Изобретение относится к применению производных теофиллина по меньшей мере с функцией простого эфира в структурно модифицированном 1-положении метилового остатка для получения лекарственных средств для лечения и профилактики состояний шока, к новым соединениям ксантина с вышеупомянутыми замещениями и к способам их получения.
Шок определяют как появляющееся острое состояние неадекватной питательной перфузии жизненно важных органов, которое всегда означает наибольшую опасность для жизни (Med. Мо. Pharm. 1989, 12/9:279-282).
Причины шока разнообразные. Так, сердечный шок вызывается первичной сердечной недостаточностью вследствие инфаркта миокарда, тяжелыми нарушениями сердечного ритма, недостаточностью сердечной мышцы или другими заболеваниями сердца, гиповолемический шок (геморрагический и травматический шок, а также ожоговый и обезвоживающий шок) вызывается потерями или смещениями жидкости, септический шок вызывается систематическим наносом микробов (грамотрицательные и грамположительные бактерии, грибки, вирусы, простейшие организмы и т.д.) или токсинами и, наконец, анафилактический шок вызывается генерализированными реакциями антиген-антитело. Однако, несмотря на многообразие причин, патогенез и клиническая картина различных форм шока оказываются довольно однообразными (Пширембел, Клинический словарь, Вальтер де Грутер-издательство, 255-е издание, 1986, стр.1513). Ключевую роль всегда играет нарушение функции клеток вследствие недостаточного снабжения ткани кислородом и субстратами (ишемия) и недостаточного удаления токсических продуктов обмена веществ (Medwelt 1989, 40:519-522). Шок является динамическим явлением, течение которого зависит в значительной степени от продолжительности ишемии.
В первой, компенсированной фазе шока организм реагирует нейронно и гормонально управляемой централизацией кровообращения, в результате которой защищаются центральные органы организма (сердце, головной мозг, легкие, печень, почки). Клиническая картина определяется тахикардией, еще нормальным или лишь незначительно пониженным кровяным давлением, гипервентиляцией с дыхательным алкалозом и, как правило, бледной, холодной и влажной кожей; при септическом шоке появляется также лихорадка, связанная иногда с ознобом. Когда исчерпываются механизмы компенсации, то в возрастающей степени ухудшается также капиллярная перфузия центральных органов. Это переходит во вторую, декомпенсированную фазу шока, которая отличается прогрессирующей гибелью клеток и потерей их функций. Состояние шока становится необратимым. Резкое повышение проницаемости стенок сосудов в области микроциркуляции приводит благодаря потере жидкости к увеличению гематокрита, к интерстициальным отекам и к высвобождению медиаторов, которые вызывают, между прочим, диссеминированное, внутрисосудистое свертывание, например, в форме длительной коагулопатии с обтурационными фибринными тромбами в конечном русле. Систематическое снижение сердечного объема в единицу времени и кровяного давления переходит к полному нарушению кровообращения. В конце шокового каскада происходит смерть в результате острого отказа сердца, печени, почек или легких (синдрома удушья, называемого также ARDS=Acute Respiratory Distress Syndrome) или в результате отказа многих органов (MOF=Multi-Organ Failure), если многие органы одновременно теряют свою функцию.
Традиционная терапия ориентируется на клиническую симптоматику и включает срочные меры для устранения угрозы для жизни, как замещение объема (например, крови), искусственное дыхание для профилактики ARDS, назначение вазоактивных фармакологических средств для поддерживания кровообращения, анальгезия и покой, исправление нарушений в кислотно-щелочном балансе; прием гепарина для предотвращения длительной коагулопатии и лечение кортикостероидами для снижения проницаемости мембраны. В зависимости от причины шока показаны и другие меры терапии, например, операция и остановка кровотечения при геморрагическом шоке, удаление очага инфекции и лечение антибиотиками при септическом шоке и возможное лечение при помощи электростимулятора и аортальная противо-пульсация с резиновым баллончиком при кардиогенном шоке. Однако, несмотря на все эти терапевтические меры, результат лечения остается чрезвычайно неудовлетворительным. Так, смертность составляет, например, при кардиогенном шоке по причине инфаркта миокарда 90% и при септическом шоке, который во всем мире чаще всего является причиной смерти в отделениях интенсивной терапии, более 50%.
Это делает понятным требование клиник к более причинно ориентированной программе терапии, которая позволяет преждевременно прерывать по возможности шоковый каскад и улучшать тем самым заметно шанс выживания. Обнадеживающими основаниями для этого являются комплексные патофизиологические процессы, которые лежат в основе прогрессивного течения шокового заболевания. По современному уровню знаний как при септических, так и при асептических формах шока (N. Engl. J. Med. 1993, 328/20: 1471-1477) в результате соответствующего патологического стимула активируется большинство систем медиаторов клеток, ответственных за воспаления, и благодаря этому провоцируется эндотелиальное воспаление с диффузными воспалительными процессами, которые называют также SIRS (Systemic Inflammatory Response Syndrome) (J. Amer. med. Ass. 1992, 268:3452). В центре этого синдрома находится генерализованиое патологическое взаимодействие между активированными гранулоцитами и клетками эндотелия через комплементарные молекулы сцепления, которое при прогрессирующем поражении сосудов приводит к нарушениям в микроциркуляции и к повреждениям органов с возрастающим функциональным ущербом и, наконец, к отказу многих органов. С провоцированием ассоциированных со стенками сосудов воспалительных процессов благодаря гранулоцитноэндотелиальному взаимодействию возникают септические и асептические состояния общего патогенетического конечного звена при развитии шока. Кроме того, имеются обоснованные указания на то, что в течение асептических форм шока очень часто вызванное не микробами начальное нарушение барьерной функции в легком и особенно в желудочно-кишечном тракте приводит к бактериальной транслокации, называемой инвазией бактерий, или их токсических продуктов в кровоток, так что асептические и септические явления наслаиваются (Medwelt 1989, 40:525-532).
Последние опыты по причинному терапевтическому вмешательству направлены теперь на специфические вмешательства в поддерживаемый воспалительными медиаторами болезненный процесс, чтобы как можно раньше прервать патологическую сигнальную цепочку и тем самым своевременно предупредить развитие поражений органов. В широкомасштабных клинических работах были исследованы, например, моноклональные антитела мыши и человека по отношению к эндотоксину (LPS=липополисахариды) из стенок клеток грамотрицательных бактерий, рекомбинантные человека и моноклональные антитела как мыши, так и человека против цитокина TNF (фактор некроза опухоли), полученные генной технологией растворимые TNF-рецепторы и другие TNF-связанные протеины, полученный рекомбинацией, проявляющийся физиологически интерлейкин-1-рецептор-антагонист Антрил (IL-1-RA) и брадикинин-антагонист Брадикор, причем до сих пор не намечается прорыва в терапии (Scrip Magazine, декабрь 1994:50-52). Поэтому продолжаются интенсивные поиски эффективных блокаторов исключительно комплексного патологического процесса, причем все больше приходят к выводу о том, что исключение специфического медиатора с широким сигнальным каскадом имеет лишь небольшие шансы на успех и что прогресса в терапии можно ожидать скорее всего от многофункционального вмешательства, будь то результат комбинации различных избирательно действующих фармацевтических средств или, предпочтительнее, результат монофармацевтических средств с широким фармакологическим спектром действия.
Для испытания препаратов на антишоковое действие были разработаны различные экспериментальные модели животных. Особенно практичную, хорошо стандартизированную и убедительную модель (Proc. Natl. Acad. Sci. США 1979, 76/11: 5939-5943) представляет вызванный эндотоксином (LPS) шок на С 57 ВL/6-мышах, который регулирует клиническую ситуацию настолько реалистично, что одновременным введением галактозамина (GalN) так сильно повышают чувствительность животных относительно LPS, что здесь достаточна сравнительно низкая летальная доза для человека для провоцирования смертельного шокового действия (DN& P 1993, 6/9:641-646; Биоспектр 1995, 1/5:46-52). В этой модели теофиллин (1,3-диметилксантин) при дозах до предела переносимости не проявляет никакого существенного защитного действия.
Неожиданно было найдено, что введение заместителей по меньшей мере с функцией простого эфира в 1-положение метилового остатка молекулы теофиллина дает очень эффективные препараты при одновременно значительно улучшенной переносимости. Известны три соединения этого структурного типа, а именно 3-п-пропилксантин с 2-метоксиэтил-, 2-этоксиэтил- или 3-метоксипропильной группой в 1-положении (J. Med. Chem. 1993, 36/10: 1380-1386), которые на основании бронхорасширительных свойств следует применять для лечения острых астматических недугов, но ссылки на их применение в качестве антишоковых средств отсутствуют.
Изобретение относится к применению по меньшей мере одного соединения формулы I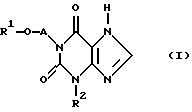
причем
R1 обозначает
a) (C1-C5)-алкил с линейной или разветвленной цепью,
b) (C1-C2)-алкокси-(C1-С3)-алкил или
c) фенил или фенил-(С1-С2)-алкил, где фениловые остатки не замещены или замещены соответственно одним или двумя атомами галогена,
А обозначает
(C1-C4)-алкиленовый мостик с неразветвленной цепью или разветвленной цепью и
R2 обозначает
a) (C1-C5)-алкил с линейной или разветвленной цепью,
b) (С3-C6)-циклоалкил,
c) (С4-С8)-циклоалкил-алкил,
d) фенил или
e) фенил-(C1-C2)-алкил,
для получения лекарственного средства для лечения и профилактики шоковых заболеваний, особенно синдрома SIRS, сепсиса, синдрома сепсиса, септического шока, отказа многих органов (МОF), синдрома ARDS, геморрагического и травматического шока, а также ожогового шока и обезвоживающего шока и подобных шоку осложнений при реперфузионном синдроме и экстракорпоральном кровообращении.
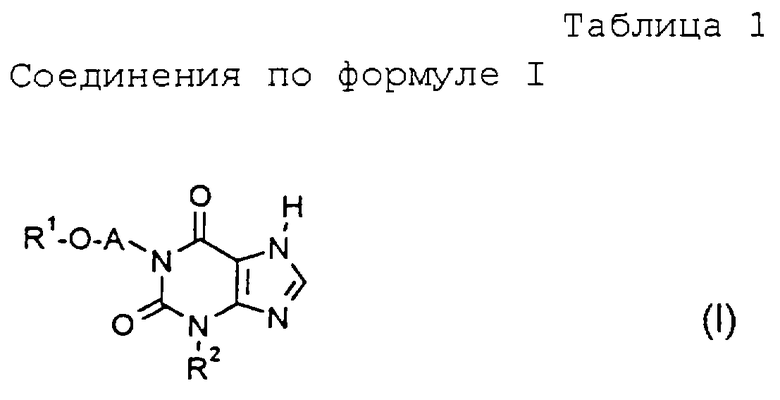
Применяют предпочтительно соединения формулы I, в которой
R1 обозначает
a) (C1-C4)-алкил с линейной или разветвленной цепью,
b) метоксиметил,
c) метоксиэтил,
d) фенил,
e) 4-хлорфенил,
f) бензил или
g) 4-хлорбензил,
А обозначает
(C1-С3)-алкиленовый мостик с неразветвленной цепью и
R2 обозначает
a) (C1-C4)-алкил с прямой или разветвленной цепью,
b) циклопропил,
c) циклопропилметил,
d) фенил или
e) бензил.
Далее предпочитают применять соединения формулы I, в которой
R1 обозначает
(C1-C4)-алкил с прямой или разветвленной цепью,
А обозначает
(C1-С3)-алкиленовый мостик с неразветвленной цепью и
R2 обозначает
(C1-C4)-алкил с прямой или разветвленной цепью, циклопропил или циклопропилметил.
