Данное изобретение относится к способу получения N-гидроксиалкиламинометилфосфоновой кислоты или ее солей. В соответствии с одним аспектом, данное изобретение относится к новому и полезному способу получения N-гидроксиалкиламинометилфосфоновой кислоты или ее солей из алканоламина, формальдегида и диалкилфосфита. В соответствии с другим аспектом, данное изобретение относится к способу получения N-фосфонометиламинокарбоновой кислоты или ее солей с помощью каталитического окисления N-гидроксиалкиламинометилфосфоновой кислоты или ее солей. Согласно еще одному аспекту, данное изобретение относится к способу получения N-фосфонометилглицина, полезного в качестве гербицида.
N-гидроксиалкиламинометилфосфоновая кислота или ее соли являются полезными в качестве сырья при производстве сельскохозяйственных химикатов.
N-фосфонометиламинокарбоновые кислоты или их соли являются полезными в качестве сельскохозяйственных химикатов. В частности, N-фосфонометилглицин, известный также под обычным названием глифосфат, является высокоэффективным и промышленно важным фитотоксическим агентом, полезным для борьбы с широким рядом сорняков и культур. Он применяется по отношению к листве очень широкого спектра многолетних и однолетних трав и широколистных растений для достижения желательного их контроля или подавления. Промышленное использование включает борьбу с сорняками на обочинах дорог, судоходных путях, линиях передач, на складских площадях и на других несельскохозяйственных угодьях. Обычно глифосат преобразуется в гербицидные композиции в форме своих различных солей, которые сохраняют анионную форму глифосфата в растворе, предпочтительно в воде.
Взаимодействие первичного амина с альдегидом и сложным фосфитным диэфиром описано в Fields, "The Synthesis of Esters of Substituted Amino Phosphonic Acids", J.Am. Chem. Soc., vol. 74, стр. 1528-1531 (1952). Однако продукт реакции содержит значительные количества нежелательного бис-фосфометилированного продукта. Аналогичным образом, взаимодействие первичного амина с формальдегидром и фосфористой кислотой описано Moedritzer and Irani, "The Direct Synthesis of -Aminomethylphonic Acids. Mannich- Type Reactions with Orthophosphorous Acid", J. Org. Chem., vol. 31, стр. 1603-1607 (1966). Как и в случае Filds, продуктом реакции является преимущественно бис-фосфометилированный продукт.
Барсуков и др. "Синтез новых комплексонов и их производных", Журнал Общей Химии, том 53, N 6, стр. 1243-1249 (1983) и Барсуков и др. "Синтез новых комплексонов алифатического ряда и исследования механизма кислотной диссоциации", Журнал Общей Химии, том 55, N 7, стр. 1594-1600 (1985) описывают взаимодействие этаноламина с параформальдегидом и кислым диметилфосфитом при мольном соотношении амин/фосфит 1.0 и мольном соотношении формальдегид/амин 1.0. Несмотря на то, что эти статьи показывают, что продукт является моно-фосфомитилированным соединением, воспроизводство реакции, описанной в экспериментальном разделе, не привело в результате к получению моно-фосфометилированного продукта, т.е. 31Р-ЯМР анализ материала в примере Барсукова показал 0% выхода N-(2- гидpoкcиэтил)aминo-метилфосфоновой кислоты (см. пример 3 далее). Процесс, описанный у Барсукова и в других статьях, следовательно, является неспособным давать N-гидроксиалкиламинометилфосфоновую кислоту промышленно практикуемым способом.
Способ получения N-гидроксиалкиламинометилфосфоновой кислоты или ее солей, который является экономичным, промышленно приемлемым и может по существу давать только моно-фосфометилированный продукт, является весьма желательным.
Краткое изложение изобретения
Целью изобретения является представление эффективного и экономичного способа получения N- гидроксиалкиламинометилфосфоновой кислоты или ее солей, который является промышленно приемлемым. Дальнейшей целью изобретения является представление способа получения N- гидроксиалкиламинометилфосфоновой кислоты или ее солей для использования в производстве N-фосфонометила-минокарбоновой кислоты или ее солей. Еще одной целью изобретения является представление эффективного и экономичного способа получения N- фосфонометиламинокарбоновой кислоты или ее солей для использования в качестве сельскохозяйственных химикатов. Следующей целью изобретения является представление эффективного и экономичного способа получения N-фосфонометилглицина, который является промышленно приемлемым.
Согласно изобретению предоставляется способ получения N-гидроксиалкиламинометилфосфоновой кислоты или ее солей, который включает контактирование алканоламина, формальдегида и диалкилфосфита в присутствии спирта при подходящих условиях реакции с получением реакционной смеси, и гидролиз реакционной смеси в кислых или щелочных условиях. Согласно одному воплощению изобретения N-гидроксиалкиламинометилфосфоновая кислота или ее соль каталитически окисляется с получением N- фосфонометиламинокарбоновой кислоты или ее соли.
Далее согласно изобретению предоставляется способ получения N-фосфонометилглицина или его солей, который включает контактирование этаноламина, формальдегида и диалкилфосфита в присутствии спирта в подходящих условиях реакции с получением реакционной смеси, гидролиз реакционной смеси в кислых или щелочных условиях с получением N-гидроксиэтиламинометилфосфоновой кислоты или ее солей, и каталитическое окисление N-гидроксиэтиламинометилфосфоновой кислоты или ее солей.
