Данное изобретение относится к получению и использованию в диагностике по изображению, в частности по рентгеновскому изображению, контрастных агентов, включающих многоядерные частицы, и к контрастным средам, содержащим подобные частицы.
Все диагностические изображения основаны на достижении различных уровней сигнала, получаемого от различных структур, находящихся внутри тела. Так, например, в рентгеновском изображении для того, чтобы данная структура тела была видна на изображении, ослабление рентгеновских лучей этой структурой должно отличаться от ослабления рентгеновских лучей окружающими тканями. Различие между сигналами от данной структуры тела и от ее окружения часто называют контрастом, и были затрачены большие усилия на поиски средств, позволяющих улучшить контраст в диагностическом изображении, поскольку чем выше контраст между структурой тела и ее окружением, тем выше качество изображений и больше их значение для врача, ставящего диагноз. Кроме того, чем больше контраст, тем меньше те структуры тела, которые можно увидеть в процессе получения изображения, то есть повышенный контраст может привести к улучшенному пространственному разрешению.
Диагностическое качество изображений в значительной степени зависит от уровня шумов, присущих способу получения изображения, и, таким образом, коэффициент эффективного качества диагноза для диагностического изображения, очевидно, представляет собой соотношение уровня контраста и уровня шумов.
Улучшение этого коэффициента качества диагноза было в течение длительного времени и остается до сих пор важной задачей. В таких методах, как рентген и ультразвук, одним из способов улучшения коэффициента качества диагноза является введение улучшающих контраст веществ, или контрастных агентов, в область тела, изображение которой собираются получить.
Так, в случае рентгена одним из ранних примеров контрастных агентов были нерастворимые неорганические соли бария, которые улучшали ослабление рентгеновских лучей в тех областях тела, где эти соли были распределены. Позже в качестве контрастных агентов для рентгеновских изображений стали преобладать растворимые иодсодержащие соединения, например, выпускаемые фирмой Nycomed AS под торговыми названиями Омнипак и Амипак.
Работы, проводившиеся в последнее время в области контрастных агентов для рентгеновских изображений, были в значительной степени сосредоточены на хелатах аминополикарбоновых кислот (АПКК) с ионами тяжелых металлов, и после выяснения того, что эффективное изображение многих участков тела требует локализации на этих участках относительно высоких концентраций ионов металла, были сделаны предположения, что для достижения этого можно использовать полихелаты, то есть вещества, содержащие более одной хелатной группировки.
Еще позже было обнаружено, что особенно эффективно улучшение контраста может быть достигнуто при использовании многоядерных комплексов, то есть комплексов, в которых сама связанная в комплексе группировка включает два или более улучшающих контраст атомов, или, для рентгена, два или более атомов тяжелых металлов. См. W091/14460 и W092/17215.
Для большей ясности слово "атом" относится к ионно или ковалентно связанным формам, а не просто к изолированным незаряженным атомам. Более того, следует понимать, что связанная в комплекс группировка, хотя и является многоядерной, не столь велика, как можно было бы предположить о самих частицах. Так, обычно она имеет максимальные размеры около 80 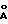 или менее, в основном 40
или менее, в основном 40 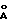 или менее.
или менее.
Данное изобретение относится к усовершенствованиям в области таких многоядерных группировок и обеспечивает в первую очередь способ получения соединений формулы
MnBuAv [I]
(где MnBuAv является многоядерной частицей; каждое М представляет собой атом тяжелого металла из группы W, Mo, Ta, Nb и Hf; каждый атом металла M ковалентно связан по меньшей мере с одним, предпочтительно с 2-6 атомами; каждое В представляет собой атом, образующий мостиковую связь, ковалентно связанный по меньшей мере с двумя, предпочтительно с 2 или 3 атомами металла M; каждое A, которые могут быть одинаковыми или разными, представляет собой атом, не образующий мостиковой связи, ковалентно связанный с атомом металла M; n и u - положительные целые числа, имеющие значение 2 или более; v представляет собой нуль или положительное целое число) или солей этих соединений, особенно физиологически приемлемых солей, включающий взаимодействие M(CO)6 и I2 в течение времени и при температуре, достаточных для образования кластера металл/иод и, возможно, замещения одного или более не образующих или образующих мостиковые связи атомов иода в указанном кластере одним или более других атомов или радикалов с образованием модифицированного кластера и/или для образования соли указанного кластера или модифицированного кластера.
В вышеприведенной формуле 1 n, u и v равны предпочтительно от 2 до 30, более конкретно от 2 до 10, в частности от 2 до 8; для n более предпочтительны значения от 2 до 6.
Некоторые из кластеров, которые могут быть получены указанным способом, являются новыми веществами и сами представляют собой аспект данного изобретения. Так, в изобретении предложены соединения формулы
MnBuAv [II]
где n равно 3, 4 или 5, предпочтительно 4 или 5; M, A, n и v определены выше, В представляет собой иод; а также соли, предпочтительно физиологически приемлемые соли данных соединений. Известны комплексы, в которых n равно 6.
M в формулах (I) и (II) предпочтительно является атомом молибдена, а особенно предпочтительно - атомом вольфрама. А и В предпочтительно представляют собой иод, но могут также быть и другими галогенами, такими как фтор, хлор или бром; атомом кислорода, например, в составе молекулы воды, молекулы спирта (например, этанола), сульфоната или ацетата трифторметана; атомом серы, например в составе изотиоцианата; атомом азота, например в составе аминного радикала или аминокислоты; или атомом фосфора, например в составе фосфорсодержащего радикала. Когда А и В являются кислородом, серой, азотом или фосфором, они могут образовывать часть большего лиганда, например, лиганда, образующего хелаты, как более подробно обсуждается далее. Если В представляет собой углерод, кислород, серу, азот или фосфор, он не обязательно должен являться частью большего лиганда; в этом случае он просто будет образовывать мостиковую структуру.
