Изобретение относится к новым гетероциклическим замещенным феноксиалкилтиадиазолам, способам их получения и применения в качестве противопикорнавирусного средства.
Краткое описание изобретения
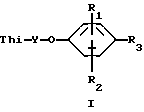
Было установлено, что соединения формулы 1 являются эффективным противопикорнавирусным средством. Соответственно данное изобретение относится к соединениям формулы: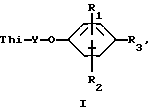
где:
Thi - представляет собой тиадиазолил или замещенный тиадиазолил, содержащий в качестве заместителя алкоксигруппу, фторметил, дифторметил, трифторметил, 1,1-дифторэтил, галоген, алкил, циклоалкил, гидроксиалкил или алкоксиалкил;
Y - алкиленовый мостик, содержащий 3-9 атомов углерода;
R1 и R2 каждый независимо выбирают из группы, включающей водород, галоген, алкил, алкенил, амино-, алкилтио-, гидроксильную группу, гидроксиалкил, алкоксиалкил, алкилтиоалкил, алкилсульфинилалкил, алкилсульфонилалкил, алкокси-, нитрогруппу, карбокси, алкоксикарбонил, диалкиламиноалкил, алкиламиноалкил, аминоалкил, дифторметил, трифторметил или цианогруппу;
R3 представляет собой алкоксикарбонил, фенил, алкилтетразолил или гетероциклический радикал, который выбирают из группы, включающей бензоксазолил, бензотиазолил, тиадиазолил, имидазолил, дигидроимидазолил, оксазолил, тиазолил, оксадиазолил, пиразолил, изоксазолил, изотиазолил, фурил, триазолил, тиенил, пиридил, пиримидинил, пиразинил, пиридазинил, или замещенный фенил или замещенный гетероцикличесий радикал, содержащий в качестве заместителя алкил, алкоксиалкил, циклоалкил, галогеналкил, гидроксиалкил, алкоксигруппу, гидроксильную группу, фурил, тиенил и фторалкил;
или их фармацевтически приемлемым кислотно-аддитивным солям.
Изобретение относится также к композициям для борьбы с пикорнавирусами, включающим соединение формулы 1 в количестве, обеспечивающем эффективное антипикорнавирусное действие, и подходящий носитель или наполнитель, и к способам борьбы с пикорнавирусами при помощи этих композиций, включая системное лечение пикорнавирусных инфекций в организме-хозяине млекопитающего.
Подробное описание предпочтительных воплощений
Соединения формулы 1 полезны для применения в качестве противопикорнавирусного средства и далее описываются подробно.
Термины "алкил" и "алкокси" означают алифатические радикалы, в том числе разветвленные, содержащие от одного до пяти атомов углерода. Таким образом, алкильные фрагменты таких радикалов представляют собой, например, метил, этил, пропил, изопропил, н-бутил, втор-бутил, трет-бутил, пентил и т. п.
Термин "циклоалкил" означает алициклический радикал, содержащий от трех до семи атомов углерода, например, циклопропил, циклобутил, циклопентил, циклогексил.
Термин "галоген" означает бром, хлор, йод или фтор.
Термин "гетероциклический радикал" или Het относится к гетероциклическому радикалу, содержащему 5 или 6 атомов углерода и от одного до четырех атомов азота и/или один атом кислорода или серы, при условии, что в гетероцикле нет двух соседних атомов кислорода и/или серы. Примерами гетероциклических радикалов являются фурил, оксазолил, изоксазолил, пиразил, имидазолил, тиазолил, тетразолил, тиенил, пиридил, оксадиазолил, тиадиазолил, триазинил, пиримидинил и т.п. Термин "гетероцикл" относится к соответствующим соединениям.
Термин "гетероциклический радикал" включает все известные изомерные радикалы описываемого гетероцикла, если не указано другого, например, термин "тиадиазолил", включает 1,3,4-тиадиазол-2-ил, 1,2,4-тиадиазол-5-ил и 1,2,4-тиадиазол-3-ил, термин "тиазолил" включает 2-тиазолил, 4-тиазолил и 5-тиазолил и другие известные видоизменения известных гетероциклических радикалов. Таким образом, подразумевают любой изомер указанного гетероциклического радикала. Эти гетероциклические радикалы могут присоединяться через любой доступный атом азота или углерода, например, термин "тетразолил" включает 5-тетразолил или тетразолил, присоединенный через любой доступный атом азота тетразолинового цикла, термин "фурил" включает фурил, присоединенный через любой доступный атом углерода и т.д. Получение таких изомеров достаточно хорошо описано и известно квалифицированному фармацевту или специалисту в области органической химии.
Некоторые гетероциклы могут существовать в виде таутомеров, и указанные соединения, несмотря на то что не перечислены все и каждая таутомерная форма, включают все таутомерные формы. Например, пиридинон и его таутомер гидроксипиридин рассматриваются как один и тот же фрагмент. Ввиду того, что гетероциклические фрагменты соединений данного изобретения могут содержать в качестве заместителей гидроксильные группы, следует учитывать, что под гидрокси-замещенными гетероциклами подразумеваются все соответствующие таутомеры.
При использовании терминов "гидроксилалкил" и "алкоксиалкил" следует учитывать, что гидроксильная и алкокси- группы могут находиться в любом возможном положении алкила. Таким образом, термины "гидроксиалкил" и "алкоксиалкил" включают, например, гидроксиметил, 1-гидроксиэтил, 2-гидроксиэтил, 2-гидроксипропил, 2-гидроксиизопропил, 2-, 3-, 4- и 5-гидроксипентил и т.п., термин "алкоксигруппа" относится к соответствующим группам алкиловых эфиров.
При использовании термина "гидроксиалкокси" следует учитывать, что гидроксильная группа может находиться в любом возможном положении алкоксигруппы, кроме положения C-1 (геминальное положение). Таким образом, термин "гидроксиалкокси" включает, например, 2-гидроксиэтокси-, 2-гидроксипропокси-, 2-гидроксиизопропокси-, 5-гидроксипентокси- группу и т.п.
Термин "алкилен" относится к линейному или разветвленному двухвалентному углеводородному радикалу, содержащему от 1 до примерно 5 атомов углерода, такому как метилен, 1,2-этилен, 1,3-пропилен, 1,4-бутилен, 1,5-пентилен, 1,4-(2-метил) бутилен и т.п. Алкилен может также содержать алкенильные или алкинильные связи.
Термин "галоген" относится к галогенам: фтору, хлору, брому и йоду.
Термин "галогеналкил" в данном описании относится к галоген-замещенному алкилу, такому как фторалкил, хлорфторалкил, бромхлоралкил, бромфторалкил, бромалкил, йодалкил, хлоралкил, и другим подобным фрагментам, в которых галогеналкил содержит один или большее количество одинаковых или разных атомов галогенов, заместивших водород. Например, термин "галогеналкил" включает хлордифторметил, 1-хлорэтил, 2,2,2-трихлорэтил, 1,1-дихлорэтил, 2-хлор-1,1,1,2-тетрафторэтил, бромэтил и т.п.
Термин "фторалкил" в данном описании означает предпочтительный подкласс галогеналкилов и относится к фторированному и перфторированному алкилу, такому как например, фторметил, дифторметил, трифторметил, 2,2,2-трифторэтил, 1,2- дифторэтил, 1,1,2,3-тетрафторбутил и т.п.
Соединения формулы 1, где R3 представляет собой азотсодержащий гетероцикл, обладают достаточной основностью для образования кислотно-аддитивных солей и полезны как в форме свободного основания, так и в форме кислотно-аддитивных солей, причем обе формы составляют область данного изобретения. Кислотно-аддитивные соли в некоторых случаях являются формой, более удобной для применения, и на практике применение солевой формы соответственно равно применению основной формы. Кислоты, которые могут использоваться для получения кислотно-аддитивных солей, предпочтительно включают такие кислоты, которые при соединении со свободным основанием дают фармацевтически приемлемые соли, то есть соли, анионы которых относительно безвредны для организма животного в фармацевтических дозах, таким образом полезные свойства, присущие свободному основанию, не утрачиваются в результате побочных действий, приписываемых анионам. Примеры подходящих кислотно-аддитивных солей включают гидрохлорид, гидробромид, сульфат, кислый сульфат, малеат, цитрат, тартрат, метансульфонат, п-толуолсульфонат, дидецилсульфат, циклогексансульфамат и т. п. Однако в область данного изобретения включаются и другие подходящие фармацевтически приемлемые соли, производные других минеральных и органических кислот. Кислотно- аддитивные соли основных соединений могут быть получены посредством растворения свободного основания в водно-спиртовом растворе, содержащем соответствующую кислоту, с последующим выделением соли посредством упаривания раствора, или в результате взаимодействия свободного основания и кислоты в органическом растворителе, в этом случае соль выделяется непосредственно в процессе реакции или ее осаждают вторым органическим растворителем или выделяют в результате концентрирования раствора или любым другим из хорошо известных способов. Несмотря на то, что фармацевтически приемлемые соли основных соединений являются предпочтительными, область данного изобретения включает все кислотно-аддитивные соли. Все кислотно-аддитивные соли полезны в качестве исходного вещества для получения свободного основания, даже если конкретная соль необходимая только в качестве промежуточного продукта, как например, в том случае, когда соль получают только с целью очистки или идентификации либо когда она используется в качестве промежуточного продукта при получении фармацевтически приемлемой соли посредством ионного обмена.
