Изобретение относится к способу получения ксилита из материала, содержащего ксилозу и ксилоновую кислоту. Ксилоза и ксилоновая кислота содержатся, например, в варочной жидкости сульфитной варки.
Сульфитный варочный раствор или варочную жидкость можно получать при обработке древесной массы сульфитным способом. Сульфитный варочный раствор содержит нерастворимое вещество древесины, лигнин, сахара, гексозу и пентозу, органические кислоты и варочные химические реагенты. Эти варочные растворы ранее сбрасывали в системы водоснабжения. Однако недавно это было запрещено на основании свода законов об охране окружающей среды, и поэтому началась разработка способов утилизации сульфитного варочного раствора. После варки сульфитный варочный раствор можно упаривать и сжигать, получая тепловую энергию, а серу и щелочь из раствора можно возвращать в варочный процесс. Однако эта технология создает новые опасности для окружающей среды из-за высокого содержания серы в сульфитном варочном растворе. Для эффективного использования необходимо, чтобы сульфитный варочный раствор фракционировали на составляющие компоненты.
Кроме ксилозы сульфитный варочный раствор также содержит ксилоновую кислоту, которая затрудняет выделение ксилозы из этого раствора. Однако было бы предпочтительным использовать ксилоновую кислоту в качестве сырьевого материала при получении ксилита.
Ксилит представляет собой природный сахарный спирт, получаемый восстановлением ксилозы, его сладкий вкус соответствует вкусу обычного сахара, но калорийность (2.4 ккал/кг) ниже, чем у сахара. Ксилит обнаружен в небольших количествах в различных фруктах и овощах, и он также образуется в теле человека как нормальный продукт метаболизма. Благодаря определенным метаболическим, стоматологическим и техническим свойствам ксилит является очень хорошим специальным подсластителем для различных целей, таких как жевательная резинка, конфеты, кондитерские изделия и т.д. Можно отметить в качестве примера, что метаболизм ксилита не зависит от инсулинового метаболизма, и, таким образом, диабетики также могут использовать ксилит. Ксилит также замедляет сокращение кишечника и может, следовательно, использоваться при диетическом питании. Было также установлено, что ксилит не вызывает кариес и даже обладает антикариесным действием.
Несмотря на многочисленные преимущества ксилита его применение достаточно ограничено. Это связано с относительно высокой стоимостью ксилита, которая в свою очередь определяется трудностями производства ксилита в промышленном масштабе.
Ранее ксилит получали гидролизом ксилансодержащих материалов. Он приводит, например, к смеси моносахаридов, содержащих ксилозу. Ксилозу затем восстанавливают до ксилита путем каталитического восстановления (гидрогенизация) обычно в присутствии никелевого катализатора, такого как никель Ренея. Техническая литература описывает несколько способов получения ксилозы и/или ксилита из ксилансодержащих материалов. Примеры включают патент США 3784408 (Jaffe et al.), патент США 4066711 (Melaja et al.), патент США 4075406 (Melaja et al.), патент США 4008285 (Melaja et al.) и патент США 3586537 (Steiner et al.).
На нескольких производствах большая часть гемицеллюлозы представляет собой ксилан, который может быть гидролизован до ксилозы. Сырьевым исходным материалом для получения ксилана является гемицеллюлоза лиственных деревьев, которая главным образом состоит из ксилана. Использование ксилана и ксилозы, получаемых в качестве побочных продуктов целлюлозно-бумажной промышленности, с недавних пор становится предметом все большего внимания. Ксилоза образуется, например, при кислотной сульфитной варке, где обычные основания включают Mg2+, Ca2+, NH4 + и Na+. Исходным материалом может также быть варочный раствор нейтральной сульфитной варки после гидролиза ксило-олигомеров ксилана. В варочных растворах кислотной сульфитной варки гемицеллюлозы уже находятся в моносахаридной форме. Термин "варочный раствор" относится в данной связи к раствору, используемому при варке или полученному после варки, или к его части. Известные каталитические способы восстановления ксилозы, используемые при получении ксилита, обычно требуют, чтобы восстанавливаемая ксилоза не содержала вредных примесей. Очистка является очень требовательной и нуждается в многостадийном способе (смотри патент США 4631129, Heikkila и PCT/F195/00224, Heikkila et al.), так как, например, катализаторы, используемые при восстановлении ксилозы, очень чувствительны к примесям (смотри Harkonen М. and Nuojua P., Kemia-Kemi, No 3 (1980) pp. 98-100).
Когда сульфитный варочный раствор используют в качестве сырья для ксилозы, то проблемой является изменение условий варки. В зависимости от условий гемицеллюлоза растворяется в различной степени и дает большее или меньшее количество ксилозы. В условиях варки, где образуется только небольшое количество ксилозы, могут также образовываться значительные количества ксилоновой кислоты. Если ксилоза должна быть чистой, то трудно выделять ксилозу, находящуюся в таком продукте, из раствора, содержащего ксилоновую кислоту, например, посредством хроматографии. Ксилоновая кислота, присутствующая в растворе, затрудняет отделение ксилозы и, следовательно, снижает выход ксилозы при кристаллизации. Однако, было бы предпочтительным иметь возможность использовать ксилоновую кислоту в качестве сырья для ксилита (смотри WO 93/19030, Vuorinen). Выделение ксилозы из сульфитного варочного раствора известно, например, из WO 94/26380, Heikkila et al., и WO 95/29002, Heikkila et al.
Настоящее изобретение относится к способу получения ксилита из материала, содержащего ксилозу и ксилоновую кислоту. Способ отличается тем, что ксилоновую кислоту отделяют из материала, содержащего ксилозу и ксилоновую кислоту, после чего отделенную ксилоновую кислоту восстанавливают. В предпочтительном выполнении изобретения также отделяется ксилоза. Ксилозу и ксилоновую кислоту предпочтительно отделяют хроматографией, экстракцией, ионообменом или кристаллизацией.
