Это изобретение относится к системам подачи лекарств с непрерывным выделением и, в частности, к способу получения микрочастиц ионного конъюгата с непрерывным выделением.
Уровень техники
Биологически разлагаемые полимерные составы для подачи лекарств были разработаны и применяются для контролируемого выделения лекарств in vivo. Смотрите, например, патенты США 3773919 и 4767628. Такие биологически разлагаемые полимерные составы предназначены для того, чтобы позволить захваченному лекарству медленно диффундировать через полимерную матрицу или покрытие, когда биологически разлагаемый полимер деполимеризируется.
В международной публикации WO 94/15587 описываются молекулярные ионные конъюгаты сложных полиэфиров с непрерывным выделением и подачи лекарств. Поскольку разложение сложных полиэфиров является ключевым этапом в процессе выделения, площадь поверхности частиц конъюгата может контролировать профиль выделения лекарства из конъюгата. Таким образом, частицы конъюгата должны иметь сходные размеры и форму, чтобы обеспечить как минимальную, так и воспроизводимую площадь поверхности, например микросферы.
Сущность изобретения
В одном своем аспекте это изобретение характеризует способ получения микрочастиц ионного конъюгата с непрерывным выделением, содержащего биологически разлагаемый полимер, включающий в себя свободную карбоксильную группу (сложный полиэфир, образованный из мономеров, таких, как молочная кислота, e-капроловая кислота, гликолевая кислота, карбонат триметилена или p-диоксанон; или их сополимер; мономеры могут быть оптически изомерами или рацематами) и лекарство, содержащее свободную аминовую группу (например, пептидное лекарство, такое, как соматостатин или LHRH), которые связаны друг с другом ионной связью. Способ включает в себя следующие этапы: (1) получение первого раствора, в котором растворен конъюгат; (2) перемешивание первого раствора (добавляемого маленькими капельками, например, через распыляющее сопло, такое, как звуковое сопло, пневматическое сопло, центробежный распылитель или сопло высокого давления) с первой жидкостью для образования первой дисперсии, где первая жидкость может смешиваться с первым раствором, а конъюгат не растворяется в первой жидкости и осаждается из первой дисперсии; и (3) выделение конъюгата от первой дисперсии.
В одном примере выполнения изобретения лекарство растворимо в первой жидкости, которая может быть спиртом (например, этаноловым или изопропиловым спиртом), гексаном или водой, или их смесью. Когда этанол используется в качестве первой жидкости, его при использовании можно поддерживать при температуре примерно от 0 до -30oC. При использовании изопропилового спирта его можно поддерживать при температуре примерно от 0 до -70oC, например, при охлаждении добавками сухого льда.
Первый раствор, который может содержать ацетон, дихлорметан, ацетонитрил, этиловый ацетат, тетрагидрофуран или глим, или их смеси, можно получить (1) растворением биологически разлагаемого полимера во второй жидкости (например, ацетоне, тетрагидрофуране, гликоне, этиловом ацетате, метиловом ацетате, ацетонитриле, формате этила или глиме, или в их смесях) для образования второго раствора; (2) растворением лекарства в третьей жидкости (например, воде или ацетоне, или в их смеси) с образованием третьего раствора, где третья жидкость может смешиваться с первой жидкостью и второй жидкостью; и (3) смешиванием второго раствора и третьего раствора с образованием первого раствора, причем перемешивание заставляет лекарство образовывать ионную связь с биологически разлагаемым полимером и образовывать конъюгат в первом растворе. Первый раствор может содержать до 40% по массе конъюгата (например, от 25 до 35% по массе конъюгата). В одном примере щелочь, например NaOH или КОН, можно добавить во второй раствор до перемешивания второго раствора и третьего раствора. Нейтрализация карбоксильных групп биологически разлагаемого полимера в щелочи облегчает образование ионного конъюгата.
