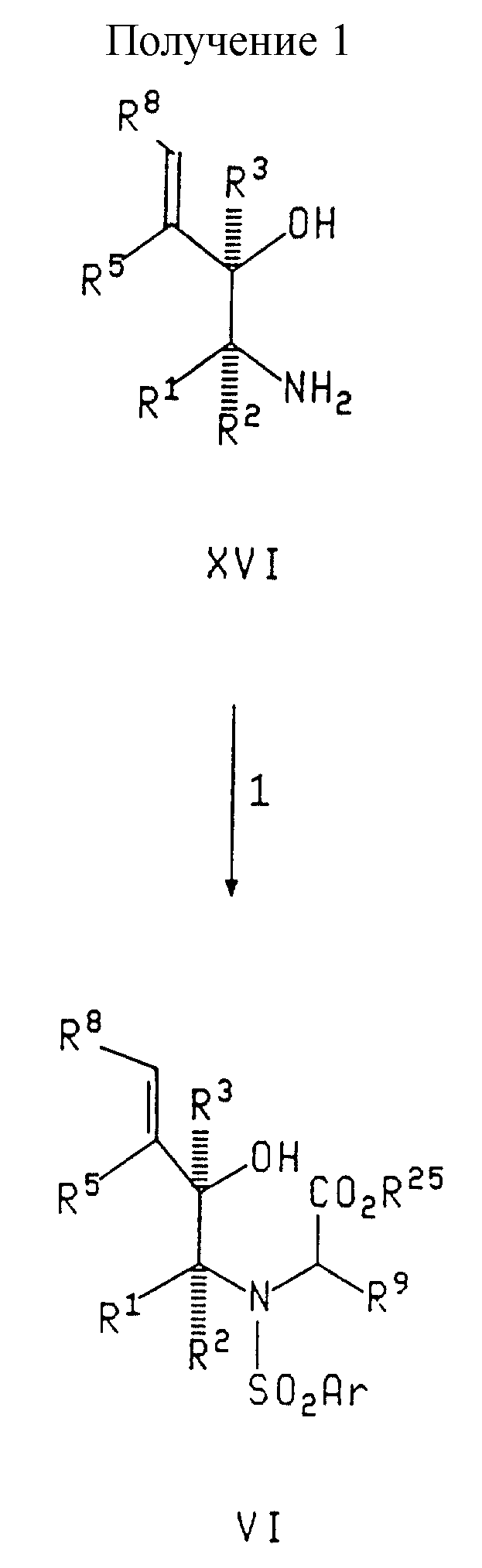
Схемы следующих реакций (см. получения 1 и 2, схемы 1 - 5 в конце описания) иллюстрируют получение соединений данного изобретения. Если не оговорено особо, то R1, R2, R3, R4, R5, R6, R7, R8 и R9, n и Ar в схемах реакций и в последующем обсуждении такие, как определено выше.
В реакции 1 Получения 1, соединение формулы XVI превращают соответствующее соединение - гидрокси сложный эфир формулы VI сначала путем взаимодействия XVI с арилсульфонилгалогенидом в присутствии триэтиламина и апротонного растворителя, такого как метиленхлорид, тетрагидрофуран или диоксан, при температуре между около 20oC до около 30oC, предпочтительно при комнатной температуре. Полученное таким образом соединение далее подвергают взаимодействию с соединением формулы
где R25 представляет собой карбобензилокси, (C1-C6)алкил, бензил, аллил или трет-бутил, в присутствии гексаметилдисилазана натрия и смеси растворителей тетрагидрофуран-диметилформамид при температуре между около -20oC до около 20oC, предпочтительно около 0oC, с образованием гидрокси эфира формулы VI.
В реакции 1 Получения 2, амино-соединение формулы XVIII, в котором R25 такой, как определено выше, превращают в соответствующий арилсульфониламин формулы XVII путем
(1) взаимодействия XVIII с арилсульфонилгалогенидом в присутствии триэтиламина и апротонного растворителя, такого как метиленхлорид, тетрагидрофуран, или диоксан, при температуре между около 20oC до около 30oC, предпочтительно при комнатной температуре,
(2) взаимодействия полученного таким образом соединения с соединением формулы
в присутствии гексаметилдисилазана натрия и смеси растворителей тетрагидрофуран-диметилформамид при температуре между от -20oC до около 20oC, предпочтительно около 0oC, и
(3) дальнейшего взаимодействия полученного таким путем соединения с озоном в растворе метиленхлорид-метанол при температуре между около -90oC до около -70oC, предпочтительно около -78oC. Полученный таким образом нестабильный озонид затем подвергают взаимодействию с трифенилфосфином с образованием арилсульфониламина формулы XVII.
В реакции 2 Получения 2, арилсульфониламин формулы XVII превращают в соответствующее соединение - гидроксиэфир формулы VI путем взаимодействия XVII с соединением формулы
где W - литий, магний, медь или хром.
В реакции 1 Схемы 1, соединение формулы VI, в котором R25 защитная группа представляет собой карбобензилокси, (C1-C6)алкил, бензил, аллил или трет-бутил, превращают в соответствующий морфолинон формулы V путем лактонизации и последующей перегруппировки Кляйзена соединения формулы VI. Реакцию завершают удалением R25 защитной группы из соединения формулы VI, которую проводят в условиях, соответствующих удалению конкретной используемой R25 защитной группы. Такие условия включают:
(а) обработку водородом в присутствии катализатора гидрирования, такого как 10% палладий на углероде, когда R25 представляет карбобензилокси,
(b) омыление, когда R25 - низший алкил,
(c) гидрогенолиз, когда R25 - бензил,
(d) обработку сильной кислотой, такой как трифторуксусная кислота или хлористоводородная кислота, когда R25 - трет- бутил, или
(e) обработку трибутилоловогидридом и уксусной кислотой в присутствии каталитического бис(трифенилфосфин) палладий (II) хлорида, когда R25 является аллилом.
В реакции 2 Схемы 1, морфолинон формулы V превращают в карбоновую кислоту формулы IV путем взаимодействия V с гексаметилдисилазаном лития в апротонном растворителе, таком как тетрагидрофуран, при температуре между около -90oC до около -70oC, предпочтительно около -78oC. Затем к реакционной смеси добавляют триметилсилил хлорид и растворитель, тетрагидрофуран, удаляют в вакууме и заменяют на толуол. Образующуюся реакционную смесь нагревают до температуры между около 100oC до около 120oC, предпочтительно около 110oC, и обрабатывают хлористоводородной кислотой с образованием карбоновой кислоты формулы IV.
В реакции 3 Схемы 1, карбоновую кислоту формулы IV превращают в соответствующую гидроксамовую кислоту формулы III обработкой IV 1-(3-диметиламинопропил)-3-этилкарбодиимидом и 1- гидроксибензтриазолом в полярном растворителе, таком как диметилформамид, с последующим добавлением гидроксиламина к реакционной смеси по истечении от около 15 минут до около 1 часа, предпочтительно через около 30 минут. Гидроксиламин предпочтительно получают in situ из солевой формы, такой как гидрохлорид гидроксиламина, в присутствии основания, такого как N-метилморфолин. Альтернативно, защищенное производное гидроксиламина или его солевую форму, где гидроксильная группа защищена в виде трет-бутилового, бензилового или аллилового эфира, может быть использовано в присутствии (бензотриазол-1- илокси)трис(диметиламино) фосфоний гексафторфосфата и основания, такого как N-метилморфолин. Удаление группы, защищающей гидроксиламин, проводят путем гидрогенолиза для бензил защитной группы или обработкой сильной кислотой, такой как трифторуксусная кислота, для трет-бутил защитной группы. Аллил защитную группу можно удалить путем обработки трибутилоловогидридом и уксусной кислотой в присутствии каталитического бис(трифенилфосфин) палладий (II) хлорида. Кроме того, N,O-бис(4-метоксибензил)гидроксиламин может быть использован в качестве защищенного производного гидроксиламина, где удаление защиты достигают, используя смесь метансульфоновой кислоты и фторуксусной кислоты.
