Предпосылки изобретения
Мультикаталитическая протеиназа, или протеасома, представляет собой высококонсервативную клеточную структуру, которая ответственна за АТФ-зависимый протеолиз большинства клеточных белков (Соuх, О., Tanaka, К. and Goldberg, A. 1996 Ann. Rev. Biochem. 65, 801-847). 20S протеасома содержит каталитическое ядро комплекса и была выкристаллизована из архебактерии Thennoplasma acidophilum (Lowe, J., Stock, D., Jap, В., Zwicki, P., Bauminster, W. and Huber, R. 1995 Science 268, 533-539) и из дрожжей Saccharomyces cerevisiae (Groll, M., Ditzel, L., Lowe, J., Stock, D., Bochtler, M., Bartunik, HD and Huber, R. 1997 Nature 386, 463-471). В отличие от архебактериальной протеасомы, которая прежде всего показывает химотрипсинподобную протеолитическую активность (Dahhnann, В., Корр, F., Kuehn, L., Niedel. В., Pfeifer, G. 1989 FEBS Lett. 251, 125-131; Seemuller, E., Lupas, A., Zuw, F., Zwicki, P and Baumeister, W. FEBS Lett. 359, 173, (1995), эукариотическая протеасома осуществляет, по крайней мере, пять идентифицируемых протеолитических активностей. Три из этих активностей подобны по специфичности химотрипсину, трипсину и пептидилглютамилпептидазе. Остальные две описанные активности проявляют предпочтение к расщеплению пептидных связей на карбоксильном конце аминокислот с разветвленной цепью (ВrААР) и по отношению к пептидным связям между нейтральными аминокислотами с короткой цепью (SnAAP) (Orlowski, M. 1990 Biochemistry 29, 10289-10297).
Хотя 20S протеасома содержит протеолитическое ядро, оно не разрушает белки in vivo, если не находится в виде комплекса с 19S "кэпом" с обоих концов ее структуры, который сам проявляет многочисленные АТФ-азные активности. Данная более крупная структура известна как 26S протеасома, и она быстро разрушает белки, подлежащие разрушению, при добавлении множества молекул полипептидов массой 8,5 кДа, убиквитина (представлено в Соuх, О., Tanaka, К. and Goldberg, A. 1996 Ann. Rev. Biochem. 65, 801-847).
Большое количество функциональных групп субстрат использовалось в качестве возможных ингибиторов сериновых и тиоловых протеаз. Несколько из указанных мотивов были описаны в качестве ингибиторов протеасомы. Они включают альдегиды пептидов (Vinitsky, A., Michaud, С, Powers, J. and Orlowski, M. 1992 Biochemistry 31, 9421-9428; Tsubuki, 3., Hiroshi, К., Saito, Y. Miyashita, N., Inomata, M. and Kawashima, S. 1993 Biochem. Biophys. Res. Commun. 196, 1195-1201; Rock, К, I., Gramm, C., Rothstein, L., Clark, K., Stein, R., Dick, L.,Hwang, D. and Goldberg, A.L. (1994) Cell 78, 761-771)), N-ацетил-L-лейцинил-L-лейцинил-L-норлейциналь (ALLN) и N-ацетил-L-лейцинил-1-лейцинилметиональ (LLM), причем наиболее мощным ингибитором данного типа является N-карбобензоксил-1-L-лейцинил-L-лейцинил-L-норвалиналь (MG115). В других сообщениях описан ряд дипептидных ингибиторов, которые имеют значения IC50 в интервале от 10 до 100 нм (Iqbal, M., Chatterjee S., Kauer, J.C., Das, M., Messina, P. , Freed, В. , Biazzo, W. and Siman, R. 1995 I-Med. Chem. 38, 2276-2277). В качестве сильных ингибиторов протеасомы также был описан ряд α-кетокарбонильных и борноэфирных производных дипептидов (Iqbai, M., Chatterjee, S., Kauer, J.C., Mallamo, J.P., Messina, P.A., Reiboldt, A. and Siman, R. 1996 Bloorg. Med-Chem. Lett 6, 287-290) и эпоксикетонов (Spattenstein, A. , Leban, J.J., Huang, J.J., Reinhardt, K.R., Viveros, O.H., Sigafoos, J. and Crouch, R. 1996 Tet. Lett. 37, 1434-1346).
Другим соединением, которое проявляет специфичность при ингибировании протеасомной активности, является лактацистин (Fenteany, G., Standaert, R.F. , Lane, W. S., Choi, S., Corey, E.J. and Schreiber, S.L. 1995 Science 268, 726-731), который представляет собой метаболит Streptomyces. Эта молекула была первоначально обнаружена по ее способности стимулировать рост аксонов в клеточной линии нейробластомы (Omura, S., Matsuzaki, К., Fujimoto, Т., Kosuge, К., Furuya, Т., Fujita, S. and Nakagawa, A. 1991 J. Antibiot. 44, 117-118), а позже было продемонстрировано, что она для ингибирует пролиферацию некоторых клеточных типов (Fentcany, G., Standaert, R.F., Reichard, G.A., Corcy, E.J. and Schrcibcr, S.L. 1994 Proc. Nat'1. Acad. Sci. USA 91, 3358-3362).
Сейчас ясно установлено, что протеасома является главной экстрализосомальной протеолитической системой, включенной в протеолитические каскады, необходимые для разнообразных клеточных функций, таких как клеточное деление, процессинг антигенов и деградацию короткоживущих регуляторных белков, таких как продукты онкогенов, циклины и факторы транскрипции (Ciechanover, А. (1994) Cell 79, 13-21; Palombell, V.J., Rando, O.J., Goldberg, A.L. and Maniaus. T. 1994 Cell 78, 773-785). Например, активная форма NF-kВ представляет собой гетеродимер, содержащий субъединицы р65 и р50. Последний представлен в цитозоле в виде неактивного предшественника (р105). Протеолитический процессинг р105 с образованием р50 протекает по убиквитин-протеосомальному пути. Дополнительно, процессированные р50 и р65 содержатся в цитозоле в виде неактивного комплекса, связанного с ингибиторным белком IкВ. Воспалительные стимулы, такие как LPS, активируют NF-кВ путем инициирования сигнального пути, который ведет к разрушению IкВ. Данные сигналы также стимулируют процессинг р105 в р50. Таким образом, два протеолитических процесса, оба управляемые убиквитин-протеасомальным путем, необходимы для индуцируемой сигналом активации NF-кВ.
Наблюдение, что убиквитин-опосредованный протеасомальный протеолиз играет решающую роль в активации NF-кВ, могло бы быть использовано в клинике путем использования ингибиторов, направленных на протеасому. Аномальная активация NF-кВ, сопровождаемая стимуляцией синтеза цитокинов, наблюдалась при различных воспалительных и инфекционных заболеваниях. Активация NF-кВ также необходима для ангиогенеза и для экспрессии молекул адгезии (САМ и селектиков), таким образом, ингибиторы протеасом могут также быть полезны при лечении заболеваний, связанных с сосудистой системой.
Достоверно подтверждено, что убиквитин-протеасомальный путь является решающим для регулируемого разрушения циклинов, что обуславливает выход из митоза и позволяет клеткам входить в следующую фазу клеточного цикла (Glotzer, М. , Murray, A.W. and Kirschncr, M.W. (1991) Nature 349, 132-138). Таким образом, ингибирование разрушения циклинов путем применения протеасомных ингибиторов вызывает остановку роста. Поэтому другим возможным применением ингибиторов протеасом является их использование в лечении заболеваний, вызванных аномальным клеточным делением.
Несколько классов пептидных ингибиторов 20S протеазы были описаны в современной литературе. α-Кетоамидная группа использовалась в протеазных ингибиторах по многим причинам. В особенности, ряд α-кетокарбонильных и борноэфирных производных дипептидов (Iqbal, M., Chatterjee, S., Kauer, J.C., Mallamo, J. P. , Messina, P.A., Reiboldt, A. and Siman, R. 1996 Bioorg. Med. Chem. Lett 6, 287-290) были описаны как мощные ингибиторы 20S протеасомальной функции. Производные 3-индолпировиноградной кислоты были заявлены как фармацевтически активные соединения для лечения расстройств центральной нервной системы (De Luca, et al, WO 88/09789) посредством механизма, который регулирует содержание кинуреновой кислоты в головном мозге.
Несмотря на то, что описаны различные композиции, которые в некоторой степени ингибируют пролиферацию клеток, сохраняется потребность в более эффективных соединениях, которые ингибируют пролиферацию клеток через 20S протеасому.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Объектом данного изобретения является способ ингибирования клеточной пролиферации у млекопитающих, в котором используется терапевтически эффективное количество композиции, неизвестной ранее для ингибирования этих клеточных пролиферативных свойств.
Также объектом данного изобретения является способ лечения заболеваний, на которые можно воздействовать ингибированием протеасомальной функции.
Далее, объектом данного изобретения является способ лечения пролиферативных заболеваний, которые вызваны ингибированием протеасомальной функции.
Другим объектом данного изобретения является применение терапевтически эффективного количества композиций, описанных здесь, для подавления нарушений клеточной пролиферации у людей.
Еще одним объектом данного изобретения является применение терапевтически эффективного количества композиций, описанных здесь, для ингибирования протеасомальной функции.
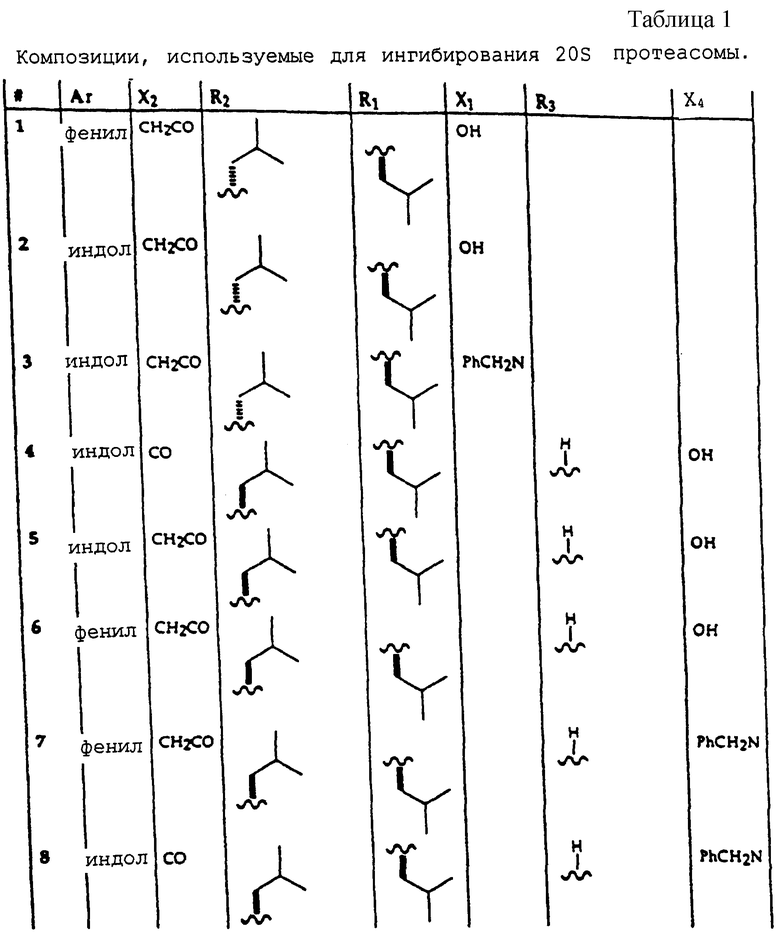
В одном воплощении, данное изобретение относится к соединению, имеющему формулу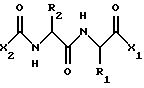
где X2 представляет собой Аr или Аr-Х3, где Х3 является -С= O или -СН2СО-, и где Аr представляет собой фенил, замещенный фенил, индол, замещенные индолы и любые другие гетероарилы; R1 и R2 каждый независимо выбран из боковых цепей известных природных α-аминокислот и не природных аминокислот, водорода, 1-10 углеродного линейного и разветвленного алкила, 1-10 углеродного линейного и разветвленного замещенного алкила, арила, замещенного арила, 1-10 углеродного линейного, разветвленного замещенного арила, алкоксиарила, 3-8 углеродного циклоалкила, гетероцикла, замещенного гетероцикла, гетероарила и замещенного гетероарила; X1 выбран из гидроксида, моноалкиламино, диалкиламино, алкоксида, арилалкоксида и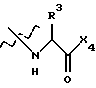
где Х4 представляет собой гидроксид, ариламино, моноалкиламино, диалкиламино, алкоксид или арилалкоксид и
R3 выбран из известных природных α-аминокислот, не природных аминокислот, водорода, 1-10 углеродного линейного и разветвленного алкила, 1-10 углеродного линейного и разветвленного замещенного алкила, арила, замещенного арила, 1-10 углеродного линейного и разветвленного замещенного арила, алкоксиарила, 3-8 углеродного циклоалкила, гетероцикла, замещенного гетероцикла, гетероарила и замещенного гетероарила.
В другом воплощении данное изобретение относится к способу ингибирования протеасомального протеазного фактора у людей, включающему введение терапевтически эффективного количества композиции, описанной выше, человеку.
В еще одном воплощении данное изобретение относится к фармацевтической композиции, содержащей соединение, описанное выше, и один или несколько фармацевтических эксципиентов.
