Изобретение относится к технологии производства ядерных материалов и может быть использовано для получения порошка керамической двуокиси урана, обогащенной по изотопу 235U, порошка керамической двуокиси урана, содержащей выгорающий поглотитель, квазигомогенной смеси порошков оксидов урана и плутония, урана и тория для производства МОХ-топлива, переводу высокообогащенного урана в низкообогащенный или для извлечения фтора из "отвального" гексафторида урана.
Известны две группы способов перевода гексафторида урана в оксиды урана, в частности в двуокись урана, - водные и неводные (сухие, газовые) [1, с. 79; 2, с. 69, рис. 4.5, 4.6, 4.7]. Первая группа включает процессы гидролиза UF6 водой, осаждение из гидролизата (NН4)2U2O7 или (NН4)4UO2(CO3)3, сушку и разложение последних до порошка оксида урана (UO2+х) (соответственно, ADU- и AUC-процессы). При прокаливании порошка в атмосфере водорода получают UO2. Вторая группа основана на реакции пирогидролиза паров UF6, т. е. взаимодействия газообразного гексафторида урана с парами Н2О в присутствии водорода. Достоинство неводных способов - простота, отсутствие больших объемов жидких радиоактивных отходов, сравнительно небольшой расход реагентов.
Превращение UF6 в UО2 по газовым способам обычно осуществляют в две стадии [2, рис. 4.7]. На первой проводят гидролиз паров UF6 водяным паром при 150-300oС, где в результате экзотермической реакции
UF6(г)+избыток Н2О(г) -->UО2F2(тв)+4НF(г)+остаточная Н2О(г) (1)
образуется уранил-фторид и разбавленная фтористо-водородная кислота. На второй стадии порошок UО2F2 подвергают термической обработке при температуре выше 550oС в аппаратах псевдоожиженного слоя или вращающихся печах водородом или его смесью с водяным паром для получения частиц оксида урана с мольным отношением O/U от 2,06-2,07 до 2,17-2,18 [3, с. 24].
Недостатком двухстадийных газовых способов является наличие гомогенных и гетерогенных стадий процесса, разделенных во времени и пространстве (реализация процесса осуществляется в нескольких технологических аппаратах), что усложняет аппаратурное оформление и технологическое проведение низкотемпературного пирогидролиза [1, с. 82]. Процесс продолжителен во времени - особенно его вторая стадия. Время пребывания твердого дисперсного материала в печах на второй стадии колеблется от 0,25 часа до нескольких часов [1, с. 98, 103].
Низкотемпературный пирогидролиз приводит к безвозвратным потерям фтора в виде разбавленной фтористо-водородной кислоты.
С точки зрения времени и протекания реакции конверсии непосредственно до двуокиси урана необходимо стремиться к образованию оксида урана в газовой фазе с последующей конденсацией UO2+х в условиях, исключающих реакции обратного частичного фторирования [3, с. 98; 4, с. 190]. Реакция пирогидролиза паров UF6 водяным паром идет с образованием UO2+х при температуре выше 1000oС.
Для конверсии гексафторида урана в двуокись урана и фтористый водород парогазовой смесью также известен одностадийный способ, заключающийся в быстром и полном превращении паров UF6 в UO2 в кислородно-водородном пламени за счет высокой температуры и ионизации вещества [3, с. 108]. Суммарная реакция восстановительного пирогидролиза в кислородно-водородном пламени может быть записана в следующем виде:
UF6(г)+избыток Н2+избыток О2 -->UO2+х(тв)+6HF(г)+остаточная Н2О(г) (2)
По патенту [5] в реакционную зону пламенного реактора вводят газовый поток, представляющий собой смесь паров UF6 и О2-содержащего газа-носителя. Сюда же раздельно подают поток газа-восстановителя. Между этими двумя потоками вдувают третий поток защитного газа-разделителя, который препятствует началу реакции до тех пор, пока оба потока реагентов не перемешаются тщательно в реакционной зоне. В результате реакций, протекающих в кислородно-водородном пламени, зажигаемом в реакционной зоне, образуются твердые частицы оксидной композиции, обогащенной UO2, и газообразные продукты. В эту газовую смесь вводят дополнительный поток О2-содержащего газа. При этом газообразные продукты реакции переходят в окисленную форму, а UO2 - в оксиды высшей валентности. Для отвода избытка тепла, выделяющегося при окислении, и снижения скорости обратных реакций в поток О2-содержащего газа впрыскивают диспергированную жидкость с большой скрытой теплотой испарения, например воду, жидкий азот и др. Впрыскивание производят в область реакционной зоны с наиболее высокой температурой.