Другой аспект данного изобретения обеспечивает контрастную среду для диагностического изображения, включающую соединение формулы (I), полученное вышеуказанным способом, или соединение формулы (II), образующее комплекс с одной или более молекулами лиганда.
В следующем аспекте данного изобретения предложены соединения формулы (I), полученные вышеуказанным способом, и соединения формулы (II) для использования в качестве контрастных агентов для диагностического изображения.
Еще в одном аспекте данного изобретения предложена контрастная среда для диагностического изображения, включающая соединение формулы (I), полученное вышеуказанным способом, или соединение формулы (II), образующее комплекс с одной или более молекулами лиганда, совместно по меньшей мере с одним стерильным фармацевтическим носителем или инертным наполнителем.
Еще в одном аспекте данного изобретения предложен способ получения изображения тела человека или животного, предпочтительно млекопитающего, который включает введение в упомянутое тело физиологически переносимого количества улучшающего контраст соединения формулы (I), полученного вышеуказанным способом, или соединения формулы (II), образующего комплекс с одной или более молекулами лиганда, и получение изображения, предпочтительно рентгеновского, по меньшей мере указанной части упомянутого тела.
Соединения металл/иод формулы (I) и (II) особенно предпочтительны для контрастной среды, так как они содержат два компонента, хорошо рассеивающих рентгеновские лучи - тяжелый металл и иод. Таким образом, основанная на этих комплексах контрастная среда является уникальной, обеспечивая радиологу возможность выбора энергий рентгеновского излучения, которые можно использовать, обеспечивая таким образом оптимизацию радиологической процедуры.
Твердофазную реакцию между M(CO6) и I2 для получения соединений согласно данному изобретению осуществляют путем нагревания реагентов, например при 140oC, чтобы удалить газообразный CO, с последующим нагреванием аморфной смеси для получения бинарных кластерных фаз. Использование более низких температур (140-220oC) приводит к образованию твердых фаз, содержащих от 3 до 4 атомов металла на кластер; увеличение температуры (250-550oC) дает фазы пятиядерных и шестиядерных кластеров. Дискретные молекулярные кластеры получают путем пространственного уменьшения и/или непосредственного перевода в раствор этих фаз кластеров.
Замещение атомов, не образующих мостиковые связи, другими атомами/группами с целью получения модифицированного кластера можно осуществить известными способами.
Например, ранее было предложено осуществить реакцию W(CO)6 и I2 для получения смешанных соединений вольфрам/иод. Однако все полученные соединения были твердыми фазами, а не дискретными кластерами. См. J.Less Common Metals 22, 136 (1970); Z. Anorg. Allg. Chem. 516. 196 (1984); Virmani et al. J. Chem. Soc. Dalton Trans., 399 (1974); Djordjevic et al., J. Chem. Soc. (A), 16 (1966); и Inorg. Chem. 12, 2356 (1973).
Соединения с формулами (I) и (II) могут иметь следующие предпочтительные структуры: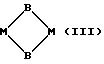
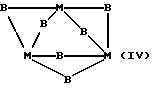
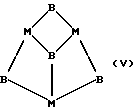
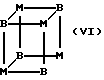
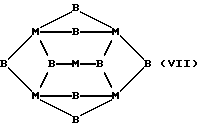
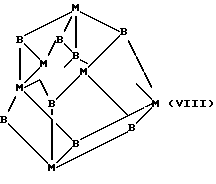
где каждое В, которые могут быть одинаковыми или различными, представляет собой образующий мостиковую связь атом иода, а каждое M - атом металла, и где другие не образующие мостиковые связи атомы, ковалентно связанные с атомами металла M, опущены из соображений наглядности.
В случае мостиковых структур этих формул структурные формулы можно записать, соответственно, M2(μ2B)2,M3(μ2B)6,M4(μ3B)4 и M6(μ3B)8(μ3B указывает, например, что В является образующим мостиковую связь атомом, связанным с 3 металлами). Как указано выше, особенно предпочтительно, чтобы эти соединения образовывали хелатные комплексы, и чрезвычайно предпочтительно, чтобы для координации по меньшей мере двух, а предпочтительно всех центров, входящих в состав лигандов, был использован один полидентатный хелатообразующий агент.
Предложенным в изобретении способом при использовании W(CO)6 можно получить бинарные фазы и/или молекулярные кластеры с центральными элементами [W3I6] 2+, [W4I7]3+, [W5I8]3+,4+, [W5(C)I8]4+. Ряд таких соединений получен непосредственно в результате перевода в раствор двух фаз, которые мы обозначаем фаза А и фаза В. Эти материалы ранее не были исследованы, хотя, как упомянуто выше, реакции между исходными веществами, W(CO)6 и иодом, были исследованы при различных условиях. Фаза А может быть получена, когда W(CO)6 и 7 эквивалентов иода нагревают при 140oC, с выделением CO; промывкой этого твердого вещества, например эфиром, удаляют непрореагировавший иод и получают отмытую фазу А. Остаточный CO присутствует в слишком малых концентрациях, чтобы быть зафиксированным с помощью инфракрасной спектроскопии. Как установлено с помощью дифракции рентгеновских лучей на порошке, отмытая фаза А обычно содержит небольшое количество нерастворимого кристаллического W4I13, в то время как ее основные компоненты аморфны. Фазу В получают непосредственно из фазы А путем нагревания в запаянной трубке, например, в течение 50 часов при 200oC. Черно-серый продукт промывают (и здесь применим эфир) для удаления непрореагировавшего иода. И здесь рентгеновская дифракция на порошке показала, что единственным кристаллическим компонентом фазы В являются случайные следы W4I13; главная составляющая аморфна.
