Настоящее изобретение относится к способам получения хлорфторциклопентенов и октафторциклопентена, которые можно использовать как промежуточные продукты для получения 1,2,3,3,4,4,5,5-октафторциклопентана, используемого в качестве фторсодержащего моющего средства, фторсодержащего осушающего растворителя или тому подобного, а также в качестве промежуточных продуктов при получении различных фторсодержащих соединений.
Известно, что фторированные циклопентаны получают, во-первых, фторированием хлорированных циклоалкенов, соответствующих фторированным циклопентанам, для получения производных вицинально хлорированного фторциклопентена, а затем фторированием и гидрированием этих производных. Известно, что производные вицинально хлорированного фторциклопентена производят первым способом, в котором используют перхлорциклоолефины, или вторым способом, в котором используют перхлорированные циклосопряженные диены. В качестве примера первого способа в J. Am. Chem. Soc., 67, 1235 (1945) описан способ получения 1,2-дихлоргексафторциклопентена реакцией октахлорциклопентена со смесью трифторида сурьмы и трифтордихлорида сурьмы. Далее, в патенте Германии 3935493 раскрыт способ производства 1,2-дихлоргексафторциклопентена посредством реакции октахлорциклопентена с хлором и фторидом водорода в присутствии пентахлорида сурьмы. В качестве примера второго способа первая публикация японского патента JP-A-8333285 описывает способ производства 1,2-дихлор-3,3,4,4,5,5-гексафторциклопентена первоначальной реакцией гексахлорциклопентадиена с хлором в присутствии трихлорида сурьмы для соответствующего превращения таким образом гексахлорциклопентадиена и трихлорида сурьмы в октахлорциклопентен и пентахлорид сурьмы с последующим добавлением фторида водорода. С другой стороны, в патенте США 2459783 раскрыт способ производства 1,2-дихлоргексафторциклопентена реакцией гексахлорциклопентена с пентафторидом сурьмы. Патент США 2449233 раскрывает способ получения 1,2-дихлоргексафторциклопентена реакцией гексахлорциклопентадиена с фторидом водорода в присутствии пентахлорида сурьмы.
В каждом из вышеупомянутых известных способов используют реакцию в жидкой фазе в присутствии катализатора на основе галогенида сурьмы. Поэтому коррозионная активность галогенида сурьмы может вызвать проблемы. Более того, в случае применения фторида водорода в качестве агента фторирования при производстве в промышленном масштабе давление реакции может составлять 10-30 кг/см2. Это может вызвать некоторые ограничения в выборе оборудования.
Задачей изобретения является создание способа получения пергалоидированного циклопентена, в частности октафторциклопентена, который был бы пригоден для производства указанного вещества в промышленных масштабах.
Согласно настоящему изобретению предложен способ получения первого пергалоидированного циклопентена, представленного общей формулой С5СlВF8-В, в которой В является целым числом от 0 до 7. Способ включает стадию (а) фторирования второго пергалоидированного циклопентена фторидом водорода в газовой фазе в присутствии катализатора фторирования. Второй пергалоидированный циклопентен представлен общей формулой C5ClAF8-A, в которой А является целым числом от 1 до 8, и А не меньше В. При помощи этого способа первый пергалоидированный циклопентен (например, 1,2-дихлор-3,3,4,4,5,5-гексафторциклопентен, другой хлорфторированный циклопентен или октафторциклопентен) можно легко получать непрерывно, например, из октахлорциклопентена, получаемого хлорированием гексахлорциклопентадиена, который легко доступен. Следовательно, вышеуказанный способ очень полезен в качестве способа промышленного производства.
Согласно настоящему изобретению в случае, когда первый пергалоидированный циклопентен является октафторциклопентеном, указанный выше способ может дополнительно включать стадию (b) фторирования продукта реакции стадии (а) способом, отличающимся от способа стадии (а), чтобы таким образом превратить продукт реакции стадии (а) в октафторпентен. Продуктом реакции стадии (а) является пергалоидированный циклопентен, представленный общей формулой С5СlВF8-В, в которой В является целым числом от 0 до 7, или общей формулой С5СlАF8-А, в которой А является целым числом от 1 до 8 и А не меньше В.
Согласно настоящему изобретению вышеуказанную стадию (а) можно осуществлять многостадийной реакцией, в которой имеется "m" реакционных зон, где m составляет целое число от 2 до 10. Реакционные зоны располагают последовательно и таким образом, чтобы температурой реакции в каждой реакционной зоне можно было управлять независимо. В такой многостадийной реакции становится возможным уменьшить производство смолистых веществ и таким образом достигнуть длительного срока службы катализатора фторирования. Поэтому становится возможным производить первый пергалоидированный циклопентен непрерывно в течение длительного времени без прерывания процесса.
В настоящем изобретении 1,2-дихлор-3,3,4,4,5,5,-гексафтор-циклопентен-1 является 1,2-дихлоргексафторциклопентеном или 1,2-дихлор-3,3,4,4,5,5-гексафторциклопентеном. Ниже, если другое не оговорено, октахлорциклопентен, используемый в качестве сырья в способе по настоящему изобретению, может быть заменен на хлорфторциклопентен, получаемый частичным фторированием октахлорциклопентена. Далее, 1,2-дихлоргексафторциклопентен как продукт реакции может включать другие хлорфторциклопентены, хлоргептафторциклопентен и октафторциклопентен.
Как определено выше, первый пергалоидированный циклопентен, целевой продукт по настоящему изобретению, представлен общей формулой С5СlВF8-В, в которой В является целым числом от 0 до 7. Другими словами, количество атомов хлора в этом циклопентене является целым числом от 0 до 7, а количество атомов фтора, следовательно, является целым числом от 1 до 8, и общее количество атомов хлора и атомов фтора равно 8. В этом циклопентене атом галогена может быть связан с любым углеродным атомом. Первый пергалоидированный циклопентен ничем особенным не ограничен, и не ограничивающими примерами его являются октафторциклопентен, 1-хлоргептафторциклопентен, 1,2-дихлор-3,3,4,4,5,5-гексафторциклопентен, 1,2,4-трихлор-3,3,4,5,5-пентафторциклопентен, 1,2,3,4-тетрахлор-3,4,5,5-тетрафтороциклопентен, 1,2,3,4,4-пентахлор-3,5,5-трифтороциклопентен, гексахлор-3,3-дифторциклопентен, гексахлор-4,4-дифторциклопентен и гептахлор-5-фторциклопентен.
Как было определено выше, второй пергалоидированный циклопентен - сырье по настоящему изобретению и представлен общей формулой С5СlАF8-А, где А является целым числом от 1 до 8. Другими словами, количество атомов хлора в этом циклопентене является целым числом от 1 до 8, а количество атомов фтора, следовательно, является целым числом от 0 до 7, и общее количество атомов хлора и атомов фтора составляет 8. В этом циклопентене атом галогена может быть связан с любым углеродным атомом. Второй пергалоидированный циклопентен ничем особенно не ограничен, и не ограничивающими примерами его являются 1-хлор-гептафторциклопентен, 1,2-дихлор-3,3,4,4,5,5-гексафторциклопентен, 1,2,4-трихлор-3,3,4,5,5-пентафторциклопентен, 1,2,3,4-тетрахлор-3,4,5,5-тетрафторциклопентен, 1,2,3,4,4-пентахлор-3,5,5-трифторциклопентен, гексахлор-3,3-дифторциклопентен, гексахлор-4,4-дифторциклопентен, гептахлор-5-фторциклопентен и октахлорциклопентен.
Первый или второй пергалоидированный циклопентен в соответствии с настоящим изобретением может быть синтезирован известным способом. Например, Newcomer; McBee, J.Amer.Chem.Soc., 71 (1949) 946, 950 описывает способ получения октахлорциклопентена хлорированием гексахлорциклопентадиена хлором в присутствии катализатора (например, хлорида металла). Кроме того, Henne et al. , J. Am. Chem. Soc., 67, 1235 (1945) описывает реакцию октахлорциклопентена со смесью трифторида сурьмы и трифтордихлорида сурьмы с получением таким образом 1,2-дихлоргексафторциклопентена, 1,2,4-трихлор-3,3,4,5,5-пентафторциклопентена, 1,2,3,4-тетрахлор-3,4,5,5-тетрафторциклопентена.
По настоящему изобретению предпочтительно, чтобы катализатор фторирования включал по меньшей мере один металл, выбранный из металлов 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 и 15 групп периодической системы. Этими металлами предпочтительно являются хром, марганец, кобальт, никель, железо, молибден, ниобий, алюминий, цинк, медь, сурьма, титан, олово и тантал. Из них наиболее предпочтительными являются хром и сурьма. В случае, если по меньшей мере два металла используют для катализатора фторирования, одним из этих по меньшей мере двух металлов предпочтительно является хром или сурьма. Далее, катализатор фторирования может быть по меньшей мере катализатором, выбранным из группы, состоящей из оксидов металлов, фторидов металлов (фторированные металлы), хлоридов металлов (хлорированные металлы), фторхлоридов металлов (фторированные и хлорированные металлы), оксифторидов металлов (частично фторированные оксиды металлов), оксихлоридов металлов (частично хлорированные оксиды металлов) и оксифторхлоридов металлов (частично фторированные и хлорированные оксиды металлов). По меньшей мере один металл, используемый в катализаторе фторирования, может быть нанесен на обычном носителе. Носитель катализатора фторирования ничем особенно не лимитирован, если только он стабилен при фторировании по настоящему изобретению. Примерами носителей являются активированный уголь, металлический алюминий, диоксид циркония, оксид магния, диоксид титана, модифицированные глины, получаемые удалением из них диоксида кремния, например фторидом водорода, оксид алюминия (глинозем), фторид алюминия, хлорид алюминия, фторхлорид алюминия, оксифторид алюминия (частично фторированный оксид алюминия), оксихлорид алюминия и оксифторхлорид алюминия. Наиболее предпочтительными примерами катализатора фторирования являются хром на носителе из активированного угля, хром на носителе из оксида алюминия, оксид хрома на активированном угле, частично фторированный оксид хрома на активированном угле и сурьма на носителе из активированного угля.