Подробное описание изобретения
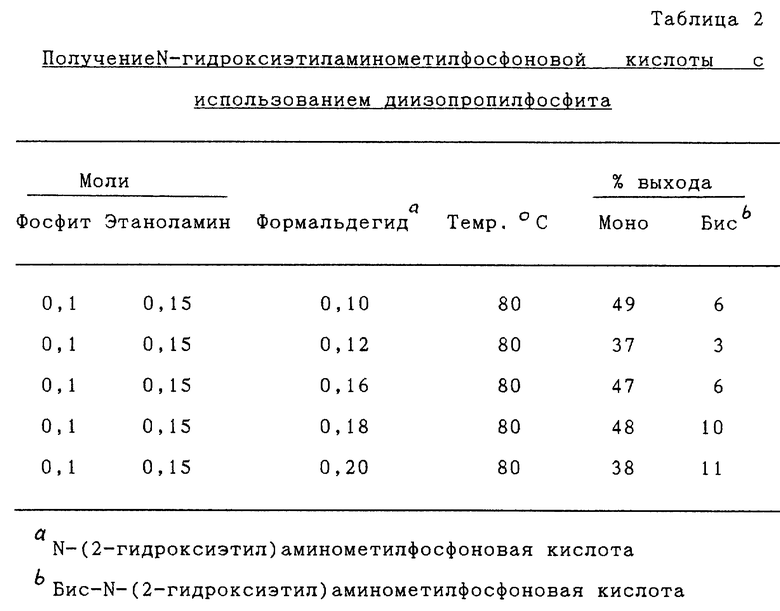
Изобретение относится к способу получения N-гидроксиалкиламинометилфосфоновой кислоты, представленной формулой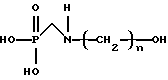
или ее солей, включающему (а) контактирование алканоламина, представленного формулой  где n имеет значения от 2 до 6, формальдегида и диалкилфосфита в присутствии спирта, представленного формулой R(OH)m, где R представляет алкильную группу, имеющую от 1 до примерно 18 атомов углерода и m имеет значения от 1 до 3, подходящих условиях времени и температуры с получением реакционной смеси, при котором молярное отношение алканоламина к фосфиту составляет примерно от 1:1 до 8:1 и молярное отношение формальдегида к алканоламину составляет примерно от 0,125:1 до 3:1, и (b) гидролиз реакционной смеси в кислых или щелочных условиях.
где n имеет значения от 2 до 6, формальдегида и диалкилфосфита в присутствии спирта, представленного формулой R(OH)m, где R представляет алкильную группу, имеющую от 1 до примерно 18 атомов углерода и m имеет значения от 1 до 3, подходящих условиях времени и температуры с получением реакционной смеси, при котором молярное отношение алканоламина к фосфиту составляет примерно от 1:1 до 8:1 и молярное отношение формальдегида к алканоламину составляет примерно от 0,125:1 до 3:1, и (b) гидролиз реакционной смеси в кислых или щелочных условиях.
Для получения N-фосфонометиламинокарбоновой кислоты, представленной формулой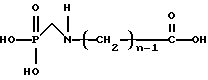
или ее солей, способы изобретения далее включают (с) каталитическое N-гидроксиалкиламинометилфосфоновой кислоты или ее солей.
Изобретение далее относится к способу получения N-фосфонометилглицина или его солей, включающему (а) контактирование этаноламина, формальдегида и диалкилфосфита в присутствии спирта, представленного формулой R(OH)m, где R представляет алкильную группу, имеющую от 1 до примерно 18 атомов углерода и m равно от 1 до 3, в подходящих условиях в смысле времени и температуры с получением реакционной смеси, при котором молярное отношение этаноламина к фосфиту составляет примерно от 1:1 до 8:1 и молярное отношение формальдегида к алканоламину составляет примерно от 0,125:1 до 3:1, (b) гидролиз реакционной смеси в кислых или щелочных условиях с получением N-гидроксиэтиламинометилфосфоновой кислоты или ее солей, и (с) каталитическое окисление N- гидроксиэтиламинометилфосфоновой кислоты или ее солей.
Используемый здесь термин "соли N-гидроксиалкиламинометилфосфоновой кислоты" означает - соли щелочных металлов или щелочноземельных металлов N-гидроксиалкиламинометилфосфоновой кислоты, а термин "соли N-фосфонометиламинокарбоновой кислоты" означает соли щелочных металлов или щелочноземельных металлов N- фосфонометиламинокарбоновой кислоты. Таким образом, продукты гидролиза и реакций окисления могут включать кислоту, ее соли или любую их комбинацию, в зависимости от конкретной реакции и выбранных условий реакции.
Алканоламины, которые могут применяться в соответствии с изобретением, представлены формулой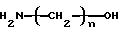
где n имеет значение от 2 до 6. Примеры алканоламинов включают этаноламин, 3-амино-1-пропанол, 4-амино-1-бутанол, 5- амино-1-пентанол, 6-амино-1-гексанол и их смеси. Предпочитаемым в настоящее время алканоламином является этаноламин вследствие свободной доступности и промышленного значения продукта, получаемого с использованием этаноламина в качестве исходного материала.
Формальдегид может применяться в соответствии с изобретением в виде параформальдегида или водного раствора формальдегида. Водный формальдегид является промышленно доступным в виде 37-50 процентных по весу водных растворов, которые могут содержать метанол, этанол или п-бутанол.
