Настоящее изобретение относится к нешипучей прессованной твердой дозированной форме, предназначенной для орального применения, способу приготовления такой дозированной формы и ее терапевтическому применению.
Ибупрофен (Ibuprofenum), а именно 2-(4-изобутилфенил)пропионовая кислота, представляет собой хорошо известный лекарственный препарат, обладающий анальгетической, противовоспалительной и жаропонижающей активностью. В продажу обычно поступает в виде рацемата (равные количества энантиомеров S(+)-ибупрофена и R(-)-ибупрофена). Также может применяться в виде любого очищенного энантиомера, особенно в форме S(+)-ибупрофена, который, как известно, является активной формой рацемата ибупрофена. Ибупрофен также используется в виде соли, например натриевой соли ибупрофена. В Великобритании ибупрофен применяется как рецепторное средство (например, Brufen (RTM)), преимущественно при лечении болезненных и воспалительных нарушений, включая ревматоидный артрит, анкилозирующий спондилит, остеоартрит, послеоперационные и постродовые боли и травматические повреждения мягких тканей, обычно в суточных дозировках до 3200 мг. Ибупрофен в Великобритании используется также как безрецептурное средство (например, Nurofen (RTM)), главным образом при лечении симптомов боли и лихорадки, включая головную боль, мигрень, ревматическую боль, мышечную боль, радикулит, невралгию, дисменорею, зубную боль и простуды, и грипп, обычно в суточных дозировках вплоть до 1200 мг. Разовая доза ибупрофена или его производного обычно эквивалентна 200 мг, 400 мг, 600 мг или 800 мг рацемического ибупрофена.
Основной проблемой в связи с указанными выше заболеваниями является улучшение начального действия ибупрофена, особенно при лечений боли. Можно думать, что быстрая дезинтеграция лекарственной формы обеспечивает быстрое поступление лекарства в тело пациента, приводя к более быстрому началу терапевтического действия по сравнению со стандартной формой выпуска. Соответственно желательно готовить твердые формы выпуска с дозированным содержанием для орального применения, которые могут быстро дезинтегрироваться в желудочно-кишечном тракте. Также предпочтительно, чтобы дозированную форму получали путем прессования на стандартных таблетирующих машинах с предшествующими таблетированию произвольно стадиями гранулирования и сушки. Однако возникает ряд проблем, связанных с обеспечением быстрого распада твердой дозированной формы, содержащей препарат ибупрофен. Одна из таких проблем заключается в том, что для достижения терапевтической дозы твердые составы обычно включают высокую дозу лекарства, например 200 - 800 мг ибупрофена, которая тем самым составляет значительную долю дозированной формы, то есть более 35 мас. %. Таким образом, существует проблема не только включения препарата ибупрoфен вместе с наполнителями, используемыми для приготовления таблетированной дозированной формы и обеспечивающими быструю дезинтеграцию, но также приготовления таблеток, которые и не слишком большие по размерам для приема внутрь, и могут быть произведены в соответствии со стандартными способами. Кроме того, твердые дозированные формы должны обладать достаточной прочностью, чтобы противостоять жестким воздействиям в процессе производства, например на стадии покрытия оболочкой в перфорированном вращающемся барабане и при упаковке и т.д., а также должны обладать подходящими показателями, характеризующими процесс дезинтеграции, чтобы обеспечить быстрое выделение лекарства из формы выпуска. Также предпочтительно, чтобы при прессовании состава, включающего смесь требуемых компонентов, не происходило его прилипание к пуансонам таблетирующей машины.
Ранее было установлено, что незначительное повышение давления прессования с целью улучшения прочностных показателей вызывает значительное увеличение длительности дезинтеграции получаемых таблеток. Таким образом, при прессовании компонентов, используя стандартные давления сжатия таблетирующей машины, трудно обеспечить подходящее время дезинтегрирования таблеток и сохранить приемлемые размеры таблеток достаточной прочности.
Немецкая патентная заявка 3922441 А относится к поиску улучшения показателей таблетирования ибупрофеновых составов и в ней раскрывается, что указанное улучшение может быть достигнуто путем полного или частичного превращения ибупрофена в соль кальция и использования ее для таблетирования. Утверждается, что составы могут по усмотрению содержать ибупрофен, S(+)-ибупрофен или их аммониевые, натриевые или калиевые соли. В таблетки могут включаться соль кальция и произвольные другие активные формы ибупрофена как отдельно получаемые соединения, или соли могут получаться in situ в ходе приготовления таблеток по реакции между ибупрофеном (кислотным лекарством) с раствором или суспензией реагента, содержащего один или несколько соединений, таких как СаО, Са(ОН)2, СаСО3, NaOH, КОН, NH40H, Nа2СО3, NаНСО3, К2СО3, КНСО3, (NН4)2СО3, NH4HC03 (в количестве 25% - 110% от эквивалентного количества ибупрофена). Получаемую смесь затем гранулируют, если необходимо, сушат и затем таблетируют после произвольного включения других наполнителей. В описании сообщается, что в зависимости от соотношения других солей, используемых вместе с солью кальция, соли аммония и щелочных металлов улучшают растворимость составов, содержащих соли кальция, и таким образом осуществляют контроль за биодоступностью, а также увеличивают гигроскопичность и липкость. Обе эти характеристки являются нежелательными с точки зрения оптимального таблетирования.
Нами установлено, что путем включения в состав для прессования карбоната или бикарбоната щелочного металла можно производить твердые дозированные формы лекарственного ибупрофена приемлемых размеров с коротким временем дезинтеграции и удовлетворительной прочности. В основу настоящего изобретения положен установленный факт, что добавка карбоната или бикарбоната щелочного металла увеличивает прессуемость состава, содержащего сжимающийся наполнитель в смеси с дезинтегрирующим компонентом, который обеспечивает приготовление твердой дозированной формы, обладающей ценными показателями твердости и дезинтегрируемости. Раскрытие в Немецкой патентной заявке 3922441А составов, включающих соль кальция произвольно вместе с натриевой или калиевой солью ибупрофена, приготовленной in situ в присутствии жидкости при получении таблеток, не входит в объем притязаний по настоящему изобретению.
Соответственно настоящим изобретением предлагается твердая нешипучая прессованная дозированная форма, содержащая ибупрофеновый лекарственный ингредиент и носитель, включающий прессуемый наполнитель вместе с дезинтегрирующим компонентом, в которой ибупрофеновый лекарственный ингредиент присутствует в количестве 35 мас.% или более от массы дозированной формы, отличающаяся тем, что носитель включает карбонат или бикарбонат щелочного металла в количестве, при котором дозированная форма имеет прочность на разрушение в пределах 2,95-6,80 т (6,5-15 Кр) и период дезинтеграции менее 10 мин, при условии, что ибупрофеновый лекарственный ингредиент не содержит кальциевой соли ибупрофена в смеси с солью щелочного металла ибупрофена.
Выражение "ибупрофеновый лекарственный ингредиент" включает ибупрофен, его S(+) и R(-)-энантиомеры и их смеси, соли, гидраты и другие производные.
Прочность на разрушение служит мерой твердости прессованной дозированной формы. Оно означает давление, которое требуется для разрушения таблетки. Прочность на разрушение твердой дозированной формы может измеряться на любой машине, предназначенной для проведения указанных испытаний, то есть путем продавливания дозированной формы выпуска между двумя кулачками и измерение усилия, требуемого для разлома таблетки по диаметру. Приемлемыми измерителями прочности на раздавливание являются ручные и автоматические приборы, поставляемые компаниями Монсанто, Эрвека и Шлойнигер. Период дезинтеграции означает временной интервал, в течение которого происходит дезинтеграция таблетки в водной среде при проведении испытаний по методике, определенной в Европейской Фармакопее 1986. Ссылка V. 5.1.1 (в редакции 1995 г.).