Молекулярный [W3I9] 1- можно получить непосредственно из отмытой фазы А путем перемешивания ее в ТГФ в течение 24 часов и добавления (Bu4N)I к фильтрату. Полученное твердое вещество можно промыть этанолом для удаления трииодидных солей; эту процедуру лучше всего проводить быстро, так как сам кластер слабо растворим в этаноле. Конечный продукт можно перекристаллизовать из дихлорметана. Масс-спектроскопия при помощи метода FAB (бомбардировка быстрыми атомами) показала основные пики, соответствующие [W6I18] 1-, [W6I17]1-, и [W6I16]1-, с предположением, что твердое вещество, исходное для [W3I9]1-, содержит кластер в форме димера.
В ходе реакции между W(CO)6 и иодом при температурах в интервале 140-200oC получали переменные количества W4I13. Другие продукты, образовавшиеся при этой температуре, легко удалялись повторной промывкой эфиром или ТГФ. Оставшееся черное твердое вещество является кристаллическим W4I13, которое, как показано, не растворяется в обычных растворителях и кислотах. Это соединение образует молекулярные кристаллы, в которых два элемента [W4Ii7Ia2]1+ связаны двумя трииодидными ионами. Две половины такого двойного кластера существуют в асимметричной ячейке; половины каждого двойного кластера связаны центром инверсии.
Когда фаза В полностью растворена в этаноле по истечении 18 часов, новый кластер, [W5I13]1-, может быть выделен в молекулярной форме, например в виде его (Pr4N)+ соли с 31% выходом очищенного вещества (в расчете на W(CO)6). Двойную фазу W5I16 наблюдали, когда фазу А нагревали при несколько более высоких температурах (250-300oC), чем при получении фазы В. Появление этой фазы при более высоких температурах предполагает ее присутствие в аморфном виде в случае получения при более низкой температуре. Мы рассматриваем W5I16 (или возможно W5I14·I2) как логического предшественника [W5I13]1-; обе частицы содержат вольфрам в средней степени окисления 2,4+. Действительно, параллельно с фазой В материал, содержащий W5I16, также дает [W5I13]1- при обработке этанолом и иодидом.
Соединение (Pr4N) [W5I13] растворимо в ТГФ, дихлорметане и ацетоне. Если ярко-зеленый раствор в дихлорметане обработать металлическим цинком в течение 24 часов, раствор приобретает коричнево-красную окраску. При добавлении избытка катиона можно выделить соединение (Pr4N)2[W5I13]. Это соединение в твердом виде стабильно на воздухе; оно растворимо в ТГФ, дихлорметане, ацетоне и ацетонитриле, но его растворы медленно окисляются на воздухе до [W5I13]1-.
Когда фаза А реагирует с избытком CsI при 300oC в течение 50 часов, образуется коричнево-черная твердая смесь, содержащая [W6I14]2-, иодид цезия и CsW6CI16. Кристаллическая структура этой фазы показывает, что она является молекулярной по своей природе и содержит индивидуальные кластеры [W5(C)I13] 1-; асимметричная ячейка содержит один кластер, один Cs+ и полторы молекулы иода.
Когда твердую смесь экстрагируют ацетонитрилом и экстракт обрабатывают (Bu4N)I, (Bu4N)+ соли [W6I14]2- и [W5(C)I13)1- соосаждаются. Экстракция этого материала тетрагидрофураном с последующей перекристаллизацией остатка из ТГФ/гексана дает чистый (Bu4N) [W5(C)I13]. Очевидно, некоторое количество остаточного CO восстановительно расщепляется в ходе образования [W5(C)I13] 1-. Возможным промежуточным соединением является [W5(CO)I13]z-, образующееся в реакционной трубке при высоком давлении СО. Это соединение растворимо в ТГФ, дихлорметане и ацетоне.
Выдерживание фазы В при 550oC в течение 50 часов приводит к осаждению W6I12 в виде оранжевого твердого вещества на одном конце реакционной трубки с 25% выходом (в расчете на W(CO)6). Структура W6I12 доказывает его двумерную природу и наличие связи [W6Ii8Ia2Ia-a4/2. Ядро [W6I8]4+ имеет знакомую гранецентрированную октаэдрическую структуру, обнаруженную для ячеек [M6X8]2+ с М = Mo и W.
Когда неотмытую фазу В нагревают при 550oC в течение 50 часов, а полученное твердое вещество промывают эфиром, то получают красно-коричневый W6I16. Структура этой фазы имеет такие же двумерные связи, как W6I12, содержит между слоями две молекулы иода на кластер. Обогащенная иодом природа этой фазы объясняется присутствием свободного иода в неотмытой фазе В.
