Ссылка на родственные заявки
Данная заявка является частичным продолжением одновременно рассматриваемой заявки №08/867308, поданной 2 июня 1997 г., по которой выдан патент США №5863985 26 января 1999 г., которая является частичным продолжением заявки №08/464735, поданной 29 июня 1995 г., по которой выдан патент США №5672659 30 сентября 1997 г., которая является заявкой, перешедшей в Национальную Фазу PCT/US 94/00148, поданной 5 января 1994 г., которая является РСТ фазой заявки Ирландии №930005, поданной 6 января 1993 г.
Предпосылки создания изобретения
Данное изобретение относится к непрерывному высвобождению биологически активных полипептидов.
Многочисленные системы доставки лекарственных средств разработаны, исследованы и использованы для регулируемого высвобождения in vivo фармацевтических композиций. Например, сложные полиэфиры, такие как поли(DL-молочная кислота), поли(гликолевая кислота), поли(ε-капролактон) и различные другие сополимеры использованы для высвобождения биологически активных молекул, таких как прогестерон; такие соединения имели форму микрокапсул, пленок или палочек (Pitt CG, Marks ТА and Schindler A. 1980). При имплантации композиции полимер/терапевтический агент, например, подкожно или внутримышечно терапевтический агент высвобождался в течение определенного периода времени. Такие биосовместимые, биодеградируемые полимерные системы предназначены для того, чтобы позволить заключенному в композицию терапевтическому агенту диффундировать из полимерной матрицы. При высвобождении терапевтического агента полимер деградирует in vivo, что позволяет избежать хирургического удаления имплантата. Хотя факторы, которые приводят к деградации полимера, не установлены, полагают, что такую деградацию полимеров можно регулировать доступностью эфирных связей для неэнзиматического автокаталитического гидролиза полимерных компонентов.
Некоторые публикации ЕРО и патенты США посвящены конструкции полимерной матрицы и ее роли в регулировании скорости и степени высвобождения терапевтического агента in vivo.
Например, Deluca (публикация ЕРО 0467389 A2/Univ of Kentucky) описано физическое взаимодействие между гидрофобным биодеградируемым полимером и белком или полипептидом. Полученная композиция была смесью терапевтического агента и гидрофобного полимера, что поддерживало диффузионное высвобождение терапевтического агента из матрицы после введения субъекту.
Hutchison (патент США 4767628/ICI) наблюдал высвобождение терапевтического агента посредством равномерного распределения в полимерном устройстве. Установлено, что данная композиция предусматривает регулируемое непрерывное высвобождение посредством перекрытия двух фаз: первой, зависимого от диффузии выщелачивания лекарственного средства с поверхности композиции, и второй, индуцированного деградацией полимера высвобождения по водным каналам.
Краткое изложение изобретения
В общих чертах в изобретении представлена фармацевтическая композиция непрерывного высвобождения, состоящая из сложного полиэфира, содержащего свободные СООН-группы, ионно сопряженные с биологически активным полипептидом, содержащим, по крайней мере, один эффективный ионогенный амин, в которой, по крайней мере, 50 мас.% полипептида, присутствующего в композиции, являются ионно конъюгированными со сложным полиэфиром.
В предпочтительных вариантах осуществления сложный полиэфир модифицирован с тем, чтобы увеличить соотношение концевых групп, карбоксильных к гидроксильным, от более единицы до приближающегося к бесконечности, то есть все из гидроксильных групп могут быть замещены карбоксилами. Примерами подходящих сложных полиэфиров являются полиэфиры, производные таких соединений, как L-молочная кислота, D-молочная кислота, DL-молочная кислота, ε-капролактон, п-диоксанон, ε-капроновая кислота, замещенный и незамещенный триметиленкарбонат (ТМС), 1,5-диоксепан-2-он, 1,4-диоксепан-2-он, гликолид, гликолевая кислота, L-лактид, D-лактид, DL-лактид, мезолактид, оксалат алкилена, оксалат циклоалкилена, сукцинат алкилена, (β-гидроксибутират) и оптически активные изомеры, рацематы или сополимеры любого из вышеперечисленных, в которых замещенный ТМС замещен (C1-C4)алкилом, предпочтительно метилом. Также могут быть использованы другие полимеры с гетероцепью, родственные традиционным сложным полиэфирам (например, полиортоэфиры, полиортокарбонаты и полиацетали).
Предпочтительно получают поликарбоксильные сложные полиэфиры путем реакции с яблочной кислотой, лимонной кислотой или винной кислотой.
В предпочтительных вариантах осуществления сложный полиэфир частично содержит концевые кислотные остатки за счет глутарового ангидрида. В других предпочтительных вариантах сложный полиэфир полностью содержит концевые кислотные остатки за счет глутарового ангидрида. Предпочтительно сложный полиэфир характеризуется средней степенью полимеризации между 10 и 300 и более предпочтительно между 20 и 50.
Ионные молекулярные конъюгаты по изобретению предпочтительно получают из сложных полиэфиров с концевыми остатками поликарбоновой кислоты, сопряженных с одноосновными и многоосновными биологически активными полипептидами, содержащими, по крайней мере, одну эффективную ионогенную аминную группу. Альтернативно любой сложный полиэфир может быть использован для образования ионного молекулярного конъюгата по изобретению при условии, что он предварительно был обработан подходящим основанием, например NaOH. Кроме того, может быть использован любой кислотостабильный пептид, например пептид, освобождающий гормон роста (GHRP), гормон, высвобождающий лютеинизирующий гормон (LHRH), соматостатин, бомбезин, пептид, высвобождающий гастрин (GRP), кальцитонин, брадикинин, галанин, меланоцитстимулирующий гормон (MSH), релизинг-фактор гормона роста (GRF), амилин, тахикинины, секретин, паратиреоидный гормон (РТН), энкефалин, эндотелин, пептид, освобождающий ген кальцитонина (CGRP), нейромедины, белок, родственный паратиреоидному гормону (РТНrР), глюкагон, нейротензин, адренокортикотропный гормон (АСТН), пептид YY (PYY), пептид, высвобождающий глюкагон (GLP), вазоактивный интестинальный пептид (VIP), пептид, активирующий аденилатциклазу гипофиза (РАСАР), мотилин, субстанция Р, нейропептид Y (NPY), TSH (тиреотропный гормон) и их аналоги и фрагменты. Такие ионные молекулярные конъюгаты способны высвобождать in vivo биоактивные компоненты с предопределенными скоростями, установленными по химической структуре, молекулярной массе и рКа обоих компонентов полученных конъюгатов. Механизм высвобождения лекарственного средства вызывает превращение нерастворимой формы конъюгата до компонентов, растворимых в воде, отчасти посредством гидролиза гидрофобного сложного полиэфира. Таким образом, высвобождение биоактивного полипептида независимо увеличивается с (а) уменьшением разницы в рКа биоактивного полипептида и сложного полиэфира, (b) химической реактивностью цепи сложного полиэфира, что отражается в нуклеофильности карбонила, (с) уменьшением плотности полиэфира, что связано с температурой стеклования и приуменьшенной кристаллизуемостью, и (d) увеличением гидрофильности матрицы.
В предпочтительных направлениях полипептид составляет 1 до 50 мас.% от общей массы ионного молекулярного конъюгата и предпочтительно более 85%, более предпочтительно 95% и наиболее предпочтительно 99% полипептида, присутствующего в композиции, оказывается ионно конъюгированным со сложным полиэфиром; полиэфирный компонент ионного молекулярного конъюгата характеризуется вязкостью приблизительно от 0,05 до 0,7 дл/г в хлороформе; средняя молекулярная масса полиэфира составляет приблизительно 1200-40000.
Полимерные ионные молекулярные конъюгаты изобретения могут быть легко получены в инъецируемых микросферах или микрочастицах и имплантируемых пленках или палочках без необходимости применения способа, который вызывает образование мультифазных эмульсий или неводных двухфазных систем. Предпочтительно микрочастицы производят посредством (а) растворения композиции в апротонном, смешивающемся с водой органическом растворителе; (b) смешивания органического растворителя в воде и (с) выделения микрочастиц из воды. В предпочтительных вариантах осуществления органический растворитель выбирают из группы, состоящей из ацетона, ацетонитрила, тетрагидрофурана, диметилформамида и диметоксиэтиленгликоля.
В других предпочтительных вариантах осуществления ионный молекулярный конъюгат сложный полиэфир/полипептид способен высвобождать in vivo терапевтически эффективную дозу биологически активного полипептида в течение периода, по крайней мере, 20 дней и более предпочтительно в течение вплоть до 95 дней, но не менее 7 дней. В некоторых других предпочтительных вариантах осуществления высвобождение терапевтического ионного молекулярного конъюгата по существу является монофазным.
В данном изобретении предпочтительно, что композиции непрерывного высвобождения получают посредством (а) получения сложного полиэфира, имеющего свободные СООН-группы, и биоактивного полипептида, содержащего, по крайней мере, один эффективный ионогенный амин, и (b) ионного конъюгирования сложного полиэфира с полипептидом с образованием ионного молекулярного конъюгата, в котором, по крайней мере, 85 мас.% полипептида, присутствующего в композиции, оказывается ионнно конъюгированными со сложным полиэфиром. Сложный полиэфир может быть полиэфиром, который, прежде всего, содержит достаточное количество свободных СООН-групп, или, если вначале имеется недостаточное количество таких групп для требуемого уровня пептидного присоединения, сложный полиэфир может (1) вступать в реакцию, например, с яблочной, лимонной или винной кислотой посредством этерификации или функционального обмена или (2) иметь на конце кислотные остатки за счет, например, глутарового ангидрида или (3) полиэфир может быть обработан основанием, например NaOH, чтобы экспонировать кислотные группы. Наконец, ионный молекулярный конъюгат сложный полиэфир/полипептид может быть превращен в имплантируемые пленки или палочки, или инъецируемые микросферы или микрочастицы, способные высвобождать in vivo полипептид.
Предпочтительно сложный полиэфир синтезируют в результате катализируемой или автокатализируемой прямой конденсации одной или более гидроксикислот, например гликолевой кислоты и молочной кислоты, в присутствии предопределенной концентрации поликарбоновой гидроксикислоты, например яблочной кислоты, лимонной кислоты или винной кислоты. Образованные таким образом сложные полиэфиры содержат гидроксильные концевые группы с присоединенными к ним кислотными остатками, причем гидроксильные группы предпочтительно имеют частично или полностью концевые кислотные остатки.
Сложные полиэфиры также могут быть синтезированы в результате катализуемой полимеризации лактонов с раскрытием кольца или в результате полимеризации циклических мономеров, таких как ε-капролактон, п-диоксанон, триметиленкарбонат, 1,5-диоксепан-2-он или 1,4-диоксепан-2-он, в присутствии инициатора цепи, например поликарбоновой гидроксикислоты.