Карбонаты и бикарбонаты щелочных металлов обычно не применяются в качестве прессуемых материалов. Никто не мог ожидать, что частичная замена в составе прессуемого компонента наполнителя на практически непрессующиеся карбонаты или бикарбонаты щелочных металлов позволяет получить твердую дозированную форму выпуска, обладающую как хорошей прочностью на разрушение, так и хорошими дезинтегрирующими свойствами. Также установлено, что другие растворимые материалы, такие как лактоза, сахароза, маннит, цитрат натрия и хлорид натрия, не обеспечивают получение таблеток, сочетающих удовлетворительные показатели прессуемости, прочности на разрушение с показателями дезинтеграции и приемлемыми размерами, как это достигается при применении карбонатов и бикарбонатов щелочных металлов в дозированной форме по настоящему изобретению.
Карбонаты и бикарбонаты щелочных металлов являются растворимыми веществами, и ранее были предложены в качестве компонентов шипучих таблеток, например, для реакции с кислым компонентом шипучей пары (см., например, WO 94/10994) или для предотвращения начала шипучей реакции, например, при хранении. Шипучие таблетки распадаются благодаря реакции между кислотой и основанием, особенно в присутствии воды, приводящей к образованию углекислого газа. Система дезинтеграции нешипучих дозированных форм по настоящему изобретению, которая предназначена для приема внутрь и для которой шипучая реакция нежелательна, отличается от шипучих систем. Настоящая дозированная форма не содержит какого-либо растворимого кислотного компонента, с которым карбонат или бикарбонат щелочного металла может реагировать по шипучей реакции.
Также известно использование бикарбоната натрия в качестве кислотного нейтрализатора, и он ранее применялся вместе с ибупрофеном в таблетированной форме для означенной цели, например, в Японской патентной заявке 631986 А. Однако в данной ссылке не раскрывается включение ибупрофена и бикарбоната натрия в таблетку с прессуемым компонентом наполнителя в смеси с дезинтегрирующим компонентом или получение твердых дозированных форм, обладающих прочностью на разрушение и дезинтегрирующими свойствами, которые являются характеристиками настоящего изобретения.
Также известно использование бикарбоната натрия в водорастворимом составе, из которого может быть приготовлен жидкий продукт приемлемого вкуса, включающем ибупрофен (33-46 мас.%), L-аргинин (34-51%) и бикарбонат натрия (9-29%) (патент США 4834966). Однако в этом источнике не раскрываются другие ингредиенты формулы, обеспечивающие достижение показателей прочности на разрушение и дезинтеграции по настоящему изобретению.
Патент США 4873231 относится к уменьшению токсичности ибупрофеновой соли путем объединения соли с 1-5 молярными избытками бикарбоната или карбоната.
Пример 13 раскрывает прессование натрийибупрофена в таблетки с одним эквивалентом бикарбоната натрия или калия при дозировке 200 или 400 мг ибупрофена. В ссылке не раскрываются дополнительные детали формы выпуска и, следовательно, нет возможности извлечь информацию, касающуюся получения дозированной твердой формы, имеющей показатели прочности на разрушение и дезинтеграцию, которые являются характерными для настоящего изобретения.
В Европейской патентной заявке 418043 А раскрывается, что хотя для подавления вкусовых качеств растворимой в воде соли ибупрофена в растворе могут применяться такие соединения, как бикарбонаты щелочных металлов, двузамещенные фосфаты щелочных металлов и трехосновные цитраты щелочных металлов, другие соединения, в том числе карбонаты щелочных металлов, не могут применяться, поскольку в эффективных для изменения вкусовых свойств количествах получаемые водные растворы имеют неприемлемо высокий рН для орального приема внутрь. Составы, используемые в ней, обычно находятся в форме свободно текущего порошка, помещенного в пакетики единичной дозы. Однако также указывается, что состав может быть в любой другой форме, например в виде водорастворимых таблеток, пригодных для растворения в воде, которые могут содержать небольшое количество шипучей пары для диспергирования таблетки после добавлении к воде. Однако в заявке не раскрывается нешипучая твердая форма выпуска, обладающая прочностью на разрушение и эффективностью дезинтеграции по настоящему изобретению.
Настоящее изобретение открывает возможность приготовления ибупрофенового лекарственного препарата в твердой дозированной форме, используя в качестве носителя вещество, общее для всех ибупрофеновых препаратов. В силу различий в свойствах разных ибупрофеновых препаратов, например точек плавления, кристаллических форм, размеров частиц, пределов текучести и т.д., трудно подобрать единый носитель, который позволял бы получать твердые дозированные формы из всех форм ибупрофена прессованием. Соответственно там, где известные технические решения, в частности, касаются характеристик формул выпуска, требуемых для дозированных форм ибупрофена, и (или) относятся к прессованию в твердые дозированные формы, во многих случаях изобретение конкретно относится или к ибупрофену, или к соли ибупрофена. Например, Европейская заявка на патент 298666 А, WO 90/08542, WO 89/02266 и патент США 4609675, все они относятся к непосредственно прессуемым рецептурам, содержащим ибупрофен в качестве активного ингредиента, но не распространяются на его соли. Так, существенно важно, что дозированная форма по настоящему изобретению может включать как ибупрофен, так и его соль, предпочтительно натриевую соль, когда получение дозированной формы особенно затруднено.
Карбонат или бикарбонат щелочного металла усиливают прессуемость прессуемого наполнителя в сочетании с ибупрофеновым лекарственным ингредиентом. Так, применение карбоната или бикарбоната щелочного металла позволяет уменьшить количество сжимаемого наполнителя, что обычно требуется для достижения удовлетворительной сжимаемости составов. Это является преимуществом, так как ибупрофеновые лекарственные ингредиенты обычно принимают в больших дозах. Поэтому уменьшение количества наполнителей рассматривается как достижение, так как позволяет получать дозированные формы приемлемых размеров. В соответствии с настоящим изобретением суммарное количество используемого прессуемого наполнителя и карбоната или бикарбоната щелочного металла меньше, чем количество прессуемого компонента наполнителя в сочетании с дезинтегрирующим компонентом, который необходим в отсутствии карбоната или бикарбоната щелочного металла, для получения дозированной формы с приемлемыми показателями твердости и характеристиками дезинтеграции.
Твердые дозированные формы выпуска по настоящему изобретению пригодны для прямого введения больному и для достижения желаемого терапевтического эффекта. Они не требуют предварительного растворения или диспергирования в воде до приема. Более того, прессованные дозированные формы по настоящему изобретению не нуждаются в дополнительной обработке после прессования состава из смеси компонентов для получения твердой дозированной формы.