Структуры соединений данного изобретения были установлены методом синтеза, элементным анализом и с помощью ИК-, УФ-, ЯМР-спектров и масс-спектроскопии. Контроль протекания реакций, идентификацию продуктов и оценку их гомогенности осуществляли методом тонкослойной хроматографии (ТСХ), газожидкостной хроматографии (ГЖХ) или другими способами контроля химического взаимодействия органических веществ.
В качестве нереагирующего (инертного) растворителя в данном изобретении может использоваться N-метилпирролидон (NMP), метиленхлорид (CH2Cl2), тетрагидрофуран (ТГФ), бензол или любой другой растворитель, который не будет принимать участие в реакции. В предпочтительном способе реакции получения соединений данного изобретения проводят в сухих растворителях под инертной атмосферой. Некоторые реагенты, используемые в примере, обозначаются аббревиатурами: трифенилфосфин (TPP), триэтиламин (TEA), диизопропилэтиламин (D1PEA), диэтиловый эфир азодикарбоновой кислоты (DEAD). Эфир представляет собой диэтиловый эфир, если не указано другого значения.
Соединения формулы 1 можно получить несколькими различными способами.
Соединения формулы 1 могут быть получены реакцией подходящего гидрокси-Y-тиадиазола и подходящего R1-R2-R3-фенола, как описывается в патенте США 5242924, включенном в данную заявку в качестве ссылки.
Соединения формулы 1 могут быть получены взаимодействием подходящего R1-R2-R3-фенола и подходящего галоген-Y-тиадиазола в соответствии с реакцией, описанной в патенте США 4942241, включенном в заявку в качестве ссылки.
Соединения формулы 1 могут быть получены также в результате сложного синтеза с образованием тиадиазольного (Thi) фрагмента на конечных стадиях синтеза.
Для получения соединений формулы 1, где Thi представляет собой 1,2,4-тиадиазолил, используются соединения структуры X-Y-O(R1-R2-4-R3-фенил), где X - функциональная группа, замещаемая 1,2,4-тиадиазолом, содержащим подходящую функциональную группу. Соединения структуры X-Y-O-(R1-R2-4-R3-фенил) получают из R1-R2-4-R3-фенолов и соединений структуры гидрокси-Y-X или галоген-Y-X теми же способами, которые применяют для получения соединений формулы 1, описанной выше. Обычно X находится в ω-положении фрагмента (т.е. в наиболее удаленном от фенокси-фрагмента положении на алкиленовом мостике). X может быть введен в соединение Y-O-(R1-R2-R3-фенол) перед взаимодействием с тиадиазолом, содержащим функциональную группу. Например, в том случае, когда Y содержит ω-алкен или алкин, соединение может взаимодействовать с подходящим олово-содержащим производным, образуя соединение, в котором X представляет собой, например, трибутил. Соединение структуры олово-Y-O-(R1-R2-R3-фенил) затем взаимодействует с галоген-1,2,4-тиадиазолом, предпочтительно с йод-1,2,4-тиадиазолом с получением соединения формулы 1.
В соответствии с другим способом 1,2,4-тиадиазол может быть образован из функциональной группы, присоединенной к Y, обычно в ω-положении, как описано выше. Этот способ получения 1,2,4-тиадиазолом хорошо известен: см. например Katrisky and Rees, Comprehensive Heterocyclic. Chemistry (1985).
Для получения соединений формулы 1, где Thi представляет собой 1,3,4-тиадиазол, предпочтительно образование 1,3,4-тиадиазола на конечной стадии из функциональной группы при Y. Например, соединение структуры (алкоксикарбонил)-Y- O-(R1-R2-4-R3-фенил) может реагировать с получением карбазида, который затем взаимодействует с активированным соединением серы, таким как реагент Лавссона (Lawesson's reagent) P4S10 или аналогичным соединением с образованием 1,3,4-тиадиазола.
Получение соединения структуры X-Y-O-(R1-R2-R3-фенил), где X - функциональная группа, описано выше.
В соответствии с еще одним способом соединение формулы 1, где T представляет собой 1,3,4-тиадиазол, может быть получено взаимодействием 1,3,4-тиадиазола, содержащего подходящую функциональную группу, с соединением X-Y-O-(R1-R2-R3-фенил), в котором X представляет собой функциональную группу, замещаемую 1,3,4-тиадиазолом.
Соединения формулы 1, где R3-фенил или гетероциклический радикал, могут быть получены взаимодействием гидрокси-Y-тиадиазола или галоген-Y-тиадиазола с R1-R2-4-(функциональная группа) фенолом с последующим замещением функциональной группы фенилом или гетероциклической группой, такой как пиридил, фурил, и т. п. на конечной стадии. Например, Thi-Y-O-(R1-R2-фенил) борат, может взаимодействовать с галогенпиридином с образованием соединения 1, где R3 представляет собой пиридил. В соответствии с другим способом, некоторые гетероциклические R3 легче получают как таковые посредством образования гетероцикла из функциональной группы на фенильном кольце. Этот способ предпочтителен для гетероциклов, содержащих два или более гетероатомов, таких как тиазолил, оксадидазолил, оксазолил и т.п.
Например, в том случае, когда R3 представляет собой гетероцикл, гетероциклическое кольцо соединения формулы 1 может быть получено из подходящего R1-R2-(функциональная группа) фенокси-Y-тиадиазола, (или ZO-R1-R24-(функциональная группа) фенола, где Z представляет собой (Thi)-Y-). В этом способе гетероцикл, появляется на кольце фенокси-фрагмента на конечной стадии синтеза, как описывается в патенте США 5075187, введенном в список ссылок данного изобретения. Подбор подходящего заместителя для введения в 4-фенокси-положение будет зависеть от того, какой гетероцикл должен содержаться в конечном продукте, например, в том случае, когда Het представляет собой 1,2,4-оксадиазолил: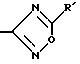
соединения 1 получают либо из подходящего 4-Z-O-R1-R2-бензонитрила, где Z - - Y-тиадиазол, реакцией, например, с гидроксиламином гидрохлоридом предпочтительно в инертном растворителе, предпочтительно алканоле, например, метаноле, этаноле, н-бутаноле и т.п. Полученный таким образом продукт взаимодействует с ангидридом кислоты формулы (R'CO)2O, где R'- алкил, галогеналкил и т.п., или ортоформиатом или сложным эфиром формиата, если R' гидроксильная или алкоксигруппа. R' появляется на P3-гетероцикле конечного продукта. Реакция протекает в интервале температур от комнатной до температуры кипения реакционной смеси в основном растворителе, таком как пиридин. Продукт представляет собой соединение формулы 1, где R3=5-R'-1,2,4- ксадиазолил, другие соединения получают аналогично.
R1-R2-R3-фенолы, используемые для получения соединений формулы 1, являются хорошо известными соединениями. Обычно их получают взаимодействием соответственно защищенного фенола, содержащего в положении 4 функциональную группу, такую как цианидная группа, альдегидная группа, галогенид, хлорид, как описано в патентах США NN 4942241, 4945164, 5051437, 5002960, 5110821, 4939267, 4861971, 4857539, 5242924 или 4843087, которые введены в список ссылок данного изобретения, для получения соответствующего подходящим образом защищенного гетероциклилфенола, защитную группу которого затем удаляют известными способами. Для получения соединений формулы 1 могут аналогично использоваться и другие известные фенолы, например, любые 4-фенилфенолы, 4-алкоксикарбонилфенолы, замещенные или незамещенные, в соответствии с приведенным выше описанием.
Предполагается, что любой R1-R2-R3-фенол может взаимодействовать в гидрокси-Y-тиадиазолом или давать соединения формулы 1.
R' может подвергаться конверсии любым способом превращения боковых групп гетероцикла, например замещением гидроксильной группы хлором, расщеплением эфирной группы с образованием гидроксильной группы и другими способами.
Следует учитывать, что ни время образования гетероциклических заместителей или пиридазина, ни порядок получения промежуточных продуктов не являются определяющими факторами для успешного синтеза соединений формулы 1. Таким образом, с помощью разумного подбора реагентов можно получить соединения формулы 1.