Диалкилфосфиты, полезные в способе изобретения, являются промышленно доступными или свободно приготавливаются обычными методами, такими как взаимодействие PCl3 и P4 со спиртом, включая многоатомные спирты. Если многоатомный спирт используется как реагент, фосфитный продукт может иметь циклическую структуру, т. е. быть циклическим фосфитным сложным эфиром. Смотри, например, Ford-Moore et al., Org. Syn., Coll., vol. IV, стр. 955 и Cook et al. J. Chem. Soc., 635 (1949) в отношении способов использования PCl3и США патент N 2661364 в отношении метода использования P4. Алкильные группы диалкилфосфитов являются прямыми или разветвленными алкильными группами, имеющими 1-18 атомов углерода и являются необязательно замещенными -OH группами. Предпочтительными алкильными группами являются те, которые являются разветвленными, или пространственно затрудненными, или замещенными -OH группой.
Примеры подходящих диалкилфосфитов включают, но не ограничиваются ими, диэтилфосфит, диизопропилфосфит, ди(2-бутил)фосфит, дибутилфосфит, диметилфосфит, дидецилфосфит, диизооктилфосфит, дилаурилфосфит, ди(1,2-дигидроксиэтан)фосфит, ди(1, 2-дигидроксипропан) фосфит, ди(1,3-дигидроксипропан) фосфит, 5,5-диметил-1,3,2- диоксафосфоринан-2-оксид и их смеси. Количество олканоламина, используемого в способе изобретения, может быть выражено в виде молярного отношения алканоламинового исходного материала к фосфитному исходному материалу. В общих чертах, молярное отношение алканоламина к фосфиту составляет от 1:1 до примерно 8:1, предпочтительно от 1:1 до примерно 3:1 и еще более предпочтительно от 1:1 до 1,5:1.
Количество формальдегида, используемого в способе изобретения, может быть выражено в виде молярного отношения исходного формальдегида к исходному алканоламину. В общих чертах, молярное отношение формальдегида к алканоламину составляет примерно от 0,125:1 до 3:1, предпочтительно примерно от 0,66:1 до 2,5:1 и более предпочтительно примерно от 1,7:1 до 2,3:1.
Реакция алканоламина, формальдегида и фосфита проводится при подходящей температуре, которая может меняться в широких пределах. Температура реакции обычно составляет в пределах примерно от 50 до 150oC, предпочтительно примерно от 60 до 120oC и более предпочтительно примерно от 70 до 110oC. Реакция алканоламина, формальдегида и фосфита проводится в течение походящего периода времени, который может меняться в широких пределах, в зависимости от разных параметров, например, от температуры реакции. Обычно продолжительность реакции находится в пределах от периода времени, необходимого для потребления фосфита, до примерно 16 часов, предпочтительно примерно от 2 часов до 16 часов и более предпочтительно примерно от 4 часов до 6 часов.
Реакция алканоламина, формальдегида и фосфита проводится в присутствии спиртового растворителя, где спирт представлен формулой R(OH)m и R представляет алкильную группу, имеющую примерно от 1 до 18 атомов углерода, и m имеет значения от 1 до 3. Алкильная группа, R, может быть прямой или разветвленной и предпочтительно является той же алкильной группой, которая используется в диалкилфосфитном исходном материале.
Примеры подходящих спиртов включают, но не ограничиваются ими, метанол, этанол, изопропанол, н-бутанол, 2-бутанол, изооктанол, дециловый спирт, изодециловый спирт, лауриловый спирт, этиленгликоль, 1,2- пропиленгликоль, 1,3-пропиленгликоль, глицерин, 2-гидроксиметил-2- метил-1,3-пропандиол, 1,3,5-тригидроксициклогексан и их смеси.
Реакция гидролиза может проводиться в кислотных или щелочных условиях с использованием любого из нескольких общепринятых способов, известных специалистам в данной отрасли.
Когда реакция гидролиза проводится в кислотных условиях, предпочтительным способом является удаление избытка алканоламина из реакционной смеси вместе с любым необязательно присутствующим спиртовым сорастворителем с последующим гидролизом реакционной смеси в соляной кислоте. Концентрация соляной кислоты предпочтительно составляет в пределах от 6 норм. HCl до 12 норм. HCl (концентрация HCl). Температура реакции кислотного гидролиза находится, главным образом, в пределах от точки кипения HCl, до примерно 250oC, предпочтительно 80-120oC. В основном, время реакции составляет в пределах от периода времени, необходимого для осуществления гидролиза, до примерно 24 часов, предпочтительно примерно от 2 часов до 16 часов. После завершения реакции гидролиза N-гидроксилалкиламинометилфосфоновая кислота может быть выделена с помощью любого общепринятого метода, известного специалистам в данной отрасли.
Когда реакция гидролиза проводится в щелочных условиях, предпочтительным способом является контактирование реакционной смеси с гидроокисью щелочного металла или гидроокисью щелочноземельного металла. Концентрация гидроокиси щелочного металла или щелочноземельного металла широко варьирует в пределах примерно от 15% по весу до 90% по весу, предпочтительно примерно от 40% по весу до 60% по весу, и наиболее предпочтительно составляет примерно 50% по весу. Количество гидроокиси щелочного металла или гидроокиси щелочноземельного металла, используемой в реакции гидролиза, может быть выражено в виде отношения эквивалентов гидроокиси к молям фосфитного исходного материала. В широком смысле, данное соотношение составляет примерно от 2:1 до 5:1, предпочтительно примерно от 2,5:1 до 4:1, и наиболее предпочтительно примерно 3:1.