Другой способ синтеза включает в себя реакцию гидроксикислоты с циклическим димером с последующей конденсацией системы открытых цепей в присутствии поликарбоновой кислоты.
Еще один способ синтеза включает в себя реакцию органической поликарбоновой кислоты с предварительно образованным сложным полиэфиром.
В вышеупомянутых предпочтительных вариантах осуществления сложный полиэфир с присоединенными на концах кислотными остатками имеет соотношение концевых групп, карбоксильных к гидроксильным, более единицы и приближающееся к бесконечности (то есть удаление всех гидроксильных групп) со средней степенью полимеризации между 10 и 300 и, в особо предпочтительных вариантах осуществления, между 20 и 50.
Альтернативно сложный полиэфир оказывается способным образовывать ионный молекулярный конъюгат с биоактивным полипептидом в результате обработки основанием, например NaOH.
Предпочтительно ионный молекулярный конъюгат сложный полиэфир/полипептид синтезируют посредством прямого взаимодействия между сложным полиэфиром, например, в свободной форме и полипептидом, например, в свободной форме в подходящей жидкой среде. В других предпочтительных вариантах осуществления изобретения подходящими растворителями для образования конъюгата могут быть смесь апротонного растворителя (например, ацетона, тетрагидрофурана (THF) или диметилового эфира этиленгликоля) и подходящего растворителя для пептида (например, вода) в таких пропорциях, что две системы оказываются совместимыми. Предпочтительно полипептид является солью монокарбоновой кислоты, имеющей рКа выше или равной 3,5. Предпочтительно полипептид содержит, по крайней мере, одну эффективную ионогенную аминную группу.
В предпочтительных вариантах осуществления полипептид составляет от 1 до 50 мас.% и предпочтительно 10 до 20 мас.% ионного молекулярного конъюгата сложный полиэфир/полипептид. В предпочтительных направлениях доступные карбоксильные группы сложного полиэфира частично нейтрализуют ионами щелочных металлов или органическими основаниями. В других предпочтительных вариантах осуществления щелочная обработка обеспечивает диссоциацию цепи сложного полиэфира и образование низкомолекулярных участков связывания.
В другом аспекте данное изобретение относится к сложному полиэфиру (обозначен как сложный полиэфир А), содержащему одну или более свободных СООН-групп и характеризующемуся соотношением карбоксила и гидроксила более единицы, причем названный полиэфир содержит элемент, выбранный из группы, состоящей из L-молочной кислоты, D-молочной кислоты, DL-молочной кислоты, яблочной кислоты, лимонной кислоты, ε-капролактона, п-диоксанона, ε-капроновой кислоты, оксалата алкилена, оксалата циклоалкилена, сукцината алкилена, β-гидроксибутирата, замещенного или незамещенного триметиленкарбоната, 1,5-диоксепан-2-она, 1,4-диоксепан-2-она, гликолида, гликолевой кислоты, L-лактида, D-лактида, DL-лактида, мезолактида и их любых оптически активных изомеров, рацематов или сополимеров при условии, что лимонная кислота, ε-капролактон и гликолид являются элементами сложного полиэфира. Предпочтительным вариантом вышеописанного сложного полиэфира (обозначен как полиэфир В) является сложный полиэфир, который содержит лимонную кислоту, ε-капролактон и гликолид. Предпочтительным вариантом непосредственно вышеописанного сложного полиэфира (обозначенного как полиэфир С) является то, что соотношение ε-капролактона и гликолида в полиэфире составляет от 90 ε-капролактона : 10 гликолида до 99 ε-капролактона : 1 гликолида. Предпочтительным сложным полиэфиром (обозначен как полиэфир D) является такой, где соотношение ε-капролактона и гликолида в полиэфире составляет 97 ε-капролактона : 3 гликолида.
В еще одном аспекте данное изобретение направлено на композицию, содержащую полиэфир А, полиэфир В, полиэфир С или полиэфир D, ионно конъюгированный с одним или более биологически активными полипептидами, включающими в себя, по крайней мере, один эффективный ионогенный амин, в которой, по крайней мере, 50 мас.% полипептида, присутствующего в композиции, оказываются ионно конъюгированными с полиэфиром.
Предпочтительный вариант осуществления непосредственно вышеописанной композиции заключается в том, что биологически активный полипептид выбирают из группы, состоящей из LHRH, соматостатина, бомбезина/GRP, кальцитонина, брадикинина, галанина, MSH, GRF, амилина, тахикининов, секретина, РТН, CGRP, нейромединов, РТНrР, глюкагона, нейротензина, АСТН, GHRP, GLP, VIP, РАСАР, энкефалина, PYY, мотилина, субстанции Р, NPY, TSH и их аналогов или фрагментов.
Предпочтительный вариант осуществления описанной непосредственно выше композиции заключается в том, что биологически активный полипептид выбирают из группы, состоящей из LHRH, соматостатина и их аналогов или фрагментов.
Предпочтительный вариант осуществления непосредственно вышеописанной композиции заключается в том, что аналог LHRH является пептидом формулы pGlu-His-Trp-Ser-Tyr-D-Trp-Leu-Arg-Pro-Gly-NH2 и аналог соматостатина является пептидом формулы H2N-β-D-Nal-Cys-Tyr-Trp-Lys-Val-Cys-Thr-NH2, в котором два остатка Суs аналога соматостатина связаны друг с другом.
Предпочтительный вариант осуществления непосредственно вышеописанной композиции заключается в том, что композиция существует в форме палочки.
Предпочтительный вариант осуществления непосредственно вышеописанной композиции заключается в том, что палочка имеет покрытие из сложного полиэфира.
Предпочтительный вариант осуществления непосредственно вышеописанной композиции заключается в том, что полиэфирное покрытие палочки представляет собой абсорбируемый сложный полиэфир.
Предпочтительный вариант осуществления непосредственно вышеописанной композиции заключается в том, что абсорбируемый сложный полиэфир содержит одну или больше свободных СООН-групп и характеризуется соотношением карбоксила и гидроксила больше единицы, в которой вышеназванный сложный полиэфир содержит элемент, выбранный из группы, состоящей из L-молочной кислоты, D-молочной кислоты, DL-молочной кислоты, яблочной кислоты, лимонной кислоты, винной кислоты, ε-капролактона, п-диоксанона, ε-капроновой кислоты, оксалата алкилена, оксалата циклоалкилена, сукцината алкилена, β-гидроксибутирата, замещенного или незамещенного триметиленкарбоната, 1,3-диоксепан-2-она, 1,4-диоксепан-2-она, гликолида, гликолевой кислоты, L-лактида, D-лактида, DL-лактида, мезолактида и их любых оптически активных изомеров, рацематов или сополимеров.
Предпочтительный вариант осуществления непосредственно вышеописанной композиции заключается в том, что абсорбируемое полиэфирное покрытие палочки является таким же, как сложный полиэфир, включенный в композицию.
В еще одном аспекте данное изобретение направлено на сложный полиэфир (обозначенный как полиэфир Е), содержащий одну или более свободных СООН-групп и характеризующийся соотношением карбоксила и гидроксила более единицы, причем названный сложный полиэфир содержит элемент, выбранный из группы, состоящей из L-молочной кислоты, D-молочной кислоты, DL-молочной кислоты, яблочной кислоты, лимонной кислоты, винной кислоты, ε-капролактона, п-диоксанона, ε-капроновой кислоты, оксалата алкилена, оксалата циклоалкилена, сукцината алкилена, β-гидроксибутирата, замещенного или незамещенного триметиленкарбоната, 1,5-диоксепан-2-она, 1,4-диоксепан-2-она, гликолида, гликолевой кислоты, L-лактида, D-лактида, DL-лактида, мезолактида и их любых оптически активных изомеров, рацематов или сополимеров при условии, что винная кислота является элементом сложного полиэфира. Предпочтительный вариант осуществления вышеописанного сложного полиэфира (обозначен как полиэфир F) заключается в том, что полиэфир содержит L-молочную кислоту или D-молочную кислоту, или в том, что сложный полиэфир содержит L-молочную кислоту или D-молочную кислоту и гликолевую кислоту. Другой предпочтительный вариант осуществления сложного полиэфира Е (обозначен как полиэфир G) заключается в том, что полиэфир содержит винную кислоту, ε-капролактон и триметиленкарбонат. Предпочтительный вариант осуществления непосредственно вышеописанного полиэфира (обозначен как полиэфир Н) заключается в том, что соотношение ε-капролактона и триметиленкарбоната в полиэфире составляет от 90 ε-капролактона : 10 триметиленкарбоната до 99 ε-капролактона : 1 триметиленкарбоната. Предпочтительный вариант осуществления непосредственно вышеописанного сложного полиэфира (обозначен как полиэфир I) заключается в том, что соотношение ε-капролактона и триметиленкарбоната в полиэфире составляет 98 ε-капролактона : 2 триметиленкарбоната.
В еще одном аспекте данное изобретение направлено на композицию, содержащую полиэфир Е, полиэфир F, полиэфир G, полиэфир Н или полиэфир I, ионно конъюгированный с одним или более биоактивных полипептидов, содержащих, по крайней мере, один эффективный ионогенный амин, в которой, по крайней мере, 50 мас.% полипептида, присутствующего в композиции, ионно конъюгировано со сложным полиэфиром.
Предпочтительный вариант осуществления непосредственно вышеописанной композиции заключается в том, что биоактивный полипептид выбран из группы, состоящей из LHRH, соматостатина, бомбезина/GRP, кальцитонина, брадикинина, галанина, MSH, GRF, амилина, тахикининов, секретина, РТН, CGRP, нейромединов, РТНrР, глюкагона, нейротензина, АСТН, GHRP, GLP, VIP, РАСАР, энкефалина, PYY, мотилина, субстанции Р, NPY, TSH и их аналогов или фрагментов.
Предпочтительный вариант осуществления непосредственно вышеописанной композиции заключается в том, что биоактивный полипептид выбран из группы, состоящей из LHRH, соматостатина и их аналогов или фрагментов.
Предпочтительный вариант осуществления непосредственно вышеописанной композиции заключается в том, что аналог LHRH является пептидом формулы pGlu-His-Trp-Ser-Tyr-D-Trp-Leu-Arg-Pro-Gly-NH2 и аналог соматостатина является пептидом формулы H2N-β-D-Nal-Cys-Tyr-Trp-Lys-Val-Cys-Thr-NH2, в котором два остатка Cys аналога соматостатина связаны друг с другом.
