Область изобретения
Изобретение относится к лиофилизированным противоопухолевым препаратам для парентерального введения в форме водного раствора. Более конкретно, изобретение относится к лиофилизированному составу, содержащему в качестве противоопухолевого действующего вещества тиоксантенон, и способу лечения с применением этого состава.
Известный уровень техники
Многочисленные традиционные лекарственные вещества и белки, предназначенные для терапевтического или диагностического использования, обладают нестабильной формой в водном растворе и требуют превращения их в твердые продукты. В технологии производства фармацевтических продуктов лиофильная сушка является одним из наиболее широко используемых методов для обеспечения необходимой стабильности препарата.
По различным причинам лиофильную сушку биоактивных веществ редко осуществляют в их чистом виде. В процессе получения препарата обычно для достижения определенных целей прибавляют другие химические компоненты, например введения буферных веществ для обеспечения необходимого показателя pH, повышения растворимости или обеспечения осмолярного равновесия. При разработке способа лиофильной сушки, состав препарата в целом оказывает влияние главным образом на параметры цикла. Таким образом, любое изменение в составе, не только содержание самого активного вещества обычно требует последующей модификации технологического цикла. Также как и наполнители, вводимые по вышеупомянутым причинам, лиофильная сушка обычно требует еще и введения вспомогательных добавок для облегчения самого процесса лиофилизации или обеспечения механической прочности "пробке" лиофилизированного препарата во время последующего его хранения и транспортировки. Такие наполнители называются лиопротекторами или стабилизаторами. Использование стабилизаторов иллюстрируется следующими ссылками.
В Международной заявке PCT/US89/04099 (WO 90/03784) раскрывается лиофилизированный фармацевтический состав, содержащий полипептид и стабилизирующее или солюбилизующее количество циклодекстрина, выбранного из группы, состоящей из гидроксипропильного, гидроксиэтильного, глюкозильного, мальтозильного, мальтотриозильного производных β- и γ-циклодекстрина.
В патенте США N 4983586 раскрывается способ снижения частоты преципитации липофильного и/или гидрофильного лекарственного средства в месте его инъекции при парентеральном введении, заключающийся в том, что лекарственный препарат вводят в форме водного раствора, содержащего примерно от 20% до 50% гидроксипропил-β-циклодекстрина. В объем заявленных лекарственных препаратов: противоопухолевые средства, седативные средства, транквилизаторы, противосудорожные средства, антидепрессанты, снотворные средства, миорелаксанты, спазмолитики, противовоспалительные средства, антикоагулянты, кардиотонические средства, сосудорасширяющие средства и антиаритмические средства.
В патенте США N 5298410 раскрываются лиофилизированные фармацевтические составы биологически активных веществ, в которых в качестве стабилизатора используют циклодекстриновое производное, буфер, например как фосфат натрия, ацетат натрия и карбонат натрия. Указанные составы могут содержать, при желании, сахарозу или трегалозу.
В качестве исходного материала для лиофилизации обычно используют ненасыщенный водный раствор, и конечный продукт получают в виде твердого вещества. Весь технологический процесс состоит из стадий, во время которых происходит удаление до > 99% воды. Во время стадии охлаждения, водные растворы превращаются в лиофилизированный концентрат, при этом одновременно удаляют воду в виде льда. Весь технологический процесс состоит из нескольких фазовых превращений, например как фазовый переход жидкость - твердое вещество и твердое вещество-газ, которые необходимо принимать во внимание для обеспечения эффективной переработки и стабильности получаемых продуктов. По мере понижения температуры, раствор обычно вначале претерпевает частичное охлаждение (то есть охлаждение до температуры ниже равновесной температуры замерзания) до начала спонтанного образования зародышей льда. Образование зародышей льда и центров роста кристаллов представляют собой сложные реакционные процессы, скорости протекания которых зависят от скорости охлаждения, концентрации раствора и других факторов. Эта стадия процесса в значительной степени определяет строение целевого лиофилизированного продукта. В процессе лиофильной сушки, растворенное вещество удерживается в фазе остатка жидкости в форме с повышающейся концентрацией, при этом степень его концентрации регулируется по диаграмме фазового равновесия. В конечном счете, раствор становится насыщенным, и одновременно в этой точке образуется твердая фаза растворенного вещества. Полученная система на следующей стадии состоит из смеси кристаллов растворенного вещества и льда.
Наполнители, прибавляемые, главным образом, для облегчения процесса лиофилизации, обычно служат для обеспечения одной из двух функций. Наполнители используются непосредственно для увеличения суммарного содержания твердого осадка с целью достижения механически более стабильного лиофилизированного продукта. Такие наполнители должны обладать способностью к кристаллизации из раствора в течение всего процесса лиофилизации, предпочтительно на стадии замораживания, поскольку именно только в виде самостоятельной фазы они будут оказывать нейтральный эффект на стабильность продукта. Стабилизаторы, с другой стороны, обеспечивают химическую защиту во время концентрирования вымораживанием и облегчают переход в стеклообразное состояние; они также обеспечивают физическую прочность лиофилизированного продукта. Температура стеклования представляет собой зависимость (функцию) химического состава от суммарного содержания твердого вещества.
В то время как в прототипе внимание в большей степени было сконцентрировано на физико-химических параметрах для коррекции состава продуктов для лиофилизации (см., например, статью Franks, F, Freeze Drying: a combination physics, chemistry, engineering and economics в журнале, Jap. J. Freeze Drying, вып. 38, стр. 5-16 (1992), таких параметров недостаточно, чтобы специалист в данной области техники смог получить конечные продукты, которые отвечали бы требуемым целям. Таким образом, все еще существует необходимость проведения тщательного исследования и/или выявления неожиданного открытия, на основе которых можно получить требуемые продукты, которые станут очевидны специалистам из описания настоящего изобретения.
В процессе создания настоящего изобретения авторы в результате проведения кропотливой научно-исследовательской работы установили, что производные тиоксантенона, обладающие противоопухолевой активностью, при доставке их в организм в традиционных фармацевтических носителях, например в форме таблетки и капсулы для перорального приема, обычно не отвечают в должной мере требованиям эффективного лекарственного средства. Возникла проблема создания лекарственной формы для производных тиоксантенона, отвечающей требованиям эффективного лекарственного средства.
Краткое изложение сущности изобретения
В основу изобретения положена задача создания препарата с действующим веществом тиоксантенон в виде лиофилизированного состава, который, после восстановления его влагосодержания, превращается в форму, пригодную для парентерального введения, и который обладает стабильностью и не подвержен какой-либо деградации или изменению во время продолжительного хранения.
Задача решена тем, что заявляемый лиофилизированный состав для лечения опухолей у млекопитающего, восстановленный по влагосодержанию, согласно изобретению, содержит:
а) от 1 до 50 мг/мл противоопухолевого средства на основе соединения формулы (I)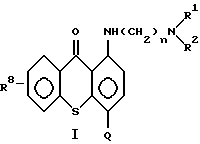
где n означает 2 или 3;
R1 и R2 каждый независимо означает низший алкил;
Q означает углеводородный остаток, выбранный из группы, состоящей из CH2NHR3,
CH2N(R4)SO2R7, CH2NHCHO, CH=N-Ar, C(O)NR5R6, CH2N(R4)C(O)R7, CH2N(C2H5)CHO,
CH2N(R4)P(O)(O - низший алкил)2, CH2N=CH-N(R9)(R10), CH2N(R4)C(O)CF3 и
CH2N(R4)C(O)OR7;
R3 означает водород или низший алкил;
R4 означает водород, низший алкил или Ar;
R5 означает водород, низший алкил или Ar;
R6 означает водород, низший алкил;
R7 означает низший алкил или Ar;
R8 означает водород, низший алкил, низший алкокси или гидроксил;
Ar означает фенил или фенил, замещенный метилом, метоксилом, гидроксилом, галогеном или нитрогруппой, при условии, что когда n равно 2, то R1 и R2 означают этил,
R8 означает водород, a Q означает CH2NHSO2Ar, при этом группа Ar может быть 4-монозамещенной метилом или галогеном; и
R9 и R10 каждый независимо означает низший алкил;
или его фармацевтически приемлемая соль присоединения кислоты или сольват;
b) от 10 мг/мл до 125 мг/мл стабилизатора, выбранного из группы, состоящей из маннита и сахарозы; и
c) от 0,025 до 0,25 М лактатного буфера, причем указанный состав имеет pH от 3,0 до 4,5. Заявляемый лиофилизированный состав может содержать также от 1,0 до 10,0 мг/мл хлорида натрия, а в качестве лактатного буфера предпочтительно содержит лактат натрия.
Лиофилизированный состав, восстановленный по влагосодержанию, для лечения опухолей у млекопитающего может также содержать:
а) от 1 до 20 мг/мл противоопухолевого средства на основе соединения формулы (II):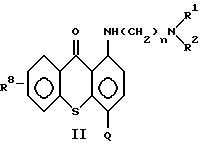
где n означает 2 или 3;
R1 и R2 каждый независимо означает низший алкил;
Q означает углеводородный остаток, выбранный из группы, состоящей из CH2NHR3, CH2NHCHO, CH= N-Ar, C(O)NR5R6, CH2N(R4)C(O)R7, CH2N(C2H5)CHO,
CH2N(R4)P(O)(O - низший алкил)2, CH2N=CH-N(R9)(R10), CH2N(R4)C(O)CF3 и CH2N(R4)C(O)OR7;
R3 означает водород или низший алкил;
R4 означает водород, низший алкил или Ar;
R5означает водород, низший алкил или Ar;
R6 означает водород, низший алкил;
R7 означает низший алкил или Ar;
R8 означает водород, низший алкил, низший алкокси или гидроксил;
Ar означает фенил или фенил, замещенный метилом, метоксилом, гидроксилом, галогеном или нитрогруппой, и
R9 и R10 каждый независимо означает низший алкил;
или их фармацевтически приемлемые соли присоединения кислот или сольват;
(b) от 30 мг/мл до 100 мг/мл стабилизатора, выбранного из группы, состоящей из маннита и сахарозы; и
c) от 0,025 до 0,25 М лактатного буфера, причем указанный состав имеет pH от 3,0 до 4,5.
Указанный лиофилизированный состав может содержать также от 1,0 до 10,0 мг/мл хлорида натрия, а в качестве лактатного буфера предпочтительно содержит лактат натрия.
Предпочтительный вариант лиофилизированного состава, восстановленного по влагосодержанию, для лечения опухолей у млекопитающего содержит: от 1 до 50 мг/мл N-[[1-[[2-(диэтиламино)этил] амино]-9-оксотиоксантен-4-ил]метил]- метансульфонамида;
b) от 0,025 до 0,25 М натрий-лактатного буфера;
с) от 10 до 125 мг сахарозы;
d) от 1,0 до 10,0 мг хлорида натрия;
e) остальное составляет вода в объеме до 1,0 мл.
Другой предпочтительный вариант лиофилизированного состава, восстановленного по влагосодержанию, для лечения опухолей у млекопитающего содержит:
от 1 до 50 мг/мл N-[[1-[[2-(диэтиламино)этил]амино]-7-метокси-9- оксотиоксантен-4-ил]метил]-формамид;
b) от 0,025 до 0,25 М натрий-лактатного буфера;
с) от 10 до 125 мг сахарозы;
d) от 1,0 до 10,0 мг хлорида натрия;
e) остальное составляет вода в объеме до 1,0 мл.
Восстановленные по влагосодержанию лиофилизированные составы настоящего изобретения предназначены для лечения злокачественных опухолей у млекопитающих.
Способ лечения предполагаемой опухоли у млекопитающего, согласно изобретению, заключается в том, что указанному млекопитающему вводят эффективную дозу состава (выбранного из вышеперечисленных), обеспечивающую уменьшение размера указанной опухоли.
Подробное описание изобретения
Лиофилизированный состав предлагаемого изобретения включает: противоопухолевое средство, содержащее действующее вещество тиоксантенон и водный носитель.
Противоопухолевые средства
В качестве противоопухолевого средства настоящего изобретения используют соединения формулы (I) в соответствии с патентом США N 5346917, включенного в полном объеме в данное описание в качестве ссылки: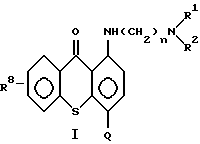
где n означает 2 или 3;
R1 и R2 каждый независимо означает низший алкил;
Q означает углеводородный остаток, выбранный из группы, состоящей из CH2NHR3, CH2N(R4)SO2R7, CH2NHCHO, CH= N-Ar, C(O)NR5R6, CH2N(R4)C(O)R7, CH2N(C2H5)CHO, CH2N(R4)P(O)(O - низший алкил)2, CH2N= CH-N(R9)(R10), CH2N(R4)C(O)CF3 и CH2N(R4)C(O)OR7;
R3 означает водород или низший алкил;
R4 означает водород, низший алкил или Ar;
R5 означает водород, низший алкил или Ar;
R6 означает водород, низший алкил;
R7 означает низший алкил или Ar;
R8 означает водород, низший алкил, низший алкокси или гидроксил;
Ar означает фенил или фенил, замещенный метилом, метоксилом, гидроксилом, галогеном или нитрогруппой, при условии, что когда n равно 2, то R1 и R2 означают этил,
R8 означает водород, a Q означает CH2NHSO2Ar, при этом группа Ar может быть 4-монозамещенной метилом или галогеном; и
R9 и R10 каждый независимо означает низший алкил;
или его фармацевтически приемлемая соль присоединения кислоты или сольват.
Указанные соединения можно использовать для лечения онкологических заболеваний у млекопитающих.
Предпочтительные противоопухолевые средства предлагаемого изобретения представляют собой соединения формулы (II):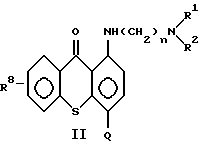
где n означает 2 или 3;
R1 и R2 каждый независимо означает низший алкил;
Q означает углеводородный остаток, выбранный из группы, состоящей из CH2NHR3, CH2NHCHO, CH= N-Ar, C(O)NR5R6, CH2N(R4)C(O)R7, CH2N(C2H5)CHO,
CH2N(R4)P(O)(O - низший алкил)2, CH2N=CH-N(R9)(R10), CH2N(R4)C(O)CF3 и CH2N(R4)C(O)OR7;
R3 означает водород или низший алкил;
R4 означает водород, низший алкил или Ar;
R5 означает водород, низший алкил или Ar;
R6 означает водород, низший алкил;
R7 означает низший алкил или Ar;
R8 означает водород, низший алкил, низший алкокси или гидроксил;
Ar означает фенил или фенил, замещенный метилом, метоксилом, гидроксилом, галогеном или нитрогруппой, и
R9 и R10 каждый независимо означает низший алкил; или их фармацевтически приемлемые соли присоединения кислот или сольват. Типичные представители соединений в соответствии с настоящим изобретением приведены в нижеследующих примерах.
Пример 1
Получение 1-[[2-(Диэтиламино)этил]амино]-4-(N-фенилформимидоил)- тиоксантен-9-она
(I: R1=R2=Et; Q=CH=N-C6H5; R8=H; n=2).
Смесь, состоящую из 17,7 г (50 ммоль) 1-[[2-(диэтиламино)-этил]амино]- 9-оксо-тиоксантен-4-карбоксальдегида и 15,1 г (150 ммоль) анилина в 100 мл толуола кипятят с обратным холодильником в течение 8 часов с использованием конденсационного горшка Dean-Stark. Результаты тонкослойной хроматографии на двуокиси алюминия при использовании смеси хлороформ/гексан/изопропиламин, 10:10:2, свидетельствовали о неполном завершении реакции. Толуол отгоняют, к реакционной массе прибавляют 25 мл анилина, и затем кипятят с обратным холодильником в течение 4 часов. После этого к реакционной массе прибавляют 40 мл ксилола и смесь кипятят с обратным холодильником еще 3 часа. Растворитель и избыток анилина упаривают в вакууме, и остаток перекристаллизовывают из бензола с выходом 19, 9 г неочищенного продукта. После перекристаллизации полученного кристаллического продукта из примерно 1,5 л гексана получают 15,8 г (86%) названного соединения, т.пл 125-126oC.