Молекула ибупрофена существует в двух энантиомерных формах, и используемый в описании термин "ибупрофеновое лекарственное средство" охватывает индивидуальные энантиомеры, особенно S(+)-ибупрофен, и их смеси в любой пропорции, в том числе 1:1, которая представляет собой рацемат ибупрофена. Ибупрофеновый лекарственный ингредиент также может присутствовать в виде любой соли или иного производного ибупрофена или его энантиомеров. При необходимости ибупрофеновый лекарственный ингредиент может включать одно или несколько ибупрофеновых активных компонентов, таких как рацемический ибупрофен и S(+)-ибупрофен в определенных сочетаниях. Однако авторы изобретения считают предпочтительным применение ибупрофенового лекарственного средства в виде индивидуального ибупрофенового активного ингредиента. Ибупрофеновое лекарственное средство также может находиться в разной степени гидратации. Настоящее изобретение применимо как к безводной, так и к гидратным формам, например, моногидрату или дигидрату. Обычно используются наиболее стабильные безводная или гидратированная форма. Предпочтительно ибупрофеновый активный компонент находится в форме соли рацемического или S(+)-ибупрофена. Примерами могут служить соли щелочных металлов, например соли натрия или калия ибупрофена; соли щелочноземельных металлов, например кальциевые или магниевые соли ибупрофена; соли металлов, например алюминиевая соль ибупрофена; соли аминокислот, например соли лизина или аргинина и ибупрофена; или соли аминов, например меглуминовая соль ибупрофена. Предпочтительно ибупрофеновый препарат включает единственную соль, выбираемую из числа солей щелочных металлов, солей аминокислот и аминосолей. Наилучшие результаты достигаются в соответствии с настоящим изобретением при применении растворимых солей ибупрофена, например солей щелочных металлов, таких как натрий и калий, так как эти соединения слабо прессуются. Также представляется затруднительным осуществлять предварительное гранулирование натриевой соли с другим инертным наполнителем перед прессованием в таблетированную форму. Обычно для получения удовлетворительных таблеток вначале требуется провести стадию обработки, такую как измельчение. В соответствии с настоящим изобретением, однако, не требуется никакой предварительной обработки натриевой соли. Также дополнительным преимуществом изобретения является использование ибупрофена натрия из заводского процесса производства при приготовлении сырья. Эти растворимые соли ибупрофена также обладают тем преимуществом, что в силу большей растворимости в водной среде при их выделении из состава улучшается абсорбция, тем самым вызывая улучшение начала действия по сравнению с существенно нерастворимыми формами ибупрофена. Особо предпочтительна натриевая соль ибупрофена, еще более предпочтительны соли натрия рацемического ибупрофена. Установлено, что дигидрат соли натрия рацемического ибупрофена является особенно стабильным гидратом и соответственно предпочтительнее использовать дигидрат соли натрия в прессованной дозированной форме выпуска в соответствии с настоящим изобретением.
Размер частиц ибупрофенового препарата должен облегчить процесс производства, например, обеспечивая текучесть в процессе производства и тем самым упрощая процесс прессования. Соответственно предпочтителен средний размер частиц в пределах 25-600 мкм, более предпочтителен - в пределах 50-300 мкм и, наиболее предпочтителен в пределах 15-250 мкм.
Обычно предпочтительны дозированные формы с высоким содержанием ибупрофенового лекарственного ингредиента, чтобы уменьшить размеры твердой дозированной формы. Типичные дозированные формы обычно содержат ибупрофеновый ингредиент в количестве, составляющем 35-90 мас.% ибупрофена в расчете на формулу, предпочтительно 35-75 мас.%, более предпочтительно 40-60 мас.% и наиболее предпочтительно 45-55 мас.%. Дозированные формы могут содержать активный ибупрофеновый ингредиент в количестве 50 мг, 100 мг, 150 мг, 200 мг, 250 мг, 300 мг, 350 мг, 400 мг, 500 мг, 600 мг и 800 мг. При использовании солей и иных производных обычно подбирают точные единичные формы, эквивалентные дозам ибупрофена, перечисленным выше, например 256 мг дигидрата соли натрия или 342 мг соли DL-лизина эквивалентны дозе ибупрофена 200 мг.
Карбонаты или бикарбонаты щелочных металлов облегчают получение твердых дозированных форм, характеризуемых прочностью на разрушение и дезинтегрирующими свойствами, приведенными выше. Карбонаты или бикарбонаты щелочных металлов предпочтительно включаются в дозированные формы в твердом виде. Нет необходимости растворять их в растворителе, например в воде, на стадии гранулирования перед прессованием в твердую дозированную форму. Такие показатели, как прочность на разрушение и дезинтегрируемость дозированной формы обеспечиваются присутствием карбоната или бикарбоната щелочного металла в гомогенной смеси с ибупрофеновым лекарственным ингредиентом и прессующимся наполнителем вместе с дезинтегрирующим компонентом. Особо предпочтительно тщательное смешение ибупрофенового лекарственного вещества с карбонатом или бикарбонатом щелочного металла.
Используемые в соответствии с настоящим изобретением карбонаты или бикарбонаты щелочных металлов могут включать карбонат или бикарбонат натрия или карбонат или бикарбонат калия либо в виде индивидуального соединения, либо в смеси. Предпочтительно щелочным металлом является натрий, и следовательно, предпочтительными компонентами являются карбонат и бикарбонат натрия. Карбонаты щелочных металлов могут применяться в безводном виде или в различной степени гидратации, например в форме моногидрата или декагидрата. Возможно применение обеих этих форм. Однако предпочтительнее применять безводную форму. Предпочтительным карбонатом щелочного металла при осуществлении настоящего изобретения является безводный карбонат натрия.
Карбонат или бикарбонат щелочного металла включают с целью облегчения создания дозированной формы ибупрофенового лекарственного ингредиента и получения твердой дозированной формы, имеющей прочность на разрушение в пределах 2,95-6,80 т (6,5-15 Кр) и период дезинтеграции менее 10 мин. Карбонат или бикарбонат щелочного металла может включаться в дозированную форму в количестве 3-20 мас. % от этой дозированной формы, предпочтительно 4-16 мас.%, более предпочтительно 5-15 мас.% и наиболее предпочтительно 6-10 мас. % от дозированной формы. Карбонат или бикарбонат щелочного металла предпочтительно имеет размер частиц в диапазоне 25-600 мкм, более предпочтительно 50-100 мкм. В предпочтительных дозированных формах отношение массы карбоната или бикарбоната натрия к ибупрофеновому лекарственному средству составляет от 1: 2 до 1: 10 мас. ч. В соответствии с особо предпочтительным аспектом настоящего изобретения дозированная форма представляет собой непосредственно прессованную таблетку, содержащую 40-85 мас.% натриевой соли ибупрофена и 5-15 мас.% карбоната или бикарбоната натрия.
Носитель может составлять до 65 мас.% дозированной формы. Предпочтительные дозированные формы включают 25-65 мас.% носителя, более предпочтительно 40-60 мас.% и наиболее предпочтительно 45-55 мас.%, носителя. В более предпочтительной дозированной форме отношение ибупрофенового лекарственного ингредиента к наполнителю равно 2:1 - 1:2 мас.ч., и носитель включает 5-20 мас.% карбоната или бикарбоната натрия.
Носитель включает прессуемый наполнитель, который применяется в достаточном количестве вместе с карбонатом или бикарбонатом щелочного металла, гарантирующим, что состав, содержащий ибупрофеновый лекарственный ингредиент, может быть сформован предпочтительно прямым прессованием в твердую дозированную форму, имеющую прочность на разрушение в пределах 2,95-6,80 т (6,5-15 Кр) и период дезинтеграции менее 10 мин. Ингредиенты обычно прессуются из сухой порошковой смеси. Смесь может содержать предварительно гранулированный продукт, например, полученный влажным или сухим гранулированием и произвольно содержащий ибупрофеновый активный компонент, и полученные сухие гранулы при необходимости могут смешиваться с другими сухими порошковыми ингредиентами и прессоваться в твердые дозированные формы. Обычно на любой стадии влажного предварительного гранулирования ибупрофеновый лекарственный ингредиент находится в гранулах. Перед прессованием карбонат или бикарбонат щелочного металла могут добавляться к сформованным гранулам с произвольными другими наполнителями, такими как смазочные вещества. Однако предпочтительно не добавлять в состав никакой жидкости (то есть, воду) на любой произвольной стадии предварительного гранулирования или перед прессованием. Вполне очевидно, что прямое прессование предпочтительнее, так как это означает проведение более эффективного процесса таблетирования, а именно осуществление только смешения ингредиентов и затем их прессования без стадий промежуточного гранулирования и сушки, которые необходимы при других процессах изготовления таблеток.