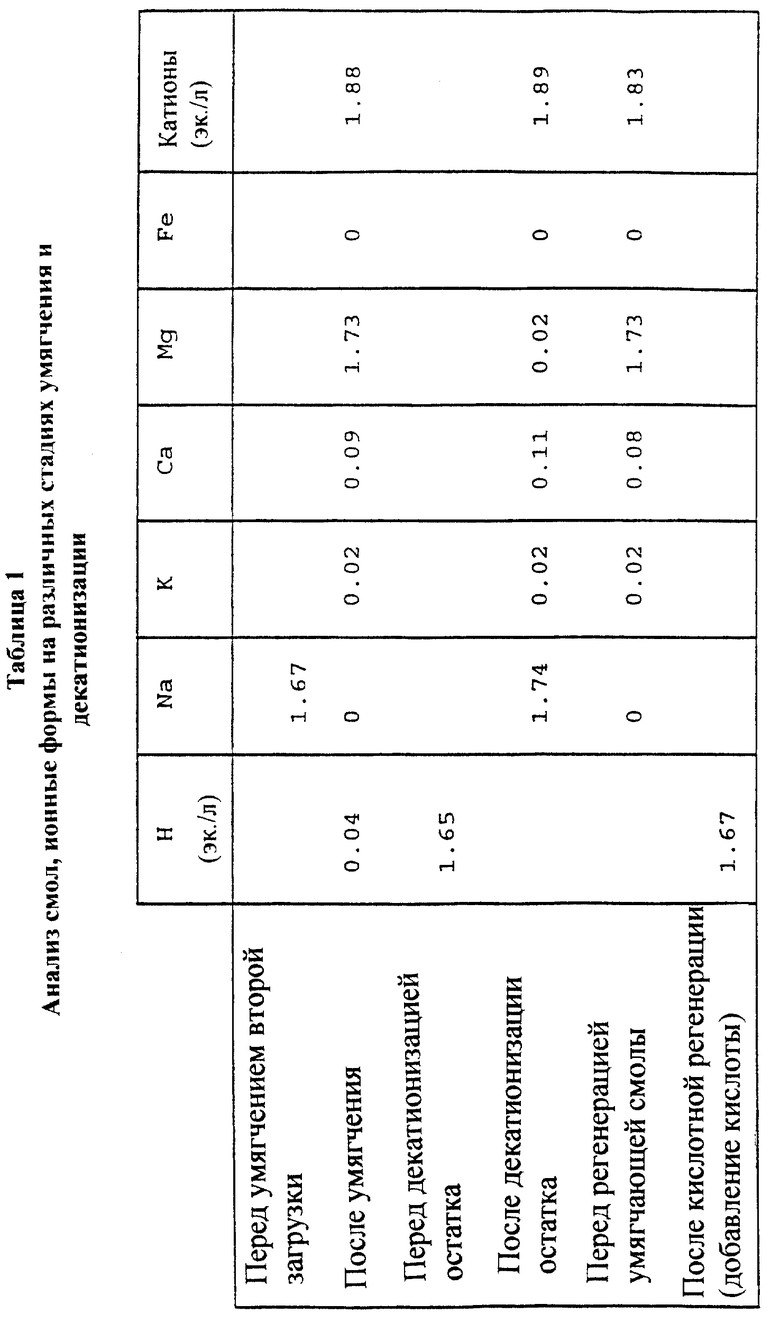
Материал, содержащий ксилозу и ксилоновую кислоту, является предпочтительно варочным раствором сульфитной варки, таким как варочный раствор магниевой сульфитной варки. Получение ксилозы хроматографическим путем из магниевого варочного раствора ограничено из-за того, что вряд ли любые другие катионы, за исключением магния, могут быть возвращены в химическую циркуляцию целлюлозного производства. Хроматографические смолы, используемые в способе, должны предпочтительно находиться в магниевой ионной форме (Mg-ионное разделение), и в этом случае анионы, возвращаемые из процессов разделения, представляют собой магниевые соли. Однако разделительные (хроматографические) смолы, используемые в двухвалентной форме, отделяют ксилозу менее эффективно, чем разделительные смолы в одновалентной форме, смотри, например, патент Финляндии 78734 (Heikkila). При применении изобретения можно использовать вместо двух успешных магниево-ионных разделений одно магниево-ионное разделение и одно натриево-ионное разделение, между которыми осуществляют умягчающий процесс, в котором магниевые ионы меняют на ионы натрия. Доминирующим катионом во фракции первого разделения является магний и он может быть возвращен как таковой на циркуляцию химических реагентов целлюлозного производства. Остаточная фракция второго разделения не может быть возвращена в циркуляцию целлюлозного производства (на выделение химических реагентов, получение энергии), так как она содержит посторонние катионы (натрий). Согласно изобретению всю остаточную фракцию второго разделения пропускают через катионобменник в водородной форме. Ионная сила фракции, которую получают при втором разделении и которую подвергают катионному обмену, соответствует относительно сильной кислоте (низкий pH) после ионного обмена и, следовательно, она может быть использована для регенерации водородной формы катионообменной смолы, используемой для умягчения загрузочного сырья на вторую стадию разделения. Одновременно ионы магния, оставшиеся в катионообменной смоле во время умягчения, могут быть возвращены в циркуляцию целлюлозного производства.
Смола, используемая для катионного обмена остаточной фракции, находится главным образом в натриевой форме после обмена ионов. Эта смола регенерируется кислотой обратно в водородную форму, и натрий, получаемый с колонки в начале регенерации, возвращают в умягчающую колонну для приведения ее в соответствующую (натриевую) форму (из водородной формы, в которую она была предварительно превращена). Кислота, которая выделяется в начале из водородной формы умягчающей колонны (натрий меняют на водород), может быть, в свою очередь, использована для регенерации водородной формы смолы, используемой для катионного обмена остаточной фракции.
С другой стороны, было установлено, что двухстадийное Mg-Mg-разделение также может быть использовано выгодным образом для отделения ксилозы и ксилоновой кислоты согласно изобретению. В хроматографическом Mg2+ втором отделении ксилозы из раствора с неотрегулированным значением pH (pH 1,5 до 2,5) ксилоновая кислота элюируется почти одновременно с ксилозой, что, в свою очередь, снижает чистоту ксилозной фракции. Когда pH загрузочного раствора увеличивают с помощью MgO до значения pH около 3 или более, ксилоновая кислота элюируется позднее, что увеличивает чистоту ксилозной фракции и широту ксилозной фракции. pH также может быть отрегулирован на первой стадии разделения, но предпочтительным является регулирование только на второй стадии разделения, учитывая расход химических реагентов.