В соответствии с другим способом, когда используется 4-ZO-R1-R2-бензонитрил, в котором Z является защитной группой, продукт после снятия защитной группы представляет собой R1-R2-R3-(гетероциклил) фенол. Этот фенол взаимодействует с тиадиазолилалкилгалогенидом или тиадиазолилалканолом или соединением структуры галоген-Y-X или гидрокси-Y-X, где тиадиазол вводят замещением или он образуется на последней стадии синтеза соединения формулы 1.
Гидрокси-Y-тиадиазолы, используемые в данном изобретении, хорошо известны и коммерчески доступны или их можно получить известными способами. Например, коммерчески доступные галоген-1,2,4-тиадиазолы могут соединяться с ω-галогеналкеновым сложным эфиром или галогеналкиновым сложным эфиром при использовании стандартных способов, таких как взаимодействие с йодидом олова, предпочтительно с последующим восстановлением до алканола известными способами.
В соответствии с еще одним способом 1,3,4-тиадиазолилалкилгалогениды, 1,3,4- тиадиазолилалканолы или R1-R2-R3-фенокси-Y-1,3,4-тиадиазолы могут быть получены взаимодействием подходящего фенокси-Y-карбазида, например с реактивом Лавссена в стандартных условиях, как описано выше для получения соединений формулы 1. Карбазид может быть получен в результате реакции известного феноксиалкилгалогенида кислоты или феноксиалкилового сложного эфира с R'-гидразидом (где R' замещается тиадиазольным циклом или образует прекурсором тиадиазольного цикла).
Простые химические превращения, общепринятые и хорошо известные квалифицированному специалисту данной области химии, могут использоваться для эффективных изменений в функциональных группах соединений данного изобретения. Например, если необходимо, можно осуществлять: ацилирование гидрокси- или амино-замещенных производных для получения соответствующих сложных эфиров или амидов, соответственно, алкилирование фенильного или фурфурилового заместителей, расщепление алкиловых или бензиловых эфиров для получения соответствующих спиртов или фенолов, гидролиз сложных эфиров или амидов с получением соответствующих кислот, спиртов или аминов, получение ангидридов, галогенидов кислот, альдегидов, простое ароматическое алкилирование, сульфонирование карбазидов, образование хлор- или фторалкилов из гидроксиалкилов или кето- соединений, замещение гидроксильной группы на галоген в гетероциклах и образование других гетероциклов и т.п.
Для получения полного представления о реакциях, используемых в химии гетероциклических соединение см., например, Katritzky and Ress Comprehensive Heterocyclic Chemistry или Castl Heteroclic Compouds, или любые другие научные труды или учебные издания в этой области.
Кроме того, следует представлять, что получение необходимого продукта с помощью некоторых реакций будет протекать с лучшим выходом при защите некоторых функциональных групп или при их превращении в не реакционноспособные группы. Такая практика введения защитных групп хорошо изучена и описана, например, в монографии Theodora Greene, Protective Groups in Organic Synthesis (1991). To есть, когда условия реакции таковы, что они могут вызывать побочные реакции с другими частями молекул, квалифицированный специалист может определить, что необходима защита этих реакционноспособных участков молекулы и принять соответствующие меры.
Исходные вещества, используемые для получения соединений формулы 1, коммерчески доступны, известны или могут быть получены известными способами. Большое количество способов получения исходных веществ приведены в патентах, включенных в список ссылок данного изобретения.
Экспериментальная часть описания
В данном описании R1, R2, R3, R4, X, Y и Het в промежуточных продуктах принимают те же значения, что и в соединениях формулы 1.
Для введения названия заместителей в формуле 1 атомы фенильного цикла любого соединения формулы 1. будут пронумерованы следующим образом: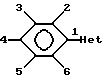
Таким образом, когда соединение формулы 1 содержит заместитель в фенильном цикле, он будет назван в соответствии с этой системой нумерации, не обращая внимание на то, как это соединение называется в действительности. Например, если соединение получают и называют R1, R2 = 3,5-диметил, это означает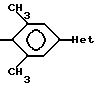
не зависимо от того, является ли этот фрагмент в соединении 3,5-диметилфенилом или 2,6-диметилфенилом.
Для введения названия заместителей в соединениях формулы 1 атомы 1,3,5- тиадиазольного цикла, описанного в данном изобретении, будут пронумерованы следующим образом: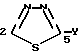
независимо от заместителя, который находится в положении 2 1,3,4-тиадиазолила для того, чтобы предупредить любые затруднения у читателя, который может быть не сведущим в части номенклатуры химических соединений. Поэтому: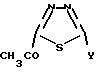
в описании назван как 2-ацетил-1,2,4-тиадиазол-5-ил, а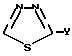
в описании назван как 1,3,4-тиадиазол-5-ил, хотя в соответствии с правилами номенклатуры эти радикалы могут называться иначе.
Пример 1.
A. 4 ((4-Циано-2,6-диметил)фенокси) масляная кислота
К раствору 5 г (34 ммоля) 4-циано-2,6-диметилфенола в 120 мл N-метилпирролидона добавляют 5,86 г (42 ммоля) карбоната калия, 0,58 г (3 ммоля) йодида калия и 4,8 мл (34 ммоля) этилового эфира 4-броммасляной кислоты, полученную смесь выдерживают при температуре 60oC в течение 24 часов. Реакционную смесь охлаждают, разбавляют водой, фильтруют, полученный твердый белый осадок промывают водой, в результате получают 8,9 г (количественный выход) этилового эфира 4-((4-циано-2,6-диметил)фенокси) масляной кислоты. Указанный эфир смешивают при комнатной температуре со 120 мл смеси этанол/вода (4:1), содержащей 820 мг (34 ммоля) Li ОН, этанол отгоняют под вакуумом, после чего водный слой промывают водой. Водный слой подкисляют, образующееся твердое белое вещество отфильтровывают и сушат, в результате получают 6,93 г (88%) 4- ((4-циано-2,6-диметил)фенокси) масляной кислоты.
B. трет-Бутиловый эфир N-(4-((4-циано-2,6-диметил) -фенокси)бутирил) карбазиновой кислоты
К раствору 4-((4-циано-2,6-диметил)фенокси) масляной кислоты (654 мг, 2,81 ммоля) в 15 мл метиленхлорида добавляют 1,2 мл (16,86 ммоля) тионилхлорида, полученную смесь кипятят с обратным холодильником в течение 3 часов. После этого смесь упаривают под вакуумом, остаток в виде масла бледно-желтого цвета в 20 мл ТГФ смешивают с 409 мг (3,09 ммоля) трет-бутилового эфира карбазиновой кислоты и добавляют несколько капель триэтиламина, после чего смесь кипятят с обратным холодильником в течение 1,5 часов. Реакционную смесь охлаждают, упаривают под вакуумом, разбавляют водой и экстрагируют метиленхлоридом (x3). Соединенные органические фракции промывают рассолом, сушат над сульфатом натрия и упаривают под вакуумом, в результате получают 899 мг (92%) трет-бутилового эфира N-(4-((4-циано-2,6-диметил)фенокси)бурил) карбазиновой кислоты
C. N-(4-((4-Циано-2,6-диметил)фенокси)бутирил)-гидразин
Смесь 6,73 г (19,4 ммоля) трет-бутилового эфира
N-(4-((4-циано-2,6-диметил)фенокси)бутирил) карбазиновой кислоты и 25 мл трифторуксусной кислоты в 100 мл метиленхлорида перемешивают при температуре 0oC в течение 1 часа, затем упаривают под вакуумом досуха. Остаток растворяют в воде, промывают эфиром и водный слой подщелачивают (до pH 9) раствором гидроксида натрия. Осадок твердого белого вещества выделяют фильтрованием, промывают водой и сушат под вакуумом, в результате получают 3,65 г (76,2%) N-(4-((4-циано-2,6-диметил)фенокси)бутирил)- гидразина.
D. N-Ацетил-N'-(4-((4-циано-2,6-диметил)фенокси)- бутирил)гидразин
К раствору 4-((4-циано-2,6-диметил)фенокси) масляной кислоты (3,9 г, 16,74 ммоля) в 120 мл метиленхлорида добавляют 6 мл тионилхлорида, полученную смесь кипятят с обратным холодильником в течение 3 часов, затем охлаждают и упаривают под вакуумом с получением масла желтого цвета. К полученному маслу добавляют 120 мл ТГФ, 1,22 г (16,74 ммоля) ацетилгидразида и 5 капель триэтиламина, смесь кипятят с обратным холодильником в течение 3 часов. После этого смесь охлаждают, белый осадок твердого вещества отфильтровывают, промывают водой и сушат под вакуумом, в результате получают 3,5 г (43%) N-ацетил-N'(4-((4- циано-2,6-диметил)фенокси)бутирил) гидразина.