В предпочтительном аспекте проведения гидролиза в щелочных условиях, спирт, образуемый в течение гидролиза, т.е. спирт, соответствующий алкильным группам в диалкилфосфите, удаляется из реакционной смеси в течение гидролиза таким способом, как, например, с помощью перегонки. Например, когда в способе используется диизопропилфосфит, во время гидролиза удаляется изопропиловый спирт.
Гидроокиси щелочных металлов для использования в процессе изобретения включают гидроокись лития, гидроокись натрия, гидроокись калия, гидроокись рубидия и гидроокись цезия. Вследствие их свободной доступности и легкости в обращении, предпочтительными являются гидроокись натрия и гидроокись калия, и особенно предпочтительной является гидроокись натрия.
Гидроокиси щелочных металлов для использования в процессе изобретения включают гидроокись бериллия, гидроокись магния, гидроокись кальция, гидроокись стронция и гидроокись бария. Гидроокись кальция является более предпочтительной вследствие ее свободной доступности.
Температура реакции щелочного (основного) гидролиза находится обычно в пределах примерно от 80 до 250oC, предпочтительно примерно от 80 до 180oC, более предпочтительно примерно от 120C до 150oC. Обычно продолжительность реакции составляет в пределах от периода времени, необходимого для осуществления гидролиза, до примерно 48 часов, предпочтительно от 2 часов до примерно 24 часов и более предпочтительно от 2 часов до примерно 16 часов. После завершения реакции гидролиза, N-гидроксилалкиламинометилфосфоновая кислота или ее соли могут быть выделены с помощью любого общепринятого метода, известного специалистам в данной отрасли.
Окисление N- гидроксилалкиламинометилфосфоновой кислоты или ее солей осуществляется в присутствии катализатора. Подходящие катализаторы окисления хорошо известны специалистам в данной отрасли и включают такие, как медь Ренея и те, что описаны в патентах США 4810426 и 5292936, которые включены для сведения.
В патенте США 4810426 окисление N-(2-гидроксиэтил)аминометилфосфоновой кислоты проводится с помощью гидроокиси щелочного металла в присутствии воды и подходящего катализатора, выбранного из кадмия, цинка, меди, палладия и платины и их соответствующих оксидов, гидроокисей и солей. Реакция окисления проводится при температуре от 200 до 300oC.
В патенте США 5292936 окисление N-(2-гидроксиэтил) аминометилфосфоновой кислоты проводится гидроокисью щелочного металла в присутствии эффективного количества медного катализатора Ренея, содержащего примерно от 50 частей на миллион до 10000 частей на миллион элемента, выбранного из группы, состоящей из хрома, титана, ниобия, тантала, циркония, ванадия, молибдена, марганца, вольфрама, кобальта, никеля и их смесей. Предпочтительными из вышеуказанных элементов являются хром, молибден и смеси хрома и молибдена. Реакция окисления проводится при температуре примерно между 120 и 220oC.
Другими особенно применимыми типами катализаторов окисления являются смешанные металлические катализаторы на носителе, такие как в заявке США N 08/269722 и последующей заявке с частичным продолжением N 08/407723, которые приведены здесь для сведения. Смешанный металлический катализатор на носителе настоящего изобретения приготавливается путем осаждения примерно от 1 до 50% по весу в расчете на общий вес катализатора элемента, выбранного из группы, состоящей из меди, кобальта, никеля, кадмия и их смесей, на носитель устойчивый к гидроокиси, имеющий примерно от 0,05 до 10% по весу якорного металла, выбранного из группы, состоящей из платины, палладия, рутения, серебра, золота и их смесей.
Подходящие устойчивые к гидроокиси носители включают оксид титана, оксид циркония и уголь. Уголь является предпочтительным. Активированный уголь даже более предпочтителен.
В частности, якорным металлом, осажденным на носителе, устойчивым к гидроокиси, может быть благородный металл. Под благородным металлом имеется в виду золото, серебро, платина, палладий, рутений или их смеси. Предпочтительными являются платина или палладий. Платина является наиболее предпочтительной. Количество якорного металла, осаждаемого на носитель, устойчивый к гидроокиси, может варьировать примерно от 0,05 до 10% по весу в расчете на общий вес катализатора. Когда на носитель, устойчивый к гидроокиси, осаждается меньше чем 0,05% якорного металла, тогда его недостаточно для объединения с медью, кобальтом, никелем и/или кадмием, чтобы получить удовлетворительный катализатор. С другой стороны, когда на носитель осаждается больше чем 10% по весу якорного металла в расчете на общий вес катализатора, размеры кристаллов платированного металла имеют тенденцию к увеличению. Более крупные размеры кристаллов платированного металла иногда ведут к пониженным каталитическим эксплуатационным характеристикам. Предпочтительно использовать примерно от 0,1 до 5% по весу якорного металла в расчете на общий вес катализатора. В настоящее время предпочтительным смешанным металлическим катализатором на носителе является смесь меди и платины или палладия на угле.
Подходящие носители, устойчивые к гидроокиси, содержащие подходящий якорный металл, могут быть получены промышленным путем.