Понятие "(С4-С8)-циклоалкил-алкил" определяет такие алкильные остатки, которые замещены (С3-С6)-циклоалкилом, причем сумма всех С-атомов меньше или равна 8. Они представляют от циклопропил-метила до -пентила, от циклобутил-метила до -бутила, от циклопентил-метила до -пропила, а также циклогексил-метильный и этильный остатки. Атомы галогена представляют собой йод, бром, фтор и, предпочтительно, хлор.
Изобретение относится также к новым соединениям формулы I, в которой
R1 обозначает
a) (C1-C5)-алкил с линейной или разветвленной цепью,
b) (C1-C2)-алкокси-(C1-С3)-алкил или
c) фенил или фенил-(C1-C2)-алкил, где фенильные остатки не замещены или замещены соответственно одним или двумя атомами галогена,
А обозначает
(C1-C4)-алкиленовый мостик с неразветвленной или разветвленной цепью и
R2 обозначает
a) (C1-C5)-алкил с прямой или разветвленной цепью,
b) (С3-C6)-циклоалкил,
c) (С4-С8)-циклоалкил-алкил,
d) фенил или
е) фенил-(С1-С2)-алкил,
причем соединения формулы I, где a) R2 обозначает n-пропил, R1 обозначает метил или этил и А обозначает этиленовый мостик или б) R2 обозначает n-пропил, R1 обозначает метил и А обозначает n-пропиленовый мостик, исключаются.
Предпочитают соединения формулы I, в которой
R1 обозначает
a) (C1-C4)-алкил с линейной или разветвленной цепью,
b) метоксиметил,
c) метоксиэтил,
d) фенил,
e) 4-хлорфенил,
f) бензил или
g) 4-хлорбензил,
А обозначает
(C1-С3)-алкиленовый мостик с неразветвленной цепью и
R2 обозначает
a) (C1-C4)-алкил с линейной или разветвленной цепью,
b) циклопропил,
c) циклопропилметил,
d) фенил или
е) бензил,
причем соединения формулы I, где a) R2 обозначает n-пропил, R1 обозначает метил или этил и А обозначает этиленовый мостик или б) R2 обозначает n-пропил, R1 обозначает метил и А обозначает n-пропиленовый мостик, исключаются.
Далее предпочитают соединения формулы I, в которой
R1 обозначает
(C1-C4)-алкил с линейной или разветвленной цепью,
А обозначает
(C1-С3)-алкиленовый мостик с неразветвленной цепью и
R2 обозначает
(C1-C4)-алкил с линейной или разветвленной цепью, циклопропил или циклопропилметил,
причем соединения формулы I, где а) R2 обозначает n-пропил, R1 обозначает метил или этил и А обозначает этиленовый мостик или б) R2 обозначает n-пропил, R1 обозначает метил и А обозначает n-пропиленовый мостик, исключаются.
Соединения формулы I подвергают депротонированию в 7-положении и поэтому образовывают с основными реактивами соли и сольваты. Для этого принимают в расчет преимущественно фармацевтически приемлемые соли щелочных и щелочно-земельных металлов и соли и сольваты с органическими основаниями, например, этилендиамин или основные аминокислоты лизин, орнитин и аргинин. Таким образом, изобретение относится также к физиологически приемлемым солям и/или к сольватам 1,3-двузамещенных ксантинов согласно формуле I и к их применению в качестве активных веществ в антишоковых средствах.
Соединения формулы I с несимметрично разветвленным алкильным остатком в положении R1 и/или R2 и/или с несимметрично разветвленным алкиленовым мостиком А имеют один или несколько асимметричных С-атомов и могут существовать, следовательно, в стереоизомерных формах. Изобретение включает поэтому как чистые стереоизомерные соединения, так и их смеси и их применение в качестве активных веществ в антишоковых средствах.
Изобретение относится далее к способу-аналогу для получения новых соединений по формуле I, принципиальные варианты осуществления которого описаны в WO 87/00523. Например, действуют таким образом, что
а) 3-замещенный ксантин формулы II,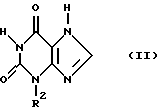
где R2 определен как в формуле I, без агента конденсации или в присутствии основного агента конденсации или в форме его солей подвергают взаимодействию с реагентом формулы III,
Ra - X, (III)
где Ra обозначает легко удаляемую группу, например, удаляемые восстановлением или гидролитически группы бензила, бензгидрила или тритила с незамещенными или замещенными фениловыми кольцами, и Х обозначает галоген, предпочтительно хлор, бром или йод, или альтернативно сложноэфирную сульфокислотную группу или сложноэфирную группу фосфорной кислоты, или
б) 7-замещенный ксантин формулы IV,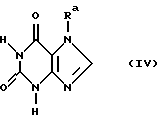
где Ra обозначает бензил с незамещенным или замещенным фенильным остатком, без агента конденсации или в присутствии основного агента конденсации, или в форме его солей подвергают взаимодействию с реагентом формулы V
R2 - X, (V)
где R2 определен как в формуле I и Х определен как в формуле III,
до 3,7-двузамещенного ксантина формулы VI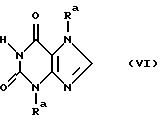
где R2 определен как в формуле I и Ra определен как в формуле III или IV, затем соединение формулы VI без агента конденсации или в присутствии основного агента конденсации или в форме его солей подвергают взаимодействию с агентом алкилирования формулы VII
R1-O-A-X, (VII)
где R1 и А определен как в формуле I и Х определен как в формуле III, до 1,3,7-тризамещенного ксантина формулы VIII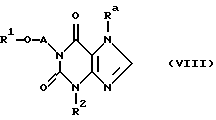
где R1, А и R2 определены как в формуле I и Ra определен как в формуле III или IV,
и последующим удалением отходящей группы Rа из промежуточного продукта формулы VIII получают соединение согласно изобретению формулы I, и это соединение, в случае необходимости, после разделения стереоизомерных форм, переводят, если требуется, в физиологически приемлемую соль.
Применяемые при этом в качестве исходных веществ монозамещенные ксантины формул II и IV и алкилирующие агенты формул III, V и VII большей частью известны и их можно легко получить известными способами. Так, например, 7-бензилксантины формулы IV получают из гуанозина бензилированием, удалением гидролизом остатка сахара и последующим превращением структуры гуанозина в структуру ксантина (Synth. Commun. 1990, 20:2459-2467).
Среди алкилирующих агентов формулы VII, пригодных для введения R1-O-А-боковой цепи в 1-положении структуры ксантина, особое место занимают те соединения, в которых А обозначает группу метилена (А=-СН2-), галогениды которых хотя и применяются с успехом в качестве реагирующих веществ, но при промышленном применении могут возникать, по меньшей мере, проблемы токсикологии. Поэтому в этом специальном случае может быть предпочтительным применение соответствующих сульфонатов, которые можно легко получать, например, взаимодействием смешанных ангидридов алифатических карбоновых кислот и алифатических или ароматических сульфокислот (J. Org. Chem. 1971, 36:528-531) с двузамещенными формальдегидацеталями формулы IX в результате очевидной и почти полностью протекающей реакции (J. Amer. Chem. Soc. 1969, 91:5663-5665):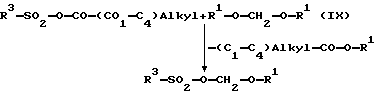
При этом R3 представляет алифатический остаток, как метил, этил, или трифторметил или ароматический остаток, например, фенил, 4-толил или 4-бромфенил, но, предпочтительно, метил или 4-толил, и R1 имеет определенные в формуле I значения.
Реакцию можно проводить как в субстрате, так и в безводном инертном относительно компонентов реакции апротонном растворителе при температурах между -20oС и +40oС, предпочтительно между 0oС и 20oС. Промежуточное выделение высокореактивных, чувствительных к гидролизу и неустойчивых к нагреву сульфонатов не требуется; их целесообразно применять непосредственно в качестве сырья для алкилирования ксантинов VI по атому азота в 1-положении, причем часто обычная добавка основного агента конденсации оказывается излишней.
Взаимодействие моно- и двузамещенных производных ксантина II, IV и VI с соответствующими алкилирующими агентами формулы III, V или VII осуществляют обычно в инертных относительно компонентов реакции диспергаторах или растворителях. В качестве таковых принимают в расчет прежде всего диполярные, апротонные растворители, например, диметилформамид, диметилацетамид, N-метилпирролидон, тетраметилмочевину, триамид гексаметилфосфорной кислоты или диметилсульфоксид; но можно применять также формамид, ацетонитрил, ацетон, бутанон или спирты, как метанол, этиленгликоль и его моно- или ди(С1-С4) простые алкиловые эфиры, этанол, пропанол, изопропанол и различные бутанолы; углеводороды, как бензол, толуол или ксилол; галогенированные углеводороды, как дихлорметан или хлороформ; пиридин и смеси названных растворителей или их смеси с водой. Реакции алкилирования целесообразно проводить в присутствии основного агента конденсации. Для этого применяются, например, гидроокиси, карбонаты, гидриды и алкоголяты щелочных и щелочно-земельных металлов и органические основания, как триалкиламины, например, триэтил- или трибутиламин, гидроокиси четвертичного аммония или фосфония и сшитые смолы с фиксированными, в случае необходимости, замещенными группами аммония или фосфония. Но производные ксантина можно применять также непосредственно в виде их отдельно полученных солей, например, солей щелочных, щелочно-земельных металлов или, в случае необходимости, замещенных солей аммония или фосфония. Далее, соединения ксантина можно легко алкилировать в присутствии вышеназванных, неорганических агентов конденсации, а также в форме их солей щелочных или щелочно-земельных металлов при помощи так называемых катализаторов фазового перехода, например, третичных аминов, четвертичных солей аммония или фосфония или также краун-эфиров, предпочтительно в двухфазной системе в условиях катализа фазового перехода. Подходящими, чаще всего имеющимися в продаже катализаторами фазового перехода являются, в частности, соли тетра(C1-C4)-алкил- и метилтриоктиламмония и -фосфония, метил-, миристил-, фенил- и бензил-три(C1-C4) алкил- и цетилтриметиламмоний-, а также (C1-C12)алкил- и бензил- соли трифенилфосфония, причем более эффективными оказываются, как правило, те соединения, которые имеют больший и симметричный по структуре катион.
Описанные выше способы осуществляют, в общем, при температуре между 0oС и точкой кипения применяемой реакционной среды, предпочтительно между 20oС и 130oС, в случае необходимости, при повышенном или пониженном давлении, но обычно при атмосферном давлении, причем время реакции может составлять от менее одного часа до нескольких часов.
Удаление группы Ra из соединений формулы VIII происходит при образовании ксантинов формулы I согласно изобретению в стандартных условиях, которые были разработаны прежде всего в рамках техники защитных групп при синтезах алкалоидов и пептидов, и, следовательно, могут считаться известными.
Затем, в случае необходимости, в фенильных кольцах отщепляют замещенную группу бензила, бензгидрила или тритила предпочтительно восстановлением. Для этого наряду с химическим восстановлением, особенно для бензиловых соединений с натрием в жидком аммиаке принимают во внимание удаление трех вышеназванных аралкильных групп путем каталитического гидрирования при помощи катализатора на основе благородного металла, причем часто оказывается пригодной замена молекулярного водорода формиатом аммония в качестве донора водорода. При этом реакционной средой служит обычно низший спирт, в случае необходимости, при добавке муравьиной кислоты или также аммиака, можно применять апротонный растворитель, как диметилформамид, или особенно ледяную уксусную кислоту, также их смеси с водой. Подходящими катализаторами гидрирования являются, главным образом, палладиевая чернь и палладий на активном угле или сульфат бария, в то время как другие благородные металлы, как, например, платина, родий и рутений по причине конкурирующего гидрирования ядра ведут к побочным реакциям и поэтому имеют ограниченное применение. Гидрирование целесообразно проводить при температурах между 20oС и 100oС и при атмосферном давлении, предпочтительно, при небольшом избыточном давлении приблизительно до 10 бар, причем, как правило, требуется время реакции от нескольких минут до нескольких часов.