Вышесказанное касательно Mg-разделения также может использоваться при обработке Ca-сульфитного варочного раствора.
Согласно изобретению разделение осуществляют предпочтительно следующим образом:
а) варочную жидкость магниевой сульфитной варки подвергают хроматографическому разделению путем использования магниевой разделительной смолы с получением фракции, обогащенной ксилозой, смешанной фракции и остаточной фракции в магниевой форме, и
b) полученную фракцию, обогащенную ксилозой, хроматографируют с использованием разделительной смолы, находящейся в магниевой форме или в форме одновалентного металла, и выделяют фракцию, концентрированную в отношении ксилозы, и выделяют фракцию, концентрированную относительно ксилоновой кислоты, находящейся в виде соли.
Когда разделительную смолу, которая находится в форме одновалентного металла, предпочтительно натрия, используют на стадии b), то полученную на стадии а) фракцию, содержащую ксилозу, умягчают перед стадией b). Если смола на стадии b) находится в магниевой форме, то pH фракции, полученной на стадии а), доводят предпочтительно до значения от 3.0 до 6.5, особенно 5.5.
Разделение также может быть осуществлено посредством экстракции, предпочтительно этанолом, или путем ионного обмена или кристаллизации Mg-ксилоната.
Оказалось, что отделенная ксилоза и ксилоновая кислота могут быть восстановлены каталитически или путем использования реагентов гидридов металлов, таких как боргидрид натрия. Восстановление осуществляют подходящим образом каталитически, в этом случае предпочтительные катализаторы включают катализаторы типа никеля Ренея и катализаторы из благородных металлов, таких как Ru, Pd, Rh и Pt, особенно Ru.
Подходящая температура восстановления при каталитическом восстановлении составляет от 70 до 150oC, предпочтительно от 100 до 130oC, и восстановление осуществляют подходящим образом при давлении от 5000 до 20000 кПа, предпочтительно от 10000 до 13000 кПа. pH восстанавливаемой ксилозы предпочтительно составляет между 5 и 7, а pH ксилоновой кислоты предпочтительно составляет между 1.5 и 2.5.
Фракции ксилозы и ксилоновой кислоты, которые были выделены перед восстановлением, должны быть по возможности очищены, например, ионным обменом. Фракции ксилозы и ксилоновой кислоты могут потребовать дополнительной очистки посредством нейтрализации/осаждения/фильтрации и/или обработки углем или адсорбентом. Также можно использовать двухстадийную гидрогенизацию, что означает, что на первой стадии гидрогенизацию осуществляют, например, с катализатором из никеля Ренея, а на второй стадии, например, с катализатором из благородного металла. Вариант выполнения изобретения предусматривает предварительное окисление, и в этом случае отделение концентрированной ксилоновой кислоты из раствора становится более эффективным. Ксилит, получаемый по изобретению, предпочтительно выделяют из полученного при восстановлении раствора хроматографическим путем или предпочтительно кристаллизацией.
Способ по изобретению позволяет значительно снизить стоимость производства ксилита. Способ обеспечивает, например, лучшее использование сырьевого материала (для получения ксилита достаточно примерно двойного количества сырьевого материала).
Следующие примеры иллюстрируют изобретение.
Пример 1
Mg-Na-разделение
Образец, полученный из варочной жидкости магниевой сульфитной варки, хроматографировали известным способом (WO 94/26380) посредством использования сильнокислой катионообменной смолы (полистирольная основа, активированная сульфонированием) в магниевой форме, получая фракцию, обогащенную ксилозой, смешанную фракцию и содержащую магний остаточную фракцию, которую возвращали на приготовление варочной кислоты для магниевой сульфитной варки. Фракцию, обогащенную ксилозой, умягчали или декатионизировали путем использования сильной катионной смолы (DOW 88), которая находилась в H+-форме и была регенерирована серной кислотой. Температура составляла 50oC, и скорость потока была 2 массовых объема в час. Рефрактометрическое содержание сухого твердого вещества (RDS) в загрузке составляло 20% по весу, а длительность цикла (по расходу) составляла 0,6 кг сухого вещества/0.5 л смолы.
pH полученного умягченного (декатионизированного) сиропа доводили NaOH до 5.5, и содержащиеся там катионы были, главным образом, катионами натрия, и вряд ли сироп содержал какие-либо двухвалентные катионы. Сироп хроматографировали с использованием сильнокислой катионообменной смолы (Finez VO 9 СTM) в натриевой форме, получая фракцию, сконцентрированную относительно ксилозы, которую выделяли, и остаточную фракцию в солевой форме (солевая фракция), содержавшую большое количество натрия. Количество ксилозы в загружаемой фракции составляло 37.2% (по сухому веществу). Количество ксилозы в конечной фракции, полученной после выделения, составляло 55% (по сухому веществу), а в остаточной фракции содержалось 2% (по сухому веществу).
Солевую фракцию декатионизировали посредством использования сильнокислой катионообменной смолы в H+-форме (DOW 88TM). Скорость потока составляла 2 массовых объема в час, температура составляла 50oC и содержание сухого вещества (RDS) в загрузке составляло 5.6% по весу. Общая длительность цикла (по расходу) составляла 7.2 л/1 л смолы. Уровень pH декатионизированной солевой фракции был около 1, и она содержала 0.252 H+ эквивалента/л. Вся декатионизированная солевая фракция (7.2 л) была использована для регенерации умягчающей смолы (500 мл), содержащей, главным образом, ионы магния. Регенерацию осуществляли таким образом, что кислая фракция, получаемая в начале цикла, и дополнительная кислота использовались для перевода умягчающей смолы из магниевой формы в H+-форму. Na-содержащая фракция декатионизированной смолы, полученная в начале регенерационного цикла, использовалась для перевода умягчающей смолы в Na-форму. Емкости и содержание катионов в смолах на различных стадиях умягчения и декатионизации показаны в табл. 1.
Пример 2
Mg-Mg-разделение
Второе Mg-разделение варочного раствора (стадия b) осуществляли на хроматографической колонке с Mg2+ разделительной смолой при следующих условиях:
Разделительная смола: Finex V 11 СTM, DVB* 6.5%
Температура: 65oC
Скорость потока: 0.9 м/ч
*Сульфонированная смола с полистирольной основой, сшитая дивинилбензолом.