В качестве альтернативы первый раствор получается растворением биологически разлагаемого полимера и лекарства во второй жидкости (например, ацетоне или смеси ацетона и воды) с образованием конъюгата в первом растворе. Согласно этому способу, биологически разлагаемый полимер можно сначала растворить во второй жидкости затем щелочь добавляется ко второму раствору, и затем лекарство растворяется во второй жидкости. Также, при желании, первый раствор можно частично или полностью испарить из первой дисперсии до изолирования конъюгата. Обработанный конъюгат можно удобно изолировать путем центрифугирования или фильтрации первой дисперсии, и изолированный конъюгат можно перемешать с водным раствором маннита до вакуумной сушки (т.е. лиофилирования). Затем изолированному конъюгату можно придать форму пленки или стержня. Изолированному конъюгату также можно придать форму сфер, т.е. микросфер со средним диаметром от 5 до 200 мкм, например, как описано здесь. Под "приданием формы сферы" имеется в виду обработка микрочастицы таким образом, чтобы она приняла форму, близкую к форме сферы.
В другом своем аспекте настоящее изобретение характеризуется способом придания сферической формы ионному конъюгату с непрерывным выделением, как описано выше. Этот способ включает в себя следующие этапы: (1) перемешивание конъюгата с первой жидкостью (например, с маслом, таким, как силиконовое масло, минеральное масло, кунжутное масло или растительное масло) с образованием первой дисперсии, причем конъюгат имеет форму микрочастицы и нерастворим в первой жидкости; (2) нагревание первой дисперсии до температуры выше, чем Tg или Tm конъюгата; (3) охлаждение первой дисперсии ниже Tg или Tm конъюгата; (4) перемешивание первой дисперсии со второй жидкостью (например, гексаном, гептаном, миристатом изопропила или спиртом, таким, как этаноловый или изопропиловый спирт) с образованием второй дисперсии, причем вторая жидкость может смешиваться с первой жидкостью, а конъюгат не растворим во второй жидкости; и (5) изолирование конъюгата от второй дисперсии. Конъюгат может иметь форму микрокапсулы со средними диаметром примерно от 5 до 200 мкм до его перемешивания с первой жидкостью, и образованную таким образом дисперсию энергично перемешивают с одновременным нагреванием, чтобы помочь разделению частиц. После того, как конъюгат изолирован, его можно промыть второй жидкостью и высушить в вакууме. По желанию, его также можно перемешать с водным раствором маннита до сушки в вакууме.
Третий аспект настоящего изобретения характеризуется способом придания сферической формы вышеописанному ионному конъюгату с непрерывным выделением (например, микрокапсуле со средним диаметром примерно от 5 до 200 мкм). Этот способ включает в себя следующие этапы: (1) перемешивание конъюгата в первой жидкости (например, воде) с образованием первой дисперсии, причем конъюгат имеет форму микрочастицы и не растворим в первой жидкости; (2) перемешивание первой дисперсии; (3) перемешивание взболтанной дисперсии со второй жидкостью (например, дихлорметаном или хлороформом) в таком количестве, что она абсорбируется конъюгатом, но не придает конъюгату растворимость, причем вторая жидкость может смешиваться с первой жидкостью; (4) выпаривание второй жидкости из первой дисперсии; и (5) изолирование осажденного конъюгата из первой дисперсии. При необходимости этот способ может, кроме того, включать в себя этап добавления поверхностно-активного вещества (например, лецитина, Tween 20, полисорбата или сульфата лаурила) к первой дисперсии, чтобы помочь стабилизации первой дисперсии, и изолированный конъюгат можно промыть первой жидкостью и высушить в вакууме. Опять же изолированный конъюгат можно перемешать с водным раствором маннита до вакуумной сушки.
В еще одном аспекте настоящее изобретение характеризуется способом придания сферической формы вышеописанному ионному конъюгату с непрерывным выделением. Способ включает в себя следующие этапы: (1) растворение конъюгата в первой жидкости (например, ацетонитриле) с образованием первого раствора; (2) перемешивание первого раствора со второй жидкостью (например, маслом) с образованием первой дисперсии, причем вторая жидкость не может смешиваться с первым раствором; (3) выпаривание первой жидкости из первой дисперсии, чтобы осадить конъюгат из первой дисперсии; и (4) выделение осадка конъюгата из первой дисперсии. На этапе перемешивания первый раствор может добавляться ко второй жидкости маленькими капельками.