ОПИСАНИЕ ПРОВЕДЕНИЯ ОСУЩЕСТВЛЕНИЯ
Изобретение относится к способу подавления нарушений клеточной пролиферации, инфекционных заболеваний и иммунологических заболеваний у млекопитающих, в особенности у людей, путем использования соединений следующей общей формулы: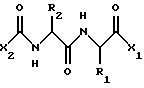
где Х2 представляет собой Аr или Аr-Х3, где Х3 включает в себя -С=О, -СН2СО- или (СН2)n, где n=0-2, и где Аr представляет собой фенил, замещенный фенил, индол, замещенные индолы и любой другой гетероарил;
R1 и R2 независимо выбраны из боковых цепей известных природных α-аминокислот и не природных аминокислот, водорода, 1-10 углеродного линейного и разветвленного алкила, 1-10 углеродного линейного и разветвленного замещенного алкила, арила и замещенного арила, 1-10 углеродного линейного и разветвленного замещенного арила, алкоксиарила, 3-8 углеродного циклоалкила, гетероцикла и замещенного гетероцикла, или гетероарила и замещенного гетероарила. R2 представляет собой предпочтительно биарил или бифенил. R1 представляет собой предпочтительно изобутил. X1 выбран из -ОН, моно- или диалкиламино, алкоксида, арилалкоксида и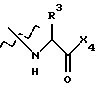
где Х4 представляет собой -ОН, ариламино, моно или диалкиламино, алкоксид или арилалкоксид, предпочтительно -ОН;
R3 выбран из боковых цепей известных природных α-аминокислот и не природных аминокислот, водорода, 1-10 углеродных линейных алкильных и разветвленных алкильных заместителей, 1-10 углеродного линейного и разветвленного замещенного алкила, арила и замещенного арила, 1-10 углеродного линейного, разветвленного замещенного арила, алкоксиарила, 3-8 углеродного циклоалкила, гетероцикла и замещенного гетероцикла, или гетероарила и замещенного гетероарила;
R3 предпочтительно является СО2Н, CH2CO2H, (CH2)2CО2H, Аrg, Lys, Asn, Gln, Asp, Glu, Phe и Nle.
Нижеследующие перечисления представляют определения для некоторых терминов, используемых здесь.
"Галоген" относится к атомам фтора, брома, хлора и йода.
"Гидроксил" относится к группе -ОН.
"Тиол" или "меркапто" относятся к группе -SH.
"Алкил" относится к циклической, разветвленной или прямой цепи алкильной группы с углеродными атомами от одного до десяти. Этот термин в дальнейшем иллюстрируется такими группами как метил, этил, н-пропил, изо-пропил, н-бутил, трет-бутил, изо-бутил (или 3-метилпропил), циклопропилметил, изо-амил, н-амил, н-гексил и тому подобное.
"Замещенный алкил" относится к низшим алкилам, таким как только что были описаны, включая одну или несколько групп, таких как гидроксил, тиол, алкилтиол, галоген, алкокси, амино, амидо, карбоксил, циклоалкил, замещенный циклоалкил, гетероцикл, циклогетероалкил, замещенный циклогетероалкил, ацил, карбоксил, арил, замещенный арил, арилокси, гетарил, замещенный гетарил, аралкил, гетероаралкил, алкилалкенил, алкилалкинил, алкилциклоалкил, алкилциклогетероалкил, циано. Эти группы могут быть присоединены к любому углеродному атому низшей алкильной группы.
"Арилокси" обозначает группы -ОАr, где Аr представляет собой арил, замещенный арил, гетероарил или замещенную гетероарильную группу, как определено ниже.
"Амино" обозначает группу NRR', где R и R' могут независимо быть водородом, низшим алкилом, замещенным низшим алкилом, арилом, замещенный арилом, гетарилом или замещенным гетарилом, как определено ниже, или арилом.