E. 2-Метил-5-(3-(4-циано-2,6-диметилфенокси)пропил)- 1,3,4-тиадиазол
К раствору 2,79 г (6,92 ммоля) реактива Лавссона в 150 мл ТГФ добавляют 1,57 г (5,43 ммоля) N-ацетил-N'-(4- ((4-циано-2,6-диметил)фенокси)бутирил) гидразина, смесь кипятят с обратным холодильником в течение 3 часов, а затем выдерживают при температуре 60oC в течение ночи. После этого реакционную смесь упаривают под вакуумом, остаток очищают методом быстрой колоночной хроматографии на силикагеле (60% этилацетат/гексан), в результате получают 700 мг масла желтого цвета, после перекристаллизации которого и очистки методом быстрой хроматографии (гексан/этилацетат) получают 750 мг (48%) 2-метил-5-(3-(4-циано- 2,6-диметилфенокси)пропил)-1,3,4-тиадиазола.
F. 2-Метил-5-(3-(4-аминогидроксииминометил-2,6- диметилфенокси)пропил)-1,3,4-тиадиазол
К раствору 2-метил-5-(3-(4-циано-2,6-диметилфенокси)пропил)-1,3,4-тиадиазола (0,69 г, 2,4 ммоля) в 75 мл этанола добавляют 1,5 г (12 ммоля) карбоната калия и 0,34 г (12 ммоля) гидроксиламина гидрохлорида, полученную смесь перемешивают при температуре 50oC в течение 14 часов. Затем смесь фильтруют, осадок несколько раз промывают теплым этанолом, фильтрат упаривают под вакуумом, в результате получают 0,98 г 2-метил-5-(3-(4-аминогидроксииминометил-2,6-диметилфенокси) пропил)-1,3,4-тиадиазола, т.пл. 79-80oC.
G. 2-метил-5-(3-(4-(5-метил-1,2,4-оксадиазол-3-ил)-2,6-диметилфенокси) пропил)-1,3,4-тиадиазол (Y = 1,3- пропилен, R1=R2 3,5-диметил, Thi = 2-метил-1,3,4-тиадиазол-5-ил, R3 = 5-метил- 1,2, 4-оксадиазолил)
К раствору 2-метил-5-(3- (4-аминогидроксииминометил-2,6-диметилфенокси)пропил)-1,3,4- тиадиазола (980 мг) в 10 мл пиридина добавляют 0,3 мл (4,2 ммоля) ацетилхлорида, полученную смесь кипятят с обратным холодильником в течение 1 часа, затем охлаждают и разбавляют водой. Смесь экстрагируют этилацетатом (x4), органическую фракцию промывают водным раствором соляной кислоты и рассолом, затем сушат над сульфатом натрия. Органическую фракцию упаривают под вакуумом и остаток в виде желтого масла очищают методом жидкостной хроматографии (MPLC)
(75% этилацетат в гексане), в результате получают 342 мг (42%) 2-этил-5-(3-(4-(5-метил-1,2,4-оксадиазол-3-ил)-2,6- диметилфенокск)пропил)-1,3,4-тиадиазола в виде твердого белого кристаллического вещества, т.пл. 83-84oC (из смеси эфир/пентан).
Пример 2
A. N-Пропионил-N'-(4-((4-циано-2,6- диметил)фенокси)-бутирил) гидразин
К раствору N-(4-((4-циано- 2,6-диметил)фенокси)бутирил)гидразина (2,3 г, 9,31 ммоля, получен в соответствии со способом примера 1) в ТГФ добавляют 0,81 мл (9,31 ммоля) пропиенилхлорида и 1 мл триэтиламина и полученную смесь перемешивают при комнатной температуре в течение 2 часов. После этого смесь упаривают под вакуумом, полученный твердый белый продукт растирают с водой, фильтруют, промывают эфиром и сушат, в результате получают 2,569 г (91%)
N-пропионил-N'-(4-((4-циано- 2,6-диметил)фенокси)бутирил) гидразина.
B. 2-Этил-5-(3-(4-циано-2,6-диметилфенокси) пропил)-1,3,4-тиадиазол
К суспензии 2,58 г (8,51 ммоля) N- пропионил-N'-(4-((4-циано-2,6-диметил)фенокси)бутирил) гидразина в 200 мл сухого ТГФ добавляют 3,44 г (8,51 ммоля) реактива Лавессона и полученную смесь кипятят с обратным холодильником в течение 20 часов. Реакционную смесь упаривают под вакуумом, остаток в виде масла желтого цвета очищают методом быстрой хроматографии в колонке с силикагелем (гексан/этилацетат, 2:1) и методом MP C (гексан/этилацетат, 1: 1), в результате получают 2,09 г (82%) 2-этил-5-(3-(4-циано-2,6-диметилфенокси) пропил)-1,3,4-тиадиазола.
C. 2-Этил-5-(3-(4-аминогидроксииминометил-2,6- диметилфенокси)пропил)-1,3,4-тиадиазол
К раствору 2-этил-5- (3-(4-циано-2,6-диметилфенокси)-пропил)-1,3,4-тиадиазола (1,6 г, 5,32 ммоля) в этаноле добавляют 3,67 г (26,58 ммоля) карбоната калия и 1,85 г (26,58 ммоля) гидроксиламина гидрохлорида, полученную смесь перемешивают при комнатной температуре в течение 1,5 дней. После этого смесь фильтруют, фильтрат упаривают под вакуумом, в результате получают 1,12 г 2-этил-5-(3-(4- аминогидроксииминометил-2,6-диметилфенокси)пропил)-1,3,4- тиадиазола, т.пл. 158-160oC.
D. 2-Этил-5-(3-(4-(5-дифторметил-1,2,4- оксадиазол-3-ил)-2,6-диметилфенокси)пропил)-1,3,4-тиадиазол (Y = 1,3-пропилен, R1 = R2 = 3,5-диметил, Thi = 2-этил-1,3,4-тиадиазол, R3 = 5-дифиорметил-1,2,4-оксадиазол -3-ил)
К раствору 2-Этил-5-(3-(4-аминогидроксииминометил-2,6- диметилфенокси)пропил)-1,3,4-триадиазола (800 мг, 2,4 ммоля) в N-метилпирролидоне (3 мл) добавляют 1,44 мл (14,46 ммоля) этилового эфира дифторуксусной кислоты, полученную смесь нагревают до 95oC и выдерживают при этой температуре в течение 4 часов, затем охлаждают и разбавляют водой. Смесь экстрагируют этилацетатом (x4), органическую фракцию промывают водой, рассолом и сушат над сульфатом натрия. Органическую фракцию упаривают под вакуумом и остаток очищают методом MPLC (25-40% этилацетат в гексане), в результате получают 500 мг (55%) 2-Этил-5-(3-(4-(5- дифторметил-1,2,4-оксадиазол-3-ил)-2,6-диметилфенокси) пропил)-1,3,4-тиадиазола в виде твердого белого кристаллического вещества, т.пл. 83-84oC (из метиленхлорида и гексана).
Пример 3
A. К раствору этилсукцинилхлорида (25 г) в 300 мл CH2Cl2, охлажденному до 0oC, по каплям добавляют 13,2 г пропионилгидразида в смеси 100 мл CH2Cl2 и 27,4 мл диизопропил-этиламина. Смесь перемешивают при комнатной температуре в течение 2 часов, затем гасят водой, экстрагируют этиленхлоридом, органический слой сушат и упаривают под вакуумом. Твердый остаток перекристаллизовывают из смеси этилацетат/гексан (5:1), в результате получают N-пропионил-N'-(этил) сукцинил-гидразид.
B. 28,1 г продукта примера 3А растворяют в 2 л ТГФ, добавляют 89,8 г P4S10 и смесь кипятят с обратным холодильником в течение 2 часов. После охлаждения добавляют 800 мл 5% раствора карбоната натрия и 1 л эфира и смесь фильтруют. Фильтрат разделяют и водную фракцию экстрагируют 750 мл Et2O. Органическую фракцию сушат над сульфатом магния и упаривают под вакуумом, в результате получают 22,7 г (53%) этилового эфира 3-(5-этил-1,3,4-тиадиазол-2-ил) пропионовой кислоты.
C. 112 мл 1 мкМ LAH (в эфире) до - 20oC под атмосферой азота. По каплям в виде суспензии (в эфире) добавляют эквимолярное количество (24 г) эфира пропионовой кислоты, полученного методом 3B и полученную смесь перемешивают в течение 15 минут. Реакцию гасят водой и основанием. В результате получают 3-(5-этил-1,3,4- тиадиазол-2-ил) пропанол (13,46 г) с выходом 78%, перед использованием на следующей стадии продукт перегоняют под вакуумом (0,1 мм Hg), собирая фракцию с температурой кипения 130 - 140oC.