Указанный способ может, кроме того, включать в себя этап промывки изолированного конъюгата третьей жидкостью (например, гексаном, гептаном или октаном), которая может смешиваться со второй жидкостью и не является растворителем для выделенного конъюгата. При желании, изолированный конъюгат можно смешать с водным раствором маннита до сушки в вакууме.
Биологически разлагаемый полимер в вышеописанном конъюгате может содержать, как минимум, одну свободную карбоксильную группу (например, от двух до десяти свободных карбоксильных групп на полимерную цепь). Примерами биологически разлагаемых полимеров, содержащих карбоновые кислоты, являются сложные полиэфиры, содержащие элементарные звенья молочной кислоты, e-капроловой кислоты, p-диоксанона, e-каприоновой кислоты, замещенного и незамещенного карбоната триметилена, 1,5-диоксепана-2-один, 1,4- диоксепана-2-один, гликолевой кислоты, оксилата алкилена, циклоалкилена, оксилата циклоалкилена, сукцината алкилена, или 3- гидрокси бутирата в оптически активных формах или как рацематы; или сополимеров любого из указанных. Дополнительные свободные группы карбоновой кислоты можно включать в биологически разлагаемый сложный полиэфир посредством реакции, например, раскрывающей кольца полимеризации или поликонденсации, с поликарбоновыми кислотами, такими, как яблочная кислота, винная кислота, памовая кислота, лимонная кислота, сукциновый ангидрид и глутаровый ангидрид. Таким образом, биологически разлагаемый полимер может быть не растворимым в воде сложным полиэфиром, включающим элементарные звенья молочной кислоты с элементарными звеньями гликолевой кислоты или без них, можно также применять другие биологически разлагаемые полимеры, такие, как полиорто- сложные эфиры, полиортокарбонаты и полианталы. Биологически разлагаемый полимер может иметь среднюю степень полимеризации, например среднее количество мономеров на полимерную цепь от 10 до 300.
Лекарство имеет одну или несколько (например, от одной до десяти) свободных аминных групп. В одном примере выполнения лекарство представляет собой стабильный к кислотам пептид. В качестве примера к пригодным стабильным к кислотам пептидам относятся выделяющий гормоны роста пептид (GHRP), лютеинизирующий выделяющий гормоны гормон (LHRH), адреномедуллин, гормон роста, соматостатин, бомбесин, выделяющий гастрин пептид (GRP), кальцитонин, брадикинин, галанин, меланоцит стимулирующий гормон (MSH), фактор, выделяющий гормоны роста (GRF), амилин, адреномедуллин, тахикинины, секретин, гормон паратироид (PTH), энкефалин, эндотелин, пептид, выделяющий ген кальцитонина (CGRP), нейромедины, белок, связанный с гормоном паратироидом (PTHrP), глукагон, нейротенсин, адренокортикотрофный гормон (ACTH), пептид YY (PYY), выделяющий глукагон пептид (GLP), вазоактивный кишечный пептид (VIP), пептид, активирующий гипофизарную аденилатциклазу (PACAP), мотилин, вещество P, нейропептид Y (NPY), TSH и их аналоги и фрагменты. Лекарство может быть растворимым (например, более чем 0,1 мг/мл); предпочтительно более чем 1,0 мг/мл в первой жидкости.
Другие признаки и преимущества данного изобретения станут очевидными из подробного описания и из формулы изобретения.
Считается, что специалист сможет на основе приведенного здесь описания использовать данное изобретение в его максимальном объеме, поэтому следующие конкретные примеры выполнения изобретения должны рассматриваться как лишь иллюстративные, а не ограничивающие каким-либо образом остальную часть изложенного.
Если не определено иначе, все технические и научные термины, используемые здесь, имеют то же самое значение, как оно обычно понимается специалистом в области, к которой относится это изобретение.