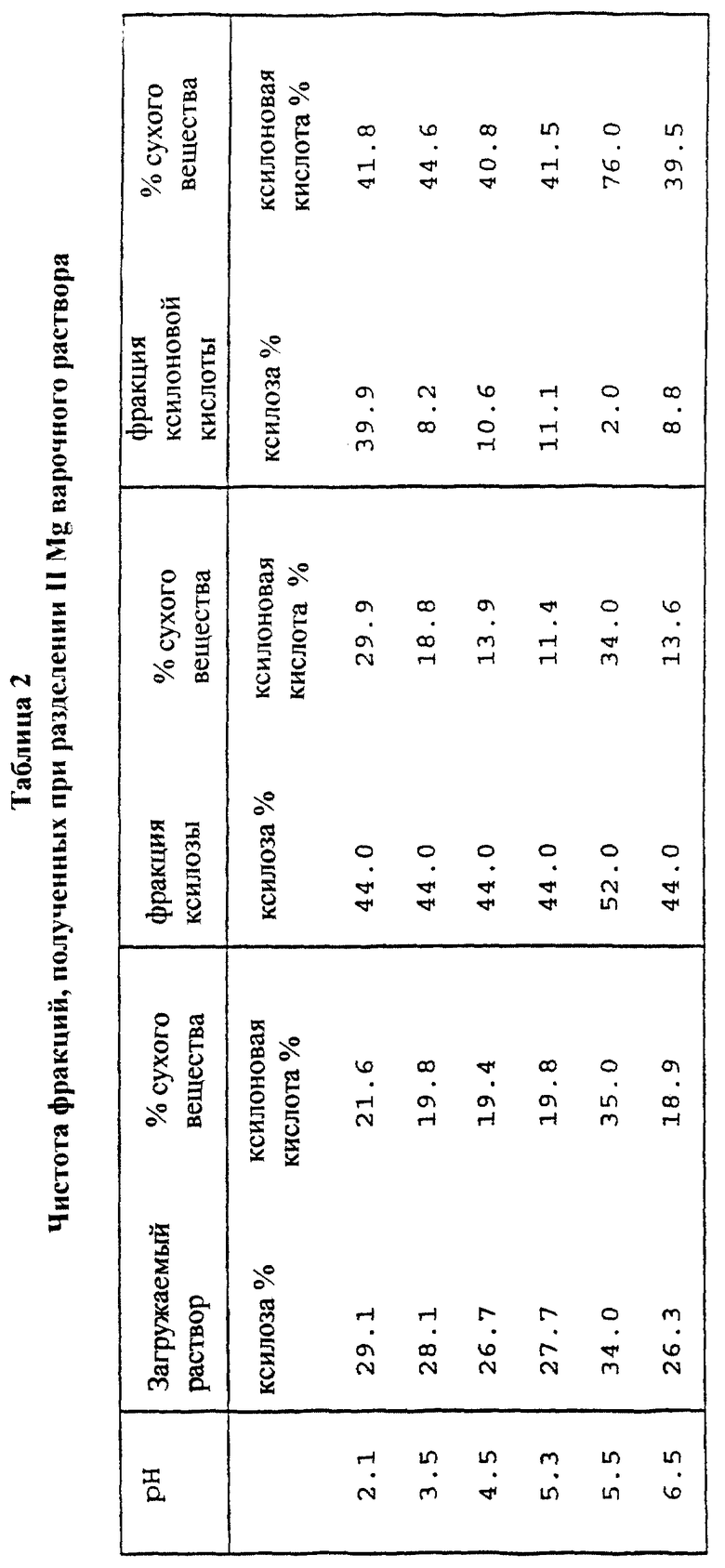
Чистота ксилозы в загрузке составляла 29.1% от сухого вещества, а чистота ксилоновой кислоты составляла 21.6% от сухого вещества. Выделяли три фракции. Чистота ксилозы в ксилозной фракции составляла примерно 44% от сухого вещества и содержание ксилоновой кислоты во фракции ксилоновой кислоты составляло примерно 41% от сухого вещества. Порядок элюирования выделенных фракций: соль, ксилоза, ксилоновая кислота.
При разделении раствора с неотрегулированным значением pH (pH 2.1) производительность ксилозной фракции составляла 5.2 кг сухого вещества/ч/м3. Когда же pH варочного раствора доводили перед разделением с помощью MgO до 3.5, 4.5, 5.3 и 6.5, то производительность ксилозы составляла 11.8, 10.7, 11.6 и 10.7 кг сухого вещества/ч/м3 соответственно. При повышении pH было получено лучшее разделение благодаря сильному удерживанию смолой ксилоновой кислоты.
Пример 3
Mg-Mg-разделение
Второе разделение Mg-варочного раствора по примеру 2 проводили на хроматографической колонке с разделительной смолой в Mg2+-форме при условиях, описанных в примере 2. Фракции ксилозы и ксилоновой кислоты имели чистоту фракций, представленную в табл. 2.