D. 9,7 г 2,6-диметил-4-(5-дифторметил-1,2,4-оксадиазол-3- ил) фенола (описан в Заявке США 07/869287, включенной в список ссылок данного изобретения) 15,7 г трифенилфосфина и 6,9 г пропанола помещают в 80 мл ТГФ. Смесь охлаждают до < 5oC, затем по каплям добавляют 10,4 г DEAD в 80 мл ТГФ под атмосферой азота и полученную смесь перемешивают в течение 1 часа. Раствор выливают в гексан и перемешивают до образования смолисто-твердой смеси. Смесь фильтруют для удаления твердого вещества. Раствор упаривают под вакуумом до получения твердого остатка бледно-желтого цвета. Технический продукт очищают методом колоночной хроматографии, в результате получают соединение формулы 1, 2-этил-5-(5-(4-(5-дифторметил-1,2,4-оксадиазол-3-ил) -2,6- диметилфенокси)пропил)-1,3,4-тиадиазол (Y = 1,3-пропилен, R1 = R2 = 3,5-диметил, Thi = 2-этил-1,3,4-тиадиазол-5-ил, R3 = 5- дифторметил-1,2,4-оксадиазол-3-ил), т.пл. 84oC.
Пример 4
A. Метиловый эфир N-(4-((4-циано-2-6- диметил)фенокси)-бутироил) карбазиновой кислоты
К раствору 4-((4- циано-2,6-диметил)фенокси) масляной кислоты (247 мл, 1,06 ммоля) в 15 мл метиленхлорида добавляют 0,4 мл (5,48 ммоля) тионилхлорида, полученную смесь кипятят с обратным холодильником в течение 3 часов. Смесь упаривают под вакуумом, остаток в виде масла в 20 мл ТГФ смешивают со 104 мл (1,16 ммоля) метилкарбозоата и добавляют 3 капли триэтиламина, затем смесь кипятят с обратным холодильником в течение 2 часов. Реакционную смесь охлаждают, упаривают под вакуумом, разбавляют водой, и отфильтровывают образующийся осадок твердого белого вещества, в результате получают 275 мг метилового эфира N-(4- ((4-циано-2,6-диметил)фенокси)бутироил) карбазиновой кислоты, т.пл. 154-155oC.
B. 2-Оксо-5-(3-(4-циано-2,6-диметилфенокси)пропил)-2,3 -дигидро-1,3,4-тиадиазол
К раствору 1,72 г (5,65 ммоля) метилового эфира N-(4-((4-циано-2,6-диметил)фенокси)бутироил) карбазиновой кислоты в 100 мл ТГФ добавляют 2,22 г (5,50 ммоля) реактива Лавссона и смесь кипятят с обратным холодильником в течение ночи. После этого реакционную смесь упаривают под вакуумом и остаток в виде масла желтого цвета очищают методом MP C (гексан/этилацетат, 1:1), в результате получают 0,406 г (24,5%) 2-Оксо-5-(3-(4-циано-2,6-диметилфенокси)пропил)-1,3, 4-тиадиазола-3H-2-она.
C. 2-Оксо-5-(3-4-аминогидроксииминометил-2,6-диметилфенокси)пропил)-1,3,4-тиадиазол-3H-2-он
К раствору 2-оксо-5- (3-(4-циано-2,6-диметилфенокси)-пропил)-2, З-дигидро-1,3,4-тиадиазол (801 мг, 2,77 ммоля) в 75 мл этанола добавляют 963 мг (13,86 ммоля) гидроксиламина гидрохлорида и 191,3 мг (13,86 ммоля) карбоната калия, полученную смесь перемешивают при комнатной температуре в течение ночи. Смесь фильтруют и фильтрат упаривают под вакуумом, в результате получают 826 мг (93%) 2-оксо-5-(3-(4- аминогидроксииминометил-2,6-диметилфенокси)пропил)-1,2- дигидро-1,3,4-тиадиазола.
D. 2-Оксо-5-(3-(4-(5-дифторметил-1,2,4-оксадиазол-3-ил)-2,6-диметилфенокси)пропил)-1,2-дигидро-1,3,4-тиадиазол (Thi = 2-гидрокси-1,3,4-тиадиазол-5-ил, R1 = R2 = 3,5-диметил, Y = 1,3-пропилен, R3 = 5-дифторметил-1,2,4-оксадиазол-3-ил)
К раствору 2-оксо-5-(3-(4- аминогидроксииминометил-2,6-диметилфенокси)пропил)-1,2- дигидро-1,3,4-тиадиазола (700 мг, 2,17 ммоля) в N- метилпирролидиноне (3 мл) добавляют 1,3 мл (13,02 ммоля) этилового эфира дифторуксусной кислоты, полученную смесь нагревают до 90oC и выдерживают при этой температуре в течение ночи. После этого смесь охлаждают, разбавляют водой и экстрагируют метиленхлоридом (x4). Органическую фракцию промывают рассолом и сушат над сульфатом натрия. Органическую фракцию упаривают под вакуумом и остаток в виде масла коричневого цвета очищают методом MPLC (25-35% этилацетат в гексане), в результате получают 637 мг (67%) 2-оксо- 5-(3-(4-(5-дифторметил-1,2,4-оксадиазол-3-ил)-2,6- диметилфенокси)пропил)-1,2-дигидро-1,3,4-тиадиазола, т.пл. 110-111oC (перекристаллизация: метиленхлорид/гексан).
Пример 5
Следующие соединения получают в соответствии с описанными выше способами:
Формула 1a
где Y - 1,3-пропилен, R1 = R2 = 3,5-диметил, R3 = 5-R5-1,2,4-оксадиазол-3-ил, Thi = 2-R4-1,3,4- тиадиазолил) (см. табл. 1).
Получают следующие соединения общей формулы 1b: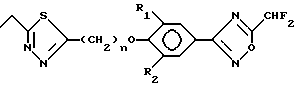
Формула 1b (см. табл. 2).
Пример 6
A. 3-Метил-5-трибутилолово-1, 2, 4-тиадиазол
К охлажденному (-95oC, под жидк. N2 и гексаном) раствору 3-метил-5-бром-1,2,4-тиадиазола (9,4 г, 52,5 ммоля) в 200 мл ТГФ добавляют по каплям 61,8 мл (105 ммоля) 1,7 N-бутиллития при температуре - 90oC. Полученный раствор розового цвета дополнительно перемешивают в течение 15 минут, а затем по каплям добавляют 17,8 г (55 ммоля) хлорида трибутилолова при температуре -90oC . Раствору дают нагреться до 0oC, после чего реакцию гасят раствором хлорида аммония. Реакционную смесь экстрагируют эфиром, органическую фракцию сушат над сульфатом натрия и упаривают под вакуумом, в результате получают 3-метил-5-трибутил-1,2, 4-тиадиазол.
B. Этиловый эфир β-(3-метил-1,2,4-тиадиазол-5-ил)- акриловой кислоты
К раствору 3-метил-5-трибутилолово-1,2,3- тиадиазола (49 ммоля) в 160 мл ксилола добавляют 11 г (49 ммоля) этилового эфира β (йод)акриловой кислоты, а затем Pd(PPh3)4 2,2 г, 2,45 ммоля). Смесь нагревают до 120oC и выдерживают при этой температуре в течение 18 часов, затем охлаждают и добавляют насыщенный водный раствор KF. Смесь фильтруют (фильтровальная бумага), осадок промывают этилацетатом, водную фракцию экстрагируют этилацетатом (x3). Соединенные органическую фракцию сушат над сульфатом натрия и упаривают под вакуумом. Остаток очищают методом хроматографии (колонка 10 см, силикагель, метиленхлорид/ацетон от 15/1 до 1/0) и повторно хроматографируют (колонка 10 см, силикагель, этилацетат/гексан 1/5), в результате получают 2 г (21%) этилового эфира β-(3-метил-1,2,4-тиадиазол-5-ил) акриловой кислоты в виде твердого белого вещества (перекристаллизация из смеси этилацетат/гексан). Затем эфир акриловой кислоты восстанавливают до спирта с помощью
AH и с помощью палладия на углероде и водорода получают насыщенный алкил.
C. 3-Метил-5-(3-(4-(5-дифторметил-1,2,4-оксадиазол-3-ил)-2,6-диметилфенокси)пропил)-1,2,4-тиадиазол (1, Thi = 3- метил-1,2,4-тиадиазол-5-ил, Y = 1,3-пропилен, R1 = R2 = метил, R3 = 5-дифторметил-1,2,4-оксадиазол-3-ил).