Успешному проведению процесса способствуют низкое давление в пламенном реакторе, высокая температура (достаточная для предотвращения образования или конденсации слаболетучих промежуточных продуктов, в частности UF4 и UО2F2), избыток Н2 и Н2О.
Способ позволяет производить переработку больших объемов сырья на малых производственных площадях [2, с. 70].
К недостаткам этого способа следует отнести использование для нагрева и конверсии паров UF6 в UO2+х и НF взрывоопасной кислородно-водородной смеси, для нормального горения которой объемное содержание UF в газе не должно превышать 10 об. % [1, с.112]. В результате в объеме пламенного реактора создается пароводяная среда с малой концентрацией НF, из которой при совместной или дробной конденсации Н2О и HF образуется малоценная разбавленная фтористо-водородная кислота. Дополнительное разбавление Н2F2 происходит при впрыскивании в реактор в качестве диспергированной жидкости воды.
Общим недостатком газовых способов конверсии UF6 является их плохое сочетание с переработкой брака, отходов и оборотов производства ядерного топлива (порошка UO2+х, таблеток, изготовления твэлов и сборок) [3, с. 113]. Большая часть брака и отходов перерабатывается через операции растворения и экстракционной перечистки с последующим получением порошков по водной технологии, т. е. через осаждение и прокалку урановых солей. Свойства порошков, получаемых на цепочке регенерации урана, в той или иной мере отличаются от свойств порошков, получаемых в основной схеме. Такие порошки требуют либо специальной подготовки на стадии получения таблеток, либо должны перерабатываться по отдельной технологии.
Другой недостаток - невозможность получения в газовых способах смеси оксидов урана с оксидами других металлов, например порошков для уран-гадолиниевого топлива (UO2-Gd2О3), где гадолиний используется в качестве выгорающего поглотителя нейтронов, порошков UO2-PuO2 или UO2-ТhО2 для МОХ-топлива. Такие порошки получают либо механическим смешением порции порошка UO2 c порошком оксида другого металла, либо методом соосаждения из растворов. В последнем случае из нитратных растворов посредством AUG- или ADU-процессов получают смесь солей, при последующем прокаливании которых при температуре 500-650oС образуются порошки стехиометрического твердого раствора оксидов металлов [6, с. 9-10; 7, с. 126].
Способ соосаждения требует или предварительной операции перевода UF6 в азотнокислый раствор уранила (для UO2-Gd2О3 или UО2-ThO2-порошков), или использования раствора природного (регенерированного) урана (для UO2-РuО2-порошков).
Поэтому наиболее часто для изготовления как (U, Gd)O2-таблеток, так и МОХ-топлива, используют сухое механическое смешивание и измельчение порошков UО2 с Gd2О3, РuО2 или другими оксидами [6, с, 8-9; 8, с. 9; 9, с. 13]. При этом наилучший путь получения гомогенного таблетирующегося порошка - двухступенчатый процесс, включающий измельчение до микронных размеров и гомогенизацию порошка UО2 с оксидом другого металла, а затем разбавление смешанного порошка диоксидом урана до требуемой концентрации примесного металла [6, с. 8-9; 8, с. 9].
Для механического смешения порошков исходный UF6 предварительно переводят в UО2+х по принятой у данного производителя технологии, а сама технологическая схема приготовления смеси существенно зависит от свойств (главным образом, текучести) порошка UО2 [6, с.8].