Пример 2
Получение N-[[1-[[2-(Диэтиламино)этил] амино]-9-оксотиоксантен-4-ил]метил]-формамида
(I: R1=R2=Et; Q=CH2NHCHO; R8=H; n=2).
Раствор, содержащий 35,4 г (0,1 моль) 1-[[2-(диэтиламино)-этил]амино]-9- оксотиоксантен-4-карбоксальдегида, 420 мл формамида и 50 мл (1 моль) уксусной кислоты, кипятят при 160oC в течение 1 часа. Реакционную смесь охлаждают, выливают в 2 л воды и затем подщелачивают путем прибавления примерно 50 мл 35% раствора гидроксида натрия. Выпавший в осадок смолянистый продукт отфильтровывают и сушат в вакууме. Высушенный осадок растворяют примерно в 1,5 л горячего этилацетата, очищают древесным углем и кристаллизуют при охлаждении. Полученный продукт отфильтровывают, промывают этилацетатом и после сушки получают 29,0 г (75%) названного соединения, т.пл. 154-155oC.
Пример 3
Получение N-[[1-[2- (Диэтиламино)этил]амино]-9-оксотиоксантен-4-ил]метил]-N- метилформамида
(IV: R1=R2=Et; R4=Me; R8=H; n=2).
По аналогичной методике, приведенной в примере 2, из смеси, содержащей 35,4 г (0,1 моль) 1-[[2(диэтиламино)этил]амино]-9-оксо-тиоксантен-4- карбоксальдегида, 394 г N-метилформамида и 50 мл уксусной кислоты, получают 24,6 г N-метилформамида, который затем перекристаллизовывают из 150 мл ацетона с получением названного продукта с т.пл 127-130oC.
Пример 4
Получение 4-(Аминометил)-1-[[2-(диэтиламино)этил]амино]-тиоксантен- 9-она
(I: R1=R2=Et; Q=CH2NH2; R8H; n=2),
Раствор, содержащий 24,4 г (64 ммоль) формамида, полученного в примере 2 в 240 мл 2 н. соляной кислоты, кипятят на паровой бане в течение 1 часа. Реакционную смесь охлаждают до комнатной температуры, подщелачивают путем прибавления 35% водного раствора гидроокиси натрия, и выпавший желтый осадок собирают фильтрованием. Полученный продукт растворяют в бензоле, очищают древесным углем, сушат сульфатом магния, фильтруют и отгоняют азеотропной перегонкой для удаления следового количества воды. Высушенный остаток кристаллизуют из метанола и изопропанола с добавлением этерифицированного хлористого водорода. Полученный твердый продукт перекристаллизовывают несколькими порциями из метанола с выделением 10,6 г продукта, т.пл. 270-272oC, в виде двусолянокислой соли.
Пример 5
Получение [[2-(диэтиламино)этил] амино]-4-[(метиламино)-метил]тиоксантен-9-она
(I: R1=R2=Et; Q=CH2NHCH3; R8=H; n=2).
По аналогичной методике, приведенной в примере 4, из 14,6 г (37 ммоль) N-метилформамида примера 3 и 50 мл 2н соляной кислоты получают 10,5 г метиламина в виде дигидрохлорида полугидрата. Полученный продукт плавится при температуре 241-243oC.
Пример 6
Получение N-[[1-[[2- (Диэтиламино)этил]амино]-9-оксотиоксантен-4-ил]-метил]- метансульфонамида
(I: R1=R2=Et; Q=CH2NHSO2CH3; R8=H; n=2).
Раствор, содержащий 10,65 г (30 ммоль) свободного основания амина, полученного в примере 4, в 100 мл пиридина охлаждают на ледяной бане и затем прибавляют одной порцией 4 г (35 ммоль) метансульфонилхлорида. Смесь перемешивают 2 часа при комнатной температуре и выливают в 750 мл воды, содержащей 2 г гидроокиси натрия. Выпавшее в осадок темно-желтое вещество собирают, промывают водой и сушат в вакууме в течение ночи. Вторую порцию продукта получают прибавлением избытка гидроокиси натрия к фильтрату с последующим фильтрованием полученного твердого вещества. Объединенные порции выпавшего продукта после сушки перекристаллизовывают из бензола. Получают 6,4 г метансульфонамида, т.пл. 169-170oC.
Пример 7
Получение 1-[[2'-(диэтиламино)этил] амино] -9-оксотиоксантен- 4-карбоксамида
(1: R1=R2=Et; Q=CONH2; R8=H; n=2).
Суспензию, приготовленную из 74 г (0,23 моль) 1-[[2- (диэтиламино)-этил] амино]-9-оксо-тиоксантен-4-карбоксальдегида и 74 г (1,06 моль) солянокислого гидроксиламина в смеси 400 мл пиридина и 400 мл этанола кипятят с обратным холодильником в течение 0,5 часа и затем прибавляют 70 мл воды для получения гомогенного раствора. Реакционный раствор кипятят в течение еще 2 часов и затем выдерживают при комнатной температуре 14 часов. Полученный кристаллический оксим отфильтровывают с количественным его выходом, т.пл. 215-218oC.
123 г оксима быстро нагревают на паровой бане в 180 мл уксусного ангидрида для полного его растворения. Полученный раствор охлаждают, затем прибавляют 100 мл 1,8М HCl в простом эфире, и полученную суспензию разбавляют 500 мл простого эфира. Суспензию выдерживают в течение 14 часов при 0oС и затем фильтруют. Остаток (123 г, т.пл. 109-112oС) суспендируют в 250 мл ксилола и кипятят с обратным холодильником в течение 20 мин. Реакционную массу охлаждают, и отфильтровывают 71,3 г нитрила, т.пл. 265oC.
Десять грамм нитрила перемешивают в 200 мл концентрированной H2SO4 при комнатной температуре в течение 3 дней. Реакционную массу затем нейтрализуют концентрированным раствором NH4OH, осадок отфильтровывают и получают перевар в теплой смеси EtOAc/EtOH, который затем фильтруют и перекристаллизовывают из быстро охлажденного раствора, т.пл. 241-243oС. Полученное кристаллическое вещество растворяют в этаноле, и затем прибавляют 1 эквивалент HCl в этаноле. Получают 6 грамм солянокислого амида, т.пл. 271-272oC.
Пример 8
Получение N-[[1-[(2-(диэтиламино)этил)амино]-9- оксотиоксантен-4-ил]метил]-N-метилметансульфонамида
(I: R1=R2=Et; Q=CH2N(CH3)SO2CH3; R8=H; n-2).
Раствор, содержащий 1,5 г (3,5 ммоль) метансульфонамида из примера 6 в тетрагидрофуране (THF) (60 мл), охлаждают до 0oC на ледяной бане, а затем прибавляют 0,16 г (4,0 ммоль) NaH. Реакционную массу доводят до комнатной температуры, перемешивают в течение 10 мин, а затем прибавляют 0,25 мл (4,0 ммоль) йодистого метила. Реакционную массу перемешивают при комнатной температуре в течение 24 часов, и растворитель упаривают в вакууме. Остаток очищают колоночной хроматографией на кремнеземе при элюировании сначала хлороформом (100%), а затем смесью 1% изопропиламин/хлороформ с выходом 1,15 г (74%) N-метилметансульфонамида в виде желтого порошка, т.пл. 175-177oC. Свободное основание также очищают раствором метансульфокислоты в метаноле с выходом метансульфонатной соли, т.пл. 194-195,5oC (обозначенной как продукт примера 8а, упоминаемого далее в описании).
Пример 9
Получение N-[[l-([2-(диэтиламино)этил] амино] -9-оксотиоксантен-4-ил] метил]фенилсульфонамида
(I: R1=R2=Et; Q=CH2NHSO2Ph; R8=H; n=2).
По методике, аналогичной примеру 6, из смеси, содержащей 2,54 г (7, 5 ммоль) свободного основания амина, полученного в примере 4, пиридина (50 мл) и бензолсульфонилхлорида (1,1 мл, 8,62 ммоль), получают 2,4 г (57%) фенилсульфонамида в виде соли метансульфокислоты, которую затем обрабатывают раствором метансульфокислоты в метаноле. Полученный продукт перекристаллизовывают из этанола.
Пример 10
Получение N-[[1-[[2-(Диэтиламино)этил]амино]-9- оксотиоксантен-4-ил]метил]-ацетамида
(I: R1=R2=Et; Q=CH2NHC; R8=H; n=2).
По методике, аналогичной примеру 6, из смеси, содержащей 4,15 г (11,7 ммоль) свободного основания амина, полученного в примере 4, пиридина (60 мл) и хлористого ацетила (0,82 мл, 11,53 ммоль), получают 2,3 г (52%) названного ацетамида в виде оранжевого твердого вещества, которое перекристаллизовывают из ацетона. Выделенное кристаллическое вещество имеет т.пл. 182-183oC.
Пример 11
Получение N-[[1-[[2-(Диэтиламино)этил]амино]-9-оксотиоксантен-4-ил] метил]бензамида
(I: R1=R2=Et; Q=CH2NHC(O)Ph; R8=H; n=2).
По методике, аналогичной примеру 6, из смеси, содержащей 1,17 г (3,29 ммоль) свободного основания амина, полученного в примере 4, пиридина (25 мл) и бензоилхлорида (0,42 мл, 3,62 ммоль), получают 1,02 г (68%) названного бензамида в виде желтого порошка. Продукт очищают колоночной хроматографией на кремнеземе при элюировании последовательно хлороформом (100%), а затем смесью 1% изопропил/хлороформ. Выделенный продукт перекристаллизовывают из этанола и расплавляют при температуре 161-163oC.
Пример 12
Получение N-[[1-[[2-(Диэтиламино)этил]амино]-9-оксо-тиоксантен-4- ил]диэтил]-фосфорамида
(I: R1=R2=Et; Q=CH2NHP(O)(OEt)2; R8=H; n=2).
Раствор, содержащий 2,28 г (6,41 ммоль) свободного основания амина из примера 4, CH2Cl2 (50 мл) и триэтиламин (2 мл), при 0oC обрабатывают диэтилфосфорохлоридатом (1,0 мл, 6,9 ммоль). Реакционную массу перемешивают при 0oC в течение 2 часов, затем при комнатной температуре 1 час. Растворитель упаривают в вакууме, и остаток очищают колоночной хроматографией на кремнеземе, вымывая последовательно этилацетатом (100%), смесью 5% метанол/этилацетат и смесью метанол/изопропиламин/этилацетат с выделением 2,28 г (72%) диэтилфосфорамида в виде желтого твердого вещества, которое после перекристаллизации из этилацетата имеет т.пл. 108-110oC.
Пример 13
Получение N-[[1-[[2-(диэтиламино)этил]амино]-9- оксотиоксантен-4-ил]метил]-N-этилформамида
(IV: R1=R2=Et; R2=Et; R2=H; n=2).
Раствор, содержащий 2,0 г (5,6 ммоль) 1-[(2-(диэтиламино)этил]амино]-9-оксотиоксантен-4-карбоксальдегида, N-этилформамид (24,0 мл) и муравьиную кислоту (3,0 мл, 79,5 ммоль), кипятят при 170oC в течение 4 часов. Реакционную смесь охлаждают, выливают в воду и подщелачивают путем прибавления 10% раствора NaOH. Выпавшее в осадок твердое вещество собирают фильтрованием и промывают водой. Твердый осадок растворяют в смеси хлороформ/вода, органический слой отделяют и сушат Na2SO4. Растворитель упаривают в вакууме и остаток очищают радиальной хроматографией при элюировании смесью изопропиламин/метанол/этилацетата (0,5: 1:98,5) с выходом 1,32 г (57%) N-этилформамида в виде оранжевого твердого вещества, т.пл. 75-77oC.
Пример 14
Получение 1-[[2-(Диэтиламино)этил]амино]-4-[(этиламино)-метил]тиоксантен-9-она
(I: R1=R2=Et; Q=CH2NHC2H5; R8=H; n=2).
По методике, аналогичной примеру 4, из смеси, содержащей 1,3 г (3,2 ммоль) свободного основания амина, полученного в примере 4, N-этилформамида примера 13 и 10,8 мл 2 н. соляной кислоты получают 1,29 г (92%) этиламина в виде дигидрохлорида. Полученный продукт перекристаллизовывают из этанола/тетрагидрофурана и расплавляют при температуре 160oC (разл.).
Пример 15
Получение 1-[[2-(диэтиламино)] этил]амино]-4- (диметиламинометилен-аминометил)-тиоксантен-9-она тригидрохлорида
(I: R1=R2=Et; Q=CH2N=CHN(Me)2; R8=H; n=2).
N-[[1-[[2-(Диэтиламино)]этил]амино]-9-оксотиоксантен-4-ил]метил]формамид (3 г) разбавляют 50 мл 2 н. HCl, и реакционный раствор кипятят на паровой бане в течение 90 мин. Смесь охлаждают, подщелачивают до pH 10 35% раствором NaOH и экстрагируют в хлороформе. Органический слой отделяют, фильтруют через K2CO3 и упаривают в вакууме. Полученный неочищенный продукт подвергают взаимодействию с диметилформамида диметилацеталем в течение ночи при 60oC. Избыток ДМФ-диметилацеталя удаляют в вакууме, и полученное названное соединение очищают флэш-хроматографией (силакагель; смесь хлороформ /iPrNH2/MeOH (98: 1:1). Полученный продукт растворяют в 2,5 М растворе HCl/EtOH (100 мл), затем охлаждают на ледяной бане, фильтруют и после сушки получают 2,38 г 1-([2-(диэтиламино)] этил]амино]-4-(диметиламинометиленаминометил)- тиоксантен-9-она тригидрохлорид в виде оранжевого твердого вещества, т.пл. 258-260oC.
Пример 16
Получение N-[[1-[[2-(Диэтиламино)этил] амино] -9-оксотиоксантен-4-ил] метил]-трифторацетамида
(I: R1=R2=Et; Q=CH2NHC(O)CF3; R8=H; n=2).
Раствор, содержащий 4-(аминометил)-1-[[2-(диэтиламино)этил] - амино]-тиоксантен-9-она (2,91 г; 8,19 ммоль) в 80 мл хлористого метилена при 0oC обрабатывают трифторацетилхлоридом (14,75 мл 0,61М раствора в толуоле; 9,0 ммоль), и реакционную смесь перемешивают при 0oC в течение 90 мин. Смесь затем упаривают в вакууме, остаток очищают флэш-хроматографией (силакагель; EtOAc (100%), затем смесью 2% изопропиламин/EtOAc) и после перекристаллизации из этилацетата получают 2,52 г (68%) продукта в виде свободного основания, т.пл. 189-190oC (пример 16). Полученное свободное основание растворяют в метаноле, обрабатывают метансульфокислотой (0,55 г, 5,72 ммоль) и после перекристаллизации из ацетона получают метансульфонатную соль, т.пл. 152-154oC (пример 16а).
Пример 17
(а)
Смесь, состоящую из тиосалициловой кислоты (50,14 г, 0,33 моль) и ацетата меди (2) (5,0 г) в диметилсульфоксиде (500 мл), нагревают с обратным холодильником и прибавляют одной порцией (54,3 г) карбонат калия. Затем шприцем к реакционной массе прибавляют 3-бромхлорбензол (42 мл, 0,36 моль), и полученную смесь кипятят с обратным холодильником в течение 3 часов. Реакционную смесь выливают в воду, очищают древесным углем и фильтруют через целит. Фильтрат подкисляют концентрированной HCl, выпавший осадок собирают фильтрованием, промывают водой и после сушки в вакууме при температуре 60oC получают 75,01 г (85%) 2-[(3-хлорфенил)тио]бензойной кислоты.