Предпочтительный вариант осуществления непосредственно вышеописанной композиции заключается в том, что композиция находится в форме палочки.
Предпочтительный вариант осуществления непосредственно вышеописанной композиции заключается в том, что палочка имеет покрытие из сложного полиэфира.
Предпочтительный вариант осуществления непосредственно вышеописанной композиции заключается в том, что абсорбируемый сложный полиэфир содержит одну или больше свободных СООН-групп и характеризуется соотношением карбоксила и гидроксила более единицы, причем названный сложный полиэфир содержит элемент, выбранный из группы, состоящей из L-молочной кислоты, D-молочной кислоты, DL-молочной кислоты, яблочной кислоты, лимонной кислоты, винной кислоты, ε-капролактона, п-диоксанона, ε-капроновой кислоты, оксалата алкилена, оксалата циклоалкилена, сукцината алкилена, β-гидроксибутирата, замещенного или незамещенного триметиленкарбоната, 1,5-диоксепан-2-она, 1,4-диоксепан-2-она, гликолида, гликолевой кислоты, L-лактида, D-лактида, DL-лактида, мезолактида и их любых оптически активных изомеров, рацематов или сополимеров.
Предпочтительный аспект непосредственно вышеописанной композиции заключается в том, что абсорбируемое полиэфирное покрытие палочки представляет собой тот же самый сложный полиэфир, включенный в композицию.
Используемый термин "полипептид" относится к белку, пептиду, олигопептиду или синтетическому олигопептиду.
Используемый термин "поликарбоксильный" относится к соединениям, имеющим более одной карбоксильной группы, например яблочной кислоте, лимонной кислоте и винной кислоте.
Используемый термин "средняя степень полимеризации" относится к количеству повторных мономерных последовательностей.
Термин "эффективный ионогенный амин" относится к полипептиду, который содержит, по крайней мере, одну группу амина, способную образовывать ион при широко распространенных условиях.
Термин "содержит концевые кислотные остатки" относится к соединениям, имеющим на конце кислоту.
Термин "частично содержит концевые кислотные остатки" относится к соединениям, в которых 1-99% их гидроксильных концевых групп имеют присоединенные кислотные остатки.
Термин "полностью содержит концевые кислотные остатки" относится к соединениям, в которых 99,9% их гидроксильных концевых групп имеют присоединенные кислотные остатки.
Термин "гидроксикислоты" относится к любому соединению, содержащему гидроксильные и карбоксильные группы.
Термин "монокарбоновая гидроксикислота" относится к органической кислоте с одной карбоксильной группой и одной или более гидроксильных групп.
Термин "поликарбоновая гидроксикислота" относится к гидроксикислоте, имеющей больше одной карбоксильной группы.
Термин "органический азеотропно-дистилляционный вытеснитель" относится к органическим жидкостям, которые совместно перегоняются с водой.
Термин "биоактивный" относится к молекуле, которая вызывает биологическое явление или оказывает влияние на биологическое событие.
Термин "ациклизация" относится к химической реакции, которая происходит посредством раскрытия кольца.
Термин "поликонденсация" относится к образованию сложного полиэфира в результате конденсации двух или более молекул.
Используемый в описании термин "абсорбируемый" сложный полиэфир относится к нерастворимому в воде сложному полиэфиру, который претерпевает диссоциацию цепи в биологической среде с образованием растворимых в воде побочных продуктов.
Данное изобретение относится к новой фармацевтической композиции, в которой химически соединен биосовместимый, биодеградируемый сложный полиэфир с олигопептидами, полипептидами, пептидами и/или белками в виде гомогенного ионного продукта. В результате химического связывания сложных полиэфиров с различными молекулярными массами с терапевтическими агентами химические характеристики композиции могут быть более точно подобраны для того, чтобы удовлетворить требования контролируемого монофазного высвобождения биологически активной полипептидной молекулы in vivo. Кроме того, композиции изобретения легко оптимизируются для приобретения функциональных свойств для большей загрузки терапевтически активного полипептида.
Другие особенности и преимущества изобретения станут очевидными из следующего подробного описания предпочтительных вариантов осуществления и из формулы изобретения.
Краткое описание чертежей
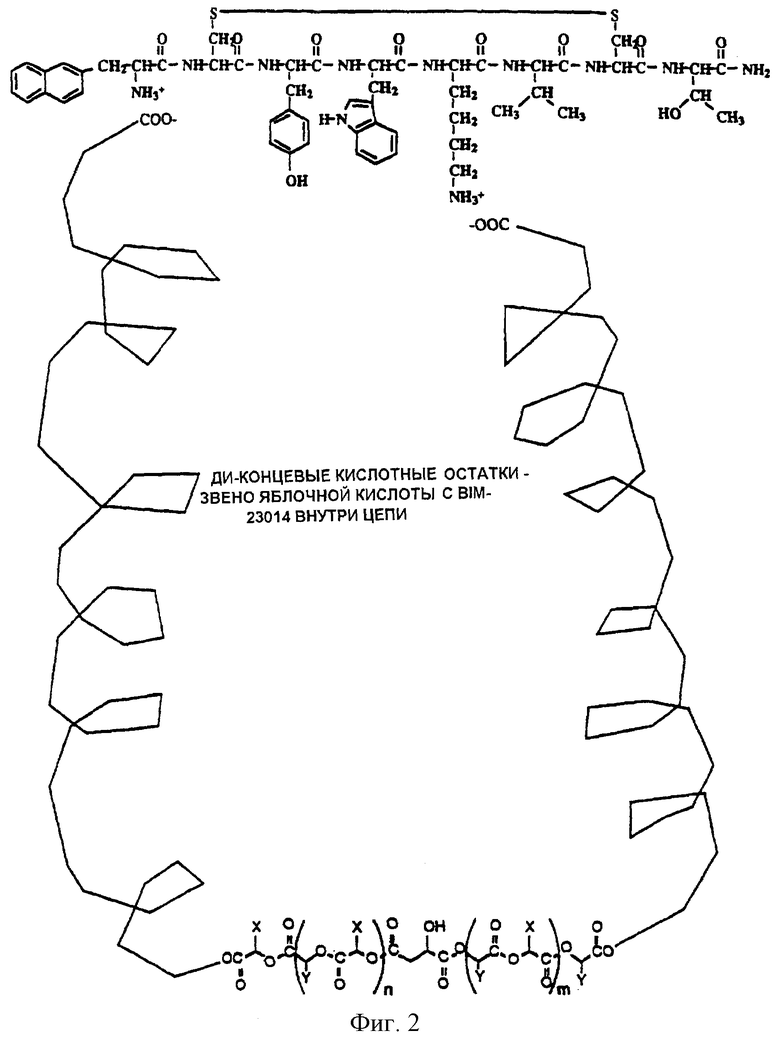
Фиг.1 является иллюстрацией, представляющей изомеры сополимера лактид/гликолид (яблочный тип) с концевыми остатками поликарбоновой кислоты.
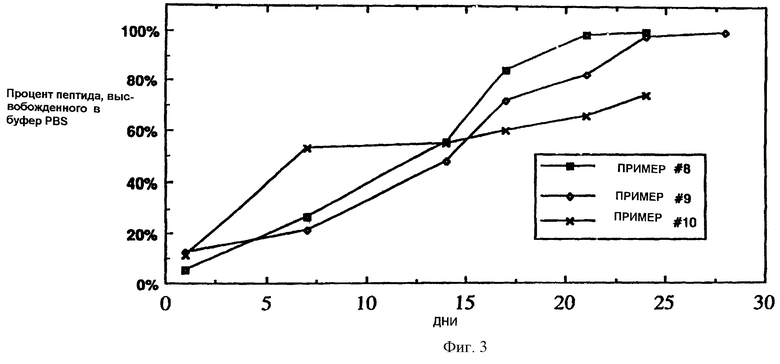
Фиг.2 является изображением ионного молекулярного конъюгата, описывающим химические взаимодействия между сополимером лактид/гликолид (яблочный тип) и Соматилином (BIM-23014).
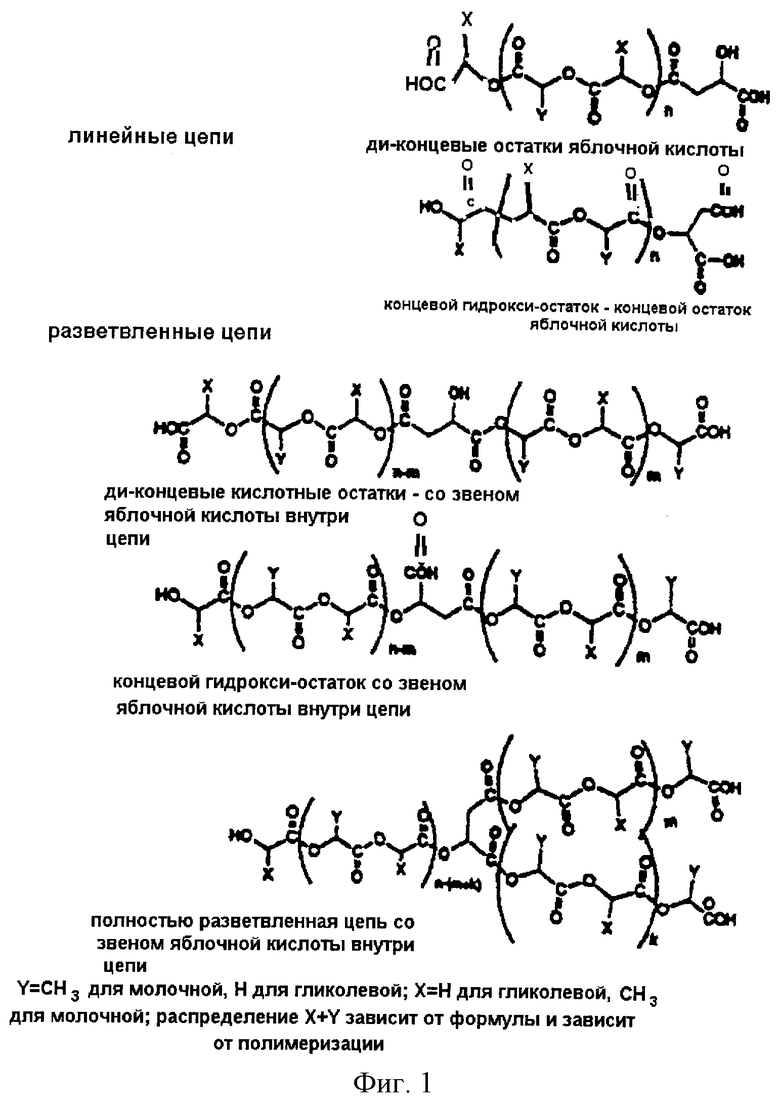
Фиг.3 является графическим изображением процента пептида, высвобожденного из ионных молекулярных конъюгатов в буфер PBS при 37°С в течение 28-дневного периода.