Наиболее близким техническим решением к предлагаемому является способ конверсии гексафторида урана в оксиды урана и фтористый водород [10, прототип], согласно которому превращение UF6 осуществляют в объеме активного кислородно-водородного теплоносителя в реакционной зоне пламенного реактора путем подачи в реактор первичной газовой смеси, состоящей из паров UF6 и кислородсодержащего газа-носителя, и вторичной смеси, содержащей газ-восстановитель. Газовые реакционные смеси разделены друг от друга струей защитного инертного газа, который предохраняет их от смешения на начальном участке. В результате реакции, протекающей в объеме факела кислородно-водородного пламени, зажигаемого в реакционной зоне, образуются твердые частицы оксидной композиции, обогащенной UO2, и газообразные продукты. Процесс ведут при избытке водорода. Для более высокой степени окисления урана и перевода остатков газа-восстановителя в окисленную форму в реактор вводят дополнительный поток кислородсодержащего газа. При этом газообразные продукты реакции переходят в окисленную форму, а UО2 - в оксиды высшей валентности. Охлаждение продуктов реакции проводят путем распыления в реакционную зону пламенного реактора жидкости с высокой скрытой теплотой парообразования, в частности деионизированной воды, жидкого азота или жидкого оксида углерода, которые подают под давлением не менее 2,1 МПа. Указанные жидкости вводят отдельно либо совместно с газомносителем в первичное или вторичное пламя, образующиеся в реакторе, либо в область, расположенную под вторичным пламенем.
Данный способ обладает всеми вышеперечисленными недостатками, присущими газопламенной технологии конверсии гексафторида урана в высокотемпературном кислородно-водородном теплоносителе.
Задачей заявленного технического решения является упрощение технологии получения смеси порошка UО2 с оксидами других металлов при высокотемпературной конверсии UF6.
Для решения этой задачи в известном способе конверсии гексафторида в оксиды урана и фтористый водород водяным паром, заключающемся в смешении первичной газовой смеси, состоящей из паров гексафторида урана и газа-теплоносителя, и вторичной газовой смеси, содержащей пары воды, и включающем впрыск диспергированной жидкости в зону конверсии, в качестве жидкости используют водный раствор по крайней мере одной соли по крайней мере одного элемента из ряда U, Pu, Th, Eu, Gd и Еr. При этом водный раствор дополнительно содержит соль аммония и/или карбамид. Кроме того, в зону конверсии дополнительно впрыскивают водный раствор аммиака, раствор соли аммония и/или карбамида. В качестве газа-теплоносителя используют азот и/или аргон, нагретые до состояния низкотемпературной плазмы.
Достижение поставленной задачи обеспечивается прежде всего тем, что при нагреве паров гексафторида урана в низкотемпературной плазме азота, аргона или их смеси образуется первичная высокотемпературная смесь газа-теплоносителя с продуктами диссоциации UF6 (UF5, UF4, UF3 и F), которая не содержит аэрозольных частиц соединений урана. При смешении первичной высокотемпературной газовой смеси с диспергированным водным раствором соли по крайней мере одного элемента из ряда U, Pu, Th, Eu, Gd и Еr происходят процессы нагрева и испарения капель раствора с последующим термическим разложением частиц солевого остатка, сопровождающиеся обогащением газовой фазы водяным паром и образованием высокодисперсных частиц оксида соответствующего металла. В результате взаимодействия паров воды с продуктами термической диссоциации UF6 в газовой фазе образуются НF и пары UO2+х. Последние кристаллизуются путем конденсации на имеющейся в зоне конверсии затравке твердой фазы. Роль затравки выполняют частицы оксидов, образовавшиеся за счет термического разложения диспергированного раствора солей. В результате на выходе зоны конверсии образуется квазигомогенная смесь UO2+х из UF6 с оксидами элементов из водного раствора солей.
Конверсия UF6 парами воды при температуре выше 1000oС сопровождается образованием оксидной композиции с мольным отношением O/U от 2,25 до 2,67 (в основном U4О9 и U3О8). Введение в водный раствор соли аммония и/или карбамида (по другому варианту - дополнительное впрыскивание (распыление) в зону конверсии водного раствора аммиака, раствора соли аммония и/или карбамида) создает в зоне конверсии газовую фазу, обогащенную водородом, поскольку при температуре выше 400oС и давлении, близком к атмосферному, аммиак полностью диссоциирует на азот и водород [11, с. 200].