(b)
К перемешанному раствору концентрированной H2SO4 при 0oC прибавляют 2-[(3-хлорфенил)тио)бензойную кислоту (75,00 г, 0,28 моль) порциями в течение 1 часа. Смесь перемешивают в течение 2 часов, выливают в концентрированный раствор NH4OH (500 мл) в воде (2,5 л). Выпавший осадок собирают фильтрованием, промывают водой и сушат в вакууме при температуре 60oC с выходом 65,9 г (95%) смеси 1-хлор- и 3-хлортиоксантен-9-она.
(с)
Смесь, состоящую из 1-хлор и 3-хлортиоксантен-9-она (14,01 г, 56,8 ммоль), пиридина (20 мл) и диэтиламинопропиламина (5,13 г, 39,4 ммоль), кипятят с обратным холодильником до полного завершения реакции. После отведения тепла, растворитель упаривают в вакууме, остаток растворяют в хлороформе и очищают колоночной хроматографией на кремнеземе, вымывая сначала хлороформом для удаления непрореагировавшего 3-хлорзамещенного изомера, а затем смесью 5% изопропиламин/хлороформ с выделением 5,10 г (54%) 1-[(3-диэтиламино)пропил]амино]-тиоксантен-9-она в виде оранжевой смолы.
(d)
Смесь, состоящую из 1-[[3-(диэтиламино)пропил]амино]-тиоксантен- 9-она (5,10 г, 15,0 ммоль), формалина (160 мл) и 5 н. уксусной кислоты (0,8 мл), нагревают до температуры 90oC в течение 16 часов, затем прибавляют дополнительную порцию 5 н. уксусной кислоты (0,20 мл), а затем формалина (50 мл), и реакционную массу кипятят при 90oС в течение примерно 57 часов. Смесь разбавляют водой, подщелачивают 5 н. раствором NaOH и экстрагируют из хлороформа. Органический слой сушат на Na2SO4, пропускают через колонку с кремнеземом и извлекают при элюировании сначала смесью 2% метанол/хлороформ, а затем изопропиламин/метанол/хлороформ (2:2:96) 3,82 г (69%) 1-[[3-(диэтиламино)пропил] амино] -4- (гидроксиметил)-тиоксантен-9-она в виде оранжевой или коричневой смолы.
(e)
Получение 1-[[3-(диэтиламино)пропил] амино]-9-оксотиоксантен-4- карбоксальдегида
(II: R1=R2=Et; R8=H; n=3).
Смесь, состоящую из 1-[[3-(диэтиламино)пропил]амино]-4- (гидроксиметил)-тиоксантен-9-она (3,82 г), толуола (60 мл) и окиси марганца (7,5 г), кипятят с обратным
холодильником в течение 6,5 часов. Смесь охлаждают до комнатной температуры, фильтруют через целит, и фильтрат упаривают в вакууме. Получают 3,3 г (87%) 1-[[3-(диэтиламино)пропил]амино]-9-оксотиоксантен-4- карбоксальдегида в виде коричневого масла.
(f)
Получение 1-[[3-(диэтиламино)пропил]амино]-4- (метиламинометил)тиоксантен-9-он-дигидрохлорида 3/2 гидрата
(I: R1=R2=Et; Q=CH2NHMe; R8=H; n=3).
Раствор, содержащий 1-[[3-(диэтиламино)пропил]амино]-9-оксотиоксантен-4-карбоксальдегида (3,3 г, 8.96 ммоль) и 3 г уксусной кислоты в 50 мл N-метилформамида, кипятят с обратным холодильником в течение 2 часов. Смесь подщелачивают 5 мл 5 н. раствора NaOH и экстрагируют в хлороформе (3 х 150 мл). Органический слой сушат на сульфате натрия, упаривают в вакууме. Полученное масло растворяют в 3 н. водном растворе HCl (50 мл) и нагревают на паровой бане в течение 3 часов. Вышеуказанную реакционную массу подщелачивают 30 мл 35% раствора NaOH и экстрагируют в хлороформе (3 х 150 мл). Органический слой сушат на сульфате натрия, упаривают в вакууме с выходом коричневого масла, которое очищают флэш-хроматографией (силакагель; элюирование сначала смесью 5% триэтиламин/EtO2, затем 5% Et3N/EtOAc и затем смесью этиламин/метанол/EtOAc (5: 5:90)) с выделением 1,1 г 1-[[3-(диэтиламино)-пропил] амино]-4-(метил аминометил)тиоксантен-9-она в виде прозрачной оранжевой смолы. Названный смолянистый продукт превращают в соответствующую двусолянокислую соль путем обработки 6 н. HCl в простом эфире с выходом 1,04 г дигидрохлорида 3/2 гидрата в виде желтого порошка, т.пл. 222-224oC.
Пример 18
(а)
Получение 4-(аминометил)-1-[[2-(диметиламино)этил] амино] -9- оксотиоксантен-9-она-дигидрохлорида 1/2 гидрата
(I: R1=R2=Me; Q=CH2NH2; R8=H; n=2).
Смесь, состоящую из N-1[1-[[2-(диметиламино)этил] амино]-9- оксотиоксантен-4-ил]метил]формамида (6,2 г) и 2 н. HCl (52 мл), нагревают до 100oC в течение 1,5-2 часов. Реакционную смесь затем выливают в ледяную воду, подщелачивают 35% раствором NaOH и экстрагируют хлороформом. Органический слой промывают водой (2х), затем солевым раствором (1х), сушат на Na2SO4 и упаривают в вакууме. Остаток очищают колоночной хроматографией на кремнеземе и извлекают при элюировании последовательно этилацетатом, смесью 0,5% триэтиламин/EtOAc, смесью 2% триэтиламин/EtOAc, CHCl3/ 1-2% изопропиламин и CHCl3/ 1-2% изопропиламин/ 2% MeOH 3,3 г (58%) требуемого продукта в виде свободного основания. Порцию свободного основания (1,25 г) растворяют в метаноле и обрабатывают концентрированной HCl (3,3 мл) в MeOH (6 мл) с выходом 1,2 г продукта в виде дигидрохлорида 1/2 гидрата, т.пл. 213oC (разл.).
(b)
Получение N-[[1-[[2-(диметиламино)этил]амино]-9- оксотиоксантен-4-ил]-метил]метаносульфонамида метансульфоната
(I: R1=R2=Me; Q=CH2NHSO2Me; R8=H; n=2).
4-(Аминометил)-1-[[2-(диметиламино)]этил]амино]-тиоксантен-9-она (2 г, 6 ммоль) в 30 мл сухого пиридина в атмосфере азота перемешивают при комнатной температуре до полного растворения реагентов. Реакционный раствор быстро охлаждают на ледяной бане и прибавляют по каплям раствор, содержащий 0,52 мл (6,7 ммоль) метансульфонилхлорида в пиридине. Реакционную массу перемешивают в течение 1 часа при комнатной температуре. Реакционную смесь затем выливают в 500 мл воды, содержащей 0,51 г гидроокиси натрия, экстрагируют в хлороформе, органический слой промывают водой (2х) и солевым раствором и сушат на безводном сульфате натрия. Смесь фильтруют и упаривают в вакууме. Остаток (2,5 г) перемешивают в простом эфире, фильтруют и после сушки фильтрата получают 2 г N-[[1-[[2- (диметиламино)этил]амино]-9-оксотиоксантен-4- ил)метил] метансульфонамида, т.пл. 126-127oC. Полученное свободное основание растворяют в MeOH и обрабатывают метансульфокислотой (0,48 г). Получают 2,0 г (67%) требуемого продукта в виде метансульфонатной соли, т.пл. 168oC (разл.).
(с)
Смесь, состоящую из 1-[[2-(диметиламино)этил]амино]-4- (гидроксиметил)-тиоксантен-9-она (9,2 г, 0,028 моль) в толуоле (322 мл) нагревают до примерно 60oC, а затем прибавляют окись марганца (MnO2, 16 г). Реакционную массу нагревают при 60oC в течение 1 часа. Смесь затем фильтруют и после упаривания фильтрата в вакууме получают 7,9 г (87%) 1-([2-(диметиламино)-этил]амино]-9- оксотиоксантен-4-карбоксальдегида (Формула II: R1=R2=Me; R8=H; n=2).
(d)
Получение N-[[1-[(2-(диметиламино)этил] амино] -9-оксотиоксантен-4-ил] метил]-формамида
(I: R1=R2=Me; Q=CH2NHCHO; R8=H; n=2).
Смесь, состоящую из 1-[[2-(диметиламино)этил]амино]- 9-оксотиоксантен-4-карбоксальдегида (4,75 г), формамида (66,5 мл) и муравьиной кислоты (7,6 мл), кипятят при 170oC в течение 4 часов. Смесь was выливают в воду со льдом (250 мл), подщелачивают 35% NaOH и экстрагируют в хлороформе. Органический слой промывают водой (2х), затем солевым раствором (1х), сушат на Na2SO4 и упаривают в вакууме с выходом 6,3 г N-[[1-[[2-(диметиламино)этил]амино]-9- оксотиоксантен-4-ил]метил]формамида.
(e)
Получение 4-(Аминометил)-1-[[2-диметиламино)этил] амино] -9-оксотиоксантен-9-она дигидрохлорида 1/2 гидрата
(I: R1=R2Me; Q=CH2NH2; R8=H; n=2).
Смесь, состоящую из N-[[l-[[2- (диметиламино)этил]амино]-9-оксотиоксантен-4-ил] метил]формамида (6,2 г) и 2 н. HCl (52 мл) нагревают до 100oC в течение 1,5-2 часов. Реакционную смесь выливают в воду со льдом, подщелачивают 35% раствором NaOH и экстрагируют в хлороформе. Органический слой промывают водой (2х), затем солевым раствором (1х), сушат Na2SO4 и сгущают в вакууме. Остаток очищают колоночной хроматографией на кремнеземе при элюировании сначала этилацетатом, затем последовательно смесью 0,5% триэтиламин/EtOAc, 2% триэтиламин/EtOAc, CHCl3/1-2% изопропиламин и CHCl3/1-2% изопропиламин/2% MeOH с выходом 3,3 г (58%) продукта в виде свободного основания. Порцию свободного основания (1,25 г) растворяют в метаноле и обрабатывают концентрированной HCl (3,3 мл) в MeOH (6 мл). Получают 1,2 г названного продукта в виде дигидрохлорида полугидрата, т.пл. 213oC (разл.).
(f)
Получение N-[[1-[[2-(диметиламино)этил] амино] -9-оксотиоксантен-4-ил]- метил]метаносульфонамида метансульфоната
(I: R1=R2=Me; Q-CH2NHSO2Me; R8=H; n=2).
4-(Аминометил)-1-[[2-(диметиламино)] этил]амино]-тиоксантен-9-он (2 г, 6 ммоль) в 30 мл сухого пиридина в атмосфере азота перемешивают при комнатной температуре до полного растворения реагентов. Реакционный раствор быстро охлаждают на ледяной бане и прибавляют по каплям раствор, содержащий 0,52 мл (6,7 ммоль) метансульфонилхлорида в охлажденном пиридине, и реакционную массу перемешивают в течение 1 часа при комнатной температуре. Реакционную массу затем выливают в 500 мл воды, содержащей 0,51 г гидроокиси натрия, экстрагируют в хлороформе, органический слой промывают водой (2х) и солевым раствором и сушат на безводном сульфате натрия. Смесь фильтруют и упаривают в вакууме. Остаток (2,5 г) перемешивают в простом эфире, фильтруют и после сушки фильтрата получают 2 г N-[[1-[[2-(диметиламино)этил]амино]-9- оксотиоксантен-4-ил)метил] метансульфонамида, т.пл. 126-127oC. Полученное свободное основание растворяют в MeOH и обрабатывают метансульфокислотой (0,48 г). Получают 2,0 г (67%) требуемого продукта в виде метансульфонатной соли, т.пл. 168oC (разл.).
Пример 19
По методике, аналогичной примеру 17(c), из смеси 1-хлор и 3-хлортиоксантен-9-она (15,15 г, 61,4 ммоль), пиридина (20 мл) и диметиламинопропиламина (6,01 г, 58,7 ммоль) получают 6,83 г 1-[[3-(диметиламинопропил]амино] тиоксантен-9-она.
(b)
По методике, аналогичной примеру 17 (d), из смеси 1-[[3-(диметиламино)-пропил] -амино] -4-тиоксантен-9-она (6,8 г, 21,8 ммоль), формалина (175 мл) и ледяной/уксусной кислоты получают 6,74 г (90%) 1-[[3-(диметиламино)пропил]амино]-4-(гидроксиметил)- тиоксантен-9-она.
(c)
По методике, аналогичной примеру 17 (e), из смеси 1-[[3-(диметиламино)пропил] амино] -4-(гидроксиметил) тиоксантен-9-она (6,7 г), толуола (80 мл) и MnO2 (12,15 г) получают 4,2 г 1-[[3-(диметиламино)пропил] амино] -9-оксотиоксантен-4- карбоксальдегида (Формула II: R1=R2=Me; R8=H; n= 2). Продукт очищают колоночной хроматографией на кремнеземе при элюировании CHCl3 (1,00%) до 1% изопропиламин/ CHCl3.
(d)
Смесь, состоящую из N-метилформамида (50 мл), муравьиной кислоты (5,2 г) и 1-[[3-(диметиламино)пропил] амино] -9- оксотиоксантен-4-карбоксальдегида (4,4 г, 12,16 ммоль), кипятят с обратным холодильником в течение 3 часов. Смесь разбавляют водой (250 мл), подщелачивают 35% раствором NaOH и экстрагируют CHCl3 (3 х 150 мл). Органический слой сушат Na2SO4, пропускают через мембранный фильтр из кремнезема и извлекают при элюировании сначала CHCl3 (100%), а затем смесью 2% изопропиламин/CHCl3 3,93 г (84%) N-[[1-[[3-(диметиламино)-пропил] амино] -9-оксотиоксантен-4- ил]метил]-N-метилформамида (Формула IV: R1=R2=Me; R4=Me; R8=H; n-3).
(e)
Получение 1-[[3-(диметиламино)пропил]амино]-4- [(метиламинометил]тиоксантен-9-она дигидрохлорида моногидрата (R1=R2=Me; Q=CH2NHMe; R8=H; n=2).
Раствор, содержащий вышеуказанный N-метилформамид (3,83 г; 10 ммоль) в 40 мл 3 н. HCl, нагревают на паровой бане в течение 4 часов; нейтрализуют 35% раствором NaOH и быстро охлаждают на льду в течение 1 часа. Жидкий слой декантируют и неочищенный продукт растворяют в хлороформ, после фильтрования через силакагель (элюент: хлороформ; смесь 1% изопропиламин/хлороформ) выделяют 2,38 г требуемого амина в виде оранжевой смолы. Полученный продукт превращают в соответствующую солянокислую соль путем растворения его в MeOH и после обработки концентрированной HCl получают 0,98 г дигидрохлорида моногидрат, т.пл. 228-229oC.
Пример 20
(а)
Смесь, состоящую из 1-[[2-(диметиламино)этил]амино]-9- оксотиоксантен-4-карбоксальдегида (4,75 г, 0,15 моль), N-метилформамида (48 мл) и муравьиной кислоты (3,9 мл), кипятят при 170oC в течение 4,5 часов, а затем выдерживают при комнатной температуре в течение примерно 64 часов. Реакционную смесь выливают в воду (250 мл), подщелачивают 35% раствором NaOH, экстрагируют CHCl3 (3х), органический слой отделяют, промывают водой (2х), затем солевым раствором (1 х) и сушат Na2SO4. После отгонки растворителя в вакууме получают 5,75 г N-[[1-[[2- (диметиламино)этил]-амино]-9-оксотиоксантен-4-ил] метил]-N- метилформамида [Формула IV: R1=R2=Me; R4=Me; R8=H; n=3).