Прессуемый наполнитель может содержаться в количестве 10-50 мас.% в расчете на дозированную форму, предпочтительно 20-50 мас.%, более предпочтительно 27-45 мас.%, наиболее предпочтительно 30-40 мас.% от дозированной формы. Отношение карбоната или бикарбоната щелочного металла к наполнителю, который может прессоваться, предпочтительно находится в пределах от 2:1 до 1:10 мас.ч.
Примерами прессуемых наполнителей могут служить одно или несколько производных целлюлозы, крахмал и его производные (например, предварительно желатинизированный крахмал), растворимые сахара (например, лактоза, сахароза, декстрин), хлорид натрия, фосфат кальция, сульфат кальция, маннит, сорбит, циклодекстрин, мальтодекстрин. Предпочтительно прессуемый наполнитель включает производное целлюлозы. Примерами приемлемых производных целлюлозы могут служить метилцеллюлоза, оксиметилцеллюлоза, оксиэтилцеллюлоза, оксипропилцеллюлоза, оксипропилметилцеллюлоза, фталат оксипропилметилцеллюлозы и микрокристаллическая целлюлоза. Предпочтительным производным целлюлозы в соответствии с настоящим изобретением является микрокристаллическая целлюлоза. Кроме того, предпочтительно производное целлюлозы с размером частиц примерно 100 мкм, предпочтительно 100-150 мкм.
В предпочтительных дозированных формах производное целлюлозы составляет 50-100 мас.% от прессуемого наполнителя, более предпочтительно 70-100 мас.% и, наиболее предпочтительно 90-100 мас.% прессуемого наполнителя. Остаток прессуемого наполнителя может приходится на долю других наполнителей, хорошо известных в технике, в том числе тех, которые перечислены выше. Предпочтительные прессуемые наполнители включают одно или несколько веществ, выбираемых из числа таких как микрокристаллическая целлюлоза, лактоза и маннит. В соответствии с предпочтительным аспектом настоящего изобретения, в котором содержание производного целлюлозы составляет 50-100 мас.% прессуемого компонента, отношение карбоната или бикарбоната щелочного металла к производному целлюлозы может составлять от 2:1 до 1:10, более предпочтительно от 1:1 до 1: 9, и наиболее предпочтительно от 1:3 до 1:8 мас. ч. В соответствии с другим предпочтительным аспектом настоящего изобретения суммарное массовое отношение производного целлюлозы и карбоната или бикарбоната щелочного металла к ибупрофеновому лекарственному ингредиенту составляет 1:10-2:1 мас. ч. , более предпочтительно 1:4-2:1 мас. ч., наиболее предпочтительно 1:1-1:2 мас. ч.
Прессуемый наполнитель объединяют с дезинтегрирующим компонентом. Примеры дезинтегрирующих компонентов включают одно или несколько веществ, таких как крахмал пшеницы, кукурузный крахмал, картофельный крахмал, крахмал гликолат натрия, низкозамещенная оксипропилцеллюлоза, альгиновая кислота, сетчатый поливинилпирролидон, силикат магния и алюминия и натрийкроскармелоза. Предпочтительные дезинтеграторы включают одно или несколько веществ из группы, содержащей натрийкроскармелозу и крахмалгликолат натрия. При использовании таких дезинтегрирующих компонентов они могут составлять до 15 мас.% дозированной формы, например 1-10 мас.%, предпочтительно 5-15 мас.% дозированной формы. Некоторые прессуемые наполнители обладают способностью к дезинтеграции, как, например, микрокристаллическая целлюлоза и (или) оксипропилметилцеллюлоза и, следовательно, отпадает необходимость в дискретном дезинтегрирующем веществе, когда прессуемый наполнитель объединяют с дезинтегрирующим компонентом. Однако предпочтительно использовать прессуемый наполнитель (который может обладать дезинтегрирующими свойствами) и дискретный дезинтегрирующий компонент как раздельные компоненты, смешанные в состав.
В особо предпочтительной дозированной форме носитель включает 8-80 мас.% прессуемого наполнителя (более предпочтительно 50-75 мас.%), 8-40 мас.% карбоната или бикарбоната щелочного металла (более предпочтительно 10-20 мас. %), 10-20 мас. % дезинтегратора (более предпочтительно 12-18 мас.%). Особо предпочтителен носитель, включающий 50-75% микрокристаллической целлюлозы, 12-18% кроскармелозы натрия и 8-20% карбоната или бикарбоната натрия. Желательно, чтобы отношение прессуемый наполнитель : карбонат или бикарбонат щелочного металла : дезинтегрирующий компонент было равно 1-9 : 1 : 0,5-2 - мас. ч., предпочтительно 2,5-6 : 1 : 0,8-1,4 мас. ч.
Прессованная дозированная форма выпуска также может включать один или несколько инертных разбавителей (которые не обладают способностью к прессованию) по усмотрению специалистов в данной области. Инертный разбавитель может составлять вплоть до 20 мас.%, формулы, предпочтительно 0-10 мас.%.
Твердая дозированная форма также может включать обладающее текучестью вспомогательное вещество, такое как тальк или коллоидный кремнезем, предпочтительно в количестве до 4 мас.% состава, например 0,5-2,0 мас.% состава. В состав дозированной формы также могут входить смазочные вещества, такие как стеариновая кислота, лаурилсульфат натрия, полиэтиленгликоль, гидрированное растительное масло, стеарат кальция, стеарилфумарат натрия или стеарат магния. Они могут составлять вплоть до 4 мас.% дозированной формы выпуска, например 0,5-2 мас. % дозированной формы. Дополнительно могут включаться вещества, препятствующие прилипанию, такие как тальк, в количестве вплоть до 4 мас.% дозированной формы, например 0,5-2 мас.% дозированной формы.
Твердая дозированная форма может выпускаться в оболочке, например, из сахара или в виде пленочного покрытия, которое оказывает минимальный эффект на период дезинтеграции. Предпочтительная твердая дозированная форма выпуска по настоящему изобретению, например таблетка, представляет собой таблетки с оболочкой, нанесенной, например, струевым способом раствора, содержащего оксипропилметилцеллюлозу и пластификатор, такой как пропиленгликоль, полиэтиленгликоль и (или) тальк в одной или нескольких оболочках.
Предпочтительная дозированная форма выпуска включает:
(a) 40-60 мас.% натриевой соли ибупрофена (более предпочтительно 45-55 мас.%);
(b) 20-50 мас.% прессуемого наполнителя, например микрокристаллической целлюлозы (более предпочтительно 30-40 мас.%);
(c) 4-16 мас.% карбоната натрия или бикарбоната натрия (более предпочтительно 5-10 мас.%;
(d) до 10 мас.% дезинтегратора, например кроскармелоза натрия или крахмалгликолат натрия (более предпочтительно 5-10 мас.%);
(e) до 4 мас. % смазывающего вещества, например стеариновой кислоты (более предпочтительно 0,5-2,0 мас.%); и
(f) до 2 мас.% текучего вещества, например коллоидный кремнезем (более предпочтительно 0,5-1 мас.%).
В следующей предпочтительной дозированной форме отношение ибупрофенового лекарственного ингредиента к носителю равно 1:2 - 2:1 мас. ч., предпочтительно 2:3 - 3:2 мас. ч., и отношение целлюлозного производного прессуемого компонента наполнителя к карбонату или бикарбонату щелочного металла равно в пределах 9:1 - 1:1, предпочтительно 5:1 - 3:1 мас.
