Область, к которой относится изобретение
Настоящее изобретение относится к серии аминовых и амидных производных, к фармацевтическим композициям, содержащим эти производные, и к промежуточным соединениям, используемым для их получения. Соединения настоящего изобретения представляют собой лиганды для рецептора Y5 нейропептида Y (NPY5), то есть рецептора, который ассоциируется с рядом расстройств центральной нервной системы и патологических состояний. Кроме того, многие соединения настоящего изобретения способствуют снижению потребления пищи у грызунов, используемых в качестве модели питания.
Предпосылки изобретения
Регуляция и функция центральной нервной системы млекопитающих осуществляется под контролем серии независимых друг от друга рецепторов, нейронов, нейротрансмиттеров и белков. Нейроны играют жизненно важную роль в этой системе, и в ответ на внешнюю или внутреннюю стимуляцию они реагируют путем высвобождения нейротрансмиттеров, которые связываются co специфическими белками. Типичные примеры эндогенных нейротрансмиттеров с небольшим размером молекул, таких как ацетилхолин, адреналин, норэпинефрин, допамин, серотонин, глутамат и гамма-аминомасляная кислота, хорошо известны, поскольку они являются специфическими рецепторами, распознающими указанные соединения в качестве лигандов ("The Biochemical Basis of Neuropharmacology", Sixth Edition, Cooper, J.R.; Bloom, F.E.; Roth, R.H. Eds., Oxford University Press, New York, NY 1991).
Появляется все больше свидетельств, что, помимо эндогенных нейротрансмиттеров с небольшим размером молекул, существенную роль в функционировании нейронов играют нейропептиды. В настоящее время предполагается, что нейропептиды локализуются, вероятно, вместе с более чем половиной из 100 миллиардов нейронов центральной нервной системы человека. Кроме человека, нейропептиды были обнаружены у некоторых других видов животных. В некоторых случаях, состав этих пептидов является достаточно однородным у разных видов животных. Обнаружение этого факта дает основание предположить, что функция нейропептидов является жизненно важной и не подвергается эволюционным изменениям. Кроме того, нейропептиды, в отличие от нейротрансмиттеров с небольшим размером молекул, обычно синтезируются нейронной рибосомой. В некоторых случаях, активные нейропептиды продуцируются как часть более крупного белка, которая ферментативно процессируется с образованием активного вещества. Исходя из указанных отличий по сравнению с нейротрансмиттерами небольших размеров, можно разработать новые стратегии на основе нейропептидов, направленные на лечение заболеваний и расстройств ЦНС. В частности, агенты, которые влияют на связывание нейропептидов с их соответствующими рецепторами или способствуют усилению ответов, опосредованных нейропептидами, являются потенциальными средствами для лечения заболеваний, ассоциированных с нейропептидами.
Существует ряд заболеваний, ассоциируемых с комплексной системой независимых друг от друга рецепторов и лигандов в центральной нервной системе, и такими заболеваниями являются нейродегенеративные болезни, патологические состояния, такие как тревога, депрессия, боли и шизофрения, и патологические состояния, которые включают метаболический компонент, а именно ожирение. Указанные состояния, расстройства и заболевания подвергаются лечению небольшими молекулами и пептидами, которые модулируют ответ нейронов на эндогенные нейротрансмиттеры.
Одним из примеров нейропептидов такого класса является нейропептид Y (NPY). NPY впервые был выделен из головного мозга свиньи (Tatemoto, К. et al. Nature 1982, 296, 659) и было показано, что он является структурно сходным с другими членами семейства панкреатических полипептидов (РР), таких как пептид YY, который синтезируется, главным образом, эндокринными клетками кишечника, и панкреатический полипептид, который синтезируется поджелудочной железой. Нейропептид Y представляет собой белок из одного пептида, который состоит из тридцати шести аминокислот, содержащих амидированный С-конец. Аналогично другим членам семейства панкреатических полипептидов, NPY имеет отличающуюся конформацию, которая состоит из N-концевой полипролиновой спиральной области и амфифильной α-спирали, соединенные характерной РР-складкой (Vladimir, S. et al. Biochemistry 1990, 20, 4509). Кроме того, были определены последовательности NPY у разных видов животных, и все они обнаруживали высокую степень аминокислотной гомологии с белком человека (>94% у крыс, собак, кроликов, свиней, коров, овец) (см. Larhammar, D. in "The Biology of Neuropeptide Y and Related Peptides", Colmers, W.F. & Wahlestedt, C. Eds., Humana Press, Totowa, NJ 1993).
Были идентифицированы и классифицированы эндогенные рецепторные белки, которые связываются с NPY и с родственными пептидами в качестве лигандов, и некоторые из этих белков были клонированы и экспрессированы. В настоящее время было выявлено шесть различных подтипов рецепторов [Y1, Y2, Y3, Y4 (PP), Y5, Y6 (ранее называемых рецептором Y5)], отличающихся по своему профилю связывания, фармакологическому применению и/или составу, если они известны (Wahlestedt, С. et al. Ann. NY Acad. Sci. 1990, 611, 7; Larhammar, D. et al. J. Biol. Chem. 1992, 267, 10935; Wahlestedt, С. et al. Regul. Pept. 1986, 13, 307; Fuhlendorff, J.U. et al. Proc. Natl. Acad. Sci. USA 1990, 87, 182; Grundemar, L. et al. J. Pharmacol. Exp. Ther. 1991, 258, 633; Laburthe, M. et al. Endocrinology 1986, 118, 1910; Castan, I. et al. Endocrinology 1992, 131, 1970; Gerald С. et al. Nature 1996, 382, 168; Weinberg, D.H. et al. Journal of Biological Chemistry 1996, 271, 16435; Gehlert, D. et al. Current Pharmaceutical Design 1995, 1, 295; Lundberg J.M. et al. Trends in Pharmaceuticals Sciences 1996, 17, 301). Большинство, а вероятно, и все белки рецепторов NPY принадлежат к семейству так называемых связанных с G-белком рецепторов (GPCR). Рецептор нейропептида Y5, т.е. предполагаемый GPCR, связывается, под действием аденилатциклазы, с циклическим монофосфатом аденозина (сАМР), негативно регулируя его клеточные уровни (Gerald С. et al. Nature 1996, 382, 168; Gerald, С. et al. PCT WO 96/16542). Так, например, NPY ингибирует форсколин-стимулированное продуцирование/уровни сАМР в нейробластомной клеточной линии. Лиганд Y5, который имитирует NPY этим способом, является агонистом, а лиганд, который конкурентно отменяет NPY-ингибирование форсколин-стимулируемого продуцирования сАМР, является антагонистом.
Сам нейропептид Y является архетипичным субстратом для рецепторов NPY и их связывание может помочь выявить ряд фармакологических и биологических эффектов in vitro и in vivo. При введении в головной мозг живых животных (интрацеребровентрикулярно (i.c.v.) или в миндалевидное тело), NPY вызывает анксиолитические эффекты у животных с разработанными моделями тревожных состояний, такими как каскадный плюс-лабиринт, наказание по Фогелю с лишением питья и парадигм конфликта "запрещение-давление" Геллера-Сейфтера (Heilig M. et al. Psychopharmacology 1989, 98, 524; Heilig M. et al. Reg. Peptides 1992, 41, 61; Heilig M. et al. Neuropsycho-pharmacology 1993, 8, 357). Было предположено, что такие соединения, которые имитируют NPY, могут быть использованы для лечения анксиолитических расстройств.
Иммунореактивность нейропептида Y заметно снижается в цереброспинальной жидкости у пациентов с доминирующей депрессией и у пациентов, склонных к суициду (Widdowson P.S. et al. Journal of Neurochemistry 1992, 59, 73), и крысы, обработанные трициклическими антидепрессантами, обнаруживают значительное увеличение уровней NPY по сравнению с контрольной группой (Heilig M. et al. European Journal of Pharmacology 1988, 147, 465). Обнаружение этого факта дает основание предположить, что неадекватный NPY-ответ может играть определенную роль в некоторых депрессивных состояниях и что соединения, которые регулируют NPY-эргическую систему, могут быть использованы для лечения депрессии.
Нейропептид Y способствует улучшению памяти и работоспособности, как было показано на модели обучения животных (Flood J.F. et al. Brain Research 1987, 421, 280), а потому он может служить в качестве стимулятора познавательной способности для лечения нейродегенеративных заболеваний, таких как болезнь Альцгеймера (БА), а также ассоциированных со СПИД’ом и старческого слабоумия.
Повышенные уровни NPY в плазме присутствовали у животных и человека, испытывающих эпизоды высокой активности симпатических нервов, такие как хирургическая операция, роды и геморрагия (Morris, M.J. et al. Journal of Autonomic Nervous System 1986, 17, 143). Таким образом, химические вещества, которые приводят к изменению NPY-эргической системы, могут быть использованы для ослабления мигрени, болей и стрессовых состояний.
Нейропептид Y также опосредует эндокринные функции, такие как высвобождение лютенизирующего гормона (ЛГ) у грызунов (Kalra, S.P. et al. Frontiers in Neuroendocrinology 1992, 13, 1). Поскольку ЛГ имеет жизненно важное значение для овуляции у млекопитающего, то соединение, которое имитирует действие NPY, может быть использовано для лечения бесплодия, особенно у женщин с дефектами так называемой лютеиновой фазы.
Нейропептид Y является сильным стимулятором потребления пищи, и его инъекция, в количестве, по крайней мере, одной биллионной грамма, непосредственно в ЦНС вызывает у крыс повышение аппетита и переедание (Clark, J.T. et al. Endocrinology 1984, 115, 427; Levine, A.S. et al. Peptides 1984, 5, 1025; Stanley, B.G. et al. Life Sci. 1984, 35, 2635; Stanley, B.G. et al. Proc. Natl. Acad. Sci. USA 1985, 82, 3940). Таким образом, NPY обладает действием, возбуждающим аппетит у грызунов, но он не обладает анксиогенным действием при интрацеребровентрикулярном введении, и такой антагонизм нейропептидных рецепторов может быть использован для лечения диабета и расстройств аппетита, таких как ожирение, нервно-психическая анорексия и нервно-психическая булимия.
В последние годы был обнаружен и разработан ряд сильных структурно отличающихся низкомолекулярных антагонистов Y1 (Hipskind, P.A. et al. Annu. Rep. Med. Chem. 1996, 31, 1-10; Rudolf, К. et al. Eur. J. Pharmacol. 1994, 271, R11; Serradeil-Le Gal, C. et al. FEBS Lett. 1995, 362, 192; Wright, J. et al. Bioorg. Med. Chem. Lett. 1996, 6, 1809; Poindexter G.S. et al., патент США №5668151; Peterson, J.M. et al. WO 9614307 (1996)). Однако, несмотря на заявленную активность у грызунов, используемых в качестве модели потребления пищи, остается неясным, можно ли ингибирование реакции потребления пищи отнести за счет антагонизма рецептора Y1.
Некоторые фундаментальные исследования дают все основания предположить, что за индуцирование NPY-стимулируемого потребления пищи у животных ответственен не классический рецептор Y1, а "атипический" рецептор Y1 и/или рецептор Y5. Было показано, что несмотря на недостаточное связывание с классическим рецептором Y1, фрагмент NPY, NPY2-36 является сильным индуктором потребления пищи (Stanley, E.G. et al. Peptides 1992, 13, 581). И наоборот, сообщалось, что сильный и селективный агонист Y1 является неактивным в стимуляции потребления пищи у животных (Kirby, D.A. et al. J. Med. Chem. 1995, 38, 4579). Сообщалось также, что селективный активатор рецептора Y5, [D-Trp32] NPY, который имеет большее отношение к описанному здесь изобретению, стимулирует потребление пищи при его инъекции в гипоталамус крыс (Gerald, С. et al. Nature 1996, 382, 168). Поскольку очевидно, что [D-Trp32] NPY является полным агонистом рецептора Y5 и при этом не обладает заметной Y1-активностью, было высказано предположение, что рецептор Y5 ответственен за реакцию в виде повышенного потребления пищи. В соответствии с этим соединения, которые являются антагонистами рецептора Y5, должны эффективно ингибировать потребление пищи, особенно стимулируемое NPY.
Ряд структурно отличающихся соединений, которые являются антагонистами рецептора Y5, были описаны в различных публикациях. В РСТ WO 97/19682 описаны арилсульфонамиды и сульфамиды, полученные из арилалкиламинов и являющиеся антагонистами Y5, и сообщалось, что они снижают потребление пищи у животных. В РСТ WO 97/20820, РСТ WO 97/20822 и в РСТ 97/20823 также заявлены сульфонамиды, которые содержат гетероциклические системы, такие как хиназолин-2,4-диазирины, и которые, как сообщалось, являются антагонистами Y5 и снижают потребление пищи. В РСТ WO 99/10330 заявлена серия гетероциклических кетонов, являющихся антагонистами рецептора Y5 NPY. В РСТ WO 99/01128 заявлены некоторые производные диарилимидазола, представляющие собой новый класс специфических лигандов NPY. В РСТ WO 98/35944 заявлена серия α-алкокси- и α-тиоалкоксиамидов, являющихся антагонистами рецептора Y5 NPY. В РСТ WO 98/35957 заявлена серия амидных производных, являющихся селективными антагонистами рецептора нейропептида Y, однако, эти соединения структурно отличаются от соединений настоящего изобретения. Описанные амиды и амины настоящего изобретения являются новыми молекулами, которые могут иметь мотивы связывания и отличаются от этих и других лигандов Y5, описанных в патентных заявках или публикациях.
Краткое описание изобретения
Настоящее изобретение относится к соединениям формулы А
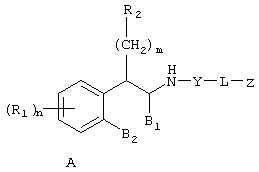
где R1 независимо выбран из группы, состоящей из водорода; гидрокси; галогена; C1-8алкила; замещенного C1-8алкила, где заместитель выбран из галогена, такого как хлор, бром, фтор и иод; C1-8алкокси; замещенного C1-8алкокси, где заместитель выбран из галогена, такого как хлор, бром, фтор и иод; трифторалкила; C1-8алкилтио; замещенного C1-8алкилтио, где заместитель выбран из галогена, такого как хлор, бром, фтор и иод, трифторС1-8алкила и C1-8алкокси; С3-6циклоалкила; С3-8циклоалкокси; нитро; амино; C1-6алкиламино; С1-8диалкиламино; С4-8циклоалкиламино; циано; карбокси; С1-5алкоксикарбонила; С1-5алкилкарбонилокси; формила; карбамоила; фенила и замещенного фенила, где заместитель выбран из галогена, гидроксила, нитро, амино и циано;
n равно 1-2;
B1 представляет водород;
B2 представляет водород;
либо B1 и B2 могут представлять метилен и, взятые вместе, образовывать пяти- или шестичленное кольцо;
m равно 0-3;
R2 независимо выбран из группы, состоящей из водорода; гидрокси; C1-6алкила; С2-6алкенила; галогена, такого как фтор и хлор; С3-7циклоалкила; фенила; замещенного фенила, где заместитель выбран из галогена, C1-6алкила, C1-6алкокси, трифторС1-8алкила, циано, нитро, амино, C1-6алкиламино и C1-6диалкиламино; нафтила; замещенного нафтила, где заместитель выбран из галогена, C1-6алкила, C1-6алкокси, трифторС1-6алкила, циано, нитро, амино, C1-6алкиламино и C1-6диалкиламино; фенокси; замещенного фенокси, где заместитель выбран из галогена, C1-6алкила, C1-6алкокси, трифторС1-6алкила, циано и нитро; гетероарильной группы, такой как пиридил, пиримидил, фурил, тиенил и имидазолил; замещенного гетероариала, где заместитель выбран из C1-6алкила и галогена; и гетероциклоалкила, такого как пирролидино или пиперидино;
Y представляет метилен (-CH2-) или карбонил (С=О);
L выбран из группы, состоящей из
C1-8алкилена; С2-10алкенилена; С2-10алкинилена;
С3-7циклоалкилена;
С3-7циклоалкилС1-4алкилена;
арилС1-4алкилена;
α-аминоС4-7алкилена;
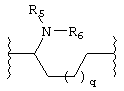
(N-метилен)пиперидин-4-ила;
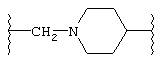
(N-метилен)пиперазин-4-ила;
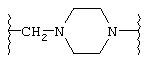
(N-метилен)пирролидин-3-ила;
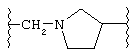
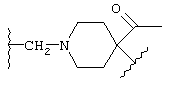
(N-метилен)-4-ацетилпиперидин-4-ила;
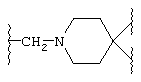
и (N-метилен)пиперидин-4,4-диила;
Z выбран из группы, состоящей из
арила;
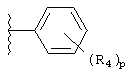
N-сульфонамидо;
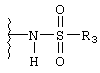
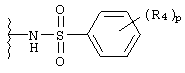
N-(арил)сульфонамидо;
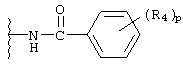
ариламидо;
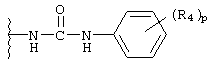
арилуреидо;
арилацетамидо;
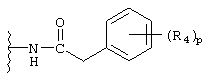
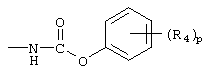
(арилокси)карбониламино;
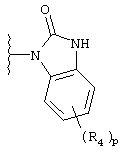
2,3-дигидpo-2-oкco-1H-бeнзимидaзoл-1-илa;
и 1-арил-2,3-дигидро-4-оксоимидазол-5,5-диила;
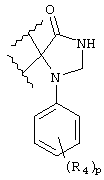
В каждом случае арильная группа может быть замещена, как указано.
R3 независимо выбран из группы, состоящей из C1-8алкила; замещенного C1-8алкила, где заместитель выбран из C1-8алкокси и галогена; циклоалкила; замещенного циклоалкила, где заместитель выбран из C1-8алкокси и галогена; нафтила; замещенного нафтила, где заместитель выбран из галогена, нитро, амино и циано; гетероарила, где гетероарильная группа выбрана из пиридила, пиримидила, фурила, тиенила и имидазолила; и замещенного гетероарила, где заместитель выбран из галогена, нитро, амино и циано;
R4 независимо выбран из группы, состоящей из водорода; C1-8алкила; замещенного C1-8алкила, где заместитель выбран из алкокси и галогена; гидрокси; галогена; циано; нитро; амино; C1-8алкиламино и С1-8диалкиламино; C1-8алкокси; замещенного C1-8алкокси, где заместителем является галоген; гидрокси; галогена; циано; нитро; амино; C1-8алкиламино и С1-8диалкиламино;
R5 независимо выбран из группы, состоящей из водорода; C1-8алкила; С1-8алкилкарбонила; ароила; карбамоила; амидино; (С1-8алкиламино)карбонила; (ариламино)карбонила и арилС1-8алкилкарбонила;
R6 независимо выбран из группы, состоящей из водорода и С1-8алкила;
p равно 1-3;
q равно 1-3;
и к их энантиомерам, диастереомерам и фармацевтически приемлемым солям, при условии, что
если L представляет С1-8алкилен; С2-10алкенилен; С2-10алкинилен; С3-7циклоалкилен; С3-7циклоалкилС1-4алкилен; арилС1-4алкилен или α-аминоалкилен,
то Z представляет фенил, N-сульфонамидо или N-(арил)сульфонамидо;
если L представляет (N-метилен)пиперазин-4-ил,
то Z представляет фенил или нафтил;
если L представляет (N-метилен)пирролидин-3-ил или (N-метилен)пиперидин-4-ил,
то Z представляет N-сульфонамидо, N-(арил)сульфонамидо, 2,3-дигидро-2-оксо-1Н-бензимидазол-1-ил; бензамидо, фенилуреидо, фенилацетамидо или (фенокси)карбониламино;
если L представляет (N-метилен)-4-ацетилпиперидин-4-ил,
то Z представляет фенил или нафтил, a Y представляет карбонил;
если L представляет (N-метилен)пиперидин-4,4-диил,
то Z представляет 1-арил-2,3-дигидро-4-оксоимидазол-5,5-диил, а Y представляет карбонил;
а если B1 и B2 оба представляют метилен, образуя тем самым шестичленное кольцо (аминотетралин), и если L выбран из группы, состоящей из C1-8алкилена; С2-10алкенилена; С2-10алкинилена или арилС1-4алкилена,
то Z не может представлять N-сульфонамидо, N-(арил)сульфонамидо или фенил;
все энантиомеры и диастереомеры соединений формулы А являются частью настоящего изобретения, как и их фармацевтически приемлемые соли.