Но можно на выбор осуществлять отщепление защитной группы Rа, как, например, 4-метоксибензил-, бензгидрил- или тритил-остатка, также гидролитически при обычном протонном катализе.
Получение соединений формулы I по изобретению в чистой стереоизомерной форме осуществляют предпочтительно дополнительным разделением стереоизомерных форм при помощи известных методов. Так как диастереомеры в противоположность энантиомерам имеют различные физические и химические свойства, разделение их смесей, например, дробной кристаллизацией или хроматографическими способами, не доставляет, как правило, никаких трудностей. Напротив, физическое расщепление рацемата на энантиомерные формы (антиподы) требует дополнительных мер предосторожности; так дробная кристаллизация удается лишь после образования диастереомерных солей с оптически активным основанием и разделение методом хроматографии удается только при применении хиральных стационарных фаз, которые проявляют различное пространственное сродство к энантиомерам.
1,2,7-тризамещенные ксантины формулы VIII представляют собой ценные промежуточные продукты для получения соединений формулы I по изобретению, проявляют, кроме того, особенно если Rа обозначает бензил, такое же фармакологическое действие как конечные продукты формулы I, и принадлежат поэтому к области притязаний настоящего изобретения, хотя по причине более низкой растворимости в воде их парентеральное применение затруднено.
Новые соединения формулы I по изобретению благодаря своим ценным фармакологическим свойствам отлично подходят для применения в качестве активных веществ в лекарственных средствах, особенно в таких, которые обеспечивают эффективное лечебное и профилактическое действие при шоковых заболеваниях и, следовательно, представляют существенный интерес для фармацевтической промышленности. Их можно назначать или индивидуально, например, в форме микрокапсул, в смесях друг с другом, или в комбинации с подходящими веществами-носителями.
Поэтому изобретение относится также к лекарственным средствам, которые в качестве активного вещества содержат по меньшей мере одно соединение формулы I, при этом исключаются в качестве активных веществ для лекарственных средств с другими показаниями описанные 3-n-пропилксантины с 2-метоксиэтилом, 2-этоксиэтилом или 3-метоксипропилом в 1-положении.
Другим аспектом настоящего изобретения является применение соединения формулы I для получения фармацевтических лекарственных форм для парентерального и орального, в случае необходимости, также ректального, трансдермального или ингаляционного назначения при шоковых заболеваниях. Подходящими твердыми или жидкими галеновыми препаратами являются, например, грануляты, порошки, таблетки, драже, микрокапсулы, сиропы, эмульсии, суспензии, гели, препараты с пролонгированным выделением активного вещества, свечи, выделяющие активное вещество, пластыри, аэрозоли, капли и, прежде всего, растворы для инъекций в форме ампул или бутылочек для инъекций для длительного внутривенного вливания, при их изготовлении обычно применяют вспомогательные вещества, как вещества-носители, разбрызгивающиеся вещества, связующие, оболочки, агенты для набухания, смазывающие средства или добавки для улучшения переработки, вкусовые добавки, сладкие вещества или растворители. В качестве часто применяемых вспомогательных веществ следует назвать, например, карбонат магния, двуокись титана, лактозу, маннит и другие сахара, тальк, молочный белок, желатину, крахмал, витамины, целлюлозу и их производные, животные и растительные масла, полиэтиленгликоли и растворители, как, например, стерильная вода, физиологический раствор хлористого натрия, спирты, глицерин и другие многоатомные спирты.
Преимущественно фармацевтические препараты изготовляют и назначают в дозированных единицах, причем каждая единица в качестве активной компоненты содержит определенную дозу соединения по формуле I. В твердых дозированных единицах, как таблетки, капсулы и свечи, эта доза может составлять до 1000 мг, однако, предпочтительно от 100 до 600 мг, и в растворах для инъекций в форме ампул до 300 мг, но предпочтительно от 20 до 200 мг.
Для лечения взрослого пациента - в зависимости от эффективности соединений по формуле I и от степени тяжести угрожающего жизни заболевания - показаны суточные дозы от 100 до 5000 мг активного вещества, предпочтительно от 300 до 3000 мг, при оральном назначении и от 30 до 3000 мг, предпочтительно от 50 до 2000 мг, при внутривенном применении. Суточные дозы можно назначать как однократным приемом в форме отдельной дозированной единицы или же нескольких меньших дозированных единиц, так и многократным приемом разделенных доз в определенные интервалы времени.
При длительном внутривенном вливании суточная доза составляет от 100 до 5000 мг, предпочтительно от 500 до 2000 мг, соответственно при скорости вливания от 0,1 до 3 мг на 1 кг веса тела в течение 1 часа, предпочтительно 0,3 до 1 мг/кг/час.
Однако, при всех формах применения можно при известных условиях устанавливать также более высокие или более низкие суточные дозы.
Наконец, соединения формулы I, если имеются клинические показания, можно назначать вместе с другими подходящими активными веществами, особенно с такими, которые регулируют сигнальный каскад шокового состояния; например, с антителами против энтеро- и эндотоксинов (LPS), с моноцитарным LPS-рецептором CD 14 или с LPS-связывающим протеином LBP; с модуляторами цитокин-сетчатой структурой как анти-ТNF-антителами, с растворимыми TNP-рецепторами и другими TNF-связывающими протеинами, с ингибиторами продуцирования и освобождения интерлейцина-1 (IL-1) и/или TNF, а также с TNF- и IL-1-рецептор-антагонистами; с ингибиторами обмена арахидоновой кислоты, а также дополнительными каскадами и каскадами коагуляции, как ингибиторы фосфолипазы А2, циклооксигеназы и липоксигеназы (например, стероиды и нестероидальные противовоспалительные средства, как ибупрофен), PAF (активирующий тромбоциты фактор)-, лейкотриен-, тромбоксан-, тромбин-, фибрин-, брадикинин и серотонин-антагонисты и анти-С5а- или -С3а-антитела; с антикоагулянтами и с ингибиторами агрегации тромбоцитов, как антитромбин III, с плазминогеновым активатором ткани tPA-1, гепарин, а также простациклин и его устойчивые синтетические производные; с ингибиторами освобождения и/или биологического действия литического энзима; с соединениями, связывающими кислородные радикалы, как супероксид дисмутазы, каталаза, альфа-токоферол или N-ацетилцистеин; с хелаторами тяжелых металлов, как дефероксамин, с ингибиторами межклеточной адгезии, как фибропектин или антитела против адгезии молекул ELAM-1, ICAM-1, VCAM-1 и CD11/CD18; или также с антибиотиками или при получении вышеназванных галеновых лекарственных форм.
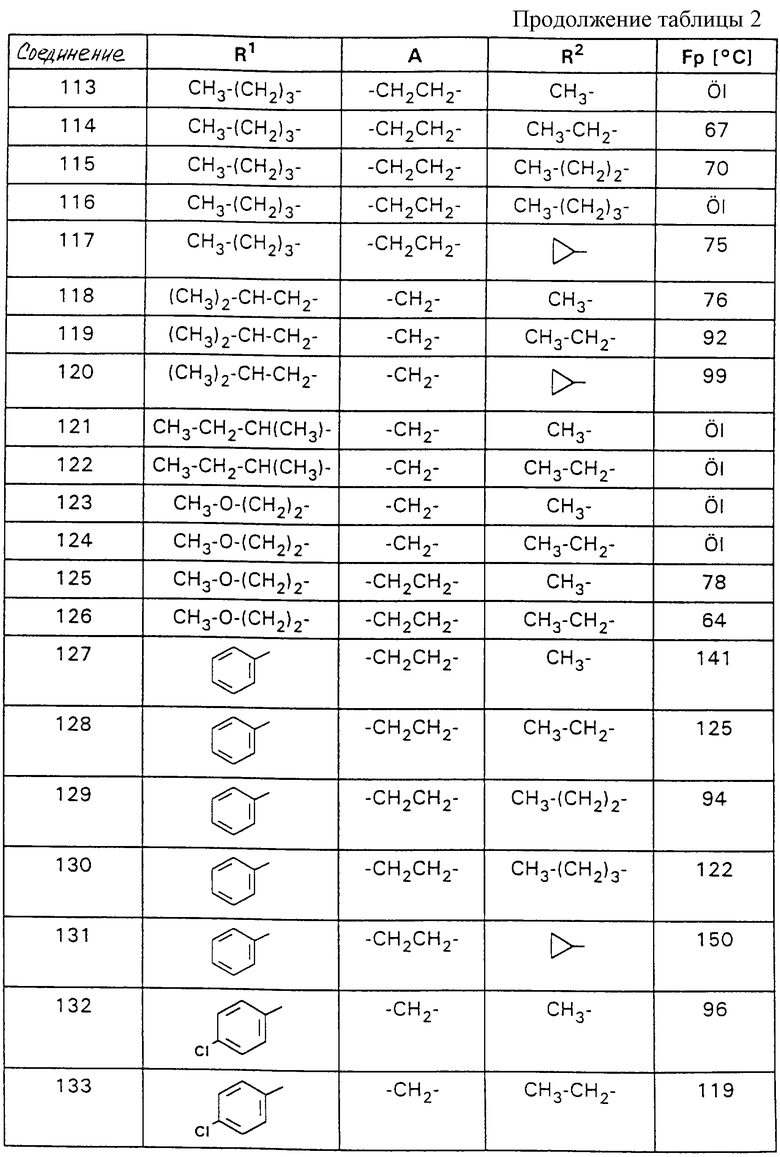
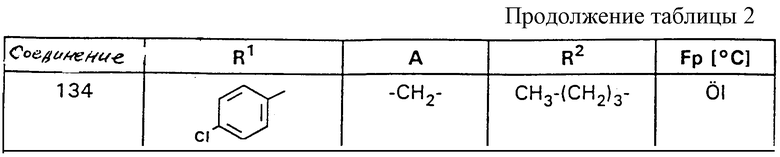
Ниже на основании представленных примеров поясняется подробнее синтез, с точки зрения структуры, приведенных в таблице 1 соединений по формуле I. В таблице 2 представлены в таком же порядке ценные промежуточные соединения формулы VIII. Для всех полученных в качестве препаратов промежуточных и конечных продуктов подтверждена структура как 1Н-ЯМР-спектроскопией, так и элементным анализом или масс-спектрометрией.
Примеры, получения
Пример 1:
1-метоксиметил-3-метилксантин (соединение 1)
а) 7-бензил-3-метилксантин
К суспензии 83 г (0,5 мол) 3-метилксантина в 500 мл метанола добавляют 20 г (0,5 мол) растворенной в 200 мл воды гидроокиси натрия и перемешивают один час при 70oС, затем смешивают при той же температуре по каплям с 85,5 г (0,5 мол) бензилбромида и выдерживают в течение 5 часов между 70 и 80oС. Потом охлаждают, отделяют на нутч-фильтре в холодном состоянии, промывают продукт на нутч-фильтре водой, растворяют в горячем состоянии в 1000 мл 1 н. гидроокиси натрия, фильтруют и при перемешивании с 4 н. соляной кислотой медленно доводят до рН 9,5. Отфильтровывают кристаллизат от еще теплого раствора, промывают водой для освобождения от хлорида и сушат в вакуумном сушильном шкафу.
Выход: 81,7 г (63,8% от теории);
точка плавления: 263oС;
C13H12N4O2 (молекулярный вес MG=256,2 г/мол).