Описание предпочтительных вариантов осуществления изобретения
Синтез
Биодеградируемые или абсорбируемые сложные полиэфиры специально подготавливают для создания требуемой химической реактивности, чтобы обеспечить контролируемую гидролизуемость цепей и проявление максимальной способности связываться с олигопептидами, полипептидами или белками, имеющими общий положительный заряд при физиологическом рН, в результате надлежащей селекции составляющих мономеров, сомономеров или сомеров с образованием цепей с предварительно установленными составами и молекулярными массами.
Для получения композиций данного изобретения применяли схему синтеза, состоящую из трех частей, которая соответствует компетенции специалистов в данной области. Стадии включают в себя (1) синтез сложных полиэфиров с концевыми остатками поликарбоновой кислоты; (2) синтез ионного конъюгата сложный полиэфир/полипептид посредством ионного взаимодействия сложных полиэфиров с концевыми остатками поликарбоновой кислоты (или сложного полиэфира, обработанного основанием) и биологически активных полипептидов и (3) превращение ионных конъюгатов в имплантаты, палочки, микросферы или микрочастицы, способные высвобождать in vivo терапевтический агент в течение 7 дней.
(1) Синтез сложных полиэфиров с концевыми остатками поликарбоновой кислоты
Цепи сложных полиэфиров с концевыми остатками поликарбоновой кислоты по изобретению синтезировали согласно способам, таким как прямая конденсация 2-гидроксикислоты и поликарбоновой органической кислоты, ступенчатая полимеризация ациклизованных продуктов, полимеризация с раскрытием кольца лактона или смеси лактонов или функциональный обмен поликарбоновой органической кислоты с предварительно полученными высокомолекулярными сложными полиэфирами (см. фиг.1). Описания синтеза сложных полиэфиров с концевыми остатками поликарбоновой кислоты согласно вышеупомянутым способам следуют.
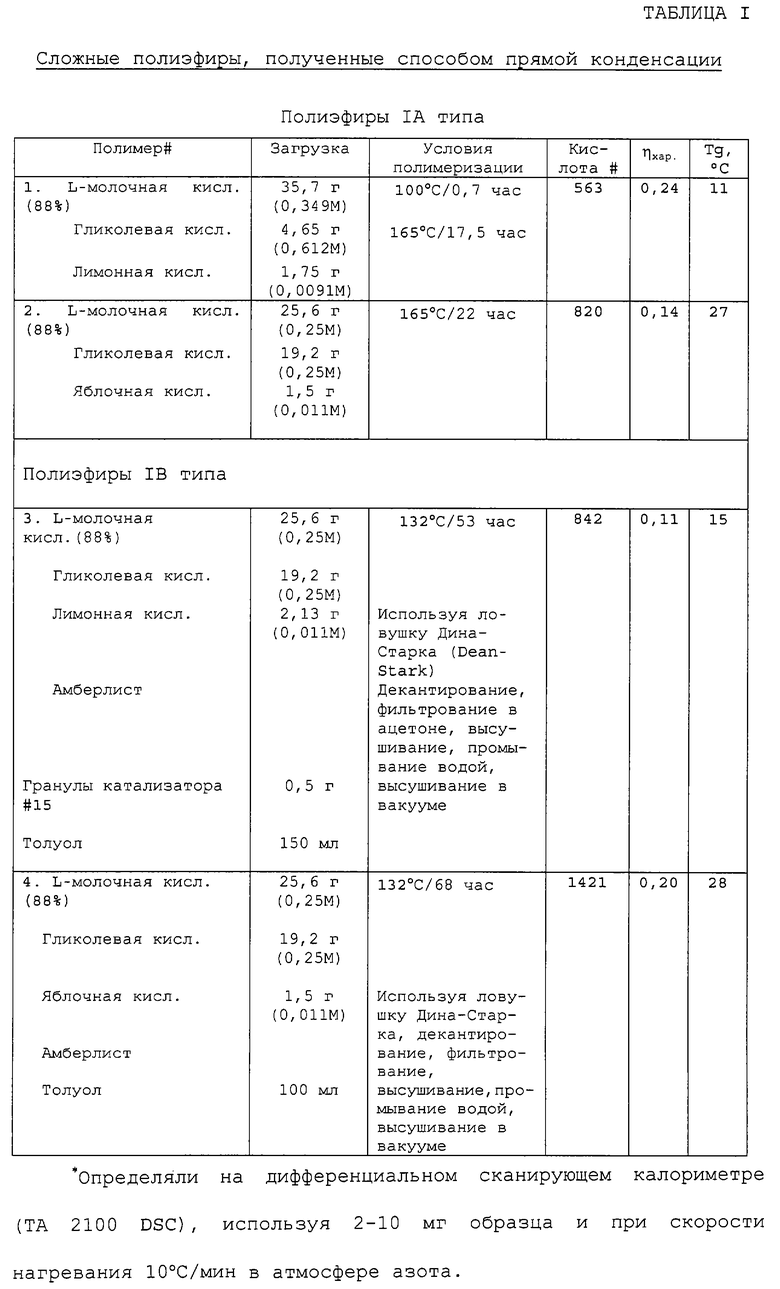
Прямую конденсацию 2-гидроксикислот в оптически активной и/или неактивной форме и предварительно определенного количества поликарбоновой органической кислоты в присутствии и в отсутствие неорганического или металлоорганического катализатора, например конденсацию гликолевой кислоты, DL-молочной кислоты и DL-яблочной кислоты обычно осуществляли нагреванием монокарбоновых гидроксикислот или смеси двух или более монокарбоновых гидроксикислот в присутствии фракции поликарбоновой гидроксикислоты в стеклянном реакторе, оборудованном для обеспечения тока сухого азота и перемешивания массы (обозначен как полиэфир IA типа, см. таблицу I). Обычно поликонденсацию проводили при 150-170°С в течение от 4 до 72 часов. Перемешивание реакционной смеси может быть обеспечено с помощью магнитной мешалки или барботирования газообразного азота через полиэфирную массу. Полимеризацию продолжали до тех пор, пока не были достигнуты требуемая средняя молекулярная масса (определенная в зависимости от вязкости раствора) и/или количество кислотных остатков (определенное титрованием концевых групп). Анализ полиэфира титрованием концевых групп проводили следующим образом. Образцы полиэфиров (300-500 мг) осторожно взвешивали и растворяли в минимальном количестве (10-30 мл) ацетона. После растворения растворы разбавляли до 100 мл бензиловым спиртом (Mallinnckrodt, Analytical Reagent) и титровали до слабо-розового цвета в конечной точке (фенолфталеин), используя гидроокись калия в растворе бензилового спирта (устанавливали нормальность против стандарта НС1). Для определения количества кислотных остатков объем основного раствора, использованного как образец (ΔVs), сравнивали с объемом основания, использованного в качестве контрольного растворителя (ΔVо).
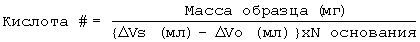
По окончании полимеризации сложный полиэфир выделяли и экстрагировали водой или разбавленным водным раствором гидроокиси натрия из соответствующего органического раствора, чтобы удалить растворимые в воде или солюбилизированные низкомолекулярные цепи.
Анализ полиэфира с помощью GPC (ГПХ) проводили следующим образом. Средние молекулярные массы (MW; М.м.) полиэфиров определяли посредством GPC, используя насос, подающий растворитель, модель 6000 Waters, и детектор, модель UV-D Dynamax (Rainin). Опыты проводили в тетрагидрофуране (Burdick & Jackson UV grade), используя колонку 50 см×10 мм, Jordi Gel DVB 1000 (Jordi Associates) при скорости 1,2 мл/мин при 25°С. Определение пика проводили при 220 нм и 1,0 AUFS. Колонку калибровали, используя ссылочные стандарты узких зон полистирола (Polysciences Inc.) с М.м.=4000; 9200 и 25000.
(Jordi Associates) при скорости 1,2 мл/мин при 25°С. Определение пика проводили при 220 нм и 1,0 AUFS. Колонку калибровали, используя ссылочные стандарты узких зон полистирола (Polysciences Inc.) с М.м.=4000; 9200 и 25000.
Модификация способа прямой конденсации влечет за собой применение органического азеотропно-дистилляционного вытеснителя и катионообменной смолы как катализатора конденсации (обозначен как полиэфир IB типа, см. таблицу I). В данном способе необходимы фильтрование и стадия выпаривания для удаления катализатора и азеотропно-дистилляционного вытеснителя соответственно. Типичные примеры сложных полиэфиров, полученных согласно описанным способам, и соответствующие результаты анализа представлены в таблице I.
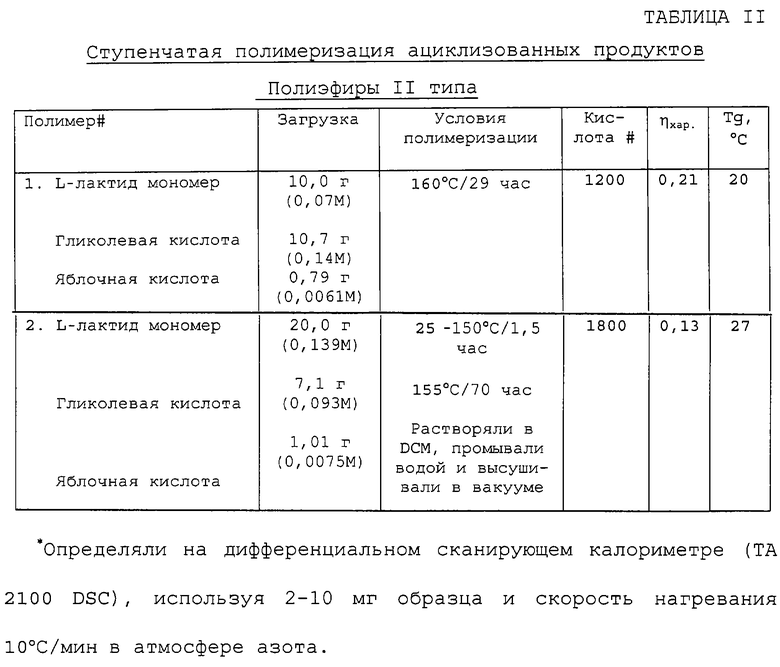
Ступенчатая полимеризация ациклизованных продуктов, при которой гидроксикислота взаимодействует с циклическими димерами и последующая конденсация полученной системы открытых цепей в присутствии предварительно установленного количества поликарбоновой кислоты и в присутствии или в отсутствие подходящего катализатора конденсации, например гликолевой кислоты, L-лактида и DL-яблочной кислоты, по существу были такими же, как способ конденсации, описанный выше, за исключением того, что при полимеризации использовали смесь монокарбоновой гидроксикислоты, циклического димера второй гидроксикислоты и поликарбоновую гидроксикислоту. Примеры сложных полиэфиров, полученных согласно данному способу, и подходящие результаты анализа суммированы в таблице II. Если циклический димер предварительно обрабатывали водой, то систему рассматривали как простую ступенчатую полимеризицию.