Смесь 5-(3-гидроксипропил)-3-метил-1,2,4-тиадиазола (242 мг, 1,53 ммоля), 4-(5-дифторметил-1,2,4-оксадидазол-3-ил)-2,6-диметилфенола (описан в заявке США 07/869287, введенной в список ссылок данного изобретения) (400 мг, 1,67 ммоля) и DEAD (290 мг, 1,67 ммоля) растворяют в 16 мл ТГФ. В указанный раствор добавляют трифенилфосфин (438 мг, 1,67 ммоля) при температуре 0oC и смесь оставляют на ночь при 20oC. Растворитель упаривают под вакуумом, добавляют водный раствор бикарбоната натрия, смесь экстрагируют метиленхлоридом (x7). Органическую фракцию сушат над сульфатом натрия и упаривают под вакуумом. Остаток очищают методом колоночной хроматографии (силикагель, колонка 10 см, этилацетат/гексан от 1/6 до 1/4), в результате получают 471 мг (81%) 3-Метил-5-(3-(4-(5-дифторметил- 1,2,4-оксадиазол-3-ил)-2,6-диметилфенокси)пропил)-1,2,4- тиадиазола в виде твердого белого кристаллического вещества, т. пл. 62-64oC.
D. 3-Метил-5-(3-(4-(5-метил-1,2,4-оксадиазол-3-ил)-2,6-диметилфенокси) пропил)-1,2,4-тиадиазол (1, Thi = 3-метил- 1,2,4-тиадидазол-5-ил, Y = 1,3-пропилен, R1 = R2 = метил, R3 = 5-метил-1,2,4-оксадиазол-3-ил).
Смесь 5-(3-гидроксипропил)-3-метил-1,2,4-тиадиазола (66 мг, 0,42 ммоля), 4-(5-метил-1,2,4- оксадиазол-3-ил)-2,6-диметилфенола (94 мг, 0,46 ммоля) и DEAD (80 мг, 0,465 ммоля) растворяют в 5 мл ТГФ. К раствору, охлажденному до 0oC, добавляют трифенилфосфин (120 мг, 0,46 ммоля) и смеси дают нагреться до 20oC. Растворитель отгоняют под вакуумом, добавляют водный раствор бикарбоната натрия и экстрагируют метиленхлоридом (x3). Органическую фракцию сушат над сульфатом натрия и упаривают под вакуумом. Остаток очищают методом колоночной хроматографии (силикагель, колонка 20 см, этилацетат/гексан, от 1/6 до 1/4) с последующей перекристаллизацией из смеси этилацетат/гексан, в результате получают 88 мг (61%) 3-метил-5- (3-(4-(5-метил-1,2,4-оксадиазол-3-ил)-2,6-диметилфенокси) пропил)-1,2,4-тиадизола в виде твердого белого кристаллического вещества, т.пл. 67-71oC.
Пример 7
Как дополнительные примеры, фенолы, описанные только в общем виде, могут далее реагировать с любым известным тиадиазолилалканолом, тиадиазолилалкилгалогенидом или с любым из указанных соединений при применении описанных выше способов получения соединений формулы 1. Предполагается, что из любого фенола, описанного в заявке 07/869287, приведенной в качестве ссылки, в результате сложных превращений в соответствии с описанными выше способами образуется тиадиазол формулы 1. Для удобства читателей используется та же номенклатура, что и для соединений формулы 1, и приведены литературные ссылки, в которых описываются известные фенолы (см. табл. 3).
Пример 8
Предполагают, что 4-гидрокси-3,5-диметилбензонитрил может реагировать с гидроксиламином гидрохлоридом в условиях примера 2C и образующийся продукт может затем взаимодействовать с этиловым эфиром хлормуравьиной кислоты и ацетоном. В результате получают соединение, где Y = 1,3-пропилен, дающее фенол, в котором R1 и R2 3,5-диметил, R3 = 5-гидрокси-1,2,4-оксадиазол-3-ил. Этот фенол может реагировать с любым из ранее полученных тиадиазолилалканолов с образованием соединений формулы 1.
B. Соединение, описанное выше, может взаимодействовать с оксилхлоридпиридином фосфора (через акцептор основания) при кипячении с обратным холодильником в течение приблизительно 4 часов, в результате получают 5-хлор-1,2,4-оксазол-3-ил формулы 1.
Биологические испытания
Биологические испытания представленных соединений формулы 1 показывают, что они обладают противопикорнавирусной активностью. Они полезны в ингибировании пикорнавирусной репродукции in vitro и главным образом активны против пикорнавирусов, включая энтеровирусы, ЕСНО-вирус и коксако-вирус и особенно риновирусы. Испытания in vitro соединений данного изобретения показали, что размножение вируса ингибировалось при минимальных ингибирующих концентрациях (МИК) в интервале от 0,05 до 7,4 микрограмм на миллилитр (мкг/мл).
Величины МИК определяют автоматизированной оценкой дозы защищающей на 50% инфицированные культуры тканей. Клетки вида HeLa в монослоях в 96-ячеечных платах инфицируют разведенными пикорнавирусами, которые в отсутствие лекарства демонстрируют эмпирически получение 80-100 фагоцитарного эффекта (ФЦЭ). Испытываемые соединения серийно разбавляют по 10 2-кратными циклами и добавляют к инфицированным клеткам. После трехдневного инкубирования при 33oC и 2,5% содержании диоксида углерода клетки фиксируют 5%-ным раствором альдегида глутаровой кислоты с последующим окрашиванием 0,25%-ным раствором фиолетового красителя в воде. После этого платы промывают, сушат и измерением оптической плотности определяют количество красителя, остающегося в ячейке (мера неповрежденных клеток). Определяют МИК, представляющую собой концентрацию соединения, при которой происходит защита 50% клеток от пикорнавирус-индуцированного ФЦЭ по сравнению с необработанным контролем.
В соответствии с описанной выше методикой представленные соединения формулы 1 испытывают против 10 H- риновирусных (HRV) серотипов, а именно: HRV -3, -4, -5, -9, -16, -18, -38, -66, -75 и -67 (отмечен в таблице как группа B) и для каждого риновирусного серотипа каждого пикорнавируса определяют величину МИК, выраженную в микромолярной концентрации. Затем определяют величину МИК50 и МИК80, которые представляют собой минимальные концентрации соединения, необходимые для 50% и 80%-ного ингибирования испытываемых серотипов, соответственно. Испытываемые соединения, как установлено, проявляют противопикорнавирусную активность против одного или более этих серотипов.
В таблице 4, помещенной ниже, приведены результаты испытаний представленных соединений данного изобретения. Группа пикорнавирусов, используемая в опыте, указана перед значениями МИК50 и МИК80, а количество серотипов, против которых испытывалось соединение (N) указано после значений МИК50 и МИК80.
Соединение примера 3(d) испытывают также против 101-H-риновирусов: 1а, 1о и 3-100 (за исключением HRV 74) по методике, описанной выше. МИК50 и МИК80 для примера 3d составляют 0,04 мкМ и 0,19 мкМ, соответственно.
Данные предварительных испытаний показывают, что соединение примера 3(о) обеспечивает in vitro и in vivo прекрасную защиту от коксаковируса B3. В соответствии с методикой, описанной выше, МИК50 in vitro для соединения примера 3(d) составляет 0,001 мкг/мл.
Данные предварительных испытаний показывают, что величина PD50 (защитная доза для предотвращения гибели 50% инфицированных мышей) заключается в области, которая указывает на полезность образца для профилактики коксаковирусной инфекции, а у инфицированных млекопитающих - для предотвращения гибели вследствие заражения.
Данные предварительных испытаний по изучению биодоступности, полученные на собаках, подтверждают, что соединение примера 3 (d) обладает очень хорошей биодоступностью. Его растворимость в искусственном желудочном соке составляет 1,1 мг/мл, а в искусственной тканевой жидкости - 0,63 мг/мл.
Рецептуры
Соединения формулы 1 могут вводиться в композиции (рецептуры) любой общеизвестной формы, включая композиции длительного высвобождения, в сочетании с одним или большим количеством нетоксичных физиологически приемлемых носителей, адъювантов или наполнителей, которые вместе определяют как носители. Композиции изготавливают с применением удобных и хорошо известных квалифицированному фрагменту методов и технологий приготовления рецептур, предназначенных для лечения инфекций и для профилактического применения: для перорального или назального введения, в твердой или жидкой форме, для ректального или местного применения и т.п.
Композиции могут вводиться человеку и животным перорально, ректально, парентерально (внутривенно, внутримышечно или подкожно), интрацистенально, интравагинально, интраперитонеально, локально (порошки, мази или капли) или в виде аэрозоля, например в виде назального или трансбуккального спрея.
Композиции для парентерального введения могут включать физиологически приемлемые стерильные водные или неводные растворы, дисперсии, суспензии или эмульсии и стерильные порошки для приготовления стерильных растворов или дисперсий для инъекций. Примерами подходящих водных и неводных носителей, наполнителей, растворителей или разбавителей являются вода, этанол, полиолы (пропиленгликоль, полиэтиленгликоль, глицерин, полиалкиленгликоли и т.п.), их подходящие смеси, растительные масла (такие как оливковое масло), органические эфиры, пригодные для инъекций, такие как этиловый эфир олеиновой кислоты. Подходящая текучесть может поддерживаться с помощью применения покрытия, такого как лицитин, сохранением необходимого размера частиц в случае дисперсий и применением поверхностно-активных веществ.