(b)
Получение 1-[2-(диметиламино)этил]амино]-4- [(метиламино)-метил]тиоксантен-9-она дигидрохлорида • 5/4 гидрата
(I: R1=R2=Me; Q=CH2NHMe; R8=H; n=2).
Аналогичным способом, описанным в примере 19Е, из смеси, содержащей 5,7 г (15,4 ммоль) соответствующего N-метилформамида примера 20 (а) и 50 мл 2н HCl, получают 1,8 г 1-[[2-(диметиламино)этил]амино]-4- [(метиламино)метил] тиоксантен-9-она в виде свободного основания после очистки флэш-хроматографией (силакагель; элюент: хлороформ; 0,5% изопропиламин/ хлороформ; 1% изопропиламин/хлороформ). Свободное основание превращают в соответствующую его соль путем обработки концентрированным раствором HCl в метаноле с получением 1,8 г (30%) продукта в виде дигидрохлорида 5/4 гидрата, т.пл. 177oC (разл.).
Пример 21
(а)
Раствор, содержащий [[3-(диметиламино)пропил]амино]-9- оксотиоксантен-4-карбоксальдегида (3,6 г; 10,57 ммоль) в 50 мл формамида, содержащего 3,6 г уксусной кислоты, кипятят с обратным холодильником в течение 1,5 часов и затем выдерживают при комнатной температуре в течение ночи. Реакционную смесь разбавляют водой (400 мл), подщелачивают 3 мл 5 н. раствором NaOH, быстро перемешивают в течение 30 мин, и выпавшее твердое вещество отфильтровывают, промывают водой и после сушки получают 3,1 г (79%) N-[[1-[[3-(диметиламино)пропил] амино] -9- оксотиоксантен-4-ил]метил]-N-метилформамида (Формула I: R1=R2=Me; Q=CH2NHCHO; R8=H; n=3), в виде желтого порошка.
Раствор формамида, полученного в примере 21 (а) (2,98 г; 8,07 ммоль), в 40 мл 3 н. HCl нагревают на паровой бане в течение 4 часов, доводят до комнатной температуры, быстро охлаждают на льду и нейтрализуют до pH 8 путем прибавления 5 н. NaoH. Полученную гетерогенную смесь экстрагируют в хлороформе (5х100 мл). Органический слой сушат на сульфате натрия и фильтруют через мембрану из силикагеля (элюент: смесь 5% триэтиламин/простой эфир, 1-5% изопропиламин/ хлороформ) с выделением 2,3 г (83%) 4-(аминометил)-1-[[3-(диметиламино)пропил] амино] тиоксантен-9-она (Формула I: R1=R2=Me; Q= CH2NH2; R8=H; n=3).
(c)
Получение N-[[1-[[3-(диметиламино)пропил]амино]-9- оксотиоксантен-4-ил] метил]-метансульфонамида метансульфоната 1/2 гидрата
(I: R1=R2=Me; Q=CH2NHSO2Me; R8H; n=3).
К охлажденному на ледяной бане раствору амина из примера 21(b) (2,2 г; 6,44 ммоль) в пиридине прибавляют метансульфонилхлорид (0,51 мл; 6,59 ммоль) и полученную смесь перемешивают при комнатной температуре в течение ночи. Реакционную массу разбавляют хлороформом и пропускают через крупнопористую мембрану из силикагеля с извлечением при элюировании смесью 5% триэтиламин/EtOAc 1,32 г N-[[1-[[3-(диметиламино)пропил]амино]-9-оксотиоксантен-4- ил] метил]-метансульфонамида в виде желтого порошка. Свободное основание растворяют в метаноле (10 мл) и обрабатывают раствором метансульфокислоты (0,31 г, 1 экв. ) в метаноле с получением 1,36 г метансульфонатной соли в форме 1/2 гидрата в виде оранжевого твердого вещества, т.пл. > 107oC.
Пример 22
Получение N-[[1-[[2- (диэтиламино)этил]амино]-9-оксотиоксантен-4-ил]метил]-N- метилэтансульфонамида метансульфоната
(I: R1=R2=Et; Q=CH2N(CH3)SO2 Et; R8=H; n=2).
Раствор, содержащий 2,03 г (5,49 ммоль) 1-[[2-(диэтиламино)этил]амино] -4- [(метиламино)метил]тиоксантен-9-она (полученного по методике, описанной в примере 5) и триэтиламин в 45 мл хлористого метилена, охлаждают до 0oC и обрабатывают этансульфонилхлоридом (0,74 г, 5,76 ммоль) с поддержанием температуры 0oC в течение 15 мин. Через 15 мин реакционную смесь охлаждают и перемешивают при комнатной температуре в течение 72 часов. Смесь упаривают в вакууме, остаток растворяют в хлороформе и после очистки при пропускании полученного раствора через колонку с набивкой из силикагеля (элюент: хлороформ; смесь 1% триэтиламин/хлороформ) выделяют 2,43 г (96%) N-[[1-[[2-диэтиламино)этил] амино] -9-оксотиоксантен-4- ил]метил]-N-метилэтансуфонамида. Названный сульфонамид перекристаллизовывают из этилацетата и после обработки метансульфокислотой в изопропаноле получают требуемый продукт в виде соли метансульфокислоты, т.пл. 159-161oC.
Пример 23
Получение N-([1-[[2-(диэтиламино)этил] амино] -9-оксотиоксантен-4- ил] метил](пара-метокси)бензолсульфонамида метансульфоната
(I: R1=R2=Et; Q=CH2NHSO2C6H4-п-OMe; R8=H; n=2).
Раствор, содержащий 1,40 г (3,94 ммоль) 4-(аминометил)-1-[[2-(диэтиламино)этил] амино]-тиоксантен-9-она (полученного по методике, описанной в примере 4) в 30 мл смеси из хлороформа и 1,5 мл триэтиламина, охлаждают до 0oC и обрабатывают пара-метоксибензолсульфонилхлоридом (0,83 г, 4,02 ммоль) с поддержанием температуры 0oC в течение 15 мин. Через 15 мин реакционную смесь охлаждают до комнатной температуры и перемешивают в течение 2 часов. Хлороформ отгоняют в вакууме, остаток растворяют в 100 мл смеси хлористого метилена с 1 мл триэтиламина и обрабатывают дополнительной порцией пара-метоксибензолсульфонилхлорида (0,85 г) при перемешивании при комнатной температуре. Затем реакционную смесь упаривают в вакууме, и остаток очищают при пропускании его через колонку с набивкой из силикагеля с выделением (при элюировании смесью 1% триэтиламин/хлороформ) 1,57 г (96%) N-[[l- [[2-(диэтиламино)этил] амино] -9-оксотиоксантен-4-ил] метил]-пара- метоксибензолсульфонамида. Указанный сульфонамид обрабатывают метансульфокислотой (0,3 г) в смеси изопропанол/изопропилацетат/метанол с получением 1,07 г требуемого продукта в виде соли метансульфокислоты, т.пл. 133-137oC.
Пример 24
Получение N-[(1-[[2-(диэтиламино)этил]амино]-9- оксотиоксантен-4-ил]метил]-этансульфонамида метансульфоната
(I: R1=R2=Et; Q=CH2NHSO2Et; R8=H; n=2).
Раствор, содержащий 2,5 г 4-(аминометил)-1-[(2-(диэтиламино)этил] амино] -тиоксантен-9-она (полученного по методике, описанной в примере 4) в 30 мл пиридина, охлаждают на ледяной бане в течение 15 мин и затем по каплям быстро прибавляют раствор, содержащий 0,95 г этансульфонилхлорида в 5 мл пиридина. Реакционную массу перемешивают при комнатной температуре в течение 1 часа. Смесь выливают в 75 мл воды, содержащей 0,75 г NaOH, экстрагируют в хлороформе, органический слой промывают последовательно водой (2х) и солевым раствором, и сушат на сульфате натрия. Затем реакционную смесь упаривают в вакууме, остаток перемешивают в простом эфире и после сушки (40oC/0,1 мм) получают 1,7 г N-[[1-[[2-(диэтиламино)этил]амино]-9- оксотиоксантен-4-ил] метил] этансульфонамида т.пл.105oC (разл.). Указанный сульфонамид растворяют в метаноле и обрабатывают раствором метансульфокислоты в метаноле с получением 1,61 г (42%) требуемого продукта в виде соли метансульфокислоты, т.пл. 135oC (разл.).
Пример 25
Получение N-[[1-([2-(диэтиламино)этил]амино]-9- оксотиоксантен-4-ил]метил]-N-этилметансульфонамида
(I: R1=R2=Et; Q=CH2N(Et)SO2Me; R8=H; n=2).
Раствор, содержащий 2,10 г (5,48 ммоль) 1-[[2-(диэтиламино)этил]амино] -4-[(этиламино)метил]тиоксантен-9-она (полученного по методике примера 14) в 30 мл хлористого метилена, охлаждают до 0oC и обрабатывают 2 мл смеси триэтиламина и метансульфонилхлорида (0,7 мл), и полученную реакционную смесь перемешивают при комнатной температуре в течение 6 часов. Растворитель отгоняют в вакууме, остаток растворяют в хлороформе, и полученный раствор очищают путем пропускания его через колонку с силикагелем (при элюировании сначала хлороформом, а затем смесью 2% триэтиламин/хлороформ). Выделенное желтое твердое вещество перекристаллизовывают из этилацетата и после сушки получают 1,11 г (44%) N-[[1-[(2- (диэтиламино)этил]амино]-9-оксотиоксантен-4-ил]метил]-N- этилметансульфонамида в виде желтого порошка, т.пл. 172-176oC.
Пример 26
Получение N-[[1-[[2-(диэтиламино)этил]амино]-9- оксотиоксантен-4-ил]метил)-3,4-дихлорбензолсульфонамида метансульфоната 1/2 гидрата
(I; R1=R2=Et; Q=CH2NHSO2C6H3-3,4-дихлор; R8=H; n=2).
К раствору 3,4-дихлорбензолсульфонилхлорида (1,84 г, 7,5 ммоль) в 35 мл сухого пиридина прибавляют 2,5 г (7 ммоль) 4-(аминометил)-1-[[2- (диэтиламино)этил] амино]-тиоксантен-9-она (полученного по методике, описанной в примере 4) в атмосфере азота, и полученную реакционную массу перемешивают при комнатной температуре в течение 15 мин, а затем выдерживают примерно в течение 72 часов. Затем реакционную смесь выливают в 75 мл воды, содержащей 0,75 г NaOH, и экстрагируют в хлороформе. Органический слой промывают водой (2х) и солевым раствором и сушат на сульфате натрия. Хлороформ отгоняют в вакууме, остаток перекристаллизовывают из этанола с получением 1,24 г N-[[1-[[2-(диэтиламино)]этил]амино]-9- оксотиоксантен-4-ил]метил]-3,4-дихлорбензолсульфонамида, т. пл. 95oC (разл. ). Свободное основание растворяют в метаноле и после обработки раствором метансульфокислоты в метаноле получают метансульфоната 1/2 гидрат, т.пл. 55oC (разл.).
Пример 27
Получение N-[[1-[[2-(диэтиламино)этил]амино]-9-оксотиоксантен-4- ил]метил]-2-фторбензолсульфонамида
(I; R1=R2=Et; Q=CH2NHSO2C6H4-2-F; R8=H; n=2).
Раствор, содержащий 1,36 г (3,83 ммоль) 4-(аминометил)-1-[[2-(диэтиламино)этил] амино]-тиоксантен-9-она (полученного по методике, описанной в примере 4) в 25 мл хлористого метилена, содержащего 1 мл триэтиламина, охлаждают до 0oC и обрабатывают 2-фторбензолсульфонилхлоридом (0,84 г; 4,32 ммоль), и затем реакционную массу перемешивают в течение нескольких часов. Растворитель отгоняют в вакууме, остаток растворяют в хлороформе и очищают флэш-хроматографией (силикагель: при элюировании хлороформом, а затем смесью 1% триэтиламин/хлороформ). Растворитель отгоняют в вакууме и после перекристаллизации полученного продукта из этилацетата получают 1,08 г (55%) N-[[1-[[2- диэтиламино)этил]амино]-9-оксотиоксантен-4-ил]метил]-2- фторбензолсульфонамида в виде оранжевого порошка, т.пл. 125-127oC.
Пример 28
Получение N-[[1-[[2-(диэтиламино)этил] амино] -9- оксотиоксантен-4-ил] метил]-N-пропилметансульфонамида
(I: R1=R2=Et; Q=CH2N(C3H7)SO2Me; R8=H; n=2).
Маслянистый продукт, полученный из 0,2 г 60% дисперсии гидрида натрия в минеральном масле, отделяют путем растирания в порошок с использованием пентана (4х). К гидриду натрия добавляют сухой ДМФ (40 мл) в атмосфере азота при перемешивании, а затем к реакционной массе добавляют 2 г N-[[1-[[2-(диэтиламино)этил] амино] -9-оксотиоксантен-4-ил] метил] метансульфонамида (пример 6) при одновременном перемешивании в атмосфере азота, и полученную смесь нагревают до 50oC в течение двух часов. Затем смесь охлаждают на ледяной бане в течение 15 мин, добавляют 0,87 г иодистого пропила, растворенного в небольшом объеме ДМФ, и реакционную смесь перемешивают при комнатной температуре в течение ночи. Смесь перемешивают с 35 мл воды, фильтруют, и остаток промывают водой и после сушки (50oC/0,1 мм /P2O5) получают 2,17 г N-[[l-[[2-(диэтиламино)этил] амино] 9-оксотиоксантен-4-ил)метил]-N- пропилметансульфонамида, т.пл. 142-143oC.
Пример 29
N-[[1-[[2- (диэтиламино)этил]амино]-9-оксотиоксантен-4-ил]метил]-N-метил-бензолсульфонамида метансульфоната
(I: R1=R2=Et; Q=CH2N (Me) SO2C6H5; R8=H; n=2).
Раствор, содержащий 5,32 г (14,4 ммоль) 1-[[2-(диэтиламино)этил]амино]- 4-[(метиламино] метил]тиоксантен-9-она (полученного по методике, описанной в примере 5) в 100 мл хлористого метилена, охлаждают до 0oC и обрабатывают триэтиламином (5 мл) и бензолсульфонилхлоридом (2 мл; 15,67 ммоль), и затем реакционную смесь перемешивают в течение 2 часов. Смесь упаривают в вакууме, и остаток очищают при пропускании его через силикагель (с элюированием последовательно хлороформом; смесью 1/2% - 1% изопропиламин/хлороформ) с извлечением 6,24 г желтого смолянистого продукта. Полученный продукт растворяют в этилацетате, и после отгонки растворителя в вакууме получают 6,06 г (83%) N-[[1-[[2-(диэтиламино)этил] амино] -9-оксотиоксантен-4- ил]метил]-N-метилбензолсульфонамида. Полученный сульфонамид (2,5 г) суспендируют в изопропаноле и после обработки метансульфокислотой (0,51 г) получают 2,63 г метансульфонатной соли, т.пл. 171-174oC.
Пример 30
(а)
К смеси м-анисовой кислоты (250 г, 1,67 моль) в уксусной кислоте (1 л) прибавляют бром (85 мл) и затем воду (1 л). Реакционную массу нагревают с обратным холодильником, охлаждают на ледяной бане, и продукт, выпавший в осадок, собирают фильтрованием и после промывания водой получают 305,7 г (79%) 2-бром-5-метоксибензойной кислоты, т.пл. 154-156oC.
(b)
К смеси, состоящей из 3-хлортиофенола (20 г, 138 моль) и ацетата меди (2) (1,8 г) в ДМФ (200 мл), прибавляют K2CO3 (23 г). Смесь нагревают до 150oC в течение 15-20 мин, затем порциями прибавляют 2-бром-5-метоксибензойную кислоту (35,8 г, 0,155 моль). Смесь нагревают в течение ночи, выливают в воду (600 мл), фильтруют, фильтрат обрабатывают древесным углем, отфильтровывают и разбавляют HCl. Выпавший осадок собирают фильтрованием, промывают водой и после сушки при температуре 50oC в вакууме на P2O5 получают 27,6 г 2-[(3-хлорфенил)тио]-5-метоксибензойной кислоты.
