Изобретение относится к новым 1,2,4-оксадиазолилфеноксиалкилизоксазолам, к способу их получения, к содержащим их композициям и способам их применения в качестве противовирусных агентов.
Известны гетероциклические феноксиалкилизоксазолы, в которых гетероциклический фрагмент является оксазолом или оксазином, которые демонстрируют противовирусную активность (патент США N 4843087).
Известны активные соединения против вирусов формулы
где
Y - алкиленовый мостик, содержащий 3 - 9 атомов углерода;
Z - N или HC;
R - водород или низший алкил, содержащий 1 - 5 атомов углерода, при условии, что если Z - это N, то R - низший алкил;
R1 и R2 - водород, галоид, низший алкил, низший алкокси, нитро, низший алкоксикарбонил или трифторметил;
Het выбирают из специфических гетероциклических групп.
В определение Het включены незамещенный 1,3,4-оксадиазол-2-ил и незамещенный 1,2,4-оксадиазол-5-ил (патент США N 485753915).
Известны активные противовирусные соединения формулы inter alia
где
Y - алкиленовый мостик, содержащий 3 - 9 атомов углерода, необязательно прерываемый одним или двумя атомами кислорода, за счет циклогексильных или олефиновых связей;
X - O, S, SO или SO2;
Z - N2 или R8C, где R8 - водород или низший алканоил;
R1 и R2 выбирают из группы, состоящей из водорода, низшего алкила, низшего алкенила, галоида, нитро, низшей алкокси, низшей алкилтио, дифторметила, трифторметила, амино, низшей алканоиламино, ди-низшей алкиламино, гидрокси, низшего алкеноила, низшего алканоила, гидроксиметила и карбокси;
R и R3 каждый является водородом или алкилом, содержащим 1 - 3 атома углерода, необязательно замещенным одним из членов группы, состоящей из гидрокси, низшей алканоилокси, низшей алкокси, галоида или N=Z', где N=Z' является амино, низшей алканоиламино, низшей алкиламино, ди-низшей алкиламино, 1-пирролидинила, 1-пиперидинила, или 4-морфолинила, при условии, что если Z является N, то R не является водородом;
Het выбирают из определенных гетероциклических групп, включающих незамещенный 1,3,4-оксадиазол-2-ил (патент США 486179129).
Известны также противовирусные активные соединения формулы
где
Y - алкиленовый мостик, содержащий 1 - 9 атомов углерода;
R' - низший алкил или гидрокси-низший алкил, содержащий 1 - 5 атомов углерода;
R1 и R2 - водород, галоид, низший алкил, низший алкокси, нитро, низший алкоксикарбонил или трифторметил;
R8 - водород или низший алкил, содержащий 1 - 5 атомов углерода.
Были раскрыты антивирусные активные соединения формулы (наряду с другими)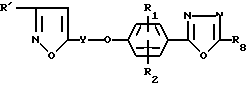
где
Y - алкиленовый мостик, содержащий 3 - 9 атомов углерода;
R' - низший алкил или гидрокси-низший алкил, содержащий 1 - 5 атомов углерода;
R1 и R2 - водород, галоид, низший алкил, низшая алкокси, нитро, низший алкоксикарбонил или трифторметил;
R8 - водород или низший алкил, содержащий 1 - 5 атомов углерода (патент США N 4945164).
Известны соединения формулы
где
Y - алкилен, содержащий 3 - 9 атомов углерода;
R1 - низший алкил, низший-алкокси-(С1-3 алкил), низший алкоксикарбонил, циклопропил или трифторметил;
R2 и R3 независимо представляют водород, низший алкил, галоид, низший-алкокси, нитро, трифторметил или гидрокси;
R4 - водород или низший алкил, где низший алкил и низший алкокси, каждый содержит 1 - 5 атомов углерода,
при условии, что если R1 является низшим алкилом, по крайней мере один из R2 и R3 представляет гидрокси (патентная заявка США N 07/731569).
В изобретении предложено соединение формулы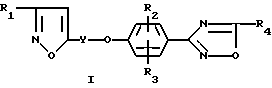
где
R1 - алкил, алкокси, гидрокси, циклоалкил, гидроксиалкил, алкоксиалкил или гидроксиалкокси;
Y - алкилен, содержащий 3 - 9 атомов углерода;
R2 и R3 независимо представляют водород, алкил, алкокси, галоид, трифторметил или нитро;
R4 - алкокси, гидрокси, галоидметил, дигалоидметил, тригалоидметил, циклоалкил, алкоксикарбонил, гидроксиалкил, алкоксиалкил, алканкарбонилоксиалкил, циано, 2,2,2-трифторэтил, (4-метилфенил)сульфонилоксиметил;
N=Q или CON=Q, где N=Q является амино, алкиламино;
или их фармацевтически приемлемые соли присоединения кислот.
В изобретении предложено также соединение формулы
где Y, R2 и R3 имеют указанные ранее значения;
R6 - алкокси, фторметил, дифторметил, тригалоидметил, циклоалкил или алкоксиалкил.
В изобретении предложено также соединение формулы
где
R2 и R3 имеют указанные ранее значения;
R7 - алкокси, фторметил, дифторметил, трифторметил, циклоалкил, алкоксиалкил или циано.
Изобретение предлагает также соединения формул XVII и XXI (см. далее).
В изобретении предложены также композиции для борьбы с пикорнавирусом, которые содержат также эффективное против вируса количество соединения формулы I в смеси с подходящим носителем или разбавителем, и способы борьбы с пикорнавирусом, которые включают борьбу с инфекцией пикорнавируса у млекопитающих.
Соединения формулы I пригодны в качестве агентов против пикорнавируса.
Соединения формул III, IV, XVII и XXI пригодны в качестве интермедиатов для получения соединений формулы I.
Предпочтительными соединениями формулы I являются соединения, в которых
R1 - C1-3-алкил, C1-3-алкокси, гидрокси, циклопропил, гидрокси-C1-3-алкил, C1-3-алкокси-C1-3-алкил или гидрокси-C1-3-алкокси;
Y - алкилен, содержащий 3 - 9 атомов углерода, особенно 3 - 5 атомов углерода;
R2 и R3 независимо являются водородом, C1-3-алкилом, C1-3-алкокси или галоидом;
R4 - C1-3-алкокси, гидрокси, галоидметил, дигалоидметил, тригалоидметил, циклопропил, C1-3-алкоксикарбонил, гидрокси-C1-3-алкил, C1-3-алкокси-C1-3-алкил, (C1-3-алкан) карбонилокси-C1-3-алкил, циано, 2,2,2-трифторэтил, 4-(метилфенил)сульфонилоксиметил, N= Q или CON=Q, где N=Q является амино, C1-3-алкиламино или ди-(C1-3-алкил)амино.
Более предпочтительными соединениями формулы I являются соединения формулы
где R1, Y, R2, R3 и R4 имеют указанные ранее для формулы I значения и особенно те, в которых R1, Y, R2, R3 и R4 имеют значения, указанные в предыдущем пункте для предпочтительных соединений формулы I.
Особенно предпочтительными являются соединения формулы I или IA, в которых R4 - C1-3-алкокси, трифторметил, дигалоидметил, тригалоидметил, циклоалкил или C1-3-алкокси C1-3-алкил, особенно трифторметил.
Следует учитывать, что предлагаемые соединения, в которых 1,2,4-оксодиазольное кольцо замещено гидрокси, амино или алкиламино, могут существовать в любой из трех таутомерных форм
где
R4 - гидрокси, амино или алкиламино,
T - O, NH или N-алкил,
и такие таутомеры входят в объем данного изобретения.
В том смысле, как здесь использовано, если нет других специальных указаний, алкил, алкан, алкокси, циклоалкил и галоид каждый имеют следующие значения:
алкил и алкокси относятся к алифатическим радикалам, включая разветвленные радикалы, содержащие 1 - 5 атомов углерода. Таким образом, алкильный фрагмент таких радикалов включает, например, метил, этил, пропил, изопропил, н-бутил, втор-бутил, трет.-бутил и пентил; алкан относится к одновалентным алифатическим радикалам, включая разветвленные радикалы, содержащие 1 - 4 атомов углерода. Так алкановый фрагмент такого радикала включает, например, метил, этил, пропил, изопропил, н-бутил и втор-бутил;
циклоалкил относится к алициклическим радикалам, содержащим 3 - 6 атомов, что иллюстрируется циклопропилом, галоид относится к брому, хлору, иоду или фтору.
В том смысле, как здесь использовано, гидроксиалкильные, алкоксиалкильные, гидрокси и алкоксигруппы могут находиться в любом доступном положении алкила. Так гидроксиалкил и алкоксиалкил включают, например, гидроксиметил, 1-гидроксиэтил, 2-гидроксиэтил, 2-гидроксипропил, 2-гидроксиизопропил, 2,3,4 и 5-гидроксипентил и т.п. и их соответствующие простые алкилэфиры.
В том смысле, как здесь использовано, в гидроксиалкокси, гидроксильная группа может находиться в любом доступном положении алкокси, отличном от положения С-1. Так гидроксиалкокси включают, например, 2-гидроксиэтокси, 2-гидроксипропокси, 2-гидроксиизопропокси, 2- и 5-гидроксипентокси и т.п.
Соединения формулы I, в которых R1 является алкилом, алкокси, циклоалкилом или алкоксиалкилом, Y, R2 и R3 имеют указанные ранее значения, а R4 является гидрокси, галоидметилом, дигалоидметилом, тригалоидметилом, циклоалкилом, алкоксикарбонилом, алкоксиалкилом, алканкарбонилоксиалкилом или 2,2,2-трифторэтилом, можно получить способом, который включает осуществление взаимодействия амидоксима (N-гидpоксикapбoкcимидaмидa) формулы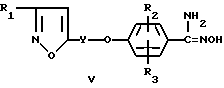
с галоидангидридом, R4COX, алкилгалоидформиатом, ROCOX в том случае, если R4 в формуле I является гидрокси, где R является метилом или этилом, или галоидангидридом, (R4CO)2O, где R1, Y, R2, R3 и R4 имеют указанные ранее значения в этом пункте, а X является бромом, хлором, фтором или медью, в безводных условиях до получения соответствующего соединения формулы I. Этот способ включает следующие методики. В одном способе амидоксим формулы V подвергают взаимодействию с галоидангидридом или ангидридом кислоты в присутствии органического или неорганического основания, например пиридина, триэтиламина или карбоната калия, в инертном растворителе, например ацетоне, метиленхлориде, хлороформе, толуоле или тетрагидрофуране, или в основании, которое также действует как растворитель, например в пиридине, при повышенной температуре (около 40 - 130oC) или при пониженной температуре (около 0 - 15oC). В последнем случае промежуточное O-ацильное производное [C(NH2= NOC(=O)-(R4 или OR)] выделяют и нагревают при температуре в интервале от около 100 - 130oC в течение промежутка времени, достаточного для циклизации до оксадиазола формулы I, что обычно происходит за промежуток времени от около 5 мин до 4 ч. В другом способе амидоксим формулы V подвергают взаимодействию с галоидангидридом или ангидридом кислоты в кислоте, которая соответствует галоидангидриду или ангидриду кислоты при повышенной температуре (около 70 - 100oC).
Соединения формулы I, в которых R1 - алкил, алкокси, циклоалкил или алкоксиалкил, Y, R2 и R3 имеют указанные ранее значения, а R4 - дигалоидметил, тригалоидметил, циклоалкил, алкоксиалкил, алканкарбонилоксиалкил или 2,2,2-трифторэтил, можно получить способом, который включает осуществление взаимодействия амидоксима формулы I с продуктом, полученным при взаимодействии карбоновой кислоты, R4CO4H, где R1, Y, R2 и R4 имеют указанные ранее в этом пункте значения, с сочетающим агентом N,N'-карбонилдиимидазолом, полученным, как указано в примерах, в инертном растворителе, например в ацетоне, тетрагидрофуране, хлороформе, метиленхлориде или толуоле, при повышенной температуре (около 40 - 80oC) до получения соответствующего соединения формулы I.
Соединения формулы I, в которых R1 - алкил, алкокси, циклоалкил или алкоксиалкил, Y, R2, и R3 имеют указанные ранее значения, а R4 является аминогруппой, можно получить способом, который включает осуществление взаимодействия амидоксима формулы V, где R1, Y, R2 и R3 имеют указанные ранее в этом пункте значения, с галоидцианом, CNX1, где X1 - бром, хлор или иод, в присутствии основания, например бикарбоната калия или натрия, в спиртовом растворителе, например этиловом спирте, при комнатной температуре до получения соединения формулы I, в котором R4 является аминогруппой.
Соединения формулы I, в которых R1 - алкил, алкокси, циклоалкил или алкоксиалкил, Y, R2 и R3 имеют указанные ранее значения, а R4 является CH2CF3, можно получить, осуществляя взаимодействие амидоксима формулы V, где R1 имеет указанные ранее в этом пункте значения, а Y, R2 и R3 имеют указанные ранее значения, с кетен-1,3-пропандитиолацеталем формулы
до получения соответствующего соединения формулы I.