Эти композиции могут также содержать адъюванты, такие как консерванты, смачивающие, эмульгирующие и диспергирующие вещества. Предохранение от воздействия микроорганизмов может достигаться с помощью различных противобактериальных и противогрибковых веществ, например, парабенов, хлорбутанола, фенола, сорбиновой кислоты и т.п. Может потребоваться включение в композицию изотонических агентов, например сахаров, хлорида натрия и т.п. Длительная абсорбция фармацевтической формы для инъекции может достигаться применением веществ, которые продлевают абсорбцию фармацевтической формы, например, моностеарата алюминия и желатина.
Твердые дозированные формы для перорального введения включают капсулы, таблетки, пилюли, порошки, лепешки и гранулы, которые могут медленно растворяться во рту для того, чтобы смыть ротовую полость и поступать далее в сочетании с раствором активного ингредиента. В таких твердых дозированных формах активное вещество находится в смеси с по меньшей мере одним обычным инертным наполнителей (или носителем), таким как цитрат натрия и вторичный кислый фосфорнокислый кальций, или (a) наполнителем или разбавителем, например, крахмалом, лактозой, сахарозой, глюкозой, маннитолом и силициловой кислотой, (b) связующими агентами, например, карбоксиметилцеллюлозой, альгинатами, желатином, поливинилпирролидоном, сахарозой и аравийской камедью, (c) увлажнителями, например, глицерином, (d) дезинтегрирующими агентами, например, агар-агаром, карбонатом кальция, картофельным крахмалом или крахмалом тапиоки, альгиновой кислотой, некоторыми комплексными соединениями кремния и карбонатом натрия, (e) замедлителями растворения, например, парафином, (f) ускорителями абсорбции, например, четвертичными аммониевыми соединениями (g) смачивающими веществами, например, цетиловым спиртом или глицеринмоностеаратом, (h) абсорбентами, такими как, например, каолин и бентонит и (i) смазывающими веществами, например, тальком, стеаратом кальция, стеаратом магния, твердыми полиэтиленгликолями, натрийлаурилсульфатом или их смесями. В случае капсул, таблеток и пилюль дозированные формы могут также включать буферные агенты.
Некоторые твердые дозированные формы могут вводиться посредством вдыхания порошка "вручную" или с помощью приспособления, такого как Spin- Haler, который используется для введения двунатриевого хромогликата (disodium cromoglycate) (Intal). При применении этого устройства порошок может быть инкапсулирован. В том случае, когда применяют жидкую композицию, лекарство может вводиться при помощи распылителя, устройства для получения аэрозоля или при помощи любого средства, которое может доставлять композицию отдельными порциями, например с помощью медицинской пипетки или аэрозольного ингалятора.
Твердые композиции аналогичного типа могут также вводиться для применения в мягкие или твердые желатиновые капсулы с использованием таких наполнителей как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и т.п.
Твердые дозированные формы, такие как таблетки, драже, капсулы, пилюли и гранулы могут изготавливаться с покрытиями и оболочками, такими как энтеросолюбильные покрытия и другие хорошо известные покрытия. Они могут содержать контрастирующие вещества, а также могут представлять собой такую композицию, в которой высвобождение действующего вещества или веществ происходит замедленным образом на определенном участке кишечного тракта.
Активные соединения могут также находиться, если это приемлемо, в микро-инкапсулированной форме, включающей один или большее количество указанных выше наполнителей.
Жидкие дозированные формы для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры. Твердые рецептуры могут приготавливаться как основа для жидких рецептур. Помимо действующих веществ, жидкие дозированные формы могут содержать инертные разбавители, обычно используемые для этих целей, такие как вода или другие растворители, солюбилизирующие вещества и эмульгаторы, например, этиловый спирт, изопропиловый спирт, этилкарбонат, этиловый эфир уксусной кислоты, бензиловый спирт, бензиловый эфир бензойной кислоты, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла, в частности масло семян хлопчатника, масло арахиса, масло семян кукурузы) оливковое масло, касторовое масло и масло кунжута, глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и сложные сорбитан-эфиры жирных кислот или смеси этих веществ и т.п. Помимо инертных наполнителей, композиция может также включать адъюванты, такие как смачивающие агенты, эмульгаторы и суспендирующие агенты, подслащивающие, вкусовые вещества и ароматизирующие добавки.
Кроме активных действующих веществ суспензии могут содержать суспендирующие вещества, например этоксилированные изостеариловые спирты, полиоксиэтиленсорбитол, полиэтиленгликоли различных молекулярных весов и сорбитан-эфиры, микрокристаллическую целлюлозу, метагидроксид алюминия, бентонит, агар-агар и трагакант или смеси этих веществ и т.п.
Композиции для ректального или вагинального введения предпочтительно представляют собой суппозитории, которые можно приготавливать смешением соединений данного изобретения с подходящими не обладающими раздражающим действием наполнителями или носителями, такими как кокосовое масло, полиэтиленгликоль или воск для суппозиториев, который при обычных температурах является твердым, но при температуре тела становится жидким и, следовательно плавится в ректальной или вагинальной полости и высвобождает активный компонент.
Композиции для введения в виде аэрозолей получают растворением соединения формулы 1 в воде или подходящем растворителе, например, спиртовом эфире (alhogol ether) или в другом инертном растворителе, смешением с летучим пропеллантом (propellant) и последующим внесением в контейнер, находящийся под давлением и содержащий дозирующий клапан для выделения вещества в виде капель нужного размера.
Пропеллант применяется в жидком (сжиженном) состоянии, как правило в единственном числе, и при атмосферном давлении имеет температуру кипения ниже комнатной температуры. Для применения в композиции, предназначенной для получения аэрозоля медицинского назначения, сжиженный пропеллант должен быть нетоксичным. Подходящими для применения в сжиженном состоянии пропеллантами, являются низшие алканы, содержащие до пяти атомов углерода, такие как бутан и пентан, или алкилхлориды, такие как метил-, этил-, или пропилхлориды. Подходящими сжиженными пропеллантами также являются фторированные и фторхлорированные алканы, выпускаемые под торговыми названиями "Фреон" (Freon) и "Генетрон" (Genetron).
Могут применяться также смеси указанных пропеллантов.
Предпочтительными сжиженными пропеллантами являются пропелланты, не содержащие хлора, например, 134a (тетрафторэтан) и 227c (гептафторпропан), которые могут использоваться как описано выше. Обычно в таких аэрозольных рецептурах используется второй растворитель, такой как эфир, спирт или гликоль.
Перечень форм единичной дозы данного изобретения определяется и непосредственно зависит от (a) конкретных характеристик активного ингредиента и конкретного действия, которое должно быть достигнуто, и от (b) ограничений, которые учитывают при получении композиций активных действующих веществ, предназначенных для людей и животных, как подробно описано в данном изобретении, причем это применение композиций является характерным признаком данного изобретения. Примерами подходящих форм, содержащих единичную дозу в соответствии с данным изобретением, являются капсулы, предназначенные для приема внутрь или аэрозоли с дозированным "выстрелом" и другие формы, представленные в данном описании.
Соединения данного изобретения полезны для профилактики и лечения инфекций, вызванных пикорнавирусной этиологией, таких как асептический менингит, инфекция верхних дыхательных путей, энтеровирусные инфекции, коксаковирусов (coxsackeivirus), энтеровирусов и т.п. Для лечения применяется эффективное, но нетоксическое количество соединения. Доза соединения, используемая для лечения, зависит от способа введения (интраназальный, интрабронхиальный, и активности конкретного соединения).
Дозированные формы для местного применения включают мази, порошки, спреи и лекарственные формы для ингаляции. Активный компонент смешивают в стерильных условиях с физиологически приемлемым носителем и любыми консервантами, буферными агентами и пропеллантамb, как это необходимо. К ним относятся также рецептуры для лечения заболеваний глаз, глазные мази, порошки и растворы.
Следует учитывать, что исходная точка для определения дозы как для профилактики, так и для лечения пикорнавирусной инфекции основывается на определении содержания соединения в плазме при приблизительно установленных в лабораторных условиях уровнях минимальной ингибирующей концентрации. Например, МИК, равная 1 мкг/мл, должна приводить к нужному исходному уровню в плазме - 0,1 мг/мл и доза для млекопитающего со средним весом 70 кг составляет примерно 5 мг. Предположительно, интервал доз может составлять от 0,01 - 1000 мг.
Величину действующей дозы активного ингредиента в композициях можно варьировать для получения количества активного ингредиента, которое дает эффективный терапевтический результат для данной композиции при данном способе введения. Следовательно, выбираемая величина дозы зависит от необходимого терапевтического эффекта, способа введения, требуемой продолжительности лечения и некоторых других факторов и легко определяется квалифицированным фармацевтом.