Количество осаждаемого металла (т.е. меди, кобальта, никеля и/или кадмия) должно быть достаточным для покрытия, по крайней мере, некоторого количества вкрапленных частиц якорного металла. В дополнение к покрытым частицам, может иметь место присутствие, по крайней мере, некоторого количества частиц платирующего металла, вкрапленного в носитель, но не прилипшего к якорному металлу. Рентгеновская фотоэлектронная спектроскопия (XPS) является методом, который может использоваться для измерения относительной концентрации поверхностных атомов в катализаторе. При использовании данного метода было обнаружено, что в катализаторах данного изобретения атомное отношение осажденного металла на поверхности к якорному металлу предпочтительно составляет более чем 2,0, и более предпочтительно, когда XPS атомное отношение на поверхности является большим, чем соответствующая величина атомного отношения в массе.
Любое число методов может использоваться для осаждения якорного металла на субстрат, устойчивый к щелочи, и для осаждения меди, кобальта, никеля и/или кадмия на якорный металл. Предпочтительно однако использовать неэлектрическое осаждение металла. Неэлектрическое осаждение металла относится к химическому осаждению прилипающего металлического покрытия на подходящий субстрат в отсутствие применяемого внешнего электрического источника.
Независимо от осаждения якорного металла на субстрат, размер металлических частиц является решающим параметром в том, что он диктует размер осаждаемых кристаллов меди, кобальта, никеля и кадмия. Размер кристаллов меди, кобальта, никеля и кадмия должен быть меньше чем примерно 500 ангстрем, а в случае меди предпочтительно, чтобы размер кристаллов был меньше чем примерно 300 ангстрем. Хотя заявители не хотят быть связаны какой-либо конкретной теорией, считается, что равномерное распределение якорного металла является наилучшим для достижения высоких реакций, но необязательно быстрого протекания реакции. Кроме того, считается, что важно иметь мелкие, хорошо восстановленные, высокодиспергированные частицы якорного металла.
На практике субстрат, содержащий якорный металл, добавляется и суспендируется в воде. Далее, платирующий раствор, например, платирующий раствор меди, приготавливается путем смешения платирующего раствора в соответствующих пропорциях, с осторожным перемешиванием суспензии субстрата и воды при температуре примерно от 0 до 30oC или выше в открытом контейнере. Платирующий раствор, содержащий комплексообразующий агент и восстановитель, добавляется к суспензии дозами при измерении pH с каждым добавлением. После соответствующего интервала времени, медленно добавляется следующая доза суспензии. Количество добавляемого платирующего раствора зависит от желаемого процентного содержания по весу каталитического металла в катализаторе. Когда осаждение каталитического металла завершается, в результате получается в основном бесцветный фильтрат. Далее полученный катализатор фильтруется и промывается дистиллированной водой. Фильтрация должна осуществляться в инертной атмосфере, такой как атмосфера азота, для избежания воздействия воздуха на катализатор. Промывка катализатора удаляет непрореагировавшие компоненты, такие как в количестве частей на миллион, примеси и непрореагировавший восстанавливающий агент, такой как формальдегид. Было найдено, что примерно от 0,5 до 1,5% по весу щелочного металла остается на катализаторе, что обычно не является вредным. Катализатор должен храниться таким образом, который позволяет избежать воздействия кислорода, предпочтительно путем содержания под водой.
В настоящее время предпочтительными катализаторами окисления для использования в способе изобретения являются катализаторы патента США 5292936 и смешанные металлические катализаторы на носителе, причем смешанные металлические катализаторы на носителе являются особенно предпочтительными.
Реакции согласно настоящему изобретению могут проводиться при атмосферном давлении или в закрытом реакционном сосуде под давлением. Когда реакции проводятся в герметичном сосуде, давление в основном должно быть давлением пара реакционной смеси в условиях реакции.
ПРИМЕРЫ
Общие процедуры: Диалкилфосфиты закупались у фирмы Aldrich Chemical, когда они были в продаже. Фосфиты, которые не были промышленно или коммерчески доступными, синтезировались из PCl3 и соответствующего спирта по методу Форд-Мура и др. Org. Syn. Coll., vol. IV, стр. 955, или по методу Кука и др. J.Chem. Soc. 635 (1949). Формальдегид закупался у фирмы Aldrich Chemical в виде 37%-ного водного раствора. Параформальдегид (91-93%) получали у фирмы Hoechst Celanese Corporation. Выход N-гидроксиалкиламинометилфосфоновых кислот или их солей определялся с помощью 31P-ЯМР в D2O с использованием метилендифосфоновой кислоты в качестве внутреннего стандарта. Типичные образцы ЯМР получались в D2O, содержащей конц. HCl так, чтобы получался образец с pH 0, 7. Спектр ЯМР получался на спектрометрах Varian VXR-300 или VXR-400. Качественный и количественный масс-спектр снимался на спектрометрах Finnigan MAT90, Finnigan 4500 и VG40-250T.
Пример 1
Данный сравнительный пример иллюстрирует использование различных диалкилфосфитов в синтезе H-(2- гидроксиэтил)аминометилфосфоновой кислоты, проводимом в отсутствие какого-либо спирта.