Амидоксим формулы V и кетен-1,3-пропандитиолацеталь подвергают взаимодействию в присутствии трифторацетата серебра в инертном растворителе, например тетрагидрофуране, диоксане, диметилформамиде или N-метилпирролидиноне, при температуре в интервале от около 60 до около 100oC. Предпочтительно вести реакцию в темноте.
Промежуточное соединение-амидоксим получают способом, изображенным на следующей схеме;
Бромфенол формулы VI взаимодействует c цианидом меди в инертном растворителе при повышенной температуре, например, в диметилформамиде при температуре кипения с обратным холодильником до получения цианофенола формулы VII. Последний подвергают взаимодействию с галоидизоксазолом формулы VIII, где X2 - хлор, бром или иод, в сухом инертном растворителе, например ацетонитриле или N-метилпирролидоне, в присутствии основания, например карбоната калия или гидроксида натрия, необязательно в присутствии каталитического количества иодида калия или иодида натрия, при повышенной температуре (50 - 120oC) до получения цианосоединения формулы IX. Цианосоединение IX реагирует с гидроксидамингидрохлоридом в присутствии основания, например карбоната калия или натрия, ацетата натрия или гидроксида натрия, в спиртовом растворителе, например этиловом спирте при повышенной температуре (50 - 150oC) до получения амидоксима формулы V.
Некоторые промежуточные соединения формулы IX, в которых R1 - алкил, циклоалкил или алкоксиалкил, а Y, R2 и R3 имеют указанные ранее значения, можно получить при взаимодействии этинильного соединения XII описанного далее, с нитрилоксидом,  , где R1 имеет указанные ранее в этом пункте значения, по способу, аналогичному тому, который описан далее для получения соединения I из этинильного соединения формулы III.
, где R1 имеет указанные ранее в этом пункте значения, по способу, аналогичному тому, который описан далее для получения соединения I из этинильного соединения формулы III.
Промежуточные бромфенолы формулы VI и цианофенолы формулы VII принадлежат к общеизвестному классу соединений и их легко получить известными способами.
Промежуточные галоидизоксазолы формулы VIII можно получить способом, описанным в патенте США 4843087, например при взаимодействии производного щелочного металла изоксазола формулы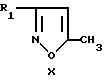
где R1 - алкил, алкокси, циклоалкил или алкоксиалкил, с дигалидом, X2 - Y' -X2, где Y' является алкиленом, содержащим 2 - 8 атомов углерода; X2 имеет указанные ранее значения. Производное щелочного металла получают in situ, обрабатывая изоксазол формулы X основанием органо-щелочного металла, таким, как бутиллитий или диизопропиламид лития в безводных условиях.
Соединения формулы I, в которых R1 - алкил, циклоалкил или алкоксиалкил, Y, R2 и R3 имеют указанные ранее значения, а R4 - алкокси, тригалоидметил циклоалкил, алкоксикарбонил, алкоксиалкил или 2, 2,2-трифторэтил, можно получить способом, который включает осуществление взаимодействия этинильного соединения формулы III, указанного ранее, где R6 имеет указанные ранее в этом пункте значения для R4, с нитрилоксидом формулы 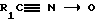 , который получают in situ из гидроксииминогалида формулы R1C(X3)=NOH, где X3 - хлор или бром, в присутствии аминового основания, например триэтиламина, пиридина или N-метилпирролидина. Гидроксииминогалиды, которые можно также получить in situ принадлежат к общеизвестному классу соединений и их легко получить обычными способами, например при взаимодействии соответствующего альдегидоксима (R1C= NOH) с галоидирующим агентом, например N-хлорсукцинимидом или бромом. Способ получения соединений формулы I за счет взаимодействия этинильного соединения формулы III, имеет место при нагревании реагентов в инертном полярном растворителе, например диметилформамиде или N-метилпирролидоне, при температуре в интервале от около 20 до около 120oC.
, который получают in situ из гидроксииминогалида формулы R1C(X3)=NOH, где X3 - хлор или бром, в присутствии аминового основания, например триэтиламина, пиридина или N-метилпирролидина. Гидроксииминогалиды, которые можно также получить in situ принадлежат к общеизвестному классу соединений и их легко получить обычными способами, например при взаимодействии соответствующего альдегидоксима (R1C= NOH) с галоидирующим агентом, например N-хлорсукцинимидом или бромом. Способ получения соединений формулы I за счет взаимодействия этинильного соединения формулы III, имеет место при нагревании реагентов в инертном полярном растворителе, например диметилформамиде или N-метилпирролидоне, при температуре в интервале от около 20 до около 120oC.
Промежуточные этинильные соединения формулы III получают по следующей схеме:
Цианофенол формулы VII подвергают взаимодействию с галоидалкином формулы XI, где X2 имеет указанные ранее значения, используя процедуру, описанную ранее для получения цианосоединения из соединений VII и VIII, до получения этинильного соединения формулы XII. Этинильное соединение XII подвергают взаимодействию с гидроксиамингидрохлоридом по способу аналогичному описанному ранее для получения амидоксима V из цианосоединения IX, до получения амидоксима формулы XIII. Амидоксим XIII подвергают взаимодействию с галоидангидридом R4COX, ангидридом кислоты (R4CO)2O, карбоновой кислотой R4CO2H, или
по способу, аналогичному описанному ранее для получения соединений формулы I из aмидoксима формулы V.
Галоидалкины формулы XI принадлежат к общеизвестному классу соединений.
Соединения формулы I, в которых R1 - алкил, алкокси, циклоалкил или алкоксиалкил, Y, R2 и R3 имеют указанные ранее значения, а R4 - алкокси, тригалоидметил, циклоалкил, алкоксикарбонил, алкоксиарил или 2,2,2-трифторэтил, можно получить способом, который включает взаимодействие фенола формулы IV, приведенной ранее, где R2 и R3 имеют указанные ранее значения, а R7 имеет указанные ранее в этом пункте значения для R4, с галоидизоксазолом формулы VIII, где R1 имеет указанные ранее в этом пункте значения, а Y и X2 имеют указанные ранее значения, до получения соответствующего соединения формулы I. Этот способ используют аналогично тому, который описан ранее для получения цианосоединения IX при взаимодействии цианофенола формулы VII и галоидизоксазола формулы VIII.
Промежуточное соединение - галоидизоксазол формулы VII можно получить, как описано ранее.
Промежуточные фенолы формулы IV можно получить при взаимодействии цианофенола формулы VII с гидроксиламингидрохлоридом, используя способ, аналогичный описанному ранее для получения амидоксима формулы V из цианосоединения IX до получения амидоксима формулы
Амидоксим формулы XIV подвергают взаимодействию с R4COX, (R4CO2)O, R4CO2H или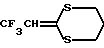
используя процедуру, аналогичную процедуре, описанной ранее для получения соединений формулы I из амидоксима формулы V до получения соответствующего фенола формулы IV.
Соединения формулы I, в которых R1 - гидроксиалкил, Y, R2 и R3 имеют указанные ранее значения, а R4 - дигалоидметил, тригалоидметил, циклоалкил, алкоксиалкил, 2,2,2-трифторэтил или аминогруппа, можно получить из соединения формулы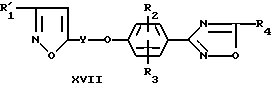
где R'1 представляет трет. -бутилдиметилсилилоксиалкил ((CH3)3CSi(Me)2-O-алкил), а Y, R2, R3 и R4 имеют указанные ранее в этом пункте значения, за счет отщепления трет.-бутилдиметилсилилового эфира.
Отщепление трет.-бутилдиметилсилилового эфира проводят, обрабатывая соединение формулы XVII сильной органической кислотой, например уксусной кислотой или трифторуксусной кислотой, или неорганической кислотой, например тетрагидрофураном или диоксаном в присутствии воды при температуре в интервале от около 20 до около 60oC.
Соединение формулы XVII, где R4 - дигалоидметил, тригалоидметил, циклоалкил, алкоксиалкил или 2,2,2-трифторэтил, можно получить способом, который включает осуществление взаимодействия фенола формулы IV, в котором R2 и R3 имеют определенные ранее значения, а R7 имеет значения, указанные ранее в этом пункте для R4, с изоксазолом формул XV и XVI
где R'1, Y и X2 имеют указанные ранее значения.
Фенол формулы IV подвергают взаимодействию с галоидизоксазолом формулы XVI, используя способ, аналогичный описанному ранее для получения цианосоединения IX из цианофенола VII и галоидизоксазола VIII.
Фенол формулы IV подвергают взаимодействию с изоксазолом формулы XV в присутствии диэтилазодикарбоксилата (ДЕАД) и трифенилфосфина в инертном растворителе, например тетрагидрофуране, хлороформе, диметилформамиде или N-метилпирролидиноне, при температуре в интервале от около -20 до около 20oC.
Промежуточное соединение - фенол формулы IV можно получить по способу, описанному ранее.
Промежуточные изоксазолы XV и XVI можно получить при взаимодействии изоксазола формулы X, где R1 - гидроксиалкил, с трет.-бутилдиметилсилилхлоридом до получения соответствующего трет.-бутилдиметилсилилового эфира формулы
где R'1 имеет указанные ранее значения, и взаимодействия производного щелочного металла соединения XVIII с этиленоксидом или X2-Y'-X2 соответственно.
Изоксазол формулы X, где R1 - гидроксиалкил, подвергают взаимодействию с трет. -бутил(диметил)силилхлоридом в присутствии 4-(диметиламино)пиридина и основания, например триэтиламина, пиридина или имидазола в сухом инертном растворителе, например метиленхлориде, хлороформе или тетрагидрофуране, при комнатной температуре до получения соединения XVIII. Изоксазол формулы XV получают при взаимодействии производного щелочного металла соединения XVIII с этиленоксидом, предпочтительно в присутствии хелатирующего агента, например N,N,N',N'-тетраметилэтилендиамина или гексаметилфосфортриамида в сухом инертном растворителе, например тетрагидрофуране, при температуре в интервале от около -78 до около 20oC. Производное щелочного металла получают in situ за счет взаимодействия соединения XVII с органическим основанием щелочного металла, например бутилллитием или литийдиизопропиламидом в безводных условиях.
Соединение формулы XVIII, где R4-дигалоидметил, тригалоидметил, циклоалкил, алоксиалкил или 2,2,2-трифторэтил, можно получить также, как и соединение формулы XVIII, где R4 является аминогруппой, в соответствии со схемой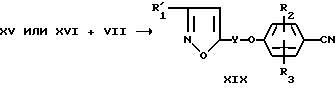

Реакцию соединения XV или XVI с цианофенолом формулы VII до получения соединения XIX ведут по способу, аналогичному описанному ранее для получения соединения XVII за счет взаимодействия фенола формулы IV с изоксазолом формулы XV или галоидизоксазолом формулы XVI, соответственно. Реакцию цианосоединения XIX с гидроксиламингидрохлоридом до получения амидоксима формулы XX и последнего с галоидангидридом, ангидридом кислоты, карбоновой кислотой, галоидцианом или кетен-1,3- пропандитиолацеталем до получения соединения XVIII можно вести способом, аналогичным описанному ранее для получения амидоксима формулы V из цианосоединения IX и для получения соединения формулы I из амидоксима формулы V.
Соединения формулы I, где R1 - гидрокси, Y, R2 и R3 имеют указанные ранее значения, а R4 - циклоалкил или алкоксиалкил, можно получить при взаимодействии соединения формулы
где R8 - алкил, а Y, R2, R3 и R4 имеют указанные ранее в этом пункте значения, с гидроксиламингидрохлоридом до получения соединения формулы I, где R1 - гидрокси.
Соединение XXI подвергают взаимодействию с гидроксиламингидрохлоридом в присутствии основания, например гидроксида натрия, и воды в спиртовом растворителе, например метиловом или этиловом спирте, при комнатной температуре в интервале температур от около 0 до около 25oC.
Промежуточные соединения формулы XXI можно получить при взаимодействии производного щелочного металла соединения III, где R6 имеет значения, указанные для R4 соединения XXI, с алкилгалоидформиатом, R8OCOX, где X имеет указанные ранее значения. Реакция идет в сухом инертном растворителе, например тетрагидрофуране или диоксане, при начальной температуре от около -78 до около -20oC с последующим нагреванием до около 20 - 25oC. Производное щелочного металла можно получить in situ при взаимодействии соединения III с органо-щелочным металлом, например бутиллитий- или литий-диизопропиламидом в безводных условиях.
Некоторые соединения формулы I являются промежуточными для других соединений формулы I, как указано ранее.
Галоидангидриды, алкилгалоидформиаты и ангидриды кислот используют в ранее описанных способах получения соединений формулы I и их промежуточных соединений, принадлежащих к хорошо известным классам соединений, и их можно легко получить известными способами.