"Амидо" обозначает группу -C(O)NRR', где R и R' могут независимо быть водородом, низшим алкилом, замещенным низшим алкилом, арилом, замещенным арилом, гетарилом, замещенным гетарилом, как определено ниже.
"Карбоксил" обозначает группу -С(О)OR, где R может независимо быть водородом, низшим алкилом, замещенным низшим алкилом, арилом, замещенным арилом, гетарилом, замещенным гетарилом и тому подобное, как определено.
"Арил" или "Аr" относят к ароматической карбоциклической группе, имеющей, по крайней мере, одно ароматическое кольцо (например, фенил или бифенил) или множественные конденсированные кольца, в которых, по крайней мере, одно кольцо является ароматическим (например, 1,2,3,4-тетрагидронафтил, нафтил, антрил или фенантрил).
"Замещенный арил" относится к арилу, необязательно замещенному одной или несколькими функциональными группами, например галогеном, низшим алкилом, низшим алкокси, алкилтио, ацетиленом, амино, амидо, карбоксилом, гидроксилом, арилом, арилокси, гетероциклом, гетарилом, замещенным гетарилом, нитро, циано, тиолом, сульфамидо и тому подобное.
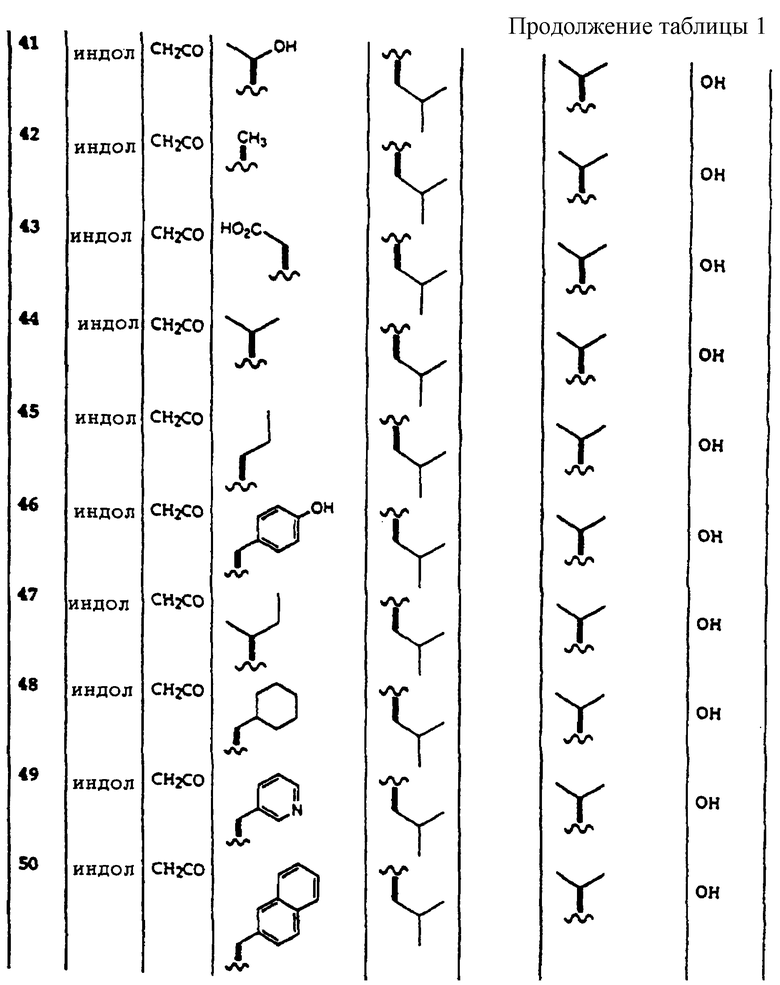
"Гетероцикл" относят к насыщенной, ненасыщенной или ароматической карбоциклической группе, имеющей единственное кольцо (например, морфолино, пиридил или фурил) или множественные конденсированные кольца (например, нафтпиридил, хиноксалил, хинолинил, индолизинил или бензо[b]тиенил) и имеющие в кольце, по крайней мере, один гетероатом, такой как N, О или S, который может быть независимо незамещенным или замещенным, например, галогеном, низшим алкилом, низшим алкокси, алкилтио, ацетиленом, амино, амидо, карбоксилом, гидроксилом, арилом, арилокси, гетероциклом, гетарилом, замещенным гетарилом, нитро, циано, тиолом, сульфамидо и тому подобное.
"Гетероарил" или "гетар" относится к гетероциклу, в котором, по крайней мере, одно гетероциклическое ядро является ароматическим. Предпочтительными гетероарилами являются фенил, замещенный фенил, индол и замещенные индолы.
"Замещенный гетероарил" относится к гетероциклу, необязательно моно- или полизамещенному одним или несколькими функциональными группами, например галогеном, низшим алкилом, низшим алкокси, алкилтио, ацетиленом, амино, амидо, карбоксилом, гидроксилом, арилом, арилокси, гетероциклом, гетарилом, замещенным гетарилом, нитро, циано, тиолом, сульфамидо и тому подобное.
"Циклоалкил" относится к дивалентной циклической или полициклической алкильной группе, содержащей от 3 до 15 атомов углерода.
"Замещенный циклоалкил" относится к циклоалкильной группе, включающей один или несколько заместителей, например галоген, низший алкил, замещенный низший алкил, алкокси, алкилтио, ацетилен, арил, арилокси, гетероцикл, гетарил, замещенный гетарил, нитро, циано, тиол, сульфамидо и тому подобное.
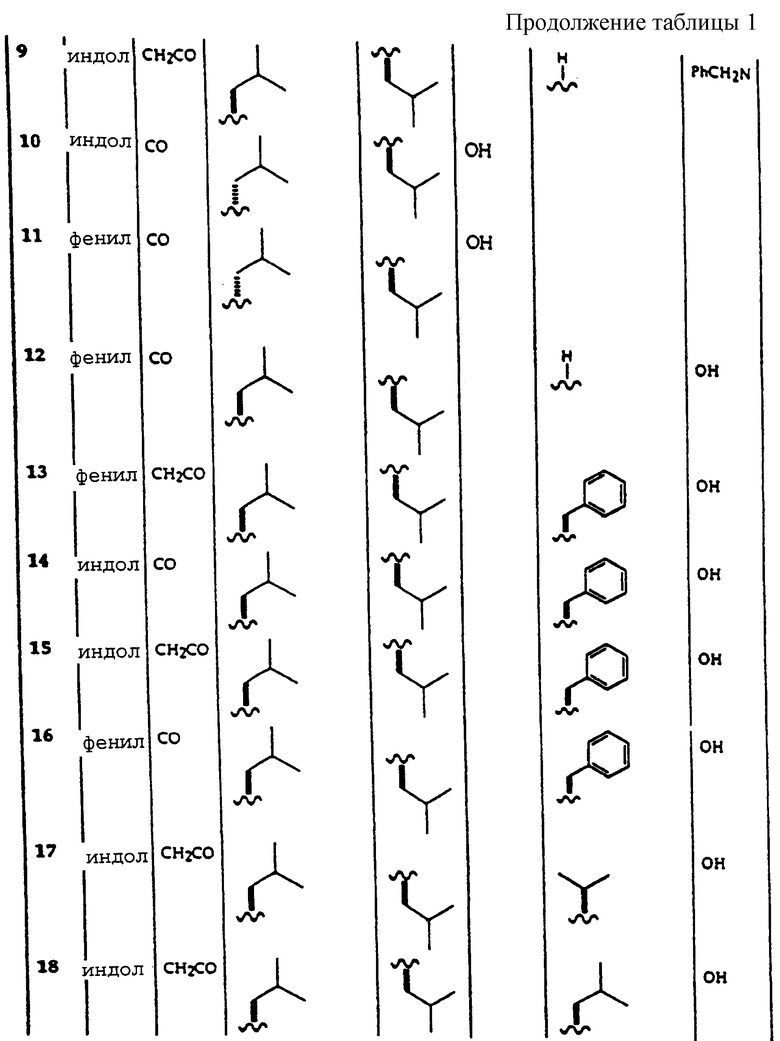
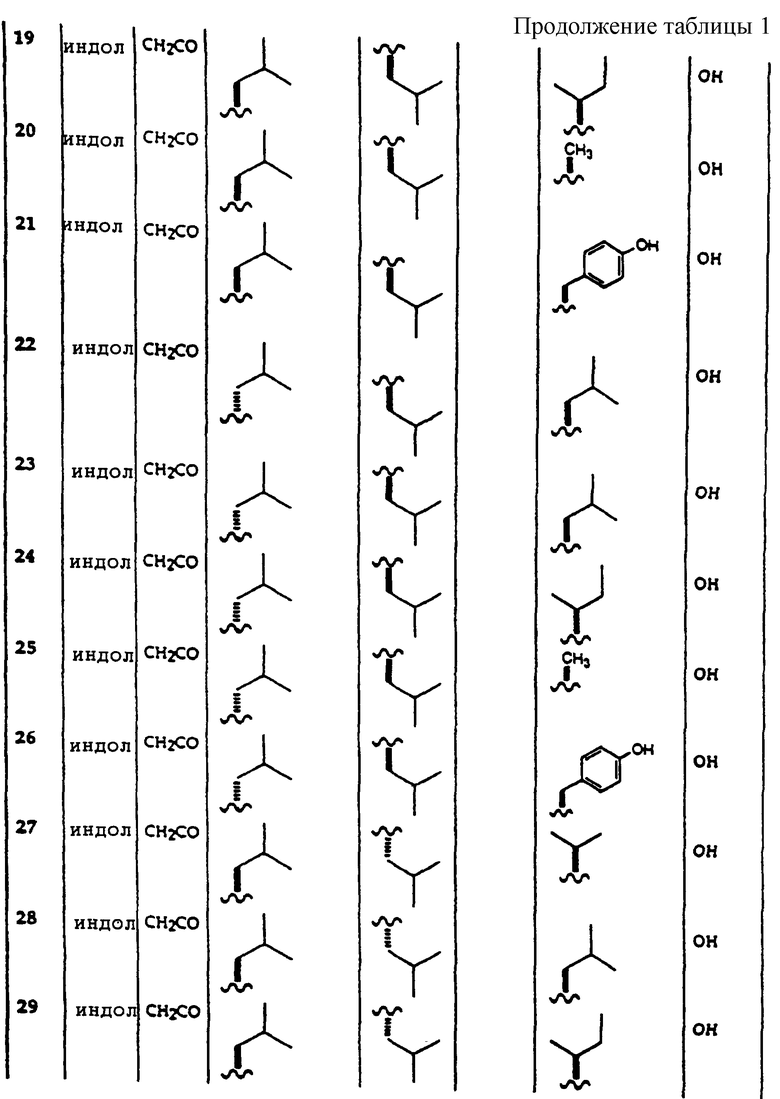
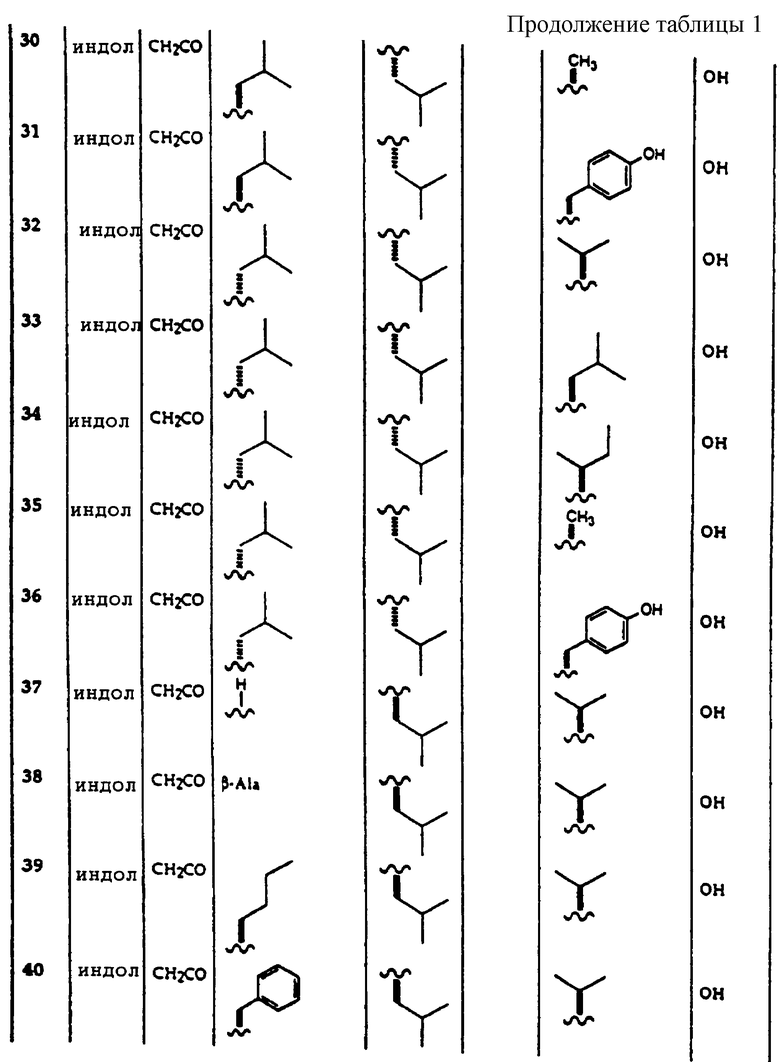
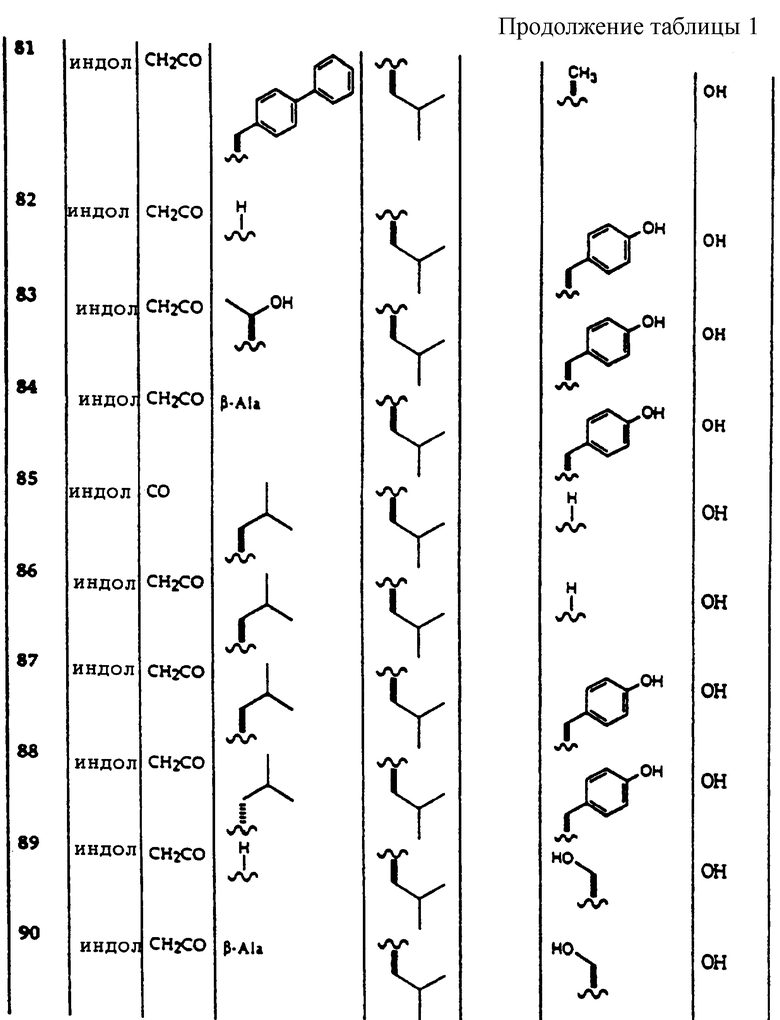
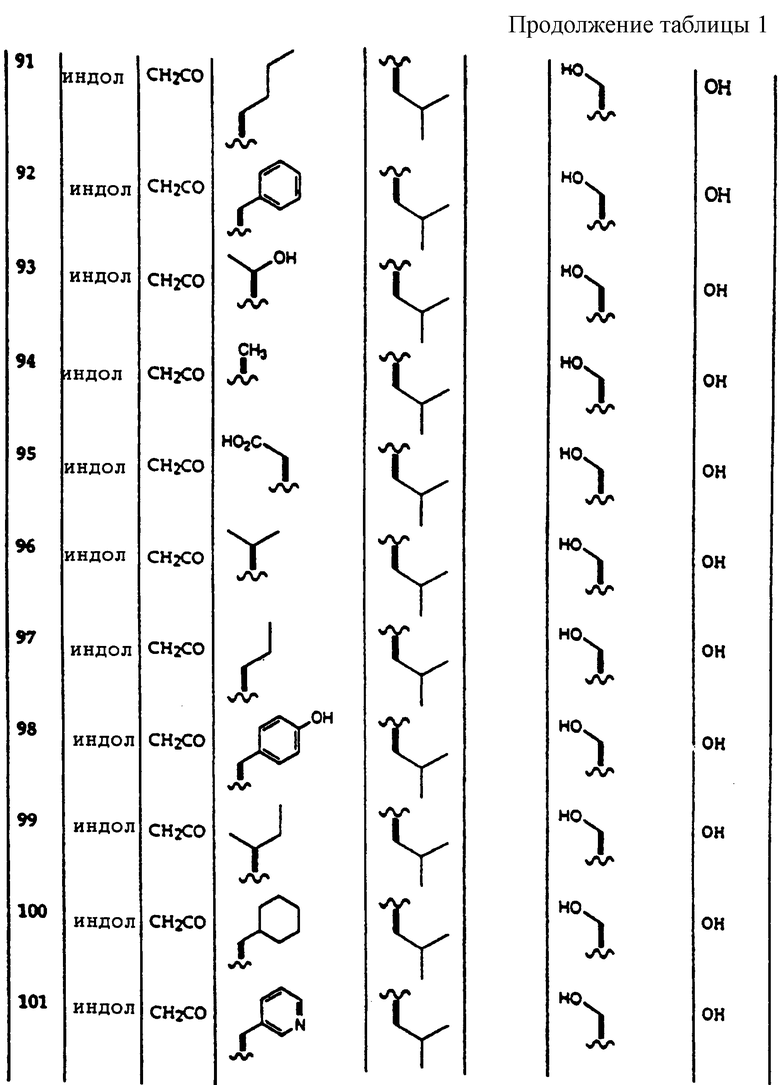
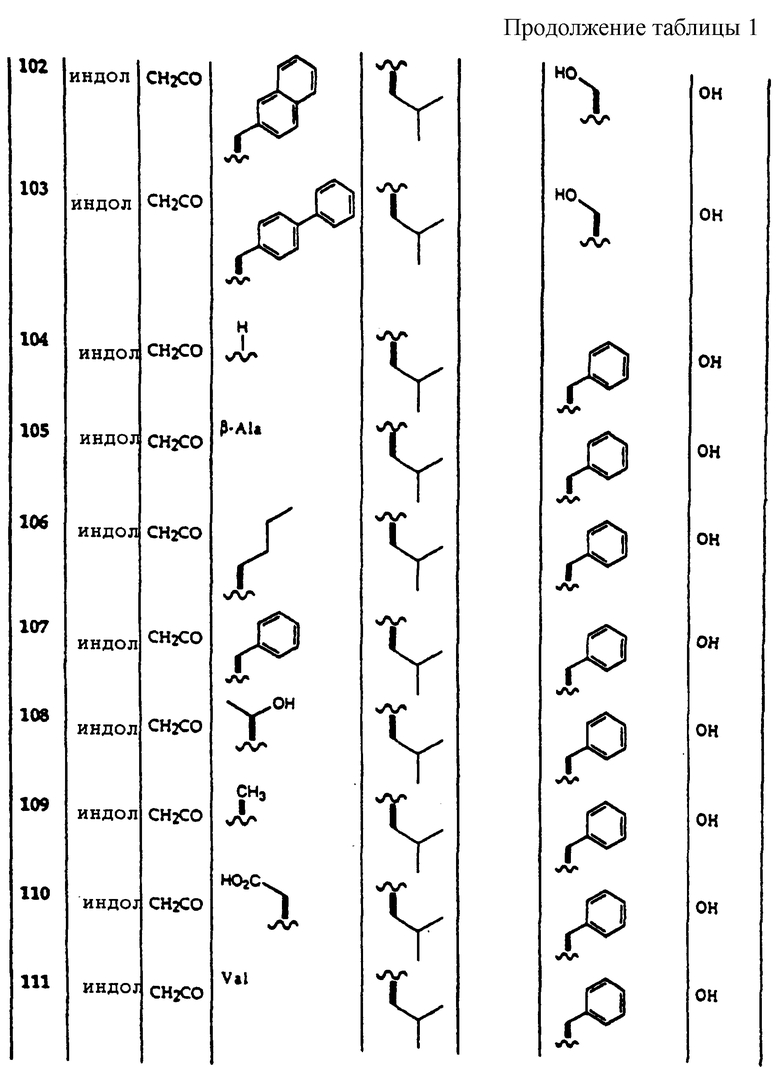
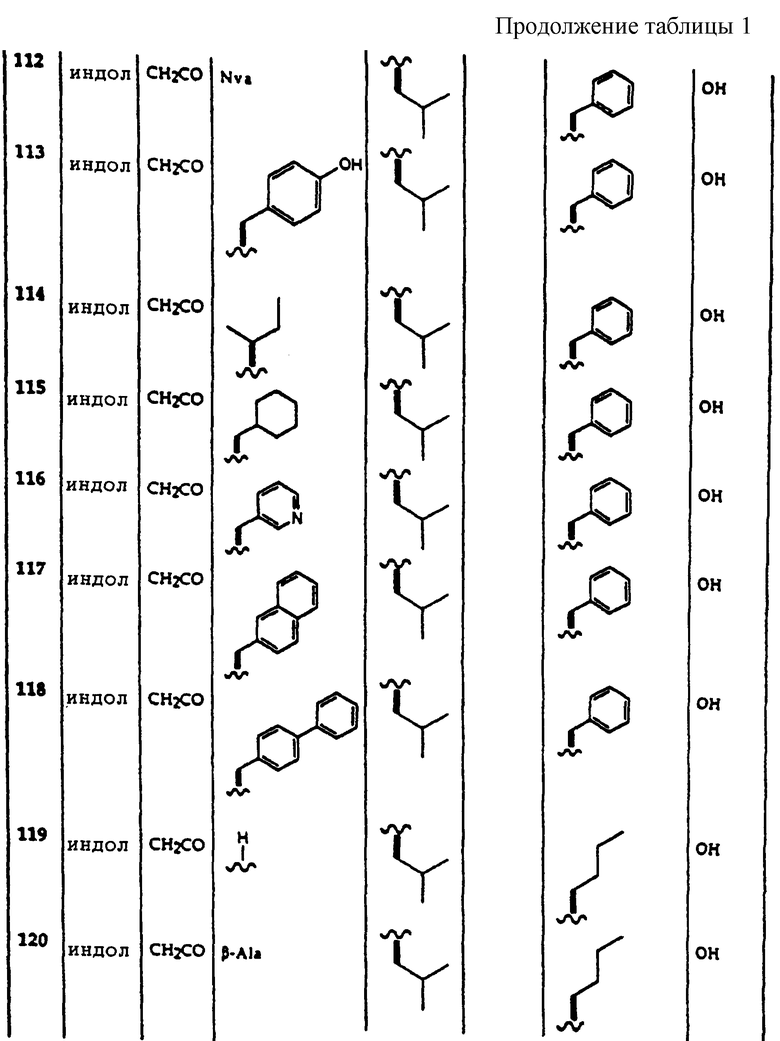
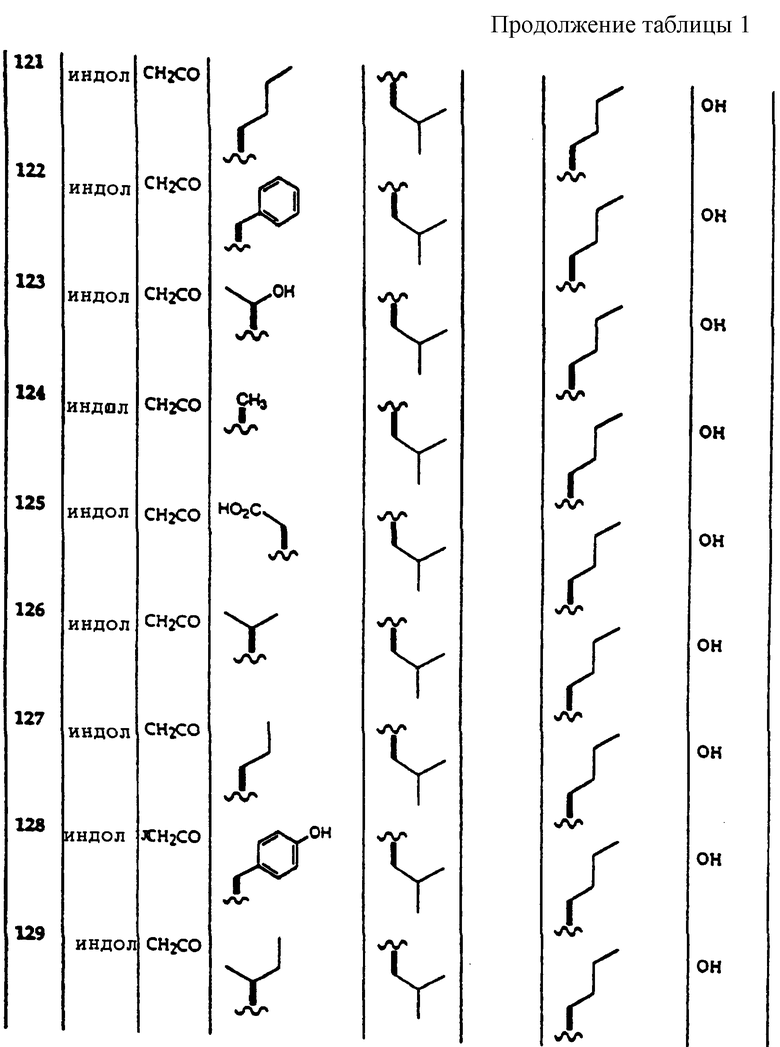
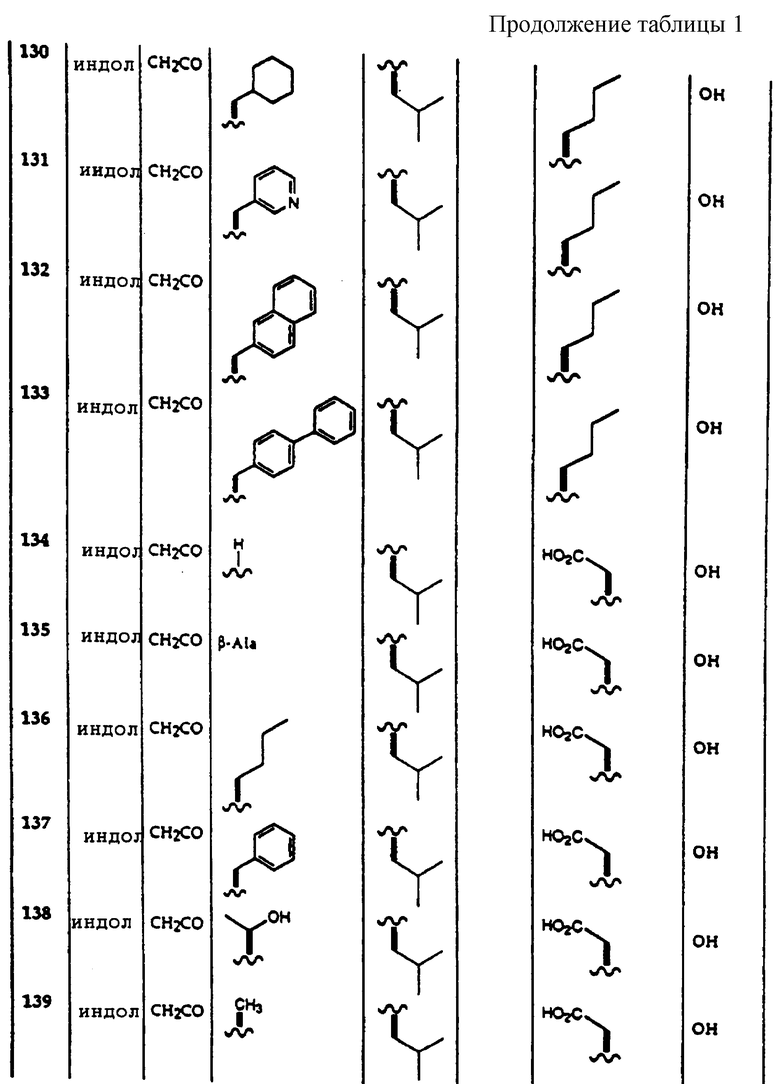