Из соединений настоящего изобретения предпочтительными являются соединения, где B1 и В2 образуют шестичленное кольцо, a m=1-3.
Если это не оговорено особо, то используемые здесь термины "алкил" и "алкокси", независимо от того, используются они отдельно или как часть группы-заместителя, означают прямые и разветвленные цепи, имеющие 1-8 атомов углерода. Так, например, алкильными радикалами являются метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, 2-метил-3-бутил, 1-метилбутил, 2-метилбутил, неопентил, гексил, 1-метилпентил, 3-метилпентил. Алкоксирадикалами являются оксиэфиры, образованные из ранее описанных прямых или разветвленных алкильных групп. Термин "арил" означает фенил и нафтил, а "ароил" означает арилацил. Термин "ацил" означает C1-8алкилкарбонил. Термин "галоген", если это не оговорено особо, означает бром, хлор, фтор и иод. Термин "циклоалкил" означает циклоалкильные группы, имеющие 3-7 атомов углерода. Что касается заместителей, то термин "независимо" означает, что если возможно присутствие более чем одного из таких заместителей, то указанные заместители могут быть одинаковыми или различными.
Соединения настоящего изобретения, которые содержат основную часть, могут быть превращены в соответствующие кислотно-аддитивные соли методами, известными специалистам. Подходящими кислотами, которые могут быть использованы для этой цели, являются хлористоводородная, бромистоводородная, иодистоводородная, перхлорная, серная, азотная, фосфорная, уксусная, пропионовая, гликолевая, молочная, пировиноградная, щавелевая, малоновая, янтарная, малеиновая, фумаровая, яблочная, винная, лимонная, бензойная, коричная, миндальная, метансульфоновая, п-толуолсульфоновая, циклогексансульфамовая, салициловая, 2-феноксибензойная, 2-ацетоксибензойная или сахарин и т.п. В основном, указанные кислотно-аддитивные соли могут быть получены реакцией свободного основания соединений формулы А с кислотой и выделения соли.
Фармацевтические композиции, содержащие одно или несколько соединений, описанных здесь в соответствии с изобретением в качестве активного ингредиента, могут быть получены путем тщательного смешивания указанного соединения или соединений с фармацевтическим носителем стандартными методами приготовления фармацевтических композиций. Носитель может иметь большое число различных форм в зависимости от требуемого способа введения (например, перорального, парентерального). Так, например, для жидких пероральных препаратов, таких как суспензии, эликсиры и растворы, подходящими носителями и добавками являются вода, гликоли, масла, спирты, отдушки, консерванты, стабилизаторы, красители и т.п.; для твердых пероральных препаратов, таких как порошки, капсулы и таблетки, подходящими носителями и добавками являются крахмалы, сахара, разбавители, гранулирующие агенты, замасливатели, связующие, дезинтеграторы и т.п. Твердые препараты для перорального введения могут быть также покрыты веществами, такими как сахара, либо они могут иметь энтеросолюбильное покрытие для модуляции главного участка абсорбции. Для парентерального введения носитель обычно состоит из стерилизованной воды, а для улучшения растворимости или консервирующих свойств в него могут быть добавлены и другие ингредиенты. Суспензии или растворы для инъекций могут быть также получены с использованием водных носителей вместе с подходящими добавками.
Для лечения расстройств центральной нервной системы описанные здесь фармацевтические композиции, в основном, содержат от 1 до около 1000 мг активного ингредиента на дозу; при этом может быть введена одна или несколько доз в день. Определение оптимальных доз и частоты введения доз для лечения конкретного патологического состояния или расстройства может быть проведено экспериментально на основе указанных данных, относящихся к лечению расстройств центральной нервной системы. Предпочтительная доза составляет 1-100 мг/кг.
В качестве модуляторов рецептора NPY5 соединения формулы А могут быть использованы для лечения расстройств питания, таких как ожирение, нервно-психическая анорексия и нервно-психическая булимия, и аномальных состояний, таких как эпилепсия, депрессия, тревожное состояние и нарушение сексуальных/репродуктивных функций, при которых может быть использована модуляция рецептора NPY5. Эти соединения конкурируют с эндогенными лигандами NPY и PYY и, возможно, с неэндогенными лигандами и связываются с рецептором NPY5. Кроме того, эти соединения демонстрируют антагонистическую активность посредством ингибирования действия NPY при связывании с рецептором Y5.
Описанные здесь соединения представляют собой лиганды рецептора NPY5, однако, они не ограничиваются лишь своим фармакологическим или биологическим действием, что обусловлено их связыванием с указанным рецептором нейропептида или с любым рецептором нейропептида, рецептором нейтротрансмиттера или рецептором, связанным с G-белком. Так, например, описанные здесь соединения могут также связываться с рецепторами допамина или серотонина. Описанные здесь соединения могут быть использованы для регуляции метаболических и эндокринных функций, а в частности функций, ассоциированных с питанием, а поэтому они могут быть использованы для лечения ожирения. Кроме того, описанные здесь соединения могут быть использованы для модуляции других эндокринных функций, а в частности функций, контролируемых гипофизом и гипоталамусом, а следовательно, они могут быть использованы для лечения ановуляции/бесплодия, обусловленных недостаточным высвобождением лютеинизирующего гормона (ЛГ) или нарушением лютеиновой фазы.
Настоящее изобретение относится к фармацевтическим композициям, содержащим одно или несколько соединений формулы А. Кроме того, настоящее изобретение относится к промежуточным соединениям, используемым при получении соединений формулы А.
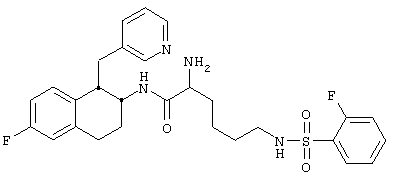
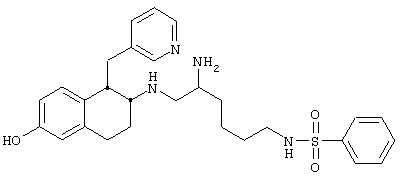
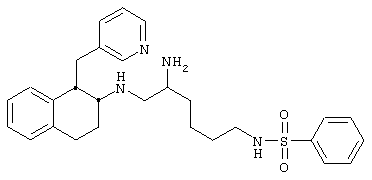
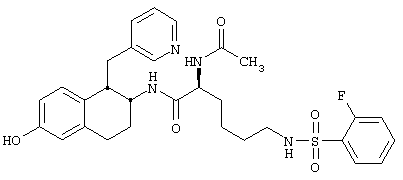
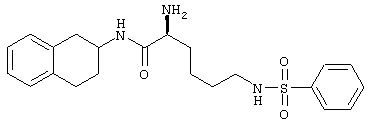
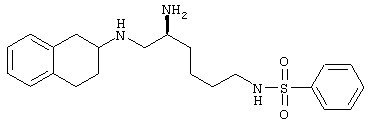
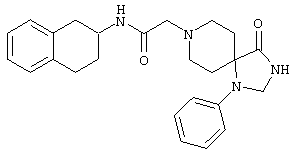
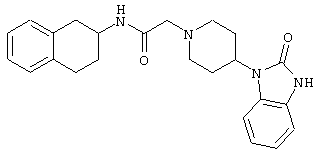
Конкретными примерами предпочтительных соединений формулы А являются:
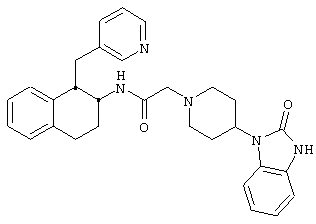
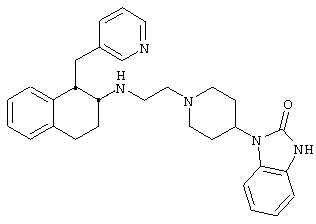
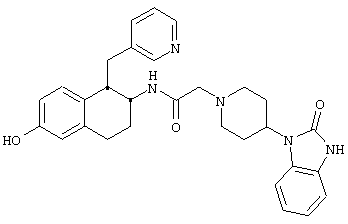
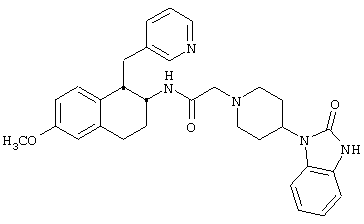
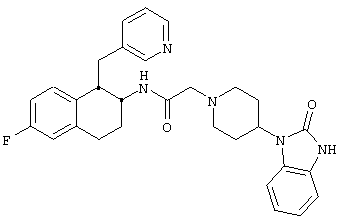
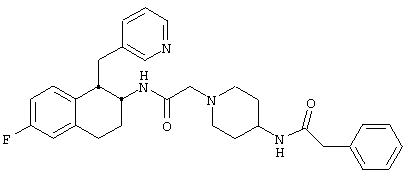
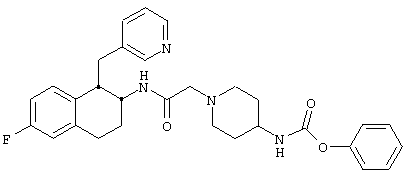
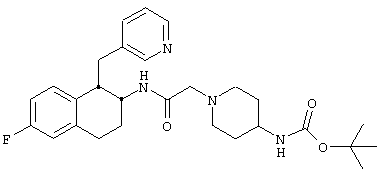
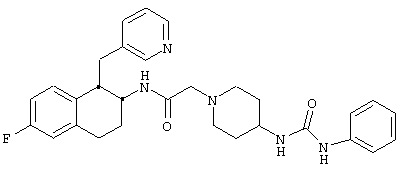
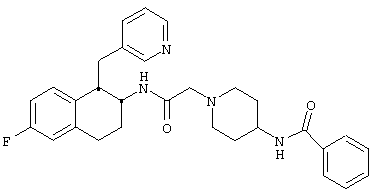
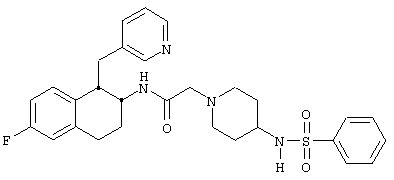
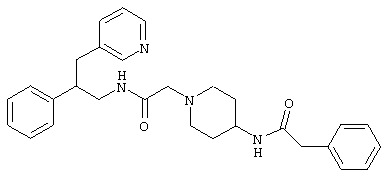
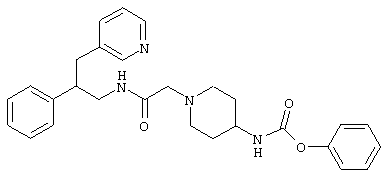
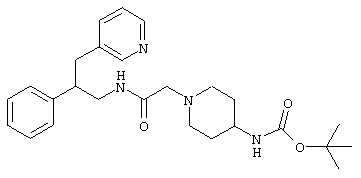
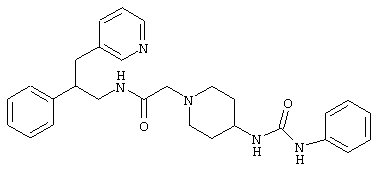
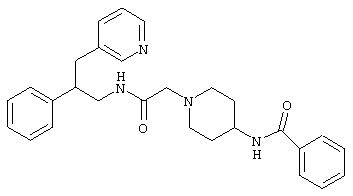
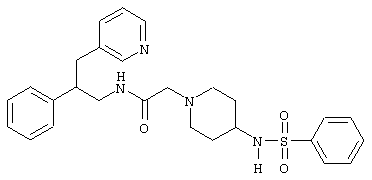
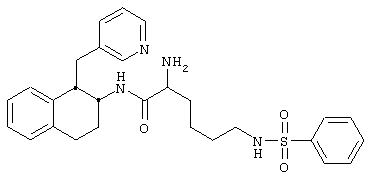
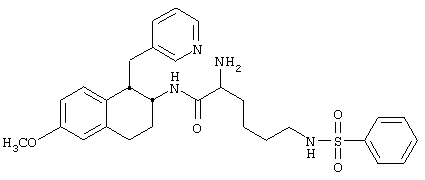
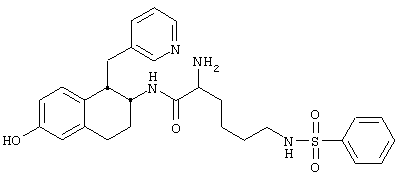
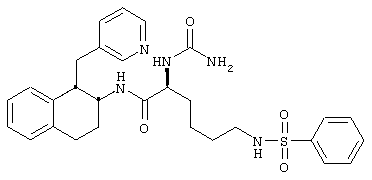
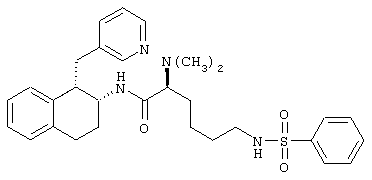
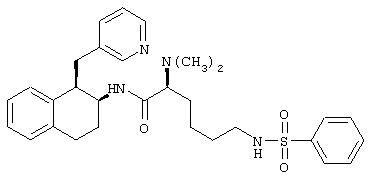
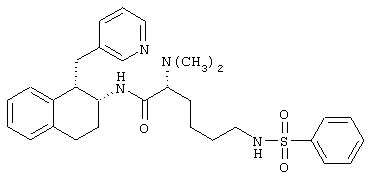
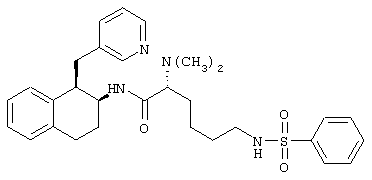
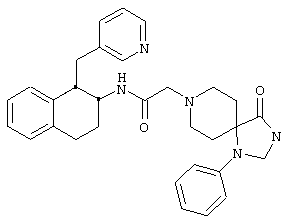
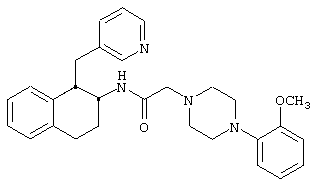
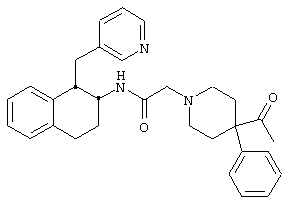
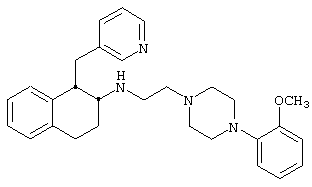
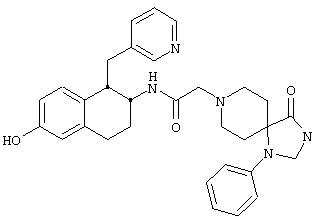
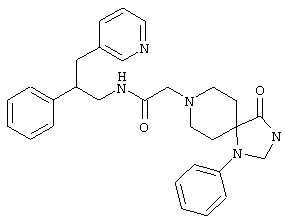
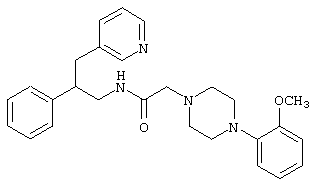
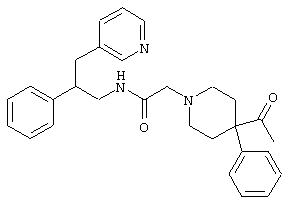
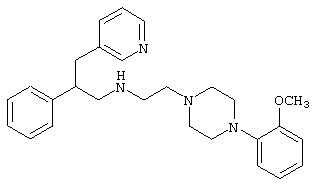
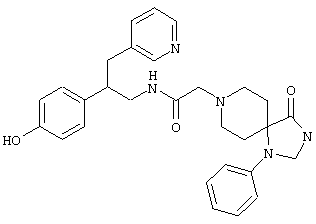
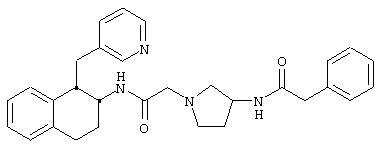
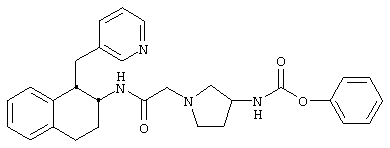
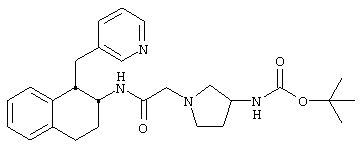
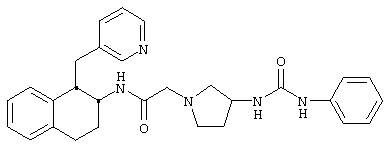
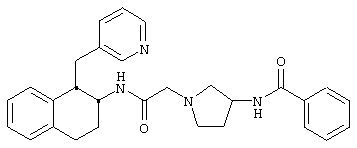
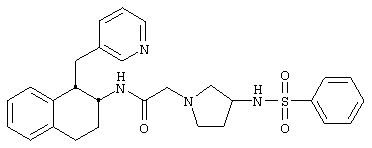
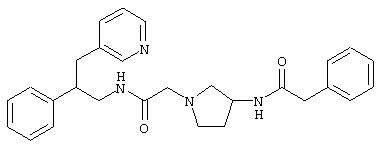
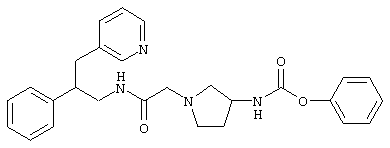
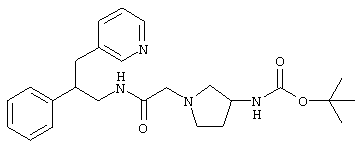
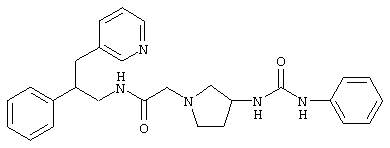
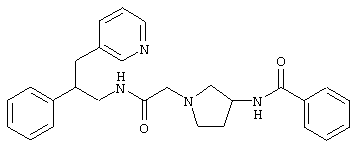
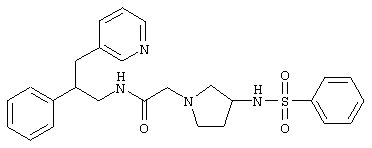
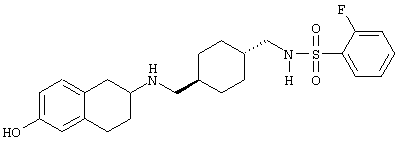
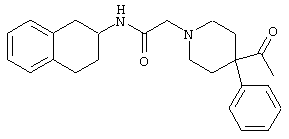
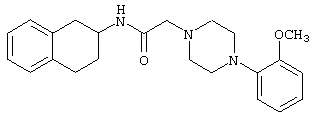
Подробное описание изобретения
Амины и амиды формулы А настоящего изобретения синтезируют несколькими различными способами химического синтеза, описанными в схемах 1-26, каждый из которых состоит из нескольких последовательных химических операций, которые могут быть проведены, в основном, как описано ниже. В случае, когда B1 и B2, взятые вместе, образуют шестичленное кольцо или пятичленное кольцо (аминотетралин или аминоиндан соответственно), общий синтез предусматривает проведение следующих стадий, таких как:
- Введение α-заместителя в ядро тетралона (или инданона)
- Превращение в соответствующий α-замещенный β-аминотетралин (или α-замещенный аминоиндан)
- Ацилирование аминотетралина (или аминоиндана) с получением амидов формулы А
- Восстановление с получением аминов формулы А.
В различных стадиях синтеза могут быть необходимы манипуляции с защитными группами.
В случае, если B1 и В2 представляют водород, общий способ синтеза состоит из следующих операций:
- Введения α-заместителя в фенилацетонитрил
- Восстановления до соответствующего β-замещенного фенетиламина
- Ацилирования указанного фенетиламина с получением амидов формулы А
- Восстановление до аминов формулы А.
В различных стадиях синтеза может потребоваться проведение манипуляций с защитными группами.