В реакции 4 Схемы 1, соединение гидроксамовой кислоты формулы III превращают, при необходимости, в соответствующее соединение пиперидина формулы II путем обработки III водородом в присутствии катализатора гидрирования, такого как 10% палладий на углероде.
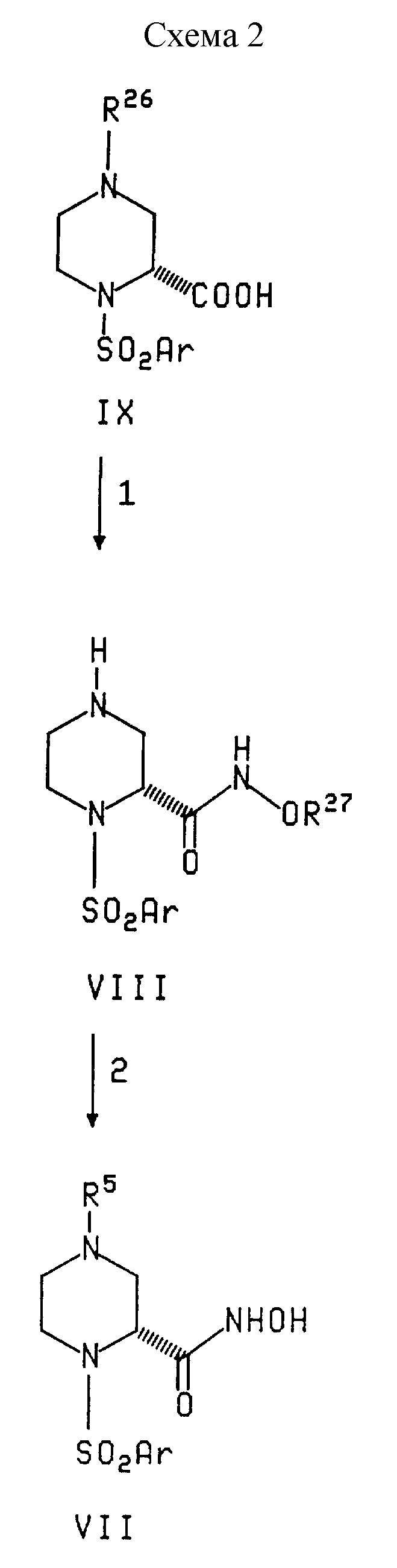
В реакции 1 Схемы 2, арилсульфонилпиперазин формулы IX, в котором R26 представляет карбобензилокси, бензил или карботретбутилокси, превращают в соединение формулы VIII взаимодействием IX с защищенным производным гидроксиламина формулы
R27ONH2•HCl
в котором R27 представляет трет-бутил, бензил или аллил, в присутствии дициклогексилкарбодиимида, диметиламинопиридина и апротонного растворителя, такого как метиленхлорид. R26 защищающую группу выбирают такой, чтобы ее можно было селективно удалить в присутствии и без потери R27 защитной группы, поэтому, R26 не может быть такой же, как R27. Удаление R26 защитной группы из соединения IX проводят в условиях, соответствующих условиям удаления конкретной используемой R26 защитной группы. Такие условия включают:
(a) обработку водородом в присутствии катализатора гидрирования, такого как 10% палладий на углероде, когда R26 представляет карбобензилокси,
(b) гидрогенолиз, когда R26 является бензилом или
(c) обработку сильной кислотой, такой как трифторуксусная кислота или хлористоводородная кислота, когда R26 является карботретбутилокси.
В реакции 2 Схемы 2, соединение формулы VIII превращают в соответствующее соединение гидроксамовой кислоты формулы VII, в котором R5 является водородом или (C1-C6)алкилом, взаимодействием, при необходимости, VIII с алкилгалогенидом, когда R5 является (C1-C6)алкилом. Последующее удаление R27 гидроксиламин защитной группы проводят путем гидрогенолиза для бензил защитной группы или обработкой сильной кислотой, такой как трифторуксусная кислота, для трет-бутил защитной группы. Аллил защитная группа может быть удалена обработкой трибутилоловогидридом и уксусной кислотой в присутствии каталитического количества бис(трифенилфосфин) палладий (II) хлорида.
В реакции 1 Схемы 3, арилсульфониламин формулы XII, в котором R25 является таким, как определено выше, превращают в соответствующее пиперазиновое соединение формулы XI взаимодействием XII с карбодиимидом и основанием, таким как триэтиламин. Соединение формулы XI далее подвергают взаимодействию с образованием соединения гидроксамовой кислоты формулы X по методике, описанной выше для реакции 3 Схемы 1.
В реакции 1 схемы 4, удаление R28 защитной группы и последующее восстановительное аминирование соединения формулы XXII, в котором Y - кислород, сера или углерод, с получением соответствующего иминового соединения формулы XXI, проводят в условиях, соответствующих условиям удаления конкретной используемой R28 защитной группы. Такие условия включают условия, используемые выше для удаления R26 защитной группы в реакции 1 Схемы 2.
В реакции 2 Схемы 4, иминовое соединение формулы XXI превращают в соответствующее пиперидиновое соединение формулы XX взаимодействием XXI с нуклеофилом формулы R2M, в котором M - литий, магний галогенид или церий галогенид. Реакцию проводят либо в эфирных растворителях, таких как диэтиловый эфир или тетрагидрофуране, при температуре между около -78oC до около 0oC, предпочтительно около -70oC.
В реакции 3 Схемы 1, сульфирование пиперидинового соединения формулы XX до соответствующего арилсульфонилпиперидинового соединения формулы XIX проводят взаимодействием XX с арилсульфонилгалогенидом в присутствии триэтиламина и апротонного растворителя, такого как метиленхлорид, тетрагидрофуран или диоксан, при температуре между около 20oC до около 30oC, предпочтительно при комнатной температуре.
В реакции 4 Схемы 4, арилсульфонилпиперидиновое соединение формулы XIX превращают в соединение гидроксамовой кислоты формулы XIX согласно способу, описанному выше в реакции 3 Схемы 1.
В реакции 1 Схемы 5, соединение формулы XXVI, в котором R29 и R31 защитные группы, каждую независимо выбирают из группы, состоящей из карбобензилокси, бензил и карботретбутилокси, и R30 - карбобензилокси, (C1-C6)алкил, бензил, аллил или трет-бутил, превращают в соответствующее иминовое соединение формулы XXV удалением R29 защитной группы и последующим восстановительным аминированием соединения формулы XXVI. R29 защитную группу выбирают такой, чтобы ее можно было селективно удалить в присутствии и без потери R31 защитной группы. Удаление R29 защитной группы из соединения формулы XXVI проводят в условиях, соответствующих условиям удаления конкретной используемой R29 защитной группы, которые не будут воздействовать на R31 защитную группу. Такие условия включают:
(a) обработку водородом в присутствии катализатора гидрирования, такого как 10% палладий на углероде, когда R29 является карбобензилокси и R31 является трет-бутилом,
(b) омыление, когда R29 является (C1-C6)алкилом и R31 является трет-бутилом,
(c) гидрогенолиз, когда R29 является бензилом и R31 является (C1-C6)алкилом или трет-бутилом,
(d) обработку сильной кислотой, такой как трифторуксусная кислота или хлористоводородная кислота, когда R29 является трет-бутилом и R31 является (C1-C6)алкилом, бензилом или аллилом, или
(e) обработку трибутилоловогидридом и уксусной кислотой в присутствии каталитического бис(трифенилфосфин) палладий (II) хлорида, когда R29 является аллилом и R31 является (C1-C6)-алкилом, бензилом или трет-бутилом.
R30 защитная группа может быть выбрана такой, чтобы ее можно было удалить на той же стадии реакции, что и R29 защитную группу.
В реакции 2 Схемы 5, иминовое соединение формулы XXV превращают в соответствующее соединение формулы XXIV взаимодействием XXV с нуклеофилом формулы R2M, в которой M - литий, магний галогенид или кальций галогенид. Реакцию проводят или в жирных растворителях, таких как диэтиловый эфир или в тетрагидрофуране при температуре между около -78oC до около 0oC, предпочтительно около -70oC.