В соответствии с настоящим изобретением активированный уголь, применяемый в качестве носителя для катализатора фторирования, не ограничивается каким-либо конкретным типом. Активированный уголь может быть приготовлен из растительного сырья, такого как древесина, опилки, древесный уголь, уголь из кокосовой скорлупы, уголь из сердцевины пальмового дерева или необработанная зола; из угля, такого как торф, лигнит, бурый уголь, битумный уголь или антрацит; нефтяного сырья, такого как мазут, сернокислый гудрон или нефтяной уголь; или сырья в виде синтетических смол. Активированный уголь можно выбирать из различных производимых в промышленности активированных углей. Примерами промышленных активированных углей, применимых в настоящем изобретении, являются активированный уголь под торговой маркой CALGON GRANULAR ACTIVATED CARBON CAL, получаемый из битумного угля компанией TOYO CALGON CO. , и уголь из кокосовой скорлупы, производимый компанией Takeda Chemical Industries, Ltd. Активированный уголь, используемый в настоящем изобретении, в основном имеет форму гранул. Форма и размеры никак особенно не ограничены и могут быть приняты в зависимости от размеров реактора. Предпочтительно, чтобы активированный уголь, используемый в изобретении, имел большую удельную поверхность. Промышленно получаемый активированный уголь будет удовлетворять изобретению в отношении удельной поверхности и объема микропор. В настоящем изобретении удельная поверхность активированного угля предпочтительно составляет больше 400 м2/г, более предпочтительно от 800 до 3000 м2/г. Кроме того, предпочтительно, если объем микропор в активированном угле превышает 0,1 см3/г, более предпочтительно составляет от 0,2 до 1,0 см3/г. В случае, если активированный уголь используют в качестве носителя в настоящем изобретении, предпочтительно активировать поверхность носителя и удалить из него золу погружением активированного угля в щелочной водный раствор гидроксида аммония, гидроксида натрия, гидроксида калия или им подобных при комнатной температуре примерно на 10 часов или более или путем предварительной обработки активированного угля кислотой, такой как азотная кислота, соляная кислота или плавиковая кислота при комнатной температуре или при нагревании. Такая предварительная обработка традиционно используется при использовании активированного угля в качестве носителя катализатора.
В соответствии с настоящим изобретением перед применением галогенида металла высокой валентности предпочтительно удалить из носителя воду, насколько это возможно, путем нагрева, вакуумирования или похожим способом, с целью предотвращения разложения галогенида, вызываемого гидролизом или подобным ему воздействием. Металл для такого галогенида выбирают из сурьмы, титана, олова, тантала или им подобных.
В соответствии с настоящим изобретением оксид алюминия, используемый в качестве носителя катализатора фторирования, не ограничен каким-либо определенным типом. Можно использовать оксид алюминия, полученный осаждением из водного раствора соли алюминия путем применения щелочного вещества, такого как аммиак, а затем формованием и сушкой осадка. На самом деле предпочтительно использовать в качестве носителя катализатора или осушителя промышленно производимый γ-оксид алюминия.
В соответствии с настоящим изобретением количество по меньшей мере одного металла, которое нанесено на носитель, предпочтительно составляет от 0,1 до 50 массовых частей, более предпочтительно от 0,5 до 50 массовых частей, еще предпочтительнее от 2 до 50 массовых частей и наиболее предпочтительно от 5 до 50 массовых частей по отношению к 100 массовым частям носителя. По мере увеличения количества по меньшей мере одного металла катализатор становится более активным. Однако, если его количество слишком большое, катализатор может стать порошкообразным и таким образом потребовать большой осторожности в обращении с ним.
В соответствии с настоящим изобретением способ приготовления катализатора фторирования никак особенно не лимитирован. Носитель катализатора фторирования может быть, например, оксидом алюминия, таким как γ-оксид алюминия, или частично фторированным оксидом алюминия, который был обработан фторидом водорода, хлоридом водорода, хлорфторзамещенным углеводородом или подобным веществом, или активированным углем. При приготовлении катализатора фторирования носитель можно погрузить в раствор по меньшей мере одного соединения или в само по меньшей мере одно соединение, если оно находится в жидкой форме, или в качестве альтернативы раствор или по меньшей мере одно само по себе соединение можно распылить на носителе. Далее носитель сушат и затем приводят в контакт с агентом фторирирования в газообразной форме (например, с фторидом водорода или хлорфторзамещенным углеводородом) в условиях нагревания, фторируя таким образом частично или полностью носитель или по меньшей мере одно соединение, нанесенное на нем. Этим завершается приготовление катализатора фторирования. В случае использования фторированного оксида алюминия в качестве носителя предпочтительно при приготовлении катализатора фторирования носитель приводить в контакт с фторидом водорода при температуре (предпочтительно около 150-800oС) не ниже, чем температура фторирования второго пергалоидированного циклопентена. Таким образом, содержание фтора в катализаторе фторирования будет стабильным во время реакции. В другом случае, когда по меньшей мере одно соединение не нанесено на носитель, это вещество может быть приготовлено следующим образом. Сначала гидроксид металла осаждают из раствора соединения по меньшей мере одного металла, используя основное вещество, такое как аммиак. Далее этот гидроксид металла превращают в оксид металла, например, спеканием, а затем этот оксид металла частично или полностью модифицируют галогеном, используя фторид водорода, хлорид водорода, хлорфторзамещенный углеводород или им подобные вещества. Например, оксид металла можно приготовить спеканием хромоксидного геля, осажденного из водного раствора нитрата хрома с использованием водного раствора аммиака. Оксид металла также может быть получен перемешиванием хромоксидного геля с гелем оксида алюминия, полученного таким же образом, как хромоксидный гель, а затем спеканием перемешанной смеси. Кроме того, оксид металла может быть получен спеканием смешанного геля, полученного соосаждением из водного раствора нитрата хрома, в котором растворен как минимум один металл (например марганец), используемый в катализаторе фторирования. Нет необходимости упоминать, что оксид металла может быть получен подобным же способом при использовании другого металла.
В соответствии с настоящим изобретением к катализатору фторирования можно при желании внести добавку, являющуюся по меньшей мере одним элементом из группы щелочноземельных металлов, таких как Мg и Са, или элементом из группы лантаноидов, таких как La и Се. Эта добавка предотвращает повторную кристаллизацию оксигалогенида, используемого в качестве соединения по меньшей мере одного металла или в качестве носителя, и поддерживает, таким образом, активность первого катализатора фторирования. Массовое соотношение по меньшей мере одного металла к добавке предпочтительно составляет от 50:50 до 99,9:0,1 и более предпочтительно от 70:30 до 99:1.
В соответствии с настоящим изобретением по меньшей мере одно соединение металла, используемое при приготовлении катализатора фторирования, может быть по меньшей мере нитратом, хлоридом, солью органической кислоты, органическим комплексом или подобным им соединением как минимум одного металла, растворимым в растворителе, таком как вода, этанол или ацетон. Кроме того, для приготовления катализатора фторирования можно растворить в неорганической кислоте, такой как соляная кислота или азотная кислота, по меньшей мере одно соединение металла, такое как оксид или гидроксид, или элементарный металл. Не ограничивающими примерами по меньшей мере одного соединения металла являются нитрат хрома, трихлорид хрома, триоксид хрома, бихромат калия, нитрат марганца, хлорид марганца, диоксид марганца, ацетат марганца, нитрат никеля, хлорид никеля, ацетат никеля, нитрат кобальта, хлорид кобальта, нитрат железа, хлорид железа, хлорид молибдена, хлорид ниобия, нитрат алюминия, хлорид алюминия, хлорид цинка, нитрат меди, хлорид меди, трихлорид сурьмы, пентахлорид сурьмы, пентафторид сурьмы, тетрахлорид титана, трихлорид титана, тетрахлорид олова и пентахлорид тантала.