(c)
К охлажденной серной кислоте (89 мл) в атмосфере азота прибавляют порциями 2-[(3-хлорфенил)тио-5-метоксибензойной кислоты (27 г, 0,092 моль) в течение 1,5-2 часов. Смесь перемешивают при комнатной температуре в течение ночи, выливают в воду (900 мл), содержащую концентрированный NH4OH (218 мл) со льдом. Выпавшее в осадок твердое вещество собирают фильтрованием и сушат при температуре 50oC в вакууме на P2O5. Получают 21 г (42%) смеси 1-хлор- и 3-хлор-7-метокси-тиоксантен-9-она.
(d)
Смесь, состоящую из 1-хлор- и 3-хлор-7-метокси-тиоксантен-9-она (20,7 г), пиридина (69 мл) и диэтиламиноэтиламина (16,1 г, 0,138 моль), нагревают при 115oC в атмосфере азота в течение 20 часов. Растворитель отгоняют в вакууме, и остаток очищают колоночной хроматографией на кремнеземе с элюированием сначала CHCl3 (100%), а затем смесью 1% изопропиламин/хлороформ, получают 11,22 г 1-[[2-(диэтиламино)этил]амино]-7- метокситиоксантен-9-она.
(e)
Смесь, состоящую из 1-[[2-(диэтиламино)этил]амино]-1- метокситиоксантен-9-она (11,2 г, 0,031 моль), 37% формальдегида (277 мл) и 5 н. уксусной кислоты (4,6 мл), нагревают при 100oC в течение 3 часов. Затем реакционную смесь охлаждают, фильтруют, фильтрат выливают в воду со льдом (600 мл) и подщелачивают 35% раствором NaOH. Смесь экстрагируют в хлороформе (3х), промывают солевым раствором и сушат Na2SO4. Растворитель отгоняют в вакууме, остаток очищают колоночной хроматографией на кремнеземе с элюированием последовательно смесью 25% CHCl3/гексан, 50%CHCl3/гексан, 75% CHCl3/гексан, 0,5% изопропиламин/CHCl3 с получением в результате 8,8 г (73%) 1-[[2-(диэтиламино)-этил]амино]-4-(гидроксиметил)-7-метокситиоксантен-9-она.
(f)
Раствор, содержащий 1-[[2-(диэтиламино)этил]амино]-4- (гидроксиметил)-7-метокситиоксантен-9-она (8,8 г, 0,023 моль) в толуоле (268 мл), нагревают до 60oC в атмосфере азота, а затем добавляют MnO2 (13,2 г). Смесь нагревают в течение ночи, фильтруют и после упаривания фильтрата в вакууме получают 7,05 г (81%) 1-[[2-(диэтиламино)этил]амино]-7- метокси-9-оксотиоксантен-4-карбоксальдегида (Формула VI: R1=R2=Et; R8=7-OCH3; n=2).
(g)
Раствор, содержащий 1-[[2-(диэтиламино)этил)амино)-7-метокси- 9-оксотиоксантен-4-карбоксальдегида (3 г, 7,8 ммоль) и 1,5 мл муравьиной кислоты в 25,5 мл N-метилформамида, нагревают до 170oC в течение 8 часов при перемешивании в атмосфере азота. Затем реакционную смесь выливают в 160 мл воды со льдом, подщелачивают 35% раствором NaOH и экстрагируют в хлороформе (3х). Органический слой промывают водой (2х), солевым раствором, сушат на сульфате натрия, и после отгонки растворителя в вакууме получают 3 г (89,9%) целевого N-[[1-[[2-(диэтиламино)этил] амино] -7-метокси-9- оксотиоксантен-4-ил]метил] -N-метилформамида (Формула IV: R1=R2=Et; R4=Me; R8=7-OCH3; n=2).
(h)
Получение 1-[[2-(диэтиламино)этил] амино] -4-[(метиламино)метил] -7-метокси-тиоксантен-9-она
(I: R1= R2=Et; Q=CH2NHMe; R8=7-OMe; n=2).
N-метилформамид примера 30(g) (3,0 г) в 2 н. водном растворе HCl (24 мл) нагревают при температуре 100oC в течение двух часов в атмосфере азота при перемешивании. Реакционную смесь затем охлаждают, выливают в 125 мл воды со льдом, подщелачивают 35% раствором NaOH, экстрагируют в хлороформе, и экстракт промывают последовательно водой (2х) и солевым раствором. Органический слой сушат на сульфате натрия и после упаривания в вакууме получают 3,1 г неочищенного продукта. Этот продукт растирают в порошок в эфире и после очистки фильтрата путем пропускания через несколько хроматографических колонок (силикагель; элюент: смесь 50% гексан/хлороформ, хлороформ, и смесь 0,25-0,5% изопропиламин/хлороформ (колонка 1); хлороформ, смесь 1% изопропиламин/1% MeOH/CHCl3 (колонка 2); и CHCl3, смесь 0,5% изопропиламин/CHCl3 (колонка 3)) получают 0,746 г 1-[[2-(диэтиламино)этил]амино]-4-[(метиламино] метил]-7-метокси- тиоксантен-9-она.
Пример 31
(а)
Получение N-[[1-[[2-(диэтиламино)этил)амино] -7-метокси- 9-оксотиоксантен-4-ил]-метил)-формамида
(I: R1=R2=Et; Q=CH2NHCHO; R8=7-OMe; n=2).
Смесь, состоящую из 1-[[2- (диэтиламино)этил]амино]-7-метокси-9-оксотиоксантен-4-карбоксальдегида (3,6 г, 0,0094 моль), формамида (48 мл) и муравьиной кислоты (6 мл), нагревают до 170oC в атмосфере азота в течение 8 часов. Смесь выливают в воду со льдом, подщелачивают 35% раствором NaOH и экстрагируют в хлороформе. Органический слой отделяют, промывают сначала водой (2х), затем солевым раствором и сушат на Na2SO4. После упаривания в вакууме получают 3,88 г N-[[1-[[2- (диэтиламино)этил]амино]-7-метокси-9-оксотиоксантен-4-ил]метил]-формамида.
(b)
Получение 4-(аминометил)-1-[[2-(диэтиламино) этил]амино]-7-метокси-тиоксантен-9-она
(I: R1=R2=Et; Q=CH2NH2; R8=7-OCH3; n=2).
Смесь, состоящую из формамида примера 31 (а) (3,88 г) и 2 н. HCl (32 мл), нагревают при 100oC в течение двух часов в атмосфере азота при перемешивании. Затем реакционную смесь охлаждают, выливают в воду, подщелачивают 10% раствором NaOH, экстрагируют в хлороформе, экстракт промывают водой, а затем солевым раствором. Органический слой сушат на сульфате натрия и после упаривания в вакууме получают 3,6 г неочищенного продукта. Этот продукт растворяют в хлороформе и после очистки флэш-хроматографией (силикагель; элюент: смесь гексан/хлороформ (50: 50) и 1% изопропиламин в гексан/хлороформ (50: 50)) получают 1,75 г целевого соединения.
(с)
Получение N-[[1-[[2-(диэтиламино)] этил] амино] -7- метокси-9-оксотиоксантен-4-ил]метил]метансульфонамида
(I: R1=R2=Et; Q=CH2NHSO2Me; R8=7-OMe; n=2).
К раствору, содержащему 1,75 г (0,0045 моль) амина примера 31(b) в 22,5 мл пиридина, охлажденному на ледяной бане, прибавляют по каплям 0,39 мл (0,005 моль) метансульфонилхлорида в небольшом количестве пиридина в атмосфере азота при перемешивании, а затем реакционную массу перемешивают при комнатной температуре в течение двух часов. Смесь выливают в 375 мл воды, содержащей 0,38 г NaOH, экстрагируют в хлороформе, и органический слой промывают водой и солевым раствором. Хлороформный слой сушат на сульфате натрия, растворитель отгоняют в вакууме, и после сушки остатка в вакууме получают 1,61 г (77%) N-[[1-[[2-(диэтиламино)-этил]амино]-7- метокси-9-оксотиоксантен-4-ил]метил]метансульфонамида, т.пл. 144oC (разл.).
Пример 32
(а)
К смеси, состоящей из 3-хлортиофенола (20 г, 0,138 моль), ацетата меди (2) (1,75 г) и ДМФ (199 мл), в атмосфере азота прибавляют порциями K2CO3 (23 г).
Реакционную массу затем нагревают до температуры 150oC и затем добавляют 2,5-дибромбензойную кислоту (43,5 г). Смесь нагревают в течение ночи, выливают в воду (600 мл), фильтруют, фильтрат обрабатывают древесным углем, и опять фильтруют. Фильтрат подкисляют концентрированной HCl, экстрагируют CHCl3, органический слой промывают солевым раствором и сушат на Na2SO4. После отгонки растворителя в вакууме получают 28,9 г 2-[(3-хлорфенил)тио]-5-бромбензойной кислоты.
(b)
Смесь, состоящую из 2-[(3-хлорфенил)тио]-5-бромбензойной кислоты (28,4 г) и концентрированной серной кислоты (80 мл), перемешивают при 0oC и затем при комнатной температуре в течение ночи. Смесь выливают в воду со льдом (850 мл), содержащую концентрированный NH4OH (199 мл), и продукт, выпавший в осадок, собирают фильтрованием и после сушки при температуре 50oC в вакууме получают 15,0 г смеси, состоящей из 1-хлор- и 3-хлор-7-бром-тиоксантен-9-она.
(c)
Смесь, состоящую из 1-хлор и 3-хлор-7-бромтиоксантен-9-она (13,6 г), пиридина (108 мл) и N,N-диэтилэтилендиамина (16,3 мл), нагревают до 115oC в течение 20 часов. Растворитель отгоняют в вакууме, и после очистки остатка колоночной хроматографией на кремнеземе при элюировании последовательно CHCl3 (100%), смесью 1% изопропиламин/CHCl3 получают 9,3 г 1-[[2-(диэтиламино)этил]амино]-7-бромтиоксантен-9-она.
(d)
Смесь, состоящую из 1-[[2-(диэтиламино)этил]амино]-7-бромтиоксантен-9-она (9,3 г, 22,9 ммоль), 203 мл 37% раствора формальдегида и 3,4 мл 5 н. раствора уксусной кислоты нагревают до 100oC в атмосфере азота в течение ночи. Смесь охлаждают до комнатной температуры, и выпавший твердый осадок отделяют фильтрованием. Фильтрат разбавляют водой, подщелачивают 35% раствором NaOH и экстрагируют в хлороформе. Органический слой промывают солевым раствором, сушат на сульфате натрия, и после отгонки растворителя в вакууме получают 10 г масла. Масло, обработанное хлористым метиленом, фильтруют, растворитель упаривают в вакууме, и после очистки полученного гидроксиметильного аналога флэш-хроматографией (силикагель; при элюировании последовательно смесью 25% хлороформ/гексан, хлороформ/гексан (1:1), 25% хлороформ/гексан, CHCl3 (100%), и 0,5-1% изопропиламин/хлороформ) получают 3,2 г 1-[[2-(диэтиламино)этил]амино]-4-(гидроксиметил)-7-бромтиоксантен-9-она.
e)
Получение 1-[[2-(диэтиламино)этил] амино)-7-бром-9- оксотиоксантен-4-карбоксальдегида
(II: R1=R2=Et; R8=7-Br; n=2).
Смесь, состоящую из 3,2 г (7,34 ммоль) спирта примера 32(d) и 4,3 г MnO2 в 85 мл толуола, нагревают при 60oC в течение 1 часа в атмосфере азота. Смесь отфильтровывают, промывают CHCl3, и объединенный фильтрат упаривают в вакууме с выходом 3 г желтого твердого продукта, который затем растирают в порошок в эфире, отфильтровывают и после сушки получают 2,7 г (94,3%) 1-[[2-(диэтиламино)этил]амино)-7-бром-9-оксотиоксантен-4- карбоксальдегида, т. пл. 145-146oC.
(f)
Получение N-[[1-[[2-(диэтиламино)этил]амино]-7-бром-9- оксотиоксантен-4-ил]метил]формамида
(I: R1=R2=Et; Q=CH2NHCHO; R8=7-Br; n=2).
Смесь, состоящую из 2,7 г (6,2 ммоль) 1-[[2- (диэтиламино)этил]амино]-7-бром-9-оксотиоксантен-4- карбоксальдегида, 31,7 мл формамида и 3,6 мл уксусной кислоты, нагревают до 170oC в атмосфере азота при перемешивании в течение 8 часов. Затем
реакционную смесь выдерживают при комнатной температуре в течение 72 часов. Смесь выливают в 150 мл воды со льдом, подщелачивают 35% раствором NaOH, и образовавшийся твердый продукт отфильтровывают, промывают водой, растворяют в хлороформе, промывают солевым раствором, сушат на сульфате натрия. После упаривания растворителя в вакууме получают 2,85 г целевого формамида в виде желтого/оранжевого твердого вещества, т.пл.132oC (разл.).
(g)
Получение 1-[[2-(диэтиламино)этил]амино]-4-(аминометил)-7- бромтиоксантен-9-она
(I: R1=R2=Et; Q=CH2NH2; R8=7-Br; n=2).
Смесь, состоящую из 2,85 г (6,6 ммоль) вышеуказанного формамида из примера 32(f)) в 26 мл 2 н. раствора HCl, нагревают до 100oC в атмосфере азота в течение 2 часов, охлаждают до комнатной температуры и выдерживают при комнатной температуре в течение ночи. Затем реакционную массу выливают в 200 мл воды со льдом, подщелачивают 35% раствором NaOH и экстрагируют в хлороформе. Органический слой промывают водой и солевым раствором, сушат на сульфате натрия, и после упаривания в вакууме получают 2,67 г темного масла. Это масло затем очищают флэш-хроматографией (силикагель; элюент: 1250 мл смеси гексан/хлороформ (1:1) 1% изопропиламин в смеси гексан/хлороформ (1:1)), в результате чего выделяют 1,87 г (70%) 1-[[2-(диэтиламино)этил] амино]-4-(аминометил)-7-бромтиоксантен-9-она, т.пл. 79-82oC.
Пример 33
Получение N-[[1-[[2-(диэтиламино)этил] амино]-7-бром-9-оксотиоксантен-4-ил] метил] метансульфонамида
(I: R1=R2=Et; Q=CH2NHSO2Me; R8=7-Br; n=2).
Раствор 1-[[2-(диэтиламино)этил]амино]-4-(аминометил)-7- бромтиоксантен-9-она (1 г, 2,3 ммоль) в 11,5 мл сухого пиридина в атмосфере азота перемешивают на ледяной бане в течение 15 мин и затем по каплям прибавляют 0,2 мл (2,6 ммоль) метансульфонилхлорида в охлажденном пиридине. Полученную смесь перемешивают при комнатной температуре. Затем реакционную смесь выливают в 200 мл воды, прибавляют 0,19 г гидроксида натрия в воде со льдом и экстрагируют в хлороформе. Органический слой промывают последовательно водой (2х) и солевым раствором и сушат на безводном сульфате натрия. Смесь фильтруют, упаривают в вакууме, остаток перемешивают в эфире, фильтруют и после сушки фильтрата получают 1,02 г N-[[1-[[2- (диэтиламино)этил]-амино] -7-бром-9-оксотиоксантен-4- ил]метил]метансульфонамида, т.пл. 134-139oC.
Пример 34
Получение метил-N-[[1-[[2-(диэтиламино)этил] амино]-9-оксотиоксантен-4- ил]метил]карбамата
(I: R1=R2=Et; Q=CH2NHCOOMe; R8=H; n=2).