Обогащение газовой фазы водородом, с одной стороны, позволяет на 200-300oС снизить начало обратной реакции фторирования оксидов урана (металлов) газообразным фтористым водородом при охлаждении пылегазовой смеси и осуществлять отделение частиц UО2+х при 500-600oС с минимальным содержанием фтора в порошке, с другой, получить порошок оксидов урана, по составу близкий к UО2.
Создание восстановительной паровой атмосферы в зоне конверсии через разложение водного раствора аммиака, раствора соли аммония и/или карбамида исключает из технологии использование взрывоопасных горючих газов.
Поскольку скорости ввода первичной газовой смеси и диспергированного водного раствора в зону конверсии реактора, как правило, существенно различаются, то за счет эффекта инжекции в реакторе образуются области возвратно-поступательного движения газа с содержащимися в нем аэрозольными частицами. В зонах циркуляции происходит агломерация частиц и их укрупнение за счет вторичной конденсации паров UO2+х на первичном аэрозоле. Рост и агломерация частиц в зоне конверсии облегчает последующее разделение пылегазовой смеси и уменьшает протекание обратных реакций фторирования UO2+х фтористым водородом на поверхности твердой фазы при охлаждении реакционного потока, поскольку суммарная скорость взаимодействия конденсированной и газовой фаз уменьшается с уменьшением площади поверхности раздела фаз.
Диапазон значений концентраций водных растворов солей U, Pu, Th, Eu, Gd и Еr и массовый расход данного раствора на конверсию UF6 определяется условиями получения порошка уран-оксидной композиции с требуемой концентрацией металла (металлов), содержащегося в водном растворе соли.
Диапазон значений концентраций соли аммония и/или карбамида в водном растворе соли U, Pu, Th, Eu, Gd и Еr определяется кристаллизационной устойчивостью соответствующего раствора.
Диапазон значений концентрации аммиака в водном растворе, дополнительно впрыскиваемом в зону конверсии, определяется использованием выпускаемой промышленностью аммиачной воды, а диапазон концентраций в этом растворе солей аммония и/или карбамида - получением порошков оксида урана с требуемым мольным отношением О/U.
Схема реализации способа показана на рисунке. Газ-теплоноситель (азот, аргон и/или их смесь) нагревают до состояния низкотемпературной плазмы (5500 - 6000oС) высокочастотным индукционным разрядом в ВЧИ-плазмотроне 1 и направляют в реакционную зону плазменного реактора 2.
Баллон 3 с гексафторидом урана нагревают в испарительной камере 4 до температуры 50-60oС. Пары UF6 компрессором 5 подают на вход плазменного реактора, где происходит его смешение с газом-теплоносителем. Молярное соотношение расходов газа-теплоносителя и гексафторида урана (N2(Ar)/UF6) поддерживают в интервале 4-10. Температура газа-теплоносителя после смешения с UF6 снижается до 2200-3200oС.
Водный раствор солей металлов или его смесь с солью аммония и/или карбамида из емкости 6 в плазменный реактор нагнетается насосом 7. Распыление раствора в камеру смешения реактора осуществляют пневматическими или пневмоцентробежными форсунками (на рисунке не показаны), обеспечивающими среднюю дисперсность распыла жидкости не более (50÷70)•10-6 м, для чего в форсунки дополнительно подводят сжатый газ (азот) из ресивера 8. Впрыск (распыление) раствора в зону конверсии ведется под углом к направлению движения газа-теплоносителя. Аналогично в плазменный реактор из емкости 9 подают водный раствор соли аммония и/или карбамида.
Во время технологического процесса давление в зоне конверсии плазменного реактора поддерживают на 10-50 кПа ниже атмосферного.
В результате термического разложения диспергированного водного раствора соли в зоне конверсии образуется аэрозоль оксидов урана с тем или иным мольным отношением О/U в зависимости от восстановительного потенциала газовой фазы или аэрозоль квазигомогенной смеси UO2+х с оксидами РuО2, ТhО2, Еu2О3, Gd2О3 и Еr2О3. Дисперсность аэрозоля ~(2-5)•10-6 м.