В соответствии с настоящим изобретением катализатор фторирования с носителем (например, активированным углем), с нанесенным на нем галогенидом металла высокой валентности, такого как сурьма, титан, олово и тантал, может быть приготовлен следующим образом. Например, такой галогенид сам по себе, если он находится в жидкой форме при комнатной температуре, постепенно добавляют к активированному углю, подвергнутому при необходимости предварительной обработке, такой как сушка, кислотная обработка и т.п. В альтернативном случае активированный уголь погружают в раствор, в котором такой галогенид растворен в инертном растворителе, с последующим нагреванием и/или вакуумированием. Примерами такого инертного растворителя являются хлорированные растворители, такие как четыреххлористый углерод, хлороформ, хлористый метилен, тетрахлорэтилен, трихлорэтилен и тетрахлорэтан; фторохлорированные растворители, такие как 2,2-дихлор-1,1,1 -трифторэтан, 1,1 -дихлор-1-фторэтан, 3,3-дихлор-1,1,1,2,2-пентафторпропан и 1,3-дихлор-1,1,2,2,3-пентафторпропан, и спирты, такие как метанол, этанол и изопропанол. Соединения сурьмы сравнительно легко окисляются. Таким образом, возможно погрузить носитель в раствор, содержащий растворенный в вышеупомянутом инертном растворителе галогенид металла низкой валентности, такой как трихлорид сурьмы, а затем превратить этот галогенид в пятивалентный галогенид, например, с помощью хлора. В этом случае принимается, что такой галогенид работает в качестве катализатора, пока сохраняется его высокая валентность. Как отмечается ниже, такой галогенид может обладать каталитической активностью при сравнительно низкой температуре. В соответствии с настоящим изобретением галогенидом металла высокой валентности может быть пентахлорид сурьмы, пентафторид сурьмы, тетрахлорид олова, тетрахлорид титана или пентахлорид тантала. Носителем для такого галогенида может служить активированный уголь. Этот галогенид может представлять собой смесь по меньшей мере двух галогенидов. На самом деле предпочтительно добавлять к пентахлориду сурьмы небольшое количество пентахлорида олова, тетрахлорида титана, пентахлорида тантала или хлорид металла, выбранного из хрома, марганца, кобальта, никеля, железа, молибдена, ниобия, алюминия, цинка и меди. Активными видами катализатора, как полагают, хотя это и не обязательно, может служить смешанный галогенид, в котором хлор частично замещен фтором.
В соответствии с настоящим изобретением изменение состава катализатора фторирования во время фторирования (т.е. во время вышеуказанной стадии (а)) можно эффективно предотвратить путем осуществления перед фторированием обработки катализатора фторирования фторирующим агентом, таким как фторид водорода, фторзамещенный углеводород или фторхлорзамещенный углеводород при температуре не ниже, чем температура реакции фторирования. Если все же активность катализатора фторирования дезактивирована реакцией, то можно реактивировать катализатор приведением дезактивированного катализатора в контакт с окисляющим веществом, таким как кислород, воздух, озон и хлор. В некоторых случаях предпочтительно постоянно или периодически подавать в реакционную систему активное вещество, такое как кислород, озон, хлор, фторид хлора, трифторид хлора, оксид азота или закись азота для поддержания срока службы катализатора.
В соответствии с настоящим изобретением температура реакции фторирования второго пергалоидированого циклопентена предпочтительно составляет от 40 до 800oС и изменяется в зависимости от типа и способа подготовки металла катализатора фторирования. Например, даже если металлом катализатора является хром, марганец, кобальт, никель, железо, молибден, ниобий, алюминий, цинк, медь или подобный им металл, или же сурьма, титан, олово, тантал или им подобный металл, температура реакции составляет предпочтительно от примерно 150 до примерно 800oС, более предпочтительно от 200 до 750oС, еще предпочтительнее от 250 до 700oС, особенно в случае, когда катализатор фторирования содержит в качестве активных центров такой металл, который находится в состоянии низкой валентности. С другой стороны, в случае, когда катализатор фторирования содержит в качестве активных центров галогенид металла высокой валентности, такого как сурьма, титан, олово, тантал или им подобный металл, температура реакции составляет предпочтительно от примерно 40 до примерно 300oС, более предпочтительно от 50 до 250oС и еще предпочтительнее от 60 до 200oС. Если температура реакции ниже, чем нижний предел (150 или 40oС), скорость реакции может стать слишком низкой и, таким образом, непрактичной. Если температура реакции слишком высокая, скорость реакции становится высокой. Однако при этом катализатор фторирования может разрушиться и, кроме того, это неэкономично, поскольку требует большого количества тепловой энергии.
В соответствии с настоящим изобретением молярное отношение второго пергалоидированного циклопентена, подаваемого в реакционную зону, к фториду водорода изменяется в зависимости от конкретных типов первого и второго пергалоидированных циклопентенов и температуры реакции. Например, если предполагают получить 1,2-дихлор-3,3,4,4,5,5-гексафторциклопентен из октахлорциклопентена, молярное соотношение (второй пергалоидированный циклопентен/фторид водорода) предпочтительно составляет от 1/60 до 1/6, более предпочтительно от 1/50 до 1/8 и еще предпочтительнее от 1/40 до 1/10. Предпочтительно надлежащим образом снизить количество фторида водорода по отношению к количеству второго пергалоидированного циклопентена в случае, когда второй пергалоидированный циклопентен находится в частично фторированной форме, или когда предполагают получать первый пергалоидированный циклопентен с более низкой степенью фторирования. Если количество фторида водорода избыточное, то производительность в единицу времени может стать слишком низкой. С другой стороны, если оно слишком мало, конверсия и выход продукта могут стать слишком низкими.
В соответствии с настоящим изобретением давление реакции фторирования ничем особенно не лимитировано. Предпочтительно, чтобы оно составляло от 1 до 10 кг/см2 в зависимости от выбора оборудования. Предпочтительно подбирать подходящие условия реакции, при которых промежуточные вещества и фторид водорода, присутствующие в реакционной системе, по существу не сжижаются, то есть они не находятся в виде капель жидкости. Время контакта при фторировании составляет предпочтительно от 0,1 до 300 с, более предпочтительно от 1 до 100 с и еще предпочтительнее от 5 до 50 с.
В соответствии с настоящим изобретением реактор, используемый при фторировании, предпочтительно выполнен из теплостойкого материала, устойчивого к коррозионному воздействию фторида водорода, хлорида водорода и им подобных веществ, такого как нержавеющая сталь, хастелой, монель-металл или платина, или материал, футерованный одним из этих металлов. В настоящем изобретении продукты реакции (первый пергалоидированный циклопентен) можно очищать традиционным способом очистки без каких-либо особых ограничений. В этом способе, например, продукты реакции совместно с хлоридом водорода и непрореагировавшим фторидом водорода выводят из реактора в форме газа или жидкости. Затем их промывают водой и/или раствором основания или подвергают обработке типа перегонки или жидкофазного разделения для удаления хлорида водорода и избыточного количества фторида водорода. Затем оставшиеся кислотные вещества удаляют основным веществом или ему подобным с последующей ректификацией, получая таким образом первый пергалоидированный циклопентен.
Как было определено выше, после стадии (а) возможно проведение стадии (b) фторирования продукта реакции стадии (а) способом, отличным от способа стадии (а). Если стадия (b) осуществляется после стадии (а), то вышеупомянутая ректификация необязательна и зависит от способа фторирования на стадии (а). На самом деле, в некоторых случаях может быть необходимо только удаление между стадиями (а) и (b) кислотных веществ из продуктов реакции. В других случаях даже удаление кислотных веществ из них может оказаться необязательным, а достаточным может быть лишь снижение содержания кислоты в продуктах реакции. Например, стадия (b) может быть фторированием, при котором атом хлора в продуктах реакции стадии (а) замещают атомом фтора из фторида металла. Этот фторид металла обычно применяют при фторировании хлорированных алканов и хлорированных алкенов. На стадии (b) можно фторировать смесь первых пергалоидированных циклопентенов. Фторид металла, используемый на стадии (b), ничем особенно не ограничен и может являться фторидом щелочного металла, таким как фторид лития, фторид натрия, фторид калия, фторид цезия и фторид рубидия. Из них наиболее предпочтительными являются фторид калия и фторид цезия. Количество молей этого фторида металла составляет предпочтительно по меньшей мере В, более предпочтительно от В до 10В и еще более предпочтительно от В до 5В на 1 моль сырьевого материала на стадии (b), то есть первого пергалоидированного циклопентена, представленного общей формулой С5СlвF8-в, где В является целым числом от 0 до 7. В соответствии с необходимостью возможно использование растворителя на стадии (b). Примерами такого растворителя являются амиды кислот, такие как формамид, ацетамид, N, N-диметилформамид, N, N-диметилацетамид и N-метилпиролидон, и сульфоксиды, такие как диметилсульфоксид и диэтилсульфоксид. При необходимости такой растворитель можно смешать с углеводородом (например, ксилолом), совместимым с этим растворителем. Температура реакции на стадии (b) предпочтительно не выше 200oС, более предпочтительно от 60 до 180oС и еще предпочтительнее от 80 до 150oС. Время реакции на стадии (b) определяют в зависимости от типа фторида металла на стадии (b), и оно может составлять до 24 часов.
В случае, когда хлоруглеводород фторируют в газовой фазе фторидом водорода в присутствии катализатора, обычно эти хлоруглеводород и фторид водорода предварительно переводят в паровую фазу нагреванием, а затем вводят в зону реакции такого фторирования. Этот нагрев обычно проводят при температуре, близкой к температуре разложения сырьевого материала для такого фторирования. В частности, хлоруглеводород, имеющий большее количество атомов хлора, имеет более высокую температуру кипения. Более того, известно, что фторид водорода сам по себе может действовать в качестве катализатора полимеризации или изомеризации. Поэтому вышеуказанный нагрев для парообразования может вызвать полимеризацию хлоруглеводорода. С течением времени полученные соединения с высокой температурой кипения могут превратиться в смолистые вещества или твердый углерод. Эти нежелательные вещества могут забить реактор и покрыть поверхность катализатора, понизив активность катализатора. Если температуру реакции на стадии (а) понизить для того, чтобы предотвратить эти проблемы, конверсия может происходить слишком медленно.