Лучшие методы выполнения изобретения
Пример 1
18,0 г 6000 г/моль 66/32/2 сополимера поли- L-молочной-со-гликолевой-со-D, L-яблочной кислоты (66% L-молочной кислоты, 32% гликолевой и 2% яблочной кислоты; кислота номер 0,373 миллиэквивалента/г) было растворено в 180 г ацетона (раствор 10% сополимера по массе). 14,4 мл 0,5 N водной NaOH было добавлено для образования карбоксилата натрия полимера. 4,28 г соли ацетата пептида Lanreotide (товарный знак) (производитель Kinerton, Дублин, Ирландия; D-Nal-c[Cys-Tyr-D-Trp-Val-Cys]-Thr-NH2; содержание ацетата = 9,60% по массе) было отдельно растворено в смеси 10 г ацетона и 10 г деионизированной воды. Количество растворенного пептида соответствовало стехиометрическому отношению кислотных групп из сополимера (например, один) и трех свободных амино-групп для пептида (например, два). Затем раствор пептида добавлялся по каплям в раствор сополимера, и получившийся раствор перемешивался в течение двух часов, чтобы осуществить солевой обмен и получить в результате состав ионного конъюгата полимер/пептид (PPIC).
Пример 2
В контролируемом по температуре реакторе с рубашкой (изготовитель Schott Glass AGB, Дублин, Ирландия) ванна на 2 л деионизированной воды была предварительно охлаждена до 0oC и энергично перемешана. Затем указанный раствор PPIC из примера 1 медленно добавлялся в реактор с использованием насоса Masterflex (изготовитель Bioblock Scientific, lllkvch, Франция), что давало расход 10-15 мл/мин, через силиконовые трубки, снабженные на конце иглой 19 калибра. Раствор PPIC подавался через иглу, которая размещалась выше водной ванны при 0oC. PPIC осаждался в ванне в виде маленьких твердых частиц. Затем твердые частицы отделялись от образовавшегося сверху в результате отстаивания раствора путем центрифугирования (30 мин при 5000 об/мин и 0-5oC), промывались пресной деионизированной водой, повторно взвешивались в воде, повторно центрифугировались и затем лиофилировались. Изолированный конъюгат фильтровался через сито в 100 мкм для удаления любых крупных частиц, которые нельзя было бы впрыскивать через иглу калибра 21. Анализ размеров получившихся частиц представлен в таблице I в конце описания.
Пример 3
Раствор PPIC из примера 1 так же осаждался, как описано выше в примере 2, за исключением того, что ванна этанола при температуре -20oC использовалась вместо водной ванны при 0oC. Анализ размеров получившихся частиц представлен в таблице I в конце описания.
Пример 4
Раствор PPIC из примера 1 также диспергировался при контролируемом расходе жидкости 4 мл/мин через распыляющее сопло, содержащее полый наконечник (Bioblock: 50 Вт, 20 кГц), над ванной этанола при -10oC в реакторе с рубашкой, контролируемом по температуре. В этом процессе пульверизации раствор сополимера высвобождался из пробы в виде мелкого тумана из маленьких капелек. Маленькие капельки падали в ванну этанола, вызывая экстрагирование из капелек деионизированной воды и ацетона. В результате капельки сополимера затвердевали в виде маленьких твердых частиц. Затем частицы извлекались центрифугированием и лиофилизировались. Анализ размеров получившихся частиц представлен в таблице 1. Под диаметром -10 (т.е. D 0,1), диаметром -50 (т.е. D 0,5) или диаметром -90 (т.е. D 0,9) имеется в виду наименьший диаметр, который больше, чем 10, 50 и 90% всех частиц соответственно. Под удельной поверхностью имеется в виду удельная поверхность получившихся частиц.
Пример 5
5,0 г PPIC, описанного выше в примере 4, было растворено в 20 г ацетона (концентрация 20% PPIC по массе). Затем раствор распылялся при расходе 4,0 мл/мин над ванной 500 мл этанола при -10oC, как описано а примере 4. После приготовления частиц PPIC в ванне 500 мл деионизированной воды добавлялось в ванну и затем ванну доводили до 0oC. Затем в ванне осуществляли перемешивание в течение 30 мин, доводилась до 20oC и перемешивалась еще в течение 30 мин. Затем частицы PPIC извлекались фильтрацией и высушивались в вакууме при комнатной температуре. Анализ получившихся частиц представлен в таблице II в конце описания.