Соединение формулы I, где R1 - алкил, циклоалкил или алкоксиалкил, Y, R2, R3 имеют указанные ранее значения, а R4 - алкокси или N=Q, где N=Q является алкиламино или диалкиламино, можно получить из соответствующих соединений формулы I, где R4 - трихлорметил. В том случае, если R4 является алкокси, трихлорметильное соединение подвергают взаимодействию с алкоксидом щелочного металла, например метоксидом натрия или этоксидом натрия, и в этом случае, если R4 является N=Q, с амином (HN=Q), в подходящем растворителе, например диметилформамиде или N-метилпирролидиноне, при комнатной температуре до получения соответствующего соединения формулы I, где R4 - алкокси, алкиламино или диалкиламино.
Соединения формулы I, где R1 - гидроксиалкил, Y, R2 и R3 имеют указанные ранее значения, а R4 - гидрокси, дигалоидметил, тригалоидметил, циклоалкил, гидроксиалкил, 2,2,2-трифторэтил или амино, можно получить из соответствующего соединения, где R1 - алкоксиалкил, за счет эфирного отщепления алкоксиалкильного фрагмента.
Алкоксиалкильное соединение обрабатывают триметилсилилиодидом в сухом инертном растворителе, например 1,2-дихлорэтане, хлороформе или ацетонитриле, при температуре в интервале от около 60 до около 80oC до получения соответствующего гидроксиалкильного соединения.
Соединения формулы I, где R1 - алкил, алкокси, циклоалкил или алкоксиалкил, Y, R2 и R3 имеют указанные ранее значения, a R4 является CON=Q, где N= Q является амино, алкиламино или диалкиламино, можно получить при взаимодействии соответствующего соединения формулы I, где R4 - алкоксикарбонил, с амином HN= Q в полярном растворителе, например этиловом спирте или N-метилпирролидиноне, при комнатной температуре до получения соответствующего соединения, где R4 является CON=Q.
Соединение формулы I, где R1 - алкокси, циклоалкил или алкоксиалкил, Y, R2 и R3 имеют указанные ранее значения, а R4 - циано, можно получить из соответствующего соединения, где R4 является CON=Q, где N=Q является амино, за счет обработки последнего трифторуксусным ангидридом в присутствии основания, например пиридина или триэтиламина, в сухом инертном растворителе, например тетрагидрофуране, хлороформе или 1,2-дихлорэтане, при температуре в интервале от около 0 до около 20oC.
Соединения формулы I, где R1 - алкокси или гидроксиалкокси, Y, R2 и R3 имеют указанные ранее значения, а R4 - алкокси, тригалоидметил, циклоалкил, алкоксиалкил, 2,2,2- трифторэтил или диалкиламино, можно получить этерификацией соответствующего соединения формулы I, где R1 - гидрокси. Этерификация протекает при взаимодействии гидроксильного соединения с алкилгалидом или гидроксиалкилгалидом, где галид является бромидом, хлоридом, или иодидом в присутствии основания, например карбоната калия или карбоната натрия, в сухом инертном растворителе, например ацетоне, бутаноне или ацетонитриле, при температуре в интервале от около 50 до около 90oC.
Соединения формулы I, где R1 - алкил, циклоалкил, гидроксиалкил, алкоксиалкил или гидроксиалкокси, Y, R2 и R3 имеют указанные ранее значения, а R4 - гидроксиалкил, можно получить трансэтерификацией соответствующего соединения формулы I, где R4 - алканкарбонилоксиалкил. Трансэтерификацию ведут, обрабатывая алканкарбонилоксиалкильное соединение неорганическим или органическим основанием, например карбонатом калия, бикарбонатом натрия или триэтиламином в спиртовом растворителе, например метиловом или этиловом спирте при комнатной температуре.
Соединения формулы I, где R1 - алкил, циклоалкил или гидроксиалкил, Y, R2 и R3 имеют указанные ранее значения, а R4 - гидроксиалкил, можно также получить эфирным отщеплением соответствующего соединения формулы I, где R4 является алкоксиалкилом. Эфирное отщепление можно вести, обрабатывая алкоксисоединение триметилсилилиодидом способом, аналогичным описанному для получения соединения формулы I, где R1 - гидроксиалкил.
Соединение формулы I, где R1 - алкил, алкокси, циклоалкил, гидроксиалкил, алкоксиалкил или гидроксиалкокси, Y, R2 и R3 имеют указанные ранее значения, а R4 - иодометил, можно получить из соответствующего соединения формулы I, где R4 - хлорметил, при взаимодействии с иодидом щелочного металла, например иодидом натрия. Реакция идет при обработке хлорметильного соединения иодидом щелочного металла, например иодидом натрия или калия, в инертном растворителе, например ацетоне или бутаноне, при температуре около 20oC.
Соединения формулы I, где R1 - алкил, алкокси, циклоалкил или алкоксиалкил, Y, R2 и R3 имеют указанные ранее значения, а R4 - (4-метилфенил)сульфонилоксиметил, получают из соответствующего соединения формулы I, где R4 гидроксиметил, при взаимодействии с (4-метилфенил)сульфонилгалоидом, где галоид является бромом, хлором или иодом, в присутствии неорганического основания, например карбоната калия или бикарбоната натрия. Реакция идет при взаимодействии реагентов в инертном растворителе, например метиленхлориде, хлороформе или 1,2-дихлорэтане, при температуре около 20oC.
Соединения формулы I являются достаточно основными для образования стабильных солей присоединения кислот, с сильными кислотами, и такие соли также входят в объем данного изобретения. Природа соли присоединения кислоты несущественна при условии, что она получена из аниона кислоты, которая практически не токсична по отношению к организмам животных. Соответствующие соли присоединения кислот включают гидрохлорид, гидробромид, сульфат, кислый сульфат, малеат, цитрат, тартрат, метансульфонат, пара-телуолсульфонат, додецилсульфат и циклогексансульфонат. Соли присоединения кислот получают обычными способами, известными специалистам.
В различных способах, описанных ранее для получения предлагаемых соединений, следует учитывать, что реакцию следует вести в течение промежутка времени, достаточного для того, чтобы обеспечить получение целевого продукта, и что для каждого конкретного типа реакции время реакции будет зависеть от одного или более из таких факторов, как, например, природа реагентов, используемый растворитель и/или температура проведения этой реакции.
Противовирусные соединения по изобретению создают для использования за счет приготовления разбавленного раствора или суспензии в фармацевтически приемлемой водной, органической или водно-органической среде для поверхностного или парантерального введения внутривенной или внутримышечной инъекцией, или офтальмологического применения или через нос, или приготавливают в виде таблеток или водных растворов с соответствующими эксцилиентами для орального приема.
Структуры предлагаемых соединений были установлены с помощью типов синтеза и элементного анализа, а также с помощью ИК спектроскопии, ЯМР и/или масс-спектров.
Далее изобретение будет описано со ссылкой на следующие примеры, которые ни коим образом не ограничивают объем изобретения.
Пример 1.
а) 3-(3-Метилизоксазол-5-ил)пропиловый спирт.
3,5-диметилизоксазол (X) (220 г, 2,27 моль) в 2,2 л тетрагидрофурана в атмосфере азота охлаждают при перемешивании до -75oC и за 1 ч добавляют 908 мл 2,5 М н-бутиллития (2,27 моль) в гексанах, поддерживая при этом температуру менее 65oC. Охлажденный раствор перемешивают в течение 30 мин после завершения добавления, а затем обрабатывают при -70oC раствором 112 г (2,54 моль) этиленоксида в 390 мл тетрагидрофурана за промежуток времени 1,5 ч, поддерживая при этом температуру около -65oC и перемешивая в течение ночи. Смесь при 8oC гасят при продолжении охлаждения в бане при 8oC, добавляя 1,2 и 2,5 М соляной кислоты за промежуток времени 20 мин, причем за это время температура повышается до 23oC, и перемешивают в течение 10 мин. Органическую фазу выделяют, промывают 500 мл воды и концентрируют до получения 147 г указанного в заглавии соединения в виде коричневого масла. Объединенные водные фазы (исходную + фазу промывки) экстрагируют метил-трет.-бутиловым эфиром (3 х 200 мл) и объединенные органические экстракты концентрируют до получения дополнительно 125 г указанного в заглавии соединения в виде коричневого масла.
b) 3-(3-Метилизоксазол-5-ил)пропилхлорид (VIII).
К продукту из части (a) (125 г, 0,885 моль) в 1225 мл метиленхлорида добавляют 192 мл 2,63 моль) тионилхлорида за промежуток времени около 1 ч, причем за это время температура повышается до 40oC при осторожном перемешивании. Нагревание при кипячении с обратным холодильником продолжают в течение 3 ч, реакционную смесь оставляют на ночь, а затем кипятят с обратным холодильником в течение 1 ч. Реакционную смесь добавляют равномерной струей к 3 кг ледяной воды при интенсивном перемешивании, причем перемешивание продолжают в течение 1 ч и выделяют водную фазу. К органической фазе добавляют 1 л воды, а затем 161 г твердого бикарбоната натрия порциями при интенсивном перемешивании. Органическую фазу выделяют и концентрируют в вакууме до получения черно-коричневого масла, которое очищают пленочной перегонкой до получения 94 г указанного в заглавии соединения в виде желтого масла, т. кип. 65oC/0,09 мм.
с) 3,5-Диметил-4-[3-(3- метилизоксазол-5-ил)пропилокси] -бензонитрил (IX).
Смесь 3,5 -диметил-4-гидроксибензонитрила (VIII) (7,36 г, 50,0 ммоль), сухого N-метил-пирролидинона (100 мл), измельченного карбоната калия (13,8 г, 100 ммоль), иодида калия (0,84 г, 5,0 ммоль) и продукта из части (b) 12,0 г, 75,0 ммоль) перемешивают при 60oC в течение 18 ч. После охлаждения до комнатной температуры полученную смесь разделяют между 200 мл воды и 100 мл этилацетата. Водный слой дважды экстрагируют порциями по 50 мл этилацетата. Объединенные органические экстракты промывают водой, рассолом, сушат над сульфатом магния и концентрируют в вакууме до получения 18,3 г желтого масла. Жидкостная хроматография среднего давления (силикагель 60, 50 х 460 мм, 25% этилацетат в гексанах) обеспечивает 12,7 г (94,1%) чистого указанного в заглавии соединения в виде твердого белого продукта 46 - 48oC (метанол).
d) 3,5-Диметил-4-[3-мeтилизoкcaзoл-5-ил)пропилокси] - N-гидроксибензолкарбоксимидамид (V).
Смесь продукта, полученного в части (c) (18,4 г, 68,1 ммоль), абсолютированного этанола (200 мл), измельченного карбоната калия (46,9 г, 0,34 моль) и гидроксиамингидрохлорида (23,6 г, 0,34 моль) кипятят с обратным холодильником в течение 18 ч. Горячую смесь фильтруют и оставшуюся твердую часть промывают горячим этанолом. Объединенные фильтраты концентрируют в вакууме до получения 19,4 г (93,9%) указанного в заглавии соединения в виде белого порошка, который был достаточной степени чистоты, чтобы быть использованным на последующих стадиях. Образец перекристаллизовывают из этанола до получения белого твердого продукта, т. пл. 129 - 130,5oC.
e) 5-{ 3-[2,6-Диметил-4- (5-трифторметил-1,2,4-оксадиазол-3-ил)фенокси] пропил} -3-метилизоксазол (I, R1 = CH3; Y = (CH2)3; R2 и R3 = 2,6-(CH3)2; R4 = CF3).
K раствору продута из части (d) (4,38 г, 14,4 ммоль) в 8,0 мл сухого пиридина добавляют 4,07 мл (28,8 ммоль) трифторуксусного ангидрида со скоростью, которая поддерживает умеренный рефлюкс. После добавления смеси дают остыть до комнатной температуры и разбавляют водой. Полученный твердый продукт промывают водой, сушат в вакууме и очищают хроматографически (силикагель 60, 15-40% этилацетат в гексанах) до получения 4,76 г чистого указанного в заглавии соединения в виде белого твердого продукта, т. пл. 61 - 62oC.
Пример 2.
a) 3,5-Дифтор-4-гидроксибензонитрил (VII).
Смесь 4-бром-2,6-дифторфенола (VI) (4,00 г, 19,0 ммоль), цианида меди (I) (1,72 г, 19,0 ммоль) и диметилформамида (40 мл) кипятят с обратным холодильником в течение 6 ч, охлаждают до комнатной температуры, разбавляют водой и фильтруют. Полученную желто-коричневую твердую часть промывают водой и оставляют. Объединенные фильтраты подкисляют (1 н. HCl) и экcтрагируют этилацетатом. Объединенные органические экстракты сушат над сульфатом магния, концентрируют в вакууме и очищают хроматографически с мгновенным испарением (силикагель 60, 20% этилацетат в гексанах) до получения 1,03 г чистого указанного в заглавии соединения в виде грязно-белого твердого продукта, т. пл. 195 - 197oC.