Диизопропилфосфит (17 г, 0,1 ммоля), параформальдегид (3,6 г, 0,12 ммоля) и этаноламин (48,8 г, 0,8 ммоля) загружались в круглодонную колбу, снабженную магнитной мешалкой и с обратным холодильником. Реакционная смесь нагревалась до 100oC в течение 16 часов. Основной гидролиз промежуточных сложных эфиров осуществлялся путем добавления 2 экв. NaOH (16 г 50%-ного раствора) и нагревания до 120oC в течение 16 часов. Реакционная смесь оставлялась до охлаждения и концентрировалась в вакууме. К реакционной смеси добавлялась вода (30 мл) и отбирался образец для анализа 31P-ЯМР. Выход N-(2-гидроксиэтил)аминометилфосфоновой кислоты (13%) рассчитывался по количеству молей загруженного фосфита. Данные таблицы 1 представляют результаты реакций, проведенных в соответствии с аналогичным ходом эксперимента, как описано выше, но с различными диалкилфосфитами и разными соотношениями реагентов.
Пример 2
Данный пример иллюстрирует преимущество использования спирта в качестве растворителя при получении N-(2-гидроксиэтил)аминометилфосфоновой кислоты из диалкилфосфитов.
Раствор параформальдегида (4,0 г, 0,13 моля), этаноламина (6 мл, 0,097 моля), диизопропилфосфита (17 г, 0,1 моля) и 50 мл изопропанола перемешивался при 120oC в течение 16 часов. Раствор концентрировался досуха, и добавлялось 17 мл 50%-ного NaOH. Раствор нагревался с обратным холодильником в течение 16 часов, охлаждался до комнатной температуры и концентрировался в вакууме. Для гомогенизации смеси добавлялось 100 мл воды. Реакционная смесь подвергалась анализу 31P-ЯМР в D2О при pH 0,8. N-(2-гидроксиэтил)аминометилфосфоновая кислота была получена с выходом 33%.
Пример 3
Ниже дан сравнительный пример, который иллюстрирует способ, описанный Барсуковым и др. "Синтез новых комплексов и их производных". Журнал Общей Химии, т. 53, N 6, стр. 12243-49 (1983) получения N-(2-гидроксиэтил)аминометилфосфоновой кислоты с использованием диметилфосфита.
Диметилфосфит (36 г, 0,3227 моля) по каплям, в течение 1 часа добавлялся к перемешиваемому раствору параформальдегида (9,8 г, 0,32 моля) и этаноламина (20 г, 0,32 моля) при температуре ниже 20oC в атмосфере азота. Раствор нагревался до 80oC в течение 1 часа и затем охлаждался до комнатной температуры. Раствор экстрагировался 350 мл бензола, как описано в статье. Бензольный раствор пропускался затем через колонку, содержащую 1 кг окиси алюминия, и колонка элюировалась 1 литром бензола. Бензольный раствор концентрировался в вакууме досуха и добавлялось 250 мл конц. HCl. Раствор нагревался до 110oC в течение 6 часов. Анализ реакционной смеси с помощью 31P-ЯМР показал 0% выхода N-(2-гидроксиэтил)аминометилфосфоновой кислоты. Данный пример демонстрирует то, что вопреки тому, что описывается в ссылке Барсукова и др., способ, предложенный Барсуковым и др., не дает N-(2-гидроксиэтил)аминометилфосфоновую кислоту.
Пример 4
Данный пример иллюстрирует использование медного катализатора Ренея, содержащего хром, патента США 5292936 для превращения N-(2-гидроксиэтил)аминометилфосфоновой кислоты в N-фосфонметилглицин.
В 160 мл никелевый автоклав, снабженный мешалкой, загружалась N-(2-гидроксиэтил)аминометилфосфоновая кислота (16,84 г, 0,11 моля), вода (11,3 мл) и 45% по весу гидроокиси калия (48,7 г, 0,39 моля) и медный катализатор Ренея, содержащий 943 частей на миллион хрома (3,53 г). Автоклав герметизировался и нагревался до 160oC при давлении 9,5 кг/см с перемешиванием жидкой фазы в автоклаве. Через 1,85 часа прекращалось выделение водорода. Выход N-фосфонометилглицина в виде его соли калия составляет 98,5%.
Пример 5
Данный пример иллюстрирует получение смешанного катализатора настоящего изобретения на основе металла на носителе.
В однолитровый стеклянный химический стакан, снабженный тефлоновым покрытием, магнитной мешалкой в виде палочки 5 см длины на магнитной пластине для перемешивания добавлялась дистиллированная вода (169 мл) и 5% по весу платины на активированном угле в порошкообразной форме, поставляемой фирмой Degussa Corporation of Pidgefield Park, N.Y., что соответствует 13,37 г на основе сухого веса. В отдельном однолитровом химическом стакане приготавливался медный платирующий раствор путем добавления следующих компонентов, большинство из которых поставляется фирмой MacDermid, Inc. of Waterbury, CT, с перемешиванием в следующей последовательности:
(1) 687 мл деионизированной воды
(2) 90 мл MACuPlex Ultra Dep 1000B*
(3) 54 мл MACuPlex Ultra Dep 1000A*
(4) 18 мл MACuPlex Ultra Dep 1000D*
(5) 5 мл 37% вес/вес формальдегида
* - продукты частной собственности MacDermid
Общий объем 854 мл
Согласно описанию продукта MacDermid для Продукта под код. N 17970, получающийся в результате водный раствор включает следующие активные ингредиенты:
Сульфат меди - 4,0 г/л
Формальдегид - 6,0 г/л
Гидроокись натрия - 9,0 г/л
Избыток хелатирующего агента EDTA - 0,06 молярн.