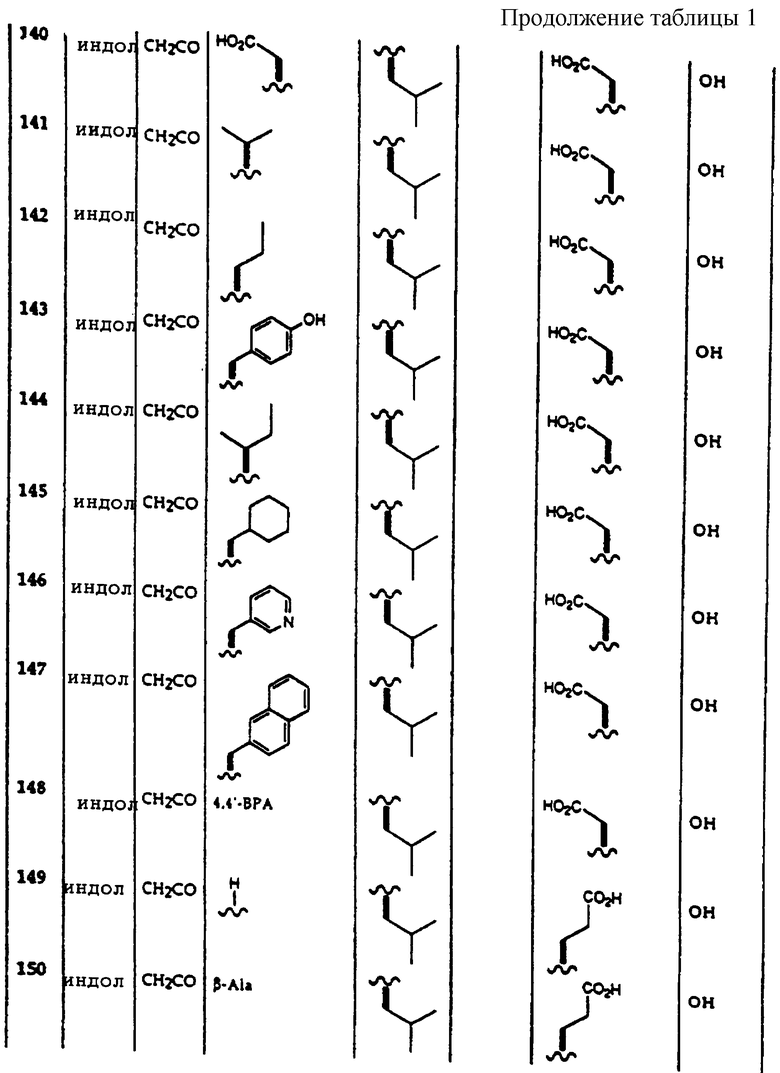
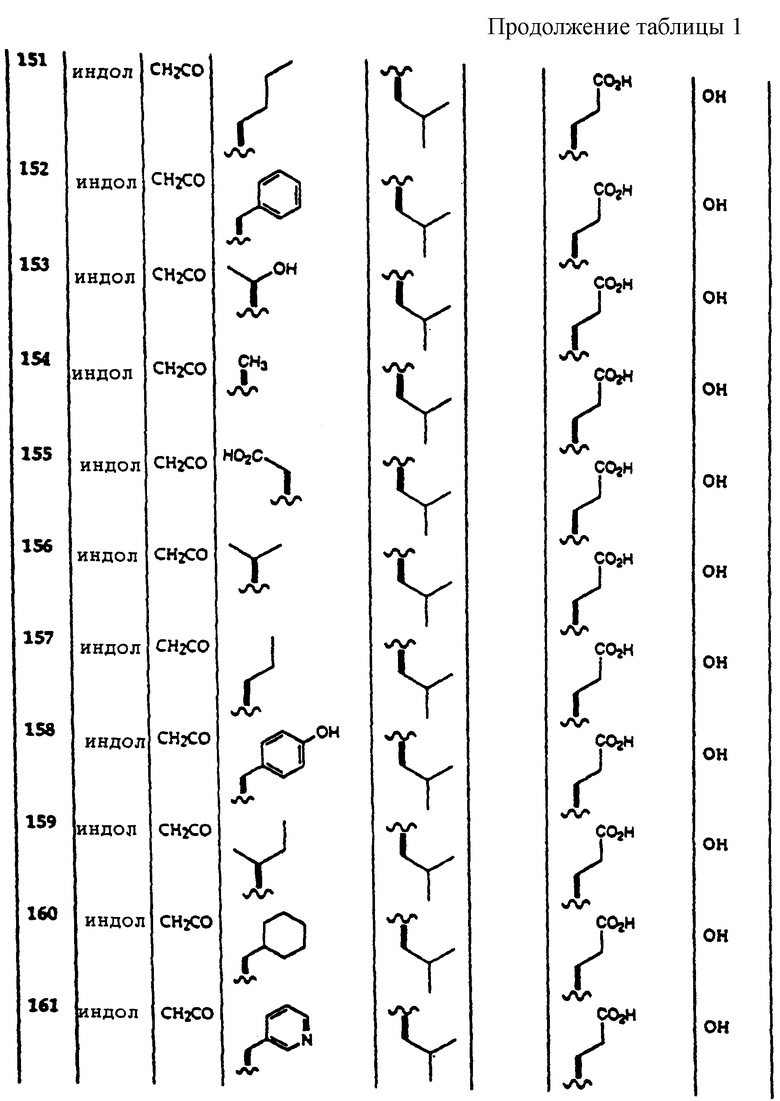
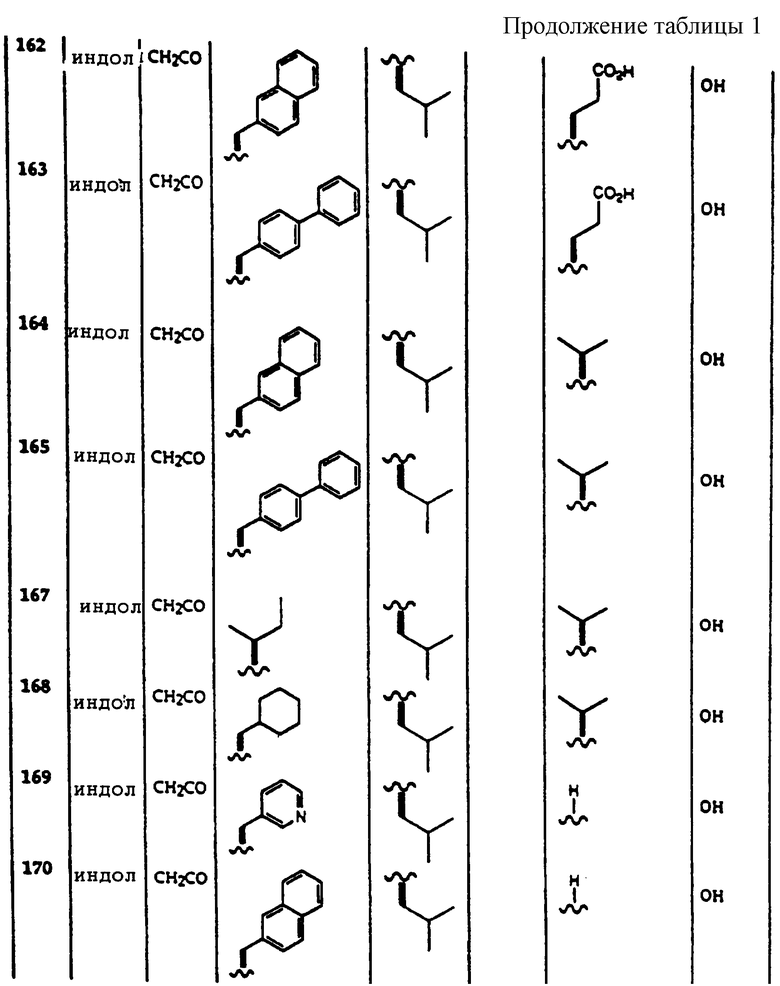
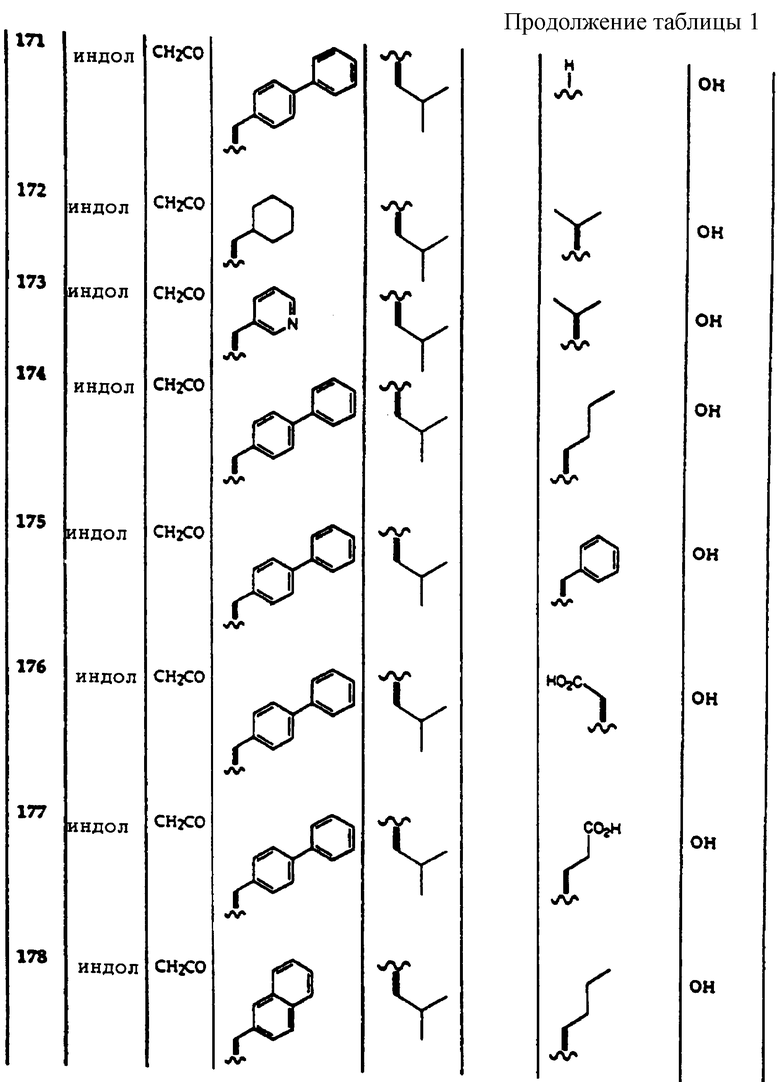
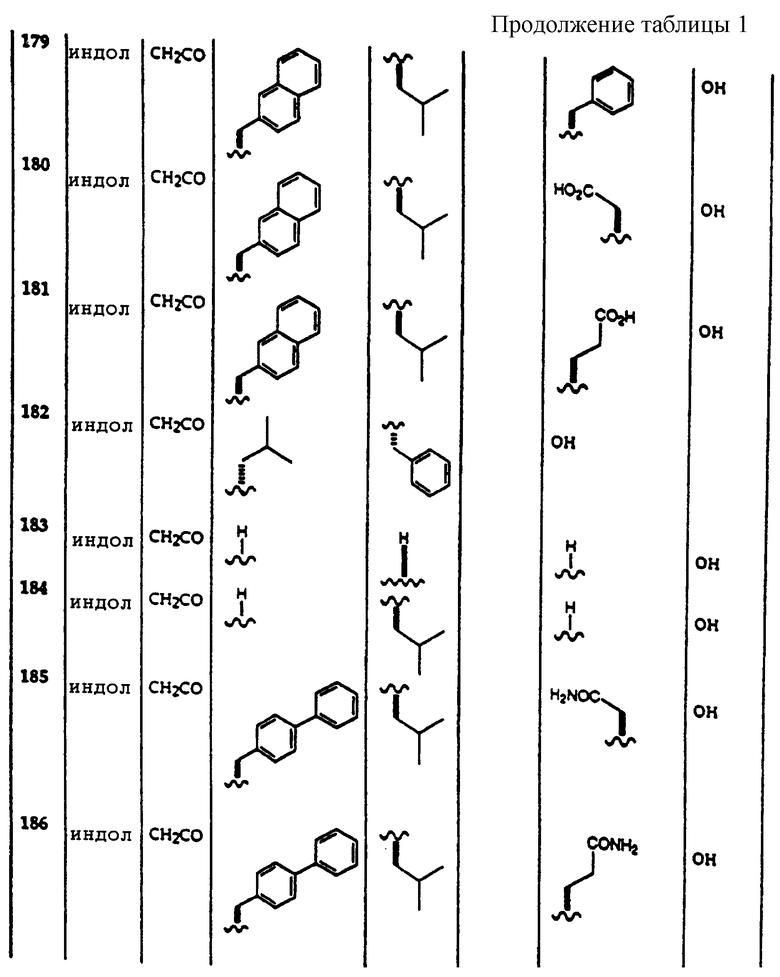
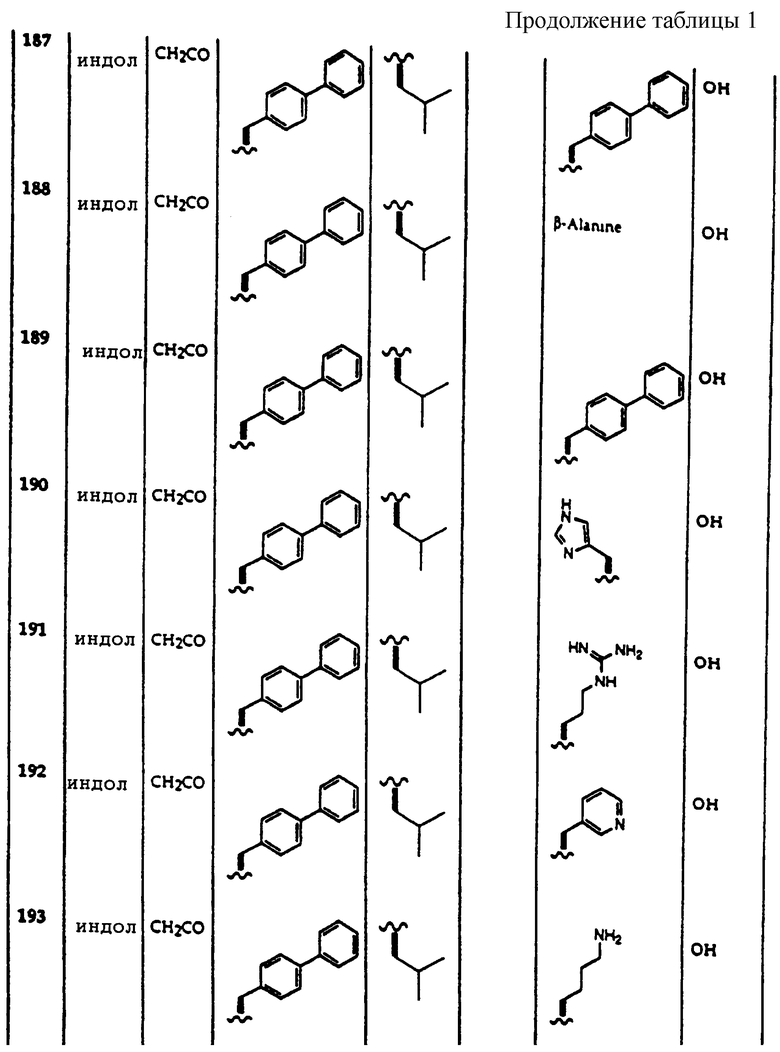
Примеры соединений, которые могут быть использованы в терапевтических методах по данному изобретению, в частности в качестве ингибиторов протеасомальной функции, представлены в таблице 1.
Соединения, описанные выше, могут использоваться для лечения заболеваний и расстройств, опосредованных 20S протеасомой, таких как антипролиферативные заболевания, рак, воспаление. Предпочтительно композиции по данному изобретению могут использоваться для лечения антипролиферативных заболеваний и воспаления. Более предпочтительно соединения по данному изобретению используются для лечения воспалительных заболеваний.
Соединения по настоящему изобретению могут использоваться для лечения заболеваний, опосредованных 20S протеасомой у млекопитающих.
Соединения по данному изобретению могут вводиться млекопитающим как для и профилактики, так и для лечения, любым способом введения, который может доставить к 20S протеасоме, по крайней мере, одно соединение по данному изобретению. Неограничивающие примеры применяемых способов введения включают пероральный, парентеральный, кожный, чрезкожный, ректальный, назальный или любой другой, подходящий для введения фармацевтической композиции способ, который известен специалистам в данной области.
Композиции по данному изобретению могут вводиться в виде подходящих фармацевтических дозированных форм. Фармацевтическая дозированная форма в значительной степени зависит от используемого способа введения. Термин фармацевтическая дозированная форма относится к объектам, таким как таблетки, капсулы, жидкости и порошки, включающие в себя ингибиторы 20S протеасомы данного изобретения, сами по себе или в присутствии одного или нескольких фармацевтических наполнителей. Выбор добавок, таких как эксципиенты и адъюванты, также в значительной степени будет зависеть от выбранного способа введения. Специалистам в фармацевтической области известно широкое разнообразие препаратов и носителей для введения композиций по данному изобретению.
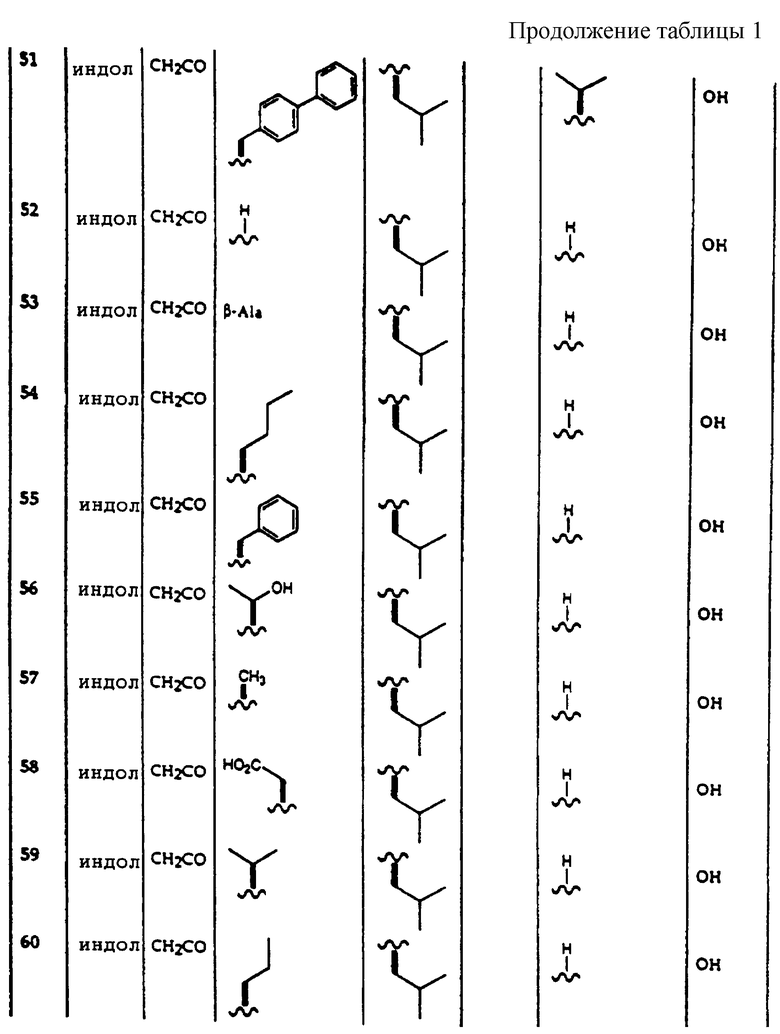
Способ введения, выбранный для соединений по данному изобретению, определяется конечной формой и композицией фармацевтической дозированной формы, содержащей ингибиторы 20S протеасомы по данному изобретению. Например, внутреннее введение соединений по данному изобретению осуществляется перорально в виде порошков, таблеток, капсул, паст, напитков, гранул или растворов, суспензий и эмульсий, которые могут быть введены перорально или в виде болюса, в содержащем лекарственное вещество пищевом продукте или в питьевой воде. Внутреннее введение может также быть выполнено, используя препарат с задерживаемым высвобождением, содержащий добавки, такие как поверхностно-активные вещества или покрытые крахмалом капсулы, или используя составы быстрого высвобождения, такие как высушенные замораживанием быстро растворяемые таблетки. Кожное введение осуществляется, например, в виде трансдермальных пластырей, распылением или "орошением" или "опрыскиванием". Парентеральное введение осуществляют, например, в виде инъекции (внутримышечно, подкожно, внутривенно, внутрибрюшинно) или путем имплантации.