Когда неотмытую фазу В нагревают в интервале 400-500oC, на горячем конце трубки образуется смесь, содержащая W6I12, W6I16 и новую фазу, черный W6I18 (WI3). Кристаллы последней фазы иногда наблюдаются при нагревании металлического вольфрама и иода при 600-800oC. И при тех, и при других условиях выход W6I18, идентифицированного по форме кристаллов (стержни) после начального определения структуры, был таким низким, что его нельзя было определить с помощью рентгеновской дифракции на порошке. В предыдущем сообщении (Z. Anorg. Allg. Chemie 516, 196 (1984)) описан синтез "серебристых игл", анализ которых дал WI3.0 при двух различных методиках получения в запаянной трубке: (1) реакция металлического вольфрама и иода при градиенте температуры 500-350oC; (2) химический перенос "WI3.3" при градиенте температуры 450-350oC. Последнее согласуется с нашим наблюдением, что реакции фазы В, проводимые в запаянной трубке в интервале приблизительно 350-400oC, дают на более холодном конце трубки серебристые иглы, состав которых по анализу точно соответствует WI3.
В прошлых сообщениях получение частиц, содержащих растворимое ядро [W6I8] 4+, в основном проводили посредством выделения из W6I12 реакционными смесями этанол/HI с образованием [W6I14]2- растворе. Хотя такими способами легко выделяют аналогичные фазы Mo6X12 X=Cl, Br, I) и W6X12 (X=Cl, Br), было доказано, что этот метод значительно менее эффективен в применении к W6I12. В соответствии с предыдущими наблюдениями мы обнаружили, что W6I12 лишь слабо растворим в смеси этанол/HI, что приводит к чрезвычайно низким выходам [W6I14]2-. Действительно, ни в одном из приведенных ранее синтезов [W6I14]2- не было приведено численного значения выхода этого продукта. Значительно лучшим путем к получению этого кластера является пространственное сокращение W6I12. В соответствии с данной процедурой внедрение KI разрывает двумерный каркас W6I12, образуя молекулярное твердое вещество возможной формулы K2W6I14 (аналогично K2Mo6Cl14), которое полностью растворяется в этаноле с образованием желаемых частиц [W6I14]2- в растворе. В данной работе это было осуществлено путем нагревания фазы В и KI в условиях, при которых в отсутствии KI получают W6I12.
Когда тесную смесь фазы В и KI нагревают при 550oC в течение 65 часов, образуется оранжево-черное твердое вещество (предположительно K2W6I14), которое содержит анион кластера [W6I14]2-. При растворении этого твердого вещества в этаноле с последующим добавлением твердого (Bu4N)I получали (Bu4N)H2[W6I14].
С точки зрения замещения одного лиганда на другой в соединениях данного изобретения кластеры претерпевают реакции замещения центрального галогенида при жестких условиях, но довольно легко вступают в реакции замещения конечного лиганда. Мы выбрали способ замещения иодида трифторметансульфонатом для того, чтобы получить кластеры с высокой лабильностью по отношению к замещению для использования в последующей работе. Следуя процедуре Shriver с сотр. [Inorg. Chem. 31, 1869 (1992)] для [Mo6Cl8(CF3SO3)6]2-, был получен (Pr4N)2[W5I13] и избыток Ag(CF3SO3) в дихлорметане. В аналогичной реакции был выделен (Bu4N)2[W6I8CF3SO3)6].
С точки зрения связывания в комплекс соединений данного изобретения и их последующего использования в качестве контрастной среды соединения формулы (I) и (II) особенно удобно представить в виде их хелатных комплексов, содержащих этилендиаминтетрауксусную кислоту (ЭДТК), диэтилентриаминпентауксусную кислоту (ДТПК) или другие аминополикарбоновые кислоты (АПКК). Такие хелатные комплексы обладают высокой стабильностью в отношениии высвобождения ионов или кластеров тяжелых металлов.
Особенно предпочтительно, чтобы электрические заряды, которые несут комплексообразующие частицы, в значительной степени или полностью уравновешивали заряды, находящиеся на связываемых в комплекс объектах; для хелатообразующих соединений типа АПКК это может быть легко достигнуто, например, путем отрыва, замещения или дезактивации (например, путем образования эфира или амида) одной или более карбоксильных групп.
Многие подходящие хелатообразующие агенты широко известны или были описаны в литературе, особенно в литературе, касающейся бифункциональных хелатообразующих веществ для агентов обезвреживания тяжелых металлов, а также контрастных агентов на основе хелатов, например, агентов, описанных в WO-A-89/00557 (Berg) и в документах, упомянутых в этой заявке и в приложенном к ней отчете о поиске, US-A-4647447 (Gries), US-A-4826673 (Dean), ЕР-А-230893 (Felder), ЕР-А-217577 (Frincke), US-A-4652519 (Warshawsky), US-A-4687659 (Quay) и многочисленных других недавних патентных публикациях Nycomed AS, Salutar Inc, Schering AG, Squibb, Bracco, Mallinckrodt, Dow и Guerbet.
Таким образом, полиамины, особенно линейные или циклические полиамины, такие как этилендиамин, 1,4,7-триазациклононан и циклен, могут быть использованы в качестве хелатообразующих агентов, так же как и АПКК, такие как ДТПК, ЭДТК и их производные, а также другие циклические и нециклические АПКК, как указано в WO-A-89/00557. Также можно использовать тридентатные трис-тиолы Хольма и др. (см. JACS 112; 8015-8023 (1990) и JACS 110; 2484-2494 (1988)).