В виду вышеизложенного в соответствии с изобретением предпочтительно при фторировании на стадии (а) обеспечить множество расположенных последовательно реакционных зон и сделать температуру реакции каждой реакционной зоны управляемой независимо. Каждая реакционная зона может представлять собой независимый реактор. Другими словами, можно обеспечить множество реакторов для проведения стадии (а). В альтернативном случае возможно обеспечение лишь одного реактора, имеющего нагревательное устройство для независимого управления температурой каждой реакционной зоны реактора. Другими словами, этот реактор имеет в своем составе множество реакционных зон. Способ проведения стадии (а) ничем особенно не ограничен и может быть способом с неподвижным слоем, способом с псевдоожиженным слоем и способом с движущимся слоем. Из них наиболее предпочтительным является способ с неподвижным слоем. Тип реактора никак особенно не ограничен, предпочтительно использовать реактор однотрубчатого или многотрубчатого типа, имеющий внешнюю нагревательную систему.
В соответствии с настоящим изобретением количество расположенных последовательно реакционных зон составляет предпочтительно по меньшей мере две, и в предпочтительном случае их не более 10, с точки зрения экономики и сложности эксплуатации. Более предпочтительно - от двух до четырех. Здесь первую реакционную зону, или реактор, определяют как зону, в которую вводят сырье стадии (а). Последующие реакционные зоны, или реакторы, до последней реакционной зоны, или реактора, расположены последовательно. Соответствующие температуры реакционных зон можно регулировать независимо друг от друга, в зависимости от сырья, катализатора в каждой реакционной зоне, условий реакции, целевых продуктов реакций. На самом деле, предпочтительно отрегулировать температуру последней зоны реакции, которая является основной реакционной зоной для проведения стадии (а), так чтобы достигнуть требуемой конверсии и селективности. Более того, предпочтительно, чтобы температуры реакционных зон от первой до последней реакционной зоны были отрегулированы по нарастающей. Другими словами, температуры реакционных зон возрастают ступенчато от первой к последней реакционной зоне. Размеры каждой реакционной зоны можно устанавливать произвольно. Фактически, основная реакционная зона в предпочтительном случае больше по размеру, чем первая реакционная зона, для более легкого управления реакцией на стадии (а). В реакционные зоны можно вводить одинаковые или разные катализаторы, но предпочтительно применять одинаковый катализатор во всех реакционных зонах. Согласно изобретению температура основной реакционной зоны связана с требуемой конверсией, селективностью и сроком службы катализатора и составляет предпочтительно от 80 до 800oС. Как было установлено ранее, температура реакции зависит в основном от типа катализатора. Если целевой продукт реакции можно получить при температуре реакции ниже 80oС, то нет необходимости создавать множество реакционных зон. Если температура реакции выше 800oС, скорость реакции может оказаться высокой. Однако срок службы катализатора может стать слишком коротким и потребуется большое количество тепловой энергии.
В соответствии с настоящим изобретением предпочтительно отрегулировать температуру реакции в первой реакционной зоне так, чтобы она была ниже температуры кипения второго пергалоидированного циклопентена. Другими словами, ожидаемая реакция, которая должна протекать в первой реакционной зоне, не обязательно требует, чтобы температура реакции была не ниже точки кипения второго пергалоидированного циклопентена при давлении в реакционной системе. При температуре реакции ниже, чем эта точка кипения, атомы хлора второго пергалоидированного циклопентена могут частично замещаться атомами фтора в первой реакционной зоне. При этом второй пергалоидированный циклопентен может превратиться в низкокипящий промежуточный продукт. Предполагают, что такое частично фторированное низкокипящее промежуточное соединение трудно поддается полимеризации, но эффективно превращается в целевой продукт реакции в последующей реакционной зоне, имеющей температуру выше, чем температура первой реакционной зоны. В результате может оказаться возможным снизить получение нежелательных перфорированных соединений и смолистых веществ, повысить выход целевого продукта реакции и продлить срок службы катализатора фторирования.
В случае, когда фторирование стадии (а) осуществляют, обеспечивая множество реакционных зон, молярное соотношение второго пергалоидированного циклопентена (например октахлорциклопентена), который подают в реакционную зону, и фторида водорода изменяют в зависимости от температуры основной или последней реакционной зоны. Молярное отношение второго пергалоидированного циклопентена к фториду водорода предпочтительно составляет от 1/60 до 1/6, более предпочтительно от 1/50 до 1/8 и еще предпочтительнее от 1/40 до 1/10. Предпочтительно в достаточной степени понизить количество фторида водорода по отношению к количеству второго пергалоидированного циклопентена в том случае, если этот второй пергалоидированный циклопентен находится в частично фторированной форме, или если предполагают получать первый пергалоидированный циклопентен с более низкой степенью фторирования. Если количество фторида водорода слишком мало, конверсия и выход могут стать слишком низкими. Даже если количество фторида водорода избыточно, он будет регенерирован и отделен после реакции и затем снова использован в качестве сырья в реакции. Таким образом, это не вызывает каких-либо проблем.
В соответствии с настоящим изобретением предпочтительно вводить в первую реакционную зону фторид водорода в количестве, большем, чем для проведения реакции в первой реакционной зоне. На самом деле температуру первой реакционной зоны можно отрегулировать до относительно низкой величины введением в нее достаточного количества фторида водорода. Этот фторид водорода может быть частично заменен на инертный газ. Однако предпочтительно избегать введения такого инертного газа, если только использование инертного газа не имеет других полезных эффектов. При исключении инертного газа отделение и очистка продукта реакции облегчаются. Так, особенно предпочтительно, чтобы инертный газ по существу не присутствовал в каждой реакционной зоне. Предпочтительно вводить общее количество фторида водорода, необходимого на стадии (а), в первую реакционную зону для того, чтобы снизить температуру реакции, хотя это и необязательно.
В соответствии с настоящим изобретением предпочтительно подогревать второй пергалоидированный циклопентен при температуре, которая примерно на 1-50oС ниже температуры кипения второго пергалоидированного циклопентена, а фторид водорода подогревать при температуре, близкой к температуре первой реакционной зоны. Хотя эти соединения можно подогревать одновременно в одном сосуде, предпочтительно подогревать эти соединения в различных сосудах.
В соответствии с настоящим изобретением предпочтительно с точки зрения проведения реакции удалять хлорид водорода, который образовался в одной реакционной зоне, до попадания его в следующую реакционную зону, поскольку этот хлорид водорода является нежелательным веществом при проведении следующей реакции в последующей реакционной зоне с точки зрения химического равновесия. Однако удаление хлорида водорода часто вызывает одновременное удаление непрореагировавшего фторида водорода. Поэтому удаление этого хлорида водорода необязательно. С другой стороны, если в каждой реакционной зоне образуются небольшие количества высококипящих органических веществ, может быть предпочтительным удаление оттуда этих органических веществ для того, чтобы продлить срок службы катализатора. Способ такого удаления никак особенно не ограничен. Например, их можно удалять адсорбцией активированным углем, абсорбцией серной кислотой или растворителем, разделением при помощи охлаждающего сжижения.
В соответствии с настоящим изобретением, когда октахлорциклопентен фторируют на стадии (а) путем обеспечения множества реакционных зон, в реакционных зонах получают промежуточные продукты или продукты реакции, такие как октафторциклопентен, 1-хлор-2,3,3,4,4,5,5-гептафторциклопентен, 1,2-дихлор-3,3,4,4,5,5-гексафторциклопентен, трихлорпентафторциклопентен, тетрахлортетрафторциклопентен, пентахлортрифторциклопентен, гексахлордифторциклопентен и гептахлормонофторциклопентен. В настоящем изобретении промежуточные соединения ничем особенно не ограничены, поскольку они являются хлорфторциклопентенами.