В реакции 3 Схемы 5, сульфирование пиперидинового соединения формулы XXIV с получением соответствующего арилсульфонилпиперидинового соединения формулы III проводят согласно методике, описанной выше в реакции 3 Схемы 4.
В реакции 4 Схемы 5, арилсульфонилпиперидин формулы XXIII превращают в гидроксамовую кислоту формулы XIV путем
(1) удаления R30, при необходимости, и R31 защитных групп из XXIII с последующим
(2) взаимодействием XXIII согласно методике, описанной выше в реакции 3 Схемы 1.
Удаление R30 и R31 защитных групп соединения формулы XXIII проводят в условиях, соответствующих условиям удаления конкретной используемой R30 и R31 защитной группы. Такие условия включают условия, используемые выше для удаления R25 защитной группы в реакции 1 Схемы 3.
Фармацевтически приемлемые соли кислых соединений изобретения являются солями, образуемыми с основаниями, а именно, катионные соли, такие как соли щелочных и щелочноземельных металлов таких, как натрий, литий, калий, кальций, магний, а также соли аммония, такие как соли аммония, триметиламмония, диэтиламмония, и трис(гидроксиметил)-метиламмония.
Аналогично, соли присоединения кислот, таких как минеральных кислот, органических карбоновых кислот и органических сульфокислот, например, хлористоводородная кислота, метансульфокислота, малеиновая кислота, также можно использовать, при условии, что основная группа, такая как пиридил, составляет часть структуры.
Способность соединений формулы I или их фармацевтически приемлемых солей (здесь и ниже обозначаемые как соединения данного изобретения) ингибировать матричные металлопротеиназы или образование фактора некроза опухоли (TNF) и, следовательно, их эффективности при лечении заболеваний, характеризуемых матричной металлопротеиназой или образованием фактора некроза опухоли, показана с помощью следующих биологических тестов in vitro.
БИОЛОГИЧЕСКИЕ ТЕСТЫ
Ингибирование коллагеназы человека (MMP-1)
Рекомбинантную коллагеназу человека активируют трипсином, используя следующее отношение: 10 мкг трипсина на 100 мкг коллагеназы. Трипсин и коллагеназу инкубируют при комнатной температуре в течение 10 минут, затем добавляют пятикратный избыток (50 мкг/10 мкг трипсина) соевого ингибитора трипсина.
Приготавливают исходные растворы ингибиторов по 10 мкМ в диметилсульфоксиде и затем разбавляют, используя следующую схему:
10 мМ ---> 120 мкМ ---> 12 мкМ ---> 1.2 мкМ ---> 0.12 мкМ
Двадцать пять микролитров раствора каждой концентрации затем добавляют в соответствующие лунки 96-луночного микрофлюоресцирующего планшета, дублируя каждую концентрацию три раза. Конечная концентрация ингибитора должна составлять 1: 4 разбавление после добавления фермента и субстрата. Положительные контроли (фермент, отсутствует ингибитор) помещают в лунки D1-D6 и контрольные пробы (отсутствует фермент, отсутствуют ингибиторы) помещают в лунки D7-D12.
Коллагеназу разбавляют до 400 нг/мл и 25 мкл затем добавляют в соответствующие лунки микрофлюоресцирующего планшета. Конечная концентрация коллагеназы в пробе составляет 100 нг/мл.
Субстрат (DNP-Pro-Cha-Gly-Cys(Me)-His-Ala-Lys (NMA)-NH2) получают в виде 5 мМ исходного раствора в диметилсульфоксиде и затем разбавляют до 20 мкМ буфером для анализа. Пробу инициируют путем добавления 50 мкл субстрата на лунку микрофлюоресцирующего планшета, получая конечную концентрацию 10 мкМ.
Измерение флюоресценции (360 нм возбуждение, 460 нм эмиссия) проводят при времени 0 и затем через 20-минутные интервалы времени. Анализ проводят при комнатной температуре при обычном времени испытания, составляющем 3 часа.
Затем строят график зависимости флюоресценции от времени как для контрольных образцов, так и для образцов, содержащих коллагеназу (данные усредняют из трех определений). Для определения IC50 значений выбирают точку времени, которая обеспечивает хороший сигнал (контрольная проба), и эта точка лежит на линейном участке кривой (обычно около 120 мин). Нулевое время используют как контроль для каждого соединения при каждой концентрации и эти значения вычитают из 120-минутных данных. Данные представляются в виде графика концентрация ингибитора - % контроля (флюоресценция ингибитора, деленная на флюоресценцию самой коллагеназы х 100). Значения IC50 определяют исходя из концентрации ингибитора, которая дает сигнал, который составляет 50% контроля.
Если оказывается, что IC50-значения < 0.03 мкМ, тогда ингибиторы испытывают при концентрации 0.3 мкМ, 0.03 мкМ, 0.03 мкМ и 0.003 мкМ.
Ингибирование желатиназы (MMP-2)
Ингибирование желатиназной активности определяют, используя Dnp-Pro-Cha-Gly-Cys(Me)-His-Ala-Lys(NMA)-NH2 субстрат (10 мкМ) в тех же самых условиях, как ингибирование коллагеназы человека (MMP-1).
72 kD желатиназы активируют 1 мМ APMA (п-аминофенилацетат ртути) в течение 15 часов при 4oC и разбавляют, получая конечную концентрацию в пробе 100 мг/мл. Ингибиторы разбавляют также, как для ингибирования коллагеназы человека (MMP-1), получая конечные концентрации в пробе 30 мкМ, 3 мкМ, 0.3 мкМ и 0.03 мкМ. Каждую концентрацию готовят трижды.
Измерение флюоресценции (360 нм возбуждение, 460 нм эмиссия) проводят при времени 0 и затем через 20-минутные интервалы времени в течение 4 часов.
Значения IC50 определяют так же, как в случае ингибирования коллагеназы человека (MMP-1). Если оказывается, что IC50-значения составляют менее, чем 0.03 мкМ, то тогда ингибиторы испытывают при конечных концентрациях 0.3 мкМ, 0.03 мкМ, 0.003 мкМ и 0.003 мкМ.
Ингибирование активности стромелизина (MMP-3)
Ингибирование активности стромелизина основано на модифицированном спектрофотометрическом анализе, описанном Weingarten и Feder (Weingarten, H. and Feder, J. , Spectrophotometric Assay for Vertebrate Collagenase, Anal. Biochem. 147, 437-440 (1985)). Гидролиз субстрата тио пептолида [Ac-Pro-Leu-Gly- SCH[CH2CH(CH3)2] CO-Leu-Gly-OC2H5] дает меркаптановый фрагмент, который можно контролировать в присутствии реагента Элмана (Ellman's reagent).
Человеческий рекомбинантный простромелизин активируют трипсином, используя соотношение: 1 мкл 10 кг/мл исходного раствора трипсина на 26 мкг стромелизина. Трипсин и стромелизин инкубируют при 37oC в течение 15 минут, затем добавляют 10 мкл 10 мг/мл соевого трипсинового ингибитора в течение 10 минут при 37oC, для блокирования трипсиновой активности.
Испытания проводят в общем объеме 250 мкл буфера для испытания (200 мМ хлорид натрия, 50 мМ MES и 10 мМ хлорид кальция, pH 6.0) в 96-луночном титрационном (microliter) микропланшете. Активированный стромелизин разбавляют буфером для анализа до 25 мкг/мл. Реагент Элмана (3-карбокси-4-нитрофенил дисульфид) полученный в виде 1М исходного раствора в диметилформамиде и разбавленный до 5 мМ буфером для анализа добавляют в количестве 50 мкл на лунку, получая конечную концентрацию 1 мМ. Получают 10 мМ исходные растворы ингибиторов в диметилсульфоксиде и разбавляют серийно буфером для анализа так, чтобы добавление 50 мкл в соответствующие лунки привело к получению конечных концентраций 3 мкМ, 0.3 мкМ, 0.003 мкМ и 0.0003 мкМ. Все концентрации готовят трижды.