Подходящие фармацевтические дозированные формы, содержащие ингибиторы 20S протеасомы по данному изобретению, включают, но не ограничиваются, растворы, такие как растворы для инъекций, пероральные растворы, концентраты для перорального приема после разведения, растворы для использования на коже или в полостях тела, препараты для "орошения" или "опрыскивания", гели; эмульсии и суспензии для перорального или кожного введения и для инъекций; полутвердые препараты; препараты, в которых активное соединение входит в основу для крема или в основу эмульсия масло-в-воде или вода-в-масле; твердые препараты, такие как порошки, предварительно приготовленные смеси или концентраты, гранулы, пилюли, таблетки, болюсы, капсулы; аэрозоли и препараты для ингаляций, и изделие, имеющее определенную форму, содержащее активное вещество.
Фармацевтические дозированные формы, которые представляют собой растворы, могут быть введены путем внутривенной, внутримышечной и подкожной инъекции. Растворы для инъекции получают путем растворения активного соединения в подходящем растворителе и, если приемлемо, путем добавления адъювантов, таких как солюбилизаторы, кислоты, основания, буферные соли, антиоксиданты и консерванты. Растворы стерильно отфильтрованы и дегазированы.
Альтернативно растворы, включающие композиции по данному изобретению, могут вводиться перорально. Концентраты композиций по данному изобретению предпочтительно вводят перорально только после разбавления концентрата до применяемой концентрации. Пероральные растворы и концентраты получают, как описано выше для случая растворов для инъекции. Растворы для использования на коже наносят по каплям, втирают, натирают, разбрызгивают или распыляют. Эти растворы получают, как описано выше для случая растворов для инъекции.
Гели наносят на кожу или вводят в полости тела. Гели получают путем обработки растворов, которые были получены, как описано для случая растворов для инъекции, с таким количеством загустителя, чтобы образовывалось прозрачное вещество сметаноподобной консистенции, или путем любых других методов, известных специалистам в данной области.
Растворы для "орошения" и "опрыскивания" выливают или разбрызгивают на ограниченные области кожи, при этом активное соединение проникает через кожу и действует системно. Растворы для "орошения" и "опрыскивания" получают путем растворения, суспендирования или эмульгирования активного соединения в подходящих растворителях или смесях растворителей, которые являются толерантными к коже. Если приемлемо, добавляют другие добавки, такие как красители, ускорители резорбции, антиоксиданты, стабилизаторы против действия света и вещества, повышающие клейкость.
Эмульсии могут вводиться перорально, чрезкожно или в виде инъекций. Эмульсии могут быть либо типа вода в масле, либо типа масло в воде. Их получают путем растворения ингибиторов 20S протеасомы либо в гидрофобной, либо в гидрофильной фазе и гомогенизируя фазу с растворителем противоположной фазы при помощи подходящих адъювантов, таких как эмульгаторы, красители, ускорители резорбции, консерванты, антиоксиданты, стабилизаторы против действия света и вещества, увеличивающие вязкость.