Твердая дозированная форма по настоящему изобретению может подвергаться прессованию, предпочтительно прямому прессованию с получением прочности на разрушение в пределах 2,95-6,80 т (6,5-15 Кр), более предпочтительно 3,63-5,44 т (8-12 Кр). Это может быть достигнуто, например, на стандартном прессе или в роторных таблетирующих аппаратах, развивающих давление сжатия в диапазоне 100-140 МПа.
Специалистам в данной области очевидно, что при различных добавках, используемых в формулах, и изменением их количества получают формы, имеющие различные показатели прочности на раздавливание и периоды дезинтеграции. Предпочтительные дозированные формы обладают прочностью на разрушение 2,95-6,80 т (6,5-15 Кр) и имеют период дезинтеграции менее 10 мин при прессовании под давлением более 80 МПа. Более предпочтительные дозированные формы проявляют прочность на раздавливание форм выпуска 2,95-6,80 т (6,5-15 Кр) и период дезинтеграции меньше 10 мин при прессовании под давлением сжатия в пределах 100-140 МПа на стандартном таблетирующем аппарате, например в роторной таблетирующей машине. Такие давления сжатия включают 110 МПа, 120 МПа и 130 МПа. Особо предпочтительные дозированные формы имеют прочность на разрушение 2,95-6,80 т (6,5 -15 Кр) и период дезинтеграции менее 10 мин при прессовании под давлениями в пределах диапазона 100-140 МПа.
Как следует из описания, необходимо, чтобы дозированная форма имела приемлемую прочность на разрушение. Необходимо, чтобы дозированная форма сохраняла свою целостность и не крошилась и (или) разламывалась в процессе производства, упаковки и транспортировки упакованного продукта. Однако также необходимо, чтобы дозированная форма не была излишне твердой, что будет препятствовать быстрому выделению из формы лекарства. Предпочтительные дозированные формы имеют прочность на разрушение - 5,44 т (-12 Кр) (не пропечатано в оригинале на с.14), более предпочтительно 3,63-5,44 т (8-12 Кр). Предпочтительно, дозированная форма имеет прочность на раздавливание в пределах 3,63-5,44 т (8 -12 Кр) при усилии прессования в диапазоне 100-140 МПа.
Период дезинтеграции таблетки, получаемой в соответствии с настоящим изобретением, меньше 10 мин. Методика измерения описана в Европейской фармакопее 1986, Ref. V. 5.1.1 (обновленная в 1995 г.) (А. Тестирование дезинтеграции таблеток и капсул). Предпочтителен период дезинтеграции менее 6 мин (например, - 6 мин), более предпочтителен период менее 5 мин (например, 1-5 мин), и наиболее предпочтителен период дезинтеграции, равный или меньше 3 мин (например, 1-3 мин).
Дозированные формы по настоящему изобретению могут быть растворимыми или нерастворимыми в воде. Установлено, что растворимость дозированной формы в воде нельзя считать критическим фактором. Некоторые из веществ, которые, как было установлено, являются наиболее пригодными в соответствии с настоящим изобретением, нерастворимы в воде. Соответственно, если одно или несколько веществ являются нерастворимыми, дозированная форма является нерастворимой в воде и, таким образом, представляет собой предпочтительную форму.
Дозированные формы, отвечающие настоящему изобретению, получают прессованием. Носитель объединяют с ибупрофеновым активным ингредиентом и прессуют (предпочтительно прямым сжатием) в твердую дозированную форму. Конечной стадии получения твердой дозированной формы (например, прессованию) может предшествовать стадия, например, предварительного влажного гранулирования или предварительного сухого гранулирования. На стадии влажного гранулирования ибупрофеновый лекарственный ингредиент обычно предварительно гранулируют связующим компонентом, таким как поливинилпирролидон в растворителе, таком как вода или углеводородный растворитель, и затем гранулы сушат. Гранулированный материал затем смешивают с другими необходимыми наполнителями и формуют в твердую дозированную форму по изобретению. В любой начальной стадии предварительного гранулирования, однако, не требуется добавлять растворитель (например, воду) на любой стадии в процессе производства, и следовательно, в предпочтительном примере осуществления изобретения не требуется никакой стадии сушки. На стадии сухого предварительного гранулирования некоторые компоненты могут прессоваться совместно, например в валках или комкованием, и гранулы затем смешивают с оставшимися добавками и прессуют в твердые дозированные формы. Дозированные формы также могут получаться просеиванием порошковых компонентов в емкость с последующим смешением всех ингредиентов для получения гомогенной смеси. Смесь затем может непосредственно прессоваться в таблетки. Этот способ представляет дополнительный аспект настоящего изобретения.
Таким образом, предлагается способ получения нешипучей твердой дозированной формы, включающей ибупрофеновый лекарственный ингредиент в количестве 35% или более мас. дозированной формы выпуска и носитель, содержащий прессуемый наполнитель вместе с дезинтегрирующим компонентом, отличающийся тем, что носитель, включающий карбонат или бикарбонат щелочного металла, объединяют с ибупрофеновым лекарственным ингредиентом с получением гомогенной твердой смеси в существенно сухих условиях, по усмотрению, с другими таблетирующими наполнителями, и прессуют смесь в одну или несколько твердых дозированных форм, имеющих прочность на разрушение в пределах 2,95-6,80 т (6,5-15 Кр) и период дезинтеграции менее 10 мин.
В соответствии с более предпочтительным способом дозированную форму получают непосредственным прессованием порошковой смеси ингредиентов и не осуществляют никакой стадии предварительного гранулирования. В таком способе ибупрофеновый лекарственный ингредиент объединяют с прессуемым наполнителем, дискретным дезинтегрирующим компонентом и карбонатом или бикарбонатом щелочного металла. Также могут добавляться и смешиваться другие необязательные добавки, такие как смазывающее вещество и вспомогательное вещество, придающее текучесть, с получением однородной смеси порошковых частиц, и, наконец, смесь непосредственно прессуют в твердые дозированные формы по настоящему изобретению.
В предпочтительном способе предлагается дозированная форма, содержащая натриевую соль ибупрофена вместе с носителем, содержащим микрокристаллическую целлюлозу и карбонат или бикарбонат натрия.
При терапии дозированные формы по настоящему изобретению принимают внутрь перорально, так как терапевтические дозированные формы существуют в виде твердых дозированных форм, предпочтительно в виде таблеток. Дозированные формы могут покрываться сахарной оболочкой или пленкой, которая существенно сразу же растворяется, как только дозированная форма входит в контакт с водной средой. Состав также может прессоваться на твердой сердцевине другого вещества с получением твердой формы, имеющей быстрорастворимую наружную оболочку. С другой стороны, прессованный состав может находиться в одном или нескольких слоях многослойной дозированной формы. В таких формулах остальные слои или сердцевина могут включать стандартные наполнители для осуществления стандартного, быстрого или медленного выделения и хорошо известны специалистам в данной области (см., например, Фармацевтические науки. Ремингтон, 17-е изд., редактор Дженнаро с сотр.).
Таким образом, в соответствии с еще одним аспектом настоящего изобретения также предлагается твердая готовая форма, содержащая слой состава, включающего ибупрофеновый лекарственный ингредиент с носителем, причем ибупрофеновый ингредиент содержится в количестве до 35 мас.% и более от состава, и носитель, включающий прессуемый наполнитель вместе с дезинтегрирующим компонентом, отличающаяся тем, что носитель включает карбонат или бикарбонат щелочного металла в таком количестве, что состав может прессоваться с получением слоя, имеющего прочность на разрушение в пределах 2,95-6,80 т (6,5-15 Кр) и период дезинтеграции менее 10 мин.