б) 7-бензил-1-метоксиметил-3-метилксантин
2,3 г (0,1 г/атом) натрия растворяют в 200 мл безводного метанола, смешивают с 25,6 г (0,1 мол) ксантина со стадии а), нагревают до прозрачного раствора при флегме, затем охлаждают, выпаривают при пониженном давлении и сушат. Полученную таким способом натриевую соль 7-бензил-3-метилксантина суспендируют в 300 мл безводного ацетонитрила, добавляют при перемешивании при 50oС раствор 8,8 г (0,11 мол) метокси-метилхлорида в 40 мл ацетонитрила по каплям и дополнительно перемешивают 8 часов при 50oС. Затем охлаждают, выпаривают при пониженном давлении, поглощают остаток хлороформом, экстрагируют путем встряхивания непрореагировавший 7-бензил-3-метилксантин с 1 н. едким натром, промывают фазу хлороформа водой до нейтрального состояния, сушат и выпаривают при пониженном давлении, причем получают 22 г (73,3% от теории) маслянистого продукта, который постепенно затвердевает и его перекристаллизовывают из сложного этилового эфира уксусной кислоты при добавлении петролейного эфира при температуре кипения.
С15Н16N4O3 (MG = 300,3 г/мол);
точка плавления: 114oС.
Введение метоксиметилгруппы в 1-положении 7-бензил-3-метилксантина производят также метоксиметил-4-толуолсульфонатом в качестве алкилирующего средства, который получают одной реакцией из хлорангидрида 4-толуолсульфокислоты и ацетата натрия или 4-толуолсульфокислоты и ацетангидрида с формальдегиддиметилацеталем в диметилформамиде (WO 87/00523) и подвергают взаимодействию in situ с 7-бензил-3-метилксантином.
с) 1-метоксиметил-3-метилксантин (соединение 1)
10,5 г (0,035 мол) 1,3,7-тризамещенного ксантина со стадии б) гидрируют в 200 мл ледяной уксусной кислоты при помощи 1,5 г палладия (10%) на активном угле при 60oС и 3,5 бар в течение 48 ч. После охлаждения перегруппировывают азотом, отфильтровывают катализатор, концентрируют при пониженном давлении и очищают твердый остаток фильтрованием через колонну с силикагелем в жидкой среде хлороформ/метанол (10/1).
Выход: 5,5 г (74,8% от теории);
точка плавления: 218oС;
C8H10N4O3 (MG=210,2 г/мол).
Анализ: вычислено, %: С 45,71, Н 4,80, N 26,66.
найдено, %: C 45,98, H 4,86, N 26,98.
Пример 2:
3-циклопропил-1-(2-метоксиэтил)-ксантин (соединение 10)
а) 7-бензил-3-циклопропилксантин
К суспензии 50 г (0,26 мол) 3-циклопропилксантина в 300 мл метанола добавляют 10,4 г (0,26 мол) растворенной в 110 мл воды гидроокиси натрия и перемешивают 1 ч при 70oС, затем смешивают при той же температуре по каплям с 44,5 г (0,26 мол) бензилбромида и выдерживают реакционную смесь в течение 4 ч между 70 и 80o С. Потом добавляют 1,04 г (0,026 мол) гидроокиси натрия и 4,45 г (0,026 мол) бензилбромида. После 1 ч охлаждают, отсасывают на нутч-фильтре в холодном состоянии и промывают продукт на нутч-фильтре водой. Полученный таким способом сырой продукт можно применять без дальнейшей очистки.
Выход: 48 г (65,4% от теории);
точка плавления: 204oС;
C15H14N4O2 (MG=282,3 г/мол).
Масс-спектр: 283 (60%, М+Н); 240 (21%), 91 (100%).
б) 7-бензил-3-циклопропил-1-(2-метоксиэтил)-ксантин
К горячему раствору с температурой 60oС 3 г (11,0 мол) 7-бензил-3-циклопропилксантина со стадии а) в диметилформамиде добавляют 2,2 г (15,9 ммол) карбоната калия и перемешивают 1 ч при 60oС. Затем добавляют по каплям 1,51 г (15,9 ммол) 2-метоксиэтилхлорида и перемешивают 6 ч при 80oС. После этого охлаждают до комнатной температуры и концентрируют при пониженном давлении. Маслянистый остаток поглощают дихлорметаном и экстрагируют 1 н. едким натром, промывают водой до нейтрального состояния, сушат над сульфатом магния, концентрируют при пониженном давлении и без дальнейшей очистки применяют на стадии с).
Выход: 2,7 г (71,7% от теории);
точка плавления: 117oС;
C18H20N4O3 (MG=340,4 г/мол);
Масс-спектр: 340 (36%, М); 282 (43%); 148 (100%); 91 (86%).
с) 3-циклопропил-1-(2-метоксиэтил)-ксантин (соединение 10)
2,2 г (6,45 ммол) 1,3,7-тризамещенного ксантина со стадии б) гидрируют в 250 мл этанола при помощи 1,05 г палладия (10%) на активном угле в течение 12 ч. Перегруппировывают азотом, отфильтровывают катализатор, концентрируют при пониженном давлении и очищают флеш-хроматографией на колонке с силикагелем, толуол/этанол (10/1).
Выход: 0,77 г (47,7% от теории);
точка плавления: 203oС.
С11Н14N4O3 (MG=250,3 г/мол).
Масс-спектр: 250 (55%, М); 192 (100%); 149 (56%); 148 (58%); 121 (82%); 120 (56%).
Пример 3:
3-бутил-1-(3-метоксипропил)-ксантин (соединение 14)
а) 7-бензил-3-бутилксантин
К суспензии 52 г (0,25 мол) 3-бутилксантина в 300 мл метанола добавляют 10 г (0,25 мол) растворенной в 100 мл воды гидроокиси натрия и перемешивают 1 ч при 70oС, затем смешивают при такой же температуре, по каплям с 42,8 г (0,25 мол) бензилбромида и выдерживают реакционную смесь в течение 5 ч между 70 и 80oС. Потом добавляют 1,0 г (0,025 мол) гидроокиси натрия и 4,28 г (0,025 мол) бензилхлорида. После 2 ч охлаждают, разбавляют 1500 мл воды, фильтруют в холодном состоянии на нутч-фильтре, промывают продукт на нутч-фильтре водой, растворяют в 1000 мл 1 н. едкого натра, фильтруют и при перемешивании с помощью концентрированной соляной кислоты медленно доводят до рН 3. Отфильтровывают кристаллизат от раствора, промывают водой для освобождения от хлорида и сушат при пониженном давлении.
Выход: 54,1 г (72,5 от теории);
точка плавления: 187oC.
C16H18N4O2 (MG=298,3 г/мол).
Масс-спектр: 298 (13%, М); 91 (100%).
б) 7-бензил-3-бутил-1-(3-метоксипропил)-ксантин
К горячему раствору с температурой 60oС 3 г (10,0 ммол) 7-бензил-3-бутилксантина со стадии а) в 90 мл диметилформамида добавляют 2,1 г (15,2 ммол) карбоната калия и перемешивают 1 ч при 60oС. Потом добавляют по каплям 1,3 г (12,0 мол) 3-метоксипропилхлорида и перемешивают 3 ч при 100oС. После этого охлаждают до комнатной температуры, смешивают с водой и экстрагируют дихлорметаном. Органическую фазу промывают водой и 1 н. едким натром, сушат при помощи сульфата натрия и концентрируют при пониженном давлении. Маслянистый остаток очищают флеш-хроматографией через колонку с силикагелем, толуол/этанол (39/1).
Выход: 3,2 г (86,5% от теории); желтое масло;
С20Н26N4O3 (MG=370,5 г/мол).
Масс-спектр: 371,3 (100%, М+Н); 339,3 (16%); 298,3 (15%);
1H-ЯМР (DMSO-d6, 200 МГц): δ= 0,90 (t, 3Н, СН2СН3); 1,30 (sixt., 2H, СН2СН2СН3), 1,63 и 1,75 (2 quint., 4Н, СН2СH2СН2); 3,32 (s, 3Н, ОСН3); 3,34 (t, 2H, ОСН2); 5,48 (s, 2H, бенз. Н); 7,25-7,40 (m, 5H, аромат. Н); 8,26 (s, 1H, N=CH).
с) 3-бутил-1-(3-метоксипропил)-ксантин (соединение 14)
0,5 г (1,35 ммол) 1,3,7-тризамещенного ксантина со стадии б) гидрируют в 50 мл этанола при помощи 0,1 г палладия (10%) на активном угле в течение 5 ч. Перегруппировывают азотом, отфильтровывают катализатор и концентрируют при пониженном давлении. Остаток можно перекристаллизовывать из метанола/простого метил-трет.-бутилового эфира.
Выход: 0,19 г (52,2% от теории);
точка плавлении: 157oС;
C13H20N4O3 (MG=280,3 г/мол).
Масс-спектр: 281,3 (М+Н, 100%); 249,2 (М-Оме, 70%).
Пример 4:
1-этоксиметил-3-пропилксантин (соединение 18)
а) 7-бензил-3-пропилксантин
К суспензии 20 г (0,103 мол) 3-пропилксантина в 112 мл метанола добавляют 4,12 г (0,103 мол) растворенной в 41 мл воды гидроокиси натрия и перемешивают 1 ч при 70oС, затем смешивают при такой же температуре по каплям с 12,23 мл (0,103 мол) бензилбромида и выдерживают реакционную смесь в течение 4 ч между 70 и 80o С. Смесь охлаждают, фильтруют в холодном состоянии на нутч-фильтре, промывают продукт на нутч-фильтре водой и сушат при пониженном давлении.
Выход: 20,3 г (69,4% от теории);
точка плавления: 186oС;
C15H16N4O2 (MG=284,3 г/мол).
Масс-спектр: 284 (18%, М); 242 (11%), 212 (13%); 91 (100%).
б) 7-бензил-1-этоксиметил-3-пропилксантин
К горячему раствору с температурой 60oС 2,2 г (7,7 ммол) 7-бензил-3-пропилксантина со стадии а) в 60 мл диметилформамида добавляют 1,71 г (12,0 ммол) карбоната калия и перемешивают 1 ч при 60oС. Затем добавляют по каплям 0,93 мл (10,0 ммол) этоксиметилхлорида и перемешивают 4,5 ч при 80oС. Добавляют еще 0,5 мл (5,3 ммол) этоксиметилхлорида и перемешивают снова 6 ч. После этого добавляют 12 мл воды и 5 мл метанола, оставляют на ночь, снова добавляют 60 мл воды и три раза экстрагируют простым метил-трет.-бутиловым эфиром. Соединенные органические фазы промывают два раза водой, сушат при помощи сульфата магния и концентрируют при пониженном давлении. Сырой продукт очищают флеш-хроматографией на колонке с силикагелем, дихлорметан/метанол (19,8/0,2).
Выход: 2,28 г (87% от теории);
точка плавления: 110oС;
С18Н22N4O3 (MG=342,4 г/мол).
Масс-спектр: 342 (7%, М); 296 (13%); 285 (33%), 91 (100%).
с) 1-этоксиметил-3-пропилксантин (соединение 18)
1,79 г (5,2 ммол) 1,3,7-тризамещенного ксантина со стадии б) гидрируют в 200 мл этанола при помощи 179 мг палладия (10%) на активном угле в течение 6,5 ч. Перегруппировывают азотом, отфильтровывают катализатор и концентрируют при пониженном давлении. Остаток очищают флеш-хроматографией через колонку с силикагелем, дихлорметан/метанол, (19,8/0,2).
Выход: 1,12 г (85% от теории);
точка плавления: 134oС;
С11Н16N4O3 (MG=252,3 г/мол).
Масс-спектр: 252 (29%, М); 208 (40%); 195 (100%); 166 (65%); 136 (50%).