При полимеризации с раскрытием кольца лактона или смеси лактонов в присутствии предварительно определенной концентрации поликарбоновой гидроксикислоты в качестве инициатора цепи и каталитических количеств металлоорганического катализатора, например смеси L-лактида, гликолида и DL-яблочной кислоты в присутствии октоата олова, использовали сухие циклические мономеры или смеси циклических мономеров, поликарбоновую гидроксикислоту и следовое количество октоата олова (использовали в виде 0,33 М раствора в толуоле), которые в атмосфере без сухого кислорода перемещали в стеклянный реактор, оборудованный для перемешивания с помощью магнитной мешалки или механического перемешивания. Реакцию полимеризации продолжали в атмосфере азота, придерживаясь подходящей схемы нагревания, до тех пор пока не была достигнута требуемая молекулярная масса (которую измеряли в зависимости от вязкости раствора). По завершении схемы полимеризации температуру снижали и непрореагировавший мономер перегоняли при пониженном давлении. Затем полиэфирную массу охлаждали и удаляли растворимые в воде низкомолекулярные фракции с помощью низкотемпературной экстракции из подходящего органического раствора. Затем раствор высушивали и растворитель удаляли. Потом определяли молекулярную массу в зависимости от имеющейся вязкости и количество кислотных остатков определяли титрованием концевых групп. Примеры полиэфиров, полученных согласно данному способу, и соответствующие результаты анализа представлены в таблице III.

Функциональный обмен поликарбоновой или многоосновной органической гидроксикислоты с предварительно полученными высокомолекулярными сложными эфирами с соотношением СООН/ОН, действительно соответствующим нулю, предпочтительно в присутствии металлоорганического катализатора, например реакция расплавления сополимера 85/15 лактид/гликолид с молекулярной массой более 5000 и СООН/ОН≤1 с DL-яблочной кислотой в присутствии октоата олова, чтобы получить низкомолекулярные полиэфиры с СООН/ОН≥1, влечет за собой нагревание высокомолекулярного полиэфира с предварительно определенным количеством поликарбоновой кислоты или поликарбоновой гидроксикислоты в присутствии следового количества металлоорганического катализатора, такого как октоат олова. Реагенты нагревали выше 150°С в атмосфере сухого азота при интенсивном перемешивании до тех пор, пока не завершался функциональный обмен (что оценивали по истощению остаточной непрореагировавшей поликарбоновой кислоты). В действительности окончание реакции определяли, контролируя молекулярную массу (в зависимости от вязкости раствора, используя капиллярный вискозиметр при 28°С) полученного низкомолекулярного полиэфира и присутствие непрореагировавшей поликарбоновой кислоты. Контроль осуществляли посредством водной экстракции образца полиэфира и анализа экстракта, используя высокоэффективную жидкостную хроматографию (HPLC; ВЭЖХ). Уровни оставшегося мономера, димера и поликарбоновой кислоты определяли с помощью ВЭЖХ, используя насос подачи растворителя, модель 6000 Waters, и детектор модели UV-D Dynamax (Rainin) (205 нм, 1,0 AUFS). Опыты проводили, используя буфер с 0,0025 N Na2PO4, pH=3,5 (изократная скорость протекания=1,0 мл/мин), используя колонку 25 см×4,6 мм с Nucleosil C18,5 um.
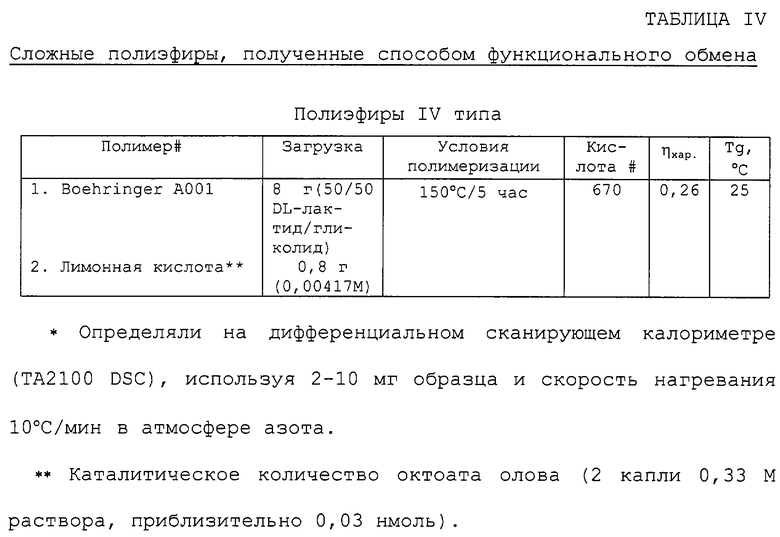
Требуемый сложный полиэфир выделяли и очищали, как описано выше для полимеризации с раскрытием кольца. Примеры полиэфиров, полученных согласно данному способу, и соответствующие результаты анализа представлены в таблице IV.
Другими мономерами, подходящими для синтеза сложных полиэфиров, примененными в изобретении, являются L-молочная кислота, DL-молочная кислота, ε-капролактон, п-диоксанон, ε-капроновая кислота, триметиленкарбонат, 1,5-диоксепан-2-он, 1,4-диоксепан-2-он, гликолид и мезолактид. Примеры использованных инициаторов цепи и/или модификаторов цепи включают в себя яблочную кислоту, лимонную кислоту и винную кислоту.
(2) Синтез ионного конъюгата сложный полиэфир/полипептид путем ионного взаимодействия сложных полиэфиров с концевыми остатками поликарбоновой кислоты и биологически активных полипептидов
Использовали вышеописанные биодеградируемые сложные полиэфиры с концевыми остатками поликарбоновой кислоты для получения ионных молекулярных конъюгатов с моно- или поликарбоксильными олигопептидами, полипептидами или белками с доступными эффективными ионогенными аминными группами (см. фиг.2). Кроме того, любой сложный полиэфир оказывался способным образовывать ионный молекулярный конъюгат с полипептидом при условии, что он был обработан основанием, например 0,1 N NaOH. Такая обработка раскрывала кислотные группы полиэфира для ионного взаимодействия с катионным полипептидом на многих участках.
Таким образом, образования таких конъюгатов достигали в результате прямого молекулярного взаимодействия компонентов в подходящем растворителе с или без предварительной обработки полиэфира неорганическим основанием, чтобы увеличить до предела его способность связывать основное лекарственное средство. Как отмечено выше, ионное взаимодействие компонентов ионного конъюгата увеличивается в пределах разницы в величинах их рКа.
Полиэфир растворяли в подходящем апротонном растворителе в диапазоне концентраций от 2 до 20% (мас./об.) Такие растворители должны растворять сложные полиэфиры, но также частично смешиваться с водой. Подходящие растворители, используемые с этой целью, включают в себя тетрагидрофуран, ацетон и диметиловый эфир этиленгликоля. К полученному раствору добавляли водный раствор основания, такого как гидроокись или карбонат натрия, калия или аммония, чтобы увеличить до предела связывающую способность полиэфира. Вообще, количество добавленного основания соответствовало количеству кислоты, представленной уровнем противоаниона основного пептида, который должен быть использован.
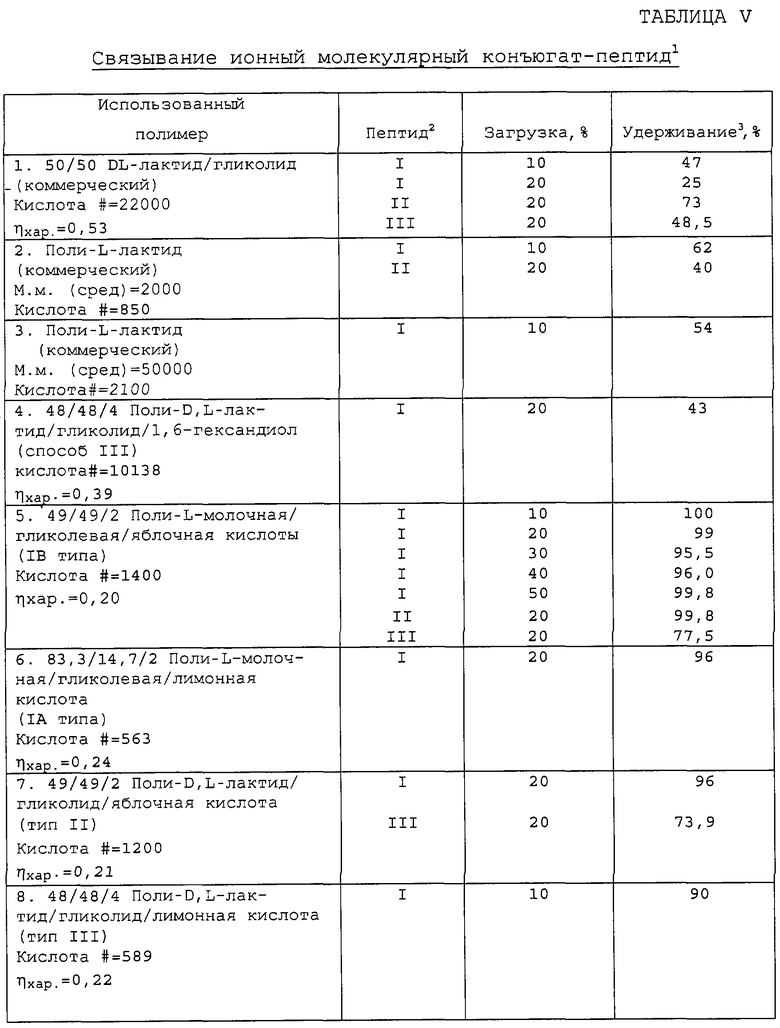
После краткого перемешивания комбинации полиэфир-основание добавляли водный раствор пептида или соли пептида при уровнях наполнения пептид/полиэфир 2 до 50 % (мас./мас.) (пептид/полиэфир). Полученную смесь перемешивали в течение периода времени (вплоть до 3 часов), а затем растворители удаляли и продукт высушивали в вакууме. Полученный материал потом можно далее переработать для дозированной композиции. Полагают, что полученные фармацевтические композиции должны быть химически стандартными композициями полностью из ионных молекулярных конъюгатов и по существу являться свободными от микроскопически или макроскопически распределенных доменов активного лекарственного средства в биодеградируемой матрице. Примеры полученных ионных молекулярных конъюгатов и соответствующие результаты анализа представлены в таблице V.