При необходимости дозированные формы по настоящему изобретению могут включать другие совместимые фармакологически активные ингредиенты (например, анальгезирующее вещество, действующее на центральную нервную систему, например, кодеин) и (или) усилители. Так, например, дозированные формы могут включать любой ингредиент, обычно используемый в лекарствах от насморка, простуды или гриппа, например кофеин, или другое производное ксантина, и (или) иное анальгезирующее, и (или) скелетно-мышечный релаксант, и (или) антигистамин, и (или) деконгестант, и (или) суппрессант от кашля, и (или) отхаркивающее средство.
Приемлемые антигистамины включают акривастин, астемизол, азатадин, азеластин, бромодифенгидрамин, бромфенирамин, карбиноксамин, цетиризин, хлорфенирамин, ципрогептадин, дексбромофенирамин, дексхлорфенирамин, дифенгидрамин, эбастин, кетотифен, лодоксамид, лоратидин, левокабастин, меквитазин, оксатомид, фениндамин, фенилтолоксамин, пириламин, сетастин, тазифиллин, темеластин, терфенадин, трипеленнамин или трипролидин. Предпочтительно используются неседативные антигистамины. Приемлемые суппрессанты от кашля включают карамифен, кодеин или декстрометорфан. Приемлемые деконгестанты включают псевдоэфедрин, фенилпропаноламин и фенилэфрин. Приемлемые отхаркивающие средства включают гуаифенезин, цитрат калия, гваяколсульфонат калия, сульфат калия и терпингидрат.
Ибупрофен и его производные, главным образом, используются как противовоспалительные, анальгезирующие и жаропонижающие средства, но также были предложены для других терапевтических целей, включая лечение периодонтальной потери костной массы, зуда и болезни Альцгеймера. Следовательно, дозированные формы по настоящему изобретению показаны при лечении всех терапевтических заболеваний, для которых ибупрофен является эффективным, включая ревматоидный артрит, остеоартроз, анкилозирующий спондилит, серонегативную артропатию, околосуставные заболевания и травматические поражения мягких тканей. Они также могут применяться при лечении послеоперационного болевого синдрома, послеродовых болей, зубной боли, дисменореи, головной боли, мигрени, ревматической боли, мышечной боли, радикулита, невралгии и мышечно-скелетной боли или боли или дискомфорта, вызываемого респираторными инфекциями, простудой или гриппом, подагры или утренней скованности.
В соответствии с еще одним аспектом настоящего изобретения предлагается способ получения нарастающей вначале анальгезирующей и (или) жаропонижающей реакции, включающей введение нешипучей прессованной твердой дозированной формы выпуска, включающей 35 мас.% или более того ибупрофенового лекарственного ингредиента вместе с носителем, содержащим прессуемый наполнитель вместе с дезинтегрирующим компонентом и карбонат или бикарбонат щелочного металла, причем дозированная форма имеет прочность на раздавливание в диапазоне 2,95-6,80 т (6,5-15 Кр) и период дезинтеграции менее 10 мин при условии, что ибупрофеновый лекарственный ингредиент не содержит кальциевой соли ибупрофена в смеси с ибупрофеновой солью щелочного металла.
В соответствии с еще одним аспектом настоящего изобретения предлагается включать карбонат или бикарбонат щелочного металла в состав носителя, содержащего прессуемый компонент наполнителя с дезинтегрирующим компонентом, причем указанный носитель подготовлен для смешения с ибупрофеновым активным ингредиентом при существенно сухих условиях и последующего прессования в твердую нешипучую дозированную форму выпуска, в которой ибупрофеновый лекарственный ингредиент составляет 35 мас.% или более дозированной формы, причем прочность на раздавливание дозированной формы составляет 2,95-6,80 т (6,5-15 Кр) и период дезинтегрирования менее 10 мин.
Получение прессованных таблеток из формул по настоящему изобретению иллюстрируется следующими примерами.
В примерах использовались рацемический ибупрофен и рацемическая натриевая соль \ S(+)-ибупрофена от фирмы Knol Pharma, Nottingham, GB; марки микрокристаллической целлюлозы от фирмы FMC Corporation, Brussels, BE под торговой маркой Avicel PH101 и РН102; кроскармелозу натрия от FMC Corporation, Brussels, BE под торговой маркой Ac-Di-Sol; коллоидный кремнезем от фирмы Degussa, Frankfurt, DE под торговой маркой Aerosil 200; гидрированное растительное масло от фирмы Karlshamn, SE под торговой маркой Sterotex; оксипропилметилцеллюлоза 2910 (50 CPs) от фирмы Colorcon, Kent, GB; оксипропилметилцеллюлоза 2910 (6 Cps) от фирмы Shin-etsu, Japan и Opaspray от фирмы Colorcon, Kent, GB; крахмалгликолат натрия от фирмы Edward Mendell, Reigate, GB, под торговой маркой Explotab; стеарилфумарат натрия от фирмы Forum Chemicals, Surrey, GB, под торговой маркой Pruv; маннит от фирмы Roquette Freres, Les-trem, Franse, под торговой маркой Pearlitol, сетчатый поливинилпирролидон от фирмы BASF, Ludwigshaven, Germany под торговой маркой Kollidon CL.
А. Способ приготовления таблеток в примерах
Таблетки готовили просеиванием всех ингредиентов и смешением до получения гомогенной смеси в стандартных смесителях. Приготовленный состав затем направляли и прессовали на одноплунжерной таблетирующей машине (Manesty F), используя усилие прессования в пределах 100-140 МПа. В некоторых примерах (примеры 1-9, 22) составы прессовали при конкретных усилиях прессования, например 100, 120, 140 МПа. В других примерах (примеры 10-21, 23-27) составы прессовали при подходящих усилиях прессования в пределах 100-140 МПа, в зависимости от применяемых ингредиентов и прочности на раздавливание и времени растворения, требуемого для полученной таблетки.
Б. Измерение свойств таблеток, приготовленных в примерах
1. Прочность на разрушение (Кр) Прочность на разрушение является мерой твердости таблетки. Она измеряется путем регистрации усилия при разрушении по диаметру в момент, когда таблетка разламывается между приводимыми в движение валками измерителя прочности на раздавливание Шленигера. Для каждой формулы примера приведены диапазон прочности на разрушение. Также для примеров 10-27 приведены средние значения прочности на разрушение.
2. Период дезинтеграции (мин)
Измерение периода дезинтеграции проводили по методике, описанной в Европейской Фармакопее 1986, разд.У.5.1.1 (измененная в 1995), используя в качестве жидкости водопроводную воду (рН примерно 7). Определяли время, за которое шесть таблеток, приготовленных из каждой формулы по примерам, полностью дезинтегрировались.