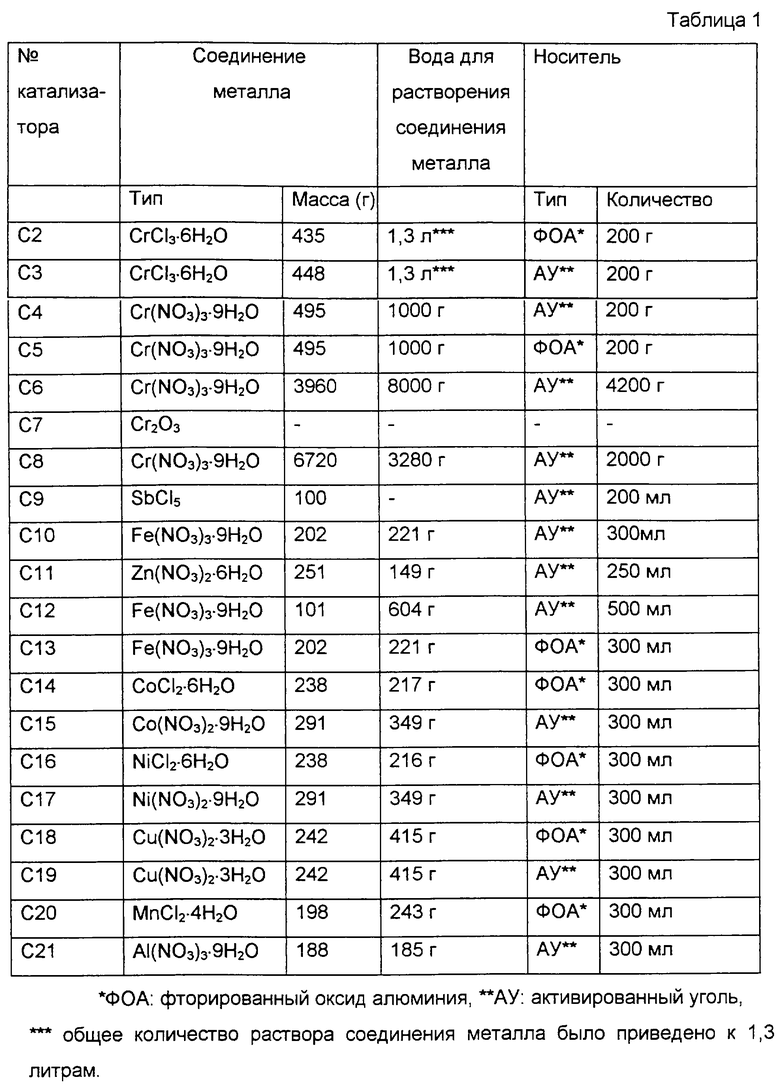
Последующие варианты приготовления катализаторов не носят ограничивающий характер и приводятся в качестве иллюстрации настоящего изобретения. Катализаторы фторирования С2-С21 готовили в соответствии с вариантами 2-21 приготовления катализаторов.
1 ВАРИАНТ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА
Сначала 800 г активированного оксида алюминия, KHS-46 (торговое наименование) SUMITOMO CHEMICAL CO., LTD с диаметром частиц 4-6 мм промыли водой для удаления пыли с его поверхности. Отдельно 306 г фторида водорода (безводной фтористоводородной кислоты) растворили в 2,760 г воды для приготовления 10% водного раствора фторида водорода. Затем этот раствор постепенно прилили к активированному оксиду алюминия с последующим перемешиванием. После этого его оставили на 3 ч, затем активированный оксид алюминия удалили из раствора, промыли его водой, потом отфильтровали, а затем сушили в течение 2 ч в электрической печи при 200oС. Высушенный активированный оксид алюминия в количестве 800 мл ввели в цилиндрическую реакционную трубку из нержавеющей стали (SUS 304), имеющую внутренний диаметр 4,2 см и продольную длину 60 см. Температуру реакционной трубки повысили в печи до 200oС, при этом через реакционную трубку пропускали азот. Затем через нее совместно с азотом пропускали фторид водорода для того, чтобы фторировать активированный оксид алюминия. Во время проведения этого фторирования температура повышалась. Однако скорости потоков азота и фторида водорода отрегулировали так, чтобы температура не была выше 400oС. После прекращения нагрева температуру в печи поддерживали при 400oС в течение 2 ч, чтобы таким образом приготовить фторированный оксид алюминия.
2 ВАРИАНТ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА
Сначала приготовили 1,3 литра раствора СrСl3 растворением 435 г СrСl3•6Н2О в чистой воде, как показано в табл.1. В этот раствор погрузили 200 г фторированного оксида алюминия, полученного в 1 варианте приготовления катализатора, как показано в табл.1, и полученный раствор оставили на один день и одну ночь. Затем оксид алюминия отделили от раствора фильтрованием, а затем сушили в течение одного дня и одной ночи при 100oС в сушильном шкафу с циркуляцией горячего воздуха. Полученный таким образом оксид алюминия на хромовом носителе в количестве 200 мл поместили в цилиндрическую реакционную трубку, снабженную электрической печью, выполненную из нержавеющей стали (SUS 304) и имеющую диаметр 2,5 см и продольную длину 40 см. Температуру реакционной трубки повысили до 300oС, при этом через нее пропускали газообразный азот. Затем, в момент, когда было установлено, что поток из нее прекратился, начали через нее пропускать поток фторида водорода совместно с газообразным азотом. Затем концентрацию фторида водорода в смеси фторида водорода и азота постепенно увеличивали. Когда участок перегрева, получаемый при фторировании оксида алюминия на хромовом носителе, достигал конца выхода из реакционной трубки, температуру реакционной трубки далее повышали до 400oС. Затем эти условия поддерживали в течение 1 ч, получая таким путем катализатор фторирования С2.
3 ВАРИАНТ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА
Сначала готовили 1,3 литра раствора СrСl3 растворением 448 г СrСl3•6Н2O в чистой воде, как показано в табл.1. В этот раствор погружали на один день и одну ночь 200 г активированного угля из гранулированной кокосовой скорлупы, приготовленного фирмой Takeda Chemical Industries, Ltd., имеющего торговое наименование GRANULAR SHIRO SAGI GX и состоящего из столбчатых углеродных зерен размером 4-6 меш. Далее активированный уголь отделяли от раствора фильтрованием и затем помещали в баклажанообразную колбу для сушки под вакуумом при 70oС с использованием выпарного аппарата. Полученный таким образом активированный оксид алюминия на хромовом носителе в количестве 200 мл помещали в цилиндрическую реакционную трубку, снабженную электрической печью, выполненную из нержавеющей стали (SUS 304) и имеющую диаметр 2,5 см и продольную длину 40 см. Температуру реакционной трубки повышали до 300oС, в то время как через нее протекал газообразный азот. Затем, в момент, когда определяли, что поток из нее прекратился, начинали пропускать через нее фторид водорода совместно с газообразным азотом. Затем концентрацию фторида водорода в смеси фторида водорода и азота постепенно увеличивали. В дальнейшем температуру реакционной трубки увеличивали до 350oС. Затем эти условия сохраняли в течение 1 ч, получая таким путем катализатор фторирования С3.
4 ВАРИАНТ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА
Сначала растворяли 495 г Сr(NО3)3•9Н2O в 1000 г чистой воды. В полученный раствор погружали на один день и одну ночь 200 г гранулированного активированного угля, такого же, как и в варианте 3 приготовления катализатора. Далее активированный уголь отделяли от раствора фильтрованием и затем помещали его в баклажанообразную колбу для сушки под вакуумом при 70oС с использованием выпарного аппарата. После сушки температуру увеличили до 150oС при атмосферном давлении для пиролиза нитрата. Этот нагрев останавливали в момент, когда прекращалось образование NО2. Полученный активированный уголь с нанесенным хромом в количестве 200 мл помещали в цилиндрическую реакционную трубку, такую же, как в примере 2 приготовления катализатора. Температуру реакционной трубки повышали до 350oС, в то время как через нее протекал газообразный азот. После этого температуру снижали до 150oС, а затем начинали пропускать через нее фторид водорода совместно с газообразным азотом. При этих условиях температуру реакционной трубки снова повышали до 350oС. Затем эти условия сохраняли в течение 1 ч, получая таким путем катализатор фторирования С4.
5 ВАРИАНТ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА
Сначала растворяли 495 г Сr(NО3)3•9Н2O в 1000 г чистой воды. В полученный раствор погружали на один день и одну ночь 200 г фторированного оксида алюминия, такого же, как и в варианте 1 приготовления катализатора. Далее оксид алюминия отделяли от раствора фильтрованием и затем помещали его в баклажанообразную колбу для сушки под вакуумом при 70oС с использованием выпарного аппарата. После сушки температуру увеличили до 150oС при атмосферном давлении для пиролиза нитрата. Это нагревание останавливали в момент, когда прекращалось образование NO2. Полученный оксид алюминия с нанесенным хромом в количестве 200 мл помещали в цилиндрическую реакционную трубку, такую же, как в примере 2 приготовления катализатора. Температуру реакционной трубки повышали до 400oС, в то время как через нее протекал газообразный азот. Затем, в момент, когда определяли, что поток из нее прекратился, температуру реакционной трубки понижали до 150oС и начинали через нее пропускать фторид водорода совместно с газообразным азотом. При этих условиях температура реакционной трубки снова повышалась до 400oС. Затем эти условия поддерживали в течение 1 ч, получая таким путем катализатор фторирования С5.
6 ВАРИАНТ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА
Сначала растворяли 3960 г Сr(NО3)3•9Н2O в 8000 г чистой воды. В полученный раствор погружали на один день и одну ночь 4200 г гранулированного активированного угля, такого же, как и в варианте 3 приготовления катализатора. Далее активированный уголь отделяли от раствора фильтрованием и затем помещали его в баклажанообразную колбу для сушки под вакуумом при 70oС с использованием выпарного аппарата. После сушки температуру увеличили до 150oС при атмосферном давлении для пиролиза нитрата. Это нагревание останавливали в момент, когда прекращалось образование NO2. Полученный активированный уголь с нанесеннным хромом в количестве 4200 мл помещали в цилиндрическую реакционную трубку, снабженную электрической печью, выполненную из нержавеющей стали (SUS 304) и имеющую диаметр 5,4 см и продольную длину 200 см. Температуру реакционной трубки повышали до 350oС, в то время как через нее пропускали газообразный азот. После этого температуру снижали до 150oС, а затем начинали пропускать через нее фторид водорода совместно с газообразным азотом. При этих условиях температуру реакционной трубки снова повышали до 350oС. Затем эти условия сохраняли в течение 1 ч, получая таким путем катализатор фторирования С6.