(3) Превращение ионных конъюгатов в имплантаты, палочки, микросферы или микрочастицы, способные высвобождать in vivo терапевтический агент, по крайней мере, в течение 20 дней с монофазным профилем
Соли ионных конъюгатов изобретения могут быть превращены в (А) стерильные инъецируемые микросферы (в присутствии от 0,1 до 10% твердого полигидридного спирта в качестве вспомогательного средства обработки или без него), содержащие от 1 до 50 мас.% полипептида, который может высвобождаться по существу согласно монофазному профилю и поддерживать фармакологическую активность в течение периода от одной до 12 недель; (В) стерильные имплантируемые пленки, полученные в результате отливки, прессования или экструзии с или без фармакологически инертного вспомогательного средства обработки и способные обеспечивать профиль высвобождения, подобный профилю, описанному в (А), и (С) стерильные инъецируемые палочки, полученные в результате экструзии или прессования, способные обеспечивать профиль высвобождения, подобный профилю, описанному в (А). К тому же палочки могут быть покрыты сложным полиэфиром, чтобы обеспечить дополнительный слой контроля за скоростью высвобождения терапевтического агента. Предпочтительно палочки покрывали абсорбируемым полиэфиром; более предпочтительно абсорбируемый полиэфир является таким, как определено в описании, и наиболее предпочтительно покрывающий абсорбируемый полиэфир является таким же, как полиэфир, заключенный в палочку.
Исследование высвобождения in vitro
Образцы материала высушенного и размолотого ионного конъюгата, весящие 50 мг каждый, помещали в сцинтилляционные флаконы диаметром 25 мм. Аликвоты по 5 мл модифицированного буфера PBS (буфер PBS: 2,87 г Na2HPO4, 0,654 г NаН2РO4, 5,9 г NaCl, 0,5 г NaN3, достаточное количество деионизованной воды до 1,0 литра; рН=7,27) добавляли в каждый флакон и флаконы помещали в вибратор Lab-Line Orbit Environ-Shaker и перемешивали вращением при 120 оборотах в минуту и 37°С. Флаконы периодически вынимали, сливали жидкость и доливали свежий раствор PBS. Количество высвобождаемого пептида определяли из декантированных растворов PBS посредством ВЭЖХ.
Экстракция пептидов из ионных конъюгатов
Образец 50 мг ионного молекулярного конъюгата смешивали в 20 мл хлористого метилена. Смесь последовательно экстрагировали порциями 50 мл, 20 мл и 20 мл 2 N уксусной кислоты. Экстракты уксусной кислоты объединяли и анализировали в отношении содержания пептидов высокоэффективной жидкостной хроматографией (HPLC; ВЭЖХ). Пептидный анализ ВЭЖХ проводили следующим образом. Анализ ВЭЖХ осуществляли, используя насос подачи растворителя, модель М-45 Waters, и детектор ЕМ Science MACS 700 при длине волны 220 нм и 1,0 AUFS. Пептиды исследовали, используя Lichrospher (ЕМ separations) C18, 100А, 5 мкм, колонку 25 см×4,6 мм и 30% ацетонитрил/0,1% TFA в качестве изократного элюирующего буфера.
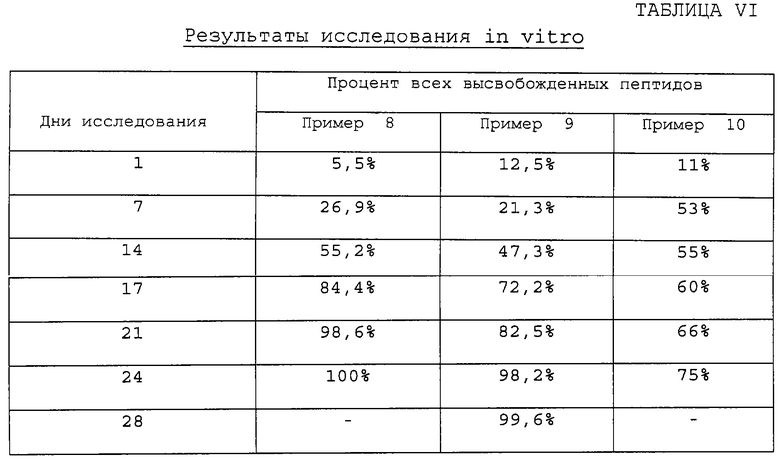
Далее представлены результаты (таблица VI) исследования in vitro, демонстрирующие количество пептида, высвобожденного в течение 28-дневного периода для ионных молекулярных конъюгатов 49:49:2 L-молочная/гликолевая/яблочная/D-Тrр6[LHRH] (пример 8), 49:49:2 L-молочная/гликолевая/яблочная/соматостатин - аналог, ингибирующий опухоль (пример 9), и 7,5:24,5:2 поли-L-лактид/гликолевая/яблочная:D-Тrр6[LHRH] (пример 10).
Количество пептидов в ионных конъюгатах
Ионно-связанные пептиды в продуктах конъюгата измеряли, растворяя 10 мг образца в 5,7 мл смеси (9:1) ацетона и 0,1 М водной трифторуксусной кислоты. Растворы перемешивали вращением приблизительно при 25°С приблизительно в течение 15-24 часов, а затем фильтровали через 5 мкм втулки тефлоновых фильтров. Затем фильтры анализировали относительно содержания пептида высокоэффективной жидкостной хроматографией (ВЭЖХ). Пептидный анализ посредством ВЭЖХ проводили, используя миллипоры, модель 777 Wisp Autosampler, насос, модель 510, и набор детекторов UV, модель 486, при 220 нм. Пептиды исследовали на Lichrospher (ЕМ Separations) колонке 25 см×4, 6 мм С18, 5 мкм 100 , скорость протекания 1,0 мл за минуту, используя 35% ацетонитрил в буфере 0,14% перхлората натрия в качестве изократной элюирующей системы. Пептиды оценивали количественно, сравнивая область точного пика в исследуемом образце с областью введенного пептидного стандарта.
, скорость протекания 1,0 мл за минуту, используя 35% ацетонитрил в буфере 0,14% перхлората натрия в качестве изократной элюирующей системы. Пептиды оценивали количественно, сравнивая область точного пика в исследуемом образце с областью введенного пептидного стандарта.
Применение
Любой из представленных в описании ионных конъюгатов кислотосодержащие полиэфиры/полипептид может быть введен реципиенту только один или в комбинации с фармацевтически приемлемой средой. Хотя конъюгат удобен для введения подкожно, внутримышечно, парентерально, посредством суппозиториев или назально, терапевтический препарат вводят в соответствии с состоянием, которое следует лечить. Концентрация композиции в препаратах изобретения изменяется в зависимости от ряда обстоятельств, включая дозу, которую следует вводить, и путь введения.
Полагают, что специалисты в данной области, используя предшествующее описание, без дальнейших разработок смогут применять данное изобретение в полном объеме. Поэтому следующие варианты осуществления должны быть истолкованы как иллюстрации, а не ограничение описания.
Пример 1. Способ прямой конденсации - синтез 50/50 поли(D,L-молочной-со-гликолевой), катализируемый амберлистом 15.
D,L-Молочную кислоту (85% водная смесь; 13,7 г, 0,13 моль) смешивали с гликолевой кислотой (10 г, 0,13 моль) в круглодонной колбе, снабженной магнитной мешалкой, ловушкой Дина-Старка (Dean-Stark) и конденсатором, охлаждаемым водой. Добавляли толуол (100 мл) и гранулы амберлиста 15 (100 мг) и смесь кипятили с обратным холодильником в атмосфере азота в течение 72 часов, удаляя воду из смеси. Смесь охлаждали, толуол декантировали из затвердевающей массы и продукт растворяли в хлористом метилене (250 мл). Раствор хлористого метилена обрабатывали активированным углем (Darco, 500 мг), фильтровали и высушивали в вакууме в роторном испарителе. Далее сложный полиэфир высушивали в высоком вакууме (1 мм рт.ст.) при 40°С для получения белого порошка, (ηхар в СНСl3=0,3, кислота #=2439, Тg=12°С).
Пример 2. Способ прямой конденсации - синтез 49/49/2 поли(L-молочная-со-гликолевая/лимонная), катализируемый амберлистом 15.
Используя систему, подобную вышеописанной, L-молочную кислоту (88% водная смесь; 25,6 г, 0,25 моль) смешивали с гликолевой кислотой (19,2 г, 0,25 моль), моногидратом лимонной кислоты (2,33 г, 0,011 моль), гранулами амберлиста 15 (500 мг) и толуолом (150 мл) в круглодонной колбе. Смесь нагревали при перемешивании до температуры кипения с обратным холодильником в течение 51 часа, удаляя воду с помощью ловушки Дина-Старка. Толуол декантировали из полутвердого продукта. Сложный полиэфир растворяли в ацетоне (300 мл) и фильтровали и высушивали на роторном испарителе. Твердый полиэфир затем повторно растворяли в хлористом метилене и промывали дважды водой (2×150 мл), чтобы удалить растворимые олигомеры. Органический раствор концентрировали на роторном испарителе, а продукт высушивали до конца в вакууме, чтобы получить белое твердое вещество (см. таблицу I, полиэфир тип IB, полимер #4) (ηхар в СНСl3=0,11, кислота #=842, Тg=15°С).
Пример 3. Способ ступенчатой полимеризации - синтез 73,5/24,5/2 поли(L-лактид-со-гликолевая/яблочная), катализируемый яблочной кислотой.
Используя цилиндрическую ампулу, емкостью 150 мл с фитингом воздушного импингера, L-лактид (20 г, 0,139 моль) смешивали с гликолевой кислотой (7,1 г, 0,093 моль) и (d,l)-яблочной кислотой (1,0 г, 0,0075 моль). Смесь перемешивали барботированием азота через входное отверстие импингера (100 мл/минуту) и нагревали от 25 до 155°С в течение 100 минут. Температуру реакции поддерживали при 155°С в течение 70 часов и воду из полимеризации удаляли в холодную ловушку на выпускном трубопроводе реактора. Через 70 часов реакционную смесь охлаждали до 100°С и вливали в охлажденный приемник из нержавеющей стали для затвердевания. Твердый полиэфир затем растворяли в хлористом метилене и дважды промывали водой (2×150 мл), чтобы удалить растворимые олигомеры. Органический раствор концентрировали на роторном испарителе, а продукт высушивали до конца в вакууме, чтобы получить белое твердое вещество (см. таблицу II, полиэфир II типа, полимер #2) (ηхар в СНСl3=0,13, кислота #=1800, Тg=27°С).