В основном, предпочтительно, чтобы соответствующий продукт каждой стадии этого способа был отделен от других компонентов реакционной смеси и подвергнут очистке, а затем использован в качестве исходного соединения в последующей стадии. Типичными методами разделения являются выпаривание, экстракция, осаждение и фильтрация. Типичными методами очистки являются колоночная хроматография (Still, W.C. et al. J. Org. Chem. 1978, 43, 2921), тонкослойная хроматография, кристаллизация и дистилляция. Структуры конечных продуктов, промежуточных соединений и исходных соединений подтверждают спектроскопическими, спектрометрическими и аналитическими методами, включая ядерный магнитный резонанс (ЯМР), масс-спектрометрию (МС) и жидкостную хроматографию (ВЭЖХ). В описании получения соединений настоящего изобретения типичными примерами эфирных растворителей являются этиловый эфир, тетрагидрофуран и диоксан; типичными углеводородными растворителями являются бензол, толуол, гексан и циклогексан, а характерными галогенуглеводородными растворителями являются дихлорметан и дихлорэтан. В тех случаях, когда продукт выделяют в виде кислотно-аддитивной соли, свободное основание может быть получено методами, известными специалистам. В тех случаях, когда данный продукт выделяют в виде кислотно-аддитивной соли, эта соль может содержать один или несколько эквивалентов кислоты.
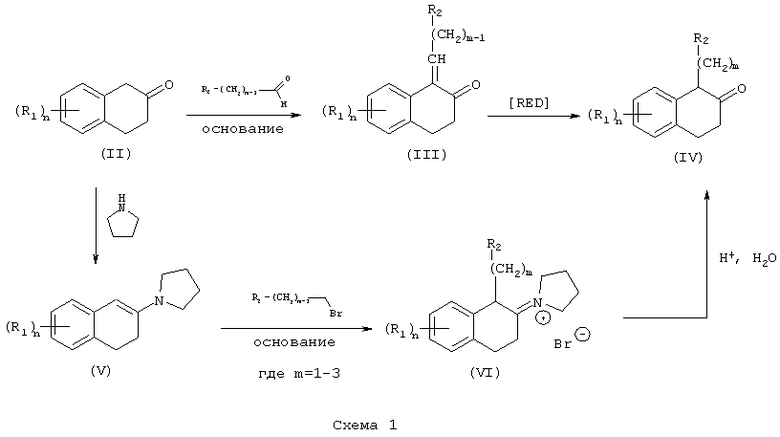
В частности, соответствующим образом замещенный β-тетралон (II) подвергают реакции с ариловым или гетероариловым альдегидом в присутствии основания, такого как пиперидин, в инертном галогенуглеводородном, эфирном или углеводородном растворителе, таком как бензол, при температуре в пределах от комнатной температуры до температуры дефлегмации, с получением соответствующего α-бензилиденил-β-тетралона или α-гетероарилметилиденил-β-тетралона (III). Этот β-тетралон (III) растворяют в инертном углеводородном, эфирном, сложноэфирном или спиртовом растворителе, таком как метанол, и подвергают реакции с газообразным водородом под давлением в пределах от атмосферного давления до 100 фунт/кв.дюйм (600 кПа), в присутствии подходящего катализатора, такого как палладий на угле. Реакцию осуществляют при температуре в пределах от комнатной температуры до температуры дефлегмации, с получением нужного α-замещенного β-тетралона (IV) (схема 1).
Альтернативный метод получения α-замещенных β-тетралонов (IV) предусматривает проведение реакции соответствующим образом замещенного β-тетралона (II) с основанием, таким как пирролидин, в инертном галогенуглеводородном растворителе, таком как дихлорметан, или в углеводородном растворителе, таком как бензол, в условиях Дина-Старка (с удалением воды) или в спиртовом растворителе, таком как метанол, при температуре в пределах от комнатной температуры до температуры дефлегмации, с получением енамина (V). Алкилирование енамина (V) осуществляют его реакцией с бензил-, гетероциклоалкил- или аллилгалогенидом в инертном растворителе, таком как ацетонитрил, при температуре в пределах от комнатной температуры до температуры дефлегмации, с получением соли α-замещенного β-иминия (VI). Гидролиз соли (VI) с получением нужного α-замещенного β-тетралонового продукта (IV) осуществляют реакцией соли (VI) с водой и неорганической или органической кислотой, такой как хлористоводородная или ледяная уксусная кислота, в инертном углеводородном, эфирном, спиртовом или галогенуглеводородном растворителе, таком как метанол и дихлорметан или в их смеси (Схема 1).
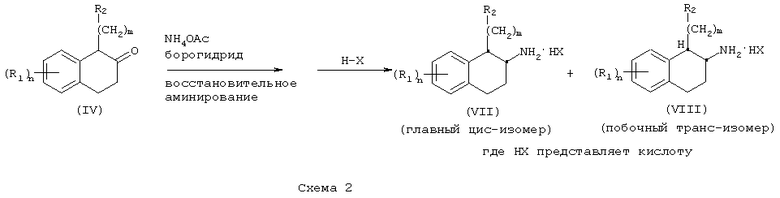
Полученные α-замещенные β-тетралоны (IV) превращают в соответствующие аминотетралины реакцией с аммониевой солью, такой как ацетат аммония, в присутствии восстановителя, такого как цианоборогидрид натрия, например, в инертном галогенуглеводородном, углеводородном, эфирном или спиртовом растворителе, таком как метанол, с получением цис-аминотетралина (VII). В некоторых случаях также образуется транс-аминотетралин (VIII) в виде побочного продукта; обе серии диастереомеров являются частью настоящего изобретения. Эти аминотетралины (VII) могут быть также выделены в виде кислотно-аддитивных солей путем обработки органической или неорганической кислотами, такой как, например, трифторуксусная кислота или хлористоводородная кислота (Схема 2).

Соединения, где m=0, получают из соответствующим образом замещенного аминотетралина (VII; m=0), используя 1-тетралоны в качестве исходных соединений, в соответствии с последовательностью синтеза, показанной на схеме 2а.
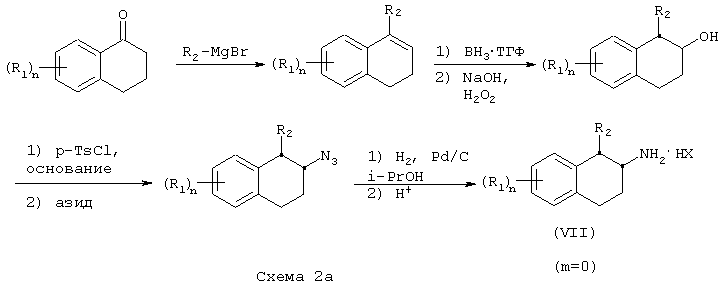
Замещенные фенетиламины (XI) получают реакцией соответствующим образом замещенного фенилацетонитрила (IX) с ариловым или гетероариловым альдегидом в присутствии основания, такого как метоксид натрия, в инертном спиртовом растворителе, таком как метанол, при температуре в пределах от комнатной до температуры дефлегмации, с получением α,β-ненасыщенного нитрила (X). После восстановления нитрила (X), например, реакцией с газообразным водородом в присутствии катализатора на основе окиси платины под давлением в пределах от атмосферного давления до около 100 фунт/кв.дюйм (600 кПа), в инертном растворителе, таком как водный спирт, при температуре в пределах от комнатной температуры до температуры дефлегмации, получают β-замещенный фенетиламин (XI). Альтернативно, реакцией фенилацетонитрила (X) с арилалкил-, гетероарилалкил- или алкилгалогенидом, например, таким как аллилбромид, в присутствии основания, такого как метоксид натрия или гидрид натрия, в инертном растворителе, таком как тетрагидрофуран или ацетонитрил соответственно, при температуре в пределах от комнатной температуры до температуры дефлегмации, получают α-замещенный фенилацетонитрил (XII). Последующее восстановление нитрила (XII), например, путем гидрогенолиза приводит к образованию β-замещенного фенетиламина (XI) (Схема 3).
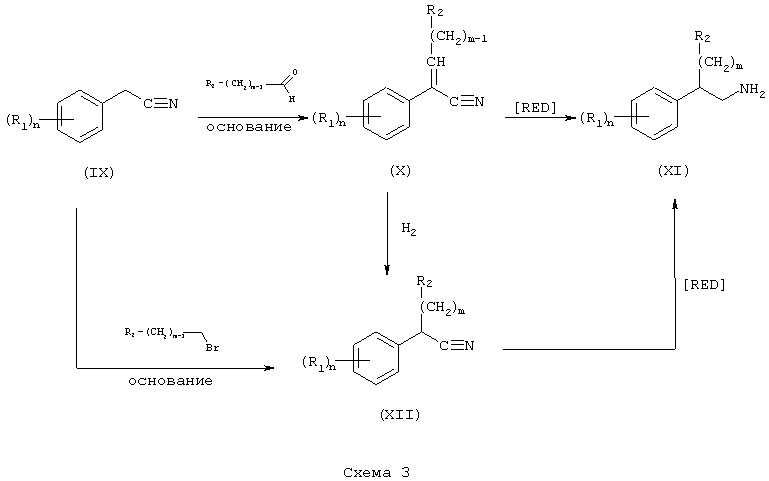
β-Аминотетралины (VII) и фенетиламины (XI), описанные выше, ацилируют подходящими методами амидирования (см. Gross & Meienhofer, Eds., "The Peptides", Vols 1-3, Academic Press, New York, NY, 1979-1981). Карбоновую кислоту превращают в активированный сложный эфир методами пептидного синтеза, известными специалистам, а затем проводят реакцию с аминотетралином (VII) или фенетиламином (XI), в результате чего получают соответствующие амиды.
Так, например, карбоновую кислоту, такую как транс-4-(2-фторбензолсульфонамидо)метилциклогексанкарбоновая кислота или 4-(трет-бутоксикарбонил)аминометилциклогексанкарбоновая кислота, подвергают реакции с HBTU (гексафторфосфат 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония) и соответствующим фенетиламином (XI) в присутствии основания, такого как диизопропилэтиламин в инертном растворителе, таком как N,N-диметилформамид, при температуре в пределах от комнатной температуры до температуры дефлегмации, в результате чего получают амид (XIII) или амид (XIV) соответственно. После отщепления защитной группы ВОС (бутоксикарбонил) от карбамата (XIV) с использованием трифторуксусной кислоты получают свободный амин, который затем подвергают сульфонилированию с получением амида (XIII).
N-Замещенные фенетиламиновые соединения А в соответствии с изобретением получают путем восстановления амида (XIII) реакцией с подходящим восстановителем, таким как боран-тетрагидрофурановый комплекс или алюмогидрид лития в инертном углеводородном растворителе, таком как толуол, или в эфирном растворителе, таком как тетрагидрофуран, при температуре в пределах от комнатной температуры до температуры дефлегмации. Конечный продукт может быть выделен в виде кислотно-аддитивной соли после обработки подходящим органическим растворителем, таким как трифторуксусная кислота, или неорганическим растворителем, таким как хлористоводородная кислота (Схема 4).
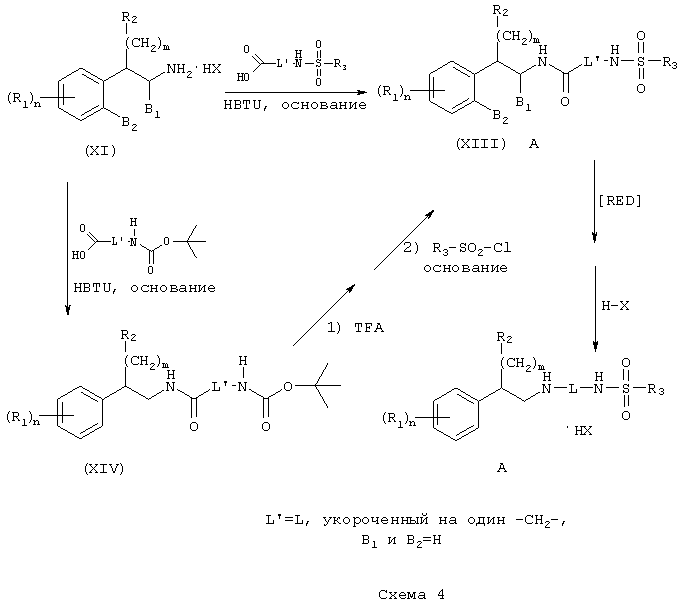
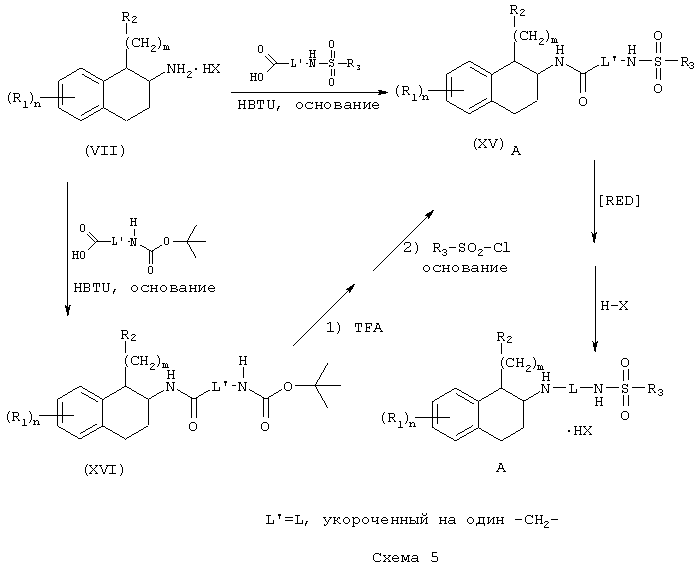
Аналоги аминотетралина (каждый из B1 и B2 представляет метилен) получают методом химического синтеза, описанным выше, за исключением того, что исходное соединение фенетиламин (XI) заменяют аминотетралином (VII) (Схема 5).
Соединения формулы А, в которых Z=2,3-дигидро-2-оксо-1Н-бензимидазол-1-ил и L=(N-метилен)пиперидин-4-ил получают из β-аминотетралинов (VII) или фенетиламинов (XI) и [4-(2-кето-1-бензимидазолинил)пиперидин-1-ил]уксусной кислоты (Схемы 6-7). Так, например, 4-(2-кето-1-бензимидазолинил)пиперидин подвергают реакции со сложным эфиром бромуксусной кислоты, таким как этилбромацетат, в присутствии аминового основания, такого как диизопропилэтиламин, в инертном растворителе, таком как ацетонитрил, при температуре в пределах от комнатной температуры до температуры дефлегмации, в результате чего получают этил[4-(2-кето-1-бензимидазолинил)пиперидин-1-ил]ацетат. Этот сложный эфир подвергают гидролизу в основных условиях, например, обработкой гидроксидом натрия в присутствии спиртового раствора, такого как водный метанол, в результате чего, после подкисления неорганической или органической кислотой, например, такой как хлористоводородная или уксусная кислота, получают [4-(2-кето-1-бензимидазолинил)пиперидин-1-ил]уксусную кислоту. Эту карбоновую кислоту подвергают реакции непосредственно с β-аминотетралинами (VII) или фенетиламинами (XI) в присутствии аминового основания и в условиях пептидного синтеза, описанного выше, в результате чего получают бензимидазолиноны (XVII) и (XVIII) формулы А, где Y=карбонил и L=(N-метилен)пиперидин-4-ил (Схемы 6-7).
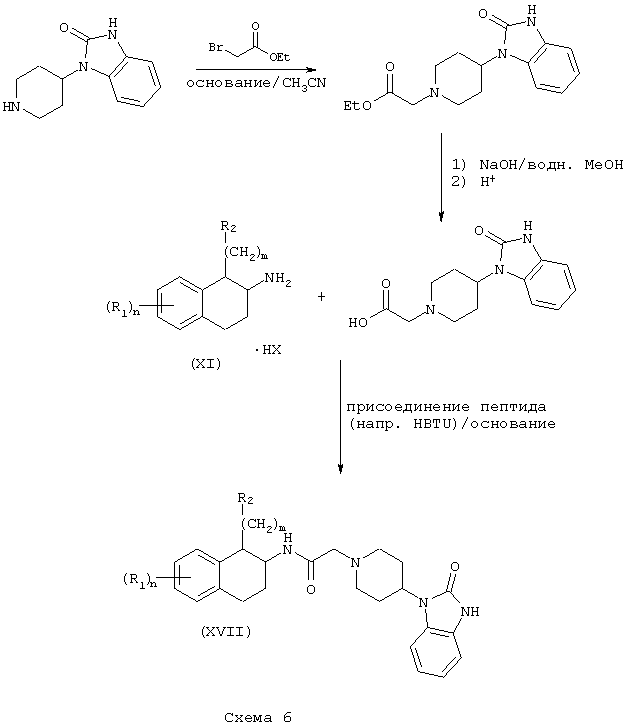
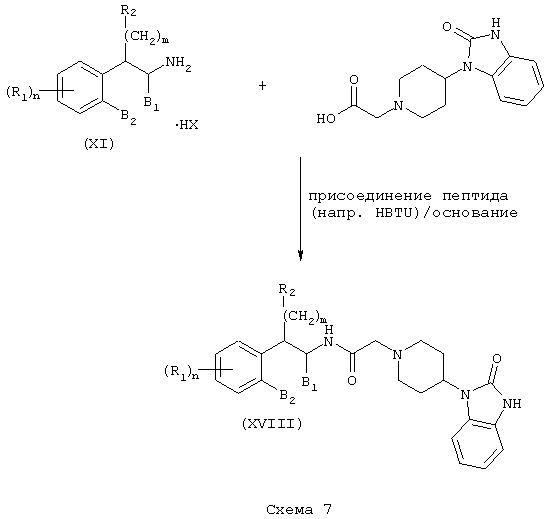
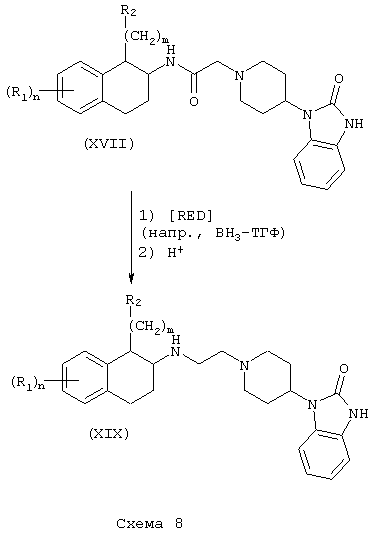
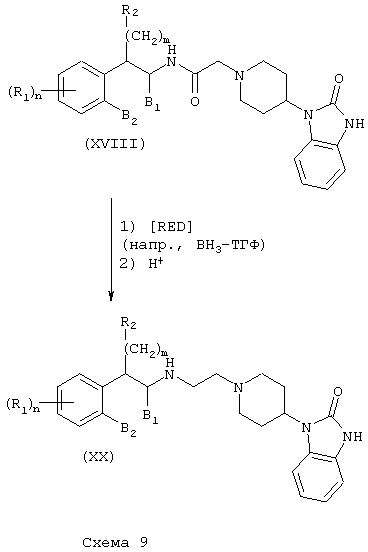
Соединения формулы А, где Y=метилен, L=(N-метилен)пиперидин-4-ил и Z=2,3-дигидро-2-оксо-1Н-бензимидазол-1-ил, получают путем восстановления амида (XVII) и амида (XVIII) реакцией с подходящим восстановителем, таким как боран-тетрагидрофурановый комплекс или алюмогидрид лития, как описано выше. С использованием аминотетралина (VII) в качестве исходного соединения получают продукты (XIX) (Схема 8), а с использованием фенетиламинов получают аналогичные амины (XX) (Схема 9).
Соединения формулы А, где Y=карбонил, L=(N-метилен)пиперазин-4-ил и Z=фенил, получают реакцией фенилпиперазина со сложным эфиром галогенуксусной кислоты, таким как, например, этилбромацетат, в присутствии аминового основания, такого как диизопропилэтиламин, в инертном растворителе, таком как ацетонитрил, при температуре в пределах от комнатной температуры до температуры дефлегмации с получением этил(4-арилпиперазин-1-ил)ацетата. Этот сложный эфир подвергают гидролизу в основных условиях, например, обработкой гидроксидом натрия в водном метаноле, в результате чего, после подкисления неорганической или органической кислотой, такой как хлористоводородная или уксусная кислота, получают (4-арилпиперазин-1-ил)уксусную кислоту. Эту карбоновую кислоту подвергают реакции с β-аминотетралинами (VII) или фенетиламинами (XI) в присутствии основания, такого как триэтиламин, в условиях пептидного синтеза, описанного выше, в результате чего получают арилпиперидины (XXI) и (XXII) соответственно формулы А, где Y=карбонил, L=(N-метилен)пиперазин-4-ил и Z=арил или замещенный арил (Схемы 10-11).