Суспензии могут вводиться перорально, чрезкожно или в виде инъекций. Их получают путем суспендирования активного соединения в жидкости, если приемлемо, с добавлением дополнительных адъювантов, таких как увлажняющие агенты, красители, ускорители резорбции, консерванты, антиоксиданты и стабилизаторы против действия света.
Фармацевтические композиции по данному изобретению могут включать одну или несколько добавок в виде фармацевтически приемлемых добавок. Применяемые добавки включают растворители, солюбилизаторы, консерванты, вещества, увеличивающие вязкость, увлажняющие агенты, красители, ускорители резорбции, антиоксиданты, стабилизаторы против действия света, вещества, повышающие клейкость, вязкость, наполнители, вкусовые агенты, смазывающие агенты и любые другие добавки для фармацевтической композиции, известные специалистам в данной области.
Добавка может быть растворителем, таким как вода, спирты, такие как этанол, бутанол, бензиловый спирт, глицерин, пропиленгликоль, полиэтиленгликоль, N-метилпирролидон, алканолы, глицерин, ароматические спирты, такие как бензиловый спирт, фенилэтанол, феноксиэтанол, сложные эфиры, такие как этилацетат, бутилацетат, бензилбензоат, простые эфиры, такие как алкиловые эфиры алкиленгликоля, такие как монометиловый эфир дипропиленгликоля, монобутиловый эфир диэтиленгликоля, кетоны, такие как ацетон, метилэтилкетон, ароматические и/или алифатические углеводороды, растительные или синтетические масла, ДМФ, диметилацетамид, N-метилпирролидон, 2,2-диметил-4-оксиметил-1,3-диоксолан.
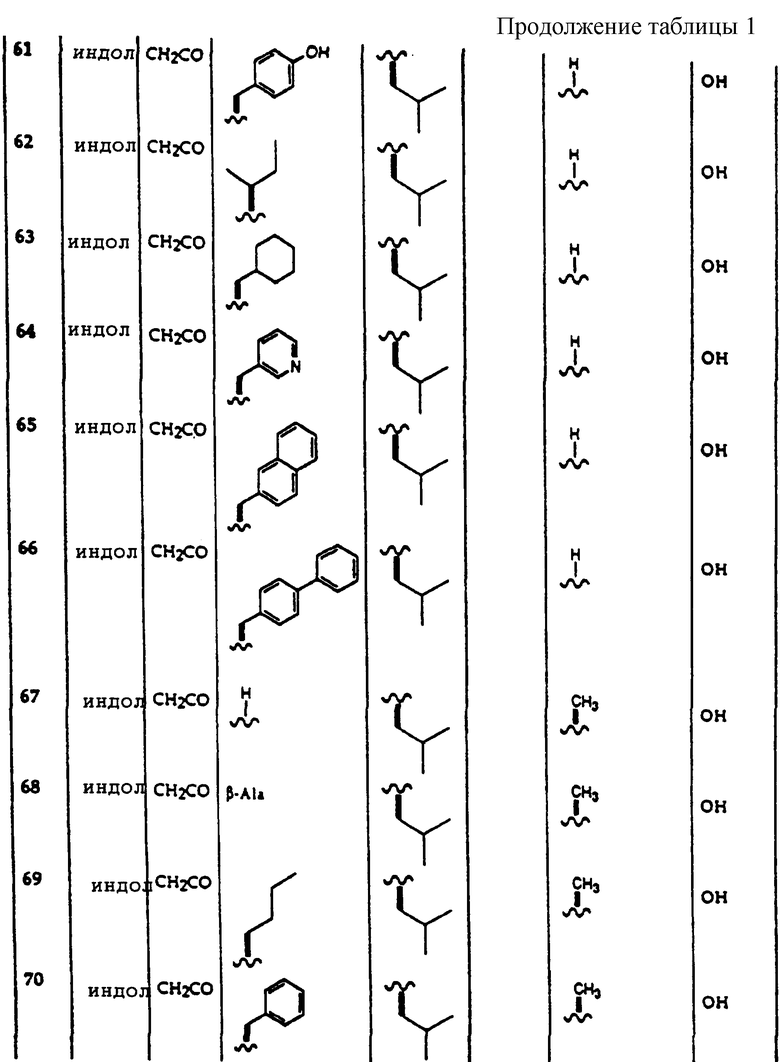
Следующие добавки могут быть использованы в качестве солюбилизаторов композиций по данному изобретению: растворители, которые улучшают растворимость активного соединения в основном растворителе или которые предотвращают его осаждение. Примерами являются поливинилпирролидон, полиоксиэтилированное касторовое масло, полиоксиэтилированные сорбитановые эфиры.
Применяемыми консервантами являются, например, бензиловый спирт, трихлорбутанол, п-оксибензойные эфиры и н-бутанол.
Применяемые загустители включают неорганические загустители, такие как бентонит, коллоидный силикагель, моностеарат алюминия, органические сгустители, такие как производные целлюлозы, поливиниловые спирты и их сополимеры, акрилаты и метакрилаты.
Другие жидкости, которые могут быть использованы в фармацевтических дозированных формах по данному изобретению, представляют собой, например, гомогенные растворители, смеси растворителей и увлажняющие агенты, которые являются обычно поверхностно-активными веществами.
Применяемые красители представляют собой все красители, которые являются нетоксичными и которые могут быть растворены или суспендированы.
Применяемые ускорители резорбции представляют собой ДМСО, текучие масла, такие как изопропилмиристат, пеларгонат дипропиленгликоля, силиконовые масла, эфиры жирных кислот, триглицериды, жирные спирты.
Применяемые антиоксиданты представляют собой сульфиты или метабисульфиты, такие как метабисульфит калия, аскорбиновую кислоту, бутилгидрокситолуол, бутилгидроксианизол, токоферол.
Применяемым стабилизатором против действия света является новантисоловая кислота.
Применяемые вещества, повышающие клейкость, включают производные целлюлозы, производные крахмала, полиакрилаты, природные полимеры, такие как альгинаты, желатин.
Применяемые эмульгаторы включают неионные поверхностно-активные вещества, такие как полиоксиэтилированное касторовое масло, полиоксиэтилированный сорбитан моноолеат, сорбитан моностеарат, моностеарат глицерина, полиоксиэтилстеарат, алкилфенольные эфиры полигликоля; амфолитические поверхностно-активные вещества, такие как Di-Na N-лаурил-бета-иминодипропионат или лецитин; анионные поверхностно-активные вещества, такие как Na-лаурилсульфат, сульфаты эфиров жирных спиртов, моноэтаноламиновая соль простого эфира моно/диалкилполигликоля сложных ортофосфорных эфиров; катионные поверхностно-активные вещества, такие как цетилтриметиламмонийхлорид.
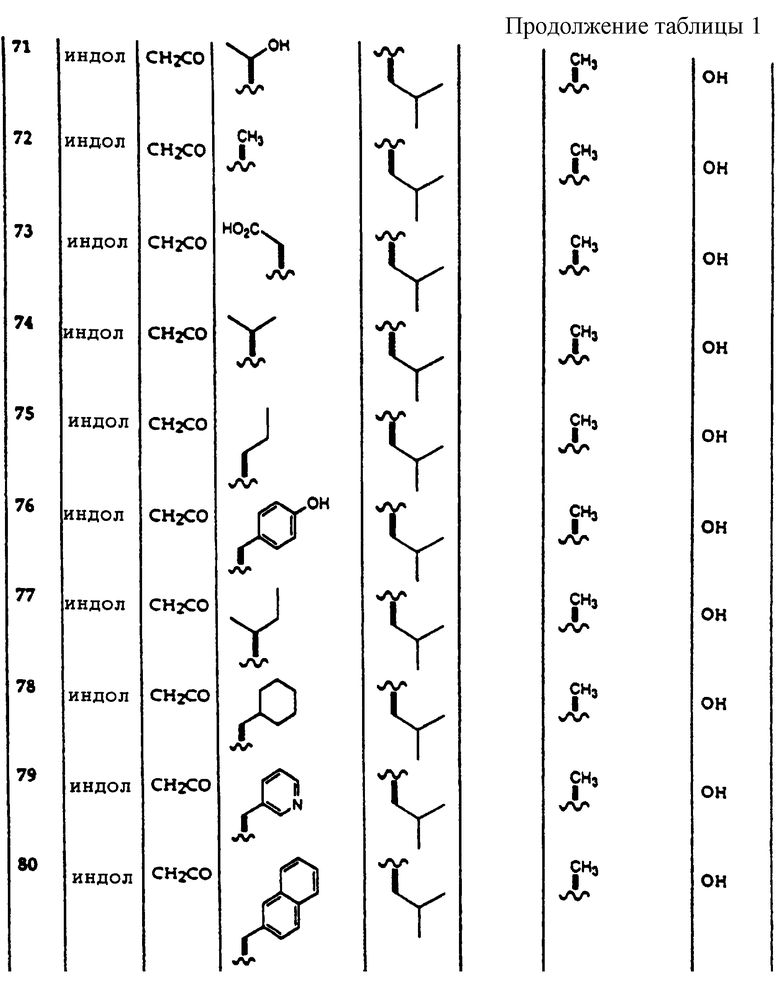
Применяемые вещества, увеличивающие вязкость, и вещества, которые стабилизируют терапевтическую эмульсию, включают карбоксиметилцеллюлозу, метилцеллюлозу и другую целлюлозу, производные крахмала, полиакрилаты, альгинаты, желатин, аравийскую камедь, поливинилпирролидон, поливиниловый спирт, сополимеры метилвинилового эфира и малеинового ангидрида, полиэтиленгликоли, воски, коллоидный силикагель или смеси упомянутых веществ.
Для получения твердых фармацевтических дозированных форм активное соединение смешивают с подходящими добавками, если целесообразно, с добавлением адъювантов и смесь составляют, как желательно. Примеры физиологически приемлемых твердых инертных добавок включают хлористый натрий, карбонаты, такие как карбонат кальция, гидрокарбонаты, оксиды алюминия, силикагели, глины, осажденный или коллоидный диоксид кремния и фосфаты. Примеры твердых органических добавок включают сахар, целлюлозу, пищевые продукты, такие как сухое молоко, животную муку, зерновую муку и грубую зерновую муку и крахмал. Другие подходящие добавки включают смазочные агенты и придающие свойство скольжения агенты, такие как стеарат магния, стеариновая кислота, тальк, бентониты; разрыхлители, такие как крахмал или поперечносвязанный поливинилпирролидон; связующие агенты, такие как крахмал, желатин или линейный поливинилпирролидон; сухие связующие агенты, такие как микрокристаллическая целлюлоза.
В фармацевтических дозированных формах, описанных здесь, активные соединения могут присутствовать в виде смеси, по крайней мере, с одним другим ингибитором 20S протеасомы. Альтернативно или кроме того, фармацевтические дозированные формы по изобретению, в дополнение к, по крайней мере, одному ингибитору 20S протеасомы, могут включать любое фармацевтическое соединение, которое способно лечить любое известное заболевание или расстройство, в тех случаях, когда введение их вместе не создает никаких недопустимых неблагоприятных эффектов.
Методы лечения заболеваний и расстройств, вызванных 20S протеасомой, включают введение эффективного количества выбранного соединения или его сочетаний, предпочтительно сосредоточенного в фармацевтической дозированной форме. Готовые к использованию фармацевтические дозированные формы по данному изобретению содержат активное соединение в концентрациях от 10 м.д. до 20 процентов по массе и предпочтительно от 0,1 до 10 процентов по массе. Фармацевтические дозированные формы по данному изобретению, которые разбавляют перед введением, предпочтительно содержат активное соединение в концентрациях от 0,5 до 90 процентов по массе и предпочтительно от 5 до 50 процентов по массе. Обычно, доказано, что для достижения эффективных результатов благоприятно вводить количества от приблизительно 0,01 мг до приблизительно 100 мг активного вещества на 1 кг веса тела в сутки.
Количество и частота введения фармацевтической дозированной формы, включающей ингибиторы 20S протеасомы по данному изобретению, будут без труда определены любым специалистом в данной области в зависимости от, среди других факторов, пути введения, возраста и состояния пациента. Эти дозированные единицы могут вводиться один или десять раз ежедневно при остром или хроническом заболевании. Никакие недопустимые токсикологические эффекты не ожидаются, когда соединения по изобретению вводятся в соответствии с существующим изобретением.
Фармацевтическую дозированную форму, содержащую ингибиторы 20S протеасомы по данному изобретению, получают в соответствии с обычными методами фармацевтики, включающими измельчение, смешивание, гранулирование и прессование при необходимости в случае таблетированных форм или измельчение, смешивание и наполнение в случае твердых желатиновых капсульных форм. Когда используется жидкая добавка, лекарственное средство будет в виде сиропа, эликсира, эмульсии или водной или неводной суспензии. Такой жидкий лекарственный препарат может вводиться непосредственно р.о. или заключен в мягкую желатиновую капсулу.
Несмотря на то, что композиции, описанные здесь, могут вводиться как описано выше, предпочтительно, чтобы способ по данному изобретению осуществлялся путем введения соединения, описанного здесь, перорально. Когда выбирается пероральный путь введения, то необходимо ввести большее количество реагирующих агентов для того же эффекта, которое достигается меньшим количеством, введенным, например, парентерально. В соответствии с широкой клинической практикой предпочтительно введение соединения по данному методу в той концентрации, которая дала бы эффективные терапевтические результаты без проявления любых вредных побочных эффектов.
Композиции по данному изобретения также имеют нетерапевтическое использование. Композиции по данному изобретению могут использоваться в качестве аналитических стандартов для исследования ингибитора 20S протеасомы.
Пример 1.
Соединения, используемые в терапевтическом методе по данному изобретению, получают обычными методами органической химии. Ссылки, к которым можно обратиться за справкой при описании синтеза этих соединений в данной области, включают Bodansky's "The Practice of Peptide Synthesis," Springer-Verlag, First Edition, 1984; "Protective Groups in Organic Synthesis", Second Edition, John Wiley and Sons, New York, 1991. Все реакции присоединения пептидов выполняются при комнатной температуре при слабом и постоянном перемешивании. Реакции сочетания пептидов и удаления защиты контролируют, используя тест Kaiser для аминов. Хаа относится к любой из коммерчески доступных аминокислот, которая может быть использована в виде предварительно присоединенной к МВНА полимеру. Yaa и Zaa относятся к любой из коммерчески доступных аминокислот.