Пример 4
Отделение ксилозы и ксилоновой кислоты из Ca-сульфитного варочного раствора путем экстракции этанолом
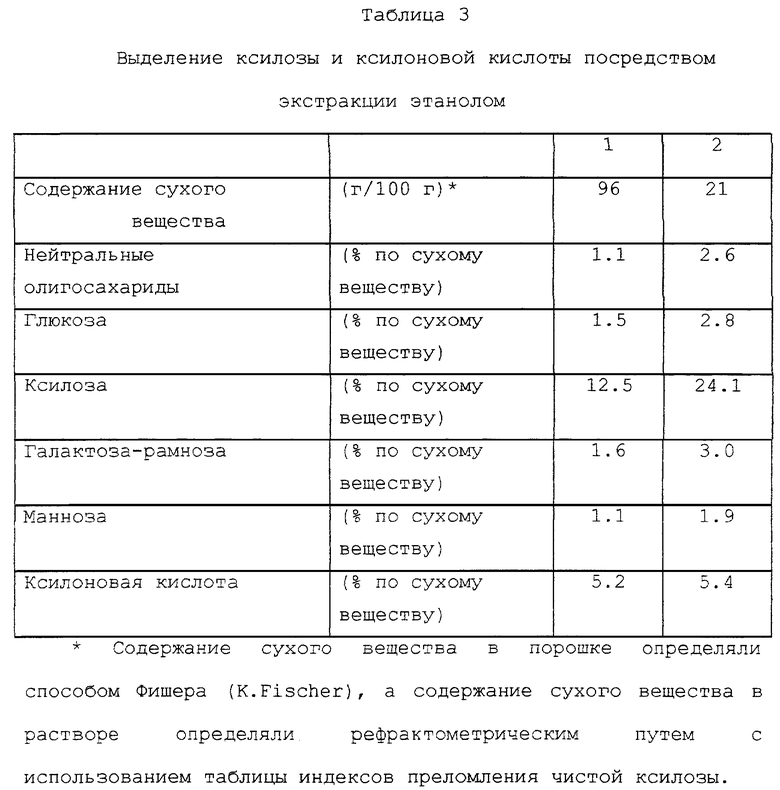
Коммерчески доступный сухой и порошкообразный продукт Ca-сульфитной варки лиственной древесины, состав которого показан в табл. 3(1), экстрагировали этанолом. Количество порошка, взятого для экстракции, составляло 1500 г, а количество 95%-ного этанола составляло 15 л. Смесь перемешивали при 50oC в течение 4 часов, после чего фильтровали и полученный осадок на фильтре высушивали. Количество растворившихся твердых веществ составляло 32%. Фильтрат выпаривали при пониженном давлении на роторном испарителе. Упаренный остаток растворяли примерно в 8 литрах воды. Состав раствора показан в табл. 3(2). Выход ксилозы составлял примерно 78%, а выход ксилоновой кислоты был примерно 43%. Выходы увеличивались до 95% и 56% соответственно при повторении экстракции этанолом.
Пример 5
Окисление ксилозы в ксилоновую кислоту
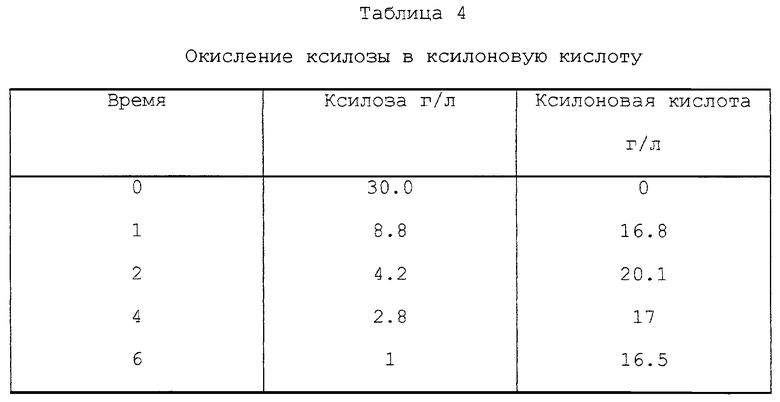
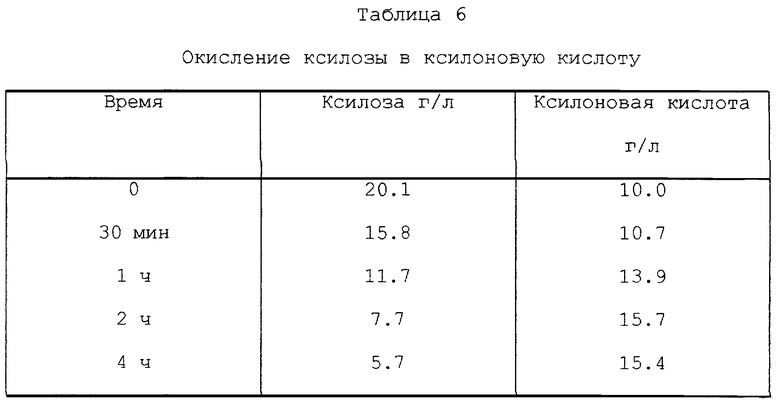
3.6 г MgO, 6 г ксилозы и 200 г раствора SO2 (концентрация от 70 до 71 г SO2/л) добавляли в автоклавы. Автоклавы закрывали и помещали в глицериновую баню при 150oC. Автоклавы выдерживали в бане в течение 1, 2, 4 и 6 ч, после чего их охлаждали. Раствор фильтровали и анализировали. За два часа 67% ксилозы окислилось до ксилоновой кислоты. Результаты представлены в табл. 4.
Пример 6
Кристаллизация Mg-ксилоната из воды
Исходный раствор представлял собой фракцию ксилоновой кислоты, выделенную из Mg-сульфитного варочного раствора. Фракцию ксилоновой кислоты кристаллизовали при 10oC и маточный раствор отделяли декантацией. Кристаллическую массу Mg-ксилоната (сухое вещество 56.2%, чистота 58.5% из расчета по сухому веществу ксилоновой кислоты) кристаллизовали из раствора этанол-вода в соотношении 120 г кристаллической массы/1 л этанола. Кристаллы сушили при комнатной температуре. Чистота кристаллов Mg-ксилоната составляла 80.9% из расчета на сухое вещество ксилоновой кислоты. Кристаллы растворяли в воде до концентрации раствора примерно 50%. Окраску раствора удаляли посредством использования угля и ускорителя фильтрования.
Раствор ксилоната магния (2040 г; содержание сухого вещества 45.8% (Karl-Fischer); чистота ксилоновой кислоты 79.3% по сухому веществу) переносили в 2-литровый сосуд (загружаемый раствор), из которого Mg-ксилонат кристаллизовали следующим образом.
В массу (Тмас 57oC) вводили затравку в виде 1 мл Mg-ксилонатного раствора. Через час после введения затравки включали линейную программу охлаждения в течение 70 часов (от 60 до 20oC). После завершения охлаждающей программы Mg-ксилонат выдерживали при постоянной температуре (20oC) в течение 72 часов. Кристаллы отфильтровывали и промывали этанолом. Кристаллы сушили при 45oC. Выход составлял 337 г.
Чистота кристаллов ксилоновой кислоты составляла 94.3% по сухому веществу.