7 ВАРИАНТ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА
Сначала гранулированный Сr2O3 в количестве 200 мл от Kojundo Kagaku Kenkyusho Co. , полученный спеканием и имеющий размер частиц не более 4 меш, помещали в цилиндрическую реакционную трубку, такую же, как в варианте 2 получения катализатора. Температуру реакционной трубки повышали до 350oС, в то время как через нее пропускали газообразный азот. После этого температуру понижали до 150oС и затем начинали пропускать через нее фторид водорода совместно с газообразным водородом. При этих условиях температуру реакционной трубки снова повышали до 350oС. Затем эти условия сохраняли в течение 1 ч, получая таким путем катализатор фторирования С7.
8 ВАРИАНТ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА
Сначала растворяли 6,72 кг Сr(NО3)3•9Н2O в 3,28 кг чистой воды. В полученный раствор погружали на один день и ночь 2,00 кг гранулированного активированного угля, такого же, как и в примере 3 приготовления катализатора. Далее активированный уголь отделяли от раствора фильтрованием и затем помещали в баклажанообразную колбу для сушки под вакуумом при 70oС с использованием выпарного аппарата. После сушки температуру увеличили до 150oС при атмосферном давлении для пиролиза нитрата. Это нагревание останавливали в момент, когда прекращалось образование NO2, получая, таким образом, катализатор фторирования С8.
9 ВАРИАНТ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА
Сначала гранулированный активированный уголь, такой же, как в варианте 3 приготовления катализатора, сушили под вакуумом при 100-120oС. Затем 200 мл этого активированного угля поместили в баклажанообразную колбу емкостью 300 мл. Затем 100 г пентахлорида сурьмы прилили по каплям в колбу при температуре не выше 50oС, при этом содержимое колбы хорошо перемешивали. Потом 200 мл полученного катализатора поместили в цилиндрическую реакционную трубку, такую же, как и в примере 2 приготовления катализатора. Затем температуру реакционной трубки повысили от комнатной температуры до 150oС и далее поддерживали при 150oС в течение 1 ч, и в это время в реакционную трубку вводили азот со скоростью 1,2 л в час и в то же самое время в реакционную трубку вводили с расходом 36 г/ч фторид водорода, приведенный в газообразное состояние в газогенераторе, находящемся в верхней части реакционной трубки. Затем подачу фторида водорода прекратили и реакционную трубку охладили до комнатной температуры. После этого, при введении в реакционную трубку хлора с расходом 300 мл/ч, температуру реакционной трубки повысили от комнатной температуры до 150oС. Затем эти условия поддерживали в течение часа, получая таким путем катализатор фторирования С9.
10, 11, 12, 15, 17, 19 И 21 ВАРИАНТЫ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРОВ
В этих примерах приготовления катализаторов повторяли вариант 4 приготовления катализатора, за исключением того, что тип и количество соединения металла, количество воды для растворения соединения металла, а также количество активированного угля по варианту 3 приготовления катализатора было изменено, как показано в табл.1. На основании этого были приготовлены катализаторы фторирования С10, С11, С12, С15, С17, С19 и С21.
13 И 18 ВАРИАНТЫ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРОВ
В этих примерах приготовления катализаторов повторяли вариант 5 приготовления катализатора, за исключением того, что тип и количество соединения металла, количество воды для растворения соединения металла, а также количество фторированного оксида алюминия по варианту 1 приготовления катализатора было изменено, как показано в табл.1. На основании этого были приготовлены катализаторы С13 и С18.
14, 16 И 20 ВАРИАНТЫ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРОВ
В этих примерах приготовления катализаторов повторяли вариант 3 приготовления катализатора, за исключением того, что тип и количество соединения металла, количество воды для растворения соединения металла, а также количество фторированного оксида алюминия по варианту 1 приготовления катализатора было изменено, как показано в табл.1. На основании этого были приготовлены катализаторы С14, С16 и С20.
Нижеследующие примеры не ограничивающего характера приводятся в качестве иллюстрации настоящего изобретения.
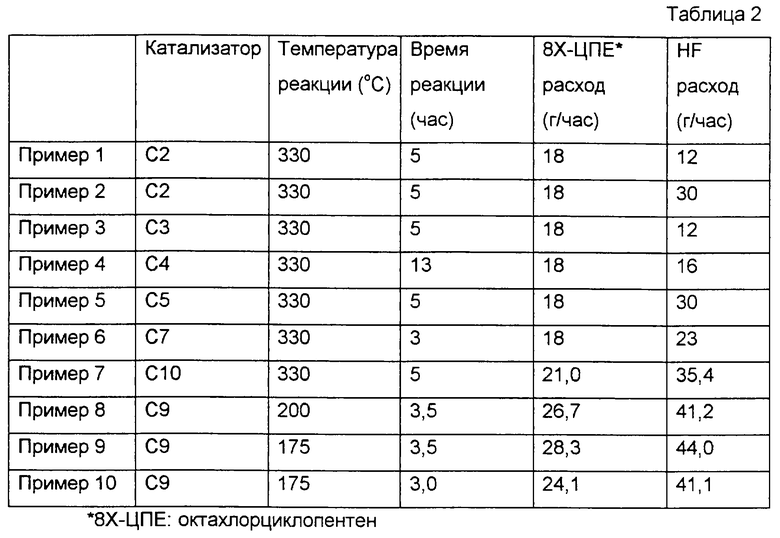
ПРИМЕР 1
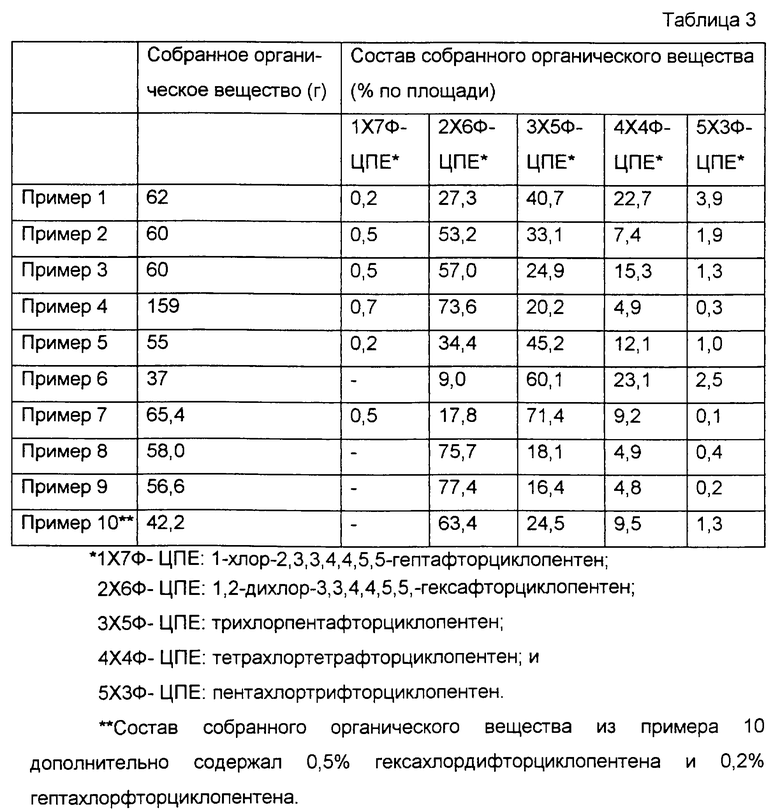
Сначала 190 мл катализатора газофазного фторирования, приготовленного по варианту 2 приготовления катализатора, поместили в цилиндрическую реакционную трубку, снабженную электрической печкой, выполненную из нержавеющей стали (SUS 304), имеющую диаметр 2,5 см и продольную длину 40 см. Затем, пока через трубку пропускали газообразный азот с расходом около 1,2 л/ч, температуру реакционной трубки повышали до 300oС. Затем вместе с газообразным азотом через трубку пропускали фторид водорода с расходом 12 г/ч. Потом температуру реакционной трубки повышали далее. Когда она достигала 330oС, поток газообразного азота останавливали. Затем, как показано в табл.2, начинали пропускание фторида водорода и октахлорциклопентена с расходами 12 г/ч и 18 г/ч соответственно. Таким образом, в сумме 90 г октахлорциклопентена было подано в реакционную трубку, а газообразный компонент, образовавшийся в реакционной трубке, был собран в ловушке с ледяной водой. Таким образом, было собрано 62 г органического вещества. При помощи анализа с использованием газовой хроматографии, как показано в табл.3, было обнаружено, что это органическое вещество содержит 27,3% 1,2-дихлор-3,3,4,4,5,5-гексафторциклопентена, 40,7% трихлорпентафторциклопентена, 22,7% тетрахлортетрафторциклопентена и 3,9% пентахлортрифторциклопентена. Эти процентные содержания определены из хроматограмм по площади, а в качестве детектора в газовой хроматографии использовали ПИД.