Полученный желто-коричневый твердый продукт суспендируют в этилацетат с небольшим количеством ацетона, фильтруют и концентрируют в вакууме. Полученный остаток разделяют между этилацетатом и 1 н. HCl. Водную фазу экстрагируют атилацетатом и объединенные органические фазы очищают, как было указано ранее, до получения дополнительных 0,43 г (49% полный выход) чистого указанного в заглавии соединения.
Следующие соединения получают способами, аналогичными способу примера 1 (с).
Пример 2b. 3,5-Дифтор-4-[3- (3-метилизоксазол-5-ил)пропилокси]-бензонитрил (IX), т. пл. 23 - 24,5oC (эфир/гексаны), полученный из 3,5-дифтор-4-гидрокси- бензонитрила (VII), и продукт примера Ib (VIII), выход 49,1%.
Пример 3a. 3,5-Дихлор -4-[3-(3-метилизоксазол-5-ил)пропилокси] -бензонитрил (IX), т. пл. 69,5 - 70,5oC (метанол) (белый твердый продукт), полученный из 3,5-дихлор-4-гидроксибензонитрила (VII), и продукт примера Ib (VIII), выход 80,7%.
Нижеследующие соединения получают способом, аналогичным способу примера 1d.
Пример 2с. 3,5-Дифтор-4-[ 3-(3-метилизоксазол-5-ил)пропилокси]-N-гидроксибензолкарбоксимидамид (V), т. пл. 122 - 124oC, получен из продукта примера 2b (IX), выход 86%. Сырой продукт очищают, суспендируя в 10% этаноле в хлороформе, фильтруя, концентрируя в вакууме и тщательно растирая полученный твердый белый продукт в холодном хлороформе.
Пример 3b. 5,5-Диxлop-4-[3-(3-мeтилизокcaзoл-5- ил)пропилокси]-N-гидроксибензолкарбоксимидамид (V) получают из продукта примера 3а (IX) (0,5 г), 0,78 продукта получают после концентрирования фильтратов в виде маслянистого твердого вещества и используют на следующей стадии.
Нижеследующие соединения получают способом, аналогичным примеру 1е.
Пример 2d. 5-{ 3-[2,6-Дифтор-4-(5-трифторметил-1,2,4- оксадиазол-3-ил)фенокси] пропил]-3-метилизоксазол (I; R1 = CH3, Y = (CH2)3; R2 и R3 = 2,6 - /F/2; R4 = CF3), т. пл. 36 - 37oC (гексаны) (белый твердый продукт) из продукта примера 2с (V) и трифторуксусного ангидрида; выход 44,5%.
Пример 3c. 5-{3-[2,6-Дихлор-4-(5-трифторметил-1,2,4-оксадиазол-3-ил) фенокси] пропил} -3-метилизоксазол (I; R1 = CH3; Y = (CH2)3; R2 и R3 = 2,6 - (Cl)2; R4-CF3), т. пл. 65 - 67oC (гексаны) (белый твердый продукт) - из продукта примера 3b (V) и трифторуксусного ангидрида; выход 80,5%.
Пример 4. 5-{ 3-[4-(5-Циклопропил-1,2,4-оксадиазол-3-ил)- 2,6-диметилфенокси] пропил} -3-метилизоксазол (I; R1 = CH3; Y = (CH2)3; R2 и R3 = 2,6-(CH3)2; R4 - циклопропил), т. пл. 85 - 88oC (метанол), (твердый белый продукт) - из продукта примера 1d (V) и циклопропанкарбонилхлорида; выход 71,0%.
Пример 5. 5-{ 3-[2,6-Диметил-4-(5-метоксиметил-1,2,4-оксадиазол-3-ил) фенокси] пропил} -3-метилизоксазол (I; R1 = CH3; Y = (CH2)3; R2 и R3 = 2,6-(CH3)2; R4 = CH2OCH3), т.пл. 63 - 64oC (эфир/гексан) (белый твердый продукт) - из продукта примера 1d (V) и метоксиацетилхлорида; выход 76,1%.
Пример 6. 5-{3-[ 2,6-Диметил-4-(5-фторметил-1,2,4-оксадиазол-3-ил)-фенокси] пропил} -3-метилизоксазол (I; R1 = CH3; Y = (CH2)3; R2 и R3 = 2,6-(CH3)2; R4 = CH2F), т.пл. 80 - 80,5oC (метанол) (белый твердый продукт) из продукта примера 1d (V) и фторацетилхлорида; выход 45,6%.
Пример 7. 5-{ 3-[2,6-Диметил-4-(5-этоксикарбонил-1,2,4- оксадиазол-3-ил)фенокси] пропил} -3-метилизоксазол (I; R1 = CH3, Y = (CH2)3, R2 и R3 = 2,6-(CH3)2; R4 = CO2CH2CH3), т.пл. 105 - 106oC (этилацетат/гексан) (белый твердый продукт) - из продукта примера 1d (V) и этилоксалилхлорида, выход 67,8%.
Пример 8. 5-{3-[2,6-Диметил-4-(5-оксо-4,5-дигидро-1,2,4- оксадиазол-3-ил)фенокси] пропил} -3-метилизоксазол (таутомер I, где R1 = CH3; Y = (CH2)3; R2 и R3 = 2,6-(CH3)2; R4 = OH).
К охлажденной (0oC) суспензии продукта примера 1d (V) (3,03 г, 10,0 ммоль), сухого ацетона (30 мл) и тонко измельченного карбоната калия (1,52 г, 11 ммоль) прикапывают раствор этилхлорформата (1,05 мл, 11,0 ммоль) в ацетоне (5,5 мл). После перемешивания при 0oC в течение часа реакционную смесь разбавляют водой (100 мл) и экстрагируют метиленхлоридом (3 х 25 мл). Объединенные органические экстракты промывают рассолом, сушат над сульфатом магния, фильтруют через короткую колонку Флорисил и концентрируют в вакууме до получения неочищенного интермедиата - О-ацильного производного в виде грязно-белого твердого продукта, который затем нагревают при 120 - 130oC в течение 45 мин до получения указанного в заглавии соединения (2,38 г, 75,4%), т. пл. 194 - 195oC (метанол), (белые иголки).
Нижеследующие соединения получают способом примера 8.
Пример 9. 5-{3-[2,6-Диметил-4-(5-метилкарбонилоксиметил-1,2,4-оксадиазол-3- ил)фенокси]пропил}-3-метилизоксазол (I; R1 = CH3; Y = (CH2)3; R2 и R3 = 2,6-(CH3)2; R4 = CH2OCOCH3), т. пл. 71 - 73oC (эфир/гексаны) (белое твердое вещество) - из продукта примера 1d (V) к ацетоксиацетилхлорида; выход 71,3%. Неочищенный продукт очищают хроматографически (силикагель 60, 35% этилацетат в гексанах).
Пример 10. 5-{3-[4-(5-Хлоpмeтил-1,2,4-oкcaдиaзoл-3-ил)-2,6- диметилфенокси] пропил} -3-метилизоксазол (I; R1 = CH3; Y = (CH2)3; R2 и R3 = 2,6- (CH3)2; R4 = CH2Cl), т. пл. 75 - 76oC (метанол), (белый твердый продукт) из продукта примера 1d (V) и хлорацетилхлорида. Неочищенный продукт очищают хроматографически (силикагель 60, 20% этилацетат в гексанах); выход 76,2%.
Пример 11. 5-{3-[2,6-Диметил-4-(5-(1-метилкарбонилоксиэтил)- 1,2,4-оксадиазол-3-ил)фенокси] пропил}-3-метилизоксазол (I; R1 = CH3; Y = (CH2)3; R2 и R3 = 2,6 -(CH3)2; R4 = CH(CH3/OCOCH3)), т. пл. 77 - 77,5oC (белый твердый продукт) - из продукта примера 1d (V) и 2-ацетоксипропионилхлорида; выход 64,6%.
Пример 12. 5-{ 3-[2,6-Диметил-4-(5-трихлорметил-1,2,4- оксадиазол-3-ил)-фенокси] пропил} -3-метилизоксазол (I; R1 = CH3; Y = (CH2)3; R2 и R3 = 2,6-(CH3)2; R4 = CCl3).
Трихлоруксусную кислоту (22,8 г, 140 ммоль) добавляют к продукту примера 1d (V) (10,6 г, 34,8 ммоль) и нагревают при 85oC до получения густого раствора. Трихлорацетилхлорид (14,5 мл, 69,6 ммоль) добавляют тремя равными порциями. После добавления первой порции начинается интенсивная реакция. Смесь нагревают в течение дополнительного часа при 94oC. Охлажденную смесь разбавляют водой и экстрагируют этилацетатом (3 х 25 мл). Объединенные органические фазы промывают насыщенным раствором бикарбоната натрия, сушат над сульфатом магния и концентрируют в вакууме до получения 10,1 г органического масла. После хроматографической обработки (силикагель 60, метиленхлорид) получают 6,94 г желтого масла, которое кристаллизуется из метанола до получения 5,03 г чистого соединения указанного в заглавии в виде белях иголок, т. пл. 77 - 77,5oC.
Пример 13. 5-{5-[4-(5-Дихлорметил-1,2,4-оксадиазол-5-ил)- 2,6-диметил-фенокси] пропил} -3-метилизоксазол (I; R1 = CH3; Y = (CH2)3; R2 и R3 = 2,6-(CH3)2; R4 = CHCl2).
Дихлоруксусную кислоту (1,24 мл, 15,0 ммоль) добавляют к продукту примера 1d (V) (1,14 г, 3,76 ммоль) и нагревают при 85oC до получения раствора. Быстро прикапывают дихлоруксусный ангидрид (1,14 мл, 7,52 ммоль) и перемешивают при 85oC в течение дополнительного часа. Посла обработки по способу примера 12 получают 1,51 г желто-коричневого масла, которое после хроматографической очистки (силикагель 60, 25% этилацетат в гексанах) дает 1,37 г (91,3%) чистого указанного в заглавии продукта в виде бледно-желтого масла, которое при стоянии отверждается, т. пл. 52 -53oC (этанол).
Пример 14. 5-{3-[4-(5-Дифтормeтил-1,2,4-oкcaдиaзoл-3-ил)- 2,6-диметилфенокси]пропил}-3-метилизоксазол (I; R1 = CH3, Y = (CH2)3; R2 и R3 2,6-(CH3)2; R4 = CHF2).
Дифторуксусную кислоту (0,31 мл, 5,0 ммоль) добавляют к холодному (-25oC) раствору N,N'-карбонилдиимидазола (0,80 г, 5,0 ммоль) в сухом тетрагидрофуране (5,0 мл). Спустя 5 мин полученную суспензию быстро прикапывают к раствору продукта примера 1d (V) в сухом тетрагидрофуране. Полученную смесь кипятят в течение 2 ч с обратным холодильником, охлаждают, разбавляют водой и экстрагируют этилацетатом (3 х). Объединенные органические фазы промывают водой, рассолом, сушат над сульфатом магния и концентрируют, в вакууме до получения 0,78 г бледно-желтого твердого вещества. После хроматографической обработки (силикагель 60, 30% этилацетат в гексанах) получают 0,55 г чистого указанного в заглавии соединения в виде бледно-желтого масла, которое при стоянии отверждается, т. пл. 70,5 - 71oC (метанол).
Пример -15. 5-{3-[4-(5-Имино-4,5-дигидро-1,2,4-оксадиазол-3- ил)-2,6-диметилфенокси] пропил} -3-метилизоксазол (таутомер I, где R1 = CH3; Y = (CH2)3; R2 и R3 = 2,6-(CH3)2; R4 = NH2).
Бромциан (1,17 г, 11,0 ммоль) добавляют порциями к смеси продукта примера 1d (V) (3,03 г, 10,0 ммоль) и бикарбонату калия (1,10 г, 11,0 ммоль) в 50% водном этаноле (8,0 мл).
Спустя 15 мин густую желтую суспензию разбавляют водой и фильтруют. Полученное твердое вещество желтого цвета промывают водой и эфиром до получения 1,48 г (45,1%) чистого указанного в заглавии соединения в виде желтого порошка, т. пл. 175 - 183oC.
Пример 16. 5-{ 3-[2,6-Диметил-4-(5-метокси-1,2,4, -оксадиазол-3-ил)фенокси] пропил} -3-метилизоксазол (I; R1 = CH3; Y = (CH2)3; R2 и R3 = 2,6-(CH3)2; R4 = OCH3).