300 мМ диметилсульфоксидный исходный раствор пептидного субстрата разбавляют до 15 мМ буфером для анализа и испытание инициируют путем добавления 50 мкл в каждую лунку, получая конечную концентрацию 3 мМ субстрата. Контрольные пробы состоят из пептидного субстрата и реагента Элмана без фермента. Образование продукта контролируют при 405 нм с помощью устройства для чтения микропланшета Molecular Devicces UVmax plate reader.
Значения IC50 определяют тем же самым способом, как для коллагеназы.
Ингибирование MMP-13
Человеческий рекомбинантный MMP-13 активируют 2 мМ APMA (п- аминофенилацетат ртути) в течение 1.5 часов, при 37oC и разбавляют до 400 мг/мл буфером для анализа (50 мМ Трис, pH 7.5, 200 мМ хлорид натрия, 5 мМ хлорид кальция, 5 мкМ хлорид цинка, 0.02% Бридж (brij)). В лунку 96-луночного микрофлюоресцирующего планшета вводят двадцать пять микролитров разбавленного фермента. Затем фермент разбавляют в соотношении 1:4 в пробе путем добавления ингибитора и субстрата, получая конечную концентрацию в пробе 100 мг/мл.
Получают 10 мМ исходные растворы ингибиторов в диметилсульфоксиде и затем разбавляют буфером для анализа так же, как в схеме разбавления ингибитора для ингибирования человеческой коллагеназы. Двадцать пять микролитров каждой концентрации вводят трижды (в трех экземплярах) в ячейки микрофлюоресцирующего планшета. Конечные концентрации в пробе составляют 30 мкМ, 3 мкМ, 0.3 мкМ и 0.03 мкМ.
Субстрат (Dnp-Pro-Cha-Gly-Cys(Me)-His-Ala-Lys(NMA)-NH2) получают так же, как для ингибирования человеческой коллагеназы (MMP-1) и в каждую лунку добавляют 50 мкл, получая конечную концентрацию 10 мкМ. Измерение флюоресценции (360 нм возбуждение, 450 нм эмиссия) проводят при времени 0 и затем через 5-минутные интервалы времени в течение 1 часа.
Положительные контроли состоят из фермента и субстрата без ингибитора, а контрольные пробы состоят только из субстрата.
Значения IC50 определяют так же, как для ингибирования человеческой коллагеназы (MMP-1). Если оказывается, что IC50- значения составляют менее, чем 0.03 мкМ, то тогда ингибиторы испытывают при конечных концентрациях 0.3 мкМ, 0.03 мкМ, 0.003 мкМ и 0.0003 мкМ.
Ингибирование образования TNF
Способность соединений или их фармацевтически приемлемых солей ингибировать образование TNF и, следовательно, демонстрация их эффективности для лечения заболеваний, включающих образование TNF, показана в следующем in vitro испытании.
Человеческие моноядерные клетки выделяют из крови человека с анти-свертывающим средством, используя одноступенчатую методику разделения Ficoll-hypaque. (2) Моноядерные клетки промывают три раза в Hanks балансированном солевом растворе (ХБСР, HBSS) с двухвалентными ионами и вновь суспендируют до плотности 2•106/мл в ХБСР (HBSS), содержащим 1% БСА (BSA). Проведенные дифференциальные подсчеты с использованием Abbott Cell Dyn 3500 анализатора показали, что содержание моноцитов колеблется в диапазоне от 17 до 24% от общего содержания клеток в этих препаратах.
180 мкл клеточную суспензию помещают в лунки с плоским дном 96-луночного планшета. Добавления соединений и LPS (100 нг/мл конечная концентрация) давали конечный объем 200 мкл. Все анализы повторяют трижды. Через четыре часа инкубации при 37oC в инкубаторе с увлажненным CO2, планшеты удаляют и центрифугируют (10 минут при приблизительно 250 х g) и надсадочную жидкость удаляют и анализируют на TNFα, используя R & D ELISA Kit.
Для введения людям, для ингибирования матричных металлопротеиназ или образования фактора некроза опухоли (TNF), может быть использован ряд обычных способов, включая пероральный, парентеральный и местный. В общем, активное соединение может быть введено перорально или парентерально в дозах от около 0.1 до 25 мг/кг веса тела субъекта, подлежащего лечению, в день, предпочтительно, от около 0.3 до 5 мг/кг. Однако некоторая вариация дозы будет обязательно иметь место в зависимости от субъекта, подлежащего лечению. Лицо, ответственное за назначение лекарственного препарата, обязано, в любом случае, оценить подходящую дозу для конкретного субъекта.
Соединения данного изобретения могут быть введены в виде целого ряда различных лекарственных форм; в общем, терапевтически эффективные соединения этого соединения присутствуют в таких лекарственных формах при уровнях концентрации, находящихся в диапазоне от около 5.0% до около 70% по весу.
Для перорального введения таблетки, содержащие различные наполнители, такие как микрокристаллическая целлюлоза, цитрат натрия, карбонат кальция, дикальций фосфат и глицин, могут применяться вместе с разрыхлителями, такими как крахмал (и предпочтительно кукурузный, картофельный крахмал или крахмал тапиоки), альгиновая кислота и некоторые сложные силикаты, наряду с связующими грануляции, такими как поливинилпирролидон, сахароза, желатин и камедь. Дополнительно, для целей таблетирования часто применяют смазывающие агенты, такие как стеарат магния, лаурил сульфат натрия и тальк. Твердые композиции похожего типа могут также использоваться в качестве наполнителей в желатиновых капсулах; предпочтительными веществами в этой связи являются лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли. Когда для перорального применения требуются водные суспензии и/или эликсиры, то активный ингредиент может быть скомбинирован с различными подслащивающими или ароматизирующими веществами, окрашивающим веществом или красителями, и, при необходимости, эмульгирующим и/или суспендирующим агентами, а также с такими разбавителями, как вода, этанол, пропиленгликоль, глицерин и различными подобными комбинациями.
Для парентерального введения (внутримышечное, внутрибрюшинное, подкожное и внутривенное использование) обычно получают стерильный инъецируемый раствор активного ингредиента. Могут также применяться растворы терапевтического соединения данного изобретения в либо кунжутном, либо в арахисовом масле или в водном пропиленгликоле. Водные растворы должны быть подходящим образом доведены и забуферены, предпочтительно до pH выше, чем 8, если необходимо, и жидкий разбавитель прежде всего придает изотоничность. Эти водные растворы пригодны для внутривенных инъекций. Масляные растворы пригодны для целей внутрисуставной, внутримышечной и подкожной инъекции. Получение всех этих растворов в стерильных условиях легко осуществимо с помощью стандартных фармацевтических методик, хорошо известных специалистам в данной области техники.
Кроме того, возможно введение соединений данного изобретения местно, например при лечении воспалительных состояний кожи, и это может быть выполнено с помощью кремов, желе, гелей, паст и мазей, в соответствии со стандартной фармацевтической практикой.
Данное изобретение иллюстрируется следующими примерами, но оно не ограничивается ими.
ПРИМЕР 1
(+)-(2R*, 3R*)-(N-гидрокси)-1-(4-метокси- бензолсульфонил)-3-метил-1,2,3,6-тетрагидропиридин-2- карбоксамид
(a) К раствору (E)-1-амино-3-пентент-2-ола (2.0 грамма, 10.0 ммол) в метиленхлориде (50 мл) добавляют триэтиламин (160 мкл, 11.0 ммол) с последующим добавлением 4-метоксибензолсульфонил хлорида (2.07 грамма, 10.0 ммол). Смесь перемешивают при комнатной температуре в течение 12 часов и разбавляют этилацетатом. Смесь промывают водой, 10% лимонной кислотой, сушат (сульфат натрия), фильтруют и концентрируют. Неочищенный продукт очищают с помощью хроматографии на силикагеле (элюирование смесью 2:1 этилацетат- гексаны) с получением (N-(2-гидроксипент-3-енил)-4- метоксибензолсульфонамида.