Пример 7
Кристаллизация Mg-ксилоната из воды
Исходный раствор представлял собой фракцию ксилоновой кислоты, выделенную из Mg-сульфитного варочного раствора. Фракцию ксилоновой кислоты кристаллизовали при 10oC и маточный раствор отделяли декантацией. Кристаллическую массу Mg-ксилоната кристаллизовали из водно-этанольного раствора в соотношении от 120 до 150 г кристаллической массы/1 л этанола. Кристаллы сушили при комнатной температуре. Чистота кристаллов Mg-ксилоната составляла от 80 до 94% по сухому веществу ксилоновой кислоты. Кристаллы растворяли в воде в примерно 35%-ном растворе, содержащем ксилоновую кислоту чистотой 71%.
18.4 кг раствора ксилоната магния (RDS 34.3%) использовали в качестве загружаемого раствора. Воду упаривали из раствора (Тмас. от 40 до 55oC). Когда RDS массы достигал 52.8%, массу переносили в 6-литровый охлаждаемый кристаллизатор (7.1 кг). Через 30 минут в массу вносили затравку 0.7 г кристаллов Mg-ксилоната при 63oC. Через час после внесения затравки включали линейную 70-часовую программу охлаждения (от 60 до 20oC). Массу, содержащую ксилоновую кислоту чистотой 72.8% по сухому веществу и содержание сухих веществ 47.9% (Karl-Fisher), центрифугировали при 4500 оборотах в минуту в течение 5 минут и сушили при 45oC. Выход составлял 1285 г кристаллов. Чистота ксилоновой кислоты кристаллов Mg-ксилоната составляла 90.2% по сухому веществу.
Пример 8
Кристаллизация Mg-ксилоната из воды
Исходный раствор представлял собой фракцию ксилоновой кислоты, выделенную из Mg-сульфитного варочного раствора.
20.6 кг раствора ксилоната магния (RDS 40.8%) использовали в качестве загружаемого раствора. Воду упаривали из раствора (Тмас. от 40 до 55oC). Когда RDS массы достигал 59.5%, начиналась самопроизвольная кристаллизация. Массу переносили в 10-литровый кристаллизатор. Через 15 минут включали линейную 18-часовую программу охлаждения (от 60 до 20oC). Массу, имеющая ксилоновую кислоту чистотой 74.2% по сухому веществу и содержание сухих веществ 51.5% (Karl-Fisher), центрифугировали при 4500 оборотах в минуту в течение 5 минут, промывали и сушили при 45oC. Выход составил 3.4 кг кристаллов. Чистота ксилоновой кислоты кристаллов Mg-ксилоната составила 82.5% по сухому веществу.
Пример 9
Адсорбция ксилоновой кислоты слабоосновной анионообменной смолой.
Загружаемый раствор представлял собой ксилозную фракцию первого разделения по примеру 1 из хроматографического разделения Mg-сульфитного варочного раствора. Для фракции обеспечивали серию ионообменников, включая сильнокислую катионообменную смолу (DOW 88TM) и две слабоосновных анионообменных смолы (DOW 66TM). Катионы связывались с катионообменной смолой и ксилоновая кислота высвобождалась из своей соли, то есть превращалась в свободную кислоту, после чего анионы сцеплялись с анионообменной смолой.
Содержание сухого вещества в загрузочном растворе составляло 32%, температура составляла 40oC и скорость потока была 2 мo/ч/колонка (мo - массовый объем). В этом эксперименте раствор обрабатывали в количестве, приблизительно соответствующем общему объему смолы.
При таком прохождении ксилоновая кислота сцеплялась с обоими анионообменными смолами: 22 г/л смолы для первой смолы и 63 г/л смолы для второй. Анализ загрузки и продукта показаны в табл. 5.
Пример 10
Отделение ксилозы посредством использования слабокислой катионообменной смолы
Хроматографическое разделение Mg-сульфитного варочного раствора осуществляли с использованием слабокислой катионообменной смолы, Finex CA 24 GCTM. Температура составляла 65oC и скорость потока была 0.19 м/ч. Уровень pH загрузочного раствора составлял 1.2, а содержание ксилозы - 9.8%. Производительность ксилозной фракции при чистоте фракции 25% составляла 9.6 кг сухого вещества/м3/ч, и максимальная чистота ксилозы при выделении составляла 31.4%. Содержание ксилоновой кислоты в загрузочном растворе составило 5.5% по сухому веществу (RDS), а в ксилозной фракции - 16.7% по сухому веществу. Порядок элюирования: большая часть соли, следом за ней почти одновременно ксилоза и ксилоновая кислота (ксилоновая кислота слегка позднее).
Пример 11
Отделение ксилозы посредством использования слабокислой катионообменной смолы
Хроматографическое разделение Mg-сульфитного варочного раствора осуществляли с использованием слабокислой катионообменной смолы, Purolite С 105TM. Температура составляла 65oC и скорость потока была 0.7 м/ч. Уровень pH загрузочного раствора составлял 4.5, а содержание ксилозы - 10.9%. Производительность ксилозной фракции при чистоте фракции 25% составляла 19.0 кг сухого вещества/м3/ч, а при чистоте фракции 40% она составляла 7.8 кг сухого вещества/м3/ч, и максимальная чистота ксилозы при выделении составляла 42.7%. Содержание ксилоновой кислоты в загрузочном растворе составило 5.5% по сухому веществу (RDS). Чистота ксилоновой кислоты в ксилозной фракции, имеющей чистоту фракции 25% по сухому веществу ксилозы, составляла 11.7% по сухому веществу, а в ксилозной фракции, имеющей чистоту фракции 40% по сухому веществу ксилозы, чистота ксилоновой кислоты составляла 18.5% по сухому веществу. Соли, ксилоза и ксилоновая кислота элюировались почти одновременно (ксилоновая кислота немного позднее).
Пример 12
Выделение ксилозы с использованием волокнистоподобной катионообменной смолы
Хроматографическое разделение жидкости Mg-сульфитной варки осуществляли с использованием волокнообразной катионообменной смолы, Smoptec 101,3TM, полистирольная основа, которая была активирована сульфоновой кислотой. Температура составляла 65oC и скорость потока была 1.8 м/ч. Уровень pH загрузочного раствора составлял 2.2, а содержание ксилозы составляло 8.9%. Максимальная чистота ксилозы при разделении составляла 23.4%. Содержание ксилоновой кислоты в загрузочном растворе составляло 5.1% по сухому веществу (RDS) и максимальная чистота ксилозной фракции составляла 15.0% по сухому веществу. Порядок элюирования: большая часть соли и ксилоза с ксилоновой кислотой (вместе).