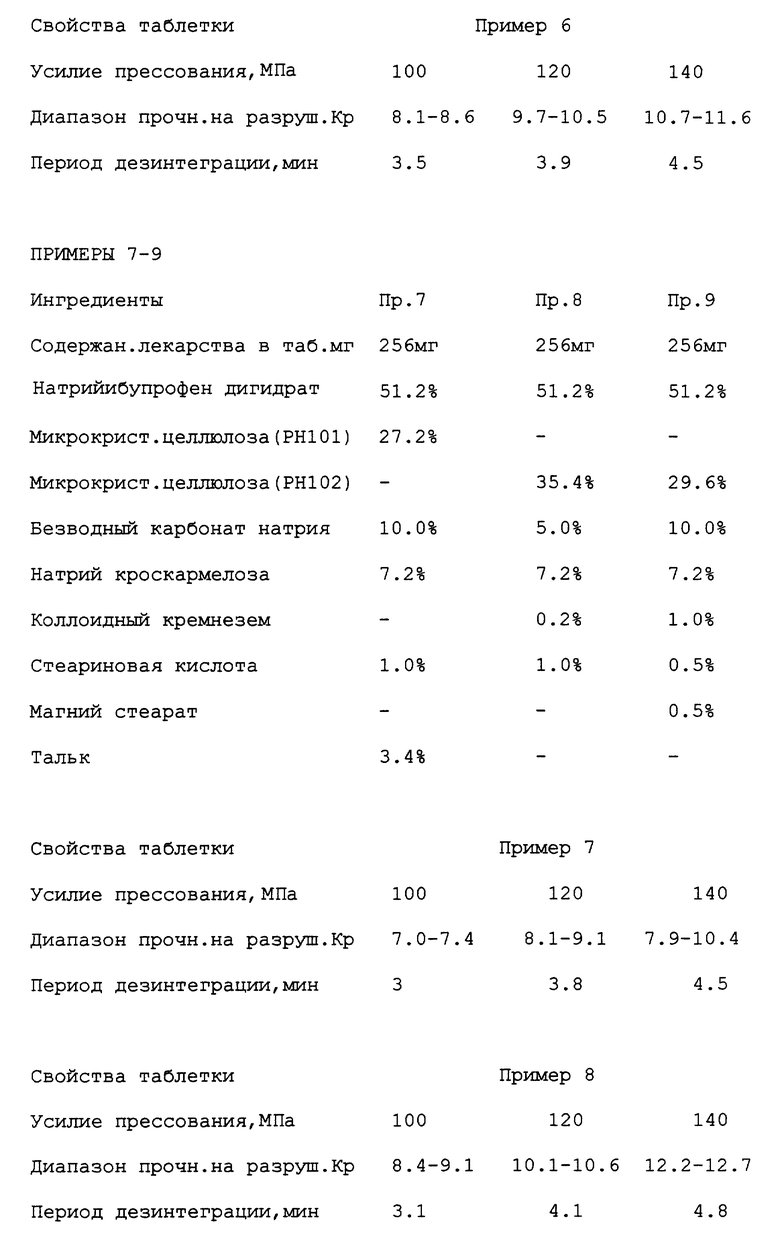
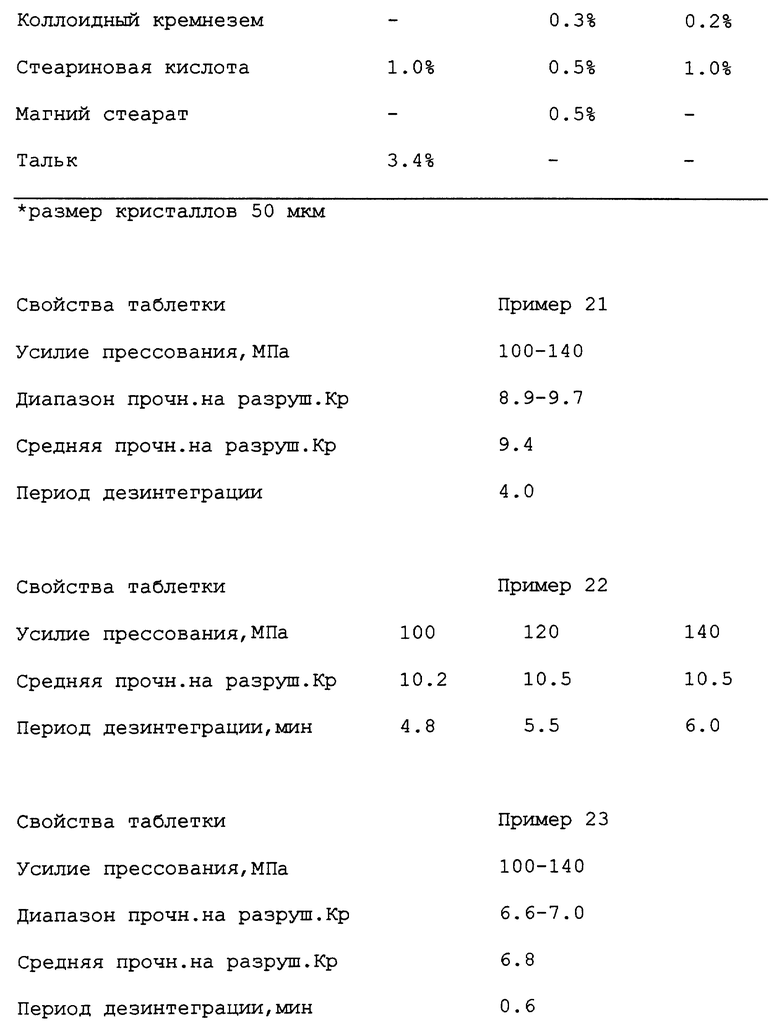
В. Таблетки по примерам и их свойства
% - массовые. Ибупрофен - рацемат за исключением случаев, где указано (см. примеры 1-11).
Ядро таблетки в примере 10 покрыто следующей оболочкой (% от массы ядра):
первая оболочка: оксипропилметилцеллюлоза 2910 (6 спуаз) (1,016%), тальк 0,204%), Opaspray White M-1-711B (0,336%).
Наружная оболочка: оксипропилметилцеллюлоза 2910 (5-0 спуаз) (0,437%), полиэтиленгликоль 6000 (0,049%), стеарат кальция (0,002%).
Период дезинтеграции оболочковой таблетки по примеру 10 составил 5,5 мин.
Сердцевины таблеток по примерам 12-14 имели такие же оболочки, что и в примере 10. Период дезинтеграции - 5.1 мин, 5.5 мин и 7.5 мин соответственно в примерах 12,13 и 14.
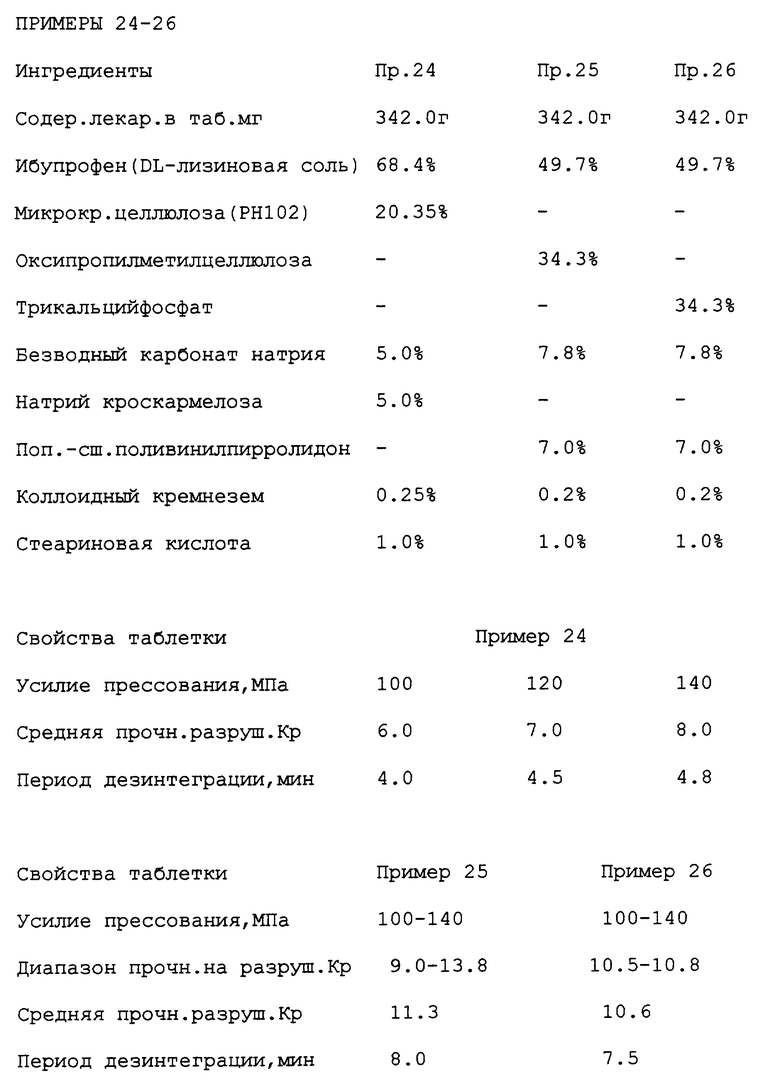
Примеры также проведены аналогично примерам 1-22, выше, содержащим натриевую соль рацемического ибупрофена в количестве 64 мг, 128 мг, 192 мг, 348 мг, 512 мг, используя те же соотношения ингредиентов, что и в примерах 1-22.