(b) К раствору (±)-(E)-N-(2-гидрокси-пент-3-енил-4-метокси- бензолсульфонамида (1.2 грамма, 4.42 ммол) в смеси тетрагидрофуран- диметилформамид (10 мл, прибл. 3:1) при 0oC добавляют натрий бис(триметилсилил)амид (4.9 мл, 1.0 М раствор в тетрагидрофуране). Через 10 минут добавляют т-бутилбромацетат (786 мл, 4.83 ммол). Смесь нагревают до комнатной температуры, перемешивают в течение 1 часа и гасят насыщенным раствором хлорида аммония. Смесь экстрагируют этилацетатом и объединенные экстракты сушат (сульфат натрия), фильтруют и концентрируют. Неочищенный продукт очищают с помощью хроматографии на силикагеле (элюирование смесью 1:1 этилацетат-гексаны) с получением т-бутилового эфира [(2-гидрокси-пент-3-енил)-(4- метоксибензолсульфонил)-амино]-уксусной кислоты.
(c) К раствору т-бутилового эфира (±)-(E)-N-[(2-гидрокси- пент-3-енил)-(4-метоксибензолсульфонил)-амино] -уксусной кислоты (900 мг, 2.43 ммол) в бензоле (10 мл) добавляют трифторуксусную кислоту (56 мкл, 0.73 ммол). Раствор нагревают до 80oC в течение 3 часов, охлаждают до комнатной температуры и концентрируют, получая (±)-(E)-4-(4-метоксибензолсульфонил)-6- пропенилморфолин-2-он, который используют без дальнейшей очистки.
(d) К раствору бис(триметилсилил)амида (2.67 ммол, 1.0 М в тетрагидрофуране) в тетрагидрофуране (5.0 мл) при -78oC добавляют раствор неочищенного (±)-(E)-4-(4-метоксибензолсульфонил)-6- пропенилморфолин-2-она с предыдущей стадии. Через 15 минут добавляют триметилсилил хлорид (1.53 мл, 12.15 ммол] и смесь нагревают до комнатной температуры. Растворитель удаляют (в вакууме) и заменяют на толуол (10 мл). Образующуюся смесь нагревают при 110oC в течение 3 часов, охлаждают до комнатной температуры и обрабатывают 1Н раствором хлористоводородной кислоты. После перемешивания в течение 10 минут смесь экстрагируют этилацетатом и объединенные экстракты сушат (сульфат натрия), фильтруют и концентрируют. Неочищенный продукт очищают с помощью хроматографии на силикагеле (элюирование смесью 2:1 этилацетат-гексаны с 1% уксусной кислоты) с получением (±)- (2R+, 3R+)-1-(4-метокси-бензолсульфонил)-3-метил-1,2, 3,6-тетрагидропиридин-2-карбоновой кислоты.
(e) К раствору (±)-(2R*,3R*)-1-(4-метокси-бензолсульфонил) -3-метил-1,2,3,6-тетрагидропиридин-2-карбоновой кислоты (100 мг, 0.36 ммол) в диметилформамиде (5 мл) добавляют гидроксибензтриазол (53 мг, 0.39 ммол) и 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид (75 мг, 0.39 ммол). Через 1 час добавляют гидроксиламин гидрохлорид (75 мг, 1.08 ммол) с последующим добавлением триэтиламина (150 мкл, 1.08 ммол). После перемешивания в течение ночи смесь разбавляют водой и экстрагируют этилацетатом. Объединенные экстракты сушат, фильтруют и концентрируют. Неочищенный продукт очищают с помощью хроматографии на силикагеле (элюирование смесью 2:1 этилацетат-гексаны с 1% уксусной кислоты) с получением (±)-(2R*, 3R*)-(N-гидрокси)-1-(4-метокси- бензолсульфонил)-3-метил- 1,2,3,6-тетрагидропиридин-2-карбоксамида в виде белого твердого вещества. Точка плавления 173oC (разл.).
Масс-спектр (термораспыление): м/з 326 (м-C(O)N(H)OH, 100%), (м, 7%), (м+H, 30%), (м+NH4, 10%).
1H ЯМР (CDCl3, 250 мГц, ппм): δ 7.72 (д, J = 8.9 Гц, 2H), 7.03 (д, J = 8.9 Гц, 2H), 5.66 (дк, J = 13.0, 2.7 Гц, 1H), 5.45 (дд, J = 13.0, 1.9 Гц), 4.37 (д, 7.0 Гц, 1H), 4.06-3.82 (м, 2H), 3.82 (с, 3H), 3.43-3.30 (м, 1H), 2.62-2.31 (м, 1H), 0.97 (д, 7.5 Гц, 2H).
ПРИМЕР 2
N-гидрокси-1-(4-метоксибензолсульфонил)-3-фенил-1,2,3,6- тетрагидропиридин-2-карбоксамид
(a) К раствору т-бутилового эфира глицина (5.0 грамм, 29.82 ммол) в метиленхлориде (50 мл) добавляют триэтиламин (6.65 мл, 62.63 ммол) с последующим добавлением 4-метоксибензолсульфонил хлорида (29.82 ммол, 6.2 грамма). Раствор перемешивают в течение 24 часов, разбавляют водой и экстрагируют этилацетатом и объединенные экстракты сушат (сульфат натрия), фильтруют и концентрируют. Неочищенный продукт очищают с помощью хроматографии на силикагеле (элюирование смесью 6:1 гексаны-этилацетат) с получением т-бутилового эфира (4-метоксибензолсульфониламино)- уксусной кислоты.
(b) К раствору т-бутилового эфира (4-метоксибензолсульфониламино) уксусной кислоты (3.0 грамма, 10 ммол) в смеси тетрагидрофуран-диметилформамид (мл, прибл. 3:1) при 0oC добавляют бис(триметилсилил)амид натрия (10.0 мл, 1.0 М раствор в тетрагидрофуране). Через 10 минут добавляют 4-бромо-2-метил-2- бутен (1.27 мкл, 11.0 ммол). Смесь нагревают до комнатной температуры, перемешивают в течение часа и гасят насыщенным раствором хлорида аммония. Смесь экстрагируют этилацетатом и объединенные экстракты сушат (сульфат натрия), фильтруют и концентрируют. Неочищенный продукт очищают с помощью хроматографии на силикагеле (элюирование смесью 1:1 этилацетат-гексаны) с получением т-бутилового эфира [(4-метоксибензолсульфонил)-(3- метил-бут-2-енил)-амино]-уксусной кислоты.
(c) Озон пропускают через раствор т-бутилового эфира [(4- метоксибензолсульфонил)-(3-метил-бут-2-енил)-амино] -уксусной кислоты (2.0 грамма, 5.4 ммол) в смеси метиленхлоридметанол (50 мл, прибл. 1:1) при -78oC до тех пор, пока раствор не приобретет устойчивую синюю окраску. Добавляют трифенилфосфин (4.24 грамма, 16.2 ммол) и полученный раствор перемешивают при комнатной температуре в течение 3 часов. Концентрированием получают сырой продукт, который очищают с помощью хроматографии на силикагеле (элюирование смесью 1: 1 этилацетат-гексаны) с получением т- бутилового эфира [(4-метоксибензолсульфонил)-(2-оксо-этил) - амино]-уксусной кислоты.
(d) К суспензии хлорида хрома (II) (1.3 грамма, 10.49 ммол) в диметилформамиде (20 мл) добавляют суспензию хлорида никеля (II) (0.026 ммол, 1 мг) в диметилформамиде (1 мл) с последующим добавлением смеси (транс)-β-иодостирола (1.20 грамма, 5.24 ммол) и т-бутилового эфира [(4-метоксибензолсуль-фонил)-2-оксо-этил)- амино] уксусной кислоты (900 мг, 2.62 ммол) в диметилформамиде (5 мл). Полученный раствор перемешивают в течение трех часов, разбавляют водой и экстрагируют этилацетатом. Объединенные экстракты промывают соляным раствором, сушат (сульфат натрия), фильтруют и концентрируют. Неочищенный продукт очищают с помощью хроматографии на силикагеле (элюирование смесью 3:2 гексан- этилацетат) с получением т-бутилового эфира (±)-(E)-[(2-гидрокси- 4-фенил-бут-3-енил)-(4-метоксибензолсульфонил)-амино] -уксусной кислоты.