Продукт примера 12 (I) (627 мг, 1,46 ммоль) добавляют к свежеприготовленному раствору метоксида натрия в метаноле (1,5 экв. натрия в 5 мл метанола) в сухом диметилформамиде (3 - 5 мл), и полученную смесь перемешивают при комнатной температуре в течение 15 - 30 мин. Реакционную смесь разбавляют водой и экстрагируют этилацетатом (3 х). Объединенные органические экстракты промывают водой, рассолом, сушат над сульфатом магния и концентрируют в вакууме. Неочищенный остаток (0,64 г) очищают хроматографически (силикагель 60, вначале 2% метанолом в метиленхлориде, а затем 5% этилацетатом в метиленхлориде) до получения чистого указанного в заглавии соединения (308 мг) в виде бесцветного масла, которое кристаллизуют из метанола; т. пл. 64,5 - 65,5oC (белое твердое вещество).
Пример 17. 5-{ 3-[2,6-Диметил-4-(5-этокси-1,2,4-оксадиазол- 3-ил)-фенокси] пропил} -3-метилизоксазол (I; R1 = CH3; Y = (CH2)3; R2 и R3 = 2,6 -(CH3)2; R4 = OCH2CH3).
По способу примера 16, но используя этоксид натрия в этаноле вместо метоксида натрия в метаноле, получают из продукта примера 12 (I) (905 мг, 2,10 ммоль) сырой остаток (0,82 г), который очищают хроматографически (силикагель 60, 2% этилацетат в метиленхлориде) до получения 0,52 г (69%) чистого указанного в заглавии соединения в виде желтого твердого продукта; т.пл. и 70 - 72,5oC (этанол).
Пример 18. 5-{3-[2,6-Диметил-4-(5-метилимино-4,5-дигидро-1,2,4-оксадиазол-3-ил) фенокси] пропил} -3-метилизоксазол(таутомер I, где R1 = CH3; Y = (CH2)3; R2 и R3 = 2,6-(CH3)2; R4 = NHCH3).
Продукт примера 12 (I) (1,00 г, 2,32 ммоль) добавляют к 5 мл 40% водного метиламина в диметилформамиде (3 - 5 мл), и полученную смесь перемешивают при комнатной температуре в течение 18 ч. Реакционную смесь разбавляют водой и экстрагируют этилацетатом (3 х). Объединенные органические экстракты промывают водой, рассолом, сушат над сульфатом магния и концентрируют в вакууме, неочищенный остаток (0,54 г) очищают хроматографически (силикагель 60, вначале 2% метанолом в метиленхлориде, а затем 50% этилацетатом в гексанах) до получения 300 мг (37,5%) чистого указанного в заглавии соединения в виде желтого твердого вещества; т. пл. 126,5 - 127oC (этанол).
Пример 19. 5-{ 3-[2,6-Диметил-4-(5-диметиламино-1,2,4- оксадиазол-3-ил)-фенокси] пропил} -3-метилизоксазол (I; R1 = CH3; Y = (CH2)3; R2 и R3 = 2,6-(CH3)2; R4 = N(CH3)2).
По способу примера 18, но используя 40% водный диметиламин вместо 40% водного метиламина и снижая время реакции до 15 - 30 мин, получают из продукта примера 12 (I) (0,97 r, 2,2 ммоль) неочищенный остаток (0,75 г), который очищают хроматографически (силикагель 60, 50% этилацетат) (гексаны) до получения 0,70 г (84 %) чистого указанного в заглавии соединения в виде бледно-желтого твердого продукта, т. пл. 123 - 124oC (этанол).
Пример 20. а) 3,5-Диметил-4-(3-этинилпропокси)бензонитрил (XII).
По способу примера 1с, используя 14,7 г (100 ммоль) 3,5-диметил-4-гидроксибензонитрил (VII) и заменяя продукт примера 1b (VIII) на 5-хлор-1-пентин (XI) (12,7 мл, 120 ммоль), получают красно-коричневое масло, которое очищают хроматографически (силикагель 60, 15% этилацетат в гексанах) до получения чистого указанного в заглавии соединения (21,2 г, 99,4%) в виде бледно-желтого масла.
b) 3,5-Диметил-4-(3-этинилпропокси)-N-гидроксибензолкарбоксимидамин (XIII).
По способу примера 1d, используя 13,0 г (61,0 ммоль) продукта из части (а), получают указанное в заглавии соединение (14,9 г, 99,3%) в виде белого твердого продукта, который имеет достаточную степень чистоты для использования на следующей стадии.
c) 3-[3,5-Диметил-4-(3-этинилпропокси)фенил] -5- трифторметил-1,2,4-оксадиазол (III).
По способу примера 1е, используя 7,40 г (30, 0 ммоль) продукта части (b), 9,0 мл сухого пиридина и 8,50 мл трифторуксусного ангидрида, получают чистое указанное в заглавии соединение (6,42 г, 65,9%) в виде бледно-желтого масла, которое кристаллизуют из метанола до получения указанного в заглавии соединения в виде белого твердого продукта, т.пл. 45,5 - 48oC.
Способ 1. Общий способ получения соединений примеров 21 - 23, 28a и 29a (далее).
К раствору N-хлорсукцинимида (NCS, 1,8 - 2,5 экв.) в сухом N,N-диметилформамиде или N-метилпирролидоне (1,6 - 3,0 мл на 1 ммоль NCS) и 1 - 2 капель пиридина, прикапывают раствор оксима (1,8 - 2,5 экв.) в том же растворителе (0,40 - 0,80 мл на 1 ммоль оксима). Внутреннюю температуру поддерживают при 25 - 30oC с помощью водной бани (25oC). Спустя час при комнатной температуре добавляют раствор соответствующего этинильного соединения (формула III или XII) (1 экв.) в том же самом растворителе (0,80 мл на ммоль этинильного соединения). Реакционную смесь нагревают до 85 - 90oC и прикапывают раствор триэтиламина (TEA, 1,8 - 2,5 экв.) в том же растворителе (0,80 - 1,6 мл на ммоль TEA) за 45 - 90 мин. Спустя еще час при 85 - 90oC полученную смесь охлаждают до комнатной температуры, разбавляют водой и экстрагируют этилацетатом (3 х). Объединенные органические фазы промывают 10% KHSO4, водой, рассолом, сушат над сульфатом (магния или натрия) и концентрируют в вакууме. Сырой продукт очищают хроматографически (силикагель 60, 15 - 40% этилацетата в гексанах).
Нижеследующие соединения получают по способу 1.
Пример 21. 5-{ 3-[2,6-Диметил-4- (трифторметил-1,2,4-оксадиазол-3-ил)фенокси] пропил}-3- (метоксиметил)изоксазол (I; R1 = CH2OCH3; Y = (CH2)3; R2 и R3 = 2,6 - (CH3)2; R4 = CF3), бесцветное масло (выход 70,1%) - из продукта примера 20с (III) (2,00 г, 6,17 ммоль) и метоксиацетальдегидоксима (1,10 г, 12,3 ммоль).
Пример 22. 5-{ 3-[2,6-Диметил-4-(5-трифторметил-1,2,4- оксадиазол-3-ил)фенокси] пропил}-3-этоксиметил)изоксазол (I; R1 = CH2OCH2CH3; Y = (CH2)3; R2 и R3 = 2,6 - (CH3)2; R4 = CF3), т.пл. 24 - 25oC (метанол) (белый порошок), выход 35,3%, из продукта примера 20с (III) (2,00 г, 6,17 ммоль) и 2-этоксиацетальдегидоксима (1,27 г, 12,3 ммоль).
Пример 23. 3-Циклопропил-5-{3-[2,6-диметил-4-(3- трифторметил-1,2,4-оксадиазол-3-ил)фенокси] пропил}-изоксазол (I; R1 = циклопропил; Y = (CH2)3; R2 и R3 = 2,6-(CH2)3; R4 = CF3) т. пл. 63,5 - 65oC (этанол) (белые иголки) (выход 82%) - из продукта примера 20с (III) (0,92 г, 2,8 ммоль) и циклопропилкарбоксальдегидоксима (048 г, 5,6 ммоль).
2-Этоксиацетальдегидоксим (использованный ранее в примере 22).
Раствор гидроксиламингидрохлорида (18,8 г, 0,270 моль), этанола (25 мл), воды (40 мл) и 1,1,2-триэтоксиэтан нагревают при 45oC в течение 30 мин, охлаждают до комнатной температуры и экстрагируют эфиром (3 х). Объединенные органические фазы сушат над сульфатом магния, концентрируют в вакууме и фильтруют через небольшие комки ваты до получения 10,1 г указанного в заглавии соединения в виде бледно-желтого масла, которое используют без дальнейшей очистки.
Пример 24. 5-Циклопропил-3-[3,5-диметил-4-(3- этинилпропокси)фенил]-2,2,4-оксадиазол (III).
По способу примера 1е, используя 5,00 г (20,3 ммоль) продукта примера 20b (XIII), 75 мл сухого пиридина и 2,77 мл (30,5 ммоль) циклопропилкарбонилхлорида, получают чистое указанное в заглавии соединение (3,98 г, 66,2%) в виде почти бесцветного масла, которое отверждается при стоянии, т. пл. 45 - 46oC (метанол).
Способ 2. Общий способ получения соединений примеров 25 - 27 (далее).
К охлажденному (0oC) раствору соответствующего альдегидоксима (2,5 экв.) в сухом диметилформамиде (ДМФ) (XX) (15 мл) добавляют 1 порцию N-хлорсукцинимида (NCS) (2,5 экв.). Спустя 1 - 2 ч продукт примера 24 (1 экв.) добавляют и все это нагревают до 80oC. Раствор триэтиламина (2,5 экв.) в сухом ДМФ (5 мл) прикапывают за 90 мин. Полученную смесь нагревают еще 18 ч. После обработки по способу примера 21 получают чистый продукт.
Нижеследующие соединения получают по способу 2.
Пример 25. 5-[3-(5-Циклопропил-1,2,4-оксадиазол-3- ил(-2,6-диметил-фенокси)пропил] -3-этилизоксазол (I; R1 = CH2CH3; Y = (CH2)3; R2 и R3 = 2,6-(CH3)2; R4 = циклопропил), бесцветное масло - из продукта 24 (III) и пропиональдегидоксима; выход 67%.
Пример 26. 5-{3-[5-(Циклoпpoпил-1,2,4-oкcaдиaзoл-3-ил- 2,6-димeтил-фенокси]пропил}-3-(метоксиметил)-изоксазол (I; R1 = CH2OCH3; Y = (CH2)3; R2 и R3 = 2,6-(CH3)2; R4= циклопропил), т.пл. 44 - 45oC (метанол) (белый твердый продукт) - из продукта примера 24 (III) и метоксиацетальдегидоксима; (выход 26,1% из комбинации двух опытов).
Пример 27. 5-{ 3-[5-(Циклопропил-1,2,4-оксадиазол-3-ил)-2,6- диметил-фенокси]пропил}-3-циклопропилизоксазол (I; R1 = R4 = циклопропил; Y= (CH2)3; R2 и R3 = 2,6-(CH3)2), т.пл. 59 - 60oC (метанол) (белый твердый продукт) - из продукта примера 24 (III) и циклопропилкарбоксальдегидоксима; выход 60,4% .
Пример 28.
а) 3,5-Диметил-4-[5-(3-этилизоксазол-5- ил)пропилокси]-бензонитрил (IX).
По способу примера 1 (приведенному ранее), но опуская пиридин и используя пропиональдегидоксим (8,6 г, 118 ммоль) и продукт примера 20а (XII) (10,1 г, 47,0 ммоль), получают 4,90 г (36,7%) чистого соединения, указанного в заглавии, т. пл. 53,5 - 54,5oC (этанол).
b) 3,5-Диметил-4-[3-(3-этилизоксазол-5-ил] пропионилокси] -N- гидроксибензолкарбоксимидамид (V).
Смесь продукта части (а) (2,01 г, 7,50 ммоль), этанола (20 мл), гидроксиламингидрохлорида (2,61 г, 37,5 ммоль) и тонко измельченного карбоната калия (5,20 г, 37,5 ммоль) кипятят с обратным холодильником в течение 18 ч. Полученную смесь фильтруют в горячем виде, фильтровальную лепешку промывают этанолом и объединенные фильтраты концентрируют в вакууме до получения 2,57 г неочищенного указанного в заглавии соединения в виде пастообразного желтого твердого продукта, который используют далее без дополнительной обработки.
с) 5-{ 3-[2,6-Диметил-4-(5-трифторметил-1,2,4-оксадиазол- 3-ил)фенокси] пропил} -3-этилизоксазол (I; R1 = CH2CH3; Y = (CH2)3; R2 и R3 = 2,6-(CH3)2; R4 = CF3).