К раствору, содержащему 2,94 г (8,27 ммоль) 4-(аминометил)-9-[[[2-(диэтиламино)] -этил] амино]-тиоксантен-9-она в 50 мл метиленхлорида, содержащего 5 мл триэтиламина, охлажденного до 0oC, прибавляют 0,7 мл (9,06 ммоль) метилхлорформиата, и полученную смесь перемешивают в течение 2,5 часов. Растворитель отгоняют в вакууме, остаток суспендируют в хлороформе и после очистки флэш-хроматографией (силикагель; элюент: хлороформ, смесь 1% изопропиламин/хлороформ) получают 2,36 г (69%) метил-N-[[1-[[2-(диэтиламино)этил)-амино] -9-оксотиоксантен-4-ил) метил]карбамата в виде желтого твердого вещества, т.пл. 129-131oC.
Пример 35
Получение 1-[[2-(диэтиламино)этил] амино]-4-[(метиламино) метил]-7-гидрокси-тиоксантен-9-она
(I: R1=R2=Et; Q=CH2NHMe; R8=7-OH; n=2).
Раствор, содержащий 1,6 г (4 ммоль) 1-[[2-(диэтиламино)этил]амино]-4-[(метиламино] метил] -7-метокси- тиоксантен-9-она (полученного по методике, описанной в примере 30(h)) в 10 мл 48% раствора HBr, нагревают до 110oC в течение 5 часов. После охлаждения, реакционную смесь нейтрализуют насыщенным раствором бикарбоната натрия и экстрагируют в хлороформе (3 х 100 мл). Полученный твердый смолянистый продукт, нерастворимый в воде или хлороформе, растворяют в метаноле и объединяют с хлороформным раствором. Растворитель упаривают в вакууме с получением 1,67 г темно-оранжевого твердого вещества. Это оранжевое твердое вещество очищают сначала флэш-хроматографией (силикагель; изопропиламин/метанол/хлороформ (1:1:98), а затем на колонке с кремнеземом при элюировании смесью изопропиламин/MeOH/CHCl3 (2:2:96)) с извлечением в результате 0,56 г (36%) 1-[[2-(диэтиламино)этил]-амино]-4-[(метиламино]метил]-7-гидрокси- тиоксантен-9-она, т.пл. 167-169oC.
Пример 36
Получение Метил-N- [[1-[[2-(диэтиламино)]этил]амино)-1-метокси-9-оксотиоксантен-4- ил]-метил]карбамата
(I: R1=R2=Et; Q=CH2NHCOOMe; R8=7-OMe; n=2).
К раствору, содержащему 1,55 г 4-(аминометил)-1- [[2-(диэтиламино)этил] амино] -7-метокситиоксантен-9-она в 40 мл хлороформа, содержащего 2 мл триэтиламина, охлажденного до 0oC, прибавляют 0,45 мл метилхлорформиата, и полученную смесь перемешивают при комнатной температуре в течение нескольких часов. Растворитель упаривают в вакууме, остаток очищают флэш-хроматографией (силикагель; элюирование последовательно хлороформом, 1% триэтиламин в смеси хлороформ/гексан (1:1)), получают 1,2 г метил N-[[1-[[2-диэтиламино)этил]амино] -7-метокси-9- оксотиоксантен-4-ил]-метил]-карбамата, и после перекристаллизации полученного продукта из этилацетата получают 0,79 г ярко-желтого твердого вещества, т.пл. 131-132oC.
Пример 37
Получение N-[[1-(2-Диэтиламино)этил] амино] -7- гидрокси-9-оксотиоксантен-4-ил]метил]метансульфонамида 3/4 гидрата
(I: R1=R2=Et; Q=CH2NHSO2CH3; R8=H; n=2).
К раствору, содержащему N-[[1-(2-(диэтиламино)этил] амино]-7-метокси-9-оксотиоксантен-4-ил] метил] метансульфонамида (0,5 г) в CH2Cl2 (45 мл) при температуре -78oС прибавляют 1 н. BBr3 в CH2Cl2 (1,75 мл). Смесь нагревают до комнатной температуры, перемешивают в течение ночи и затем выливают в воду со льдом (250 мл), содержащую 35% NaOH (8 мл). Реакционную смесь затем подкисляют, разбавляют HCl, подщелачивают кристаллами Na2CO3 и затем экстрагируют этилацетатом. Органический слой отделяют, промывают солевым раствором, сушат на Na2SO4 и упаривают в вакууме. Остаток очищают колоночной хроматографией на кремнеземе, вымывая смесью 5% MeOH/EtOAc, с извлечением 0,28 г (58%) N-[[1-(2-диэтиламино)этил] амино]-7-гидрокси-9-оксотиоксантен-4-ил]метансульфонамида 3/4 гидрата, т.пл. 78oC (разл.).
Соединения предлагаемого изобретения, приведенные в примерах, проверяли на противоопухолевую активность на мышах согласно следующей методике:
Животных объединяли в общую группу, вводили подкожно троакаром 12 размера от 30 до 60 мг фрагментов опухоли, и снова объединяли вместе перед случайным распределением их на группы, получавшие различное лечение, и группу контроля. При лечении рака на ранней стадии, химиотерапию начинали проводить с 1 по 5 день после прививки животным опухоли, то есть пока число раковых клеток было относительно небольшим (от 107 до 108 клеток). При лечении животных в запущенной стадии болезни, химиотерапию начинали проводить, пока опухоль не достигнет относительно большого размера (от 200 до 300 мг). В опухоле размером 300 мг присутствует в сумме примерно 3 х 108 раковых клеток. Во время проведения данного испытания при запущенной стадии болезни опухоли у 90% животных достигали 2,5-кратного увеличения. Замеры опухолей осуществляли штангенциркулем еженедельно (или два раза в неделю в случае наиболее быстро прогрессирующих опухолей). Мышей, у которых опухоли достигали размера 1500 мг (то есть прежде, чем они могли вызвать дискомфорт у животных), умерщвляли. Массу опухолей оценивали на основании данных, полученных при измерении двух линейных размеров.
Замеры в группах, получавших лечение и контрольной группе проводили при достижении массы опухоли у животных в контроле примерно от 700 до 1200 мг (средний показатель в группе). В каждой группе определяли среднюю массу опухоли (включая нулевые значения). Показатель T/C (масса опухолей у получавших лечение животных, сопоставимая с массой опухолей животных в Контрольной группе), выраженный в процентах, свидетельствует о наличии противоопухолевой эффективности: этот показатель T/C, равный или > 42%, свидетельствует по оценке Отдела по разработке лекарственных препаратов Отделения раковой терапии Национального онкологического института (NCI) о достоверной противоопухолевой активности. Показатель T/C < 10% считается как доказательство высокодостоверной противоопухолевой активности. Потеря веса тела (усредненное значение по группе) более 20% или гибель от препарата животных более 20% считается как свидетельство чрезмерно токсичной дозировки.
В таблице 1 приведены результаты противоопухолевой активности испытуемых соединений относительно аденокарциномы поджелудочной железы *03, а в таблице 2 - против аденокарциномы толстой кишки *38.
Соединение примера 5 испытывали на активность против множества различных опухолей при внутривенном его введении по схеме, приведенной в таблице 3, и при пероральном введении в дозе 300 мг/кг оно проявляло активность против аденокарциномы толстой кишки *38.
Соединение примера 6 испытывали на активность против множества различных опухолей при болюсном внутривенном его введении по схеме, приведенной в таблице 4.
Соединение примера 8 (а) испытывали на активность против множества различных видов опухолей в соответствии со схемой приема, приведенной в таблице 5.
Соединение примера 36 испытывали на активность против множества различных видов опухолей в соответствии со схемой приема, приведенной в таблице 6.
Типичные составы предлагаемого изобретения испытывали на активность против аденокарциномы молочной железы 16/C/RP, в соответствии со схемой приема в таблице 7.
Типичные составы предлагаемого изобретения испытывали на активность против Р388/адриамицин-устойчивого лейкоза в соответствии со схемой приема в таблице 8.
Во время проведения вышеуказанного испытания, исследователи столкнулись с трудностями, связанными с деградацией растворов для инъекции, которые не были свежеприготовленными перед их применением. Таким образом, усилия были направлены на создание растворов для инъекций, не разлагающихся при хранении, в результате чего стало бы возможным использование соединений, проявляющих противоопухолевую активность для лечения онкологических больных.
В указанных экспериментальных исследованиях использовали соединение, полученное в примере 6, имеющее следующую структурную формулу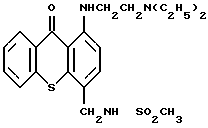
и химическое название N-[[1-[[2-(диэтиламино)этил] амино]-9- оксотиоксантен-4-ил] метил] -метансульфонамид, обладающий цитотоксической противоопухолевой активностью. Это соединение для простоты иногда упоминается в описании изобретения как WIN 33377. Активность этого соединения оценивали в клинических исследованиях с использованием лекарственного препарата в форме полностью стабилизированного раствора в ампулах при концентрации его 2,5 мг/мл в цитратном буфере (pH 5,5). Для обеспечения необходимого срока хранения, препарат держали в холодильнике (2-8oC). Хранение при более высоких температурах приводило к образованию очень слабо растворимых димеров, которые выпадали в осадок при низкой концентрации. Таким образом, существует потребность в получении лиофилизированного состава с целью создания коммерчески пригодного лекарственного препарата, который можно было бы хранить при температуре окружающей среды. С этой целью использовали три состава, приведенные в таблице 9.
Водные растворы, содержащие соединение WIN 33377, приготавливали до введения стабилизаторов.
К приготовленным растворам прибавляли при различных концентрациях три стабилизатора: маннит (фирмы Fison AR марки М/2405), декстран (фирмы Sigma Chemical Co., медицинский D-4751) и сахароза (фирмы Prolabo Normapur AR марки 27480,294). Использовали дифференциальный сканирующий калориметр фирмы Perkin-Elmer, оснащенный вспомогательной измерительной аппаратурой для работы в режиме низких температур. Данные собирали и анализировали на микрокомпьютере фирмы Dell 210 с программным обеспечением DARES. Температурную калибровку выполняли в соответствии с инструкциями изготовителя, используя в качестве референс-материалов индий и воду. Жидкие образцы для анализа хранили в больших чашках из нержавеющей стали с герметичной крышкой. Образцы загружали в калориметр при температуре 27oC и подвергали циклу охлаждение-нагревание в интервале от -53oC до 27oC. Скорость нагрева-охлаждения составляла 5oС/мин. Лиофилизированные образцы также хранили в больших чашках из нержавеющей стали с герметичной крышкой, но в сухой атмосфере азота для снижения возможности поглощения влаги лиофилизированной лепешкой. Скорость нагрева-охлаждения составляла 10oС/мин для усиления амплитуды любого сигнала по всему диапазону температур от -53oC до 127oC. Лиофильную сушку выполняли в лабораторном сублиматоре.
Оценку стабильности лиофилизированных образцов проводили методом высокоэффективной жидкостной хроматографии (ВЭЖХ), которая обеспечивала количественный анализ WIN 33377 или определение общего содержания хроматографических примесей.
Метод анализа
При использовании оборудования для ВЭЖХ (фирмы Kontron), хроматографию проводили при нижеследующих условиях:
Колонка: Partisil ODS - 3,5 мкмоль, 10 х 0,46 см;
Подвижная фаза: A: B (77:23 об./об.), где A = 0,5 М аммоний-ацетатный буферный раствор, pH 4,8 и B=ацетонитрил;
Скорость потока: 2,0 мл/мин;
Длина волны детектора 258 нм;
Температура: 40oC;
Впрыскиваемый объем 20 мкл.
Метод определения общего содержания хроматографических примесей
Для определения общего содержания хроматографических примесей использовали метод ВЭЖХ с градиентным элюированием, выполняемый при нижеследующих условиях:
Колонка:
Hypersil BDS, C18,5 мкмоль, 25 х 0,44 см (внутр. диаметр);
Подвижная фаза: A:7,71 г/л ацетата аммония + 6,0 мл/л ледяной уксусной кислоты + 10 мл/л триэтиламина с pH, доведенной до 4,8, B: ацетонитрил.
Условия градиентного элюирования представлены в табл. A.
Скорость потока: 2,0 мл/мин;
Длина волны детектора 438 нм;
Температура: 40oC;
Впрыскиваемый объем 20 мкл.
Создание препарата
Первый этап в любой разработке способа лиофильной сушки должен включать определение всех физико-химических свойств растворов до получения состава для лиофилизации. Образцы из трех приготовленных составов, приведенные в таблице 9, анализировали методом дифференциальной сканирующей калориметрии по методике, описанной выше. Релевантные данные по определению температур перехода сведены в таблице 10.
Было установлено, что эти три смеси проявляют разные физико-химические характеристики: состав на основе ацетатного буфера может быть в кристаллической форме, в цитрат содержащем составе одна его часть, как обнаружено, находится в кристаллическом состоянии, в то время как другая его часть образует стекловидную форму; и лактат содержащий состав образует стеклообразную форму.
Из этих трех типов физико-химического поведения, частичная кристаллизация создает самые большие проблемы для сублимации. Процесс частичной кристаллизации обычно нельзя предугадать, и, в случае фармацевтических препаратов, производимых в ампулах, это может привести к существенным различиям в структуре лиофилизата в ампулах. Это, в свою очередь, может привести к изменениям показателя эффективности лиофильной сушки в ампулах. Общий КПД представляет собой изменение качества продукта в ампулах, например как стабильность, восстановление влагосодержания и срок хранения в ампулах. Изменения влагосодержания препарата в ампулах самое прямое свидетельство этой проблемы.
Частичную кристаллизацию, продемонстрированную на примере цитратного состава, можно предотвратить путем добавления к смеси соответствующих стеклообразующих наполнителей. Такой наполнитель препятствует кристаллизации другого вещества (веществ) и обеспечивает переход в стекловидное состояние. Количество стеклообразующего наполнителя зависит от природы кристаллизующегося вещества и скорости кристаллизации. В данном исследовании цитратный состав сочли непригодным, главным образом, из-за того, что в цитратном буфере, лекарственное вещество, хотя и растворяется во время приготовления исходного препарата, затем начинает кристаллизоваться при хранении при температуре 4oС или 25oC в течение нескольких часов. Поскольку процесс лиофилизации не оказывает неблагоприятного влияния на растворимость лекарственного вещества, то при регидратации цитратного состава в момент его применения может возникнуть некоторая трудность в его растворении и помимо этого даже может иметь место единичная кристаллизация во время введения лекарственного препарата.
Суммарное содержание растворенных веществ во всех трех составах было низким, примерно от 1 до 2% мас./мас. Такое содержание недостаточно для обеспечения адекватной структуры "пробки" и поэтому потребовалась некоторая модификация рецептуры. В кристаллический состав 1 необходимо было добавить наполнители. В состав 3 (аморфный), с другой стороны, необходимо было ввести стеклообразующую добавку. В качестве стабилизаторов были выбраны маннит для кристаллической формы препарата и сахароза или декстран для аморфной модификации. Эти стабилизаторы вводили в растворы, приведенные в таблице 9, при концентрациях 50 мг/мл. Пробы приготовленных растворов анализировали методом дифференциальной сканирующей калориметрии (ДСК).
В результате измерений, выполненных методом ДСК, получили значения температуры стеклования для трех препаратов. Эти температуры означают максимально допустимые температуры первичной сушки при условии того, что деградация и ухудшение качества препарата сведены к минимуму. Декстран содержащая система имеет самые высокие температуры стеклования и обычно обеспечивает самый короткий цикл сушки, поскольку скорость сублимации льда растет экспоненциально с ростом температуры. Однако, соединение WIN 33377 требует многократного введения, и декстран при некоторых обстоятельствах может вызвать анафилактический шок. Поэтому этот препарат был отвергнут по клиническим соображениям.
Следующие выводы были сделаны относительно двух других составов.