Пример 5:
3-этил-1-пропоксиметилксантин (соединение 27)
а) 7-бензил-3-этилксантин
180 г (1 мол) 3-этилксантина вносят в 1000 мл диметилформамида, нагревают при перемешивании до 80oС и после внесения 88 г (0,64 мол) карбоната калия по каплям смешивают с 133 г (1,05 мол) бензилхлорида в течение 1 ч. После этого перемешивают 2 ч при 100oС, смешивают с 1000 мл воды, фильтруют осажденный продукт на нутч-фильтре, промывают водой для освобождения от соли и сушат в вакуумном шкафу при 100oС. Если требуется, проводят дальнейшую очистку вторичным осаждением из 1 н. едкого натра при помощи 4 н. соляной кислоты аналогично примеру 1 а).
Выход: 262 г (97% от теории);
точка плавления: 218oС;
C14H14N4O2 (MG=270,3 г/мол);
Анализ: вычислено, %: C 62,21, H 5,22, N 20,73.
найдено, %: C 62,07, H 5,36, N 20,84.
б) 7-бензил-3-этил-1-пропоксиметилксантин
Аналогично примеру 1 б) превращают 27 г (0,1 мол) 7-бензил-3-этилксантина в натриевую соль, затем в ацетонитриле подвергают взаимодействию с 13 г (0,12 мол) пропоксиметилхлорида (полученного с выходом 67% из 1,3,5-триоксана, 1-пропанола и хлористоводородного газа) и обрабатывают, причем 30 г (87,6% от теории) получают в виде аналитически чистого продукта, который, в случае необходимости, можно перекристаллизовывать из сложного этилового эфира уксусной кислоты.
С18Н22N4O3 (MG=342,4 г/мол);
точка плавления: 92oС;
Анализ: вычислено, %: C 63,14, H 6,48, N 16,36.
найдено, %: C 62,95, H 6,55, N 16,21.
с) 3-этил-1-пропоксиметилксантин (соединение 27)
17,1 г (0,05 мол) продукта со стадии б) и 5 г (0,08 мол) формиата аммония перемешивают в 150 мл этанола через 6 г палладия (10%) на активном угле при 35oС в течение нескольких дней, при этом целесообразно последовательное добавление остального формиата аммония до общего количества 22 г (0,35 мол). Фильтруют, концентрируют фильтрат, остаток поглощают раствором карбоната натрия, промывают хлороформом, доводят водную фазу при помощи 2 н. соляной кислоты до рН 4, экстрагируют продукт встряхиванием с хлороформом и после сушки и выпаривания перекристаллизовывают из сложного этилового эфира уксусной кислоты.
Выход: 8,6 г (68,2% от теории);
точка плавления: 159oС;
С11Н16N4O3 (MG=252,3 г/мол).
Анализ: вычислено, %: C 52,37, H 6,39, N 22,91.
найдено, %: C 52,85, H 6,88, N 22,50.
Гидролитическое дебензилирование аналогично примеру 1 с) приводит к такому же соединению с выходом 58,9%.
Пример 6:
3-изобутил-1-пропоксиметилксантин (соединение 31)
а) 7-бензилгуанин-гидрохлорид
К суспензии 40 г (0,147 мол) гуанозина в 200 мл диметилсульфоксида добавляют по каплям 40 мл (0,34 мол) бензилбромида и перемешивают 4 ч при комнатной температуре. Смешивают с 100 мл концентрированной соляной кислоты и перемешивают 30 мин при комнатной температуре. После этого добавляют в 1200 мл метанола, отсасывают осадок и промывают метанолом.
Выход: 35,9 г (92% от теории);
точка плавления: >325oС;
C12H12ClN5O (MG=277,7 г/мол);
основание: C12H11N5O (MG = 241,6 г/мол).
Масс-спектр: 242,2 (100%, М+Н).
б) 7-бензилксантин
35,9 г (0,13 мол) 7-бензилгуанингидрохлорида со стадии а) растворяют в смеси из 90 мл воды и 807 мл ледяной уксусной кислоты и нагревают до 100oС. После охлаждения до 50oС добавляют сразу раствор 35,88 г (0,52 мол) нитрита натрия в 90 мл воды. После 16 ч при комнатной температуре отсасывают образованный осадок, промывают на нутч-фильтре водой и сушат.
Выход: 26,0 г (83% от теории);
точка плавления: >266oС;
C12H10N4O2 (MG=242,5 г/мол).
Масс-спектр: 243,1 (95%, М+Н); 91 (100%).
с) 7-бензил-3-изобутилксантин
1,5 г (6,2 ммол) 7-бензилксантина со стадии б) растворяют в 50 мл диметилформамида при 50oС и смешивают частями с 0,149 г (6,2 ммол) гидрида натрия и перемешивают 1 ч при 50oС. Добавляют по каплям 0,67 мл (6,2 ммол) изобутилбромида и нагревают до 80oС. После 5 ч добавляют остальные 0,2 мл (1,86 ммол) изобутилбромида и перемешивают еще 5 ч. После этого добавляют 12 мл воды и 5 мл метанола, перемешивают 2 ч при комнатной температуре, добавляют остальные 60 мл воды и экстрагируют три раза простым метил-трет.-бутиловым эфиром. Органические фазы промывают водой, сушат сульфатом магния, сгущают при пониженном давлении и очищают флеш-хроматографией на колонке с силикагелем, дихлорметан/метанол (99/1).
Выход: 1,16 г (63% от теории);
C16H18N4O2 (MG=298,3 г/мол);
1H-ЯМР (DMSO-d6, 200 МГц): δ=0,85 (d, СН(СН3)2); 2,16 (m, 1H, СН2СН(СН3)2); 3,73 (d, 2H, СН2СН); 5,45 (s, 2H, бензил. Н); 7,23-7,40 (m, 5H, аромат. Н), 8,20 (s, 1H, N=CH); 11,13 (s.br., 1H, NH).
d) 7-бензил-3-изобутил-1-пропоксиметилксантин
К суспензии 1,16 г (3,9 ммол) 7-бензил-3-изобутилксантина со стадии с) в 60 мл диметилформамида добавляют при 60oС 0,86 г (6,2 ммол) карбоната калия и перемешивают 1 ч при этой температуре. Затем добавляют по каплям 0,56 мл (5,1 ммол) пропоксиметилхлорида и перемешивают 5,5 ч при 80oС. После этого добавляют 12 мл воды и 5 мл метанола, оставляют на ночь, снова добавляют 60 мл воды и экстрагируют четыре раза, каждый раз с 150 мл простого метил-трет. -бутилового эфира. Соединенные органические фазы промывают 200 мл воды, сушат сульфатом магния и сгущают при пониженном давлении. Сырой продукт очищают флеш-хроматографией на колонке с силикагелем, дихлорметан.
Выход: 1,2 г (83% от теории);
точка плавления: 72oС;
С20Н26N4O3 (MG=370,5 г/мол).
Масс-спектр: 370 (40%, М); 310 (55%); 299 (100%); 256 (55%), 91 (85%).
е) 3-изобутил-1-пропоксиметилксантин (соединение 31)
859 мг (2,32 ммол) 1,3,7-тризамещенного ксантина со стадии d) гидрируют в 22 мл этанола при помощи 86 мг палладия (10%) на активном угле в течение 6 ч. Перегруппировывают азотом, отфильтровывают катализатор и концентрируют при пониженном давлении. Остаток очищают флеш-хроматографией на колонке с силикагелем, дихлорметан/метанол (19/1).
Выход: 588 мг (90% от теории);
точка плавления: 141oС;
С13Н20N4O3 (MG=280,3 г/мол).
Масс-спектр: 280 (25%, М); 222 (37%); 209 (100%); 166 (85%); 136 (55%).
Пример 7:
3-фенил-1-пропоксиметилксантин (соединение 32)
а) 7-бензил-3-фенилксантин
К суспензии 3,0 г (13,2 ммол) 3-фенилксантина в 18 мл метанола добавляют раствор 0,53 г (13,2 ммол) гидроокиси натрия в 5,3 мл воды и перемешивают 1 ч при 70oС. Затем смешивают по каплям с 1,56 мл (13,2 ммол) бензилбромида, перемешивают 7 ч при 70oС, отсасывают после охлаждения осадок, промывают водой, растворяют осадок в 50 мл 1 н. едкого натра, отфильтровывают от нерастворимой части и при помощи 4 н. соляной кислоты устанавливают рН 8-9. Образованный осадок отсасывают, промывают водой и очищают флеш-хроматографией на колонке с силикагелем, дихлорметан/метанол (79/1).
Выход: 1,13 г (27% от теории);
точка плавления: 250oС;
C18H14N4O2 (MG=318,6 г/мол).
Масс-спектр: 319 (100%, М+Н); 91 (19%).
б) 7-бензил-3-фенил-1-пропоксиметилксантин
К суспензии 0,65 г (2,04 ммол) 7-бензил-3-фенилксантина со стадии а) в 20 мл диметилформамида добавляют при 60oС 0,45 г (3,26 ммол) карбоната калия и перемешивают 1 ч при этой температуре. Затем добавляют по каплям 0,29 мл (2,65 ммол) пропоксиметилхлорида и перемешивают 1,5 ч при 80oС. После этого добавляют 20 мл воды, экстрагируют три раза, каждый раз с 24 мл простого метил-трет. -бутилового эфира, промывают соединенные органические фазы, каждый раз с 12 мл воды, сушат сульфатом магния и концентрируют при пониженном давлении. Сырой продукт очищают флеш-хроматографией на колонке с силикагелем, гептан/этилацетат (5/7).
Выход: 0,69 г (87% от теории);
точка плавления: 103oС;
C22H22N4O3 (MG=390,4 г/мол).
Масс-спектр: 391,2 (100%, М+Н); 331,2 (12%); 241,1 (25%).
с) 3-фенил-1-пропоксиметилксантин (соединение 32)
535 мг (1,37 ммол) 1,3,7-тризамещенного ксантина со стадии б) гидрируют в 20 мл этанола при помощи 50 мг палладия (10%) на активном угле. Перегруппировывают азотом, отфильтровывают катализатор и концентрируют при пониженном давлении. Остаток очищают флеш-хроматографией на колонке с силикагелем, дихлорметан/метанол (19/0,3).
Выход: 232 мг (56% от теории);
точка плавления: 220oС;
С15Н16N4O3 (MG=300,3 г/мол).
Масс-спектр: 300 (23%, М); 242 (68%); 229 (55%); 185 (100%).
Пример 8:
3-циклопропилметил-1-пропоксиметил-ксантин (соединение 34)
а) 7-бензил-3-циклопропилметилксантин
Раствор 7 г (29,0 ммол) 7-бензилксантина из примера 6б) в 200 мл диметилформамида нагревают до 50oС и смешивают частями с 0,69 г (29,0 ммол) гидрида натрия и перемешивают 1 ч при 50oС. К этой суспензии добавляют 2,76 мл (29,0 ммол) циклопропилметилбромида и повышают температуру до 80oС. После выдерживания в течение 7 ч при 80oС добавляют снова 1 мл (11,0 ммол) циклопропилметилбромида. Еще через 6 ч добавляют 24 мл воды и 10 мл метанола, оставляют на ночь, смешивают снова с 120 мл воды и экстрагируют три раза, каждый раз с 300 мл простого метил-трет.-бутилового эфира. Органические фазы промывают водой, сушат сульфатом магния и концентрируют при пониженном давлении. Сырой продукт очищают флеш-хроматографией на колонке с силикагелем, дихлорметан/метанол (99%).
Выход: 4,8 г (56% от теории);
точка плавления: 185oС;
С16Н16N4O2 (MG=296,4 г/мол).