Для введения в организм человека или животного хелатные соединения формул (I) и (II) должны быть соответственным образом соединены с фармацевтическими или ветеринарными носителями или инертными наполнителями. Контрастная среда по данному изобретению может содержать фармацевтические или ветеринарные добавки, например стабилизаторы, антиоксиданты, агенты, регулирующие осмотические свойства, буферные добавки, регуляторы pH, красители, одоранты, регуляторы вязкости и т.п. Они могут находиться в формах, удобных для парентерального или энтерального введения, например для инъекций или вливаний, или для введения непосредственно в полость тела, имеющую выходящий наружу канал, например, в желудочно-кишечный тракт, мочевой пузырь и в матку. Таким образом, среда по данному изобретению может быть в виде обычных фармацевтических форм, таких как таблетки, таблетки в оболочке, капсулы, порошки, растворы, суспензии, дисперсии, сиропы, суппозитории и т.д.; однако в целом предпочтительными являются растворы, суспензии и дисперсии в физиологически приемлемой среде, например, в воде для инъекций. Если среда создается для парентерального введения, среда-носитель, содержащая многоядерный комплекс, должна быть преимущественно изотонической или несколько гипертонической. Кроме того, среда для парентерального введения должна содержать небольшие количества, например 0,01-10 мольных процентов по отношению к многоядерному комплексу, свободных хелатообразующих агентов или непрочных хелатных комплексов с физиологически переносимыми эакомплексованными частицами (например, Ca2+); предпочтительны также небольшие добавки солей натрия или кальция.
Для использования в качестве контрастной среды для рентгеновских исследований среда по данному изобретению обычно должна иметь содержание тяжелых атомов от 1 ммоль/л до 5 моль/л, предпочтительно от 0,1 до 2 моль/л. Дозировка от 0,5 до 1,5 ммоль/кг вполне достаточна для обеспечения нужного контраста, хотя обычно предпочтительны дозировки от 0,8 до 1,2 ммоль/кг.
Для сцинтиллографии дозировка радиоактивных частиц обычно ниже.
Таким образом, в данном изобретении предложены особенно эффективные средства, посредством которых можно улучшить эффективность контрастных сред путем увеличения относительной доли объема молекулы, которую занимают улучшающие контраст атомы тяжелых металлов.
Далее настоящее изобретение будет проиллюстрировано (но не ограничено) следующими примерами (все соотношения и процентные составы даны по массе, а все температуры в градусах Цельсия, если это не оговорено особо). Были использованы соединения W(CO)6 (Strem), иод (Strem), (R4N)I (R=Pr, Bu; Aldrich), (Ph4P)I (Aldrich) и Ag(CF3SO3) (Aldrich) без дополнительной очистки. W(CO)6 и иод перед использованием растирали в ступке. Растворители перегоняли из соответствующего осушающего агента и дегазировали перед использованием. Твердофазные реакции проводили в трубках из пирекса с размерами (внутренний диаметр) х (наружный диаметр) х (длина) = 13 х 19 х 260 мм. За исключением получения (Pr4N)[W5I15] ·ТГФ, (Bu4N)[W5(C)I13] и (Bu4N)2[W6I14 ], реакции в растворе проводили в атмосфере чистого азота с использованием стандартного метода Шленка.
Пример 1 - синтез "Фазы А".
Хорошо перемешанные 2,0 г (5,7 ммоль) W(CO)6 и 10 г (39 ммоль) I2 помещали в трубку из пирекса, установку дегазировали и присоединяли к масляному гидрозатвору. Реакционную смесь нагревали на масляной бане при 140oC до прекращения выделения CO (приблизительно 3 часа). Полученное черно-серое твердое вещество (Фаза А) извлекали из трубки и измельчали. Это твердое вещество неоднократно промывали эфиром до тех пор, пока фильтрат не стал бесцветным (приблизительно 300 мл), в этот момент осталось 1,8 г черного твердого вещества (отмытой фазы А).
Пример 2 - синтез (Bu4N)[W3I9].
Отмытую фазу А (1,8 г) добавили к 200 мл ТГФ и перемешивали в течение ночи. Раствор профильтровали через целит (Celite), и к темно-красному фильтрату добавили твердый (Bu4N)l (0,50 г, 1,4 ммоль). Раствор перемешивали еще в течение 8 часов, затем концентрировали приблизительно до 5 мл при пониженном давлении. Этот раствор покрыли слоем эфира 25 мл и выдерживали при -15oC в течение ночи. Полученное красно-коричневое твердое вещество отфильтровали и быстро промыли 10 мл этанола. Затем это твердое вещество промыли эфиром (3 х 10 мл), высушили под вакуумом и перекристаллизовали медленным испарением дихлорметанового раствора; получено 0,38 г (11% в расчете на W(CO)6) красно- черного кристаллического продукта. Спектр поглощения (ТГФ): λmax(∈m): 278 (30,400), 308 (15,000), 337 (sh, 12,300), 428 (sh,8130), 455 (sh, 8869), 510 (4730), 604 (sh, 1060), 721 (468) нм. Анализ: расчет для C16H36I9NW3: С, 9,93; H, 1,87; I, 58,99; N, 0,72; W, 28,49. Найдено: С, 10,03; , 1,93; I, 58,92; N, 0,75; W, 28,32.
Пример 3 - синтез W4I13.