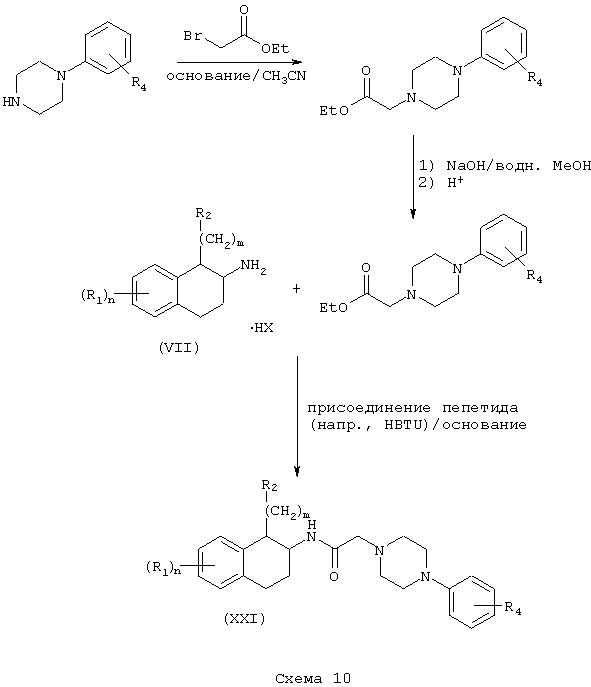
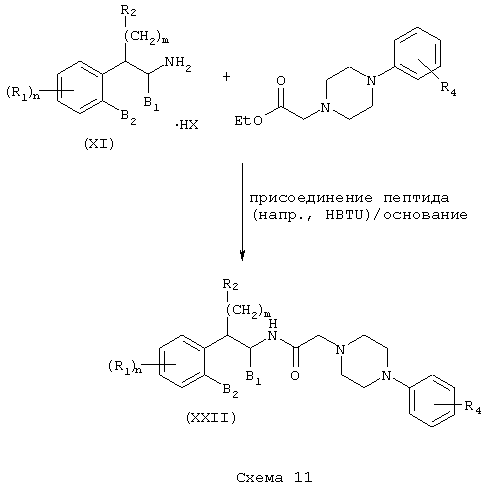
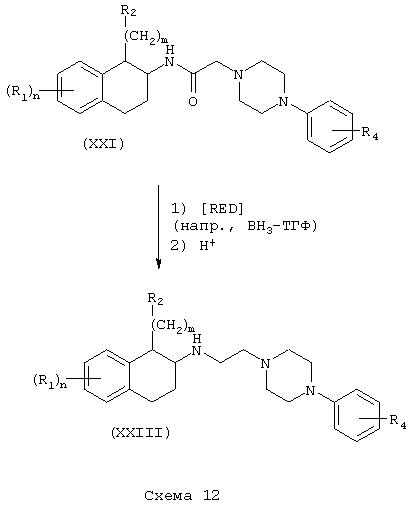
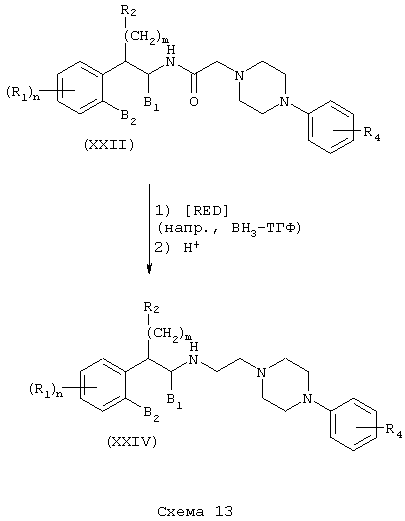
Соединения формулы А, где Y=метилен, L=(N-метилен)пиперазин-4-ил и Z=арил, получают восстановлением амидов (XXI) и (XXII) реакцией с подходящим восстановителем, таким как боран-тетрагидрофурановый комплекс или алюмогидрид лития (см. Схему 9), с получением аминотетралинов (XXIII) и фенетиламинов (XIV) соответственно (Схемы 12-13).
Заменяя 4-арилпиперазины 4-арилпиперидинами в Схемах 10 и 11, получают тетралинамиды (XXV) и фенетиламиды (XXVI) формулы А, где L=(N-метилен)пиперидин-4-ил, Z=арил и Y=карбонил (Схемы 14-15).
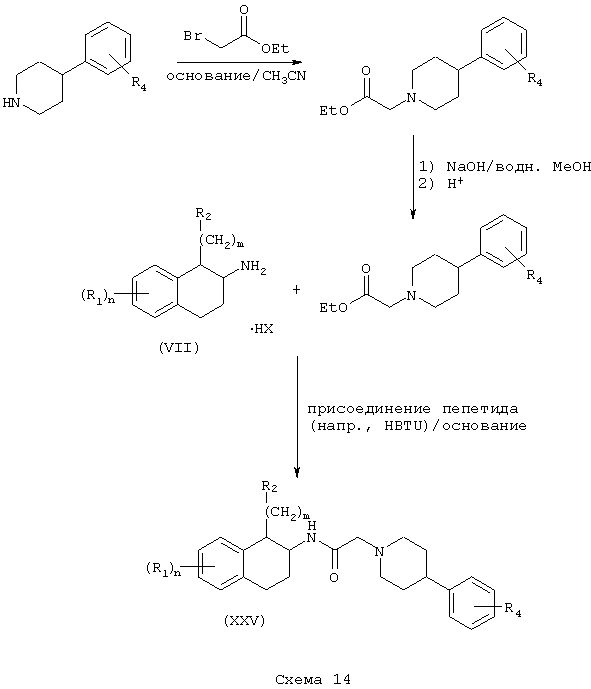
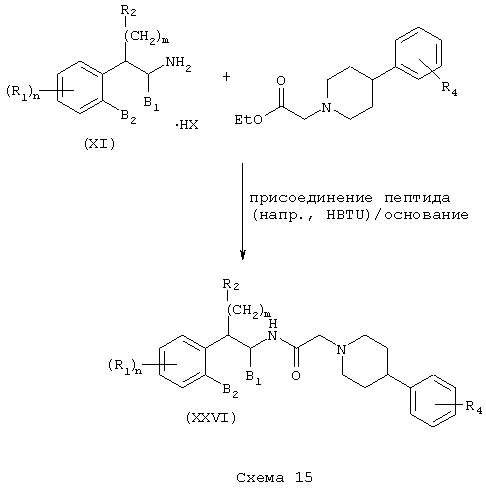
Независимо восстановлением амидов (XXV) и (XXVI) восстановителем, таким как боран-тетрагидрофурановый комплекс, получают амины (XXVII) и (XXVIII) формулы А, где L=(N-метилен)пиперидин-4-ил, Z=арил и Y=метилен (Схема 16).
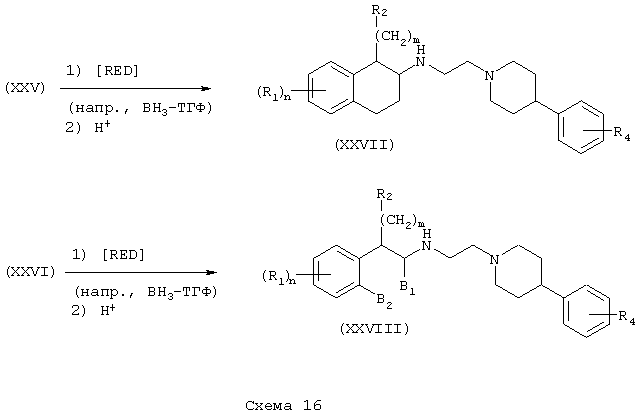
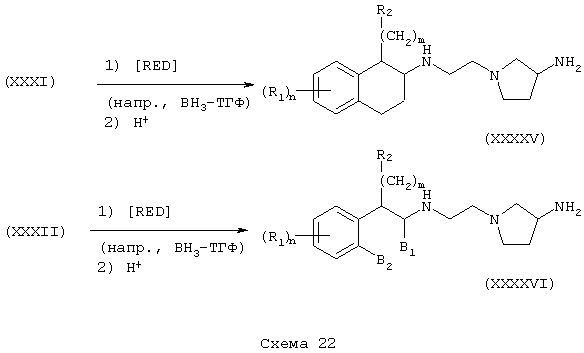
Соединения формулы А, где Y=карбонил, L=(N-метилен)пирролидин-3-ил и Z=N-(арил)сульфонамидо, получают реакцией соответствующим образом защищенного аминопирролидина, такого как (3-трет-бутоксикарбониламино)пирролидина со сложным эфиром галогенуксусной кислоты, например, таким как этилбромацетат, в присутствии аминового основания, такого как диизопропилэтиламин, в инертном растворителе, таком как ацетонитрил, при температуре в пределах от комнатной температуры до температуры дефлегмации, с получением этил[(3-трет-бутоксикарбониламино)пирролидин-1-ил]ацетата. Этот сложный эфир подвергают гидролизу в основных условиях, например, обработкой гидроксидом натрия в водном метаноле, в результате чего, после подкисления неорганической или органической кислотой, например, такой как хлористоводородная или уксусная кислота, получают [(3-трет-бутоксикарбониламино)пирролидин-1-ил]уксусную кислоту. Эту карбоновую кислоту подвергают реакции с β-аминотетралинами (VII) или фенетиламинами (XI) в присутствии основания, например, такого как триэтиламин, в условиях пептидного синтеза, описанного выше, в результате чего получают тетралинамиды (XXIX) и фенетиламиды (XXX) соответственно. После обработки органической или неорганической кислотой, например, такой как трифторуксусная кислота и хлористоводородная кислота, получают свободные конечные амины (XXXI) и (XXXII). Эти соединения подвергают сульфонилированию реакцией с сульфонилгалогенидами, например, такими как бензолсульфонилхлорид, в присутствии основания, в результате чего получают тетралинамиды (XXXIII) и фенетиламиды (XXXIV) (Схемы 17-18).
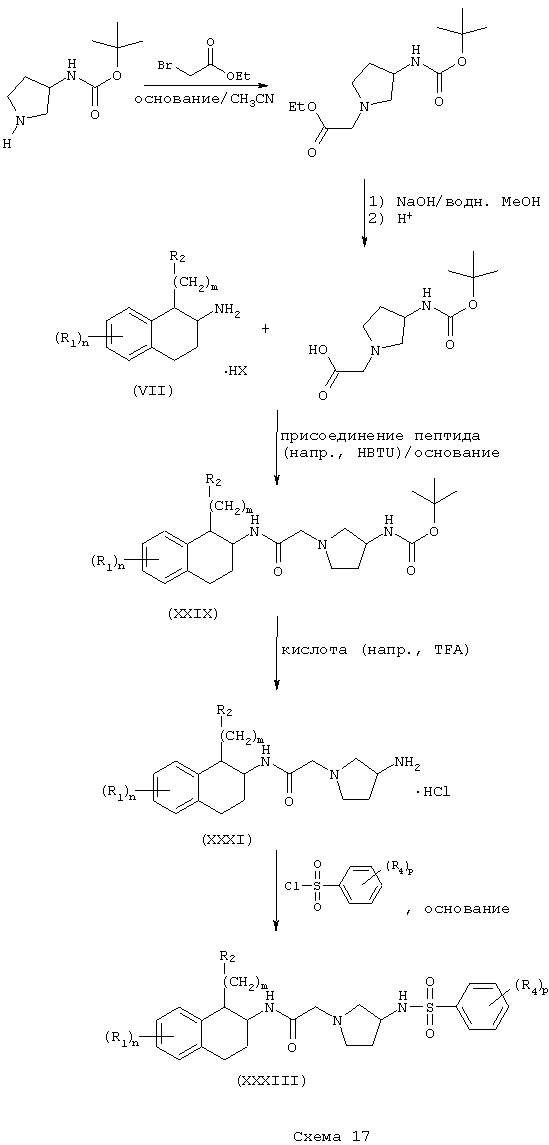
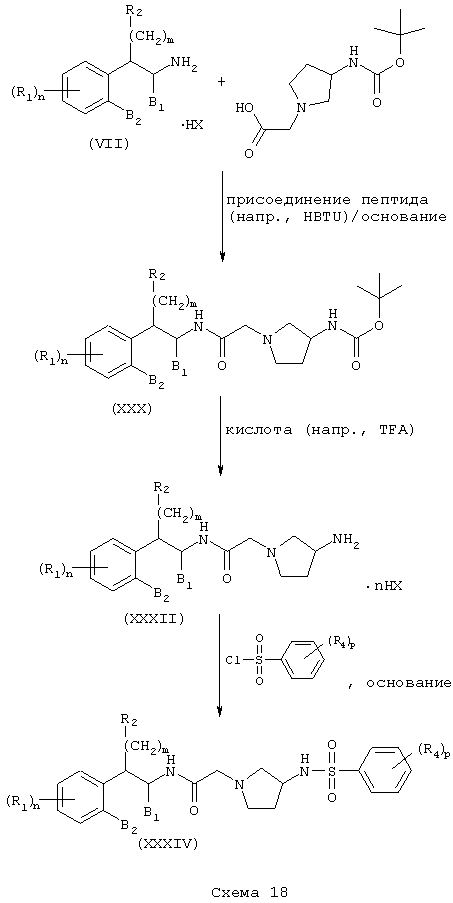
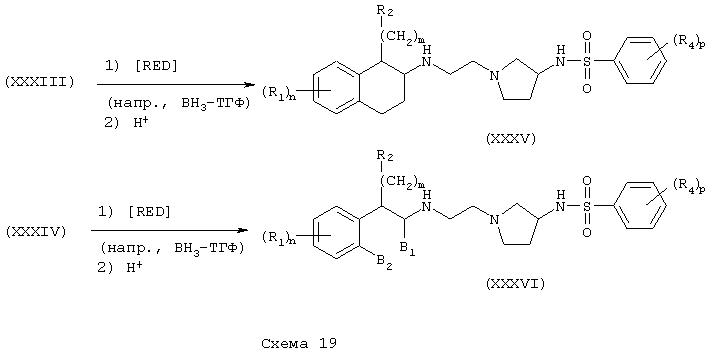
Независимо восстановлением амидов (XXXIII) и (XXXIV) восстановителем, таким как боран-тетрагидрофурановый комплекс, получают амины (XXXV) и (XXXVI) формулы А, где L=(N-метилен)пирролидин-3-ил, Z=сульфонамидо или (арил)сульфонамидо и Y=метилен (Схема 19).
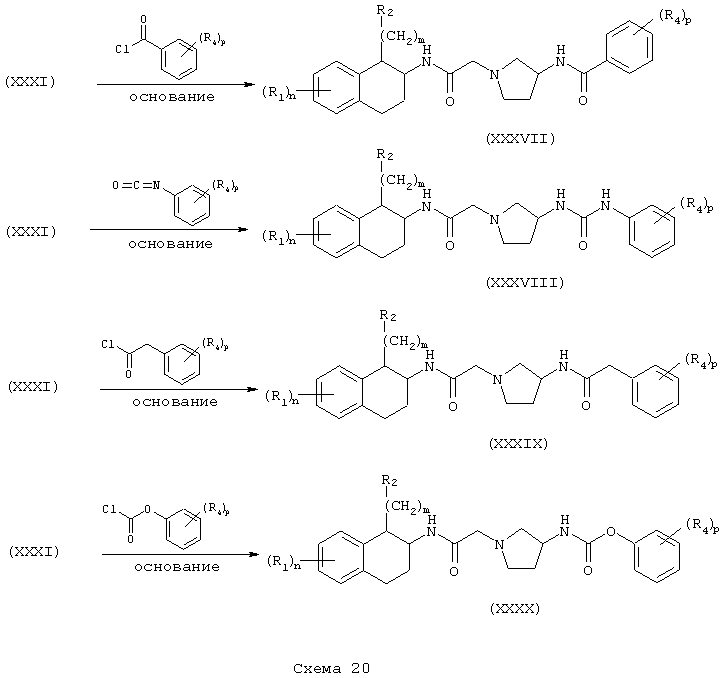
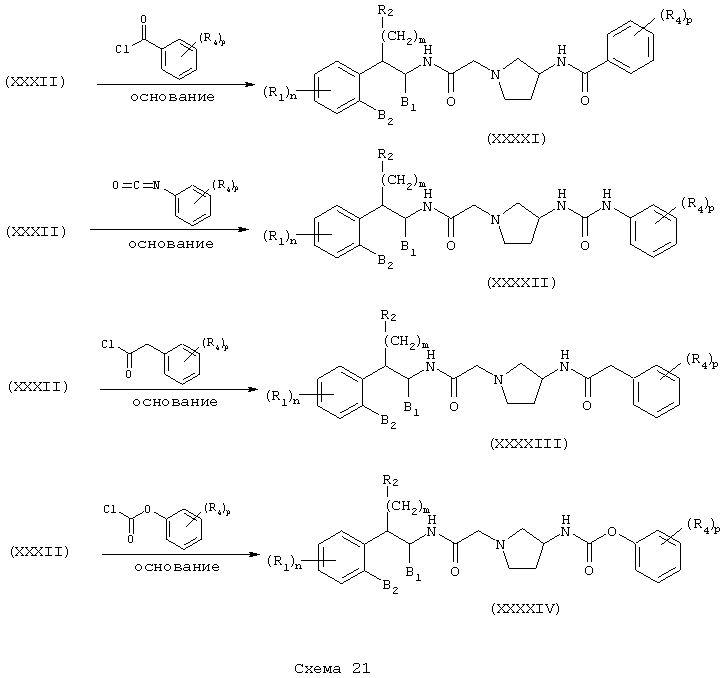
Тетралинамиды и фенетиламиды формулы А, где Y=карбонил, L=(N-метилен)пирролидин-3-ил и Z=бензамидо, фенилуреидо, фенилацетамидо и феноксикарбониламино (или бутоксикарбониламино), получают реакцией аминов (XXXI) и (XXXII) соответственно в инертном растворителе, при температуре в пределах от комнатной температуры до температуры дефлегмации, в присутствии основания, такого как амин или гидроксид, с ароилгалогенидом, арилизоцианатом, арилацетилгалогенидом или хлорформиатом, таким как фенилхлорформиат (или ди-трет-бутилдикарбонат) с получением бензамидов (XXXVII) и (XXXXI), фенилмочевин (XXXVIII) и (XXXXII), фенилацетамидов (XXXIX) и (XXXXIII) и фенилкарбамата (ХХХХ) и (XXXIV) соответственно (Схемы 20-21).
Соединения формулы А, где Y=метилен, L=(N-метилен)пирролидин-3-ил и Z=бензамидо, фенилуреидо, фенилацетамидо и феноксикарбониламино (или бутоксикарбониламино), получают восстановлением амидов (XXXI) и (XXXII) до их соответствующих аминов (XXXXV) и (XXXXVI) обработкой восстановителем, таким как боран-тетрагидрофурановый комплекс или алюмогидрид лития. Затем амины (XXXXV) и (XXXXVI) независимо подвергают реакции с ароилгалогенидом, арилизоцианатом, арилацетилгалогенидом или арилхлорформиатом (или карбонатом, таким как ди-трет-бутилкарбонат) в присутствии основания в инертном растворителе, как описано в Схемах 20-21, в результате чего получают бензамиды (XXXXVII) и (XXXXXI), фенилмочевины (XXXXVIII) и (XXXXXII), фенилацетамиды (XXXXIX) и (XXXXXIII) и фенилкарбаматы (ХХХХХ) и (XXXXXIV) соответственно (Схемы 22-24).