Выделение частичек оксидов из пылегазового потока осуществляют в системе разделения 10, состоящей из циклонного аппарата и металлокерамического фильтра, включенных последовательно, при температуре соответственно 500-600oС и 200-300oС. Порошок собирают в контейнер 11. Очищенный газ, состоящий из фторида водорода (HF), остаточного водяного пара, азота (аргона) и кислорода/водорода, направляют на совместную или дробную конденсацию паров воды и фтористого водорода в холодильник-конденсатор 12 с получением в сборнике 13 фтористоводородной кислоты различной концентрации. После ректификации кислоты в колонне 14 получают два продукта: безводный фтористый водород (99,99% НF) 15 и кубовый остаток 16 - азеотроп 40%HF - 60%H2O. Азот (аргон) и газообразный фтористый водород с примесью кислорода/водорода направляют в блок газоочистки 17 для утилизации НF. В выхлопе 18 с установки имеют газовую смесь азота с кислородом/водородом. Кубовый остаток концентрирует в себе проскок аэрозолей оксидов урана и может быть возвращен в плазменный реактор в виде раствора урана путем добавки к исходному раствору солей металлов.
Порошок оксидов подвергают термообработке в восстановительной атмосфере для удаления остаточного фтора и доведения мольного отношения О/U до 2,06÷2,18. Партии порошка UO2 анализируют на содержание в них 235U, Pu, Th, Eu, Gd или Еr и, если требуется, подвергают усреднению.
Порошок U3О8, полученный в результате конверсии гексафторида урана, обедненного по изотопу 235U, направляют на удаление остаточного фтора и складируют.
Пример 1. Переработке подвергали гексафторид урана, обедненный по изотопу 235U. В зону конверсии впрыскивали водный раствор уранилфторида или аммонийуранилпентафторида с аналогичным содержанием изотопа 235U и концентрацией по урану 1-10 г/л. Процесс использовался для извлечения фтора из "отвального" UF6. Испарение UF6 проводили из баллонов емкостью 2,5 м3 при их нагреве до 333К. Производительность испарителя - 50-60 кг UF6/ч; мощность плазмотрона - до 55 кВт; мощность, потребляемая плазменной установкой от электросети - до 90 кВт; расход газа-теплоносителя - 10-1 2 кг/ч; расход раствора - 9-11 кг/ч; расход газа на распыление - 3-4 кг/ч (пневматические форсунки); степень улавливания урановой пыли - 0,999; энергозатраты - 1,6÷1,8 кВт/кг UF6; выход фтора во фторсодержащие продукты - 93-94%; остаточное содержание фтора в оксидах урана - 0,03÷1,2 мас.%. Получили порошок оксидов урана с кислородным коэффициентом 2,69÷2,72. Часть остаточного фтора осталась в дисперсной фазе в виде UО2F2. Способ может быть использован для реализации замкнутого по урану и фтору процесса переработки "отвального" UF6.
Пример 2. Переработке подвергали гексафторид урана, обедненный по изотопу 235U. Способ реализовывали аналогично примеру 1. В зону конверсии дополнительно впрыскивали водный раствор аммонийуранилтрикарбоната в растворе карбоната аммония с концентрацией по урану до 2 г/л, полученный после регенерации и отмывки технологического оборудования. Производительность испарителя - 50-60 кг UF6/ч; мощность плазмотрона - до 55 кВт; мощность, потребляемая плазменной установкой от электросети - до 90 кВт; расход газа-теплоносителя - 10-12 кг/ч; расход раствора - 9-11 кг/ч; расход газа на распыление - 0,8-1,1 кг/ч (пневмоцентробежная форсунка); степень улавливания урановой пыли - 0,999; энергозатраты - 1,6÷1,8 кВт/кг UF6; выход фтора во фторсодержащие продукты - 93-94%; остаточное содержание фтора в оксидах урана - 0,01÷0,80 мас.%. Получили порошок оксидов урана с кислородным коэффициентом 2,61÷2,65. Способ может быть использован для совмещения процесса переработки "отвального" UF6 с утилизацией жидких отходов изотопно-разделительного производства.