Получающийся в результате платирующий раствор фильтровался и затем добавлялся к суспензии 5% платины на активированном угле посредством добавления 122 мл порций через каждые 3 минуты при 40oC. Для контроля за протеканием реакции проверялось значение pH. Время между добавлениями увеличивалось, когда выделение газа становилось слишком энергичным.
После завершения добавления плитирующего раствора, катализатор удалялся с помощью фильтрования с использованием 4-литровой вакуумной колбы. 350 мл фильтровальной воронки с грубым стеклом и стеклянного купола над верхушкой воронки с атмосферой азота. После фильтрования твердое вещество промывалось 3-4 250-миллилитровыми порциями деионизированной воды. Выход по сухому весу в данном получении составляет 18,4 г. Микроанализ катализатора показывает элементный состав - 13,4% по весу меди и 3,4% по весу платины, в расчете на общий вес катализатора. Было найдено, что средний размер кристаллов меди, определенный с помощью расширения XRD линия, составляет 157 ангстрем.
Пример 6
Данный пример показывает другой способ получения смешанного металлического катализатора настоящего изобретения на носителе.
В 2-литровый стеклянный химический стакан, содержащий полимерное тефлоновое покрытие, снабженный магнитной палочкой для перемешивания длиной 2,5 см на магнитной пластине, добавлялась дистиллированная вода (190 мл) с последующим добавлением 5% по весу платины на активированном угле, поставляемой Degassa Corporation, что соответствует 16,42 г (сухой вес). Водный платирующий медный раствор приготавливался в 4-литровом химическом стакане путем добавления при перемешивании следующих компонентов.
(1) 500 мл деионизированной воды
(2) NaKC4H4O6•4H2O (тартрат) (29,99 г, 0,106 моля); перемешивать до растворения
(3) В отдельном химическом стакане растворить 11,79 г CuSO4•SH2O (3 г Cu), (0,047 моля) в 400 мл деионизированной воды
(4) Раствор меди (3) добавить к получающемуся раствору тартрата (2)
(5) Добавить 13,60 г 50% по весу NaOH (0,17 моля)
(6) 11,35 мл 37% по весу формальдегида (0,15 моля)
Общий обьем 1125 мл
Получающийcя в результате платирующий раствор добавляется к суспензии 5% по весу платины на угле, в целом примерно в виде двенадцати 79 мл порций по одной порции через каждые 2,5 минуты. Проверялось значение pH для контроля за протеканием реакции и для задержки добавления порции во времени в том случае, когда дегазация раствора становилась слишком бурной. Катализатор, после добавления к суспензии платирующего раствора, удаляется фильтрованием, как в примере 9. Выход по сухому весу составляет 20,03 г. Состав анализируется, и найдено, что он содержит 14,5% меди и 3,8% платины в расчете на общий вес катализатора. Средний размер кристаллов меди составляет 119 ангстрем.
Пример 7
Данный сравнительный пример иллюстрирует получение N-(2-гидроксиэтил)аминометилфосфоновой кислоты с использованием диизопропилфосфита в отсутствии спирта в качестве растворителя.
Водный 37%-ный раствор формальдегида (8,2 г, 0,1 моля) и этаноламина (9 г, 0,15 моля) перемешивался при комнатной температуре между 1 и 2 часами с последующим добавлением диизопропилфосфита (17 г, 0,1 моля). Реакционный раствор нагревался при 80oC в течение 3 часов. К реакционной смеси добавлялась вода (50 мл) и 16 мл 50%-ного NaOH, и смесь перегонялась в аппарате Дина-Старка в течение 3 часов при 150oC для гидролиза промежуточных сложных эфиров с одновременным удалением воды, изопропанола и этаноламина (80-100 мл - общий удаленный объем). Образовывался белый осадок, который растворялся с помощью дополнительных 50 мл воды. Анализ реакционной смеси 31P-ЯМР показал 49% выхода N-(2-гидроксиэтил)аминометилфосфоновой кислоты и 6% выход бис-N-(2-гидроксиэтил)аминометилфосфоновой кислоты.
Пример 8
Данный сравнительный пример иллюстрирует получение N-(2-гидроксиэтил)аминометилфосфоновой кислоты, когда количество формальдегида варьируется и реакция проводится с диалкилфосфитом в отсутствии спирта в качестве растворителя.
Применяемые условия реакции являются такими же, как те, что описаны в примере 7, за исключением того, что количество используемого формальдегида варьировалось между 1 и 2 эквивалентами в расчете на фосфит. Результаты этих экспериментов представлены в таблице 2.
Пример 9
Данный пример иллюстрирует эффект от применения в реакции разного количества формальдегида, когда диалкилфосфит используется в присутствии спирта в качестве растворителя.
В типичной реакции, 37% формальдегид (17 г, 0,2 моля) добавлялся к смеси этаноламина (6,1 г, 0,1 моля) в 100 мл метанола.
Смесь нагревалась до 70oC, и в виде одной порции добавлялся диметилфосфит (11 г, 0,1 моля). Реакционная смесь перемешивалась при 70oC несколько часов. Добавлялась концентрированная HCl (50 мл), и реакционная смесь нагревалась при 120oC в течение 2 часов для гидролиза промежуточных сложных эфиров. Анализ реакционной смеси посредством 31P-ЯМР показал 74% выход N-(2-гидроксиэтил)аминометилфосфоновой кислоты. Результаты идентичных реакций, проведенных с различными количествами формальдегида, приведены в таблице 3.