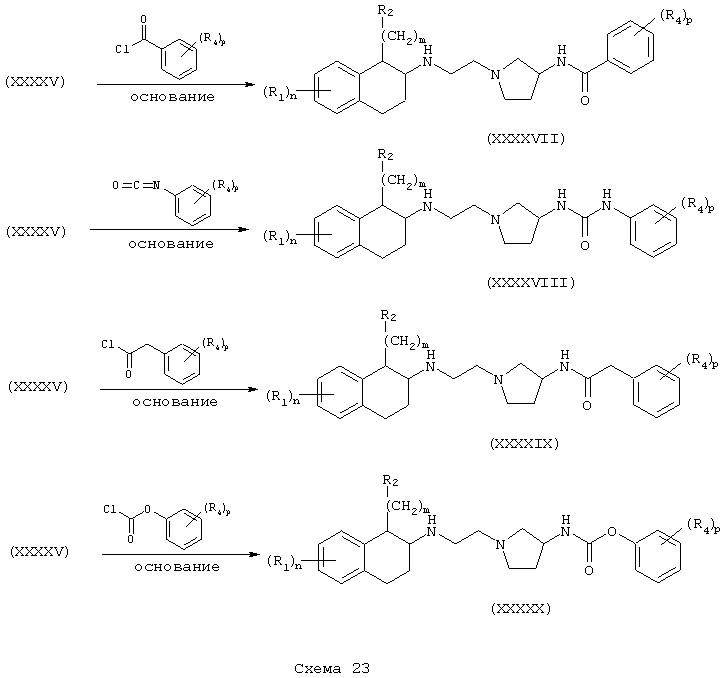
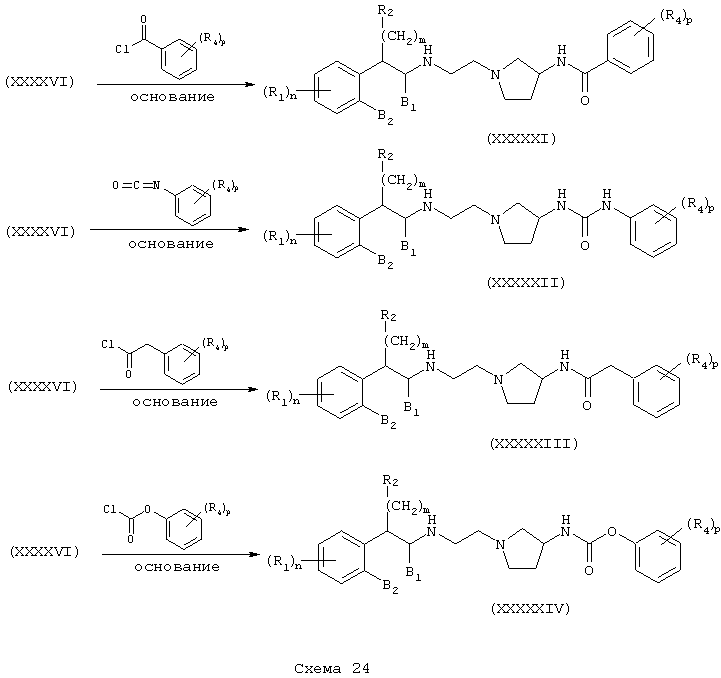
Заменяя (3-трет-бутоксикарбониламино)пирролидон на соответствующим образом защищенный аминопиперидин, такой как (4-трет-бутоксикарбониламино)пиперидин в схемах 17-24, получают соединения формулы А, где L=(N-метилен)пиперидин-4-ил, Y=метилен или карбонил и Z=N-(арил)сульфонамидо, сульфонамидо, бензамидо, фенилуреидо, фенилацетамидо или (фенокси)карбониламино.
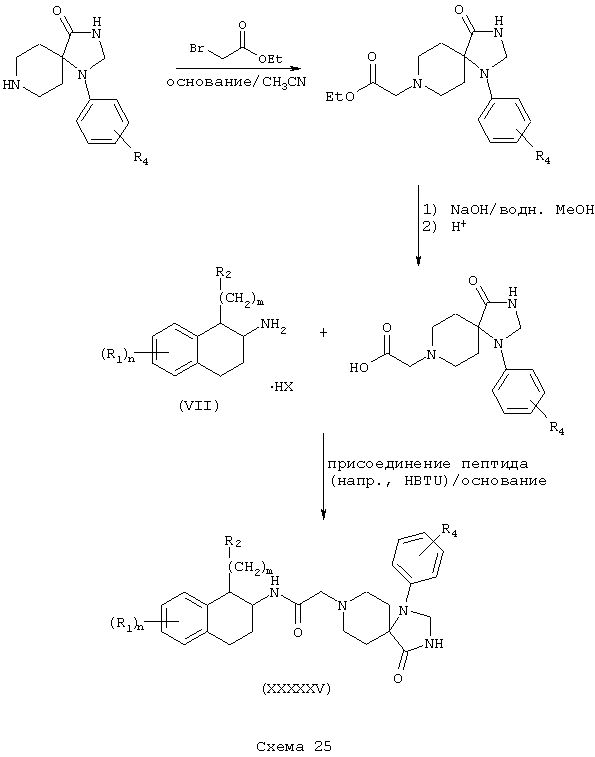
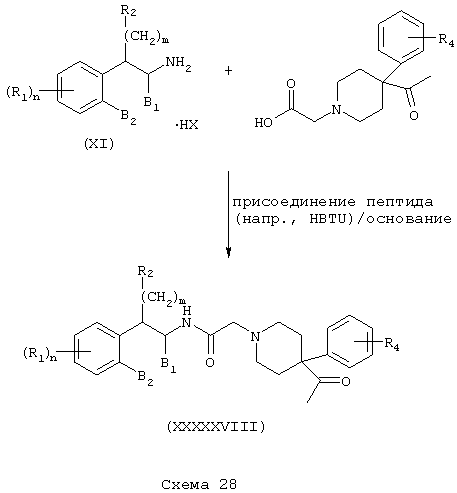
Соединения формулы А, где Y=карбонил, L=(N-метилен)пиперидин-4,4-диил и Z=1-арил-2,3-дигидро-4-оксоимидазол-5,5-диил получают реакцией 1-арил-1,3,8-триазоспиро[4,5]декан-4-она со сложным эфиром галогенуксусной кислоты, таким как этилбромацетат, в присутствии аминового основания, такого как диизопропилэтиламин, в инертном растворителе, таком как ацетонитрил, при температуре в пределах от комнатной температуры до температуры дефлегмации, с получением этил(1-арил-1,3,8-триазоспиро[4,5]декан-4-он-8-ил)ацетата. Этот сложный эфир подвергают гидролизу в основных условиях, например обработкой гидроксидом натрия в спиртовом растворе, таком как водный метанол, в результате чего, после подкисления неорганической или органической кислотой, например, такой как хлористоводородная или уксусная кислота, получают (1-арил-1,3,8-триазоспиро[4,5]декан-4-он-8-ил)уксусную кислоту. Эту карбоновую кислоту подвергают реакции непосредственно с β-тетралинами (VII) или фенетиламинами (XI) в присутствии основания, например, такого как триэтиламин, в условиях пептидного синтеза, описанного выше, в результате чего получают аминотетралинамиды (XXXXXV) и фенетиламиды (XXXXXVI) соответственно формулы А, где Y=карбонил, L=(N-метилен)пиперидин-4,4-диил и Z=1-арил-2,3-дигидро-4-оксоимидазол-5,5-диил (Схемы 25-26).
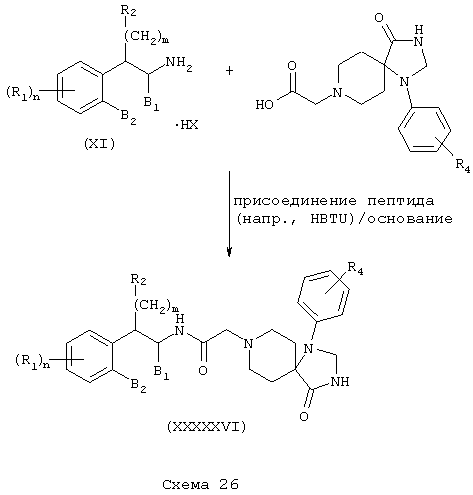
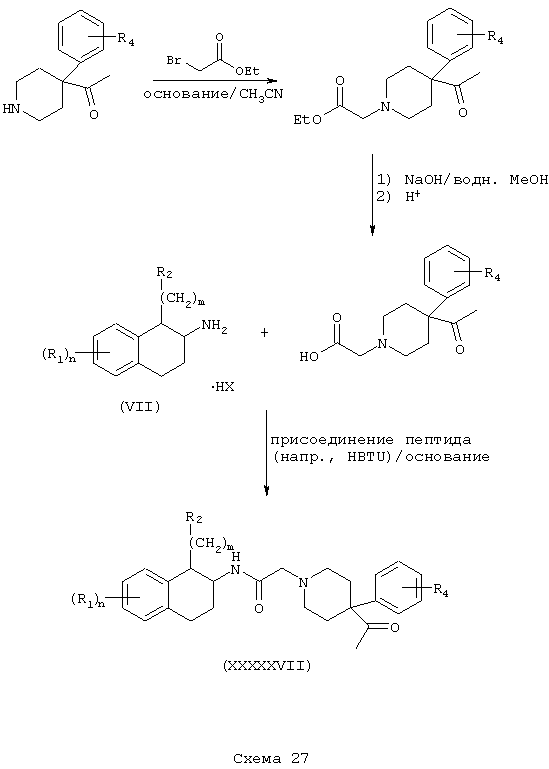
Соединения формулы А, где L=(N-метилен)-4-ацетилпиперидин-4-ил и Z=фенил, получают реакцией 4-ацетил-4-фенилпиперидина со сложным эфиром галогенуксусной кислоты, например, таким как этилбромацетат, в присутствии аминового основания, такого как диизопропилэтиламин, в инертном растворителе, таком как ацетонитрил, при температуре в пределах от комнатной температуры до температуры дефлегмации, с получением этил[(4-ацетил-4-фенилпиперидин-1-ил]ацетата. Этот сложный эфир подвергают гидролизу в основных условиях, например, обработкой гидроксидом натрия в водном метаноле, в результате чего, после подкисления неорганической или органической кислотой, например, такой как хлористоводородная или уксусная кислота, получают [(4-ацетил-4-фенилпиперидин-1-ил]уксусную кислоту. Эту карбоновую кислоту подвергают реакции с β-аминотетралинами (VII) или фенетиламинами (XI) в присутствии основания, например, такого как триэтиламин, в условиях пептидного синтеза, описанного выше, в результате чего получают (тетралинамидо)арилпиперидины (XXXXXVII) и (фенетиламидо)арилпиперидины (XXXXXVIII) соответственно формулы А, где Y=карбонил, L=(N-метилен)-4-ацетилпиперидин-4-ил и Z=фенил (Схемы 27-28).
Другие соединения настоящего изобретения, имеющие формулу А, могут быть получены описанными здесь методами, причем модификации вышеописанных экспериментальных схем известны или очевидны или могут быть внесены самим специалистом. Так, например, ряд β-тетралонов являются либо известными, либо они могут быть легко получены реакцией фенилуксусных кислот с газообразным этиленом в присутствии кислоты Льюиса (например, Stjernlof, P. et al. J. Med. Chem. 1995, 38, 2202); и эти соединения могут быть превращены непосредственно в аминотетралины (VII) путем восстановительного аминирования (Схема 2). Фенетиламиновые промежуточные соединения (XI) могут быть получены из фенилацетонитрилов методами, описанными в литературе (Journal, Hawes & Wibberley, J. Chem. Soc. С. 1966, 315 и 320; см. также J. Am. Chem. Soc. 1989, 111, 5954, и Synthesis 1997, 11, 1268), и могут быть использованы для получения соединений формулы А, в которых B1 и В2 оба представляют водород (схема 3). Соединения, в которых группа(ы) R1 может варьироваться, могут быть получены описанными выше химическими методами; в некоторых случаях используются манипуляции с защитными группами и эти манипуляции являются либо очевидными, либо известны каждому специалисту. В качестве примеров могут служить защита аминогруппы карбаматом, амидом или фталамидом, и защита гидроксильной группы эфиром или сложным эфиром. Другие заместители R1 вводят посредством манипуляции с функциональными группами, такими как, например, восстановление нитрогруппы до амина или дегидратирования амида с получением нитрила.
Варьирование группы R2 легко осуществляют с использованием замещенных бензальдегидов, нафтилальдегидов и гетероарилкарбоксальдегидов или с использованием алкил-, алкилен-, алкинил- и бензилгалогенидов или с использованием феноксиалкил- и галогеналкилгалогенидов в схемах 1 и 3. Соединения, в которых группа L варьируется, получают из пиперазинов, пиперидинов или пирролидинов, как описано в схемах 6, 10, 14, 17 и 25. Соединения, в которых L представляет алкилен, алкенилен, алкинилен, циклоалкилен или циклоалкилалкилен, получают из аминокарбоновых кислот, таких как аминогексановая кислота, аминогексеновая кислота, аминогексиновая кислота. Соединения, в которых L представляет α-аминоалкилен, получают из аминокислот, таких как лизин, которые могут быть использованы в рацемической или энантиомерной форме.
Соединения формулы А, в которых Z представляет сульфонамидо или (арил)сульфонамидо, где группа R3 или R4 варьируется, могут быть получены путем сульфонилирования, причем имеются сотни сульфонилгалогенидов или сульфоновых кислот, которые являются коммерчески доступными и многие из них являются известными. Соединения формулы А, в которых Z представляет сульфонамидо или (арил)сульфонамидо, где заместитель R3 представляет гетероарил, могут быть получены путем замены бензолсульфонамида пиридинил-, тиенил- или фурилсульфонилхлоридом, как описано в схемах 4-5. Аналогичным образом алкилсульфонил- и циклоалкилсульфонилгалогениды, взятые отдельно или в присутствии активирующего агента, такого как кислота Льюиса, могут быть использованы для получения сульфонамидов формулы А, где заместителем R3 является алкил или циклоалкил соответственно. Соединения, в которых Z представляет фенил или арил, получают непосредственно из арилпиперазинов и арилпиперидинов, как описано в схемах 10 и 14 соответственно; при этом сотни арилпиперазинов и арилпиперидинов являются известными или коммерчески доступными и могут быть использованы для получения соединений настоящего изобретения. Соединения формулы А, в которых Z представляет бензамидо, фенилуреидо, фенилацетамидо или (фенокси)карбониламино, получают из ароилгалогенидов, изоцианатов, фенилацетилгалогенидов и хлорформиатов, как описано в схемах 20-21 и 23-24, и сотни реагентов этого типа являются коммерчески доступными или известными.
Соединения формулы А, где B1 и В2, взятые вместе, образуют пятичленное кольцо (аминоиндан) получают из инданона с использованием описанных здесь химических методов. Во избежание образования региохимических изомеров, которые трудно поддаются разделению, предпочтительно использовать симметричный индан-2-он.
Примеры
Настоящее изобретение более подробно описано в нижеследующих примерах, которые приводятся лишь в целях иллюстрации, но не ограничивают изобретение. Все соединения были идентифицированы рядом методов, включая спектроскопию ядерного магнитного резонанса, масс-спектрометрию, а в некоторых случаях инфракрасную спектроскопию и элементный анализ. Данные ядерного магнитного резонанса (ЯМР 300 МГц) приведены в миллионных долях магнитного поля по отношению к тетраметилсилану. Данные масс-спектроскопии приводятся в единицах масса/заряд (m/z). Если это не оговорено особо, то материалы, используемых в этих примерах, были получены из легко доступных коммерческих источников или синтезированы стандартными методами, известными специалистам.
Примеры 1-2
2-Амино-6-[(2-фторфенилсульфонил)амино]-N-[цис-1,2,3,4-тетрагидро-6-метокси-1-(3-пиридинилметил)-2-нафтенил-(2S)-гексанамида бис-гидрохлорид 7 и
N-[5-амино-6-[[цис-1,2,3,4-тетрагидро-6-метокси-1-(3-пиридинилметил)-2-нафтил]амино]гексил-2-фторбензолсульфонамида трис-гидрохлорид 8
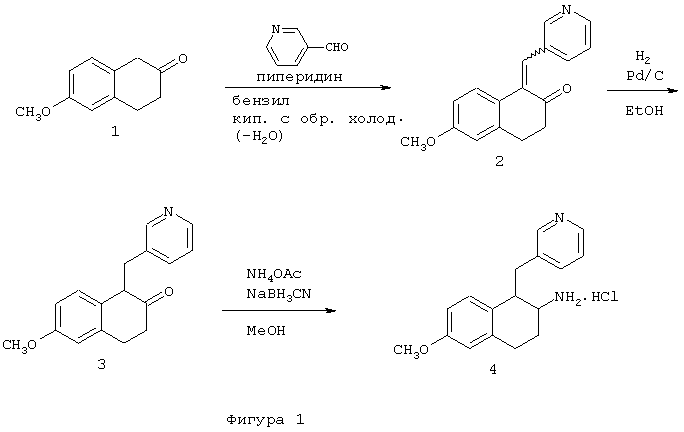
А. 6-Метокси-β-тетралон 1 (2,0 г, 11,3 ммоль) и диизопропилэтиламин (0,20 мл, 1,1 ммоль) растворяли в бензоле (60 мл) при перемешивании в круглодонной 100 мл колбе. Затем добавляли 3-пиридилкарбоксальдегид (1,1 мл, 11,7 ммоль), реакционный сосуд продували аргоном и присоединяли ловушку Дина-Старка с обратным холодильником. Смесь кипятили с обратным холодильником в течение 19 часов. После охлаждения ВЭЖХ-анализ указывал на отсутствие образования каких-либо продуктов. На этой стадии добавляли пиперидин (0,094 мл, 1,1 ммоль) и кипячение с обратным холодильником продолжали в течение 23 часов. Растворители удаляли в вакууме с получением стеклообразного оранжевого твердого вещества. Затем осуществляли хромотографическую очистку [на колонке с силикагелем (размеры 5×29 см), элюируя градиентом: 100% гексан (400 мл), 75%/25% гексан/этилацетат (об./об.) (400 мл), 50%/50% гексан/этилацетат (об./об.) (400 мл), 25%/75% гексан/этилацетат (об./об.) (400 мл) и, наконец, 100% этилацетат]. После выпаривания соответствующих фракций получали 3,4-дигидро-6-метокси-1-((3-пиридинил)метилиденил)-2-нафталинон 2 (1,484 г, 5,59 ммоль) в виде оранжевого масла, которое отверждалось после выдерживания в холодильнике.
МС (МН+) 266; 1H ЯМР (CDCl3): δ 2,67 (т, 2Н), 3,02 (т, 2Н), 3,83 (с, 3Н), 6,60 (дд, 1Н), 6,82 (д, 1Н), 7,19 (м, 2Н), 7,51 (с, 1Н), 7,71 (д, 1Н), 8,49 (дд, 1Н), 8,65 (д, 1Н).
В. Нафталин-2-он 2 (1,442 г, 5,44 ммоль), полученный, как описано выше, растворяли в абсолютном этаноле (50 мл) и переносили в 250 мл колбу Парра для гидрирования. Отдельно осторожно добавляли этанол к 10% палладию на угле (0,020 г) и эту суспензию добавляли в сосуд Парра. Смесь гидрировали под давлением 50 фунт/кв.дюйм (3,455 кПа) в течение 16 часов. Катализатор удаляли фильтрацией через целит. Спектроскопический анализ указывал на присутствие некоторого количества исходного соединения, после чего к этаноловому раствору добавляли еще некоторое количество палладиевого катализатора (0,081 г) и гидрирование повторяли в течение 20 часов. Затем катализатор удаляли фильтрацией через целит. После удаления растворителей в вакууме получали 3,4-дигидро-6-метокси-1-(3-пиридинилметил)-2(1Н)-нафталинон 3 в виде оранжевого масла, которое было использовано в последующей стадии без дополнительной очистки. МС (МН+) 268.
С. Нафталин-2-он 3, полученный, как описано выше, растворяли в метаноле (275 мл) в круглодонной 1 л колбе. К перемешиваемому метаноловому раствору добавляли ацетат аммония (4,27 г, 55,4 ммоль) и перед проведением реакции смесь оставляли для полного растворения. Затем к этому метанольному раствору добавляли цианоборогидрид натрия (1,703 г, 27,5 ммоль). Реакционный сосуд продували азотом и раствор кипятили с обратным холодильником в течение 18 часов. После этого растворители удаляли в вакууме и получали желтое твердое вещество, которое растворяли в этиловом эфире (500 мл) и 0,1 М растворе гидроксида натрия (275 мл). Органический слой удаляли и промывали дополнительным количеством 0,1 М раствора гидроксида натрия (275 мл) и воды (250 мл). Объединенные промывные воды подвергали обратной экстракции этиловым эфиром (3×100 мл). Органические экстракты объединяли и сушили над сульфатом натрия. Растворители удаляли в вакууме и остаток растворяли в этиловом эфире и в минимальном количестве дихлорметана. После добавления избытка 1 М хлористого водорода в этиловом эфире образовывался темно-коричневатый осадок. Растворители удаляли в вакууме и полученное твердое вещество растирали с эфиром и сушили в вакуумной печи с получением бис-гидрохлорида 1,2,3,4-тетрагидро-6-метокси-1-(3-пиридинилметил)-2-нафталинамина 4 в виде коричневато-оранжевого твердого вещества (1,208 г, 3,54 ммоль).
МС (МН+) 269; 1Н ЯМР (ДМСО-d6):δ 1,95-2,20 (м, 2Н), 2,68-3,29 (м, 4Н), 3,30-3,48 (м, 2Н), 3,69 (с, 3Н), 5,98 (д, 1Н), 6,41 (дд, 1Н), 6,75 (д, 1Н), 7,98 (дд, 1Н), 8,36 (д, 1Н), 8,68-8,89 (м, 5Н) (Фигура 1).