Весь продукт из части (b) растворяют в пиридине (2,3 мл) и трифторуксусном ангидриде (2,1 мл, 15 ммоль) добавляют по каплям. Полученную смесь кипятят с обратным холодильником в течение 1 ч, охлаждают до комнатной температуры, разбавляют водой и экстрагируют метиленхлоридом (3 х). Объединенные органические фаза промывают 1 н. HCl, водой, рассолом, сушат над сульфатом магния и концентрируют в вакууме, получают бледно-желтое масло (2,15 г), его обрабатывают хроматографически (силикагель 60, метиленхлорид) до получения 2,10 г (70,7%) чистого соединения, указанного в заглавии, в виде белого твердого продукта, т. пл. 137 - 138oC (метанол).
Пример 29.
a) 3,5-Димeтил-4-{3-[3-(2-мeтoкcиэтил)изoкcaзoл-5- ил]-пропилокси}бензонитрил (IX).
По способу 1 (изложенному ранее), используя 3-метокси-пропиональдегидоксим (1,94 г, 18,8 ммоль) и продукт примера 20а (XII) (2,20 г, 10,3 ммоль), получают 1,51 г (46,5%) чистого указанного в заглавии соединения в виде бесцветного масла, которое кристаллизируют из этанола в виде тонких белых игл, т.пл. 64 - 64,5oC. Выделяют 0,89 г (40,4%) исходного продукта примера 20a.
b) 5-{ 3-[2,6-Димeтил-4-(5-трифторметил-1,2,4-оксадиазол- 3-ил)фенокси] пропил} -3-(метоксиэтил)изоксазол (I; R1 = CH2CH2OCH3; R2 и R3 = 2,6-(CH3)2; Y = (CH2)3, R4 = CF3).
Натрий (442 мг, 19,2 мг-атом) растворяют в сухом метаноле (20 мл), содержащемся в дополнительной воронке. Этот раствор прикапывают к раствору гидроксиламиногидрохлорида (1,34 г 19,2 ммоль) в сухом метаноле (10 мл). Образуется тонкий0 белый осадок. Спустя 1 ч добавляют раствор продукта из части (а) (1,21 г, 3,85 ммоль) в сухом метаноле (5 мл) и полученную смесь нагревают при кипячении с обратным холодильником в течение 2,5 ч. Горячую реакционную смесь фильтруют, фильтровальную лепешку промывают метанолом и объединенные фильтраты концентрируют в вакууме, получают белое маслянистое твердое вещество, которое растворяют в пиридине (4 мл) и добавляют трифторуксусный ангидрид (1,63 мл, 11,6 ммоль) с такой скоростью, чтобы поддержать осторожное кипение с обратным холодильником, полученную смесь нагревают при кипении с обратным холодильником еще 30 мин, охлаждают до комнатной температуры, разбавляют водой и экстрагируют этилацетатом (3 х). Объединенные органические фазы промывают 10% KHSO4, водой, рассолом, сушат над сульфатом магния и концентрируют в вакууме до получения 2,27 г желтого масла. В результате хроматографической обработки (силикагель 60, 30% этилацетат в гексанах) получают 1,28 г (78%) чистого указанного в заглавии соединения в виде бесцветного масла. В результате кристаллизации из метанола получают белое твердое вещество, т.пл. 36,5 - 37oC.
3-метоксипропиональдегидоксим (использован ранее в примере 29а).
К раствору гидроксиамингидрохлорида (2,80 г, 40,2 ммоль), 10% водного ацетата натрия (4,0 мл) и воды (6 мл) добавляют 1,1,3-триметоксипропан (2,12 мл, 14,9 ммоль) и нагревают при 40-50oC в течение 30 мин. После охлаждения до комнатной температуры раствор насыщают хлоридом натрия и экстрагируют эфиром (3 х) и метиленхлоридом (3 х). Объединенные органические фазы сушат над сульфатом магния, фильтруют через Флорисил и концентрируют в вакууме до получения 1,6 г указанного в заглавии соединения в виде бесцветного масла, которое используют без дальнейшей очистки.
Способ 3. Общий способ для получения соединений примеров 30a и 30b, 31a и 31b, 32a и 32b.
Смесь соответствующего 4-гидроксибензонитрила (VII) (1 экв.), сухого этанола (3,7 - 8,9 мл на ммоль 4- гидроксибензонитрила), гидроксиамингидрохлорида (5 экв.) и тонко измельченного карбоната калия (5 экв.) кипятят с обратным холодильником при эффективном перемешивании в течение 18 ч. Горячую реакционную смесь фильтруют и фильтровальную лепешку промывают этанолом. Объединенные фильтраты концентрируют в вакууме до получения неочищенных амидоксимов (XIV), который растворяют в пиридине (1 - 2 мл на ммоль 4-гидроксибензонитрила). Добавляют трифторуксусный ангидрид (5 экв.) с такой скоростью, чтобы поддержать осторожный рефлюкс. После нагревания дополнительно 0,5 - 3 ч охлажденную реакционную смесь разбавляют этилацетатом и водой (4:1) до гомогенизации, органическую фазу экстрагируют холодным 1 н.KOH (3 х). Основные экстракты подкисляют концентрированной HCl и экстрагируют этилацетатом (3 х). Объединенные органические фазы промывают рассолом, сушат над сульфатом магния, концентрируют в вакууме. В результате хроматографической обработки (силикагель 60, этилацетат в гексанах или Флорисил, метиленхлорид) получают чистый 4- гидроксифенил-5-трифторметил-1,2,4-оксадиазол.
По способу 3 получают следующие неочищенные промежуточные амидоксимы и соответствующие 4-гидрокси-5-трифторметил-1,2,4- оксадиазолы.
Пример 30a. 3,5-Диметил-4, N -дигидроксибензолкарбоксимидамид (XIV) из 3,5-диметил-4- гидроксибензонитрила.
Пример 30b. 3-(3,5-Диметил-4-гидроксифенил)-5-трифторметил-1,2,4-оксадиазол (IV), т.пл. 114 -115oC (гексан) (белые иголки) - из продукта примера 30a; выход 75,2%.
Пример 31a. 3,5-Дихлор-4, N- дигидроксибензолкарбоксимидамид (XIV) из 3,5-дихлор-4- гидроксибензонитрила.
Пример 31b. 3-(3,5-Дихлор-4-гидроксифенил)-5-трифторметил- 1,2,4-оксадиазол (IV), т. пл. 96 - 98oC (гексан) (белые иголки)- из продукта примера 31a; выход 52,0%.
Пример 32a. 4,N-Дигидроксибензолкарбоксимидамид (XIV)- из 4- гидроксибензонитрила.
Пример 32b. 3-(4-гидроксифенил)-5-трифторметил-1,2,4- оксадиазол (IV), т. пл. 74 - 75oC (гексанов) (белые иглы) из продукта примера 32a; выход 56,4oC.
Пример 30с. 5-{ 5-[2,6-Диметил-4-(5-трифторметил-1,2,4- oкcaдиaзoл-3-ил)-фенокси] пентил} -3-метилизоксазол (I; R1 = CH3; Y = (CH2)5; R2 и R3 = 2,6- (CH3)2; R4 = CF3).
По способу примера 1c, но заменяя на продукт примера 30b (IV) (1,0 г, 3,9 ммоль) 3,5- диметил-4-гидроксибензонитрил и на 5-(3-метилизоксазол-5-ил) пентилбромид (VIII) (1,0 г, 4,3 ммоль) 3-(3-метилизоксазол-5- ил)пропилхлорид и используя 0,72 г (4,3 ммоль) иодида калия, получают 0,25 г (16%) чистого указанного в заглавии соединения в виде белого твердого вещества т. пл. 41 - 42oC (метанол).
Пример 31с. 5-{ 5-[2,6-Диxлop-4-(5-тpифтopмeтил-1,2,4- oкcaдиaзoл-3-ил)-фенокси] пентил} -3-метилизоксазол (I; R1 = CH3; Y = (CH2)5; R2 и R3 = 2,6-(Cl)2; R4 = CF3).
По способу примера 1c, но заменяя 3,5-диметил-4-гидроксибензонитрил на продукт примера 31b (IV) (0,93 г, 3,1 ммоль) и 3-(3-метилизоксазол-5-ил)пропилхлорид на 5-(3-метилизоксазол-5-ил)пептилбромид (VIII) (1,0 г, 4,3 ммоль) и используя 0,72 г (4,3 ммоль) иодида калия, получают 0,83 г (60%) чистого указанного в заглавии соединения в виде белого твердого вещества т. пл. 42 - 43oC (гексаны).
Пример 32с. 3-Метил-5-{3-[4-(5-трифторметил-1,2,4-оксадиазол-3- ил)-фенокси]пропил}изоксазол (I; R1 = CH3; Y = (CH2)3; R2 = R3=H; R4 = CF3).
По способу примера 1с, но заменяя 3,5-диметил-4-гидроксибензонитрил на продукт примера 32b (IV) (0,42 г, 1,8 ммоль) и используя 0,63 г (4,0 ммоль) продукта примера 1b (VIII) и 0,67 г (4,0 ммоль) иодида калия, получают после тщательного растирания в холодном метаноле 0,48 г (76%) чистого указанного в заглавии соединения в виде белого порошка т. пл. 68 - 69oC (метиленхлорид-гексаны).
Пример 33. 5-{ 3-[2,6-Диметил-4-(5-трифторметил-1,2,4- оксадиазол-3-ил)-фенокси] пропил} -3-(2-гидроксиэтил)изоксазол (I; R1 = CH2CH2OH; Y = (CH2)3; R2 и R3 = 2,6-(CH3)2; R4 = CF3).
Раствор продукта примера 29b (I) (1,28 г, 3,00 ммоль), сухого 1,2-дихлорэтана (9 мл) и триметилиодида (1,71 мл, 12,0 ммоль) кипятят с обратным холодильником в течение 4 ч. К охлажденной реакционной смеси добавляют метанол (8 мл). Полученную смесь разбавляют водой и экстрагируют этилацетатом (3 х). Объединенные органические фазы промывают 10% NaHSO3 насыщенным NaHCO3, рассолом, сушат над сульфатом магния и концентрируют в вакууме. В результате хроматографической обработки (силикагель 60, 50% этилацетата в гексанах) получают 1,11 г (90,2%) чистого указанного в заглавии соединения в виде бесцветного масла, которое отверждается при стоянии, т. пл. 74,5 - 75oC (метанол) (белый твердый продукт).
Пример 34.
а) 3-(трет.- Бутилдиметилсилилоксиметил)-5-метилизоксазол (XVIII).
К охлажденному (5oC) раствору 3-гидроксиметил- 5-метилизоксазола (X) (16,8 г, 148 ммоль) в трет.бутил-диметилсилилхлориде (24,6 г, 163 ммоль) в сухом метиленхлориде (100 мл) добавляют за 15 мин раствор триэтиламина (22,7 мл, 163 ммоль) в метиленхлориде (25 мл). 4-Диметиламинопиридин (1,81 г, 14,8 ммоль) добавляют и густую реакционную смесь перемешивают при комнатной температуре в течение 48 ч. Добавляют 100 мл воды и водный слой экстрагируют метиленхлоридом (3 х). Объединенные органические фазы промывают рассолом, сушат над сульфатом магния, фильтруют через фильтр, состоящий из слоя Флорисила и слоя силикагеля 60, и концентрируют в вакууме. Полученное желтое масло (36,6 г) очищают хроматографически (силикагель 60, 2% этилацетат в гексанах) до получения 27,7 г (81,9%) чистого указанного в заглавии соединения в виде бледно-желтого масла.
b) 3- 3-(трет. -Бутилдиметилсилилоксиметил)изоксазол-5-ил] -пропиловый спирт (XV).
К холодному (-78oC) раствору продукта из части (a) (13,0 г, 57,0 ммоль) и N, N, N',N'-тетраметилэтилендиамина (1,2 мл, 7,9 ммоль) в сухом тетрагидрофуране (THF) (150 мл) добавляют за 5 мин н-бутиллитий (31,3 мл, 2,0 М в гексане). Яркий оранжево-желтый анионный раствор перемешивают в течение 25 мин. Этиленоксид (50,0 мл 7,6 M раствор в сухом THF) добавляют за 10 мин. Спустя 1,5 ч добавляют насыщенный NH4Cl (30 мл). Полученную смесь оставляют нагреваться до комнатной температуры и разбавляют водой. Водный слой экстрагируют этилацетатом (3 х). Объединенные органические фазы промывают рассолом, сушат над сульфатов магния, фильтруют через силикагель 60 и концентрируют в вакууме. После хроматографической обработки (силикагель 60, 20% этилацетат в гексанах) получают 3,44 г выделенного продукта из части (а) и 8,18 г (52,7%) чистого указанного в заглавии соединения в виде бесцветного масла.
с) 3-(трет. -Бутилдиметилсилилоксиметил)-5- {3-[2,6-диметил-4-(5- трифторметил-1,2,4-оксадиазол-3-ил)-фенокси]пропил}-изоксазол (XVII).