Трубку из пирекса, содержащую фазу А, запаяли под вакуумом, нагрели в трубчатой печи при 165oC в течение 50 часов и охладили до комнатной температуры со скоростью 0,2oC/мин. Трубку вскрыли (см. Меру предосторожности 1), содержимое извлекли и промыли аликвотами эфира для удаления непрореагировавшего иода. Черное твердое вещество затем промывали ТГФ (5 х 50 мл) и снова эфиром (3 х 5 мл); осталось 0,30 г (8,8% в расчете на W(CO)6) черного микрокристаллического продукта. Анализ: расчет для I13W4: I, 69,17; W, 30,83. Найдено: I, 69,51; W, 30,41. Мера предосторожности 1. Хотя большая часть CO была удалена при синтезе фазы А, реакционная трубка при извлечении из печи находится под высоким давлением CO. Чтобы минимизировать возможность взрыва, трубку осторожно надпиливают металлическим напильником и помешают в толстую трубчатую оболочку из пористой резины. Затем трубку вскрывают в хорошо вентилируемом вытяжном шкафу с закрытой заслонкой, ударив по ней осторожно один раз молотком.
Пример 4 - синтез "Фазы В".
Трубку из пирекса, содержащую фазу А, запаяли под вакуумом и нагревали в трубчатой печи при 200oC в течение 50 часов; затем ее охладили (0,2oC/мин) до комнатной температуры. Трубку вскрыли (см. Меру предосторожности 1) и ее содержимое извлекли и измельчили. Черно-серое твердое вещество (неотмытая фаза В) было промыто порциями эфира (всего 200 мл), чтобы отмыть от непрореагировавшего иода. Когда фильтрат стал бесцветным, осталось 2,1 г темного сине-черного твердого вещества (фаза В).
Пример 5 - синтез (Pr4N)[W5I13]ТГФ.
Фазу В (1,75 г) добавили к 200 мл этанола в течение 30 минут. Темно-зеленую суспензию перемешивали в течение 18 часов при комнатной температуре, за это время все твердое вещество растворилось. Избыток (Pr4N)I (0,50 г, 1,6 ммоль) был добавлен в виде твердого вещества к раствору, что немедленно вызвало выпадение синего осадка. Раствор перемешивали в течение 30 мин; твердую фазу отделили фильтрацией и промыли этанолом (15 мл) и эфиром (2 х 10 мл). Твердое вещество высушили при вакуумировании вытяжным вентилятором и перекристаллизовали из ТГФ/гексана при -15oC с получением 0,98 г (31% в расчете на W(CO)6) синего микрокристаллического твердого вещества. Спектр поглощения (ТГФ): λmax(∈m): 298 (33,700), 347 (sh, 13,900), 370 (sh 11,800), 423 (sh, 7720), 478 (5880), 524 (4990), 588 (4190), 623 (4080), 722 (2900), 844 (2050) нм. Анализ: расчет для C16CH36I13NOW5: С, 6,79; H, 1,28; I, 58,34; N, 0,50; W, 32,53. Найдено: С, 6,48; H, 1,24; I, 58,84; N, 0,52; W, 32,79.
Пример 6 - синтез (Pr4N)2[W5I13].
В 100 мл колбу Шленка поместили 1,0 г (0,35 ммоль) (Pr4N)[W5I13]ТГФ 0,056 r (0,86 ммоль) цинковой пыли и 0,22 г (0,70 ммоль) (Pr4N)I. Содержимое колбы тщательно дегазировали и добавили 30 мл дихлорметана. Реакционную смесь перемешивали в течение 24 часов, в течение этого времени наблюдалось изменение цвета от темно-зеленого до коричнево-красного. Смесь фильтровали через Celite и концентрировали приблизительно до 3 мл под вакуумом. Раствор был покрыт слоем 10 мл эфира и выдержан при - 15oC в течение ночи. Красновато-коричневое твердое вещество было собрано путем фильтрации, промыто этанолом (10 мл) и эфиром (2 х 10 мл) и высушено под вакуумом. Этот материал был перекристаллизован из смеси дихлорметан/эфир с получением в качестве продукта 0,76 г (74%) микрокристаллического черного твердого вещества. Спектр поглощения (ТГФ): λmax(∈m): 348 (12,500), 408 (sh, 7630), 535 (3760) нм. ЭПР (CH2Cl2, 120 K): аксиально, g = 1,97, расстояние между пиками 260 G. Анализ: расчет для C24H56I13N2W5: С, 9,79; H, 1,92; I, 56,08; N, 0,95; W, 31,26. Найдено: С, 9,62; H, 1,85; I, 56,21; N, 0,92; W, 31,36.
Пример 7 - синтез (Pr4N)2[W5I8(CF3SO3)5].
Смесь 0,41 г (0,14 ммоль) (Pr4N)2[W5I13] и 0,21 г (0,78 ммоль) Ag(CF3SO3) перемешивали в 35 мл дихлорметана в течение 18 часов при отсутствии света. Происходило постепенное изменение цвета от коричнево-красного до пурпурного, и осаждался AgI. Реакционную смесь профильтровали через Celite, и фильтрат сконцентрировали до 5 мл при пониженном давлении. Эфир (25 мл) осторожно налили слоем сверху на раствор, что привело к образованию черных кристаллов на протяжении 24 часов. Кристаллический продукт был собран и промыт 2 х 10 мл эфира с получением 0,22 г (51%) чистого продукта. Спектр поглощения (ТГФ): λmax(∈m) 291 (sh 11,600), 358 (sh, 6770), 439 (3960), 598 (1850) нм. Соединение было идентифицировано с помощью рентгеноструктурного анализа на монокристалле.
Пример 8 - синтез (Bu4N)[W5(C)I13]TГФ.