Масс-спектр: 297,3 (100%, М+Н),
б) 7-бензил-3-циклопропилметил-1-пропоксиметилксантин
В раствор 1,5 г (5,06 ммол) 7-бензил-3-циклопропилметилксантина со стадии а) в 60 мл диметилформамида добавляют при 60oС 1,12 г (8,1 ммол) карбоната калия и перемешивают при этой температуре. Затем добавляют по каплям 722 мкл (6,58 ммол) пропоксиметилхлорида и перемешивают 4 ч при 80oС. Добавляют 12 мл воды и 5 мл метанола и перемешивают 2 ч при 50oС. После этого снова добавляют 60 мл воды, экстрагируют три раза простым метил-трет.-бутиловым эфиром, соединенные органические фазы промывают два раза водой, сушат сульфатом магния и концентрируют при пониженном давлении. Сырой продукт очищают флеш-хроматографией на колонке с силикагелем, дихлорметан/метанол (19,8/0,2).
Выход: 1,32 г (71% от теории);
точка плавления: 88oС,
С20Н24N4O3 (MG=368,4 г/мол).
Масс-спектр: 368 (9%, М); 310 (11%); 297 (13%); 91 (100%).
с) 3-циклопропилметил-1-пропоксиметилксантин (соединение 34)
938 мг (2,55 ммол) 1,3,7-тризамещенного ксантина со стадии б) гидрируют в 60 мл этанола при помощи 130 мг палладия (10%) на активном угле в течение 15 ч. Перегруппировывают азотом, отфильтровывают катализатор и концентрируют при пониженном давлении. Остаток очищают флеш-хроматографией на колонке с силикагелем, дихлорметан/метанол (39/1).
Выход: 671 мг (95% от теории);
точка плавления: 132oС;
C13H18N4O3 (MG=278,3 г/мол).
Масс-спектр: 278 (26%, М); 220 (80%); 207 (64%); 136 (87%); 122 (67%); 55 (100%).
Пример 9:
1-(2-пропоксиэтил)-3-пропилксантия (соединение 37)
а) 7-бензил-1-(2-пропоксиэтил)-3-пропилксантин
К суспензии 2,2 г (7,8 ммол) 7-бензил-3-пропилксантина (полученной по примеру 4 а) в 70 мл диметилформамида добавляют при 60oС 1,7 г (12,48 ммол) карбоната калия и перемешивают 1 ч при этой температуре. Затем добавляют по каплям 1,3 мл (10,14 ммол) 2-пропоксиэтилхлорида и перемешивают 10 ч при 80oС. После этого добавляют 1,2 мл метанола и 14 мл воды, оставляют на ночь, смешивают с остальными 70 мл воды и экстрагируют три раза, каждый раз с 84 мл простого метил-трет. -бутилового эфира. Соединенные органические фазы промывают два раза, каждый раз с 42 мл воды, сушат сульфатом магния и концентрируют при пониженном давлении. Сырой продукт очищают флеш-хроматографией на колонке с силикагелем, дихлорметан/метанол (19/0,1).
Выход: 2,3 г (80% от теории);
точка плавления: 55oС;
C19H24N4O3 (MG=356,4 г/мол).
Масс-спектр: 356 (10%, М); 297 (15%); 285 (38%); 91 (100%).
б) 1-(2-пропоксиэтил)-3-пропилксантин (соединение 37)
1,75 г (4,7 ммол) 1,3,7-тризамещенного ксантина со стадии а) в 75 мл этанола гидрируют при помощи 0,2 г палладия (10%) на активном угле в течение 6 ч. Перегруппировывают азотом, отфильтровывают катализатор и концентрируют при пониженном давлении. Остаток очищают флеш-хроматографией на колонке с силикагелем, дихлорметан/метанол (38/1).
Выход: 0,93 г (70% от теории);
точка плавления: 137oС;
C13H20N4O3 (MG=280,6 г/мол).
Масс-спектр: 281,3 (45%, М+Н); 221,2 (100%).
Пример 10:
1-бутоксиметил-3-изопропилксантин (соединение 42)
а) 7-бензил-3-изопропилксантин
Раствор 3,5 г (1,45 ммол) 7-бензилксантина из примера 6 б) в 60 мл диметилформамида нагревают до 50oС и смешивают частями с 0,35 г (1,45 ммол) гидрида натрия, разбавляют 20 мл диметилформамида и перемешивают 1 ч при 50oС. К этой суспензии добавляют 1,36 мл (1,45 ммол) 2-бромпропана и повышают температуру до 80oС. В ходе реакции добавляют всего 4,91 мл (52,3 ммол) 2-бромпропана. После всего 16 ч при 80oС добавляют 10 мл воды и 2 мл метанола, перемешивают 10 мин, смешивают с остальными 70 мл воды и экстрагируют три раза, каждый раз с 70 мл простого метил-трет.-бутилового эфира. Органические фазы промывают водой, сушат сульфатом магния и концентрируют при пониженном давлении. Сырой продукт очищают флеш-хроматографией на колонке с силикагелем, дихлорметан/метанол (19/0,4).
Выход: 1,17 г (29% от теории);
точка плавления: 219oС;
C15H18N4O2 (MG=286,6 г/мол).
Масс-спектр: 285,2 (100%, М+Н).
б) 7-бензил-1-бутоксиметил-3-изопропилксантин
К суспензии 0,75 г (2,64 ммол) 7-бензил-3-изопропилксантина со стадии а) в 20 мл диметилформамида добавляют при 60oС 0,583 г (4,22 ммол) карбоната калия и перемешивают 1 ч при этой температуре. Затем добавляют по каплям 0,42 г (3,43 ммол) бутоксиметилхлорида и перемешивают 6 ч при 80oС. После этого добавляют остальные 0,11 г (0,87 ммол) бутоксиметилхлорида и перемешивают снова 5 ч. Потом добавляют 20 мл воды, экстрагируют три раза, каждый раз с 30 мл простого метил-трет.-бутилового эфира, соединенные органические фазы промывают два раза, каждый раз 20 мл воды, сушат сульфатом магния и концентрируют при пониженном давлении. Сырой продукт очищают флеш-хроматографией на колонке с силикагелем, гептан/этилацетат (2/1).
Выход: 0,66 г (68% от теории); масло;
С20Н26N4O2 (MG=370,7 г/мол).
Масс-спектр: 371,4 (100%, М+Н); 297,2 (33%);
1H-ЯМР (DMSO4-d6, 200 МГц): δ= 0,82 (t, 3Н, (СН2)3); 1,48 (d, 6H, СН(СН3)2); 1,14-1,56 (m, 4H, СН2(СН2)2СН3); 3,50 (t, 2Н, ОСН2); 5,06 (m, 1H, СН(СН3)2); 5,30 (s, 2H, бензил. Н); 5,50 (s, 2H, OCH2N); 7,24-7,43 (m, 5H, аромат. Н); 8,31 (s, 1H, N=CH).
с) 1-бутоксиметил-3-изопропилксантин (соединение 42)
660 мг (1,78 ммол) 1,3,7-тризамещенного ксантина со стадии б) гидрируют в 60 мл этанола при помощи палладия (10%) на активном угле в течение 14 ч, перегруппировывают азотом, отфильтровывают катализатор и концентрируют при пониженном давлении. Остаток очищают флеш-хроматографией на колонке с силикагелем, дихлорметан/метанол (19/0,3).
Выход: 416 мг (83% от теории);
точка плавления 131oС;
С13Н20N4O3 (MG=280,3 г/мол).
Масс-спектр: 281,2 (100%, М+Н); 207,2 (30%).
Пример 11:
7-бензил-1-изобутоксиметил-3-метилксантин (48)
К суспензии 2,25 г (8,8 ммол) 7-бензил-3-метилксантина (полученного по примеру 1 а) в 50 мл N-метилпирролидона добавляют при 60oС 1,9 г (14,08 ммол) карбоната калия и перемешивают 1 ч при этой температуре. Затем добавляют по каплям 1,4 г (11,44 ммол) изобутоксиметилхлорида и перемешивают 3 ч при 80oС. Добавляют остальные 0,5 г (4,4 ммол) изобутоксиметилхлорида и снова перемешивают 2 ч. После этого добавляют 50 мл воды и экстрагируют три раза, каждый раз 60 мл простого метил-трет.-бутилового эфира. Соединенные органические фазы промывают два раза, каждый раз 30 мл воды, сушат сульфатом магния и концентрируют при пониженном давлении. Сырой продукт очищают флеш-хроматографией на колонке с силикагелем, дихлорметан/метанол (19/0,2).
Выход: 2,54 г (85% от теории);
точка плавления: 76oС;
С18Н22N4O3 (MG=342,4 г/мол).
Масс-спектр: 343,3 (100%, М+Н); 269,2 (88%); 179,1 (24%).
б) 1-изобутоксиметил-3-метилксантин (соединение 48)
2,1 г (6,14 ммол) 1,3,7-тризамещенного ксантина со стадии а) гидрируют в 50 мл этанола при помощи 0,4 г палладия (10%) на активном угле в течение 25 ч. Перегруппировывают азотом, отфильтровывают катализатор и концентрируют при пониженном давлении. Остаток очищают флеш-хроматографией на колонке с силикагелем, дихлорметан/метанол (19/0,3).
Выход: 0,59 г (38% от теории);
точка плавления: 160oС;
С11Н16N4O3 (MG=252,3 г/мол).
Масс-спектр: 252 (7%, М); 196 (10%); 180 (100%); 179 (88%); 167 (56%).
Пример 12:
1-втор.-бутоксиметил-3-этилксантин (соединение 52)
а) 7-бензил-1-втор.-бутоксиметил-3-этилксантин
К суспензии 3,0 г (11,0 ммол) 7-бензил-3-этилксантина (полученного по примеру 5 а) в 60 мл диметилформамида добавляют при 60oС 2,45 г (18,0 ммол) карбоната калия и перемешивают 1 ч при этой температуре. Потом добавляют по каплям 1,77 г (14,0 ммол) втор.-бутоксиметилхлорида и перемешивают 5 ч при 80oС. Снова добавляют 0,7 г (5,5 ммол) втор.-бутоксиметилхлорида и перемешивают еще 3 ч. После этого добавляют 12 мл воды и 5 мл метанола и перемешивают 2 ч при 50oС. Затем добавляют остальные 60 мл воды, экстрагируют три раза, каждый раз с 200 мл простого метил-трет.-бутилового эфира, промывают соединенные органические фазы 200 мл воды, сушат сульфатом магния и концентрируют при пониженном давлении. Сырой продукт очищают флеш-хроматографией на колонке с силикагелем, дихлорметан/метанол (19,8/0,2).
Выход: 3,29 г (84% от теории); масло;
C19H24N4O3 (MG=356,4 г/мол).
Масс-спектр: 356 (4%, М); 284 (71%); 271 (32%); 91 (100%);
1H-ЯМР (DMSO-d6, 200 МГц): δ=0,73 (t, 3H, СН2СН3); 0,05 (d, 3H, СНСН3); 1,21 (t, 3H, NСН2СН3); 1,35 (quint., 2H, СНСН2СН3); 3,57 (sixt., 1H, CHCH2); 4,02 (q, 2H, NСН2СН3); 5,30 (АВ-система, 2H, OCH2N); 5,50 (s, 2H, бензил. Н); 7,23-7,40 (m, 5Н, аромат. Н); 8,32 (s, 1H, N=CH).
б) 1-втор.-бутоксиметил-3-этилксантин (соединение 52)
2,73 г (7,66 ммол) 1,3,7-тризамещенного ксантина со стадии а) гидрируют в 100 мл этанола при помощи 273 мг палладия (10%) на активном угле в течение 12,5 ч. Перегруппировывают азотом, отфильтровывают катализатор и концентрируют при пониженном давлении. Остаток очищают флеш-хроматографией на колонке с силикагелем, дихлорметан/метанол (19,7/0,3).
Выход: 1,82 г (89% от теории);
точка плавления: 189oС;
C12H18N4O3 (MG=266,3 г/мол).
Масс-спектр: 266 (4%, М); 194 (87%); 193 (100%); 181 (63%); 136 (87%).