Пример 3. Конверсии подвергали гексафторид урана с обогащением по изотопу 235U 2-4%. Испарение UF6 проводили из баллонов емкостью 0,8 м3 при их нагреве до 333 К. В зону конверсии впрыскивали водный раствор уранилфторида или аммонийуранилпентафторида с ураном с аналогичного обогащения по изотопу 235U и содержанием 1-10 г/л, которые моделировали оборотные растворы таблеточного производства. Кроме того, в зону конверсии дополнительно распыляли водный раствор аммиака. Производительность испарителя - 36-41 кг UF6/ч; мощность плазмотрона - до 55 кВт; мощность, потребляемая плазменной установкой от электросети, - до 90 кВт; расход газа-теплоносителя - 10-12 кг/ч; расход раствора - 11-13 кг/ч; расход газа на распыление - 3-4 кг/ч; энергозатраты - 1,9÷2,1 кВт/кг UF6; остаточное содержание фтора в оксидах урана - 0,03÷0,3 мас.%. Получили порошок оксидов урана с кислородным коэффициентом 2,20÷2,25. Порошок оксидов урана направляли на дополнительную термообработку в водородно-паровой среде для уменьшения кислородного коэффициента и приведения остаточного содержания фтора в требования спецификации. Способ может быть использован в технологии получения порошков UO2 для реакторного топлива.
Пример 4. Конверсии подвергали гексафторид урана с обогащением по изотопу 235U 1,5%. В зону конверсии впрыскивают водный раствор уранилнитрата с ураном того же обогащения, имитирующего уран 90-93% обогащения. Концентрация урана в растворе 80-100 г/л. Раствор дополнительно содержал ацетат аммония. Производительность испарителя - 20-22 кг UF6/ч; мощность плазмотрона - до 35 кВт; мощность, потребляемая плазменной установкой от электросети, - до 70 кВт; расход газа-теплоносителя - 8-10 кг/ч; расход раствора - 5-6 кг/ч; расход газа на распыление - 3,0-3,5 кг/ч; остаточное. Процесс может быть использован в технологии уничтожения оружейного урана с переводом его в низкообогащенный для получения порошков UO2 ядерного топлива.
Пример 5. Конверсии подвергали гексафторид урана, имеющий обогащение по изотопу 235U 34%. В зону конверсии впрыскивают водный раствор нитрата гадолиния или его смесь с нитратом уранила. Концентрация гадолиния в растворе рассчитывалась из условия получения смеси оксидов, содержащей 2-5 мас.% Gd2О3. Раствор дополнительно содержал ацетат аммония при концентрации 5-30 г/л. Способ реализовали аналогично примеру 2. Процесс может быть использован для получения порошков ядерного топлива с выгорающими нейтронными поглотителями. Аналогично получали порошки оксида урана с оксидами европия и эрбия: UO2-Eu2О3 и UO2-Еr2О3.
Пример 6. Конверсии подвергают гексафторид урана с обогащением по изотопу 235U 1,5-2%. В зону конверсии впрыскивали водный раствор нитрата церия с концентрацией 5-10 г/л, имитирующего нитрат плутония. Раствор дополнительно содержал карбамид в количестве 50-70 г/л. Способ реализовывали аналогично примеру 4. Получили порошки UО2,2-СеО2. Процесс может быть использован с целью получения порошков UО2-РuО2 для ядерного топлива легководных реакторов.
Пример 7. Конверсии подвергали гексафторид урана с содержанием изотопа 235U 0,5 -1 мас.%. В зону конверсии впрыскивали водный раствор смеси нитратов урана и церия (имитатор плутония) с концентрацией 20-50 г/л. Раствор содержал также ацетат аммония. Способ реализовывали аналогично примеру 4.
Процесс может быть использован с целью получения порошков UO2-РuО2 для ядерного топлива быстрых реакторов.
Пример 8. Конверсии подвергают гексафторид урана, имеющий обогащение по изотопу 235U 15-20%. В зону конверсии впрыскивали водный раствор нитрата тория. Кроме того, в раствор дополнительно впрыскивали раствор карбамида в аммиачной воде. Способ реализовывали аналогично примеру 4.
Способ может быть использован в процессе получения порошков UO2-ТhО2 для ядерного топлива быстрых реакторов.
Понятно, что изобретение не ограничивается приведенными примерами. Возможны изменения в пределах объема предложенной формулы изобретения.