Как показано в таблице II, были получены частицы с разной морфологией. Частицы в примере 4 были крупнее и имели меньшую удельную поверхность. Как показал электронный сканирующий микроскоп, частицы, полученные в примере 4, были также более пористыми, вероятно из-за замерзшей воды, оставшейся в частицах после осаждения. Когда дисперсия в ванне была возвращена к комнатной температуре, вода растаяла и вытекла в ванну этанола, оставляя открытые канальчики в микрочастицах. Следовательно, эти частицы были более хрупкими и давали фрагменты малых размеров.
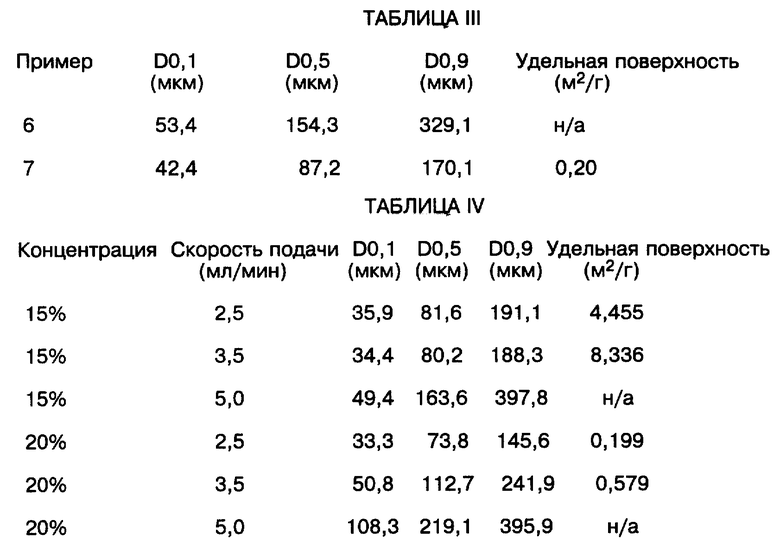
Пример 6
Раствор PPIC, описанный выше в примере 5, распылялся при 2,5 мл/мин над 1,5 л деионизированной воды при 0oC. Анализ размеров получившихся частиц представлен в таблице III в конце описания.
Пример 7
Раствор PPIC, описанный выше в примере 5, распылялся при 2,5 мл/мин над 1,5 л этанола при -10oC. Анализ размеров получившихся частиц представлен в таблице III в конце описания.
Пример 8
Два раствора PPIC готовились в ацетоне, как описано выше в примере 5. Первый раствор имел концентрацию PPIC 15%, тогда как второй раствор имел концентрацию PPIC 20%. Растворы распылялись над ванной этанола при -10oC с величинами расхода 2,5, 3,5 и 5,0 мл/мин, как описано в примере 5. Анализ размеров получившихся частиц представлен в таблице IV в конце описания.
Анализ частиц с использованием сканирующего электронного микроскопа показал, что размер частиц и удельная поверхность увеличивались с увеличением скорости подачи.
Пример 9
5,0 г микрочастиц PPIC из примера 4 было растворено в 45 г ацетона (концентрация 10% по массе). Затем раствор добавлялся по каплям в энергично перемешиваемые 500 мл n-гексана при комнатной температуре. Раствор n-гексана становился мутным по мере осаждения частиц PPIC. PPIC удалялся фильтрацией и высушивался при вакууме и при комнатной температуре.
Пример 10
В реакторе с рубашкой 3,0 г микрочастиц PPIC, описанных в примере 2, диспергировалось в энергично перемешиваемые 250 мл медицинского силиконового масла 12.500 cs (изготовитель Dow Coming, Мидленд, Мичиган) (с 1% PPIC по массе). После перемешивания смесь нагревалась до 120oC, что выше Tg в 55oC для PPIC, и держалась при этой температуре 30 мин. Во время нагревания отдельные изолированные частицы плавились с образованием сферических капелек. Затем дисперсия охлаждалась до 20oC и разводилась 1250 мл гексана. Затем микросферы затвердевали, извлекались фильтрацией, промывались свежим гексаном и окончательно высушивались в вакууме. Характеристики полученных микросфер приведены в таблице V в конце описания. Конечные микросферы имели малый диаметр по сравнению с частицами в примере 2 в результате уплотнения частиц во время их плавления.