Иодид цезия (1,25 г, 4,81 ммоль) был добавлен в трубку из пирекса, содержащую фазу А. Затем трубка была запаяна под вакуумом и нагрета в трубчатой печи со стальной футеровкой при 300oC (см. Меру предосторожности 2) в течение 50 часов; затем ее охладили (0,2oC/мин) до комнатной температуры. Затем трубку открыли (см. Меру предосторожности 1) и промыли 200 мл эфира, чтобы уда лить непрореагировавший иод. Когда фильтрат стал бесцветным, осталось 1,2 г коричнево-черного частично кристаллического твердого вещества. Масс-спектроскопия при помощи метода бомбардировки быстрыми атомами растворимой в дихлорметане части этого твердого вещества выявила присутствие (Cs[W5(C)I13], Cs2[W6I14] и иодидов цезия, которые невозможно было полностью идентифицировать. Черную твердую фазу перемешивали в течение ночи в 150 мл ацетонитрила с получением темно-оранжевого раствора. Этот раствор профильтровали и к фильтрату добавили 0,75 г (2,0 ммоль) твердого (Bu4N)I. Моментально выделившийся красный осадок был отфильтрован и промыт эфиром (2 х 10 мл). Фильтрат упарили досуха с образованием красно-оранжевого твердого остатка. Объединенные твердые вещества были частично растворены в 50 мл ТГФ, и раствор был отфильтрован. Нерастворившееся красное твердое вещество было промыто этанолом (5 х 10 мл) и перекристаллизовано из смеси ТГФ/гексан с образованием продукта в виде 0,18 г (5,5% в расчете на W(CO)6) красного микрокристаллического твердого вещества. Спектр поглощения (ТГФ):  329 (sh, 33,900), 403 (sh, 11,200), 442 (9830) нм. Анализ: расчет для C21H44I13NOW5: С, 8,71; H, 1,53; I, 56,97; N, 0,48; W, 31,75. Найдено: С, 8,54; H, 1,46; I, 57,06; N, 0,49; W, 31,68. Мера предосторожности 2. Имеется некоторый риск взрыва при нагревании фазы А в запаянной трубке из пирекса при 300oC. Следовательно, рекомендуется, чтобы трубчатая печь была расположена в хорошо вентилируемом вытяжном шкафу, сделанном во взрывобезопасном исполнении. Рекомендуется также использовать футерованную сталью трубку, чтобы минимизировать разрушение печи в случае взрыва. Нагрев до температур выше 300oC, изменение размеров трубки из пирекса или увеличение количества вещества, участвующего в реакции, также могут привести к взрыву и противопоказаны.
329 (sh, 33,900), 403 (sh, 11,200), 442 (9830) нм. Анализ: расчет для C21H44I13NOW5: С, 8,71; H, 1,53; I, 56,97; N, 0,48; W, 31,75. Найдено: С, 8,54; H, 1,46; I, 57,06; N, 0,49; W, 31,68. Мера предосторожности 2. Имеется некоторый риск взрыва при нагревании фазы А в запаянной трубке из пирекса при 300oC. Следовательно, рекомендуется, чтобы трубчатая печь была расположена в хорошо вентилируемом вытяжном шкафу, сделанном во взрывобезопасном исполнении. Рекомендуется также использовать футерованную сталью трубку, чтобы минимизировать разрушение печи в случае взрыва. Нагрев до температур выше 300oC, изменение размеров трубки из пирекса или увеличение количества вещества, участвующего в реакции, также могут привести к взрыву и противопоказаны.
Пример 9 - синтез W6I12
Фаза В (0,62 г) была запаяна в трубку из пирекса под вакуумом и нагревалась в трубчатой печи при 550oC в течение 50 часов. Трубку охладили до комнатной температуры со скоростью 0,2oC/мин и открыли. Продукт в виде 0,23 г (25% в расчете на W(CO)6) оранжевого твердого вещества отложился на одном конце трубки, а на другом конце было обнаружено некоторое количество свободного иода. Соединение было идентифицировано с помощью рентгеноструктурного определения на монокристалле, а затем с помощью дифракции рентгеновских лучей на порошке.
Пример 10 - синтез W6I16.
Неотмытая фаза В (8,0 г) была запаяна в трубку из пирекса под вакуумом. Трубку нагревали в трубчатой печи при 550oC в течение 50 часов, а затем охладили до комнатной температуры со скоростью 0,2oC/мин. Трубку вскрыли и продукт реакции промыли 100 мл эфира. Продукт получили в виде 1,3 г (44% в расчете на W(CO)6) красно-коричневого твердого вещества. Соединение было идентифицировано с помощью рентгеноструктурного анализа на монокристалле, а затем с помощью дифракции рентгеновских лучей на порошке.
Пример 11 - синтез (Bu4N)2[W6I14].
Фаза В (1,6 г) и KI (1,0 г, 6,0 ммоль) были запаяны под вакуумом в трубку из пирекса и нагревались при 550oC в течение 65 часов. Трубку охладили до комнатной температуры (0,2oC/мин) и открыли. Черно-оранжевый продукт был полностью растворен в 125 мл этанола с образованием темно-оранжевого раствора, который был профильтрован. Добавление 0,50 г (1,4 ммоль) (Bu4N)I к фильтрату вызвало мгновенное выпадение желто-оранжевого осадка. Этот материал был собран с помощью фильтрации и промыт холодным этанолом (2 х 10 мл) и эфиром (10 мл). Был получен продукт в виде 0,95 г желто-оранжевого твердого вещества; выход 30% в расчете на W(CO)6, использованный при получении aазы А. Масс-спектроскопия при помощи метода бомбардировки быстрыми атомами: m/z 3122 ([Bu4N)(W6I14]1-), m/z 2881 ([HW6I14])1-. Параметры ячейки, полученные на монокристалле, такие же, как и приведенные ранее для этого соединения.