Раствор продукта примера части (b) (XV) (1,00 г, 3,67 ммоль), продукт примера 30b (IV) (1,04 г, 4,04 ммоль) и трифенилфосфин (1,06 г, 4,04 ммоль) в сухом тетрагидрофуране (THF) (10 мл) охлаждают до 0oC. Раствор диэтилазодикарбоксилата (ДЕАД) (0,61 мл, 1,04 ммоль) в сухом THF (15 мл) прикапывают в течение 20 мин. Раствор перемешивают в течение 30 мин при 0oC и 18 ч при комнатной температуре, разбавляют водой и экстрагируют этилацетатом (2 х). Объединенные органические фазы промывают 10% NaOH, рассолом, сушат над сульфатом магния, фильтруют через силикагель 60 и концентрируют в вакууме до получения 3,44 г желтого масла, после хроматографической обработки (силикагель 60, 10% этилацетат в гексанах) получают 1,73 г (83,6%) чистого соединения, указанного в заглавии, в виде бесцветного масла.
d) 5-{ 3-[2,6-Диметил-4-(5-трифторметил-1,2,4-оксадиазол-3- ил)фенокси] пропил} -3-(гидроксиметил)изоксазол (I; R1 = CH2OH; Y = (CH2)3; R2 и R3 = 2,6-(CH3)2; R4 = CF3).
Раствор продукта из части (с) (XVII) (0,75 г, 1,5 ммоль), тетрагидрофуран (60 мл) и 1 н. HCl (7,5 мл) перемешивают при комнатной температуре в течение 18 ч и разбавляют водой (100 мл), pH устанавливают 7 (pH бумага) твердым NaHCO3 и экстрагируют этилацетатом (3 х). Объединенные органические фазы промывают рассолом, сушат над сульфатом магния и концентрируют в вакууме до получения 0,73 г желтого масла, которое очищают хроматографически (силикагель 60, 50% этилацетат в гексанах) до получения 0,58 г (100%) чистого указанного в заглавии соединения в виде белого твердого продукта, т. пл. 92 - 93oC (белые иглы из этанола).
Пример 35.
а) 3,5-Диметил-4-{ 3-[3-(трет.-бутилдиметилсилилоксиметил)- изоксазол-5-ил]пропилокси}бензонитрил (XIX.)
К охлажденному (0oC) метиленхлоридному (25 мл) раствору 3,5-диметил-4-гидроксибензонитрила (VII) (773 мг, 5,26 ммоль), продукту из примера 34b (XV) (1,43 г, 5,26 ммоль) и трифенилфосфину (1,38 г, 5,26 ммоль) прикапывают за 30 мин раствор диэтилазодикарбоксилата (ДЕАД) (915 мг, 5,26 ммоль) в метиленхлориде (5 мл). Этот раствор перемешивают при 0oC в течение 50 мин и при комнатной температуре в течение 18 ч, после чего его промывают водой, 2,5 M NaOH, рассолом, сушат над сульфатом натрия и концентрируют в вакууме. Остаток тщательно растирают в эфире до получения трифенилфосфиноксида, полученный фильтрат концентрируют в вакууме, а остаток очищают хроматографически (силикагель 60, 15% этилацетат в гексанах) до получения 1,73 г (82,2%) чистого указанного в заглавии соединения в виде бесцветного масла.
b) 3,5-Диметил-4-{ 3 -[3 -(трет.-бутилдимeтилсилилоксимeтил)- изоксазол-5-ил]пропилокси}-N- гидроксибензолкарбоксимидамид (XX).
Смесь продукта из части (а) XIX (1,22 г, 3,05 ммоль), этанола (30 мл) гидроксиламингидрохлорида (1,06 г, 15,2 ммоль) и тонкоизмельченного карбоната калия (2,10 г, 15,2 ммоль) кипятят с обратным холодильником в течение 5 ч и фильтруют. Фильтровальную лепешку промывают этанолом и объединенные фильтраты концентрируют в вакууме до получения 1,30 г белого твердого продукта. Часть этого материала (0,78 г) очищают хроматографически (силикагель, обращенная фаза, 17% вода в метаноле) до получения 0,47 г указанного в заглавии соединения, которое содержит приблизительно 5% (анализ ЯМР) десилилированного материала.
c) 5-{ 3-[4-(5-Циклопропил-1,2,4-оксадиазол-3-ил)-2,6- диметилфeнoкcи] пpoпил} -3-(гидpoксиметил)изoкcaзoл (I; R1 = =CH2OH; Y = (CH2)3; R2 и R3 = 2,6- (CH3)2, R4 = циклопропил).
K раствору очищенного продукта из части (b) (XX) (0,47 г, 1,1 ммоль) в пиридине (20 мл) добавляют циклопропилкарбонилхлорид (0,15 мл, 1,6 ммоль). Полученную смесь нагревают при 90oC в течение 26 ч. Пиридин удаляют в вакууме, а остаток разделяют между водой и этилацетатом. Водную фазу экстрагируют этилацетатом (3 х). Объединенные органические фазы промывают 3 н. HCl (2 x), рассолом, сушат над сульфатом натрия и концентрируют в вакууме до получения 0,61 г желтого масла. В результате хроматографической обработки (силикагель 60, 35% этилацетат в гексанах) получают 0,25 г (62%) чистого указанного в заглавии соединения в виде бесцветного масла. После кристаллизации из метиленхлорида и гексанов получают указанное в заглавии соединение в виде белого твердого продукта, т. пл. 80 - 81oC.
Пример 36.
а) 5-Циклопропил-3-[4-(5-этоксикарбонил-4- пентинилокси)-3,5-диметилфенил]-1,2,4-оксадиазол (XXI).
К холодному (-78oC) раствору в сухом тетрагидрофуране (20 мл) продукта примера 24 (III) (1,30 г, 4,41 моль) прикапывают за 15 мин н-бутиллитий (2,30 мл, 2,3 М в гексане). Спустя еще 30 мин при -78oC добавляют этилхлорформиат (0,63 мл, 6,6 ммоль) и полученную смесь медленно нагревают до 0oC за 2 ч. Реакцию гасят насыщенным NH4Cl и экстрагируют этилацетатом (3 х). Объединенные органические фазы промывают рассолом, сушат над сульфатом натрия и концентрируют в вакууме до получения бесцветного масла (2,05 г). После хроматографической обработки (силикагель 60, 10 - 20% этилацетат в гексанах) получают 1,38 г (85,0%) чистого соединения, указанного в заглавии, в виде бесцветного масла.
b) 5-{ 3-[4-(5-Циклопропил-1,2,4-оксадиазол-3-ил)-2,6- диметилфенокси] пропил} -3-гидроксиизоксазол (I; R1 = OH; Y = (CH2)3; R2 и R3 = 2,6-(CH3)2; R4 = циклопропил).
Смесь продукта из части (а) (XXI) (810 мг, 2,20 ммоль), 15 мл этанола, гидроксиламингидрохлорида (400 мг, 5,76 ммоль) и 10% NaOH (5 мл) перемешивают при комнатной температуре в течение 24 ч (после 8 ч получают раствор). Добавляют 6 мл воды, полученную смесь подкисляют концентрированной HCl до pH 2 (pH бумага) и экстрагируют эфиром (4 х). Объединенные органические фазы промывают рассолом, сушат над сульфатом натрия и концентрируют в вакууме до получения белого твердого продукта. После хроматографической обработки (силикагель 60, 50% этилацетат в гексанах) получают 0,55 г (70%) чистого указанного в заглавии соединения в виде белого твердого вещества, т. пл. 155 - 156oC (этилацетат и гексаны).
Пример 37. 5-{3-[4-(5-Циклопропил-1,2,4-оксадиазол-3-ил)-2,6-диметилфенокси] пропил} -3-этоксиизоксазол (I; R1 = OCH2CH3; Y = (CH2)3; R2 и R3 = 2,6-(CH3)2; R4 = циклопропил).
Смесь продукта примера 36b (I) (0,30 г, 0,85 ммоль), сухого ацетона (25 мл), тонко измельченного карбоната калия (0,24 г, 1,7 ммоль) и этилиодида (0,18 мл, 2,2 ммоль) нагревают при 50oC в течение 18 ч, фильтруют и концентрируют в вакууме до получения розоватого твердого вещества. После храматографической обработки (силикагель, 50% этилацетат в гексанах) получают 0,19 г не совсем чистого указанного в заглавии соединения и 0,12 г (37%) чистого побочного продукта (соответствующего 2,3- дигидро-2-этил-3-оксоизоксазольному соединению) в виде бесцветного масла. Чистое указанное в заглавии соединение получают после хроматографической очистки (силикагель с обращенной фазой, 20% вода в метаноле), выход 0,14 г (43%), т. пл. 70 -71oC (метанол).
Пример 38. 5-{3-[4-(5-Аминокарбонил-1,2,4-оксодиазол-3-ил)- 2,6-диметилфенокси] пропил} -3-метилизоксазол (I; R1 = CH; Y = (CH2)3; R2 и R3 = 2,6-(CH3)2; R4 = CONH2).
Тонко измельченный продукт примера 7 (I) (3,08 г, 8,00 ммоль) добавляют к 10% этанольному аммиаку (80 мл). Спустя 15 мин получают раствор и начинается образование тонкого осадка. Спустя 4 ч полученную смесь фильтруют и твердую часть промывают холодным этанолом до получения 2,35 г (82,5%) чистого указанного в заглавии соединения в виде тонкого белого порошка, т. пл. 177 -178oC (изопропилацетат).
Пример 39. 5-{3-[4-(5-Циано-1,2,4-оксадиазол-3-ил)-2,6- диметилфенокси] пропил} -3-метилизоксазол (I; R1 = CH3; Y = (CH2)3; R2 и R3 = 2,6-(CH3)2; R4 = CN).
K охлажденной (0oC) суспензии продукта примера 38 (I) (1,60 г, 4,50 ммоль) в сухом пиридине (11,2 мл) в сухом тетрагидрофуране (27 мл) добавляют трифторуксусный ангидрид (1,90 мл, 13,5 ммоль). Полученную смесь перемешивают при 0oC в течение 4 ч и при комнатной температуре в течение 18 ч, разбавляют 100 мл воды и экстрагируют этилацетатом (2 х 25 мл). Объединенные органические фазы промывают 1н. HCl (3х), рассолом, сушат над сульфатом магния и концентрируют в вакууме. Подученное твердое вещество (1,67 г) хроматографируют (силикагель 60, 20% этилацетат в гексанах) до получения 1,38 г (90,8%) чистого указанного в заглавии соединения в виде белого твердого вещества, т. пл. 93 - 94oC (этилацетат в гексанах).
Пример 40. 5-{3-[2,6-диметил-4-(5-гидроксиметил) -1,3,4-оксадиазол-3-ил)фенокси] пропил}-3-метилизоксазол (I; R1 = CH3; Y = (CH2)3; R2 и R3 = 2,6-(CH3)2; R4 = CH2OH).
Смесь продукта примера 9 (I) (4,12 г, 10,7 ммоль) и тонко измельченного карбоната калия (1,48 г, 10,7 ммоль) в 40 мл сухого метанола перемешивают при комнатной температуре в течение 15 мин и разделяют между водой (30 мл) и этилацетатом (30 мл). Водную фазу экстрагируют этилацетатом (1 х 25 м) и объединенные органические фазы промывают рассолом, сушат над сульфатом магния и концентрируют в вакууме. После хроматографической обработки (силикагель 60, 50% этилацетат в гексанах) получают 3,35 г (91,2%) чистого указанного в заглавии соединения в виде белого твердого вещества, т. пл. 116,5 - 117oC (эфир).
Пример 41. 5-{3-[2,6-Диметил-4-(5-(иодометил)-1,2,4- оксадиазол-3-ил)-фенокси] пропил}-3-метилизоксазол (I; R1 = CH3; Y = (CH2)3; R2 и R3 = 2,6-(CH3)2; R4 = CH2I).
Раствор иодида натрия (0,45 г, 3,0 ммоль) в сухом ацетоне (5 мл) прикапывают к раствору продукта примера 10 (I) (905 мг, 2,50 ммоль) в сухом ацетоне (5 мл). Спустя 4 ч полученную желтую суспензию выливают в воду (50 мл) и экстрагируют метиленхлоридом (3 х 25 мл). Объединенные органические фазы промывают рассолом, сушат над сульфатом магния и концентрируют в вакууме до получения коричневого масла (1,56 г). После фильтрования через Флорисил (метиленхлорид) получают зеленовато-желтое масло (1,43 г), которое отверждается при стоянии при 0oC. После хроматографической обработки (силикагель 60, 25% этилацетат в гексанах) получают 1,06 г (93,8%) чистого, указанного в заглавии соединения в виде бледно-желтого твердого вещества т. пл. 89 -90oC (белые иглы из смеси эфир-пентана).
Пример 42. 5-{ 3-[2,6-Диметил-4-15-(4-метилфенилсульфонилоксиметил)-1, 2-(4-оксадиазол-3-ил)фенокси] пропил} -3-метилизокcазол (I; R1 = CH3; Y = (CH2)3; R2 и R3 = 2,6-(CH3)2; R4 = 4-CH3C6H4SO2OCH2).