Пример 11
0,2 г микрочастиц PPIC, описанных в примере 2, были диспергированы в 5 мл деионизированной воды и энергично перемешивались с помощью вихревого вибратора. Затем 100 мл микролитров дихлорметана (DCM) добавлялись сверху к перемешиваемой дисперсии. Добавление небольшого количества DCM вызывало разбухание поверхности частиц PPIC. Перемешивание продолжалось при комнатной температуре в течение 4 ч, что позволяло испарить DCM с последующим затвердением раздутой поверхности частиц. Сканирующий электронный микроскоп показал, что получившиеся частицы имели сферическую форму с более гладкой поверхностью по сравнению с исходным материалом. Было сужено распределение размеров частиц и уменьшился максимальный размер частиц в результате увеличения плотности частиц.
Пример 12
1 л масла кунжутных семян (изготовитель Vitamins, Inc., Чикаго, Иллинойс) помещался в 2-литровую колбу с тремя шейками, погруженную в водную ванну. Масло перемешивалось со скоростью 600 об/мин с использованием тефлоновой перемешивающей лопатки, соединенной с расположенным выше двигателем для перемешивания. 500 мг поверхностно-активного вещества, лецитина, соевых бобов (изготовитель Sigma Chemicals, Сент-Луис, Миссури), добавлялись к маслу кунжутных семян, и смесь перемешивалась в течение 10 мин. Затем 10 г состава PPIC растворялись в 100 мл ацетонитрила, давая прозрачный раствор. Составы PPIC получались с использованием Lanreotide (товарный знак), сопряженного с одним из следующих трех полимеров: сополимер 64/34/2 поли-DL-молочной-со-гликолевой-D, L-яблочной кислоты (средний молекулярный вес 6.000) (состав 1); сополимер 74/24/2 поли-DL-молочной-со-гликолевой-D,L-яблочной кислоты (средний молекулярный вес 6.000) (состав 2); и сополимер 98/2 поли-DL- молочной-со-D,L-яблочной кислоты (состав 3).
Этот прозрачный раствор PPIC добавлялся по каплям через капельную воронку. Когда добавление было завершено, температура внешней водной ванны поднималась до 40oC, и масло перемешивалось в течение 20 ч. Затем добавлялся 1 л гексана, чтобы развести масло кунжутных семян, и масло фильтровалось через воронку со спеченной средой. Микросферы, собранные в воронке фильтра, далее промывались несколько раз 500 мл общего объема гексана. Частицы высушивались при 36oC в течение двух дней в вакууме. Характеристики полученных микросфер представлены в таблице VI в конце описания.
Пример 13
Реактор загружался мономерами гликолида (изготовитель Purac Biochem, Нидерланды, 84,83 г), лактида (Purac Biochem, 210,67 г) и L(+)-винной кислотой (изготовитель Riedel-de-Haen, Зеельце, Германия, изделие номер 33.801, 4,50 г) и содержащим двухвалентное олово 2-этил гексаноатом (Sigma, Сент-Луис, изделие номер S-3252) в растворе (0,1025 М, 4,34 мл) толуола (Riedel-de-Haen). L(+)-винная кислота предварительно высушивалась над фосфорным пентаоксидом (Riedel-de- Haen) в сушильном аппарате Abderhalden в течение 10 ч. Затем реактор (соединенный с насосом через жидкую азотную ловушку) помещался в вакуум (0,04 мбар) с перемешиванием в течение 50 мин для удаления толуола. Затем реактор в атмосфере свободного от кислорода азота (изготовитель BOC gases, Дублин, Ирландия, содержание влаги 8 VPM) погружался в масляную ванну (температура 200oC), и скорость перемешивания увеличивалась до 125 об/мин. До начала погружения нагревательная лента (типа 45500 Thermolyne, установка входного контроля = 4) помещалась на крышку реактора. Отмечалось время, требуемое для полного расплавления содержимого реактора, обычно 10 мин для загрузки 300 г при 200oC. Образцы брались каждый час во время синтеза и анализировались с помощью гельпроникающей хроматографии для определения процентного отношения остаточного мономера и для получения значений среднечисленной молекулярной массы (Mn) и средневесовой молекулярной массы (Mw). Обычное время реакции составляет порядка 6 ч.