Соединения по данному изобретению могут быть получены путем твердофазного пептидного синтеза (SPPS) обычным способом, который заключается в следующем: взвешивают Хаа-МВНА-полимер и помещают в шприц, оборудованный фриттовым фильтром. Полимеру дают предварительно набухнуть в ДМФ и затем удаляют N-концевую защитную группу путем обработки 30% пиперидином в ДМФ в течение 30 минут. Раствор со снятой защитной группой удаляют. Полимер с удаленной защитой промывают пять раз ДМФ, пять раз МеОН и затем пять раз ДМФ. Аминокислота Yaa может затем быть присоединена к полимеру с удаленной защитой, используя Yaa в ДМФ, содержащем Yaa, карбодиимидный связывающий реагент и НОВТ (гидроксибензотриазол), по 3 эквивалента каждого. Успешное связывание с растворами Yaa может быть необходимым для достижения эффективности сочетания, которое проходит тест Kaiser. Удаление N-концевой защитной группы и Yaa связывание могут быть повторены, чтобы присоединить третью аминокислоту Zaa. На последней стадии сочетания используют кетокислоту, карбодиимид и НОВТ в ДМФ и эту стадию повторяют до тех пор, пока реакция присоединения не пройдет тест Kaiser. Завершенную пептидную последовательность на полимере сушат в вакууме, по крайней мере, шесть часов и затем отщепляют обработкой в течение 2,5 часов либо 95/5 трифторуксусной кислотой/водой, либо свежеприготовленным раствором 90% трифторуксусной кислоты, 3% этандитиола, 5% тиоанизола и 2% анизола. Продукты отщепления выделяют либо лиофиллизацией из воды, либо растиранием с диэтиловым эфиром. Чистоту продуктов оценивают с помощью тонкослойной хроматографии. Выбранные пептидные образцы анализируют с помощью 1H ЯМР для подтверждения идентичности продукта.
Пример 2.
В этом примере (3'-индолпировиноградную кислоту)-N-бифенилаланин-D-Leu-Asp-OH получают согласно методу примера 1.
Fmoc-N-Asp(Ot-Bu)-МВНА-полимер (20 мг) взвешивают и помещают в шприц, оборудованный фриттовым фильтром. Полимеру дают предварительно набухнуть в 1 мл ДМФ в течение 30 минут. Удаляют Fmoc (фторенилметилоксикарбонил)защитную группу обработкой 20% пиперидином в ДМФ в течение 30 минут. Раствор с удаленной защитной группой удаляют. Полимер с удаленной защитой промывают пять раз ДМФ, пять раз МеОН и затем пять раз ДМФ. Fmoc-D-Leu-OH присоединяют к полимеру с удаленной защитой (1 экв), используя раствор Fmoc-D-Leu-OH (3 экв) в 1 мл ДМФ, содержащем карбодиимид (3 экв) и НОВТ (гидроксибензотриазол) (3 экв). Второе или третье связывание с растворами Fmoc-D-Leu-OH может быть необходимо для достижения эффективности сочетания, которое проходит тест Kaiser. Удаление защитной группы с Fmoc и стадию сочетания с аминокислотой повторяют для присоединения Fmoc-N-(4,4-бифенил)аланина. На последней стадии присоединения используют индолпировиноградную кислоту (5 экв), диизопропилкарбодиимид (5 экв) и НОВТ (5 экв) в ДМФ и эту стадию повторяют до тех пор, пока реакция сочетания не пройдет тест Kaiser. Завершенную пептидную последовательность на полимере сушат в вакууме, по крайней мере, шесть часов и затем отщепляют обработкой в течение 2,5 часов либо 95/5 трифторуксусной кислотой/водой, либо свежеприготовленным раствором 90% трифторуксусной кислоты, 3% этандитиола, 5% тиоанизола и 2% анизола. Отщепленные продукты выделяют либо лиофиллизацией из воды, либо обработкой диэтиловым эфиром. Чистоту продуктов оценивают с помощью тонкослойной хроматографии.
1H ЯМР (400 МГц, d6-ДМСО): δ 6,5-7,7 (м, 14Н), 4,5 (м, 1Н), 4,1 (м, 2Н), 3,4 (м, 2Н), 3 (м, Н), 2,7 (м, 1Н), 1,1-1,5 (м, 3Н), 0,5-0,9 (м, 6Н).
Пример 3.
В этом примере получают (3'-индолпировиноградную кислоту)-N-бифенилаланин-D-Leu-Asp-OH, используя Chiron Mimotopes Pin Technology.
Первый аминокислотный остаток Хаа присоединяют к 4-(гидроксиметил)феноксиацетамидо-маркер)полимерным иглам (5,7 ммоль/иглу) путем связывания каждой иглы в 800 мкл связывающем растворе (100 мМ аминокислоты, 100 мМ DIC, 10 мм DMAP, 1/4 ДМФ/СН2С12) в течение двух часов. Иглы затем ополаскивают 5 минут ДМФ промывкой, два раза по 5 мин МеОН промывками и 15 минут сушат на воздухе. Группы Fmoc со снятой защитой обрабатывают в течение 30 минут 800 мкл 20% пиперидина в ДМФ. Повторяют промывку игл (1 ДМФ промывка, 2 МЕОН промывки, 15 минут сушка на воздухе). Второй остаток аминокислоты Yaa связывают (100 мМ Yaa, 100 мМ DIC, 100 мМ НОВТ и индикатор бромфенол синий в ДМФ) до тех пор, пока синий цвет больше не остается на поверхности иглы. Связывание при необходимости повторяют. Цикл промывки и удаления защиты с Fmoc также повторяют. Следующую аминокислоту Zaa присоединяют путем повторения связывания и процедур промывки как в случае Yaa, повторяя реакцию присоединения в случае необходимости. Последний остаток присоединяют с индолпировиноградной кислотой 15 экв 100 мМ, 15 экв DIC, 15 экв НОВТ и индикатором бромфенолом синим в ДМФ. Реакцию присоединения при необходимости повторяют. После последней промывки оранжевые иглы удаляют с их подложек и отщепляют в отдельной 2 мл пластиковой центрифужной пробирке с 1,5 мл свежеприготовленного раствора 90% трифторуксусной кислоты, 5% тиоанизола, 3% этандитиола и 2% анизола в течение 2,5 часов. Иглы удаляют из пробирок и смесь продувают практически досуха струей азота. Растирают с Et2O и центрифугируют каждую пробирку. Эту стадию повторяют три раза для каждой пробирки. Осажденные пептиды собрирают, лиофилизируют, взвешивают и используют. Чистоту продукта оценивают тонкослойной хроматографией. Исходные вещества собирают и анализируют относительно достоверных образцов, полученных в примере 1.
Пример 4.
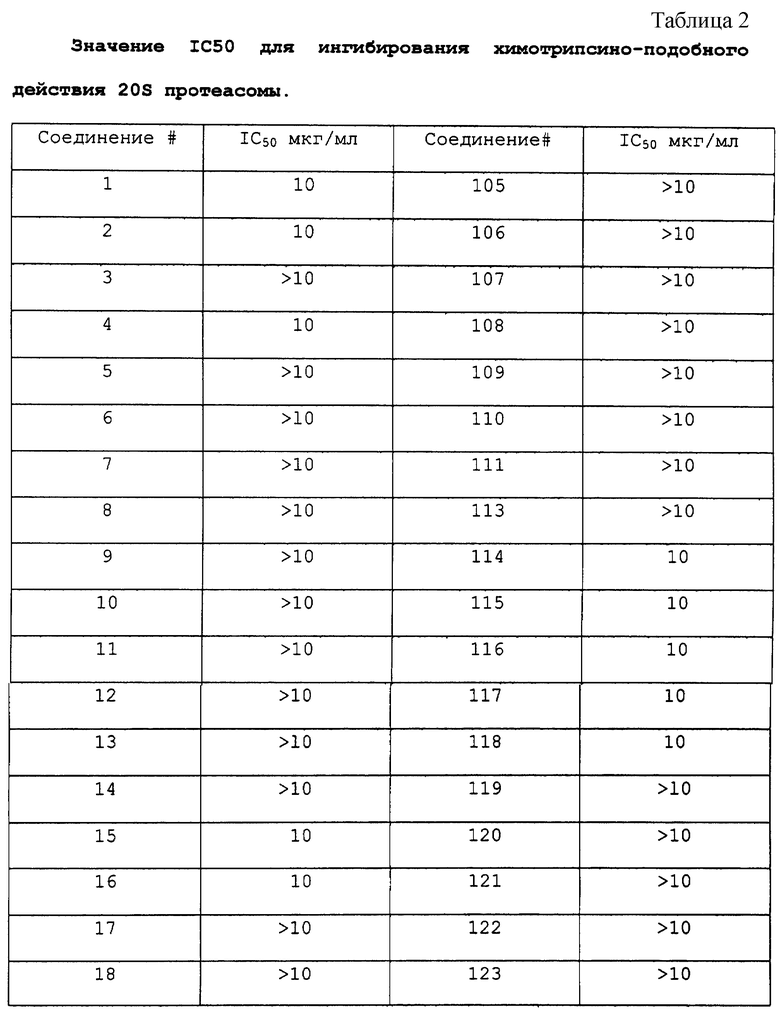
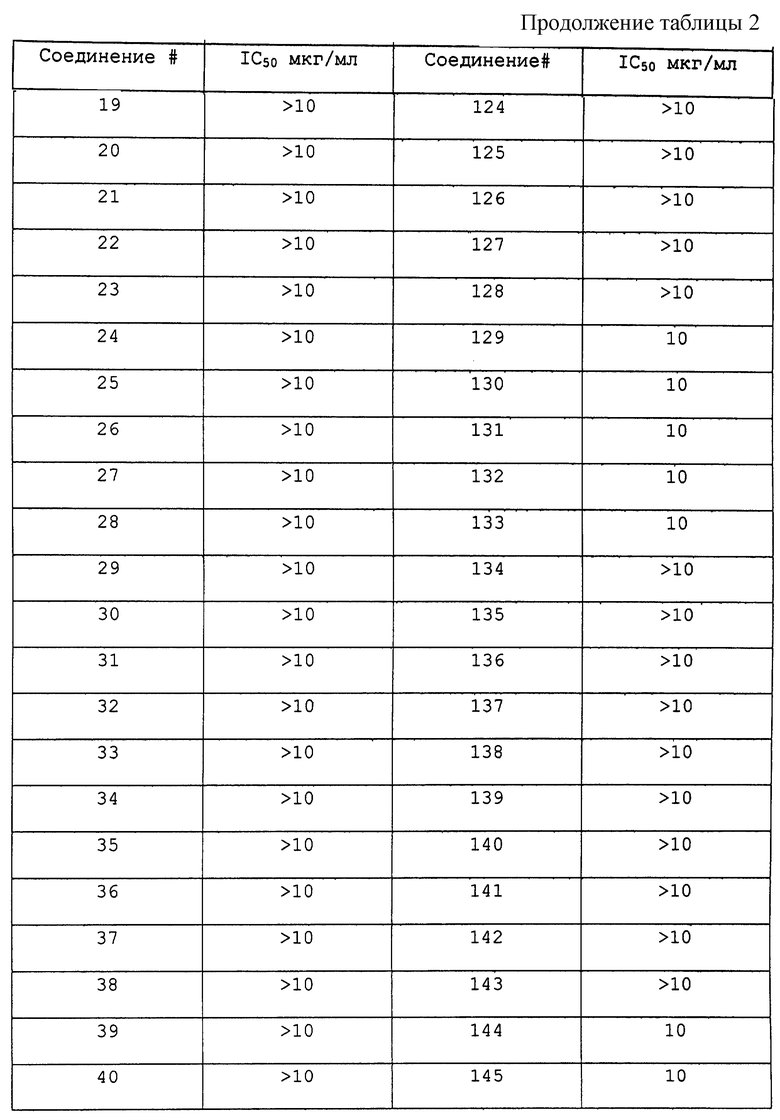
Соединения по данному изобретению, полученные в соответствии со способом примера 1, анализировали следующим образом. 20S каталитическую субъединицу протеасомы (также известную как мультикаталитический протеиназный комплекс) очищают до гомогенности из бычьего мозга согласно опубликованным методам (Wilk S. and Orlowski M. 1983, 40 842 J. Neurochem). Химотрипсическую активность комплекса измеряют путем увеличения флюоресценции после отщепления субстрата пептида сукцинил-лейцин-лейцин-валин-тирозин-7-амино-4-метилкумарин. Стандарт in vitro исследования состоит из 2 мкг 20S протеасомы, 0,1-100 мкг/мл ингибитора протеасомы в 200 мкл 50 мМ HEPES, содержащего додецилсульфат натрия 0,1%, рН 7,5. Протеолитическую реакцию инициировали добавлением 50 мкМ флуорогенного пептидного субстрата и оставляли развиваться в течение 15 минут при 37oС. Реакцию завершают добавлением 100 мкл 100 мМ ацетатного буфера, рН 4,0. Скорость протеолиза прямо пропорциональна количеству высвободившегося аминометилкумарина, который измеряют с помощью флуоресцентной спектроскопии (Ех 370 нм, ЕМ 430 нм).