Пример 13:
1-(2-метокси-этоксиметил)-3-метилксантин (соединение 53)
а) 7-бензил-1-(2-метокси-этоксиметил)-3-метил-ксантин
Смесь из 25,6 г (0,1 мол) 7-бензил-3-метилксантина (полученного по примеру 1 а), 15,2 г (0,11 мол) карбоната калия и 16,2 г (0,13 мол) 2-метокси-этоксиметил-хлорида в 500 мл ацетонитрила нагревают 5 ч при перемешивании до 50oС, после этого обрабатывают аналогично примеру 1б) и полученный маслянистый продукт очищают при помощи фильтрования на колонке с силикагелем в текучей среде хлороформ/метанол (10/1).
Выход: 22,8 г (66,2% от теории); масло;
C17H20N4O4 (MG=344,3 г/мол).
Анализ: вычислено, %: C 59,29, H 5,85, N 16,27.
найдено, %: C 59,01, H 5,93, N 16,02.
б) 1-(2-метокси-этоксиметил)-3-метилксантин (соединение 53)
Гидролитическое дебензилирование 22,7 г (0,066 мол) соединения со стадии а) по примеру 1 с) дает после хроматографической очистки и перекристаллизации из этанола 10,9 г конечного продукта (65% от теории).
С10Н14N4O4 (MG=254,3 г/мол);
точка плавления: 188oС.
Анализ: вычислено, %: C 47,24, H 5,55, N 22,04.
найдено, %: C 47,22, H 5,45, N 22,06.
Пример 14:
3-этил-1-(2-(2-метоксиэтокси-этил)-ксантин (соединение 56)
14 г (0,037 мол) 7-бензил-3-этил-1-(2-(2-метоксиэтокси)-этилксантина получают из 7-бензил-3-этилксантина (полученного по примеру 5 а) и 1-бром-2-(2-метоксиэтокси) этана (полученного по примеру 2б) с выходом 98% от теории (C19H24N4O4 (MG= 372,4 г/мол); точка плавления после перекристаллизации из простого диизопропилового эфира: 64oС;
Анализ: вычислено, %: C 61,28, H 6,50, N 15,04.
найдено, %: C 61,44, H 6,49, N 15,26.
и, аналогично примеру 1 с), подвергают гидролитическому дебензилированию. Полученный сырой продукт без хроматографической очистки на колонке перекристаллизовывают непосредственно из сложного этилового эфира уксусной кислоты.
Выход: 7,5 г (71,8% от теории);
точка плавления: 140oС;
C12H18N4O4 (MG=282,3 г/мол).
Анализ: вычислено, %: C 51,05, H 6,43, N 19,85.
найдено, %: C 51,51, H 6,37, N 19,87.
Пример 15:
3-метил-1-(2-феноксиэтил)-ксантин (соединение 60)
а) 7-бензил-3-метил-1-(2-феноксиэтил)-ксантин
К суспензии 3,0 г (11,7 ммол) 7-бензил-3-метилксантина (полученного по примеру 1 а) в 70 мл диметилформамида добавляют при 60oС 2,6 г (18,72 ммол) карбоната калия и перемешивают 1 ч при этой температуре, затем добавляют по каплям 3,1 г (15,21 ммол) 2-феноксиэтилбромида и перемешивают 5 ч при 80oС. После этого фильтруют сырую смесь, концентрируют фильтрат при пониженном давлении, поглощают дихлорметаном, промывают 1 раз 1 н. едким натром и два раза водой. Соединенные органические фазы сушат с сульфатом магния и концентрируют при пониженном давлении. Сырой продукт очищают флеш-хроматографией на колонке с силикагелем, гептан/этилацетат (1/2).
Выход: 3,52 г (80% от теории);
точка плавления: 141oС;
C21H20N4O3 (MG=376,4 г/мол).
Масс-спектр: 376 (2%, М); 283 (100%); 91 (87%).
б) 3-метил-1-(2-феноксиэтил)-ксантин (соединение 60)
3,0 г (8,0 ммол) 1,3,7-тризамещенного ксантина со стадии а) гидрируют в 500 мл этанола при помощи 0,3 г палладия (10%) на активном угле в течение 6 ч. Перегруппировывают азотом, отфильтровывают катализатор и концентрируют при пониженном давлении. Остаток очищают флеш-хроматографией на колонке с силикагелем, гептан/этилацетат (1/10).
Выход: 1,09 г (48% от теории);
точка плавления: 207oС;
C14H14N4O3 (MG=286,3 г/мол).
Масс-спектр: 287,2 (45%, М+Н); 193,1 (100%).
Пример 16:
1-(4-хлорфеноксиметил)-3-метилксантин (соединение 65)
а) 7-бензил-1-(4-хлорфеноксиметил)-3-метил-ксантин
К суспензии 3,0 г (12,0 ммол) 7-бензил-3-метилксантина (полученного по примеру 1 а), в 50 мл диметилформамида добавляют при 60oС (2,59 ммол) карбоната калия и перемешивают 1 ч при этой температуре. Затем добавляют по каплям 2,69 г (15,0 ммол) 4-хлорфеноксиметилхлорида и перемешивают 8 ч при 80oС. После этого сырую смесь фильтруют, фильтрат концентрируют при пониженном давлении, поглощают дихлорметаном, промывают 1 раз с 1 н. едким натром и два раза с водой. Органические фазы сушат с сульфатом магния и концентрируют при пониженном давлении. Сырой продукт очищают флеш-хроматографией на колонке с силикагелем, дихлорметан/метанол (19,8/0,2).
Выход: 4,15 г (87% от теории);
точка плавления: 96oС;
C20H17ClN4O3 (MG=396,8 г/мол).
Масс-спектр: 398 (2%, 37Cl, M); 396 (6%, 35Cl, M); 269 (100%); 91 (72%).
б) 1-(4-хлорфеноксиметил)-3-метилксантин (соединение 65)
3,37 г (8,5 ммол) 1,3,7-тризамещенного ксантина со стадии а) гидрируют в 450 мл этанола при помощи 0,34 г палладия (10%) на активном угле в течение 5 ч. Перегруппировывают азотом, отфильтровывают катализатор и концентрируют при пониженном давлении. Остаток очищают флеш-хроматографией на RP-18-колонне, вода/ацетонитрил (7/3).
Выход: 0,91 г (34% от теории);
точка плавления: 218oС;
С13Н11СlN4O3 (MG=306,7 г/мол).
Масс-спектр: 309,1 (6%, 37Cl, M+H); 307,1 (19%, 35Cl, M+H); 179,1 (100%); 167 (11%).
Пример 17:
1-бензилоксиметил-3-метилксантин (соединение 68)
а) 3-метил-7-тритилксантин
К суспензии 3,9 г (23,5 ммол) 3-метилксантина в 85 мл диметилформамида добавляют при 60oС частями 0,62 г (25,88 ммол) гидрида натрия, перемешивают 1,5 ч при этой температуре и нагревают до 90oС. Затем добавляют 6,6 г (23,67 ммол) тритилхлорида в 30 мл диметилформамида и перемешивают 3 ч при 90oС. После этого отсасывают в горячем состоянии и концентрируют при пониженном давлении, остаток поглощают в 1 н. едком натре, нагревают до 80oС и отсасывают. Фильтрат доводят до рН 4-5 при помощи 2 н. соляной кислоты. Образованный при этом осадок очищают флеш-хроматографией на колонке с силикагелем, дихлорметан/метанол (19/0,2).
Выход: 6,55 г (68% от теории);
точка плавления: 242oС;
C25H20N4O2 (MG=408,7 г/мол).
Масс-спектр: 409,1 (21%, М+Н); 244,2 (17%); 243,2 (100%); 167,0 (17%).
б) 1-бензилоксиметил-3-метил-тритилксантин
К раствору 2,4 г (5,9 ммол) 3-метил-7-тритилксантина со стадии а) в 50 мл диметилформамида добавляют при 60oС 1,3 г (9,44 ммол) карбоната калия и перемешивают 1 ч при этой температуре. Затем добавляют по каплям 1,06 мл (7,67 ммол) бензилоксиметилхлорида и перемешивают 7 ч при 80oС. После этого добавляют 50 мл воды и экстрагируют три раза, каждый раз с 60 мл простого метил-трет. -бутилового эфира. Соединенные органические фазы промывают два раза, каждый раз с 30 мл воды, сушат с сульфатом магния и концентрируют при пониженном давлении. Сырой продукт очищают флеш-хроматографией на колонке с силикагелем, гептан/этилацетат (3/2).
Выход: 1,57 г (51% от теории);
точка плавления: 164oС;
С33Н28N4O3 (MG=528,9 г/мол).
Масс-спектр: 535,2 (74%, M+Li); 243,1 (100%).
с) 1-бензилоксиметил-3-метилксантин (соединение 68)
К суспензии 1,2 г (2,27 ммол) 1,3,7-тризамещенного ксантина со стадии б) в 11 мл этанола добавляют смесь 1,1 мл этанола и 2,2 мл 1 н. соляной кислоты. Кипятят 1,5 ч при флегме, концентрируют при пониженном давлении и очищают флеш-хроматографией на колонке с силикагелем, дихлорметан/метанол (19/0,5).
Выход: 0,6 г (92% от теории);
точка плавления: 208oС;
C14H14N4O3 (MG=286,3 г/мол).
Масс-спектр: 287,2 (57%, М+Н); 257,1 (77%); 179,1 (100%); 91,1 (24%).
Пример 18:
1-(2-(4-хлорбензилокси)-этил)-3-метилксантин (соединение 70)
а) 1-(2-(4-хлорбензилокси)-этил)-3-метил-7-тритилксантин
К раствору 2,4 г (5,9 ммол) 3-метил-7-тритилксантина (полученного по примеру 17 а) в 50 мл N-метилпирролидона добавляют при 60oС 1,3 г (9,44 ммол) карбоната калия и перемешивают 1 ч при этой температуре. Затем добавляют по каплям 1,57 г (7,67 ммол) 2-(4-хлорбензилокси)этилхлорида и перемешивают 1 ч при 80oС. Потом добавляют остальные 1,0 г (4,9 ммол) 2-(4-хлорбензилокси)этилхлорида и перемешивают снова 1 ч. После этого добавляют 50 мл воды, экстрагируют три раза, каждый раз с 60 мл простого метил-трет.-бутилового эфира, соединенные органические фазы промывают два раза, каждый раз с 30 мл воды, сушат с сульфатом магния и концентрируют при пониженном давлении. Сырой продукт очищают флеш-хроматографией на колонке с силикагелем, гептан/этилацетат (3/2).
Выход: 2,13 г (63% от теории);
точка плавления: 179oС;
С34Н29С1N4O3 (MG=577,1 г/мол).
Масс-спектр: 585 (5%, 37Cl, M+Li); 583,2 (8%, 35Cl, M+Li); 243,1 (100%).
б) 1-(2-(4-хлорбензилокси)-этил)-3-метилксантин (соединение 70)
К суспензии 1,3 г (2,26 ммол) 1,3,7-тризамещенного ксантина со стадии а) в 14 мл этанола добавляют смесь 1,4 мл этанола и 2,8 мл 1 н. соляной кислоты. Кипятят 1 ч при флегме, концентрируют при пониженном давлении и очищают флеш-хроматографией на колонке с силикагелем, дихлорметан/метанол (19/0,5).
Выход: 0,72 г (95% от теории);
точка плавления: 152oС;
C15H15ClN4O3 (MG=334,7 г/мол).
Масс-спектр: 336 (1%, 37Cl, M); 334 (2%, 35Cl, M); 194 (100%); 179 (25%); 166 (65%).