Процесс разработки фармацевтической дозированной формы включает подбор подходящих ингредиентов для составления рецептуры и определение подходящего содержания активного ингредиента для достижения оптимальной биодоступности и наиболее продолжительного периода полураспада в плазме крови и т.п., он хорошо известен квалифицированным специалистам, которые при разработке фармацевтической композиции для терапевтического применения обычно определяют in vivо взаимосвязь "доза-эффект".
Кроме того, следует учитывать, что подходящая доза для достижения оптимальных результатов терапевтического действия составляет главный предмет исследования для специалиста, который обычно рассматривает взаимосвязь "доза-эффект" при разработке регламента для терапевтического применения. Например, специалист может рассматривать минимальные ингибирующие концентрации in vitro как основной фактор для определения эффективных уровней содержания лекарства в плазме. Однако этот и другие методы составляют область практических приемов, используемых специалистом при разработке лекарственных средств.
Следует учитывать, что величина специфической дозы для любого конкретного пациента будет зависеть от различных факторов, в том числе от веса, общего состояния здоровья, половой жизни, питания, времени и способа введения, скорости абсорбции и разрастания, сочетания с другими лекарствами и тяжести заболевания, и она легко определяется квалифицированным врачом.
При введении лекарства до заражения, то есть профилактически, предпочтительно чтобы введение происходило в интервале примерно от 0 до 48 часов до заражения животного-хозяина патогенным пикорнавирусом. При терапевтическом введении для ингибирования инфекции предпочтительно введение в пределах одного - двух дней после заражения патогенным вирусом.
Величина вводимой дозы будет зависеть от вида пикорнавируса, который необходимо ингибировать для извлечения или профилактики, типа животного, которому она вводится, его возраста, состояния здоровья, веса, степени заражения, вида другого лечения, если оно проводилось, частоты обработки и природы требуемого эффекта.
Соединения данного изобретения находят также применение для профилактики распространения пикорнавирусной инфекции. Соединения могут использоваться в аэрозольных опрыскивателях для нанесения на зараженные поверхности, на доступные для инфекции продукты, такие как ткани и т.п., используемые зараженным больным. Кроме того, соединения могут использоваться для обработки домашней утвари (тканей, других вещей, доступных для швабры, и т.п.) для предупреждения распространения инфекции посредством инактивирования пикорнавирусов.
Вследствие того, что соединения данного изобретения обладают способностью подавлять рост пикорнавирусов при добавлении их в среду, где развивается пикорнавирус, предполагается, что соединения данного изобретения могут использоваться в дезинфицирующих растворах, например, в водном растворе с поверхностно-активным веществом, для обеззараживания поверхностей, на которых присутствуют полио-, коксако-, риновирус и/или другие пикорнавирусы, такие поверхности включают, но перечень не ограничивается только ими, стеклянную посуду в больницах, столы в ресторанах, поверхности, предназначенные для продуктов питания, раковины в ванных комнатах и другие места, где могут развиваться пикорнавирусы.
Контакт рук с носовой слизью может являться наиболее важным путем передачи пикорнавируса. Стерилизация рук людей, находящихся в контакте с больными, инфицированными риновирусом, предотвращает дальнейшее распространение заболевания. Предполагается, что соединение данного изобретения при ручной стирке или применении для защиты рук и продуктов, ингибирует репродукцию риновируса и снижает вероятность распространения заболевания.
| название | год | авторы | номер документа |
|---|---|---|---|
| 1,2,4-ОКСАДИАЗОЛИЛФЕНОКСИАЛКИЛИЗОКСАЗОЛЫ И КОМПОЗИЦИЯ ПРОТИВ ПИКОРНОВИРУСОВ | 1993 |
|
RU2114112C1 |
| ПРОИЗВОДНЫЕ САХАРИНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ АДДИТИВНАЯ СОЛЬ КИСЛОТЫ ИЛИ ОСНОВАНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОЯВЛЯЮЩАЯ АКТИВНОСТЬ ИНГИБИТОРА ЭЛАСТАЗЫ | 1992 |
|
RU2114835C1 |
| ПРОИЗВОДНОЕ 2-ЗАМЕЩЕННОГО САХАРИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОЯВЛЯЮЩАЯ ИНГИБИТОРНУЮ АКТИВНОСТЬ ПРОТИВ ЭЛАСТАЗЫ | 1992 |
|
RU2101281C1 |
| НОВЫЕ ТИОФЕНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ АГОНИСТОВ S1P1/EDG1 РЕЦЕПТОРА | 2007 |
|
RU2442783C2 |
| ПРОИЗВОДНЫЕ ФЕНОКСИ- ИЛИ ФЕНОКСИАЛКИЛПИПЕРИДИНА И АНТИВИРУСНАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1993 |
|
RU2125565C1 |
| ПРОИЗВОДНЫЕ КАРБОНОВОЙ КИСЛОТЫ, СОДЕРЖАЩИЕ 2,5,7-ЗАМЕЩЕННОЕ ОКСАЗОЛОПИРИМИДИНОВОЕ КОЛЬЦО | 2011 |
|
RU2560876C2 |
| ПРОИЗВОДНЫЕ КАРБОНОВОЙ КИСЛОТЫ, СОДЕРЖАЩИЕ 2,5-ЗАМЕЩЕННОЕ ОКСАЗОЛОПИРИМИДИНОВОЕ КОЛЬЦО | 2011 |
|
RU2564018C2 |
| ПРОИЗВОДНЫЕ ПИРИДИН-4-ИЛА В КАЧЕСТВЕ ИММУНОМОДУЛИРУЮЩИХ АГЕНТОВ | 2007 |
|
RU2447071C2 |
| ПРОИЗВОДНЫЕ БЕНЗИЛГИДРОКСИДА, ИХ ПОЛУЧЕНИЕ И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2015 |
|
RU2704017C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ КАРБОНОВЫХ КИСЛОТ, ИМЕЮЩИЕ 2,5-ЗАМЕЩЕННОЕ ОКСАЗОЛОПИРИМИДИНОВОЕ КОЛЬЦО | 2011 |
|
RU2559896C2 |




Описываются новые феноксиалкилтиадиазолы общей формулы I, где Thi представляет собой тиадиазолил или замещенный тиадиазолил, содержащий в качестве заместителя алкоксигруппу, фторметил, дифторметил, трифторметил, 1,1-дифторэтил, галоген, алкил, циклоалкил, гидроксиалкил или алкоксиалкил; Y - алкиленовый мостик, содержащий 3 - 9 атомов углерода; R1 и R2 каждый независимо выбирают из водорода, галогена, алкила, гидроксила, гидроксиалкила, алкоксиалкила, алкокси, нитро, карбокси, алкоксикарбонила, дифторметила или трифторметила; R3 представляет собой фенил или гетероциклический радикал, который выбирают из группы, содержащей бензоксазолил, бензотиазолил, тиадиазолил, оксазолил, тиазолил, оксадиазолила - изоксазолил, фурил, тиенил, пиридил, или замещенный фенил, или замещенный гетероцикл, содержащий в качестве заместителя алкил, алкоксиалкил, циклоалкил, галогеналкил, гидроксиалкил, алкокси, гидрокси или фторалкил, или его фармацевтически приемлемые кислотно-аддитивные соли. Вышеуказанные соединения проявляют противопикорнавирусную активность. Описывается также способ профилактики и лечения, а также способ борьбы с пикорнавирусами. 4 с. и 19 з.п. ф-лы, 4 табл.
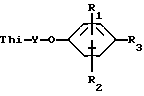
где Thi - представляет собой тиадиазолил или замещенный тиадиазолил, содержащий в качестве заместителя алкоксигруппу, фторметил, дифторметил, трифторметил, 1,1-дифторэтил, галоген, алкил, циклоалкил, гидроксиалкил или алкоксиалкил;
Y - алкиленовый мостик, содержащий 3 - 9 атомов углерода;
R1 и R2 каждый независимо выбирают из водорода, галогена, алкила, гидроксила, гидроксиалкила, алкоксиалкила, алкокси, нитро, карбокси, алкоксикарбонила, дифторметила или трифторметила;
R3 представляет собой фенил или гетероциклический радикал, который выбирают из группы, содержащей бензоксазолил, бензотиазолил, тиадиазолил, оксазолил, тиазолил, оксадиазолил, изоксазолил, фурил, тиенил, пиридил или замещенный фенил, или замещенный гетероциклический радикал, где заместитель выбирают из группы, включающей алкил, алкоксиалкил, циклоалкил, галогеналкил, гидроксиалкил, алкокси, гидрокси или фторалкил, или его фармацевтически приемлемые соли.
| EP, 0337151, A1, 1989 | |||
| Способ получения гетероциклических соединений | 1986 |
|
SU1438610A3 |
| US 4942241, OA1, 1990. | |||