Предлагаемый способ конверсии гексафторида урана обеспечивает получение на выходе плазменной установки гомогенной смеси оксидов урана с другими оксидами других металлов за одну стадию с одновременной утилизацией оборотных растворов.
Источники информации
1. Раков Э.Г., Тесленко В.В. Пирогидролиз неорганических фторидов. - М.: Энергоатомиздат, 1987. -152 с.
2. Лебедев B.М. Ядерный топливный цикл (технико-экономический анализ): Учебное пособие. - Обнинск. ЦИПК, 1991. - 137 с.
3. Майоров А. А., Браверман И.Б. Технология получения порошков керамической двуокиси урана. - М.: Энергоатомиздат, 1985. - 128 с.
4. Туманов Ю.Н. Электротермические реакции в современной химической технологии и металлургии. - М.: Энергоатомиздат, 1981. - 232 с.
5. Патент 4005042 (США), МПК С 01 G 43/02, 1975.
6. Горский В. B. Уран-гадолиниевое оксидное топливо. Ч. 3 - Технология изготовления и методы контроля (U, Gd)О2-таблеток. - Атомная техника за рубежом, 1989, 5. - с. 8-15.
7. Котельников Р.Б. и др. Высокотемпературное ядерное топливо. - М., Атомиздат, 1978. - 432 с.
8. ЛеБастар Ж. Рециклирование и приготовление смешанного оксидного топлива: достижения Франции и Бельгии. - Атомная техника за рубежом, 1995. 11, - с. 6-11.
9. Смешанное топливо для водоохлаждаемых реакторов. - Атомная техника за рубежом. 1992. 9. - с. 12-14.
10. Патент 4031029 (CШA), MПK C 01 G 43/02, 1975 г. (прототип).
11. Куликов И.С. Термодинамика карбидов и нитридов, Справ. изд. - Челябинск: Металлургия. Челябинское отд. 1988 г. - 320 с.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ИЗГОТОВЛЕНИЯ ТЕПЛОВЫДЕЛЯЮЩИХ ЭЛЕМЕНТОВ | 2003 |
|
RU2252459C2 |
| СПОСОБ ПЕРЕРАБОТКИ ГЕКСАФТОРИДА УРАНА | 1991 |
|
RU2090510C1 |
| СПОСОБ КОНВЕРСИИ ОТВАЛЬНОГО ГЕКСАФТОРИДА УРАНА В МЕТАЛЛИЧЕСКИЙ УРАН | 2010 |
|
RU2444475C1 |
| СПОСОБ ДЛЯ КОНВЕРСИИ ГЕКСАФТОРИДА УРАНА В ДИОКСИД УРАНА И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2001 |
|
RU2211184C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕКСАФТОРИДА НИЗКООБОГАЩЕННОГО УРАНА ИЗ ОРУЖЕЙНОГО ВЫСОКООБОГАЩЕННОГО УРАНА | 2005 |
|
RU2292303C2 |
| СПОСОБ КОНВЕРСИИ ОТВАЛЬНОГО ГЕКСАФТОРИДА УРАНА В МЕТАЛЛИЧЕСКИЙ УРАН | 2014 |
|