(e) т-бутиловый эфир (±)-(E)-[(2-гидрокси-4-фенил-бут-3- енил)-(4-метоксибензолсульфонил)-амино] -уксусной кислоты подвергают реакциям, описанным в Примере 1 (c). Неочищенный продукт перекристаллизовывают из хлороформа, получая (±)-(E)-4- (4-метоксибензолсульфонил)-6-стирил-морфолин-2-он.
(f) (±)-(E)-4-(4-метоксибензолсульфонил)-6-стирил-морфолин- 2-он подвергают реакциям, описанным в Примере 1 (d). Неочищенный продукт очищают с помощью хроматографии на силикагеле (элюирование смесью 2:1 гексан-этилацетат с 1% уксусной кислоты) с получением (±)-(2R*,3R*)-1-(4-метокси-бензол- сульфонил)-3-фенил-1,2,3,6-тетрагидропиридин-2-карбоновой кислоты.
(g) (±)-(2R*, 3R*)-1-(4-метокси-бензолсульфонил)- 3-фенил-1,2,3,6-тетрагидропиридин-2-карбоновую кислоту подвергают реакциям, описанным в Примере 1 (e). Неочищенный продукт очищают с помощью хроматографии на силикагеле (элюирование смесью 1:1 гексан-этилацетат с 1% уксусной кислоты) с получением N-гидрокси-1- (4-метокси-бензолсульфонил)-3-фенил-1,2,3,6-тетрагидропиридин-2- карбоксамида в виде белого твердого вещества. Точка плавления 151-154oC (разл.).
Масс-спектр [ПБМС м/Х.И. (NH3)]: м/з 388 (м+NH4, 100%).
1H ЯМР (CD3OD) δ 7.75 (д, J = 8.5 Гц, 2H), 7.38-7.12 (м, 5H), 7.04 (д, J = 8.5 Гц, 2H), 5.91 (д, J = 8.9 Гц, 1H), 5.28 (д, J = 9.9 Гц, 1H), 4.89 (с, H2O), 4.57 (д, 6.8 Гц, 1H), 4.07 (АВк, JAB = 18.0 Гц, ΔvAB = 39.1 Гц, 2H), 3.85 (о, 3H), 3.39 (шс, CD3OD).
ПРИМЕР 3
(±)-(2R*, 3R*)-N-гидpoкcи-1- (4-метоксибензолсульфонил)-3-фенилпиперидин-2-карбоксамид
(a) К раствору (±)-(2R*, 3R*)-1-(4- метоксибензолсульфонил)-3-фенил-1,2,3,6-тетрагидропиридин-2-карбоновой кислоты (65 мг, 0.17 ммол) (из Примера 20), добавляют бензилгидроксиламин гидрохлорид (32 мг, 0.20 ммол), дициклогексилкарбодиимид (41 мг, 0.20 ммол) и диметиламинопиридин (27 мг, 0.22 ммол). Образующуюся смесь перемешивают в течение ночи, разбавляют этилацетатом и фильтруют через CeliteTM и выпаривают. Неочищенный продукт очищают с помощью хроматографии элюированием смесью 1:1 гексан-этилацетат с получением (±)-(2R*, 3R*)-N-бензилокси-1- (4-метоксибензолсульфонил)-3-фенил-1,2,3,6-тетрагидропиридин-2- карбоксамида.
(b) К раствору (±)-(2R*,3R*) -N-бензилокси-1-(4-метокси-бензолсульфонил)-3-фенил-1,2,3, 6-тетрагидропиридин-2-карбоксамида (35 мг, 0.073 ммол) в этаноле (5 мл) добавляют 10% палладий на углероде (10 мг, 5 мол). Воздух откачивают под вакуумом и колбу повторно наполняют водородом (повторяют два раза). Затем реакционную смесь перемешивают в течение 1 часа, и в течение этого времени ее фильтруют через CeliteTM и концентрируют. Продукт (±)-(2R*, 3R*)-N-гидрокси- 1-(4-метоксибензолсульфонил)-3-фенил-пиперидин-2-карбоксамид собирают в виде белого твердого вещества. Точка плавления 163oC (разл. ).
Масс-спектр [ПБМС м/Х.И. (NH3)]: м/з 390 (м+H2), (м+NH4).
1H ЯМР (CD3OD) δ 7.73 (д, J = 8.9 Гц, 2H), 7.31-7.37 (м, 5H), 7.04 (д, J= 8.9 Гц, 2H), 4.89 (с, H2O), 4.34 (д, J = 5.4 Гц, 1H), 3.86 (с, 3H), 3.74-3.63 (м, 2H), 3.31 (шс, CD3OD), 2.99-2.90 (м, 1H), 2.58-2.52 (м, 1H), 1.94-1.88 (м, 1H), 1.67-1.60 (м, 2H).
ПРИМЕР 4
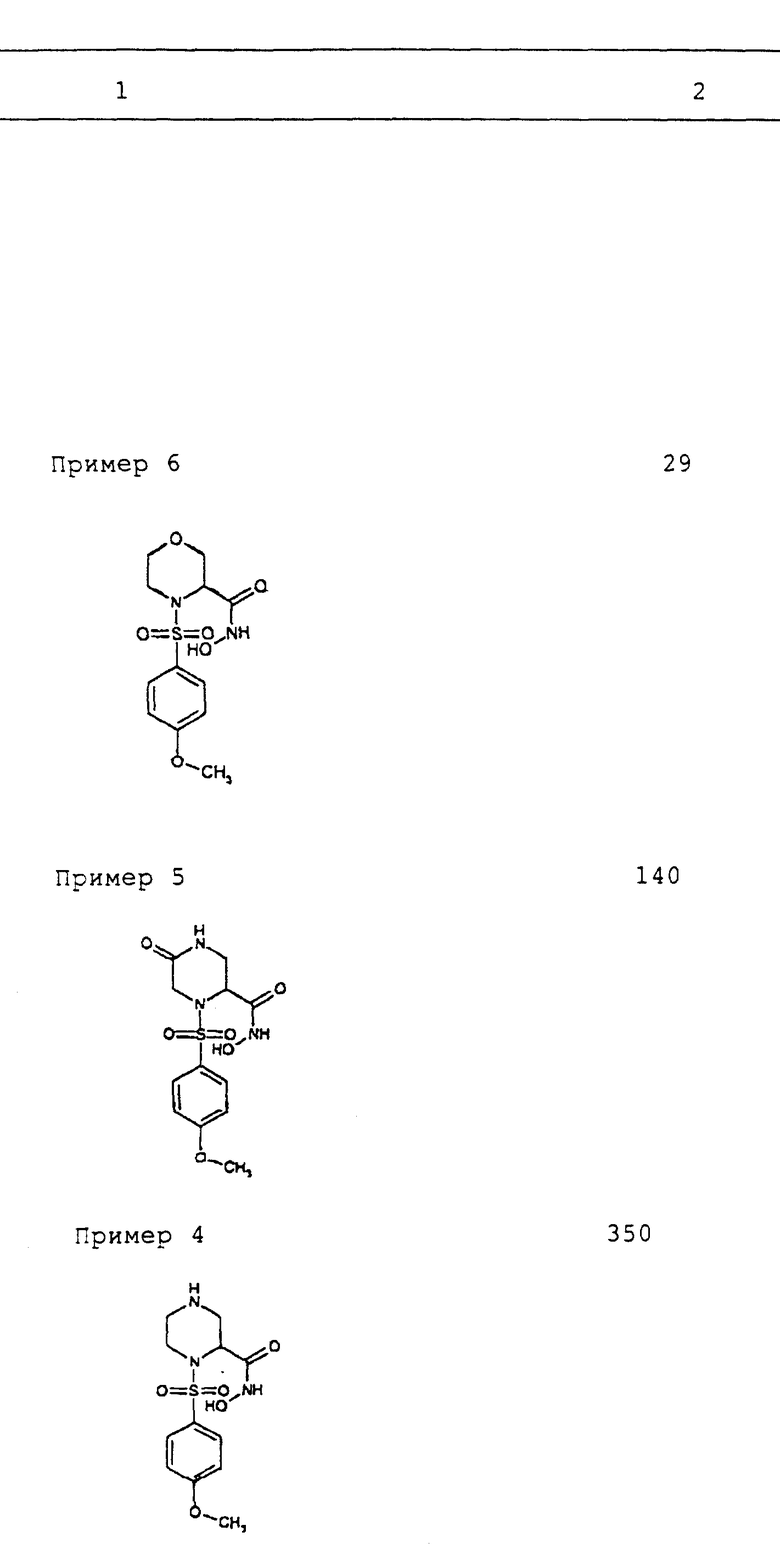
(±)-N-гидрокси-1-(4-метоксибензолсульфонил)-2- пиперазинкарбоксамид гидрохлорид
(a) К раствору (±)-4-бензилоксикарбонил-2-пиперазинкарбоновой кислоты (1.90 грамм, 7.2 ммол) в смеси диоксан-вода (10 мл, прибл. 1:1) добавляют 1N раствор гидроксида натрия (15 мл, 15 ммол) с последующим добавлением 4-метоксибензолсульфонил хлорида. Раствор перемешивают в течение 1 часа, подкисляют 1N хлористоводородной кислотой и экстрагируют этилацетатом. Объединенные экстракты промывают, сушат (сульфат натрия), фильтруют и концентрируют. Неочищенный продукт очищают с помощью хроматографии на силикагеле (элюирование смесью 2:1 гексан- этилацетат с 1% уксусной кислотой) с получением (±)-1-(4- метоксибензолсульфонил)-4-бензилоксикарбонил-2-пиперазинкарбоновой кислоты.