D. N-трет-Бутоксикарбонил-L-лизин (2,49 г, 10,1 ммоль) помещали в круглодонную 200 мл колбу. Затем в эту колбу помещали магнитный стержень для перемешивания, после чего добавляли 10 мл диоксана и 21 мл 1 н. раствора гидроксида натрия. Полученный раствор перемешивали несколько минут до полного растворения. Затем пипеткой вводили раствор 2-фторбензолсульфонилхлорида (2,00 г, 10,3 ммоль) в диоксане (11 мл). Реакционный сосуд продували аргоном, закрывали и оставляли для перемешивания при комнатной температуре примерно на 1,5 часа. Затем стержень для перемешивания удаляли и растворитель выпаривали при пониженном давлении до тех пор, пока в колбе не оставалась только вода. К этой смеси добавляли воду, объем доводили примерно до 50 мл и добавляли 1 н. хлористоводородную кислоту (22 мл), что приводило к образованию клейкого осадка. Эту смесь экстрагировали метиленхлоридом (3×50 мл) и объединенные органические экстракты промывали 1 н. хлористоводородной кислотой (1×50 мл), а затем насыщенным раствором соли (1×50 мл). Органические экстракты сушили над сульфатом магния, фильтровали и концентрировали в вакууме с получением сульфонилированного N-трет-бутоксикарбониллизина 5 (3,93 г, 9,7 ммоль) в виде не совсем белого стеклообразного полутвердого вещества.
ЯМР (d6-ДМСО): δ 12,42 (с, 1Н), 7,90 (т, 1Н), 7,79 (т, 1Н), 7,71 (м, 1Н), 7,49-7,34 (м, 2Н), 7,02 (д, 1Н), 3,78 (м, 1Н), 2,83 (м, 2Н), 1,63-1,16 (м, 15Н); МС: М-Н=403.
Е. Сульфонилированный L-лизин 5, полученный в предыдущей реакции (3,92 г, 9,69 ммоль), помещали вместе с бис-гидрохлоридом 1,2,3,4-тетрагидро-6-метокси-1-(3-пиридинилметил)-2-нафталинамина 4 (3,53 г, 10,34 ммоль) в круглодонную 300 мл колбу, снабженную стержнем для перемешивания. После этого добавляли N,N-диметилформамид (ДМФ) (50 мл), а затем диизопропилэтиламин (5,6 мл, 32,1 ммоль) и смесь перемешивали. После растворения добавляли гексафторфосфат 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (HBTU) (3,72 г, 9,81 ммоль) и колбу продували аргоном, закрывали и оставляли для перемешивания при комнатной температуре на 30 минут. Затем добавляли воду (~5 мл) для гашения реакции и растворитель удаляли в вакууме с получением коричневого масла. Этот продукт очищали колоночной хроматографией на колонках с силикагелем (размер 6×12 см), элюируюя градиентом смеси метиленхлорид-ацетон-метанол. После выпаривания соответствующих фракций получали аддукт 6 (в виде коричневато-зеленой пены, 4,63 г, 7,07 ммоль) в виде смеси диастереомеров. МС: МН+=655.
F. Сульфонилированный лизинотетралинамид 6, полученный в предыдущей реакции (4,59 г, 7,01 ммоль), помещали в круглодонную 200 мл колбу, снабженную стержнем для перемешивания, и добавляли метиленхлорид (100 мл). При перемешивании добавляли раствор 95% TFA/5% H2O (об./об.) (10 мл) и реакционную смесь оставляли для перемешивания в атмосфере азота при комнатной температуре на 3,5 часа. Реакционную смесь концентрировали в вакууме и остаток растирали с диэтиловым эфиром. Жидкость декантировали и добавляли дополнительное количество эфира. Полученное твердое вещество фильтровали и сушили в вакууме с получением нужного бис-гидрохлорида тетралинамида-лизиносульфонамида 7 (4,28 г, 5,47 ммоль) в виде смеси дистереомеров. Часть этого продукта (4,01 г) разделяли на рацемические серии диастереомеров обращенно-фазовой хроматографией на колонке (Bondapak C18, 6×(40×100 мм) с использованием градиента Н2O/СН3СN (+0,1% TFA)). Соответствующие фракции выделяли и лиофилизовали с получением диастереомера а (2,17 г, 2,77 ммоль) и диастереомера b (1,78 г, 2,27 ммоль) в виде бис-ТFА-солей (абсолютные конфигурации этих диастереомеров не были определены).
Диастереомер а: д.и.=96%; ЯМР (d6-ДМСО): δ 8,57 (м, 2Н), 8,30 (с, 1Н), 8,11 (шир., 3Н), 7,96 (т, 1Н), 7,80-7,64 (м, 3Н), 7,55 (дд, 1Н), 7,48-7,32 (м, 2Н), 6,71 (с, 1Н), 6,58-6,46 (м, 2Н), 4,03 (м, 1Н), 3,79 (м, 1Н), 3,69 (с, 3Н), 3,24 (м, 1Н), 3,03-2,73 (м, 6Н), 2,08-1,91 (м, 1Н), 1,85-1,58 (м, 3Н), 1,53-1,31 (м, 4Н); МС: МН+=555. Диастереомер b: д.и.=100%; ЯМР (d6-ДМСО): δ 8,68 (д, 1Н), 8,57 (д, 1Н), 8,49 (с, 1Н), 8,21 (шир., 3Н), 8,01 (д, 1Н), 7,93 (т, 1Н), 7,78 (дт, 1Н), 7,73 (м, 2Н), 7,52-7,37 (м, 2Н), 6,75 (с, 1Н), 6,56 (м, 2Н), 3,99 (м, 1Н), 3,85 (м, 1Н), 3,71 (с, 3Н), 3,23 (м, 1Н), 3,08-2,76 (м, 6Н), 2,00-1,59 (м, 4Н), 1,53-1,22 (м, 4Н); МС: МН+=555 (Фигура 2).
G. Диастереомер а 7, полученный в предыдущей реакции (2,02 г, 2,58 ммоль), помещали в круглодонную 200 мл колбу, снабженную стержнем для перемешивания, и добавляли ТГФ (60 мл). После перемешивания добавляли раствор борана в ТГФ (40 мл 1 М раствора, 40 ммоль), колбу продували азотом и присоединяли обратный холодильник. Смесь кипятили с обратным холодильником в течение 24 часов и на этой стадии добавляли дополнительную порцию раствора борана (10 мл). Реакционную смесь кипятили с обратным холодильником еще 14 часов. Реакционную смесь оставляли для охлаждения и осторожно добавляли воду (10 мл) для гашения реакции. Затем добавляли хлористоводородную кислоту (20 мл 1 н. раствора) и реакционную смесь кипятили с обратным холодильником в течение 2 часов. Растворители удаляли в вакууме и остаток суспендировали в воде (250 мл). Полученную смесь слегка подкисляли добавлением 1 н. хлористоводородной кислоты. Водный раствор промывали метиленхлоридом (3×250 мл) и водный слой отделяли. Затем добавляли раствор гидроксида аммония до тех пор, пока рН не становился щелочным. После этого воду удаляли в вакууме и получали белое твердое вещество. Полученное вещество растирали с метиленхлоридом и осажденные соли борана удаляли фильтрацией. Оставшиеся органические экстракты концентрировали в вакууме с получением неочищенного продукта в виде пены. Это вещество очищали флеш-хроматографией на колонке с силикагелем (размеры 6×11 см), элюируя градиентом смеси метиленхлорид-метанол-гидроксид аммония. После выпаривания соответствующих фракций остаток обрабатывали избытком смеси этанола и хлористого водорода, а затем выпаривали и сушили в вакууме, в результате получали сульфонамид аминотетралина 8 в виде желтой трис-гидрохлоридной соли (0,898 г, 1,38 ммоль).
ЯМР (d6-ДМСО): δ 10,83 (шир., 1Н), 10,08 (шир., 1Н), 8,80 (д, 1Н), 8,73 (м, 4Н), 8,43 (д, 1Н), 7,97 (м, 2Н), 7,81 (т, 1Н), 7,71 (м, 1Н), 7,51-7,33 (м, 2Н), 6,75 (с, 1Н), 6,37 (д, 1Н), 5,83 (д, 1Н), 3,80 (м, 1Н), 3,71-3,30 (м, 8Н), 3,11 (м, 1Н), 2,98-2,69 (м, 4Н), 2,34-2,13 (м, 2Н), 1,73-1,55 (м, 2Н), 1,54-1,29 (м, 4Н); МС: МН+=541 (Фигура 2).
Пример 3
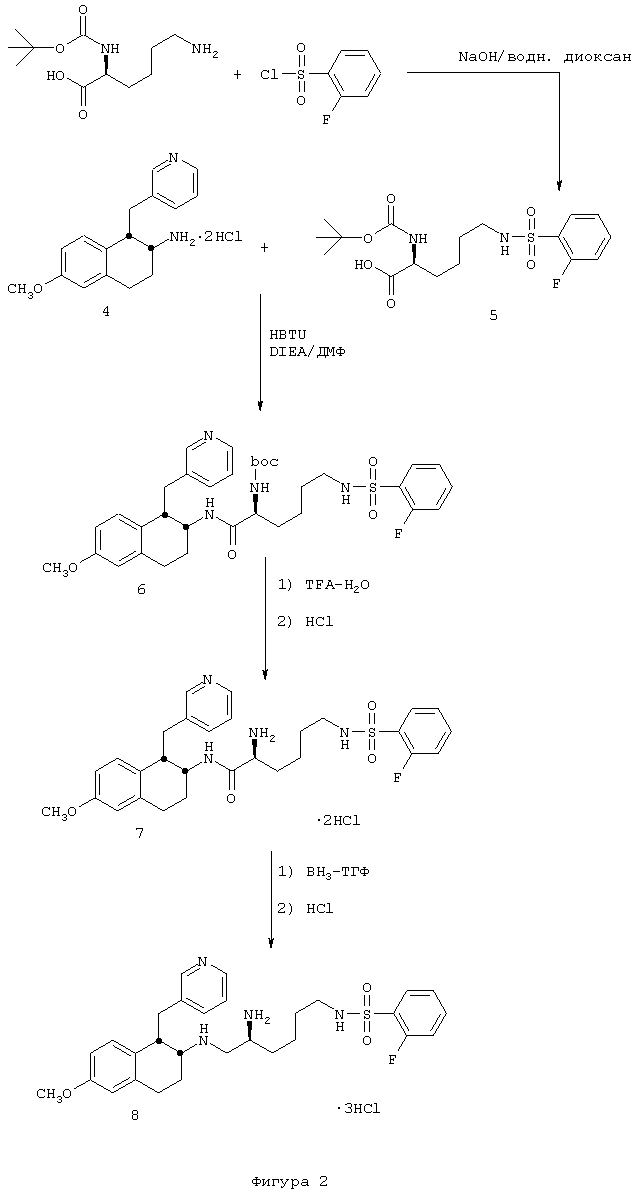
N-[5-Амино-6-[[цис-1,2,3,4-тетрагидро-6-гидрокси-1-(3-пиридинилметил)-2-нафтил]амино]гексил-2-фторбензолсульфонамида трис-гидрохлорид 9
Сульфонамид аминотетралина 8, полученный в предыдущей реакции (0,160 г, 0,246 ммоль), помещали в круглодонную 50 мл колбу, снабженную стержнем для перемешивания. Затем добавляли метиленхлорид (25 мл) и суспензию охлаждали в ледяной бане в течение нескольких минут. К реакционной смеси добавляли трибромид бора в метиленхлориде (1 М, 1,25 мл, 1,25 ммоль). Колбу продували аргоном, закрывали и оставляли для нагревания до комнатной температуры, смесь перемешивали в течение 16 часов и на этой стадии реакцию гасили добавлением метанола (1 мл). Растворители удаляли в вакууме и к полученному остатку добавляли дополнительную аликвоту метанола. После выпаривания растворителя из этой смеси получали неочищенный продукт, который был очищен обращенно-фазовой хроматографией (Bondapak C18, 3×(40×100 мм) с использованием градиента H2O/CH3CN (+0,1% TFA)). Соответствующие фракции собирали и лиофилизовали. Затем полученный продукт обрабатывали смесью этанола и хлористого водорода, после чего выпаривали и сушили в вакууме с получением фенольного продукта 9 в виде белой трис-гидрохлоридной соли (0,145 г, 0,228 ммоль).
ЯМР (d6-ДМСО): δ 10,77 (шир., 1Н), 10,01 (шир., 1Н), 9,31 (шир., 1Н), 8,79 (д, 1Н), 8,67 (м, 4Н), 8,37 (д, 1Н), 7,97 (м, 2Н), 7,81 (дт, 1Н), 7,72 (м, 1Н), 7,52-7,36 (м, 2Н), 6,57 (с, 1Н), 6,22 (дд, 1Н), 5,69 (д, 1Н), 3,79 (м, 1Н), 3,68-3,30 (м, 5Н), 3,04 (м, 1Н), 2,92-2,68 (м, 4Н), 2,33-2,10 (м, 2Н), 1,73-1,56 (м, 2Н), 1,55-1,32 (м, 4Н); МС: МН+=527 (Фигура 3).
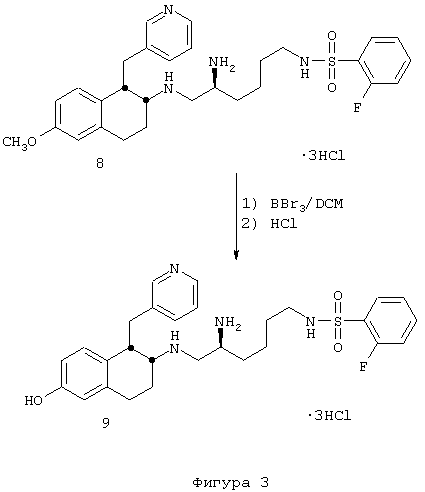
Пример 4
(2S)-2-(Ацетиламино)-6-[(2-фторфенилсульфонил)амино]-N-[цис-1,2,3,4-тетрагидро-6-метокси-1-(3-пиридинилметил)-2-нафтенил]гексанамида бис-гидрохлорид 10
Смесь диастереомеров тетралинамид-лизиносульфонамид 7 (0,195 г, 0,249 ммоль) помещали в круглодонную 50 мл колбу, снабженную стержнем для перемешивания. После этого добавляли ацетонитрил (25 мл), а затем триэтиламин (0,122 мл, 0,875 ммоль). При перемешивании добавляли ацетилхлорид (0,021 мл, 0,295 ммоль) и колбу продували аргоном, закрывали и перемешивали в течение ночи при комнатной температуре. Растворители удаляли в вакууме и остаток растворяли в метиленхлориде (75 мл). Эту смесь промывали 1 н. гидроксидом натрия (2×25 мл), а затем насыщенным раствором соли (1×25 мл). Органические слои сушили над сульфатом магния, фильтровали и концентрировали в вакууме с получением ацетатного продукта 10 в виде желтовато-коричневого твердого вещества (0,139 г, 0,233 ммоль) в качестве 1:1 смеси диастереомеров.
ЯМР (CDCl3): δ 8,52 (д, 0,5Н), 8,43 (д, 0,5Н), 8,28 (д, 1Н), 7,89 (м, 1Н), 7,57 (м, 1Н), 7,44 (д, 0,5Н), 7,39-7,13 (м, 3,5Н), 6,92 (т, 0,5Н), 6,77 (д, 0,5Н), 6,70-6,54 (м, 3Н), 6,48 (дд, 1Н), 6,34 (д, 0,5Н), 5,59 (т, 0,5Н), 4,40-4,06 (м, 2Н), 3,78 (д, 3Н), 3,29 (м, 1Н), 3,19-2,82 (м, 6Н), 2,01 (д, 3Н), 1,92-1,71 (м, 2Н), 1,72-1,32 (м, 6Н); МС: МН+=597 (Фигура 4).
Пример 5
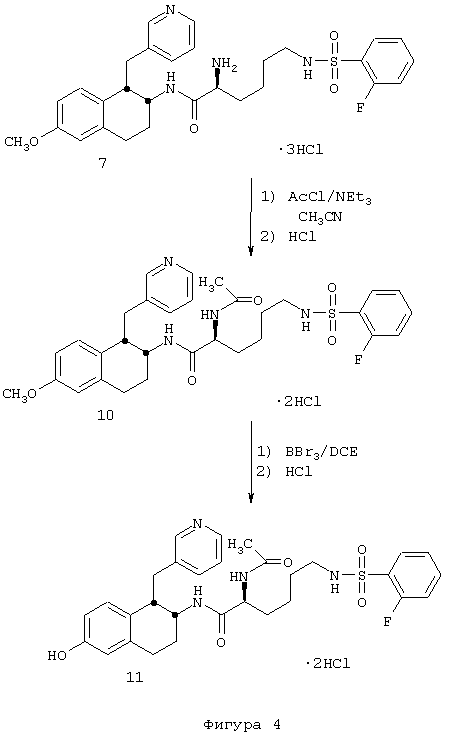
(2S)-2-(Ацетиламино)-6-[(2-фторфенилсульфонил)амино]-N-[цис-1,2,3,4-тетрагидро-6-гидрокси-1-(3-пиридинилметил)-2-нафтенил]гексанамида бис-гидрохлорид 11
Бис-амид 10, полученный в предыдущей реакции (0,114 г, 0,191 ммоль), помещали в круглодонную 50 мл колбу, снабженную стержнем для перемешивания. Затем добавляли метиленхлорид (20 мл) и раствор охлаждали в ледяной бане в течение нескольких минут. К реакционной смеси добавляли трибромид бора в метиленхлориде (1 М, 1,0 мл, 1,0 ммоль). Колбу продували аргоном, закрывали и оставляли для нагревания до комнатной температуры, смесь перемешивали в течение 16 часов, после чего реакцию гасили добавлением метанола (1 мл). Растворители удаляли в вакууме и полученный продукт обрабатывали дополнительной аликвотой метанола. Смесь выпаривали в вакууме с получением неочищенного фенольного тетралинамида 11, который был очищен обращенно-фазовой колоночной хроматографией, позволяющей разделять и очищать рацемические пары диастереомеров (Bondapak С18, 3×(40×100 мм), градиент Н2О/СН3CN (+0,1% TFA)). После лиофилизации соответствующих фракций, каждый диастереомер обрабатывали смесью этанола и хлористого водорода, подвергали выпариванию, и наконец, сушили в вакууме с получением индивидуальных диастереомеров рацемической смеси в виде желтовато-коричневых гидрохлоридных солей; диастереомера а (0,036 г, 0,058 ммоль) и диастереомера b (0,057 г, 0,092 ммоль) (абсолютные конфигурации этих диастереомеров не были определены). Диастереомер а: д.и.=100%; ЯМР (d6-ДМСО): δ 9,22 (очень шир., 1Н), 8,79 (д, 1Н), 8,48 (с, 1Н), 8,20 (д, 1Н), 8,08-7,87 (м, 4Н), 7,83-7,63 (м, 2Н), 7,50-7,33 (м, 2Н), 6,54 (с, 1Н), 6,43-6,28 (м, 2Н), 4,19 (кв., 1Н), 3,93 (м, 1Н), 3,18 (м, 1Н), 3,08-2,67 (м, 6Н), 1,92 (м, 1Н), 1,84 (с, 3Н), 1,73 (м, 1Н), 1,58-1,16 (м, 6Н); МС: МН+=583. Диастереомер b: д.и.=66%; ЯМР (d6-ДМСО): δ 9,20 (очень шир., 1Н), 8,77 (д, 1Н), 8,57 (с, 1Н), 8,28-8,14 (м, 2Н), 8,08-7,84 (м, 3Н), 7,83-7,62 (м, 2Н), 7,50-7,32 (м, 2Н), 6,54 (с, 1Н), 6,47-6,29 (м, 2Н), 4,10 (кв., 1Н), 3,85 (м, 1Н), 3,27-3,08 (м, 2Н), 3,03-2,66 (м, 5Н), 1,90 (с, 3Н), 1,87-1,63 (м, 2Н), 1,57-1,13 (м, 6Н); МС: МН+=583 (Фигура 4).