Пример 4. Способ полимеризации с раскрытием кольца - синтез 75/25 поли(L-лактид-со-гликолид), инициированный яблочной кислотой.
L-Лактид (12,0 г, 0,0833 моль), гликолид (3,21 г, 0,0277 моль), яблочную кислоту (0,3042 г, 0,00227 моль) и катализатор октоат олова (0,33 М в толуоле, 67 мкл, 0,022 ммоль) в атмосфере сухого азота вносили в стеклянную ампулу с магнитной мешалкой. Систему продували N2 и откачивали вакуумом несколько раз перед закрытием ампулы. Реактивы затем расплавляли при 140°С и расплав нагревали при 180, 190, 180 и 150°С в течение 1; 4,5; 12 и 2 часов соответственно. После охлаждения до комнатной температуры полиэфир повторно нагревали до 110°С в вакууме при давлении менее 1 мм рт.ст. приблизительно в течение одного часа для удаления мономера, повторно охлаждали при комнатной температуре, гасили в жидком азоте, выделяли и высушивали в вакууме (ηхар в СНСl3=0,20, кислота #=2560, Тg=39°С).
Пример 5. Способ полимеризации с раскрытием кольца - синтез 50/50 поли(D,L-лактид-со-гликолид), инициированный лимонной кислотой.
D,L-Лактид (10,0 г, 0,0694 моль) смешивали с гликолидом (8,06 г, 0,0694 моль), лимонной кислотой (1,07 г, 0,00555 моль) и катализатором-октоатом олова (0,33 М в толуоле, 84 мкл, 0,0278 ммоль) в атмосфере сухого азота в стеклянной ампуле, имеющей магнитную мешалку и закрытую под вакуумом. Реактивы расплавляли и нагревали при 180, 185, 195 и 120°С в течение 1; 2; 7 и 9 часов соответственно. Полиэфир охлаждали до комнатной температуры, гасили в жидком азоте, выделяли и высушивали (ηхар в СНСl3=0,26, кислота #=970, Тg=23°С).
Пример 6. Способ полимеризации с раскрытием кольца - синтез 50/50 поли(D,L-лактид-со-гликолид), инициированный 1,6-гександиолом.
Используя систему, подобную вышеописанной системе, D,L-лактид (10,0 г, 0,0694 моль), гликолид (8,06 г, 0,0694 моль), 1,6-гександиол (0,656 г, 0,00555 моль) и октоат олова (0,33 М в толуоле, 84 мкл, 0,0278 ммоль) в атмосфере сухого азота вносили в стеклянную ампулу, которую впоследствии закрывали в вакууме. Реакционную смесь нагревали при 150, 185, 150 и 120°С в течение 0,5; 4; 1; 5 и 3 часов соответственно. Полученный полиэфир извлекали и высушивали (см. таблицу III, полиэфир III типа, полимер #5) (ηхар в СНСl3=0,39, кислота #=10138, Тg=30°С).
Пример 7. Способ функционального обмена - синтез карбоксилсодержащего 50/50 поли(D,L-лактид-со-гликолид).
50/50 поли(D,L-лактид-со-гликолид) (Boehringer A001, 8 г), лимонную кислоту (0,8 г, 4,16 ммоль) и октоат олова (2 капли) добавляли в стеклянную ампулу в атмосфере сухого азота и закрывали. Смесь нагревали при 150°С в течение 4 часов, охлаждали до комнатной температуры, гасили в жидком азоте, выделяли и высушивали (см. таблицу IV, полиэфир IV типа, полимер #1) (ηхар в СНСl3=0,26, кислота #=670, Tg=23°C).
Пример 8. Синтез 49:49:2 L-молочная/гликолевая/яблочная (см. таблицу I, полимер#4) и D-Trp6[LHRH]ионного молекулярного конъюгата.
500 мг 49:49:2 L-молочная/гликолевая/яблочная кислота (синтезированные способом прямой конденсации; М.м.=9500; кислота #=1420) растворяли в 10 мл ацетона (Mallinckrodt Analytic Reagent). Добавляли порцию 0,1 N раствора гидроокиси натрия (1,14 мл) и смесь перемешивали при комнатной температуре в течение 15 минут. Раствор 100 мг D-Trp6[LHRH] (BIM-21003 пептид I; содержание основания 87%, содержание ацетона 7%) в 1,0 мл воды добавляли и смесь перемешивали в течение 1 часа при комнатной температуре. Затем растворители удаляли, сначала с помощью Ротовапа (Rotovap) при Т<40°С, а затем в эксикаторе в течение 1 часа при комнатной температуре в вакууме 1 мм рт.ст. Высушенное твердое вещество растирали и перемешивали в 100 мл деионизованной воды и выделяли фильтрованием. Водный фильтрат исследовали с помощью ВЭЖХ и обнаружили, что он содержит <1 мг растворимого пептида. Твердый материал высушивали несколько дней в вакууме, чтобы получить 540 мг белого порошка. Порошок использовали в исследованиях in vitro (см. таблицу VI, пример 8).
Пример 9. Синтез 49:49:2 L-молочная/гликолевая/яблочная (см. таблицу I, полимер 4) и ионного молекулярного конъюгата соматостатин/аналог, ингибирующий опухоль.
100 мг 49:49:2 L-молочная/гликолевая/яблочная (синтезированные способом прямой конденсации; М.м.=9500; кислота #=1420) растворяли в 2 мл ацетона (Mallinckrodt Analytic Reagent). Приливали порцию 0,1 N раствора гидроокиси натрия (0,32 мл) и смесь перемешивали при комнатной температуре в течение 15 минут. Добавляли раствор 20 мг соматостатин/аналог, ингибирующий опухоль (BIM-23014 пептид II; содержание основания 83%, содержание ацетона 9,8%), в 1,2 мл воды и смесь перемешивали в течение 1 часа при комнатной температуре. Затем удаляли растворители, сначала с помощью ротовапа при Т<40°С, а потом в эксикаторе в течение 1 часа при комнатной температуре в вакууме 1 мм рт.ст. Высушенное твердое вещество растирали и перемешивали в 20 мл деионизованной воды и выделяли фильтрованием. Водный фильтрат исследовали с помощью ВЭЖХ и обнаружили, что он содержит <0,05 мг растворимого пептида. Твердый материал высушивали несколько дней в вакууме, чтобы получить 106 мг белого порошка. Порошок размалывали и использовали при исследовании высвобождения in vitro (см. таблицу VI, пример 9).
Пример 10. Синтез 73,5:24,5:2 поли-L-лактид/гликолевая/яблочная (см. таблицу II, полимер #2) и ионного молекулярного конъюгата D-Trp6 [LHRH].
800 мг 73,5:24,5:2 поли-L-лактид/гликолевая/яблочная (синтезированных согласно способу полимеризации со ступенчатым ростом цепи ациклизованных продуктов; кислота #=1800) растворяли в ацетоне (16 мл). Приливали порцию 0,1 N раствора гидроокиси натрия (2,8 мл) и раствор перемешивали при комнатной температуре в течение 20 минут. Добавляли раствор 200 мг D-Trp6[LHRH] (BIM-21003; содержание основания 87%, содержание ацетона 7%) в 2 мл воды и смесь перемешивали в течение 90 минут. Растворители удаляли и полученное твердое вещество растирали в деионизованной воде, как в примере 8, установили, что присутствует менее 1% соли растворимого пептида. Выделенные твердые вещества высушивали 4 дня в вакууме, чтобы получить 839 мг белого порошка. Порошок размалывали и использовали для исследования высвобождения in vitro (см. таблицу VI, пример 10).
Пример 11. Образование микрочастиц 1,50 ионного конъюгата пептид-полимер из полиэфира L-лактид/гликолид/D,L-молочная кислота (65:33:2).
Конъюгаты синтезировали полимеризацией с раскрытием кольца, как описано в примере 4 (М.м.=4700, степень полидисперсности = 1,3, что определяли посредством GPC на 50×1 см колонке со смешанным линейным слоем Jordi Gel, элюент THF, детектор рассеяния света Wyatt Mini Dawn dn/dc=0,05, кислота #1475 - титрованием, Тg=42°С), и растворяли в 40 мл ацетона. Кислотные группы нейтрализовали 2,0 мл 0,5 М раствора гидроокиси натрия и перемешивали в течение 5 минут. Раствор 0,5 г BIM-23014 (содержание пептида 83,7%, содержание ацетата 11,5%) в 20 мл воды Milli-Q при перемешивании медленно добавляли к раствору полимера. Для предотвращения осаждения во время добавления пептида также добавляли порциями дополнительные 40 мл ацетона. Прозрачный бесцветный раствор перемешивали в течение одного часа, а затем упаривали в вакууме до сухого состояния. Полученное белое твердое вещество повторно растворяли в смеси 20 мл ацетона и 2 мл воды Milli-Q для образования прозрачного раствора. Полученный раствор вводили через 0,2 мкл тефлоновый фильтр в быстро перемешивающийся резервуар с 500 мл Milli-Q воды при 4°С. Фаза комплекса полимер/пептид немедленно разделялась на небольшие частицы при контакте с водой. После перемешивания суспензии в течение 30 минут при 4°С оставшийся ацетон удаляли при пониженном давлении и твердые вещества изолировали центрифугированием, повторно суспендировали с 100 мл Milli-Q воды и повторно центрифугировали. Выделенные твердые вещества высушивали лиофилизацией, чтобы получить 1530 мг белых свободно текучих порошков. Средний размер частиц 2-100 мкм. Показано, что Тg ионного конъюгата располагается при 53°С. По данным ВЭЖХ установлено, что общий остаточный (несвязанный) пептид во всех водных супернатантах составляет 63 мг. Как было определено посредством элементного азотного анализа, общее исходное содержание пептида составляет 19,9% массы. Используя способ экстракции ацетон/0,1 М TFA, обнаружили, что процент экстрагируемого из конъюгата пептида составляет 16,9 мас.%. Таким образом, полученный конъюгат сохраняет на 84,8% ионный (экстрагируемый) характер связи.
Система доставки в форме палочки 1 типа (CONC2 и CGC1)
Пример А-1. Получение сополимера 97/3 капролактон/гликолид (CGC1), инициированного лимонной кислотой.