Для состава на основе сахарозы и лактатный буфер, первичную сублимацию следует проводить при температуре примерно -40oC. Это обеспечивает запас надежности порядка 5oC (достаточный для фактически используемой сушилки), чтобы компенсировать температурные градиенты в рабочей зоне сушилки. Каждый флакон содержал по 10 мл препарата с наполнением его до уровня глубиной 1,63 см. Флакон диаметром 2,8 см обеспечивал площадь препарата, равную 6,15 см.
Скорость эффективного процесса сублимации для этих образцов рассчитывали как величину 0,226 г/ампула/час от общей массы продукта, при этом примерно 9,4 г состояло из льда, а оставшуюся часть составляло твердое вещество и не замерзшая вода.
При скорости 0,226 г/час необходимо примерно 42 часа для полной сублимации льда при температуре -40oC.
При использовании состава на основе смеси маннита и ацетата, процесс первичной сублимации необходимо было проводить при самых низких термических переходах, то есть, -30oC, как показали результаты измерений ДСК для ацетат содержащих составов. Заполняемые объемы и размеры флаконов были такие же, но сублимацию можно было проводить в этом случае при более высокой температуре; при этом расчетная эффективная скорость сублимации для этих образцов составляла 0,670 г/ампула/час. Как и в предыдущем случае, пришлось подвергнуть сублимации 9,4 г льда; время, необходимое для процесса первичной сублимации этой смеси, сократилось до 14 часов.
Поддержание продуктов в рекомендуемом интервале температур в течение вышеприведенных промежутков времени, при условии, что температуры при фронте сублимации равны температурам в центре образования зародышей льда, обеспечивает полный цикл первичной сублимационной сушки. Температуру продукта затем следует повышать постепенным линейным изменением температуры хранения для того, чтобы удалить остаточную влагу из продукта.
Приготовление лиофилизированных препаратов
В таблице 11 приведены переменные процесса необязательной лиофилизации растворов, содержащих ацетатный и лактатный буферы, которые стабилизированы, соответственно, маннитом и сахарозой.
Лиофилизацию составов настоящего изобретения проводят в соответствии со следующим способом.
Флаконы требуемых размеров для наполнения их составами настоящего изобретения подбирают в соответствии с требованиями, предъявляемыми к дозированию. При подборе флаконов для размещения необходимой дозы лекарственного препарата, объем наполнения не должен превышать высоту свободного пространства в емкости флакона. Например, не следует наполнять флакон емкостью менее 10 мл препаратом объемом по 5 мл. После наполнения препаратом, флаконы загружают в сублимационную камеру и помещают на стеллажи с морозильником, которые предварительно были охлаждены до 4oC. Термопары помещают внутрь партии флаконов для регулирования температуры состава в течение процесса лиофилизации. Флаконы после этого приводят в равновесное состояние с температурой, поддерживаемой на стеллажах (4oC), прежде чем снизить ее до -40oC в случае состава, содержащего сахарозу, и -30oC для состава на основе маннита. Сразу же как только температура состава на основе сахарозы и состава, содержащего маннит, снизится до -40oC и -30oC, соответственно, флаконы выдерживают в этих температурах еще 2 часа для обеспечения полного замораживания каждого состава (на этом этапе для маннит содержащего состава предусматривается операция сшивки). После завершения этого периода времени змеевик конденсатора охлаждают до температуры -6oC, и включают вакуумный насос для создания вакуума в конденсаторной камере с одновременным проведением процесса первичной и вторичной сублимации. Во время протекания процесса первичной лиофилизации открывают главный клапан между конденсатором и сушильной камерой, и из сушильной камеры сбрасывают давление примерно до 100 мкм с удалением газа азота. Эта часть цикла лиофилизации (первичной сушки) занимает примерно от 40 до 50 часов. Процесс первичной сушки протекает до тех пор, пока весь лед не исчезнет из замороженной биоматрицы. Во время протекания процесса вторичной лиофилизации температуру повышают в интервале от -20oC или -30oC до +25oC для удаления всей оставшейся влаги, которая не была удалена во время первичной лиофилизации. Продолжительность протекания процесса вторичной лиофилизации составляет приблизительно 15 часов. После завершения процесса вторичной лиофилизации главный клапан закрывают и сушильную камеру наполняют азотом так, чтобы в сушильной камере поддерживалось немного повышенное давление. После этого приводят в действие толкатель для закупорки пробок во флаконы. Сушильную камеру после этого приводят в равновесное состояние с атмосферным давлением, дверцы камеры открывают для извлечения из нее флаконов, которые укупоривают обжимом. Флаконы хранят затем при рекомендуемой температуре до их применения при восстановлении влагосодержания лиофилизированного продукта дистиллированной водой для инъекции.
Результаты проверки лиофилизированного продукта на стабильность
Стабильность высушенных продуктов оценивали после хранения их при температуре 30oC, 40oC и 50oC в течение до 4 недель. Исследования завершали с проведением количественного анализа взятых образцов и определения в них следов хроматографических примесей. Результаты количественного анализа состава, содержащего маннитол, свидетельствовали о том, что никаких существенных изменений в лекарственном образце не произошло после хранения продукта в течение 4 недель при температуре вплоть до 50oC. Общее содержание хроматографических примесей возросло от первоначального уровня от 0,35 до 0,54 % мас./мас. при температуре 30oC; 0,49% мас./мас. при температуре 40oC и 0,56% мас. /мас. при температуре 50oC. Влагосодержание лиофилизированного продукта составляло 2,4 %, мас./мас., и внешний вид лепешки (вакуум-осадка) был удовлетворителен, то есть, не отмечено никакой депрессии продукта. В двух из пяти флаконов, однако, не произошло регидратации продукта до получения прозрачного желтого раствора. Флаконы с лиофилизированным продуктом, влагосодержание которого можно было восстановить до получением прозрачного раствора, имели более низкий pH (5,0), чем флаконы, которые не давали прозрачные растворы (pH 5,6). Это смещение pH, как полагают, обусловлено испарением уксусной кислоты в процессе лиофилизации раствора.
Результаты анализа на стабильность состава, содержащего сахарозу (с остаточной влажностью 5%), свидетельствуют о том, что не произошло никаких существенных изменений в аналитическом образце или суммарного количества хроматографических загрязнений после хранения его при температуре 30oC и 40oC в течение 4 недель. Флаконы, которые хранили при температуре 50oC, показали снижение в аналитическом образце и увеличение суммарного содержания хроматографических примесей. Некоторую депрессию лиофилизированной лепешки наблюдали через две недели хранения при всем испытуемом диапазоне температур.