Пример 6
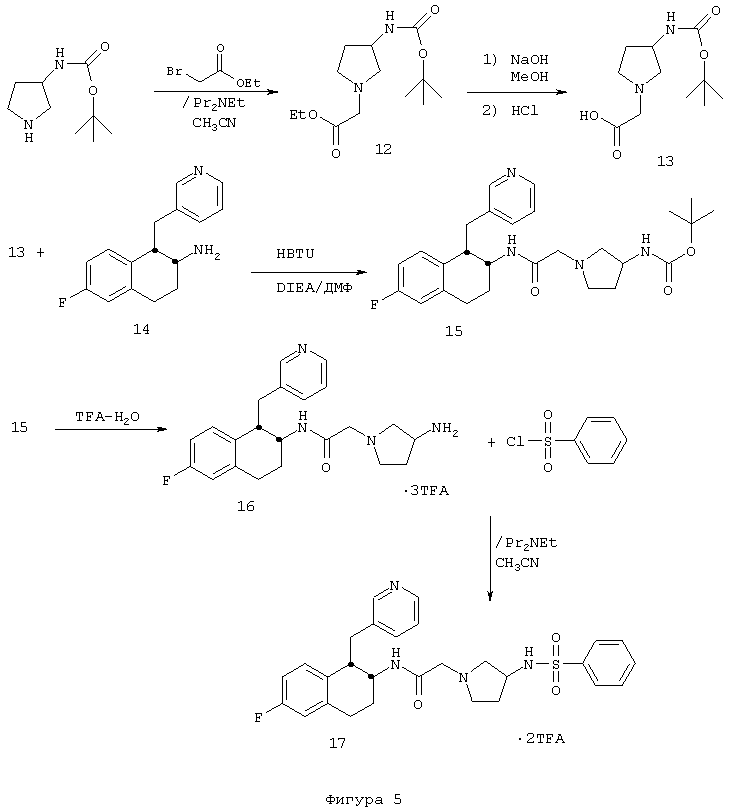
3-[(Фенилсульфонил)амино]-N-[цис-1,2,3,4-тетрагидро-6-фтор-1-(3-пиридинилметил)-2-нафтил]-1-пирролидинацетамида бис-трифторацетат 17
А. Рацемический 3-(N-бутоксикарбонил)аминопирролидин (5,13 г, 27,5 ммоль) помещали в круглодонную 300 мл колбу, снабженную стержнем для перемешивания. После этого добавляли ацетонитрил (100 мл), в результате чего получали суспензию, к которой добавляли диизопропилэтиламин (7,2 мл, 41,3 ммоль), а затем этилбромацетат (3,1 мл, 28,0 ммоль). Колбу продували азотом и подсоединяли обратный холодильник. Реакционную смесь кипятили с обратным холодильником в течение 1,5 часа, а затем оставляли для охлаждения и перемешивания при комнатной температуре на ночь. Растворители удаляли в вакууме с получением маслянистого твердого вещества. Это вещество растворяли в метиленхлориде (200 мл) и последовательно промывали раствором бикарбоната натрия (1×200 мл), водой (1×200 мл) и насыщенным раствором соли (200 мл). Органические слои сушили над сульфатом магния, фильтровали и растворители удаляли в вакууме с получением густого масла, которое медленно кристаллизовалось после отстаивания, в результате чего получали сложный эфир пирролидинилуксусной кислоты 12 (6,96 г, 25,6 ммоль).
ЯМР (CDCl3): δ 4,98 (шир. д, 1Н), 4,27-4,13 (м, 3Н), 3,33 (с, 2Н), 2,98 (м, 1Н), 2,83-2,66 (м, 2Н), 2,48 (м, 1Н), 2,27 (м, 1Н), 1,67 (м, 1Н), 1,44 (с, 9Н), 1,28 (т, 3Н).
В. Сложный эфир пирролидинуксусной кислоты 12, полученный в предыдущей реакции (6,95 г, 25,5 ммоль), помещали в круглодонную 300 мл колбу. Затем вставляли магнитный стержень для перемешивания и добавляли метанол (100 мл). Смесь перемешивали до тех пор, пока не растворится исходное вещество. Затем добавляли раствор гидроксида натрия (1 н., 75,0 мл, 75,0 ммоль). Реакционный сосуд закрывали и смесь оставляли для перемешивания в течение 20 часов, после чего добавляли раствор хлористоводородной кислоты (1 н., 75,0 мл, 75,0 ммоль). Полученную смесь оставляли для перемешивания на несколько минут. Растворители удаляли в вакууме и полученное твердое вещество обрабатывали метиленхлоридом. Органический экстракт сушили над сульфатом магния, фильтровали и концентрировали в вакууме с получением пирролидинилуксусной кислоты 13 в виде белого твердого порошка (6,30 г, 25,8 ммоль).
ЯМР (d6-ДМСО): δ 7,21 (шир. д, 1Н), 4,05 (м, 1Н), 3,38 (с, 2Н), 3,23 (м, 1Н), 3,02 (м, 2Н), 2,78 (м, 1Н), 2,12 (м, 1Н), 1,73 (м, 1Н), 1,39 (с, 9Н); МС: МН+=245.
С. 1,2,3,4-Тетрагидро-6-фтор-1-(3-пиридинилметил)-2-нафталинамина бис-гидрохлорид 14 (0,331 г, 1,01 ммоль), полученный из 6-фтор-β-тетралона с использованием химических реакций, описанных в примере 1 (фигура 1), помещали в круглодонную 25 мл колбу, снабженную стержнем для перемешивания, и добавляли ДМФ (5 мл). Затем добавляли пирролидинилуксусную кислоту 13 (0,250 г, 1,02 ммоль), полученную в предыдущей реакции, после чего добавляли диизопропилэтиламин (0,580 мл, 3,33 ммоль), а затем (HBTU) (0,387 г, 1,02 ммоль). Колбу продували аргоном, закрывали и оставляли для перемешивания при комнатной температуре на 2 часа. Реакционную смесь разбавляли насыщенным раствором соли (50 мл) и метиленхлоридом (150 мл) и слои разделяли. Органические слои еще раз промывали насыщенным раствором соли (2×50 мл). Объединенные промывные насыщенные водные растворы соли экстрагировали метиленхлоридом (2×25 мл) и объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали в вакууме с получением неочищенного продукта. Этот продукт очищали с помощью обращенно-фазовой хроматографии на колонке (Bondapak С18, 3×(40×100 мм) с использованием градиента H2О/СН3СN (+0,1% TFA)). После лиофилизации соответствующих фракций получали соль пирролидинацетамид-бис-TFA 15 в виде белого порошка (0,251 г, 0,35 ммоль); МС: МН+=483.
D. Пирролидинацетамид 15, полученный в предыдущей реакции (0,205 г, 0,288 ммоль), помещали в круглодонную 50 мл колбу, снабженную стержнем для перемешивания. После этого добавляли метиленхлорид (25 мл), а затем небольшое количество воды (~0,5 мл) и TFA (2 мл). Реакционный сосуд закрывали и оставляли для перемешивания при комнатной температуре на 19 часов, после чего растворитель удаляли в вакууме с получением трис-ТFА-соли 3-аминопирролидинацетамида 16 (0,204 г, 0,282 ммоль).
ЯМР (d6-ДМСО): δ 8,69 (д, 1Н), 8,64 (д, 1Н), 8,49 (с, 1Н), 8,36 (шир., 3Н), 7,93 (д, 1Н), 7,67 (т, 1Н), 7,02 (д, 1Н), 6,83 (м, 2Н), 4,13 (с, 2Н), 4,07-3,88 (м, 3Н), 3,87-3,22 (м, 4Н), 3,15-2,69 (м, 4Н), 2,41 (м, 1Н), 2,14-1,69 (м, 3Н); МС: МН+=383.
Е. Ацетамид аминопирролидина 16, полученный в предыдущей реакции (0,074 г, 0,102 ммоль), помещали в круглодонную 50 мл колбу, снабженную стержнем для перемешивания, и добавляли ацетонитрил (20 мл). После этого добавляли диизопропилэтиламин (0,078 мл, 0,448 ммоль), а затем бензолсульфонилхлорид (0,013 мл, 0,102 ммоль). Колбу продували аргоном, закрывали и оставляли для перемешивания при комнатной температуре на 3 часа, после чего растворители удаляли в вакууме. Остаток очищали обращенно-фазовой колоночной хроматографией (Н2O/СН3СN (+0,1% TFA)). После выделения и лиофилизации соответствующих фракций получали бис-ТFА-соль 3-[(фенилсульфонил)амино]-N-[цис-1,2,3,4-тетрагидро-6-фтор-1-(3-пиридинилметил)-2-нафтил]-1-пирролидинацетамида 17 в виде белого твердого вещества (0,067 г, 0,089 ммоль).
ЯМР (d6-ДМСО): δ 8,62 (д, 2Н), 8,47 (с, 1Н), 8,25 (м, 1Н), 7,92 (д, 1Н), 7,83 (м, 2Н), 7,66 (м, 4Н), 7,02 (д, 1Н), 6,84 (м, 2Н), 4,18-3,73 (м, 4Н), 3,72-2,72 (м, 9Н), 2,07 (м, 1Н), 1,98-1,67 (м, 3Н); МС: МН+=523 (Фигура 5).
Примеры 7-8
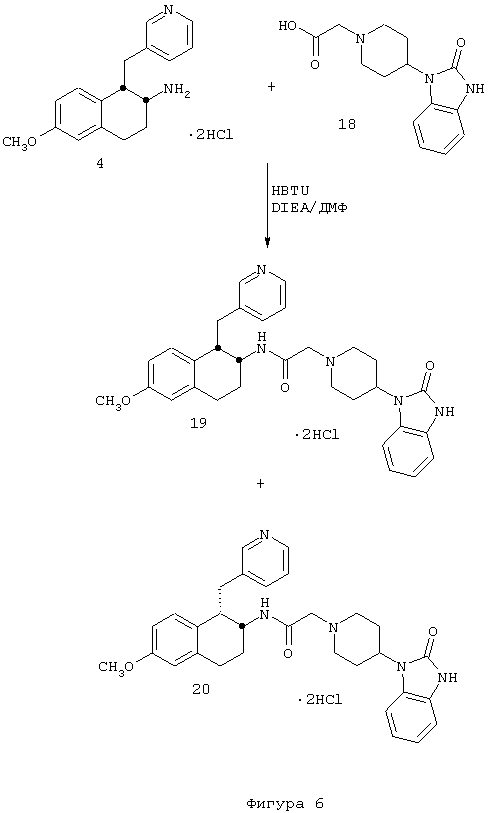
4-(2,3-Дигидро-2-оксо-1Н-бензимидазол-1-ил)-N-[цис-1,2,3,4-тетрагидро-6-метокси-1-(3-пиридинилметил)-2-нафтил]-1-пиперидинацетамида бис-гидрохлорид 19 и
4-(2,3-дигидро-2-оксо-1Н-бензимидазол-1-ил)-N-[транс-1,2,3,4-тетрагидро-6-метокси-1-(3-пиридинилметил)-2-нафтил]-1-пиперидинацетамида бис-гидрохлорид 20
Раствор гексафторфосфата 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (HBTU) (0,974 г, 2,57 ммоль), 4-(2,3-дигидро-2-оксо-1Н-бензимидазол-1-ил)-1-пиперидинуксусной кислоты (1,20 г, 2,57 ммоль) и N,N-диизопропилэтиламина (1,8 мл, 10,3 ммоль) в N,N-диметилформамиде (15 мл) перемешивали при комнатной температуре в течение 5 минут. К этой смеси добавляли бис-гидрохлорид 1,2,3,4-тетрагидро-6-метокси-1-(3-пиридинилметил)-2-нафталинамина 4 (0,80 г, 2,34 ммоль) и перемешивание продолжали в течение 18 ч. Раствор нагревали до 100°С в течение 1 часа. Полученный раствор охлаждали и выливали в насыщенный раствор водного бикарбоната натрия. Тонкодисперсный зеленый осадок отфильтровывали и твердое вещество очищали с помощью обращенно-фазовой C18-ВЭЖХ, элюируя градиентом вода/ацетонитрил/трифторуксусная кислота, 10/90/0,1→90/10/0,1. Цис-продукт 19 выделяли в виде бесцветного твердого вещества (0,386 г, 22%).
1Н ЯМР (ДМСО-d6): δ 1,76 (м, 4Н), 2,72-3,02 (м, 4Н), 3,16 (д, 2Н), 3,29-3,46 (м, 3Н), 3,54-3,75 (м, 2Н), наложенный на 3,72 (с, 3Н), 3,92-4,07 (м, 3Н), 4,53-4,65 (м, 1Н), 6,63 (д, 1Н), 6,70-6,77 (м, 2Н), 7,04 (шир. с, 3Н), 7,59 (шир. с, 1Н), 7,99 (т, 1Н), 8,37 (д, 1Н), 8,74 (м, 2Н), 8,96 (д, 1Н), 10,5-10,71 (шир. с, 1Н) и 11,03 (с, 1Н); МС m/e 512 (МН+).
Была получена смесь цис/транс-изомеров (~8/2) (0,490 г, 28%), а также очищенный транс-изомер 20 в виде бесцветного твердого вещества (0,136 г, 8%).
1Н-ЯМР (ДМСО-d6): δ 1,70 (м, 6Н), 2,63-3,81 (м, 9Н), наложенный на 3,72 (с, 3Н), 3,83-4,00 (м, 3Н), 4,47-4,60 (м, 1Н), 6,67-6,82 (м, 3Н), 7,02 (шир. с, 3Н), 7,21 (д, 1Н), 7,70 (т, 1Н), 8,14 (д, 1Н), 8,50-8,73 (м, 3Н), 9,70-10,10 (шир. с, 1Н) и 11,0 (с, 1Н); МС m/e 512 (МН+) (Фигура 6).
Пример 9
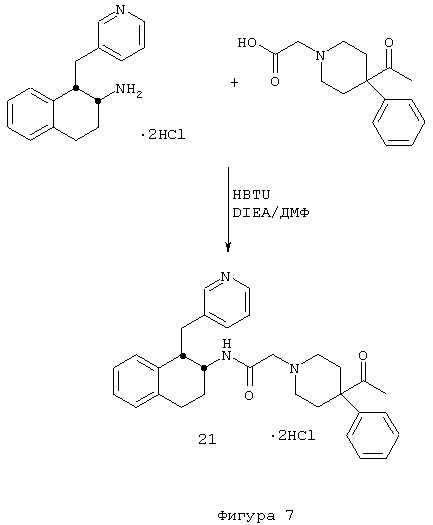
4-Ацетил-4-фенил-N-[цис-1,2,3,4-тетрагидро-1-(3-пиридинилметил)-2-нафтил]-1-пиперидинацетамида бис-гидрохлорид 21
1,2,3,4-Тетрагидро-1-(3-пиридинилметил)-2-нафталинамина бис-гидрохлорид 4 (0,75 г, 2,41 ммоль) подвергали реакции с 2-(4-ацетил-4-фенилпиперидин-1-ил)уксусной кислотой (0,86 г, 2,65 ммоль), N,N-диизопропилэтиламином (2,0 мл, 11,3 ммоль) и HBTU (1,01 г, 2,65 ммоль) в N,N-диметилформамиде (15 мл) при комнатной температуре в течение 2 часов, как описано выше в Примерах 7-8. После водной обработки продукт отфильтровывали. Этот продукт растворяли в изопропаноле (~30 мл) и обрабатывали насыщенным раствором хлористоводородной кислоты в изопропаноле (~5 мл). Растворитель выпаривали в вакууме и остаток растирали с диэтиловым эфиром, в результате чего получали бис-гидрохлорид 4-ацетил-4-фенил-N-[цис-1,2,3,4-тетрагидро-1-(3-пиридинилметил)-2-нафтил]-1-пиперидинацетамида 21 в виде аморфного светло-желтого твердого вещества (1,2 г, 90%); МС m/e 482 (МН+) (Фигура 7).
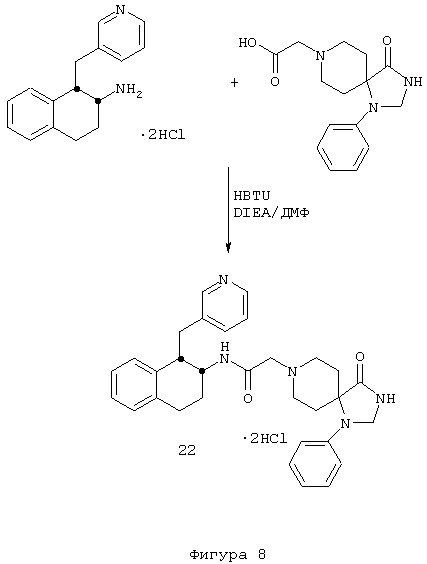
Пример 10
4-Оксо-1-фенил-N-[цис-1,2,3,4-тетрагидро-1-(3-пиридинилметил)-2-нафтил]-1,3,8-триазаспиро[4.5]декан-8-ацетамида бис-гидрохлорид 22
1,2,3,4-Тетрагидро-1-(3-пиридинилметил)-2-нафталинамина бис-гидрохлорид 4 (0,75 г, 2,41 ммоль) подвергали реакции с 2-(1-фенил-1,3,8-триазаспиро[4.5]декан-4-он)уксусной кислотой (1,12 г, 2,41 ммоль), N,N-диизопропилэтиламином (1,68 мл, 9,63 ммоль) и HBTU (0,91 г, 2,41 ммоль) в N,N-диметилформамиде (15 мл) при комнатной температуре в течение 4 часов, как описано выше в Примерах 7-8. После водной обработки продукт отфильтровывали. Этот продукт растворяли в метаноле (~30 мл) и обрабатывали концентрированной хлористоводородной кислотой (~5 мл). Растворитель выпаривали в вакууме и остаток растирали с диэтиловым эфиром, в результате чего получали бис-гидрохлорид 4-оксо-1-фенил-N-[цис-1,2,3,4-тетрагидро-1-(3-пиридинилметил)-2-нафтил]-1,3,8-триазаспиро[4.5]декан-8-ацетамида 22 в виде аморфного желтовато-коричневого твердого вещества (1 г, 81%).
1H NMR (ДМСО-d6) δ 1,93 (s, 4Н), 2,80-3,08 (м, 4Н), 3,18-3,30 (м, 2Н), 3,38-3,66 (м, 3Н), 3,70-3,89 (м, 2Н), 3,94-4,13 (м, 3Н), 4,65 (с, 2Н), 6,80 (т, 2Н), 7,00-7,29 (м, 8Н), 8,03 (т, 1Н), 8,44 (д, 1Н), 8,81 (шир. с, 2Н), 8,97 (д, 1Н), 9,16 (с, 1Н), 10,83 (шир. с, 1Н); МС m/e 510 (МН+) (Фигура 8).
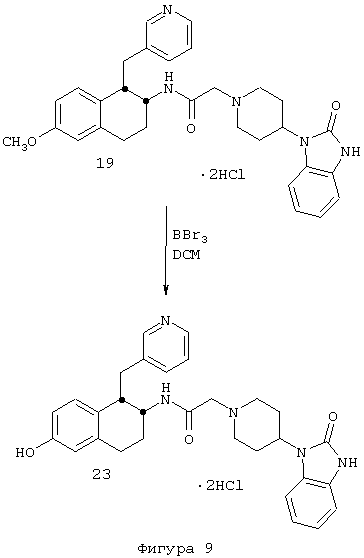
Пример 11
4-(2,3-Дигидро-2-оксо-1Н-бензимидазол-1-ил)-N-[цис-1,2,3,4-тетрагидро-6-гидрокси-1-(3-пиридинилметил)-2-нафтил]-1-пиперидинацетамида бис-гидрохлорид 23
Раствор 4-(2,3-дигидро-2-оксо-1Н-бензимидазол-1-ил)-N-[цис-1,2,3,4-тетрагидро-6-метокси-1-(3-пиридинилметил)-2-нафтил]-1-пиперидинацетамида 19 (0,28 г, 0,37 ммоль) в дихлорметане (2 мл) по каплям добавляли к раствору трибромида бора (1,8 ммоль) в дихлорметане (22 мл) при 0°С. После перемешивания полученного раствора при 0°С в течение 1,5 ч добавляли метанол (~2 мл) и перемешивание продолжали при 0°С еще в течение 0,5 ч. Растворитель выпаривали в вакууме и остаток очищали обращенно-фазовой C18-ВЭЖХ, элюируя градиентом вода/ацетонитрил/TFA, 90/10/0/1→10/90/0,1. Продукт растворяли в метаноле и обрабатывали этанольным раствором хлористоводородной кислоты. Растворитель выпаривали и этот процесс повторяли дважды, в результате чего получали бис-гидрохлоридную соль 4-(2,3-дигидро-2-оксо-1Н-бензимидазол-1-ил)-N-[цис-1,2,3,4-тетрагидро-6-гидрокси-1-(3-пиридинилметил)-2-нафтил]-1-пиперидинацетамида 23 (0,148 г, 68%) в виде бесцветного твердого вещества.