Получался аморфный сополимер, содержащий 66,21% элементарных звеньев лактида, 33,11% элементарных звеньев гликолида и 0,68% элементарных звеньев винной кислоты (66/33/1 PLGTA). Кислотное число титрования определялось как 0,303 мэкв/г (Meq/r; нормальность NaOH, умноженная на объем раствора NaOH, требуемый для нейтрализации одного грамма сложного полиэфира). Среднечисленная молекулярная масса сополимера имела величину 10.250, средневесовая молекулярная масса сополимера была 11.910, давая значение Mw/Mn равное 1,16.
41,32 г указанного 10.000 г/моль сополимера 66/32/2 поли- L-молочной-со-гликолевой-со-L(+)-винной кислоты (кислотный номер 0,303 meq/г) было растворено в 165,52 г ацетона (Riedel-de Haen) путем разрушения ультразвуком в ванне Branson для разрушения ультразвуком (изготовитель Branson, Дэнбери, Коннектикут, США) с получением раствора, имеющего концентрацию PLGTA 19,98% по массе.
К этому раствору добавлялось 37,6 мл 0,2 N карбоната натрия (изготовитель Aldrich, Гиллингем, Дорсет, Великобритания), создавая избыток в 1,2 раза натрия относительно карбоксильных групп сополимера. Раствор перемешивался в течение 30 мин для облегчения образования соли натрия. Затем он подавался в сопло распылителя с расходом 8,0 мл/мин с использованием насоса Masterflex (изготовитель Cole Parmer, Бэррингтон, Иллинойс, США). Раствор распылялся в реактор с рубашкой на 6 л, содержащий 2 л деионизированной воды, охлажденной до 2,5oC с использованием ванны с циркуляцией (изготовитель Huber, Оффенбург, Германия). Эта вода перемешивалась со скоростью 350 об/мин с использованием лопатки с 4 лопастями, соединенной с двигателем мешалки.
Когда распыление было завершено, дисперсия помещалась в 6 бутылей для центрифугирования и вращалась со скоростью 5000 об/мин в течение 30 мин в центрифуге Sorvall (изготовитель DuPont Sorvall Products, Уилмингтон, Делавар, США). Получившиеся после центрифугирования лепешки повторно взвешивались в деионизированной воде и повторно вращались. Образовавшийся сверху в результате отстаивания слой удалялся, и лепешки замораживались в морозилке на половину суток, а потом высушивались в маломасштабном лиофилизаторе (изготовитель Edwards, Кроли, Западный Сассекс, Великобритания) на следующий день. Извлекалось 33,16 г промытого сополимера, что представляет собой выход 80,24%.
4,92 г указанного 10.000 г/моль сополимера 66/33/1 поли-L-молочной-со-гликолевой-со-D,L-винной кислоты (66% L-молочной кислоты, 33% гликолевой кислоты и 1% винной кислоты) растворялось в 11,58 г ацетонитрила (Riedel-de Haen; типа, пригодного для высокоэффективной жидкостной хроматографии) путем разрушения ультразвуком в ванне для разрушения ультразвуком Branson (изготовитель Branson) и перемешивалось на смесительной пластине, давая раствор с концентрацией PLGTA 29,82% по массе.
Этот раствор сополимера/ацетонитрила подавался из стеклянного резервуара через сопло распылителя с использованием вращающегося поршневого насоса FMI (изготовитель FMI, Ойстер Бей, Нью-Йорк, США), установленного на расход 2,0 мл/мин. Выходная мощность распылителя устанавливалась на 50 Вт с амплитудой 80%. Раствор распылялся в реактор с рубашкой на 6 л, содержащий 1,5 л реактива общего назначения - изопропилового спирта (изготовитель Labscan/y6AHH, Ирландия), охлажденного до -70oC твердыми гранулами CO2 (изготовитель AIG, Дублин, Ирландия), и перемешивался со скоростью 300 об/мин лопаткой с 4 лопастями, соединенной с двигателем мешалки. Температура изопропилового спирта оставалась равной или близкой -70oC на протяжении всего распыления, которое длилось приблизительно 8 мин .