Результаты анализа на стабильность состава, содержащего WIN 33377 и маннит, приведены в таблице 12, а в таблице 13 приведены результаты анализа на стабильность состава, содержащего WIN 33377 и сахарозу.
В результате исследования был найден подходящий состав для лиофильной сушки (смесь сахароза и лактата), но его можно было далее оптимизировать, и поэтому были введены дополнительные требования для практической его реализации. Было установлено, что концентрацию лекарственного вещества в исходном растворе можно довести до 20 мг/мл, что позволило бы уменьшить наполовину объемы наполнения и сократить продолжительность процесса первичной лиофилизации. Было также важно сбалансировать изотоничность раствора путем прибавления хлорида натрия. Поскольку эта соль не оказывает никакого влияния на температуру стеклования (T'ст), скорректированные составы (таблица 14) анализировали с помощью дифференциального сканирующего калориметра. Результаты проведенных замеров температуры стеклования приведены в таблице 15.
Из данных, приведенных в таблице, видно, что введение NaCl снижает температуру стеклования в концентрате, полученном лиофилизацией. NaCl имеет температуру стеклования, которая намного ниже, чем у сахарозы (-87oC по сравнению с -32oC). Таким образом, по мере увеличения массовой доли NaCl (от A до D), температура стеклования снижается до значения чистой NaCl. Прибавление NaCl к составу стандартного 5% раствора сахарозы приводит к снижению температуры стеклования на 2oC. Снижение содержания сахарозы и компенсаторное увеличение концентрации NaCl, которая необходима для поддержания изотоничности, приводит к дальнейшему снижению температуры стеклования. Температура стеклования высушенного продукта можно рассматривать как максимальную температуру, воздействие которой должен выдержать продукт. Повышенная температура стеклования приводит к возможной депрессии пробки, превращая продукт в жидкость, где скорости диффузии быстро растут, приводя к деградации активного вещества. Температура стеклования зависит от влагосодержания; таким образом любая дисперсия значений влагосодержания в пределах партии продукта вызовет дисперсию значений температуры стеклования. Таким образом, целесообразно хранить продукт по меньшей мере при температуре на 5oC ниже измеренной температуры стеклования, что позволило бы корректировать температуру для обеспечения максимально безопасного хранения в пределах партии продукта.
Состав, содержащий раствор WIN 33 77 (100 мг) и сахарозу, во флаконе емкостью 20 мл
Наполнитель - Количество (мг на флакон)
WIN 33377 - 100,0
Сахароза - 250,0
Натрия хлорид - 5,0
0,5 М раствор молочной кислоты - 1,0 мл
1,0 М гидроокись натрия - 57,0 мкл
Вода для инъекций до - 5,0 мл
pH (от 3,70 до 4,30) - 4,00
Стабильность предпочтительного состава, содержащего сахарозу, оценивали на основе количественного анализа при хранении в течение промежутка времени до 6 месяцев при температуре 30oC и 40oC. Результаты анализа и показатели pH приведены в таблице 16. Продукт имел исходное влагосодержание 1,3% (мас. /мас.). Депрессию продукта наблюдали через 2 недели хранения при температуре 40oC. Эту депрессию ожидали, поскольку указанная температура хранения была близка к температуре стеклования для этой партии (42oC). Результаты испытания на стабильность свидетельствуют о том, что полученный продукт химически устойчив, то есть не отмечено никаких изменений в аналитических образцах или показателей pH через 6 месяцев его хранения при температуре 30oC или 40oC.
Повышенная стабильность, обеспечиваемая настоящим изобретением, позволяет хранить продукт при комнатной температуре и увеличивает срок его годности. Этот лиофилизированный продукт пригоден для упаковки как в традиционные стеклянные флаконы, так и в предварительном наполненном шприце. Составы настоящего изобретения проявляют высокую терапевтическую эффективность (имеют большую полезность) при лечении новообразований, которую не обеспечивали до настоящего времени.
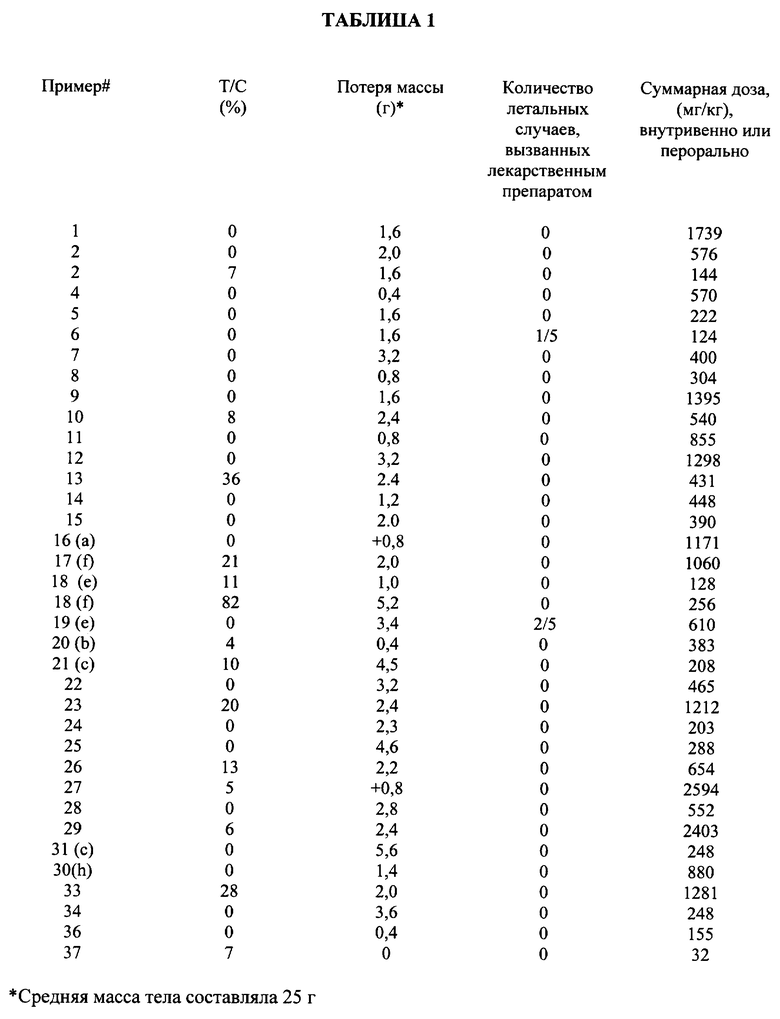

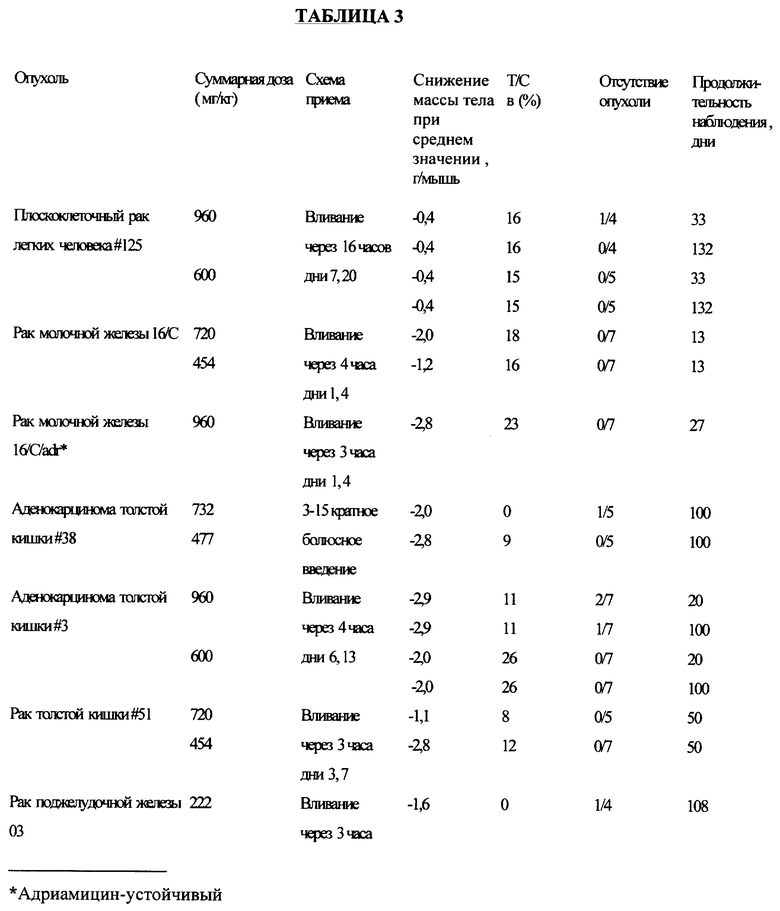
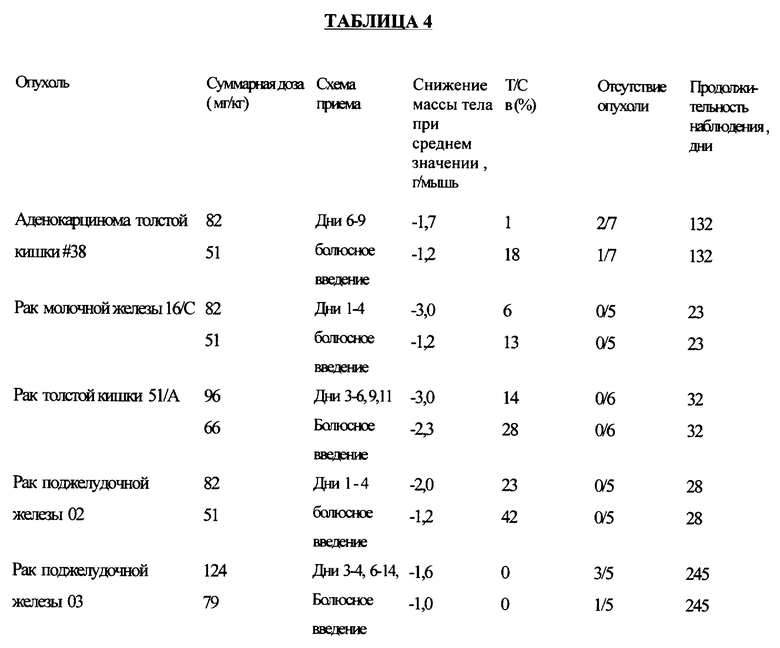
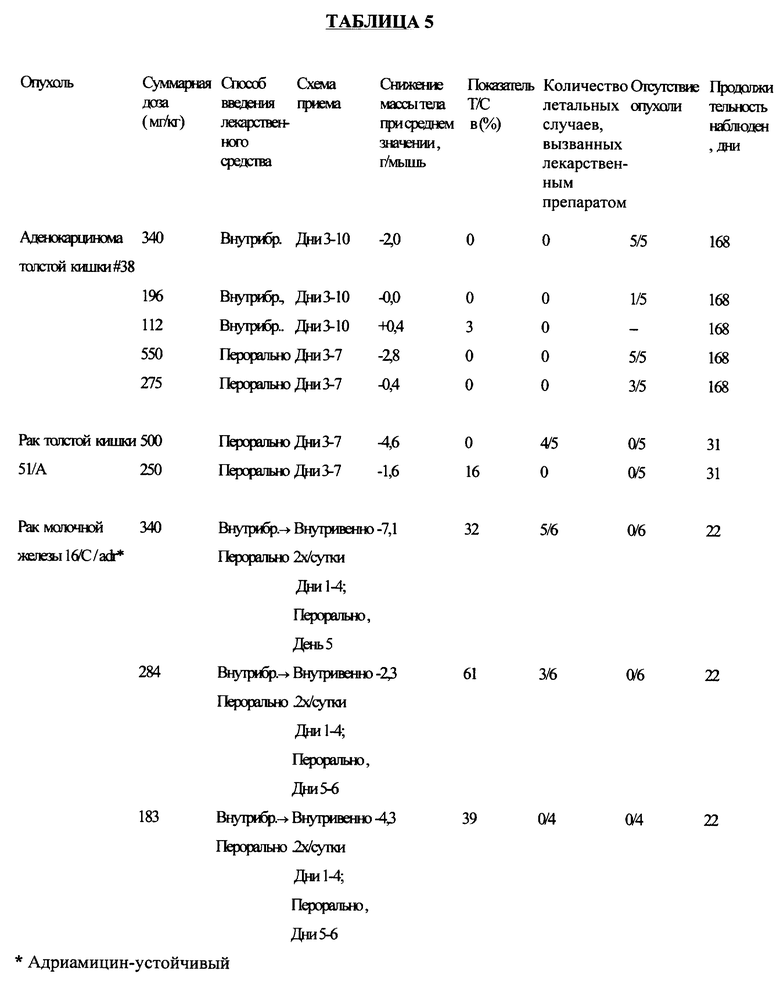
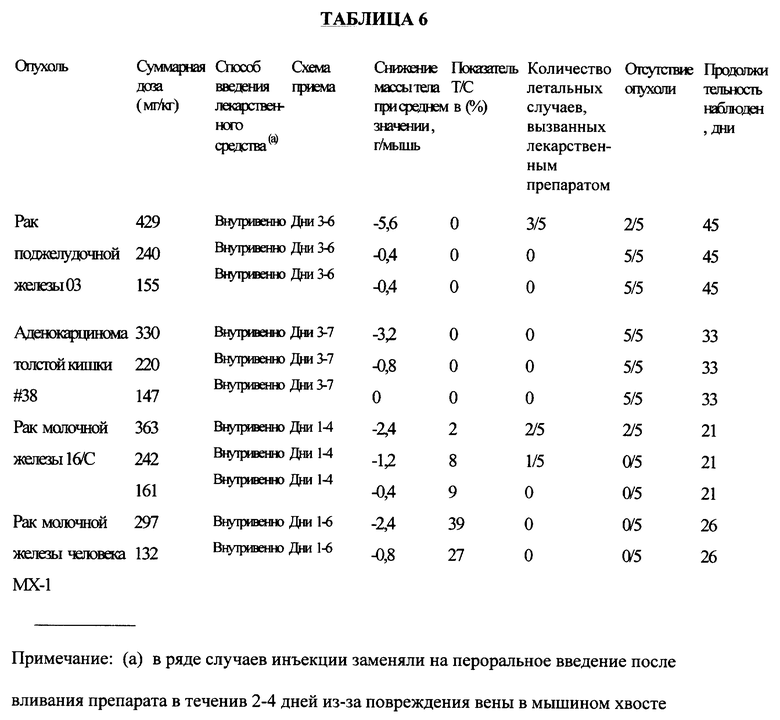

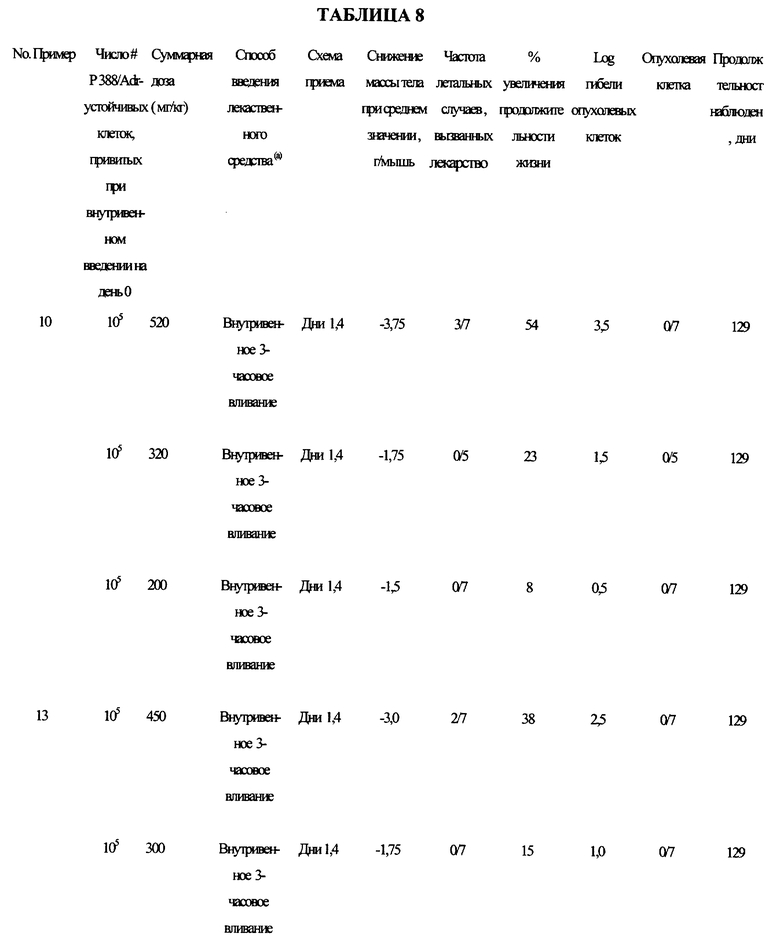
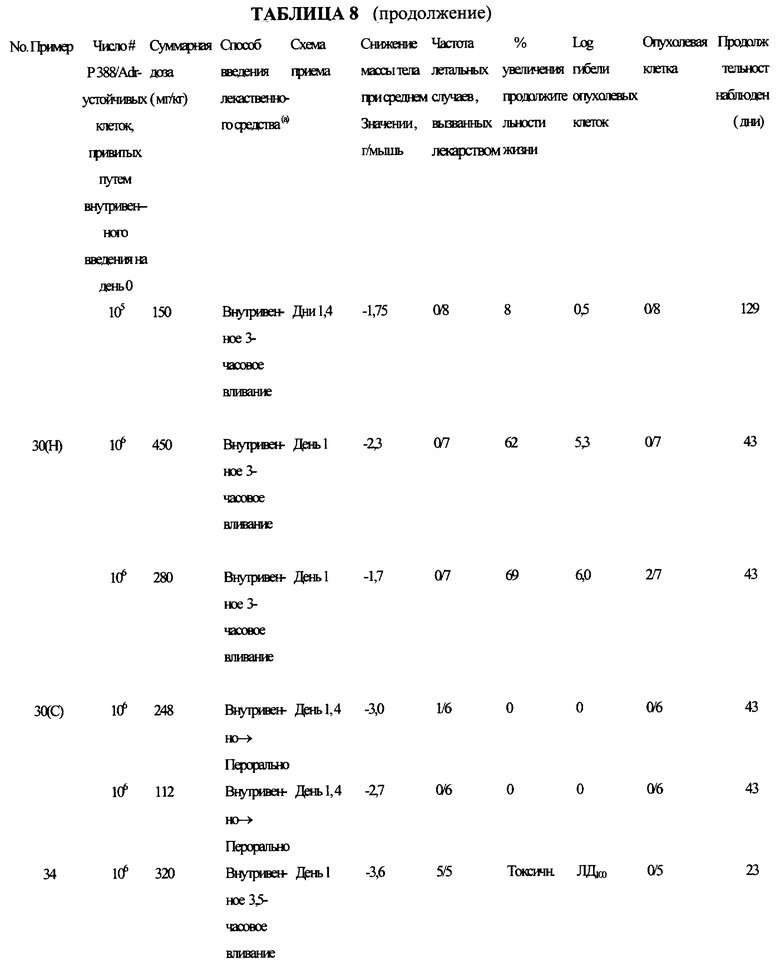


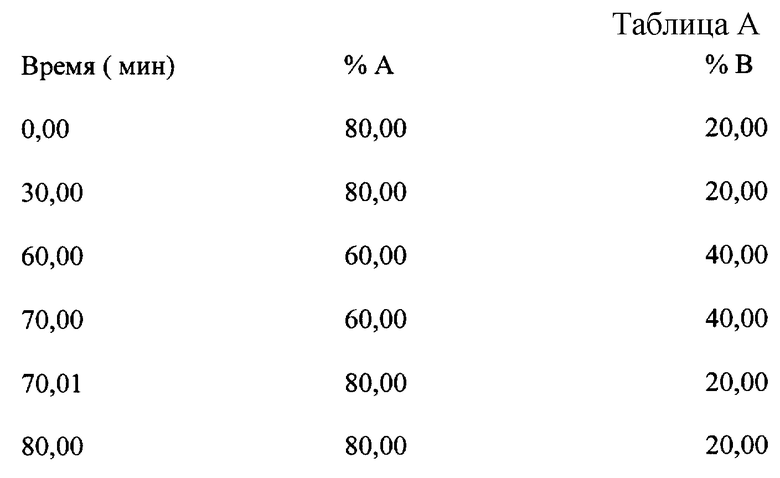





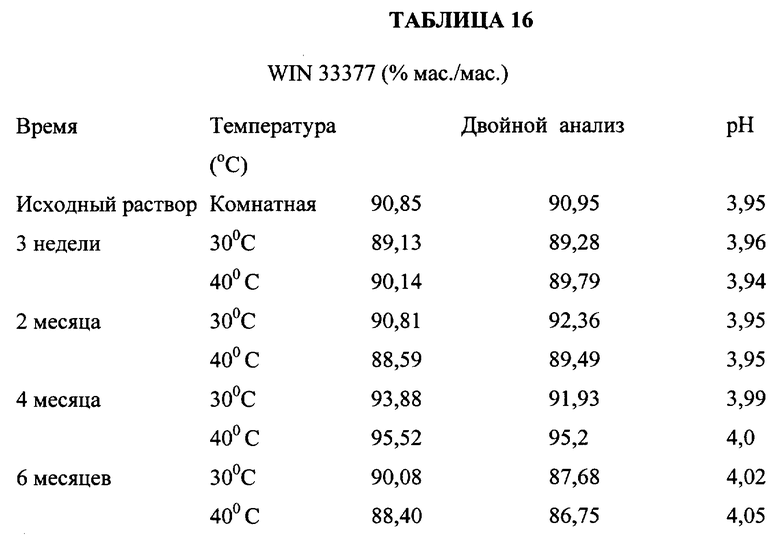
Изобретение относится к области медицины и касается лиофилизированного состава для лечения опухолей у млекопитающих, восстановленного по влагосодержанию, содержащего в качестве противоопухолевого действующего вещества тиоксантенон в сочетании с маннитом или сахарозой в качестве стабилизатора в лактатном буфере. Состав обладает повышенной стабильностью. 8 с. и 4 з.п. ф-лы, 16 табл.
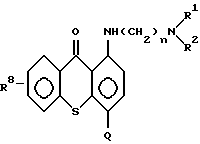
где n означает 2 или 3;
R1 и R2 каждый независимо означает низший алкил;
Q означает углеводородный остаток, выбранный из группы, состоящей из CH2NHR3, CH2N(R4)SO2R7, CH2NHCHO, CH= N-Ar, C(O)NR5R6, CH2N(R4)C(O)R7, CH2N(C2H5)CHO, CH2N(R4)P(O)(O-низший алкил)2, CH2N=CH-N(R9)(R10), CH2N(R4)C(O)CF3 и CH2N(R4)C(O)OR7;
R3 означает водород или низший алкил;
R4 означает водород, низший алкил или Аг;
R5 означает водород, низший алкил или Аг;
R6 означает водород, низший алкил;
R7 означает низший алкил или Аг;
R означает водород, низший алкил, низший алкокси или гидроксил;
Аг означает фенил или фенил, замещенный метилом, метоксилом, гидроксилом, галогеном или нитрогруппой, при условии, что когда n равно 2, то R1 и R2 означают этил;
R8 означает водород, а Q означает CH2NHSO2Ar, при этом группа Аг может быть 4-монозамещенной метилом или галогеном;
R9 и R10 каждый независимо означает низший алкил,
или его фармацевтически приемлемая соль присоединения кислоты или сольват, b) от 10 мг/мл до 125 мг/мл стабилизатора, выбранного из группы, состоящей из маннита и сахарозы, и с) от 0,025 до 0,25 М лактатного буфера, причем указанный состав имеет pH от 3,0 до 4,5.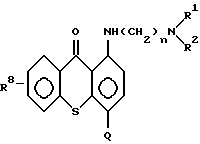 (II)
(II)
где n означает 2 или 3;
R1 и R2 каждый независимо означает низший алкил;
Q означает углеводородный остаток, выбранный из группы, состоящей из CH2NHR3, CH2NHCHO, CH= N-Ar, C(O)NR5R6, CH2N(R4)C(O)R7, CH2N(C2H5)CHO,
CH2N(R4)P(O)(O-низший алкил)2, CH2N= CH-N(R9)(R10), CH2N(R4)C(O)CF3 и CH2N(R4)C(O)OR7;
R3 означает водород или низший алкил;
R4 означает водород, низший алкил или Аг;
R5 означает водород, низший алкил или Аг;
R6 означает водород, низший алкил;
R7 означает низший алкил или Аг;
R8 означает водород, низший алкил, низший алкокси или гидроксил;
Аг означает фенил или фенил, замещенный метилом, метоксилом, гидроксилом, галогеном или нитрогруппой;
R9 и R10 каждый независимо означает низший алкил,
или их фармацевтически приемлемые соли присоединения кислот или сольват, (b) от 30 мг/мл до 100 мг/мл стабилизатора, выбранного из группы, состоящей из маннита и сахарозы, и с) от 0,025 до 0,25 М лактатного буфера, причем указанный состав имеет pH от 3,0 до 4,5.