K охлажденной (0oC) смеси продукта примера 40 (I) (343 мг, 1,00 ммоль) и тонко измельченного карбоната калия (0,28 г, 2,0 ммоль) в сухом метиленхлориде (5 мл) прикапывают отфильтрованный раствор (4-метилфенил)сульфонилхлорида (0,23 г, 1,2 ммоль) в метиленхлориде (2 мл). Полученную смесь перемешивают при комнатной температуре в течение 72 ч, после чего добавляют дополнительно 0,40 ммоль карбоната калия и (4-метилфенил)сульфонилхлорида. Спустя 24 ч полученную смесь разделяют между водой (10 мл) и этилацетатом (10 мл). Органическую фазу промывают 1 н. NaOH (1 х 5 мл), рассолом, сушат над сульфатом магния и концентрируют в вакууме. После хроматографической обработки (силикагель 60, 40% этилацетат в гексанах) получают 478 мг (96,1%) чистого указанного в заглавии соединения в виде белого твердого вещества т. пл. 97 - 98oC (эфир).
Пример 43. 5-{ 3-[2,6-Диметил-4-(5-(2,2,2-трифторэтил) -1,2,4-оксадиазол-3-ил)фенокси] пропил}-3-метилизоксазал (I; R1 = CH3; Y = (CH2)3; R2 и R3 = 2,6-(CH3)2; R4 = CH2CF3).
Смесь продукта примера 1d (V) (4,55 г, 15,0 ммоль), сухого тетрагидрофурана (45 мл), 2-трифторэтилиден-1,3-дитиана (3,60 г, 18,0 ммоль) и трифторацетата серебра (7,3 г, 33 ммоль) кипятят с обратным холодильником в темноте в течение 22 ч, охлаждают до комнатной температуры и фильтруют. Фильтровальную лепешку зеленого цвета промывают этилацетатом (4 х 20 мл). Объединенные фильтраты концентрируют в вакууме.
Полученный остаток растворяют в метиленхлориде (50 мл) и промывают водой (3 х 25 мл), 0,1 М NaHCO3 (cвежеприготовленным, 25 мл), рассолом, сушат над сульфатом магния, фильтруют через Флорисил и концентрируют в вакууме до получения 5,39 г пасты желтого цвета. После хроматографической очистки (силикагель 60, 15% этилацетат в гексанах) получают 2,22 г (37,5%) чистого указанного в заглавии соединения в виде твердого белого вещества, т. пл. 84 - 85oC (метанол) (белые пластинки).
Пример 44. 5-{3-[4-(5-Циклопропил-1,2,4-оксадиазол-3-ил)- 2,6-диметилфенокcи] пропил} -3-(2-гидроксиэтокси)изоксазол (I; R1 = HOCH2CH2O; Y = (CH2)3; R2 и R3 = 2,6-(CH3)2; R4 = циклопропил).
Смесь продукта примера 36b (I) (0,75 г, 2,1 ммоль), сухого ацетона (25 мл), тонко измельченного карбоната калия (0,32 г, 2,3 ммоль) и 2-бромэтанола (0,19 мл, 2,7 ммоль) кипятят с обратным холодильником в течение 5 ч, фильтруют и концентрируют в вакууме до получения розоватого масла. После хроматографической обработки (силикагель 60, 50% этилацетата в гексане) получают 0,51 г не совсем чистого соединения, указанного в заглавии и 1,48 г (57%) чистого побочного продукта (соответствующего 2,3-дигидро-2- (2-гидроксиэтил)-3-оксо-изоксазольному соединению) в виде твердого вещества белого цвета. Чистое указанное в заглавии a соединение (0,31 г, 37%) получают в результате градиентного элюирования (силикагель 60, гексаны до 50% этилацетат в гексанах), т. пл. 64 - 65oC (метиленхлорид в гексанах).
По способу примера 1c, но заменяя 3,5-диметил-4-гидроксибензонитрил (VII) на эквивалентные количества следующих соединений:
4-гидрокси-3-нитробензонитрила;
4-гидрокси-3,5-диметоксибензонитрила;
4-гидрокси-3-трифторметилбензонитрила,
получают, соответственно, следующие соединения формулы IX:
4-[3-(3-метилизоксазол-5-ил)пропилокси]-3-нитробензонитрил;
3,5-диметокси-4-(3-[3-метилизоксазол-5-ил]пропилокси)- бензонитрил;
4-[3-(3-метилизоксазол-5-ил)пропилокси]-3- (трифторметил)-бензонитрил.
По способу примера 1d, но заменяя продукт примера 1с (IX) на эквивалентные количества вышеуказанных соединений формулы IX можно получить следующие соединения формулы V:
4-[3-(3-метилизоксазол-5-ил)пропилокси] -3-нитро- N-гидроксибензолкарбоксимидамид;
3,5-диметокси-4-[3-(3- метилизоксазол-5-ил)пропилокси]-N-гидроксибензолкарбосимидамид;
4-[3-(3-метилизоксазол-5-ил] пропилокси)-3-трифторметил- N-гидроксибензолкарбоксимидамид.
По способу примера 1е, но заменяя продукт примера 1d (V) на эквивалентные количества вышеуказанных соединений формулы V, получают соответственно следующие соединения формулы I:
3-метил-5-{3-[2- нитро-4-(5-трифторметил-1,2,4-оксадиазол-3- ил)фенокси] пропил}изоксазол (I; R1 = CH3; Y = (CH2)3; R2 = 2-NO2; R3 = H, R4 = CF3);
5-{ 3-[2,6-диметокси-4- (5-трифторметил-1,2,4-оксадиазол-3-ил)фенокси] пропил}-3-метилизоксазол (I; R1 = CH3; Y = (CH2)3; R2 и R3 = 2,6-(OCH3)2; R4 = CF3);
3-метил-5-{ 3-[2-тpифтopмeтил-4- (5-тpифтоpметил-1,2,4-оксадиазол-3-ил)фенокси]пропил}изоксазол (I; R1 = CH3; Y = (CH2)3; R2 = 2-CF3; R3 = H; R4 = CF3).
По способу примера 20a, b и c, используя эквивалентные количества реагентов в каждом случае, но заменяя в примере 20а 5-хлор-1-пентин на 11-хлор-1-ундецин, получают следующие соединения:
3,5-диметил-4-(9-этинилнонилокси)бензонитрил (XII);
3,5-диметил-4- (9-этинилнонилокси)-N-гидрокси-бензолкарбоксимидамид (XIII);
3-[3,5-диметил-4-(9-этинилнонилокси)пентил] -5-трифторметил- - 1,2,4-оксадиазол (III).
По способу примера 1, используя эквивалентные количества ацетальдегидоксима и 3-[3,5- димeтил-4-(9-этинилнoнилокси)-фенил] -5-трифторметил-1,2,4-оксадиазола (III), можно получить 5-{9-[2,6-диметил-4-(5-трифторметил-1,2,4- окcадиазол-3-ил)фенокси]нонил}-3-метилизоксазол (I; R1 = CH3; Y = (CH2)9; R2 и R3 = 2,6-(CH3)2; R4 = CF3).
По способу 1, используя эквивалентные количества н-гексилальдегидоксима и продукта примера 20с (III), можно получить 5-{3-[2,6-диметил-4-(5-тpифтopметил)фенокси]пpoпил}-3- (н-пентил)изоксазол (I; R1 = (CH2)4CH3; Y = (CH2)3; R3 и R2 = 2,6-(CH3)2; R4 = CF3).
По способу примера 37, но заменяя на эквивалентное количество н- пентилбромида этилиодид, получают 5-{3-[4-(5-циклопропил-1,2,4- оксадиазол-3-ил)фенокси] пропил} -3-пентилоксиизоксазол (I; R1 = O(CH2)4CH3; Y = (CH2C)3; R3 и R2 = 2,6-(CH3)2; R4 = циклопропил).
По способу примера 37, но заменяя на эквивалентные количества продукта примера 40 (I) на н-пентилбромид, продукт примера 36 (b) (I) на этилиодид, соответственно, получают 5-{ 3-[2,6-диметил-4-[5-(н- пентилоксиметил)-1,2,4-оксодиазол- 3-ил] -фенокси]пропил}-3- метилизоксазол (I; R1 = CH3; Y= (CH2)3; R2 и R3 = 2,6-(CH3)2; R4 = CH2O(CH2)4CH3).
По способу примера 1е, но заменяя на эквивалентное количество циклогексилкарбонилхлорида трифторуксусный ангидрид, получают 5-{3-[4- (5-циклогексил-1,2,4-оксадиазол-3-ил)-2,6-диметилфенокси] пропил} -3- метилизоксазол (I; R1 = CH3; Y = (CH2)3; R3 и R2 = 2,6-(CH3)2; R4=циклогексил).
Биологическая оценка представительных соединений формулы I показала, что они обладают противовирусной активностью. Они пригодны для ингибирования репликации вируса in vitro и особенно активны против пикорнавируса, особенно риновирусов. Тестирование in vitro представительных соединений по изобретению против пикорновирусов показало, что репликация вирусов ингибируется при минимально ингибирующей концентрации (MIC) 0,002 - 9,608 мкг/мл.
Тестирование проводили следующим образом: MIC значения определяли в автоматическом анализе по инфицированию тканевой культуры в дозе 50% (TCID-50), HeLa (Висконсин) клетки в 96 ячеечных пластинах инфицировали разбавлениями вируса, что эмпирически дало 80 - 100% цитопатического эффекта (CPE) за 3 дня в отсутствии лекарств. Соединения, подлежащие тестированию, сериально разбавляли в циклах 10, 2 - кратных и добавляли инфицированные клетки. После 3 дней инкубирования при 33oC и 2,5% CO2 клетки фиксировали 5%-ным раствором глутаральдегида, а затем подкрашивали 0,25%-ным раствором кристаллфиолета в воде. Затем пластину споласкивали, сушили и количественно определяли оставшийся в ячейках краситель (мера интактных клеток) с помощью оптического декситометра, MIC определяли как концентрацию соединения, которая защищает 50% клеток от индуцированного вирусом CPE по отношению к необработанному контрольному вирусному препарату.
В вышеописанных тестовых процедурах представительные соединения формулы I тестировали против блока из 15 серотипов риновирусов человека (HRV), а именно HRV -2, -14, -1A, -1B, - 6, -21, -22, -15, -25, -30, -50, -67, -89, -86 и -41, и определяли значения MIC, выраженные в мкг/мл для каждого серотипа риновируса. Было обнаружено, что тестированные соединения демонстрируют противовирусную активность против одного или более из этих серотипов.
Значения MIC (мкг/мл), полученные для соединения примера 1e, вышеописанной тестовой процедуре, представлены в таблице.

Новые 1,2,4-оксадиазолилфеноксиалкилизоксазолы получают взаимодействием амидоксима (N-гидроксикарбооксиимидамида) с галоидангидридом кислоты или алкилгалоидформиатом в присутствии органического основания в инертном растворителе. 5-{ 3-[2,6-диметил-4-(5-трифторметил-1,2,4-оксадиазол-3-ил)фенокси] про- пил} -3-метилизоксазол, т.пл. 61 - 62oC. Соединения могут быть использованы в композициях против пикорновирусов. 2 с. и 6 з.п. ф-лы, 1 табл.
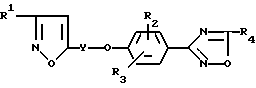
где R1 - алкил, алкокси, гидрокси, циклоалкил, гидроксиалкил, алкоксиалкил или гидроксиалкокси;
Y - алкилен, содержащий от 3 до 9 атомов углерода;
R2 и R3 - независимо водород, алкил, алкокси, галоид, трифторметил или нитро;
R4 - алкокси, гидрокси, галоидметил, дигалоидметил, тригалоидметил, циклоалкил, алкоксикарбонил, гидроксиалкил, алкоксиалкил, алканкарбонилоксиалкил, циано, 2,2,2-трифторэтил, (4-метилфенил)сульфонилоксиметил;
N=Q или CON=Q, где N=Q является амино, алкиламино или диалкиламино,
или его фармацевтически приемлемые аддитивные соли кислот.
4. Соединение по п.3, в котором R4 представляет C1-3-алкокси, фторметил, дигалоидметил, тригалоидметил, циклоалкил или C1-3-алкокси - C1-3-алкил.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| J | |||
| Heterocyclic chem., 1976, 13, 1079 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Adv | |||
| Heterocyclic Chem., 197 6, 20,65 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| SU, 1593568, C 07 D 413/12, 1990 | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| EP, 413289, C 07 D413/12 , 1991 | |||
| Кипятильник для воды | 1921 |
|
SU5A1 |
| US, 4942241, C 07 D 271/06, 1990. | |||