Когда распыление было завершено, дисперсия нагревалась до 10oC естественным путем в течение 5,5 ч. Затем она фильтровалась через фильтровальную бумагу Whatman N.1 (диаметр 9 см) с помощью вакуума. Фильтровальная бумага и лепешка помещались в эксикатор или сушильный шкаф вместе с сушильными полосками силикагеля, и воздух вытягивался через автоматическую морозильную ловушку при -110oC. Спустя 24 ч 4,24 г материала извлекалось. Анализ получившихся частиц представлен в таблице VII в конце описания.
Следует понимать, что хотя изобретение было описано в сочетании с его подробным описанием, предыдущее описание предназначено для иллюстрации, а не для ограничения объема изобретения, который определяется рамками прилагаемой формулы изобретения. Другие аспекты, преимущества и модификации находятся в рамках патентной формулы.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИОННЫЙ КОНЪЮГАТ С ДЛИТЕЛЬНЫМ ПЕРИОДОМ ВЫСВОБОЖДЕНИЯ ПЕПТИДА, СПОСОБ СИНТЕЗИРОВАНИЯ ИОННОГО КОНЪЮГАТА, СПОСОБ СИНТЕЗИРОВАНИЯ МИКРОЧАСТИЦ | 1994 |
|
RU2146128C1 |
| СЛОЖНЫЙ ПОЛИЭФИР И КОНЪЮГАТ НА ЕГО ОСНОВЕ | 1994 |
|
RU2185393C2 |
| СПОСОБ ПОЛУЧЕНИЯ СОСТАВА С ЗАМЕДЛЕННЫМ ВЫСВОБОЖДЕНИЕМ АКТИВНОГО ИНГРЕДИЕНТА | 2000 |
|
RU2211694C1 |
| БИОРАЗРУШАЕМЫЙ СЛОЖНЫЙ ПОЛИЭФИР И СПОСОБ ЕГО ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 1997 |
|
RU2165942C2 |
| ИОННЫЕ МОЛЕКУЛЯРНЫЕ КОНЪЮГАТЫ N-АЦИЛИРОВАННЫХ ПРОИЗВОДНЫХ ПОЛИ(2-АМИНО-2-ДЕОКСИ-D-ГЛЮКОЗЫ) И ПОЛИПЕПТИДОВ | 1996 |
|
RU2172323C2 |
| ИОННЫЕ МОЛЕКУЛЯРНЫЕ КОНЬЮГАТЫ БИОДЕГРАДИРУЕМЫХ СЛОЖНЫХ ПОЛИЭФИРОВ И БИОАКТИВНЫХ ПОЛИПЕПТИДОВ | 2000 |
|
RU2237681C2 |
| ФОСФОРИЛИРОВАННЫЕ ПОЛИМЕРЫ И ИХ КОНЪЮГАТЫ | 1999 |
|
RU2202563C2 |
| ПЕПТИДНАЯ КОМПОЗИЦИЯ С ЗАМЕДЛЕННЫМ ВЫСВОБОЖДЕНИЕМ | 2000 |
|
RU2237675C2 |
| НАЦЕЛЕННЫЕ КОНЪЮГАТЫ И ЧАСТИЦЫ И ИХ СОСТАВЫ | 2015 |
|
RU2695220C2 |
| ПРЕПАРАТ С ОТСРОЧЕННЫМ ВЫСВОБОЖДЕНИЕМ, СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1995 |
|
RU2181999C2 |




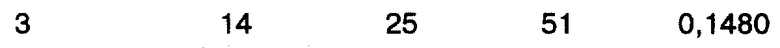

Изобретение относится к фармации и затрагивает способ получения и придания сферической формы ионному конъюгату, который содержит биологически разлагаемый полимер, содержащий свободные карбоксильные группы, и лекарство, содержащее свободные аминогруппы, причем полимер и лекарство образовали между собой ионные связи. Изобретение обеспечивает непрерывное выделение лекарства. 3 с. и 34 з.п. ф-лы, 7 табл.
| Способ получения лекарственного средства в форме шариков | 1991 |
|
SU1837872A3 |
| US 3773919 A, 20.11.1973 | |||
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| Экономайзер | 0 |
|
SU94A1 |