(b) К раствору (±)-1-(4-метоксибензолсульфонил)-4- бензилоксикарбонил-2-пиперазинкарбоновой кислоты (100 мг, 0.23 ммол) в метиленхлориде (5 мл) добавляют O-т-бутил-гидроксиламин гидрохлорид (35 мг, 0.28 ммол), диметиламинопиридин (37 мг, 0.30 ммол) и дициклогексилкарбодиимид (57 мг, 0.28 ммол). После перемешивания в течение ночи реакционную смесь разбавляют гексанами и высадившееся твердое вещество отфильтровывают. Раствор концентрируют и неочищенный продукт очищают с помощью хроматографии на силикагеле (элюирование смесью 2:1 этилацетат-гексаны с 1% уксусной кислотой) с получением (±) -N-(т-бутилокси)-1-(4-метоксибензолсульфонил)-4-бензилоксикарбонил- 2-пиперазинкарбоксамида.
(c) К раствору (±)-N-(т-бутилокси)-1-(4-метоксибензолсульфонил)- 4-бензилоксикарбонил-2-пиперазинкарбоксамида (68 мг, 0.134 ммол), в метаноле (6 мл) добавляют 10% палладий на углероде (7 мг). Воздух откачивают под вакуумом и колбу повторно наполняют водородом (повторяют 2 раза). Реакционную смесь затем перемешивают в течение 1 часа, и в это время ее фильтруют через CeliteTM и концентрируют. Продукт (±)-N-(т-бутилокси)-1- (4-метоксибензолсульфонил)-2-пиперазинкарбоксамид используют без какой-либо дополнительной очистки.
(d) К раствору (±)-N-(т-бутилокси)-1-(4- метоксибензолсульфонил)-2-пиперазинкарбоксамида (30 мг) в дихлорэтане добавляют этанол (1 капля). Раствор охлаждают до -10oC и хлористый водород в виде газа продувают через раствор в течение 5 минут. Затем реакционную смесь герметизируют и перемешивают в течение 24 часов, и в течение этого времени объем смеси уменьшают до 1/3 путем испарения, и осадившееся твердое вещество фильтруют и сушат (в вакууме), получая (±)-N-гидрокси-1-(4- метоксибензолсульфонил)-2-пиперазинкарбоксамид гидрохлорид в виде белого твердого вещества. Точка плавления 167oC (разл.).
Масс-спектр (термораспыление): м/з 343 (м+1 100%).
1H ЯМР (CD3OD, 250 мГц, ппм): δ 7.76 (д, J = 8.9 Гц, 2H), 7.07 (д, J = 8.9 Гц, 2H), 3.87 (шс, H2O), 4.19 (д, J = 3.3 Гц, 1H), 3.87 (с, 3H), 3.58 (шд, J = 6.2 Гц, 1H), 3.42 (шд, J = 6.1 Гц, 1H), 3.30 (шс, CD3OD), 3.16 (д, J = 13.5 Гц, 1H), 2.87 (шд, J = 13.3 Гц, 1H), 2.69 (дд, J = 13.3 Гц, 1H), 2.51 (дт, J = 12.5, 3.8 Гц, 1H).
ПРИМЕР 5
N-гидрокси-1-(4-метоксибензолсульфонил)-5-оксо-пиперазин-2- карбоксамид
(a) К раствору (±)-бензилоксикарбониламино-2-т-бутокси- карбониламинопропионата (2.8 грамм, 7.9 ммол) в метиленхлориде (25 мл) при 0oC добавляют раствор хлористоводородной кислоты (г), растворенной в диоксане (25 мл). Раствор перемешивают при 0oC в течение 4 часов и затем концентрируют. Неочищенный продукт гидрохлорид метилового эфира 3-бензилоксикарбониламино-2-амино- пропионовой кислоты используют без дополнительной очистки.
(b) Гидрохлорид метилового эфира 3-бензилоксикарбониламино- 2-амино-пропионовой кислоты подвергают реакциям, описанным в Примере 1 (a). Неочищенный продукт очищают с помощью хроматографии на силикагеле (элюирование смесью 1: 1 гексан-этилацетат) с получением метилового эфира (±)-3-бензилоксикарбониламино-2-(4- метоксибензолсульфониламино)-пропионовой кислоты.
(c) Метиловый эфир (±)-3-бензилоксикарбониламино-2-(4- метоксибензолсульфониламино)-пропионовой кислоты подвергают реакциям, описанным в Примере 1. Неочищенный продукт очищают с помощью хроматографии на силикагеле (элюирование смесью 3: 2 этилацетат-гексан) с получением метилового эфира (±)-3- бензилоксикарбонил-амино-2-[т-бутоксикарбонилметил-(4- метоксибензолсульфонил)-амино]-пропионовой кислоты.
(d) Метиловый эфир (±)-3-бензилоксикарбонил-амино-2-[т- бутоксикарбонилметил-(4-метоксибензолсульфонил)-амино] - пропионовой кислоты подвергают реакциям, описанным в Примере 4 (c). Продукт метиловый эфир 3-амино-2-[т-бутоксикарбонилметил-(4- метоксибензол-сульфонил)-амино]-пропионовой кислоты используют без дополнительной очистки.
(e) К раствору соли метилового эфира 3-амино-2-[т- бутоксикарбонилметил-(4-метоксибензолсульфонил)-амино] - пропионовой кислоты (2.46 грамм, 6.1 ммол) в метиленхлориде (20 мл) при 0oC добавляют трифторуксусную кислоту (5 мл). Раствор перемешивают при 0oC в течение 12 часов и затем концентрируют. Неочищенный продукт соль трифторуксусной кислоты метилового эфира 1-(4-метокси-бензолсульфонил)-5-оксо-пиперазин-2-карбоновой кислоты используют без дополнительной очистки.
(f) К раствору соли трифторуксусной кислоты метилового эфира 3-амино-2-[карбонилметил-(4-метоксибензолсульфонил)-амино] - пропионовой кислоты (2.11 грамм, 6.1 ммол) в метиленхлориде (5 мл) добавляют 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид (1.76 грамм, 9.2 ммол) и триэтиламин (3.4 мл, 24.4 ммол). Образовавшуюся смесь перемешивают в течение ночи, разбавляют этилацетатом и промывают 1N раствором хлористоводородной кислоты. Органический слой сушат (сульфат натрия), фильтруют и концентрируют. Неочищенный продукт очищают с помощью хроматографии на силикагеле (элюирование этилацетатом) с получением метилового эфира 1-(4-метоксибензолсульфонил)-5- оксопиперазин-2-карбоновой кислоты.