Пример 13
Отделение ксилозы с использованием сильнокислой катионообменной смолы
Хроматографическое разделение Mg-сульфитного варочного раствора осуществляли с использованием сильнокислой катионообменной смолы, Finex CS 11 GCTM. Температура составляла 65oC и скорость потока была 0.7 м/ч. Уровень pH загрузочного раствора составлял 1.0, а содержание ксилозы - 11.9%. Производительность ксилозной фракции при чистоте фракции 40% составляла 11.2 кг сухого вещества/м3/ч, и максимальная чистота ксилозы при выделении составляла 44.8%. Содержание ксилоновой кислоты в загрузочном растворе составило 5.5% по сухому веществу (RDS), а в ксилозной фракции - 25% по сухому веществу. Порядок элюирования: соль, ксилоза и ксилоновая кислота. Две последние фракции частично перекрывались.
Пример 14
Окисление ксилозы
2 г MgO, 62 г жидкости Mg-сульфитной варки и 140 г раствора SO2 (концентрация от 70 до 72 г SO2/л) загружали в автоклав. Автоклавы закрывали и помещали в глицериновую баню при 150oC. Автоклавы выдерживали в бане в течение 30 мин, 1 ч, 2 ч и 4 ч, после чего их охлаждали. Раствор отфильтровывали и анализировали. За четыре часа 28% ксилозы окислилось в ксилоновую кислоту. Результаты представлены в табл. 6.
Пример 15
Гидрогенизация ксилиновой кислоты
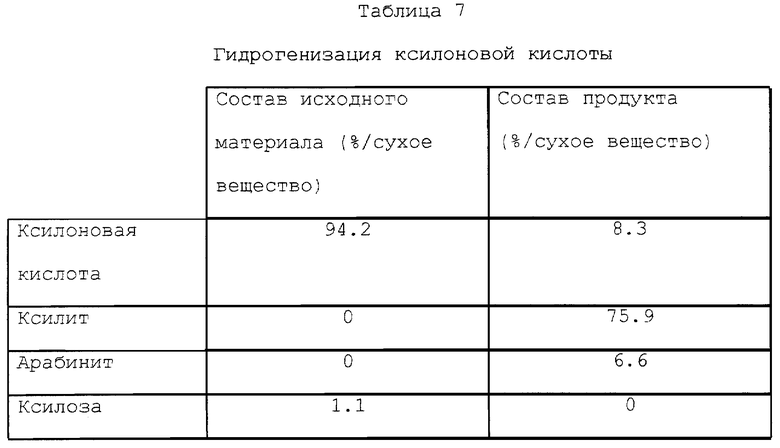
Исходным сырьем была фракция ксилоновой кислоты, которую получали, как описано в примерах 6, 7 и 8, и из которой магний был удален катионным обменом.
Гидрогенизацию осуществляли в 5-литровом автоклаве Медимекса (Medimex) (реактор периодического действия) при 110oC и при давлении 13000 кПа при использовании в качестве катализатора Ru/угле (5% Ru на угле, Engelhard CP 56xL/R/WW), количество которого составляло 18% по сухому веществу. Время гидрогенизации составляло 3 часа. В табл. 7 показаны составы исходных материалов и полученного продукта.
Пример 16
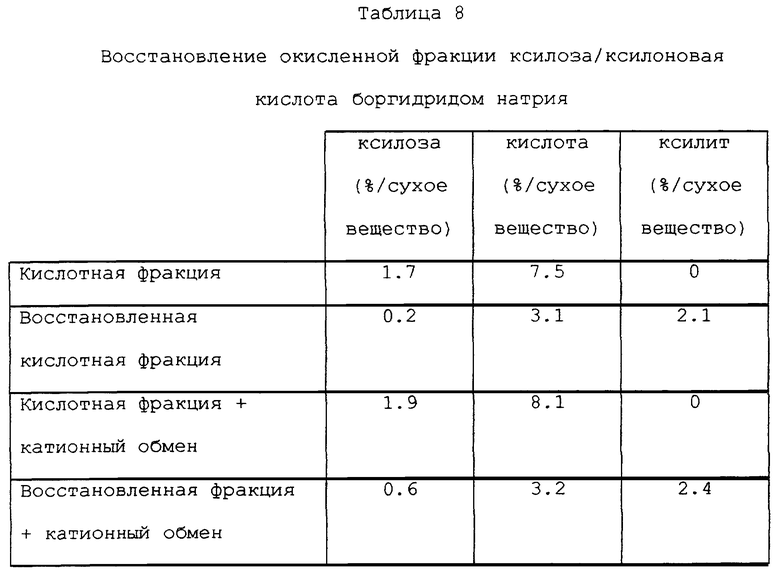
Восстановление боргидридом натрия окисленной фракции ксилоза-ксилоновая кислота, получаемой хроматографическим разделением.
Реакцию осуществляли при нормальном давлении и при комнатной температуре при перемешивании. Время реакции составляло 2 часа после добавления реагента (добавляли постепенно). Фракцию восстанавливали как таковую, а также после катионного обмена при содержании сухих веществ около 10%. Боргидрид натрия добавляли в соотношении 3 г/100 г естественного веса раствора (=10 г сухого вещества). Боргидрид натрия добавляли в виде 4% водного раствора. Реакцию прекращали подкислением раствора до pH 2 с использованием 6 н. соляной кислоты. Результаты показаны в табл. 8.
Пример 17
Гидрогенизация с никелем Ренея смеси, концентрированной относительно ксилоновой кислоты
166 г/л ксилоновой кислоты в 70%-ном метаноле гидрогенизировали в автоклаве с никелем Ренея (2 г) при 122oC и давлении 6500 кПа в течение 18 часов. Результаты представлены в табл. 9.
Пример 18
Гидрогенизация с родием смеси, концентрированной относительно ксилоновой кислоты
166 г/л ксилоновой кислоты в воде гидрогенизировали в автоклаве при использовании в качестве катализатора 0.17 г 5% Rh/Mo/Al2O3 при 140oC и давлении 6500 кПа в течение 18 часов. Результаты представлены в табл. 10.
Пример 19
Кристаллизация ксилитола
Исходным материалом был ксилит, полученный, как описано в примерах 15-18.