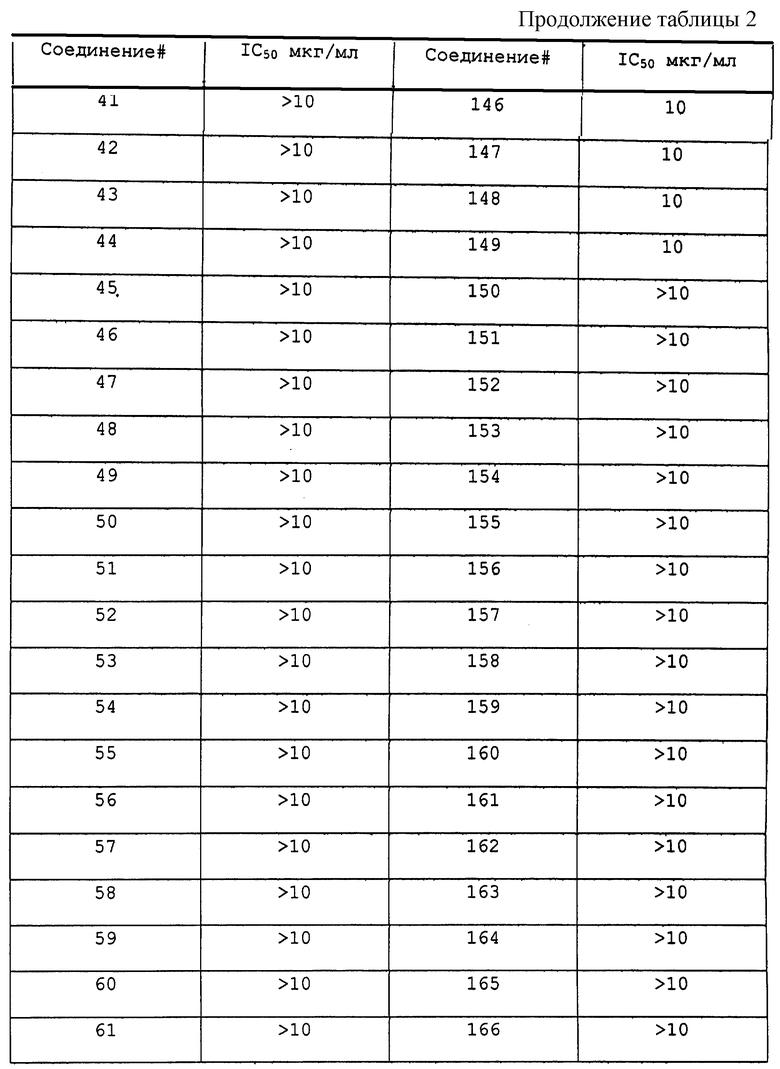
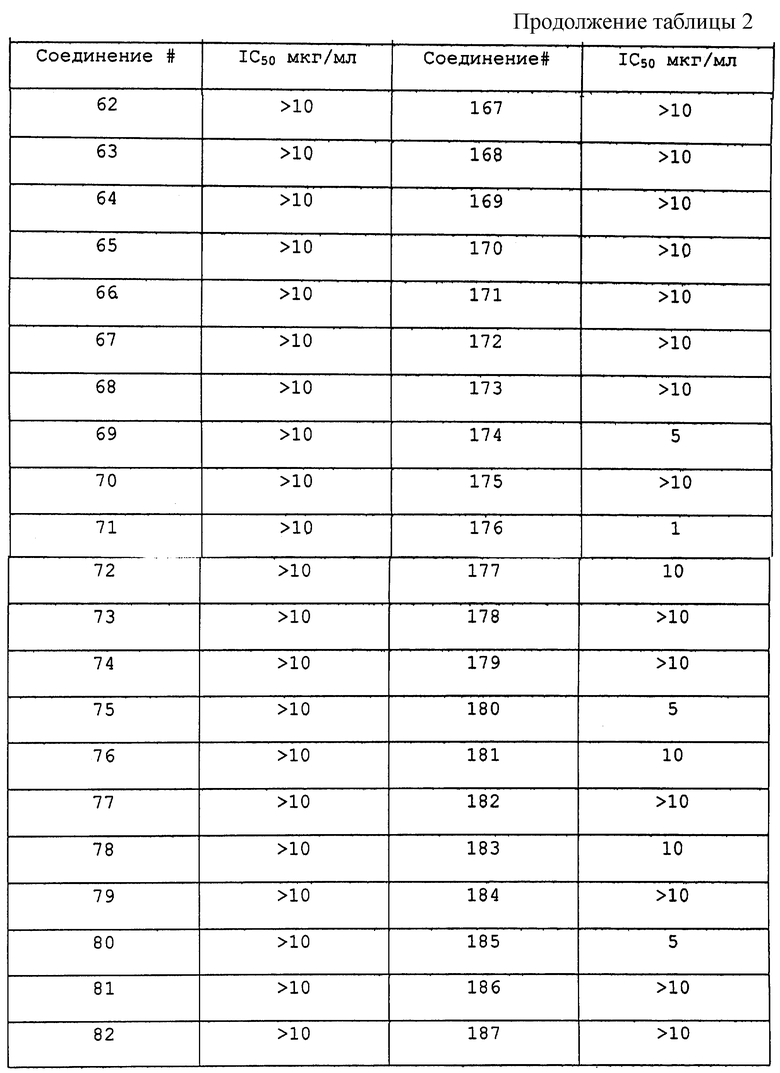
Результаты исследования ингибитора 20S протеасомы представлены в таблице 2.
Соединения по данному изобретению, полученные в соответствии со способом по примеру 1, были также исследованы следующим образом. 20S каталитическую субъединицу протеасомы (также известную, как мультикаталитический протеиназный комплекс) очищали до гомогенности из бычьего мозга согласно опубликованным методам (Wilk S. and Orlowski M. 1983, 40 842 J. Neurochem). Триптическую активность комплекса измеряют по увеличению флюоресценции после отщепления субстрата пептида CBZ-D-Leu-Arg-(7-амино-4-метилкумарин). Стандарт in vitro исследования состоит из 2 мкг 203 протеасомы, 0,1-100 мкг/мл ингибитора протеасомы в 200 мкл 50 мМ HEPES, содержащего додецилсульфат натрия 0,1%, рН 7,5. Протеолитическую реакцию инициировали добавлением 50 мкМ флуорогенного пептидного субстрата и оставляли развиваться в течение 15 минут при 37oС. Реакцию завершали добавлением 100 мкл 100 мМ ацетатного буфера, рН 4,0. Скорость протеолиза прямо пропорциональна количеству высвободившегося аминометилкумарина, который измеряли с помощью флуоресцентной спектроскопии (ЕХ 370 нм, ЕМ 430 нм). Соединения 1-207 были исследованы на ингибирование триптической активности и активность в качестве ингибиторов в > 10 мкг/мл.
Пример 5.
Соединения по данному изобретению, полученные в соответствии с методом по примеру 1, были также исследованы следующим образом. 20S каталитическую субъединицу протеасомы (также известную как мультикаталитический протеиназный комплекс) очищают до гомогенности из бычьего мозга согласно опубликованным методам (Wilk S. and Orlowski M. 1983, 40 842 J. Neurochem). Триптическую активность комплекса измеряли по увеличению флюоресценции после отщепления субстрата пептида CBZ D-Ala-Leu-Arg-(7-амино-4-метилкумарин). Стандарт in vitro исследования состоит из 2 мкг 20S протеасомы, 0,1-100 мкг/мл ингибитора протеасомы в 200 мкл 50 мМ HEPES, содержащего додецилсульфат натрия 0,1%, рН 7,5. Протеолитическую реакцию инициировали добавлением 50 мкМ флуорогенного пептидного субстрата и оставляли развиваться в течение 15 минут при 37oС. Реакцию завершают добавлением 100 мкл 100 мМ ацетатного буфера, рН 4,0. Скорость протеолиза прямо пропорциональна количеству высвободившегося аминометилкумарина, который измеряли с помощью флуоресцентной спектроскопии (ЕХ 370 нм, ЕМ 430 нм). Соединения 1-207 были исследованы на ингибирование триптической активности и активность как ингибиторов в > 10 мкг/мл.
Пример 6.
Соединения по данному изобретению, полученные в соответствии с методом по примеру 1, были также исследованы следующим образом. 20S каталитическую субъединицу протеасомы (также известную как мультикаталитический протеиназный комплекс) очищают до гомогенности из бычьего мозга согласно опубликованным методам (Wilk S. and Orlowski M. 1983, 40 842 J. Neurochem). Триптическую активность комплекса измеряют по увеличению флюоресценции после отщепления субстрата пептида CBZ D-Ala-Leu-Glu-(7-амино-4-метилкумарин). Стандарт in vitro состоит из 2 мкг 20S протеасомы, 0,1-100 мкг/мл ингибитора протеасомы в 200 мкл 50 мМ HEPES, содержащего додецилсульфат натрия 0,1%, рН 7,5. Протеолитическую реакция инициировали добавлением 50 мкМ флуорогенного пептидного субстрата и оставляли развиваться 15 минут при 37oС. Реакцию завершают добавлением 100 мкл 100 мМ ацетатного буфера, рН 4,0. Скорость протеолиза прямо пропорциональна количеству высвободившегося аминометилкумарина, который измеряли с помощью флуоресцентной спектроскопии (ЕХ 370 нм, ЕМ 430 нм). Соединения 1-207 были исследованы на ингибирование пептидилглютамиловой активности при > 10 мкг/мл. Соединение 190 было активно при 5 мкг/мл.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИРОВАНИЕ ПРОТЕАСОМ 26S И 20S ИНДАНОНАМИ | 1997 |
|
RU2195310C2 |
| ПУРИНРИБОЗИДЫ В КАЧЕСТВЕ АНТИАРИТМИЧЕСКИХ СРЕДСТВ | 2001 |
|
RU2248208C2 |
| СПОСОБ ЛЕЧЕНИЯ ДИАБЕТА | 2003 |
|
RU2320343C2 |
| АГОНИСТЫ А3 РЕЦЕПТОРОВ АДЕНОЗИНА | 2002 |
|
RU2298557C2 |
| СПОСОБ ЗАЖИВЛЕНИЯ РАН С ПРИМЕНЕНИЕМ АНТАГОНИСТОВ АДЕНОЗИНОВОГО РЕЦЕПТОРА A | 2005 |
|
RU2385322C2 |
| СПОСОБ СНИЖЕНИЯ ЧАСТОТЫ СЕРДЕЧНЫХ СОКРАЩЕНИЙ, ВКЛЮЧАЮЩИЙ ВВЕДЕНИЕ АГОНИСТА РЕЦЕПТОРА АДЕНОЗИНА А ВМЕСТЕ С БЕТА-БЛОКАТОРОМ, БЛОКАТОРОМ КАЛЬЦИЕВЫХ КАНАЛОВ ИЛИ СЕРДЕЧНЫМ ГЛИКОЗИДОМ | 2003 |
|
RU2332220C2 |
| ПРОИЗВОДНЫЕ ГЕТЕРОАРИЛАЛКИЛПИПЕРАЗИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2243970C1 |
| ПРОЛЕКАРСТВА АНТАГОНИСТОВ A РЕЦЕПТОРА АДЕНОЗИНА | 2006 |
|
RU2415858C2 |
| ПРОИЗВОДНЫЕ АДЕНОЗИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ КОРРЕКЦИИ ЭЛЕКТРИЧЕСКИХ НАРУШЕНИЙ В СЕРДЦЕ МЛЕКОПИТАЮЩЕГО | 1997 |
|
RU2172320C2 |
| ЧАСТИЧНЫЕ И ПОЛНЫЕ АГОНИСТЫ АДЕНОЗИНОВЫХ РЕЦЕПТОРОВ A | 2003 |
|
RU2340623C2 |

Описываются новые соединения общей формулы I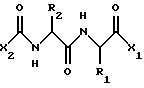
где Х2 - Ar или Ar - Х3, где Х3 представляет собой -С=О или -СН2СО- и Ar представляет собой фенил или индол; R1 и R2 каждый независимо выбран из водорода или боковых цепей природных α-аминокислот, представленных линейным или разветвленным (С1-С10)алкилом, который может быть замещен гидрокси, карбокси, амино, циклогексилом, фенилом или гидроксизамещенным фенилом, бифенилом, нафтилом или пиридилом; Х1 выбран из -ОН или группы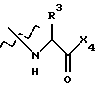
где Х4 представляет собой -ОН; R3 выбран из водорода или боковых цепей природных α-аминокислот, представленных линейным или разветвленным (С1-С10)алкилом, который может быть замещен гидрокси, фенилом или гидроксизамещенным фенилом. Кроме того, описана фармацевтическая композиция, обладающая способностью ингибировать химотрипсиноподобное действие 20S протеасомы, включающая соединение I и один или несколько фармацевтических эксципиентов, представленная в виде раствора или таблетки. 2 с. и 13 з.п.ф-лы, 2 табл.
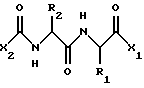
где Х2 представляет собой Аr или Аr-Х3, где Х3 представляет собой С= О или -СН2СО- и Аr представляет собой фенил или индол;
R1 и R2 каждый независимо выбраны из водорода или боковых цепей природных α-аминокислот, представленных линейным или разветвленным (С1-С10) алкилом, который может быть замещен гидрокси, карбокси, амино, циклогексилом, фенилом или гидроксизамещенным фенилом, бифенилом, нафтилом или пиридилом;
Х1 выбран из -ОН, или группы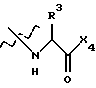
где Х4 представляет собой -ОН;
R3 выбран из водорода или боковых цепей природных α-аминокислот, представленных линейным или разветвленным (С1-С10 алкилом, который может быть замещен гидрокси, фенилом или гидроксизамещенным фенилом.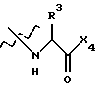
где Х4 и R3 указаны в п. 1.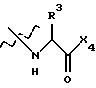
где Х4 представляет собой -ОН и R3 представляет собой Н.
| RU 94016167 А1, 10.04.1996 | |||
| US 5656604 А, 12.08.1997 | |||
| Линейный электродвигатель | 1972 |
|
SU468339A1 |
| Шланговое соединение | 0 |
|
SU88A1 |
| DA SETTIMO А | |||
| et | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Structural requirements for inverse agonist / antagonist receptor interactions" DRUG | |||
| DES | |||
| Способ изготовления фанеры-переклейки | 1921 |
|
SU1993A1 |
| ПЕЧНОЙ ЖЕЛЕЗНЫЙ РУКАВ (ТРУБА) | 1920 |
|
SU199A1 |