Круглодонную колбу, снабженную механической мешалкой, дважды высушивали пламенем и продували сухим аргоном. Колбу загружали ε-капролактоном (1,455 моль, 166 г), гликолидом (0,08865 моль, 10,3 г), лимонной кислотой (0,075 моль, 14,4 г) и октоатом олова (0,0003 моль, 375 мкл 0,8 М раствора в толуоле). Полимеризацию проводили, используя следующую схему: в атмосфере аргона загруженный материал нагревали от комнатной температуры приблизительно до 150°С в течение 1 часа и приблизительно 20 минут после расплавления с непрерывным перемешиванием (70 оборотов в минуту). Загрузку поддерживали приблизительно при 150°С в течение 11,5 часов. После завершения полимеризации небольшое количество непрореагировавшего мономера перегоняли приблизительно при 120°С в течение 15 минут в вакууме (приблизительно 0,1 мм рт.ст.). Материал вливали в банку и оставляли для охлаждения.
Полимер анализировали посредством GPC (Мn=3543, М.м.=7708), FTIR, DSC (Tn=52°C) и титрованием карбоксильных групп (средняя эквивалентная масса =623 Да).
20 г полимера растворяли в 50,0 мл ацетона и раствор осаждали при перемешивании на бане со льдом. Твердый продукт изолировали фильтрованием.
Очищенный продукт анализировали с помощью GPC (Мn=4214, М.м.=9688), DSC (Тn=45,2°С) и титрованием (средняя эквивалентная масса =780).
Пример В-1. Получение ионного конъюгата (CONC1).
1,5 г очищенного полимера (CGC1) растворяли в 7,5 мл ацетонитрила в стеклянном флаконе. В отдельном флаконе 250,0 мг LHRH-ацетата растворяли в 1,5 мл дистиллированной воды. Растворенный полимер фильтровали через 0,45 мкм шприц-фильтр Acrodisc во флакон, содержащий углекислый натрий (чтобы нейтрализовать LHRH-ацетат). Раствор LHRH добавляли по каплям к раствору профильтрованного полимера. Объединенный раствор перемешивали с магнитным стержнем приблизительно в течение 1,5 часов при комнатной температуре. Конъюгат осаждали, добавляя его по каплям в перемешивающийся изопропиловый спирт (IPA), охлажденный жидким азотом. Осадок собирали центрифугированием и высушивали в течение ночи в вакууме. Выход конъюгата составил 73,5%. Конъюгат исследовали посредством DSC (Тn=50,9°С) и FTIR. Элементный анализ материала дал 1,81% азота. На основании этого установлено, что содержание LHRH составляет 10,0%.
Пример С-1. Получение системы доставки в форме палочки.
Ионный конъюгат (0,3987 г CONC2) и полимер (1,206 г CGC1) смешивали осторожным измельчением и расплавляли вместе приблизительно при 58°С в нагревающемся блоке. Расплавленный материал смешивали, а затем всасывали в капиллярные трубки 18G и оставляли для охлаждения. Материал выдавливали и разрезали на отрезки, которые содержали надлежащую дозу лекарственного средства и помещали в стерильные спиральные иглы 10-калибра (готовые для инъекции). Все стадии примера С-1 проводили в вытяжном шкафу с ламинарным потоком. Содержание LHRH в палочках составляло 2,5%.
Система доставки в форме палочки 2 типа (CONC2 и CGC1)
Пример А-2. Получение сополимера 97/3 капролактон/гликолид (CGC1), инициированного лимонной кислотой.
В данном примере использовали тот же самый полимер (CGC1), полученный в примере А-1.
Пример В-2. Получение ионных конъюгатов (CONC2).
CONC2 получали согласно способу, описанному в примере В-1. Элементный анализ показал, что процент азота составляет 2,31%. На основании этих данных содержание LHRH оказалось 12,76%.
Пример С-2. Получение системы доставки в форме палочки.
CONC2 (0,1854 г) и 0,5565 г очищенного CGC1 механически смешивали, а затем нагревали приблизительно до 60°С. Смешанный и расплавленный материал всасывали в капиллярные трубки калибра 18 и выдавливали поршнем. Палочки разрезали на отрезки, которые содержали надлежащую дозу лекарственного средства, и помещали в стерильные спиральные иглы калибра 18 (готовые для инъекции). Все стадии примера С-2 проводили в вытяжном шкафу с ламинарным потоком. Содержание LHRH в палочках составляло 3,2%.
Система доставки в форме палочки 3 типа
Пример А-3. Получение сополимера (СТТ1) 98/2 капролактон/триметиленкарбонат (ТМС), инициированного винной кислотой.
Круглодонную колбу, снабженную механической мешалкой, три раза высушивали пламенем и продували сухим аргоном. Колбу загружали ε-капролактоном (1,47 моль, 168 г), ТМС (0,03 моль, 3,06 г), винной кислотой (0,0142 моль, 2,134 г) и октоатом олова (0,0003 моль, 375 мкл 0,8 М раствора в толуоле). Полимеризацию проводили, используя следующую схему: при продувании аргоном загруженный материал нагревали от комнатной температуры приблизительно до 150°С приблизительно в течение 1 часа, времени перемешивания расплавленной реакционной смеси (60 оборотов в минуту). Температуру поддерживали приблизительно при 150°С в течение 9 часов. Непрореагировавший мономер перегоняли приблизительно при 100°С приблизительно в течение 1 часа при пониженном давлении (0,1 мм рт.ст.). Полимер вливали в банку и оставляли для охлаждения.
Полимер анализировали посредством GPC (Мn=13221, М.м.=35602).
Пример В-3. Получение ионных конъюгатов (CONCTT1).
1,5 г очищенного полимера из примера А-3 растворяли в 7,5 мл ацетонитрила в стеклянном флаконе. В отдельном флаконе 250 мг LHRH-ацетата растворяли в 1,5 мл дистиллированной воды. Растворенный полимер фильтровали через 0,45 мкм шприц-фильтр Acrodisc во флакон, содержащий 56,5 мг карбоната натрия (для нейтрализации LHRH-ацетата). Раствор LHRH добавляли по каплям в отфильтрованный раствор полимера. Объединенный раствор перемешивали магнитом приблизительно в течение 3 часов при комнатной температуре. Конъюгат осаждали, добавляя его по каплям в перемешивающийся IPA, охлажденный жидким азотом.
Осадок собирали центрифугированием и высушивали в течение ночи в вакууме.
Выход конъюгата составил 81,1%. Элементный анализ материала показал 2,04% азота. На основании этого установлено, что содержание LHRH составляет 11,3%.
Пример С-3. Получение системы доставки в форме палочек.
СТТ1 (0,8909 г) расплавляли приблизительно при 55°С. К полученному расплаву добавляли 0,2250 г CONCTT1 и всю систему нагревали приблизительно до 65°С. Расплавленную систему затем всасывали в капиллярные трубки калибра 18 и выдавливали поршнем. Палочки разрезали на отрезки, которые содержали надлежащую дозу лекарственного средства, и помещали в стерильные спиральные иглы калибра 18 (готовые для инъекции). Все стадии примера С-3 проводили в ламинарном проточном вытяжном шкафу. Содержание LHRH в палочках составляло 2,3%.
Система доставки в форме палочек 4 типа
Пример А-4. Получение сополимера 94/6 капролактон/гликолид (CGT6), инициированного винной кислотой.
Круглодонную колбу, снабженную механической мешалкой, три раза высушивали пламенем и продували сухим аргоном. Колбу загружали ε-капролактоном (1,41 моль, 161 г), гликолидом (0,09 моль, 10,4 г), винной кислотой (0,005 моль, 0,73 г) и октоатом олова (0,0003 моль, 375 мкл 0,8 М раствора в толуоле). Полимеризацию проводили, используя следующую схему: при продувании аргоном загрузочный материал нагревали от комнатной температуры приблизительно до 150°С приблизительно в течение 1 часа, времени перемешивания расплавленной реакционной смеси (60 оборотов в минуту). Температуру поддерживали приблизительно при 150°С в течение 1 часа. Затем температуру поднимали приблизительно до 180°С приблизительно в течение четырех часов. Материал охлаждали приблизительно до 107°С и оставляли в вакууме при 1,5 мм рт.ст. приблизительно в течение 1,5 часов. Материал вливали в банку и оставляли для охлаждения.
После сбора полимер анализировали DSC (Тn=54,5°С) и GPC (Мn=26254, М.м.=68101).
Пример В-4. Получение ионных конъюгатов (CONCTT2).
CONCTT2 получали, как описано в примере В-1, но с использованием LHRH-ацетата и сополимера из примера А4.
Пример С-4. Получение системы доставки в форме палочки.
CGT6 (1,4 г) и CONCTT2 (0,4779 г) нагревали приблизительно до 57°С, охлаждали, измельчали, затем повторно нагревали до той же самой температуры. Затем расплавленную систему всасывали в капиллярные трубки калибра 18 и выдавливали поршнем. Палочки разрезали на отрезки, которые содержали надлежащую дозу лекарственного средства, и помещали в стерильные спиральные иглы калибра 18 (готовы для инъекции). Все стадии примера С-4 проводили в ламинарном проточном вытяжном шкафу. Содержание LHRH составляло 2,8%.
Пример D-4. Покрытие палочки системы С-4 с использованием инертного предшественника сополимера.
CGT6 (1,4 г) растворяли в 1,5 мл дихлорметана. Палочки из примера С-4 погружали в полученный раствор полимера, немедленно удаляли и высушивали в ламинарном проточном вытяжном шкафу в условиях окружающей среды.
Из вышеприведенного описания специалисты в данной области легко смогут удостовериться в существенных особенностях данного изобретения и без отклонения от духа и компетенции изобретения смогут производить разные изменения и модификации, чтобы адаптировать его к различным употреблениям и условиям. Таким образом, иные варианты осуществления также находятся в пределах формулы изобретения.
Изобретение относится к сложному полиэфиру, содержащему одну или более свободных карбоксильных групп и характеризующемуся соотношением карбоксильной и гидроксильной групп более единицы. Данный сложный полиэфир представляет собой сополимер ε-капролактона и гликолида при их соотношении (90-99):(1-10) и получен в присутствии инициатора - лимонной кислоты. Также изобретение относится к варианту сложного полиэфира и к вариантам композиции, содержащей сложный полиэфир. Изобретение позволяет получить композиции, в которых химически соединен биосовместимый, биодеградируемый сложный полиэфир с олигопептидами, полипептидами, пептидами и/или белками в виде гомогенного ионного продукта. Кроме того, композиции изобретения легко оптимизируются для приобретения функциональных свойств для большой загрузки терапевтически активного полипептида. 6 н. и 18 з.п. ф-лы, 6 табл., 3 ил.
| WO 9415587 A2, 21.07.1994.WO 9739738 A2, 30.10.1997.SU 1685952 A1, 23.10.1991. |