ПРИМЕРЫ 2-7
В этих примерах повторяли пример 1, за исключением того, что тип катализатора фторирования время реакции и расходы октахлорциклопентена и фторида водорода были модифицированы, как показано в табл.2. Результаты приведены в табл.3.
ПРИМЕР 8
Сначала 200 мл активированного угля по варианту 9 приготовления катализатора поместили в цилиндрическую реакционную трубку, такую же, как в примере 1. Затем, пока поток азота и фторида водорода проходил через реакционную трубку при температуре 50oС с соответствующими расходами 1,2 л/ч и 41,2 г/ч, 93,3 г октахлорциклопентена и 144,3 г фторида водорода подавали в реакционную трубку в течение 3,5 ч. Газовый компонент, образовавшийся в реакционной трубке, собрали в ловушке с ледяной водой. Полученное органическое вещество анализировали таким же способом, как в примере 1. Результаты показаны в табл.3.
ПРИМЕР 9
Сначала 100 мл активированного угля по варианту 9 приготовления катализатора поместили в цилиндрическую реакционную трубку, такую же, как в примере 1. Затем, пока поток азота, хлора и фторида водорода проходил через реакционную трубку при температуре 50oС с соответствующими расходами 1,2 л/ч, около 150 мл/ч и 44 г/ч, температуру реакционной трубки повысили до 175oС. В этот момент поток азота остановили, а затем 99,1 г октахлорциклопентена и 153,9 г фторида водорода подавали в реакционную трубку в течение 3,5 ч. Газовый компонент, образовавшийся в реакционной трубке, собрали в ловушке с ледяной водой. Полученное органическое вещество анализировали таким же способом, как в примере 1. Результаты показаны в табл.3.
ПРИМЕР 10
В этом примере повторяли пример 9 за исключением того, что условия реакции были изменены, как показано в табл.2. Результаты приведены в табл.3.
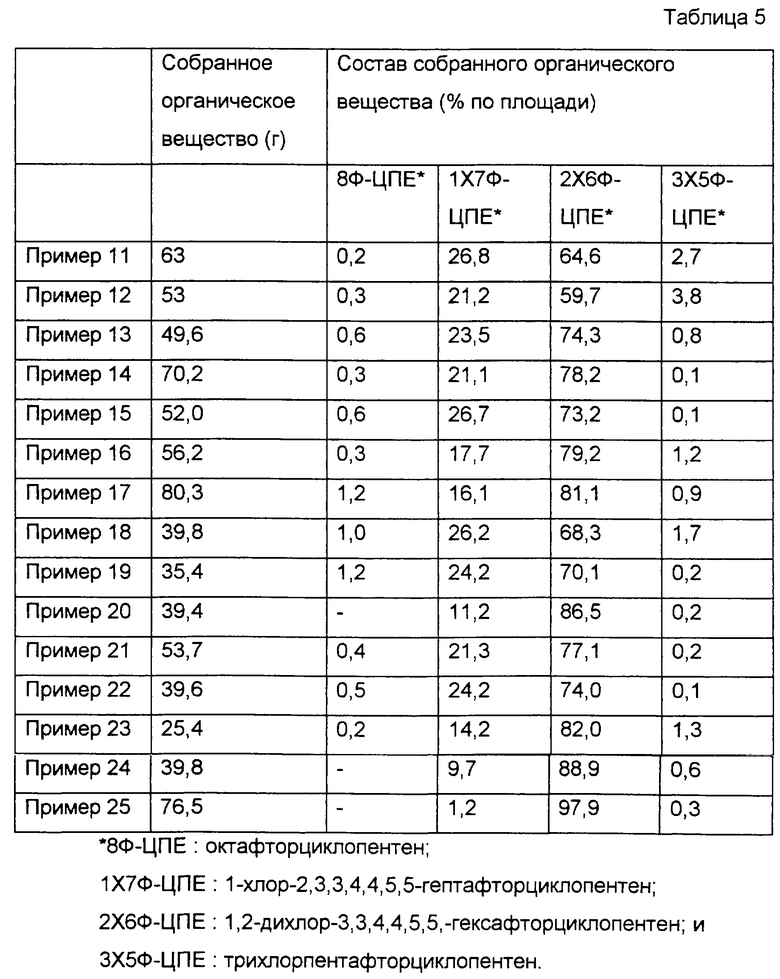
ПРИМЕР 11
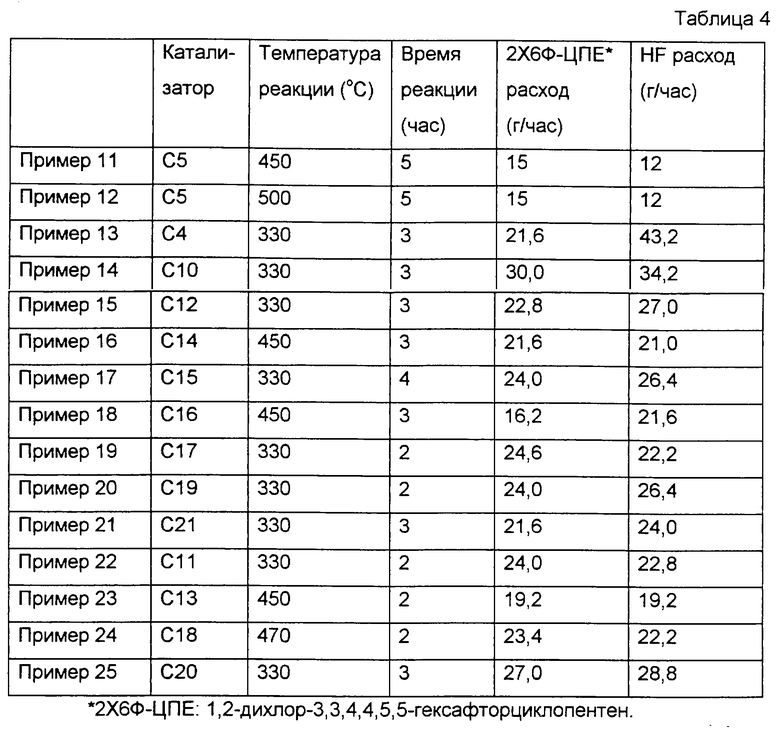
Сначала 190 мл фторированного оксида алюминия по варианту 5 приготовления катализатора поместили в цилиндрическую реакционную трубку, такую же, как в примере 1. Затем, пока через трубку пропускали поток газообразного азота с расходом около 1,2 л/ч, температуру реакционной трубки повышали до 300oС. Затем вместе с газообразным азотом через трубку начинали пропускать фторид водорода с расходом 12 г/ч. Потом температуру реакционной трубки повышали дополнительно до 450oС, и в этот момент поток газообразного азота останавливали. Затем в реакционную трубку начинали подачу фторида водорода и 1,2-дихлор-3,3,4,4,5,5-гексафторциклопентена (2Х6Ф-ЦПЕ) с соответствующими расходами 12 г/ч и 15 г/ч, как показано в табл.4. Всего вводили 75 г 2Х6Ф-ЦПЕ, а газообразный компонент, образовавшийся в реакционной трубке, собирали в ловушке с ледяной водой. Полученное органическое вещество анализировали таким же способом, как в примере 1. Результаты показаны в табл.5.
ПРИМЕРЫ 12-25
В этих примерах повторяли пример 11 за исключением того, что условия реакции были модифицированы, как показано в табл. 4. Результаты приведены в табл.5.
ПРИМЕР 26
Сначала в реактор емкостью 300 мл загрузили 59,5 г сухого фторида калия и 114 г N,N-диметилформамида с последующим нагревом до 130oС. Затем 100 г хорошо высушенных продуктов реакции из примера 4, являющихся смесью пергалоидированных циклопентенов, состав которой приведен в табл.3, добавили по каплям в реактор в течение 4,9 ч, и в то же самое время органическое вещество, получаемое в реакторе, отгоняли при температуре 27-28oС, получив таким путем 76,3 г октафторциклопентена (выход 90%).
ПРИМЕР 27
В этом примере использовали реактор фторирования, состоящий из первого и второго реакторов, соединенных друг с другом последовательно трубопроводами. В первый и второй реакторы загрузили активированный уголь (катализатор фторирования в газовой фазе) из варианта 8 приготовления катализатора в количествах 400 мл и 2,0 л соответственно. Первый реактор представлял собой цилиндрическую реакционную трубку из нержавеющей стали (SUS304) с электрическим нагревателем для ее нагрева, имеющую диаметр 4,2 см и продольную длину 40 см. Второй реактор представлял собой цилиндрическую реакционную трубку рубашечного типа, нагреваемую теплоносителем, выполненную из нержавеющей стали (SUS304) и имеющую диаметр 5,4 см и продольную длину 100 см.
Предварительное устройство первого реактора для стабилизации в нем катализатора осуществляли следующим образом. В то время как газообразный азот пропускали через первый реактор с расходом около 12 л/ч, температуру первого реактора повысили до 350oС. Затем температура первого реактора была снижена до 150oС. После этого начали пропускать через него фторид водорода вместе с газообразным азотом. Во время постепенного повышения концентрации фторида водорода температуру первого реактора повысили до 330oС. В этот момент поток газообразного азота был прекращен. Затем температуру первого реактора понизили до 250oС. После этого фторид водорода, который был нагрет до 250oС при прохождении через подогревающее устройство, подали в первый реактор с расходом 330 г/ч.