97 г ксилита (RDS 11.4%, загрузочный раствор) упаривали при 60oC до RDS 91.4%. Массу переносили в 1-литровую реакционную колбу, где в него вносили затравку 0.06 г кристаллов ксилита при 60oC. Включали линейную 49-часовую программу охлаждения (от 60.5 до 30oC). После охлаждения температуру массы увеличивали примерно на 3oC и массу центрифугировали. Чистота ксилита в массе составляла 77% по сухому веществу. Кристаллы отделяли центрифугированием (диаметр корзины 22 см, размер ячеек 0.15 мм) при 4500 оборотах в минуту в течение 5 минут и кристаллы промывали. Выход составил 30 г высушенных кристаллов. Чистота кристаллов ксилита составила 81.2% по сухому веществу.
Пример 20
Кристаллизация ксилита
Исходным материалом был ксилит, полученный, как описано в примерах 15-18.
Раствор ксилита фильтровали через мембрану 12 мкм. 170 г (сухое вещество) ксилита (RDS 19.7%, загрузочный раствор) упаривали при 60oC до RDS 91.3%. Массу переносили в 1-литровую реакционную колбу, где в нее при 60oC вносили затравку 0.05 г кристаллов ксилита. Включали линейную 41-часовую программу охлаждения (от 60.5 до 30oC). После охлаждения температуру массы увеличивали примерно на 3oC и массу центрифугировали.
Чистота ксилита в массе составляла 64.3% по сухому веществу. Кристаллы отделяли на центрифуге (диаметр корзины 22 см, размер ячеек 0.15 мм) при 4500 оборотах в минуту в течение 5 минут и промывали. Выход составил 54 г высушенных кристаллов. Чистота кристаллов ксилита составила 93.3% по сухому веществу.
Пример 21
Кристаллизация ксилита
Исходным материалом был ксилит, полученный, как описано в примерах 15-18.
Раствор ксилита фильтровали через мембрану 12 мкм. 185 г (сухое вещество) ксилита (RDS 20.9%, загружаемый раствор) упаривали при 60oC до RDS 92.2%. Массу переносили в 1-литровую реакционную колбу, где в нее при 56.5oC вносили затравку 0.05 г кристаллов ксилита. Включали линейную 69-часовую программу охлаждения (от 57 до 30oC). После охлаждения температуру массы увеличивали примерно на 3oC и массу центрифугировали. Чистота ксилита в массе составляла 56.5% сухого вещества. Кристаллы отделяли на центрифуге (диаметр корзины 22 см, размер ячеек 0.15 мм) при 4500 оборотах в минуту в течение 5 минут и промывали. Выход составлял 55 г высушенных кристаллов. Чистота кристаллов ксилита составляла 68.0% по сухому веществу.
Пример 22
Гидрогенизация ксилозы
Исходным материалом была фракция ксилозы, полученная по примеру 1 и подвергнутая обычному ионному обмену.
Гидрогенизацию осуществляли в порционном автоклаве (Medimex) при условиях, когда давление составляло 4000 кПа, температура 100oC и содержание сухого вещества в сиропе 47.8% по весу. Количество катализатора (никель Ренея, Chemcat J 10 GSTM) - 10% из расчета на сухое вещество сиропа сырой каталитической суспензии. Время реакции составляло 90 минут. Содержание ксилозы и ксилита в загрузочном и полученном растворах показаны в табл. 11.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ КСИЛИТА | 1997 |
|
RU2176995C2 |
| СПОСОБ ПОЛУЧЕНИЯ КСИЛОЗЫ | 1998 |
|
RU2211074C2 |
| СПОСОБ ФРАКЦИОНИРОВАНИЯ СУЛЬФИТНОГО ВАРОЧНОГО РАСТВОРА | 1994 |
|
RU2110317C1 |
| СПОСОБ ФРАКЦИОНИРОВАНИЯ РАСТВОРА | 1995 |
|
RU2136345C1 |
| СПОСОБ ПОЛУЧЕНИЯ КСИЛИТА ИЗ ВОДНОГО РАСТВОРА КСИЛОЗЫ | 1990 |
|
RU2108388C1 |
| СПОСОБ ИЗВЛЕЧЕНИЯ КСИЛОЗЫ ИЗ РАСТВОРОВ | 1996 |
|
RU2177038C2 |
| СПОСОБ ДЛЯ ИЗВЛЕЧЕНИЯ ОРГАНИЧЕСКОГО СОЕДИНЕНИЯ ИЗ РАСТВОРОВ | 1996 |
|
RU2184148C2 |
| СПОСОБ ПОЛУЧЕНИЯ КСИЛОЗЫ И ЦЕЛЛЮЛОЗЫ ДЛЯ ХИМИЧЕСКОЙ ПЕРЕРАБОТКИ | 2009 |
|
RU2512339C2 |
| СПОСОБ ФРАКЦИОНИРОВАНИЯ ПУТЕМ ХРОМАТОГРАФИЧЕСКОГО ПРОЦЕССА, ИМИТИРУЮЩЕГО ПОДВИЖНЫЙ СЛОЙ | 1997 |
|
RU2191617C2 |
| Способ выделения сахаров и лигносульфонатов из отработанного сульфитного щелока | 1986 |
|
SU1500164A3 |




Изобретение относится к получению ксилита. Способ осуществляют путем отделения ксилозы и ксилоновой кислоты от материала, содержащего ксилозу и ксилоновую кислоту, представляющего собой варочную жидкость сульфитной варки. Ксилозу отделяют и кристаллизуют. Ксилоновую кислоту отделяют и восстанавливают до ксилита. Восстановление осуществляют при температуре 70-150oС, под давлением от 10000 до 13000 кПа в присутствии катализатора, в состав которого входят соединения на основе Ru, Pd, Pt, Rh или Ni-Ренея, кроме того, используют боргирид натрия в качестве металлгидридного реагента. Ксилит выделяют хроматографически или путем кристаллизации. Технический результат - получение ксилита с высоким выходом из сульфитного варочного раствора. 40 з.п.ф-лы, 11 табл.
| US 4008285 А1, 15.02.1977 | |||
| СПОСОБ ПОЛУЧЕНИЯ КСИЛИТА | 0 |
|
SU165163A1 |
| GB 1302997 А1, 10.01.1973 | |||
| ЕР 0694515 А2, 31.01.1996. | |||