(g) К раствору метилового эфира 1-(4-метоксибензолсульфонил)- 5-оксо-пиперазин-2-карбоновой кислоты. (200 мг, 0.61 ммол) в смеси метанол-тетрагидрофуран-вода (5 мл, прибл. 6:2:1) при 0oC добавляют гидроксид лития (64 мл, 1.53 ммол). Образовавшуюся смесь перемешивают в течение 30 минут, подкисляют 1N хлористоводородной кислотой и экстрагируют этилацетатом. Объединенные экстракты сушат (сульфат натрия), фильтруют и концентрируют. Неочищенный продукт 1-(4-метоксибензолсульфонил)-5-оксо-пиперазин-2-карбоновую кислоту используют без дополнительной очистки.
(h) К раствору 1-(4-метоксибензолсульфонил)-5-оксо- пиперазин-2-карбоновой кислоты (166 г, 0.53 ммол) в метиленхлориде (5 мл) добавляют O-бензилгидроксиламин гидрохлорид (255 мг, 1.6 ммол), 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид (153 мг, 0.8 ммол) и триэтиламин (370 мкл, 2.65 ммол). Образовавшуюся смесь перемешивают в течение ночи, разбавляют этилацетатом и промывают 1N хлористоводородной кислотой. Органический слой сушат (сульфат натрия), фильтруют и концентрируют. Неочищенный продукт очищают с помощью хроматографии на силикагеле (элюирование 5% метанолом в метиленхлориде с получением N-(бензилокси)-1-(4-метоксибензол-сульфонил)-5-оксо- пиперазин-2-карбоксамида.
(i) N-(бензилокси)-1-(4-метоксибензолсульфонил)-5-оксо- пиперазин-2-карбоксамид подвергают реакциям, описанным в Примере 4 (с), получая N-гидрокси-1-(4-метоксибензолсульфонил)-5-оксо- пиперазин-2-карбоксамид в виде белого твердого вещества.
Масс-спектр (термораспыление): м/з 343 (м+H, 60%), (м+NH4, 17%).
1H ЯМР (CD3OD, 250 мГц, ппм) δ 7.79 (д, J = 8.9 Гц, 2H), 4.90 (с, H2O), 4.47 (дд, J = 5.0 Гц, 3.2 Гц, 1H), 4.03 (с, 2H), 3.88 (с, 3H), 3.47 (дд, J = 13.4, 3.2 Гц, 1H), 3.35-3.30 (м, 2H), 3.30 (с, CD3OD).
ПРИМЕР 6
N-гидрокси-1-(4-метоксибензолсульфонил)-морфолин-2-карбоксамид
(a) Морфолин-2-карбоновую кислоту подвергают реакциям, описанным в Примере 4 (a), получая 1-(4-метоксибензолсульфонил) - морфолин-2-карбоновую кислоту.
(b) 1-(4-метоксибензолсульфонил)-морфолин-2-карбоновую кислоту подвергают реакциям, описанным в Примере 5(h), получая N- бензилокси-1-(4-метоксибензолсульфонил)-морфолин-2-карбоксамид.
(c) N-бензилокси-1-(4-метоксибензолсульфонил)-морфолин-2- карбоксамид подвергают реакциям, описанным в Примере 4 (c), получая N-гидрокси-1-(4-метоксибензолсульфонил)-морфолин-2- карбоксамид в виде белой пены.
Масс-спектр (термораспыление): м/з 343 (м+H, 100%),
[α]D: +57o (с = 0.60, CICl3).
1H ЯМР (CDCl3, 250 мГц, ппм): δ 7.78 (шд, J = 8.0 Гц, 2H), 7.38 (шс, 1H), 7.01 (шд, J=8.0 Гц, 2H), 4.34 (шс, J= 2H), 3.87 (с, 3H), 3.85-3.30 (м, 3H), 3.30-3.15 (м, 2H).















Описываются новые производные арилсульфонилгидроксамовой кислоты формулы (I), где значения Аr, R3-R8, Y, X указаны в п.1 формулы изобретения, которые могут быть ингибиторами матричных металлопротеаз или образования фактора некроза опухоли и использоваться для лечения состояния, такого как артрит, карцинома, образование язвенной ткани, рестеноз, периодонтальная болезнь, врожденный буллезный апидермолиз, склерит и других заболеваний, характеризуемых матричной металлопротеиназной активностью, а также СПИД, сепсис, септический шок и других заболеваний, включающих образование TNF. 11 з.п. ф-лы.

или их фармацевтически приемлемая соль, где пунктирная линия представляет необязательную двойную связь;
X - углерод;
Y - углерод, кислород, сера или азот;
R3, R4, R5, R6, R7 и R8 выбирают из водорода, (C1-C6)алкила, который может быть замещен фенилом или бензилоксигруппой; (C2-C6)алкинила; фенила, необязательно замещенного хлором и фтором; пиридила; (C1-C6)ациламино; или группы формулы
в которой n = 0 - 6;
Z - гидрокси, (C1-C6)алкокси; или
R3 и R4, или R5 и R6 могут соединяться вместе с образованием карбонила;
Ar - фенил, который может быть замещен одной из групп, выбранной из (C1-C6)алкила, (C1-C6)алкокси, фенилокси или пиридилокси группы;
при условии, что R7 отличен от водорода только тогда, когда R8 отличен от водорода;
при условии, что R6 отличен от водорода только тогда, когда R5 отличен от водорода;
при условии, что R3 отличен от водорода только тогда, когда R4 отличен от водорода;
при условии, что когда Y - кислород, сера или азот и когда одна или более из группы, состоящей из R3, R4, R7 и R8, независимо является заместителем, содержащим гетероатом, то этот гетероатом не может быть непосредственно связан с 3- или 5-положениями;
при условии, что когда Y - азот, R4 отсутствует;
при условии, что когда Y - кислород, сера, R5 и R6 отсутствуют;
при условии, что когда Y - азот, R6 отсутствует;
при условии, что когда пунктирная линия представляет собой двойную связь, R4 и R6 отсутствуют;
при условии, что когда R3, R5 независимо являются заместителем, содержащим гетероатом, когда пунктирная линия представляет собой двойную связь, то гетероатом не может быть непосредственно связан с положениями X и Y;
при условии, что когда Y представляет гетероатом, то пунктирная линия не является двойной связью;
при условии, что когда R3, R4, R5, R6, R7 и R8 все определены как водород или (C1-C6)алкил, то либо Y - кислород, сера или азот, либо пунктирная линия является двойной связью.
(2R, 3S)-N-гидрокси-3-этинил-1-(4-метоксибензолсульфонил)-пиперидин-2-карбоксамида;
(+)-(2R*, 3R*)-(N-гидрокси)-1-(4-метоксибензолсульфонил)-3-метил-1,2,3,6-тетрагидропиридин-2-карбоксамида;
N-гидрокси-1-(4-метоксибензолсульфонил)-3-фенил-1,2,3,6 - тетрагидропиридин-2-карбоксамида;
(+)-(2R*, 3R*)-N-гидрокси-1-(4-метоксибензолсульфонил)-3-фенилпиперидин-2-карбоксамида;
(+)-N-гидрокси-1-(4-метоксибензолсульфонил)-2-пиперазинкарбоксамида гидрохлорида;
N-гидрокси-1-(4-метоксибензолсульфонил)-5-оксопиперазин-2-карбоксамида;
N-гидрокси-1-(4-метоксибензолсульфонил)-морфолин-2-карбоксамида.
| Способ получения 2,4-дизамещенных морфолинов или их солей | 1980 |
|
SU937453A1 |
| RU 94046364 А1, 1996 | |||
| Снегоочиститель для обыкновенных дорог | 1931 |
|
SU27695A1 |
| Планарный тензорезистор | 1976 |
|
SU577394A1 |
| 0 |
|
SU158955A1 | |