Таблетки готовили аналогично примерам 24-26, выше, с ибупрофен DL-лизиновой солью в количестве 171,0 мг, 256,6 мг и 513,0 мг, используя те же соотношения между ингредиентами, указанные в примерах 24-26.
На чертеже (фиг. 1) приведено сравнение периодов дезинтеграции:
(а) прессованной дозированной формы по настоящему изобретению, содержащей натриевую соль ибупрофена (пример 22) со сравнительным примером А (без(би)карбонатного компонента); и
(б) прессованной дозированной формы по настоящему изобретению, содержащей лизиновую соль ибупрофена (пример 24), со сравнительным примером В (без(би)карбонатного компонента).
Периоды дезинтеграции представлены в функции давления прессования.
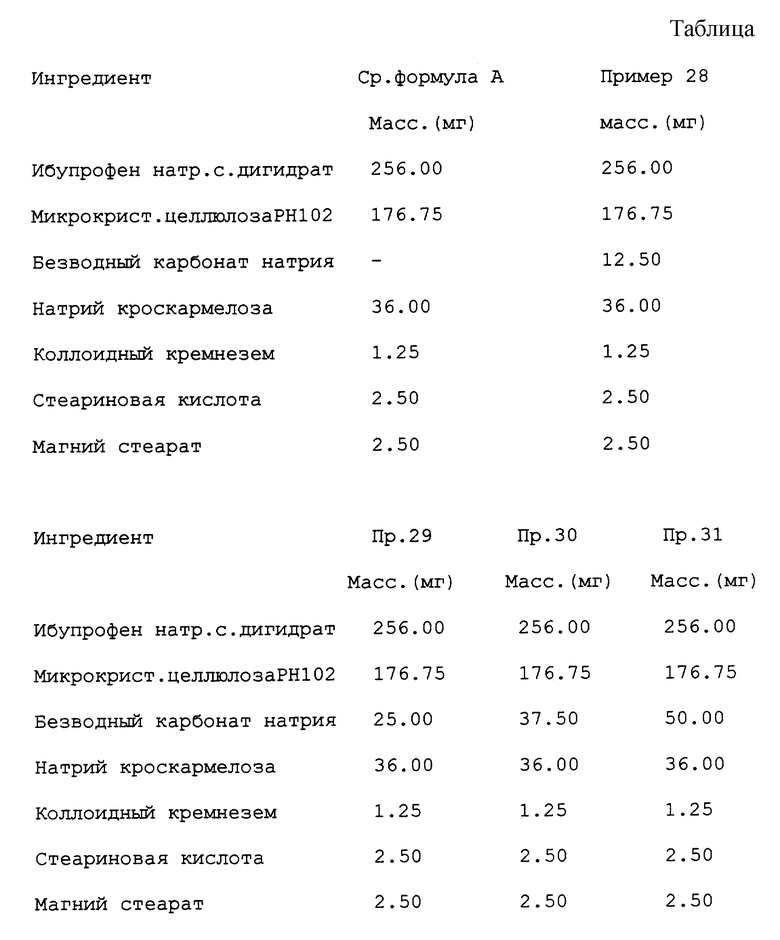
Фиг. 2 иллюстрирует сравнение свойства таблеток указанного ниже состава без карбонат натрия (сравнительная формула А) и с различными количествами карбоната натрия, дополнительно включаемых в этот пример (как показано ниже). Периоды дезинтеграции указаны в функции давления прессования. (см. таблицу).
Из фиг. 1 и 2 следует, что при стандартном осуществлении прессования в пределах 100-140 МПа период дезинтеграции таблетки, не содержащей карбоната натрия, резко увеличивается, отражая существенное увеличение периода дезинтеграции при небольшом увеличении давления прессования. Для таблеток, содержащих карбонат натрия, неожиданно оказалось, что период дезинтеграции меньше зависит от наклона усилия прессования, что обуславливает преимущества настоящего изобретения. На фиг. 2 видно, что для таблеток с карбонатом натрия периоды дезинтеграции менее 300 с, в то время как без этого компонента период дезинтеграции более 420 с.









Изобретение относится к области фармакологии и касается нешипучей прессованной твердой дозированной формы, предназначенной для орального применения. Изобретение заключается в том, что дозированная форма включает ибупрофеновый активный ингредиент и носитель, включающий прессуемый наполнитель вместе с дезинтегратором, которая содержит ибупрофеновый лекарственный ингредиент в количестве 35 мас.% или более от дозированной формы, характеризуемая тем, что носитель дополнительно включает карбонат или бикарбонат щелочного металла. Также предлагает применение и способ получения нешипучей твердой дозированной формы предложенного средства. Изобретение обеспечивает достаточную прочность и обладает подходящими показателями, обеспечивающими быстрое выделение лекарства. 3 с. и 18 з.п. ф-лы, 2 ил., 1 табл.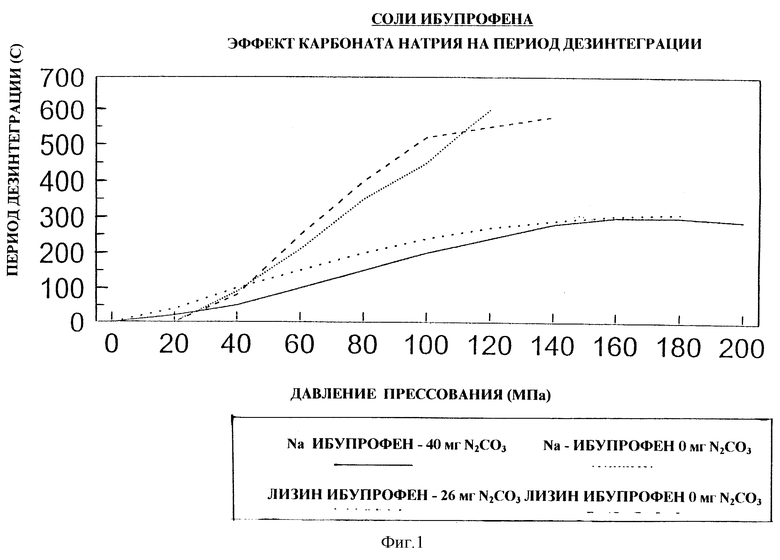
| US 4684666 А, 04.06.1987 | |||
| СПОСОБ ФЛОТАЦИИ МЕДНЫХ РУД^^^>&>&:it.^.^r '•>&••On- | 0 |
|
SU181564A1 |
| Шланговое соединение | 0 |
|
SU88A1 |
| DE 3922441 A, 17.01.1991 | |||
| US 4681897 A, 21.07.1987 | |||
| US 4788220 A, 29.11.1988. | |||