Пример 12 - синтез (Bu4N)2[W6I8(CF3SO3)6].
Смесь 250 мг (74 ммоль) (Bu4N)2[W6I14] и 135 мг (525 ммоль) Ag(CF3SO3) в 30 мл дихлорметана перемешивали в течение 18 часов в отсутствие света и отфильтровали через Celite для удаления AgI. Ярко-желтый фильтрат сконцентрировали приблизительно до 3 мл при пониженном давлении; 20 мл эфира осторожно вылили сверху на раствор. В течение нескольких часов выделились ярко-желтые кристаллы; они были отфильтрованы и промыты эфиром (3 х 10 мл) с получением 0,21 г (81%) чистого продукта. Спектр поглощения (CH2Cl2): λmax(∈m) 289 (12,000), 343 (sh, 4830) нм. Анализ: расчет для C38H72F18I8N2O18S6W6: С, 13,04; H, 2,07; F, 9,78; I, 29,03; N, 0,84; S, 5,49; W, 31,56. Найдено: С, 13,08; H, 2,05; F, 9,65; I, 28,91; N, 0,86; S, 5,38; W, 31,42.
| название | год | авторы | номер документа |
|---|---|---|---|
| ВОЛЬФРАМОВЫЕ ЧАСТИЦЫ В КАЧЕСТВЕ РЕНТГЕНОКОНТРАСТНЫХ ВЕЩЕСТВ | 2004 |
|
RU2361617C2 |
| РЕНТГЕНКОНТРАСТНАЯ КОМПОЗИЦИЯ, СПОСОБ ВИЗУАЛИЗАЦИИ ПРИ ТОМОГРАФИЧЕСКИХ ИССЛЕДОВАНИЯХ В ЭКСПЕРИМЕНТЕ И СПОСОБ ПОЛУЧЕНИЯ РЕНТГЕНКОНТРАСТНОЙ КОМПОЗИЦИИ | 1992 |
|
RU2074002C1 |
| ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ ДЛЯ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ОКСАЗОЛИДИНОНА И ХИНОЛОНА | 2004 |
|
RU2373203C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2081112C1 |
| ФОТОХИМИЧЕСКИ АКТИВНЫЕ ПОЛИМЕРНЫЕ МАТЕРИАЛЫ | 2011 |
|
RU2592545C2 |
| КАСКАДНЫЕ ПОЛИМЕРНЫЕ КОМПЛЕКСЫ, ИСХОДНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2166501C2 |
| ЙОДИРОВАННЫЕ СЛОЖНЫЕ ЭФИРЫ | 1990 |
|
RU2088579C1 |
| КОМПЛЕКСЫ КАСКАДНЫХ ПОЛИМЕРОВ, СОДЕРЖАЩЕЕ ИХ ДИАГНОСТИЧЕСКОЕ КОНТРАСТНОЕ СРЕДСТВО И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1996 |
|
RU2197495C2 |
| ИОДСОДЕРЖАЩИЕ ДЕНДРИМЕРНЫЕ ПОЛИМЕРЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ДИАГНОСТИЧЕСКОЕ СРЕДСТВО ДЛЯ РЕНТГЕНОДИАГНОСТИКИ | 1994 |
|
RU2147592C1 |
| ТИОЗАМЕЩЕННЫЕ ПИРИДИНИЛБИСФОСФОНОВЫЕ КИСЛОТЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ ИЛИ ЭФИРЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 1993 |
|
RU2124019C1 |
Изобретение относится к химико-фармацевтической промышленности и касается способа получения контрастных агентов. Изобретение заключается в способе получения многоядерных кластерных соединений путем взаимодействия карбонила металла и иода и использования этих кластеров и производных от них кластеров в диагностическом изображении. Описаны также новые многоядерные кластерные соединения, контрастная среда для диагностического изображения. Изобретение обеспечивает улучшение коэффициента эффективного качества диагноза для диагностического изображения. 6 с. и 8 з.п. ф-лы.
MnBuAv,
где MnBuAv - многоядерная частица;
каждый M - атом тяжелого металла, выбранного из группы, включающей W, Mo, Ta, Nb и Hf,
каждый атом металла M ковалентно связан по меньшей мере с одним другим атомом;
каждый B - атом, образующий мостиковую связь, ковалентно связанный по меньшей мере с двумя атомами металла М;
каждый А, которые могут быть одинаковыми или разными, является атомом, не образующим мостиковую связь, ковалентно связанным с атомом металла М;
n и u - положительные целые числа, имеющие значение 2 или более;
v = 0 или является положительным целым числом,
или его соли, включающий взаимодействие M(CO)6 и I2 в течение времени и при температуре, достаточных для образования кластера металл/иод, и, возможно, замещение одного или более образующих или не образующих мостиковые связи атомов иода в этом кластере одним или более иным атомом или радикалом с образованием модифицированного кластера и/или образование соли упомянутого кластера или модифицированного кластера.
MnBuAv,
в котором n равно 3, 4 или 5;
M, A, n, u и v определены в п.1;
B является иодом,
или соль этого соединения.
| Пожарный двухцилиндровый насос | 0 |
|
SU90A1 |
| Огнетушитель | 0 |
|
SU91A1 |
| Автоматический огнетушитель | 0 |
|
SU92A1 |