Пример 10
Данный пример иллюстрирует получение N- гидроксиэтиламинометилфосфоновой кислоты с использованием циклического 5,5-диметил-1,3,2-диоксафосфоринан-2-оксида диалкилфосфита.
Смесь п-формальдегида (6,6 г, 0,22 моля), этаноламина (6,1 г, 0,1 моля) и метанола (100 мл) перемешивалась при 80oC в течение 4 часов в атмосфере азота. Одной порцией к смеси добавлялся 5,5-диметил-1,3,2-диоксафосфоринан-2-оксид (15 г, 0,1 моля). Реакционная смесь перемешивалась при 80oC несколько часов. Добавлялась концентрированная HCl (50 мл), и смесь нагревалась с обратным холодильником в течение 16 часов. Анализ реакционной смеси посредством 31P-ЯМР показал 59% N-гидроксиэтиламинометилфосфоновой кислоты.
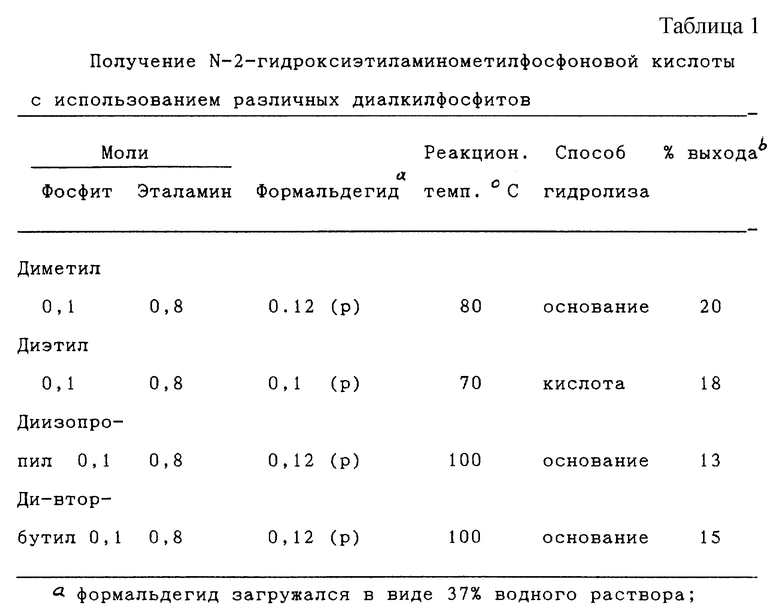


Изобретение относится к способам получения N-гидроксиалкиламинометилфосфоновой кислоты формулы (I), N-фосфонометиламинокарбоновой кислоты формулы (II), N-фосфонометилглицина (III) или их солей. Соединения (I) получают взаимодействием алканоламина формулы H2N-(CH2)n-OH, где n - число 2-6, с формальдегидом и диалкилфосфитом в присутствии спирта при 50-150°С, с последующим гидролизом образующейся реакционной смеси в кислотных или щелочных условиях. Соединения II получают каталитическим окислением N-гидроксиалкиламинометилфосфоновой кислоты или ее солей гидроокисью щелочного металла в присутствии эффективного количества медного катализатора Ренея, содержащего от 50 до 10000 ч. на миллион элемента, выбранного из группы, состоящей из хрома, титана, ниобия, тантала. N-фосфонометилглицин III получают взаимодействием этаноламина, формальдегида и диалкилфосфита в присутствии спирта при 50-150°С, с последующим гидролизом образующейся реакционной смеси в кислотных или щелочных условиях с получением соединения формулы 1, которые подвергают каталитическому окислению. Полученные соединения являются высокоэффективными гербицидами и используются в сельском хозяйстве. 3 с. и 23 з.п.ф-лы, 3 табл.
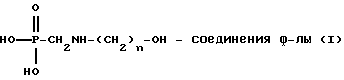
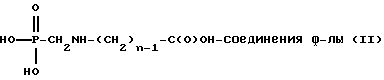
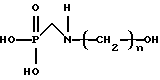
или ее солей, включающий а) контактирование алканоламина, представленного формулой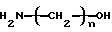
в которой n имеет значения 2 - 6,
формальдегида и диалкилфосфита в присутствии спирта при температуре 50 - 150oC с получением реакционной смеси, в которой молярное отношение алканоламина к фосфиту составляет примерно от 1:1 до 8:1, а молярное отношение формальдегида к алканоламину составляет примерно от 0,125:1 до 3:1, и b) гидролиз упомянутой реакционной смеси в кислотных или щелочных условиях.
R(OH)m,
в которой R представляет алкильную группу, имеющую от 1 до примерно 18 атомов углерода;
m имеет значение от 1 до 3.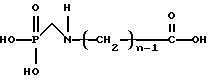
или ее солей,
где n имеет значения от 2 до 6,
включающий каталитическое окисление N-гидроксиалкиламинометилфосфоновой кислоты или ее солей, полученных согласно способу по п.1.
R(OH)m,
где R представляет алкильную группу, имеющую от 1 до примерно 18 атомов углерода;
m имеет значение от 1 до 3.
| US 4810426 A, 07.03.89 | |||
| US 4065491 A, 27.12.77 | |||
| US 5292936 A, 08.03.94 | |||
| US 3567768 A, 02.03.71 | |||
| SU 1282820 A3, 07.01.87 | |||
| ЖОХ, 1983, т.53, N 6, с.1243-1249. |