Предварительное устройство второго реактора для стабилизации в нем катализатора осуществляли следующим образом. В то время как газообразный азот пропускали через второй реактор с расходом около 12 л/ч, температуру второго реактора повысили до 350oС. Затем температура второго реактора была снижена до 150oС. После этого начали пропускать через него фторид водорода вместе с газообразным азотом. Во время постепенного повышения концентрации фторида водорода температуру второго реактора повысили до 330oС. В этот момент поток газообразного азота был прекращен. Затем фторид водорода, выходящий из первого реактора, пропускали через второй реактор.
Затем октахлорциклопентен, который был подогрет до 250oС, подали в виде жидкости в первый реактор с расходом 210 г/ч. После начала реакции на выходе из первого реактора отбирали образцы образующегося газового компонента. Затем кислотные газы (фторид водорода и хлорид водорода) удаляли из газового компонента, а полученный газовый компонент анализировали при помощи газовой хроматографии. При этом было обнаружено, что газовый компонент имеет состав:
2,3% 1,2-дихлор-3,3,4,4,5,5-гексафторциклопентена,
14,3% трихлорпентафторциклопентена,
39,9%тетрахлортетрафторциклопентена,
32,7% пентахлортрифторциклопентена,
8,0% гексахлордифторциклопентена
и 0,8% гептахлормонофторциклопентена.
Реакцию продолжали таким же образом, как описано выше. Таким образом, всего подали 42 кг октахлорциклопентена в первый реактор с расходом 210 г/ч за 200 ч, а образовавшийся газ, выходящий из второго реактора, собрали в ловушке с ледяной водой. При этом было получено 26,4 кг органического вещества. Это органическое вещество было проанализировано методом газовой хроматографии, и установлено, что оно имеет состав:
2,0% 1-хлор-2,3,3,4,4,5,5-гептафторциклопентена,
77,0% 1,2-дихлор-3,3,4,4,5,5-гексафторциклопентена,
16,3% трихлорпентафторциклопентена,
3,7%тетрахлортетрафторциклопентена
и 0,3% пентахлортрифторциклопентена.
После проведения реакции визуально было проверено состояние подогревательных устройств для октациклопентена и фторида водорода, а также состояние катализатора в непосредственной близости от входа в первый реактор. При этом органическое вещество, смолистые вещества и углеродные остатки не были обнаружены.
ПРИМЕР 28
Сначала цилиндрическую реакционную трубку, снабженную электрической печкой, выполненную из нержавеющей стали (SUS 304) и имеющую диаметр 2,5 см и продольную длину 40 см, заполнили 4,2 литрами активированного угля из варианта 6 приготовления катализатора. Затем, пока через реакционную трубку пропускали газообразный азот с расходом около 30 л/ч, температуру реакционной трубки повышали до 300oС. Затем вместе с газообразным азотом через реакционную трубку пропускали фторид водорода с расходом 501 г/ч. Температуру реакционной трубки далее повысили до 330oС, и в это время поток газообразного азота был остановлен. Затем начали пропускать октахлорциклопентен с расходом 305 г/ч, в то время как фторид водорода пропускали с расходом 501 г/ч. Таким же образом в реакционную трубку подали 82,35 кг октахлорциклопентена за 270 ч, а образовавшийся газ, выходящий из реакционной трубки, собирали в предварительно охлажденном до -15oС приемник из нержавеющей стали (SUS16). Собранные продукты реакции (фторид водорода и органическое вещество) были разделены на органический и водный слои. Отделенный органический слой промыли 10% водным раствором гидрокарбоната натрия, а затем высушили безводным хлоридом кальция. При этом было получено 51,53 кг органического вещества. Анализом при помощи газовой хроматографии было определено, что органическое вещество имеет состав:
1,37% 1-хлор-2,3,3,4,4,5,5-гептафторциклопентена,
77,6% 1,2-дихлор-3,3,4,4,5,5-гексафторциклопентена,
16,5% трихлорпентафторциклопентена,
3,6%тетрахлортетрафторциклопентена
и 0,3% пентахлортрифторциклопентена.
После проведения реакции визуально были проверены состояния подогревательных устройств для октациклопентена и фторида водорода, а также состояние катализатора в непосредственной близости от входа в реакционную трубку. При этом в устройстве для подогрева октациклопентена были обнаружены значительные количества углеродных остатков. Таким образом, предполагается, что произошло разложение октахлорциклопентена. Более того, углеродные остатки были найдены также на катализаторе в непосредственной близости ко входу в реакционную трубку.
Содержание патентов Японии 9-252222, поданного 17 сентября 1997 г, и 10-194140, поданного 9 июля 1998 г, включая описания, формулы изобретений и рефераты, полностью включено в настоящее описание путем ссылки на них.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ КОЛИЧЕСТВЕННОГО АНАЛИЗА НЕОЧИЩЕННОГО ФТОРМЕТИЛ-1,1,1,3,3,3-ГЕКСАФТОРИЗОПРОПИЛОВОГО ЭФИРА И СПОСОБ ПОЛУЧЕНИЯ ФТОРМЕТИЛ-1,1,1,3,3,3-ГЕКСАФТОРИЗОПРОПИЛОВОГО ЭФИРА | 1995 |
|
RU2132055C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1,2-ДИХЛОР-3,3,4,4,5,5-ГЕКСАФТОРЦИКЛОПЕНТЕНА | 2006 |
|
RU2318792C2 |
| Способ получения гексафторизопропанола и фторметилгексафторизопропилового эфира (севофлурана) | 2016 |
|
RU2629366C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФТОРИРОВАННОГО КЕТОНА | 2001 |
|
RU2279422C2 |
| КАТАЛИЗАТОР ДЛЯ ФТОРИРОВАНИЯ НИЗШИХ АЛИФАТИЧЕСКИХ ГАЛОИДУГЛЕВОДОРОДОВ И СПОСОБ ФТОРИРОВАНИЯ НИЗШИХ АЛИФАТИЧЕСКИХ ГАЛОИДУГЛЕВОДОРОДОВ | 1992 |
|
RU2040333C1 |
| СПОСОБ ПОЛУЧЕНИЯ ФТОРИРОВАННОГО СУЛЬФОНИЛФТОРИДА | 2004 |
|
RU2379285C2 |
| СПОСОБ ПРОИЗВОДСТВА ГАЛОГЕНИРОВАННОГО АЛКЕНОВОГО СОЕДИНЕНИЯ И ФТОРИРОВАННОГО АЛКИНОВОГО СОЕДИНЕНИЯ | 2020 |
|
RU2793785C2 |
| СПОСОБ ПОЛУЧЕНИЯ СОДЕРЖАЩЕГО АТОМ ФТОРА СУЛЬФОНИЛФТОРИДНОГО СОЕДИНЕНИЯ | 2001 |
|
RU2278854C2 |
| СОЛЬ ПЕРФТОРКАРБОНОВОЙ КИСЛОТЫ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2007 |
|
RU2453529C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФТОРИРОВАННОГО СЛОЖНОГО ЭФИРА | 2002 |
|
RU2291145C2 |
Изобретение относится к получению хлорфторциклопентенов и октафторциклопентену, используемых в качестве промежуточных продуктов для получения 1,2,3,3,4,4,5,5-октафторциклопентана. Способ получения 1,2-дихлор-3,3,4,4,5,5-гексафторциклопентана осуществляют путем фторирования октахлорциклопентена фторидом водорода в газовой фазе в присутствии катализатора фторирования. Катализатор включает соединение, выбранное из группы, состоящей из CrCl3•6Н2О, Cr(NО3)3•9Н2O, Cr2О3, SbCl5, Fe(NO3)3•9H2O,
Zn(NO3)2•6Н2О, СоСl2•6Н2О, Со(NО3)2•9Н2O, NiCl2•6Н2О, Ni(NO3)3•9H2O Cu(NО3)2•3Н2О, MnCl2•4H2O, Al(NO3)3•9H2O. Катализатор фторирования находится на носителе. В качестве носителя используют активированный уголь, оксид алюминия, частично фторированный оксид алюминия, фторированный алюминий. Температура реакции составляет 150 - 800oС, продолжительность контакта фторида водорода и октахлорциклопентана 0,1 - 300 с, реакционное давление 1 - 10 кг/см2. Продукт реакции, полученный на стадии фторирования октафторциклопентена фторидом водорода, фторируют фторидом металла, заменяя атом хлора в продукте на атом фтора. Получают октафторциклопентен. Фторид металла выбирают из группы, включающей фторид лития, фторид натрия, фторид калия, фторид цезия и фторид рубидия. Технический результат - улучшение технологических параметров процесса. 2 с. и 9 з.п. ф-лы, 5 табл.
MnCl2•4H2O, Al(NO3)3•9H2O, и (б) фторирования продукта реакции стадии (а) в указанный октафторциклопентен фторидом металла, чтобы таким образом заменить атом хлора в указанном продукте реакции стадии (а) на атом фтора указанного фторида металла.
| US 3178482 А, 13.04.1965 | |||
| US 3258500 A, 28.07.1966 | |||
| US 3859372 A, 07.01.1975 | |||
| US 3514253 A, 29.05.1970 | |||
| Ф9?1Д ЭНеПЕРТОВ | 0 |
|
SU383367A1 |