RU2562288C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОКСИДА УРАНА ИЗ РАСТВОРА УРАНИЛНИТРАТА И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2015 |
|
RU2601765C1 |
| СПОСОБ ПЕРЕРАБОТКИ ГЕКСАФТОРИДА УРАНА НА ОКСИД УРАНА И БЕЗВОДНЫЙ ФТОРИД ВОДОРОДА И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2015 |
|
RU2599528C1 |
| СПОСОБ ОБНАРУЖЕНИЯ УТЕЧКИ ГАЗООБРАЗНОГО ГЕКСАФТОРИДА УРАНА И/ИЛИ ФТОРИСТОГО ВОДОРОДА И ДЕТЕКТОР ДЛЯ ОБНАРУЖЕНИЯ УТЕЧКИ | 2013 |
|
RU2541708C1 |
| СПОСОБ КОНВЕРСИИ ГЕКСАФТОРИДА УРАНА ДО ТЕТРАФТОРИДА УРАНА И БЕЗВОДНОГО ФТОРИДА ВОДОРОДА | 2015 |
|
RU2594012C1 |
Изобретение относится к технологии производства ядерных материалов и может быть использовано для получения порошка керамической двуокиси урана, обогащенной по изотопу 235U, порошка керамической двуокиси урана, содержащей выгорающий поглотитель, квазигомогенной смеси порошков оксида урана и плутония, урана и тория для производства МОХ-топлива (металлоксидного), переводу высокообогащенного урана в низкообогащенный или для извлечения фтора из "отвального" гексафторида урана. Результат изобретения - упрощение технологии получения смеси порошка UO2 с оксидами других металлов при высокотемпературной конверсии UF6. Конверсию UF6 в UO2 и HF водяным паром проводят путем смешения первичной газовой смеси, состоящей из паров гексафторида урана и газа-теплоносителя, и вторичной газовой смеси, содержащей пары воды, при одновременном впрыске диспергированной жидкости в зону конверсии. В качестве жидкости используют водный раствор, по крайней мере, одной соли, по крайней мере, одного элемента из ряда U, Pu, Th, Eu, Gd и Ег. При этом водный раствор дополнительно содержит соль аммония и/или карбамид. Кроме того, в зону конверсии дополнительно впрыскивают водный раствор аммиака, раствор соли аммония и/или карбамида. В качестве газа-теплоносителя используют азот и/или аргон, нагретые до состояния низкотемпературной плазмы. 3 з.п. ф-лы, 1 ил.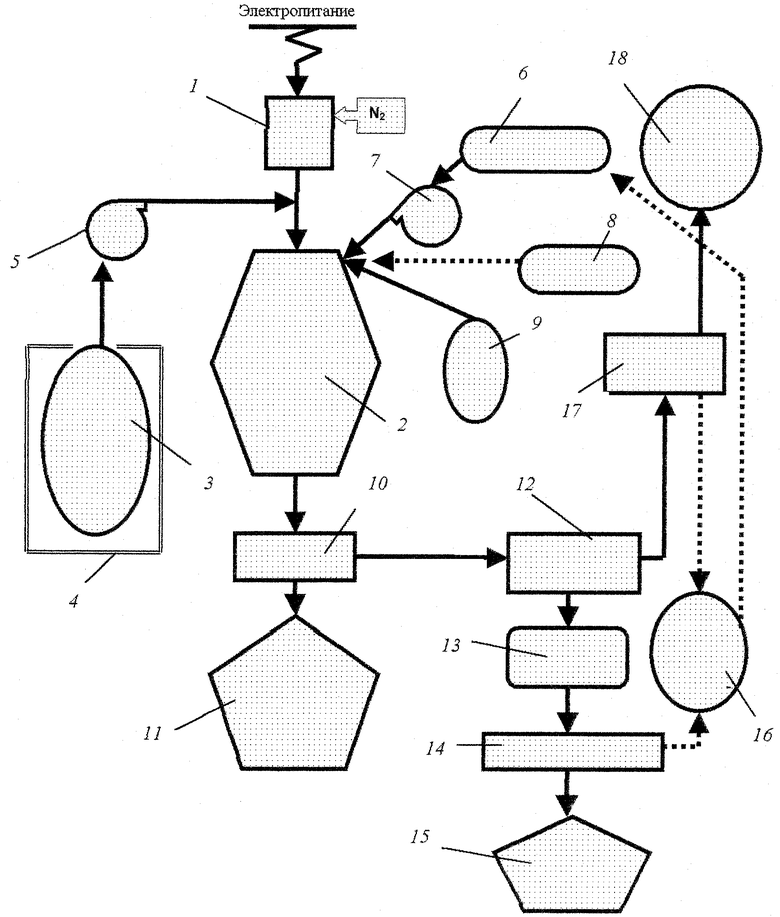
| US 4031029 А, 21.06.1977 | |||
| RU 2066299 С1, 10.09.1996 | |||
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| US 4005042 A, 25.01.1977 | |||
| ГАЛКИН Н.П | |||
| и др | |||
| Исследование процесса превращения гексафторида урана в двуокись, Атомная энергия, 1982, т.52, в.1, с.36-39. | |||