Фармакологическое испытание и результаты
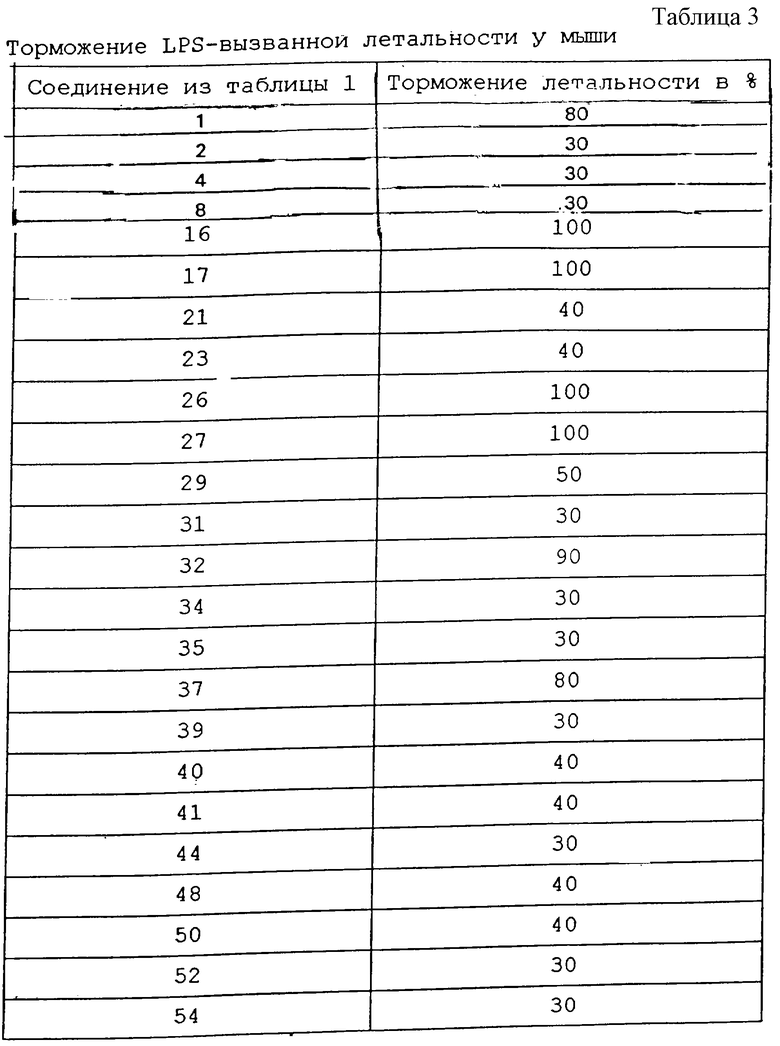
Ярко выраженное антишоковое действие соединений по формуле I демонстрировали на хорошо известной модели вызванного эндотоксином (LPS) смертельного шока на С 57 ВL/6-мышах на основании снижения смертности.
Для проведения опытов назначали внутривенной инъекцией каждому животному смесь 10 нг LPS из Salmonella abortus equi и 7,5 мг галактозамина в 0,2 мл буферного фосфатного физиологического раствора хлористого натрия; инъекция, как правило, приводила к смерти в течение 6-9 ч. Испытываемые препараты за 1 ч до LPS-провокации назначали внутрибрюшинно при дозе 100 мг/кг. Животные контрольной группы (n= 10) получали вместо этого чистый 0,9%-ный раствор хлористого натрия в качестве плацебо. Для оценки действия препарата в обработанном коллективе (n=10) определяли количество оставшихся в живых животных через 48 ч после приема LPS и отсюда, ссылаясь на смертность в контрольной группе, определяли торможение летальности в процентном отношении. Результаты опытов приведены в табл. 3.
В пределах более широкой фармакологической классификации, оказалось, кроме того, что соединения формулы I дополнительно могут продолжительное время тормозить обусловленную ишемией гибель клеток в центральной нервной системе. Поэтому они применяются также для лечения и профилактики церебрососудистых заболеваний, таких как инсульт; транзиторные ишемические приступы (TIA); слабоумие при множественном инфаркте; слабоумие смешанного типа; с сосудистой и дегенеративной (Арцхаймер) компонентой; повреждения спинного мозга; травма головного мозга вследствие ранений головы; нейрональные повреждения после остановки сердечной деятельности, (неородовая) асфиксия и реанимация, а также сосудисто-хирургические вмешательства (например, байпас-операции) в области снабжающих головной мозг основных артерий.
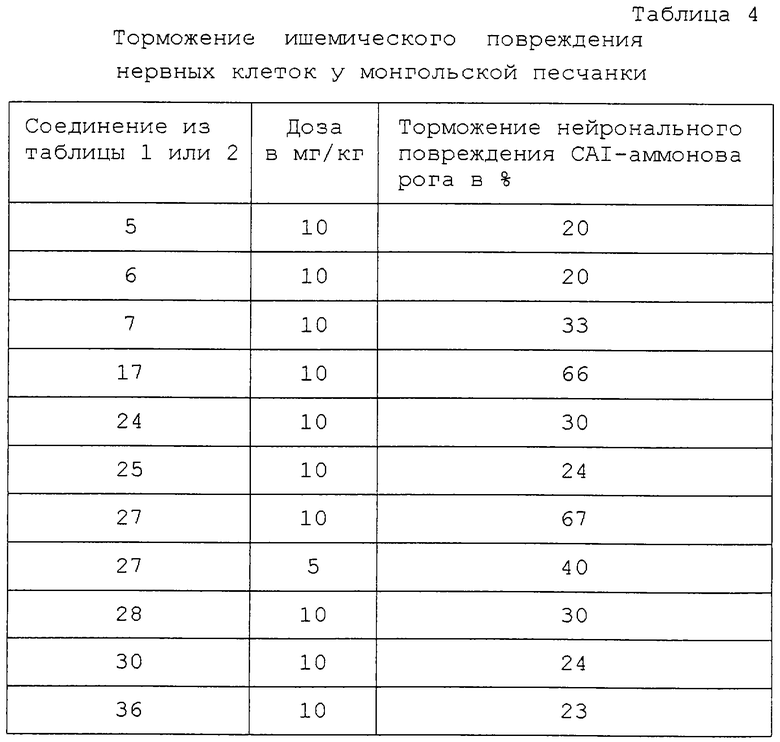
Нейрональное защитное действие производных теофиллина по формуле I можно убедительно обнаружить, между прочим, на модели промежуточной глобальной ишемии у мышей песчанок (Gerbil). И этот результат является неожиданным в том отношении, что сам теофиллин при сравниваемых опытных условиях не тормозит ишемическое повреждение нервных клеток ни у песчанок (Gerbil) (J. Gereb. Blood Flow Metab. 1987, 7/1: 74-81), ни у крысы (J. Gereb. Blood Flow Metab. 1994, 14/1: 166-173), а еще более усиливает его.
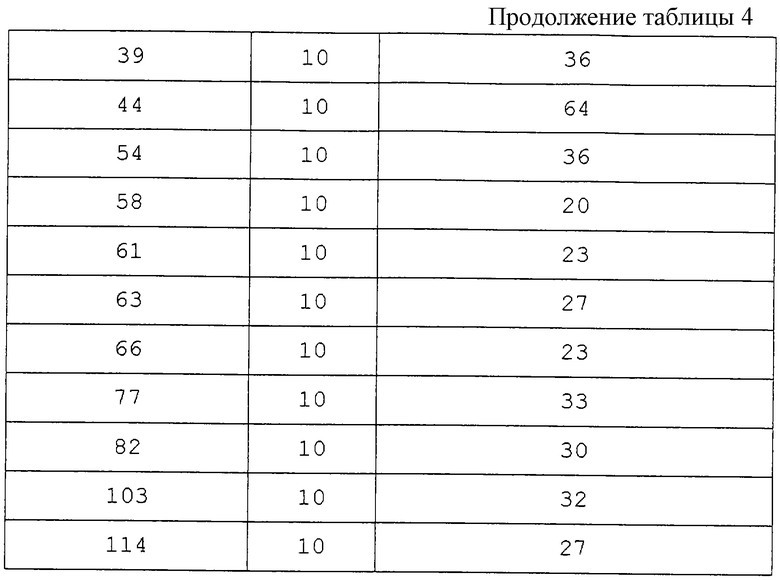
Для осуществления опытов, которые проводили в соответствии с немецким законом об охране животных, выбирали наугад 30 монгольских песчанок с весом тела между 60 и 70 г, распределяя на два коллектива соответственно по 15 животных. Животным первого коллектива назначали через 30 мин после ишемического периода соответствующее тест-вещество путем внутрибрюшинной инъекции, в то время как животные второго коллектива, который являлся необработанной контрольной группой, получали такой же объем соответствующего индифферентного лекарственного препарата. Для образования временной ишемии переднего мозга под галотан-наркозом закрепляли на обогреваемом операционном столе в положении на спине, осторожно обнажали обе сонные артерии и при помощи зажима микроаневризмы перекрывали на 3 мин. Через 7 дней после 3-минутного ишемического периода животных под галотан-наркозом декапитировали, быстро и щадяще вынимали головной мозг, сначала фиксировали погружением в раствор Карно (этанол/хлороформ/уксусная кислота = 6/3/1) и затем заливали в парафин, после этого получали коронарные срезы толщиной от 4 до 6 мкм через аммонов рог приблизительно на высоте брегмы и эти срезы окрашивали гематоксилином и эозином. После этого определяли в пределах слепого опыта при помощи светового микроскопа размер эозинофильных некрозов пирамидных клеток в CAI-области аммонова рога на основании полуколичественного гистопатологического счета (0 = нет; 1 = легкие; 2 = среднетяжелые; 3 = тяжелые и 4 = комплексные некрозы). В качестве оценочного параметра для нейрозащитного действия служило изменение в процентах среднего гистопатологического счета группы препаратов в противоположность необработанной контрольной группы. Результаты опытов представлены в табл. 4.
Изобретение относится к области медицины и касается лекарственного средства для лечения шоковых заболеваний, содержащего соединение формулы I, а также к новым соединениям общей формулы I. Средство обладает повышенной антишоковой активностью. 2 с. и 4 з.п. ф-лы, 4 табл.
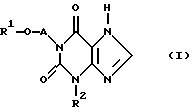
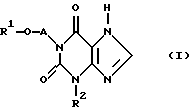
в которой R1 - (С1-С5)-алкил с линейной или разветвленной цепью, -СН3-О-(СН2)2, или фенил, или (С1-С2) - алкилфенил, где фенильные остатки могут быть заменены одним атомом хлора,
А - (С1-С4)-алкиленовый мостик с неразветвленной или разветвленной цепью;
R2 - (С1-С5)-алкил с линейной или разветвленной цепью, циклопропил, циклопропилметил, фенил или (С1-С2) - алкилфенил,
его стереоизомерные формы и физиологически приемлемые соли.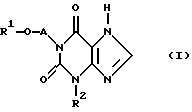
в котором R1 - (С1-С5)-алкил с линейной или разветвленной цепью, -СН3-О-(СН2)2, фенил или метилфенил, где фенильные остатки могут быть замещены одним атомом хлора;
А - (С1-С4)-алкиленовый мостик с линейной или разветвленной цепью;
R2 - (С1-С5) - алкил с линейной или разветвленной цепью, циклопропил или фенил, за исключением соединений, в которых R2 - н-пропил, R1 - метил или этил и А - этиленовый мостик или в которых когда R2 - н-пропил, R1 - метил и А - н-пропиленовый мостик.
его стереоизомерные формы и физиологически приемлемые соли.
Приоритет по пунктам:
07.06.1996 по пп.4-6;
24.07.96 по пп.1-3.
| SU 755199 A, 07.08.1980 | |||
| Способ получения производных ксантина или их кислотно-аддитивных солей | 1980 |
|
SU1079176A3 |
| Торфодобывающая машина с вращающимся измельчающим орудием | 1922 |
|
SU87A1 |
| US 4769377 A, 06.09.1988. | |||