1H ЯМР (ДМСО-d6): δ 1,73-2,03 (м, 4Н), 2,70-2,94 (м, 4Н), 3,05-3,20 (шир. с, 2Н), 3,27-3,47 (м, 3Н), 3,55-3,76 (м, 2Н), 3,92-4,15 (м, 3Н), 4,54-4,67 (м, 1Н), 6,46 (д, 1Н), 6,58 (с, 2Н), 7,05 (м, с, 3Н), 7,60 (шир. с, 1Н), 7,94 (т, 1Н), 8,30 (д, 1Н), 8,72-8,83 (м, 2Н), 8,96 (м, 1Н), 9,30 (шир. с, 1Н), 10,64 (шир. с, 1Н) и 11,05 (с, 1Н); MS m/e 512 (МН+) (Фигура 9).
Примеры 12-13
транс-N-[2-(4-Фторфенил)-3-(3-пиридинил)пропил]-4-[((2-фторфенилсульфонил)амино)метил]-1-циклогексанамида гидрохлорид 26 и транс-N-[[[2-(4-фторфенил)-3-(3-пиридинил)пропил]амино]метил]-4-циклогексил]метил]-2-фторбензолсульфонамида бис-гидрохлорид 27
А. Металлический натрий (0,71 г, 30,9 ммоль) добавляли к метанолу (75 мл) и перемешивали при комнатной температуре до тех пор, пока твердое вещество не было израсходовано. Затем добавляли 4-фторфенилацетонитрил (3,5 мл, 29,3 ммоль) и смесь перемешивали при комнатной температуре в течение 10 минут. После этого добавляли 3-пиридинкарбоксальдегид (2,77 мл, 29,3 ммоль) и полученный раствор кипятили с обратным холодильником в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и нейтрализовали 2 н. хлористоводородной кислотой (16 мл, 32 ммоль). Растворитель выпаривали в вакууме и полученный остаток распределяли между водой (~200 мл) и дихлорметаном (~200 мл). Органический слой сушили над сульфатом натрия, фильтровали и растворитель выпаривали в вакууме с получением 2-(4-фторфенил)-3-пиридин-3-ил-ацетонитрила 24 в виде бесцветного твердого вещества (6,11 г, 93%).
1Н-ЯМР (CDCl3): δ 7,16 (т, 2Н), 7,42-7,47 (м, 1Н), 7,48 (с, 1Н), 7,66-7,70 (м, 2Н), 8,47 (д, 1Н), 8,65 (д, 1Н), 8,84 (с, 1Н); МС m/e 225 (МН+).
В. Суспензию 2-(4-фторфенил)-3-пиридин-3-акрилонитрила 24 (1,5 г, 6,68 ммоль) и окиси платины (IV) (0,51 г, 2,24 ммоль) в этаноле (60 мл) и воде (15 мл) подвергали реакции с газообразным водородом под давлением 65 фунт/кв.дюйм (450 кПа) в течение 6 ч. Катализатор удаляли фильтрацией и растворитель выпаривали в вакууме. Остаток растворяли в диэтиловом эфире (50 мл) и небольшое количество нерастворившегося продукта отфильтровывали. Эфирный раствор обрабатывали 1 н. хлористым водородом в диэтиловом эфире (20 мл). Осажденное желтое твердое вещество отфильтровывали и осторожно промывали диэтиловым эфиром с получением бис-гидрохлоридной соли β-(3-пиридинилметил)-4-фторфенетиламина 25 в виде светло-желтого твердого вещества (1,67 г, 82%).
1H-ЯМР (ДМСО-d6): δ 3,03-3,21 (м, 4Н), 3,44-3,53 (м, 1Н), 7,13 (т, 2Н), 7,27-7,33 (м, 2Н), 7,93 (т, 1Н), 8,27 (д, 1Н), 8,42 (шир. с, 3Н), 8,72-8,80 (м, 2Н); МС m/e 231 (МH+).
С. Раствор гексафторфосфата 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (HBTU) (1,03 г, 2,57 ммоль), транс-4-[(2-фторфенил)сульфониламинометил]циклогексанкарбоновой кислоты (1,20 г, 2,57 ммоль) и N,N-диизопропилэтиламина (1,9 мл, 11,1 ммоль) в N,N-диметилформамиде (15 мл) перемешивали при комнатной температуре в течение 10 минут. Затем добавляли дигидрохлорид 2-(4-фторфенил)-3-пиридин-3-ил-пропиламина 25 (0,75 г, 2,47 ммоль) и полученный раствор перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь выливали в воду (~100 мл) и продукт экстрагировали дихлорметаном (~100 мл). Органический слой промывали водой (3×100 мл), концентрировали и полученный остаток очищали флэш-хроматографией с использованием в качестве элюента метанола (5-10%) и триэтиламина (0,5%) в дихлорметане, в результате чего получали нужный циклогексанамид в виде масла. Этот продукт растворяли в диэтиловом эфире (~50 мл) и обрабатывали 1 н. хлористым водородом в диэтиловом эфире. Образованное бесцветное твердое вещество отфильтровывали, промывали эфиром и сушили в вакууме, в результате чего получали гидрохлорид N-[2-(4-фторфенил)-3-(3-пиридинил)пропил]-4-[((2-фторфенилсульфонил)амино)метил]-1-циклогексанамида 26 в виде бесцветного твердого вещества.
1Н-ЯМР (ДМСО-d6):δ 0,69-0,83 (м, 2Н), 1,07-1,19 (м, 3Н), 1,52-1,71 (м, 4Н), 1,94 (т, 1Н), 2,66 (шир. с, 2Н), 2,99-3,10 (м, 1Н), 3,17-3,43 (м, 4Н), 7,07 (т, 2Н), 7,16-7,21 (м, 2Н), 7,35-7,47 (м, 2Н), 7,66-7,95 (м, 5Н), 8,28 (д, 1Н) и 8,74 (шир. с, 2Н); MS m/e 528 (МН+) (Фигура 10).
D. Гидрохлорид N-[2-(4-фторфенил)-3-(3-пиридинил)пропил]-4-[((2-фторфенилсульфонил)амино)метил]-1-циклогексанамида 26 распределяли между насыщенным раствором водного бикарбоната натрия и дихлорметаном. Органический слой сушили над сульфатом натрия и растворитель выпаривали в вакууме с получением свободного основания в виде масла. Полученное масло (0,5 г, 0,944 ммоль) растворяли в тетрагидрофуране (~20 мл) и полученный раствор по каплям добавляли к раствору борана (4,0 ммоль) в тетрагидрофуране (14 мл) при комнатной температуре. Полученный раствор кипятили с обратным холодильником в течение 2 ч. Полученную смесь охлаждали до комнатной температуры и добавляли несколько капель воды до тех пор, пока не был израсходован весь непрореагировавший боран. Затем добавляли 4 н. раствор хлористоводородной кислоты (2 мл) и раствор кипятили с обратным холодильником в течение 45 мин. После охлаждения раствора добавляли 3 н. водный гидроксид натрия (2,7 мл) и смесь концентрировали в вакууме.
Остаток распределяли между водой (~50 мл) и дихлорметаном (~50 мл). Органический слой сушили над сульфатом натрия и растворитель выпаривали в вакууме. Остаток растворяли в диэтиловом эфире (~20 мл) и обрабатывали 1 н. хлористым водородом в диэтиловом эфире (~4 мл). Бесцветный осадок отфильтровывали, осторожно промывали диэтиловым эфиром и сушили в вакууме, в результате чего получали бис-гидрохлорид транс-N-[[[2-(4-фторфенил)-3-(3-пиридинил)пропил]амино]метил]-4-циклогексил]метил]-2-фторбензолсульфонамида 27 (0,371 г, 67%).
1H-ЯМР (ДМСО-d6):δ 0,70-0,87 (м, 4Н), 1,22-1,36 (шир. с, 1Н), 1,64-1,88 (м, 6Н), 2,65-2,77 (м, 3Н), 2,99-3,33 (м, 3Н), 3,54-3,70 (м, 2Н), 7,13 (т, 2Н), 7,24-7,34 (м, 2Н), 7,37-7,48 (м, 2Н), 7,67-7,87 (м, 3Н), 7,96 (т, 1Н), 8,17 (д, 1Н), 8,68 (с, 1Н), 8,70 (с, 1Н), 9,03 (шир. с, 1Н) и 9,24 (шир. с, 1Н); МС m/e 514 (МН+) (Фигура 10).
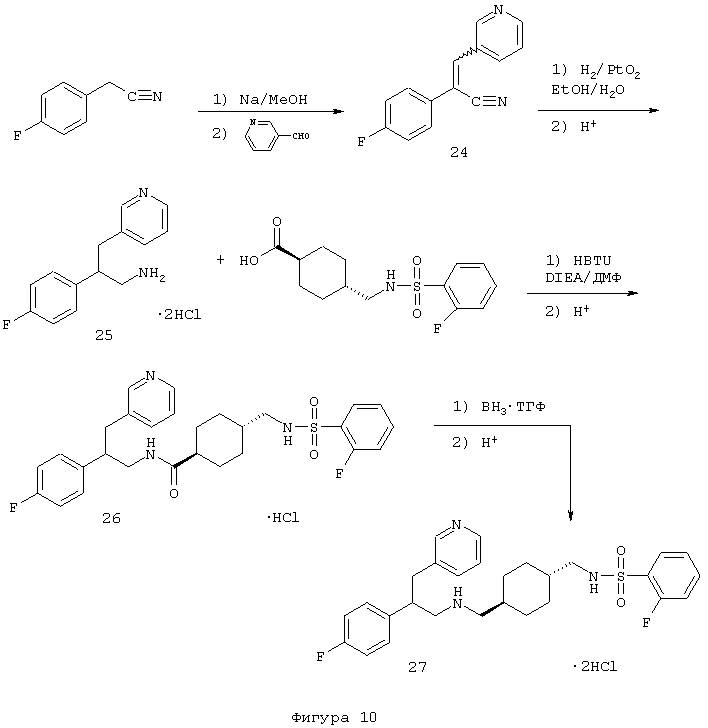
Пример 14
N-[2-(4-Фторфенил)-3-(3-пиридинил)пропил]-4-[(2-фторфенилсульфонил)амино]-1-пиперидинацетамида бис-трифторацетат 30
А. Раствор [4-(1,1-диметилэтокси)карбониламинопиперидин-1-ил]уксусной кислоты (0,5 г, 1,94 ммоль), гексафторфосфата 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (0,73 г, 1,94 ммоль) и N,N-диизопропилэтиламина (1,5 мл, 8,71 ммоль) в N,N-диметилформамиде (15 мл) перемешивали при комнатной температуре в течение 5 минут. Затем добавляли дигидрохлорид β-(3-пиридинилметил)-4-фторфенетиламина 25 (0,586 г, 1,94 ммоль) и полученный раствор перемешивали при комнатной температуре в течение 24 ч. Раствор выливали в насыщенный раствор водного бикарбоната натрия (~100 мл) и продукт экстрагировали дихлорметаном (~100 мл). Органический слой промывали водой (5×~100 мл) и сушили над сульфатом натрия. Растворитель выпаривали в вакууме с получением пиперидинацетамида 29 в виде масла, 0,52 г (57%).
1Н-ЯМР (CDCl3) δ 0,98-1,25 (м, 2Н), 1,45 (с, 9Н), 1,71-1,79 (м, 2Н), 2,05-2,17 (м, 2Н), 2,41-2,50 (м, 2Н), 2,75-3,00 (м, 3Н), 3,04-3,17 (м, 1Н), 3,33-3,47 (м, 2Н), 3,72-3,83 (м, 1Н), 4,36 (шир. с, 1Н), 6,93-7,14 (м, 7Н), 7,25 (м, 1Н), 8,24 (с, 1Н), 8,39 (д, 1Н); МС m/e 471 (МН+).
В. Раствор пиперидинацетамида 28 (0,46 г, 0,977 ммоль) в дихлорметане (6 мл) обрабатывали трифторуксусной кислотой (2 мл) и перемешивали при комнатной температуре в течение 3 ч. Растворитель выпаривали в вакууме. Остаток растворяли в 1,2-дихлорэтане (~10 мл) и растворитель выпаривали в вакууме (эту процедуру повторяли дважды для удаления трифторуксусной кислоты) и получали 4-амино-1-пиперидинацетамид 29 в виде соли трис-трифторуксусной кислоты, выделенной в виде стеклообразного вещества янтарного цвета, 0,66 г (95%).
1H-ЯМР (ДМСО-d6); МС m/e 371 (МН+).
С. 2-Фторбензолсульфонилхлорид (25 мг, 0,126 ммоль) добавляли к раствору 4-амино-1-пиперидинацетамида 29 (82 мг, 0,115 ммоль) и N,N-диизопропилэтиламина (0,10 мл, 0,575 ммоль) в ацетонитриле (1 мл) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 16 ч, а затем добавляли воду (0,30 мл) и раствор наносили на обращенно-фазовую C18-колонку для ВЭЖХ-очистки. Колонку элюировали градиентом вода/ацетонитрил/трифторуксусная кислота с получением N-[2-(4-фторфенил)-3-(3-пиридинил)пропил]-4-[(2-фторфенилсульфонил)амино]-1-пиперидинацетамида бис-трифторацетата 30 в виде бесцветного твердого вещества, 28 мг (32%).
1H-ЯМР (ДМСО-d6): δ 1,70-1,85 (м, 4Н), 2,91-3,47 (м, 10Н), 3,66-3,80 (м, 2Н), 7,07 (т, 2Н), 7,18 (м, 2Н), 7,38-7,50 (м, 2Н), 7,64 (т, 1Н), 7,71-7,85 (м, 2Н), 7,92 (д, 1Н), 8,31 (д, 1Н), 8,49 (с, 1Н), 8,57 (с, 1Н), 8,60 (с, 1Н); МС m/e 529 (МН+) (Фигура 11).
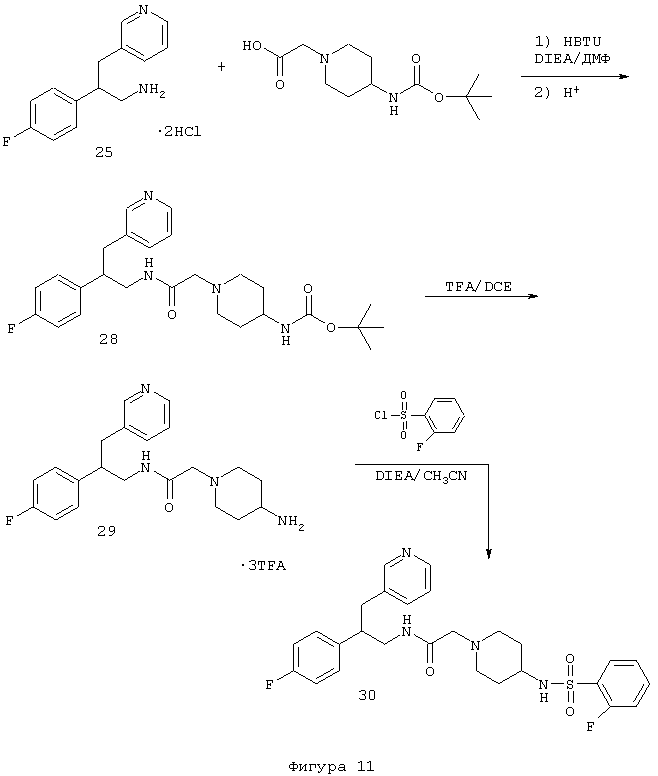
Другие соединения настоящего изобретения, которые были получены с использованием вышеописанных экспериментальных схем, приводятся в таблице 1.
Масс-спектроскопические данные соединений
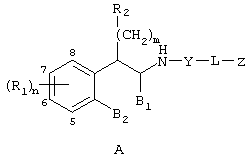
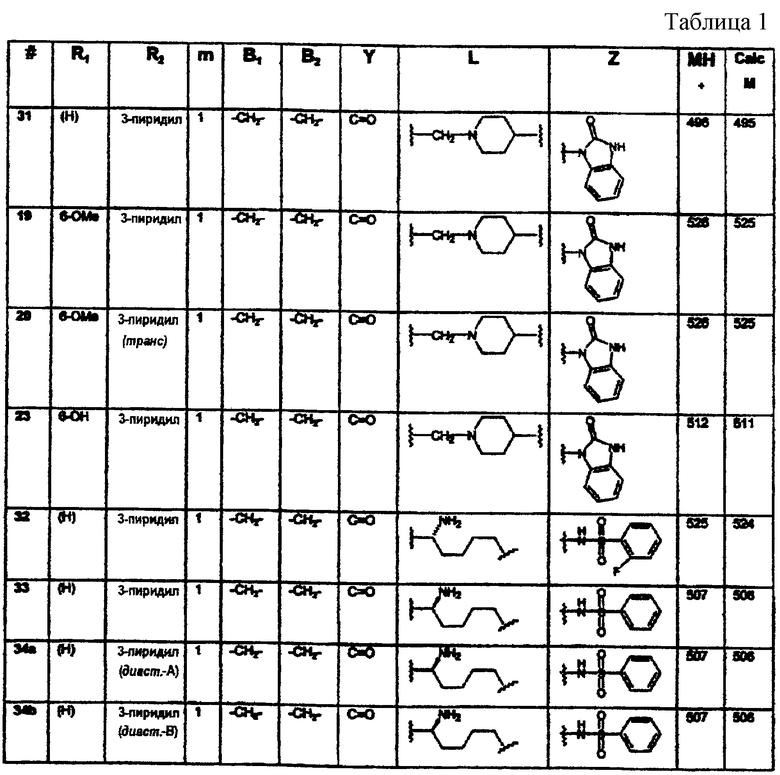
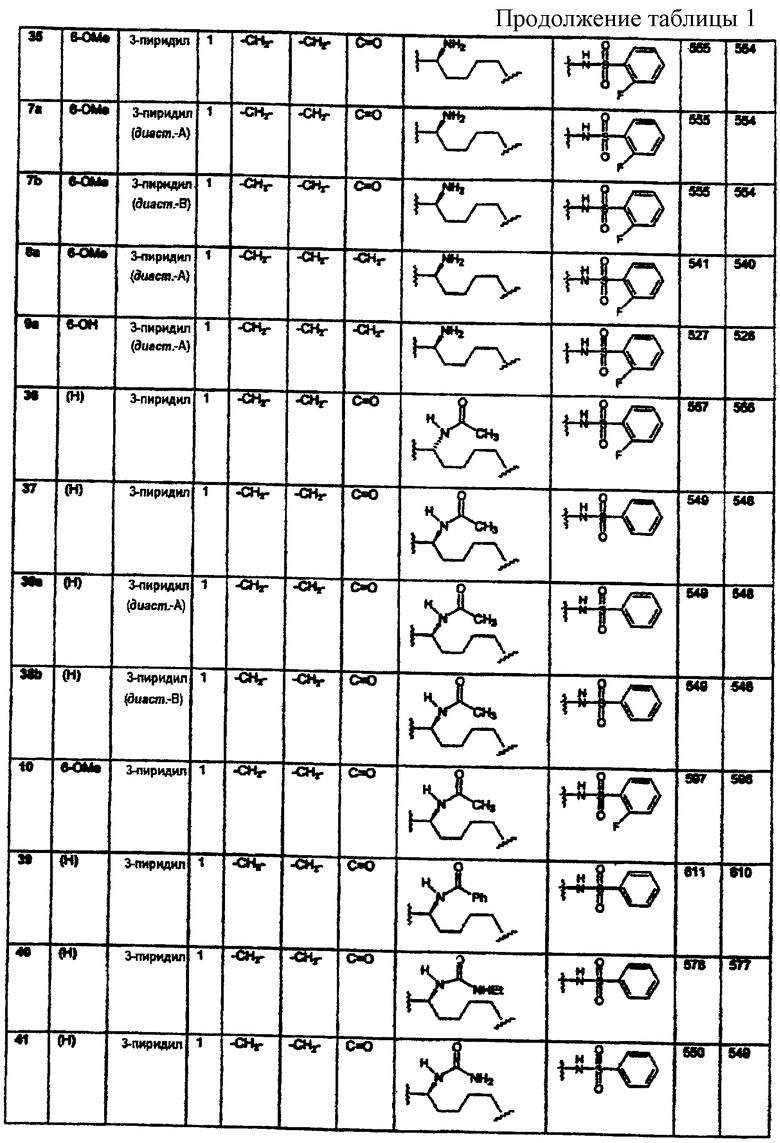
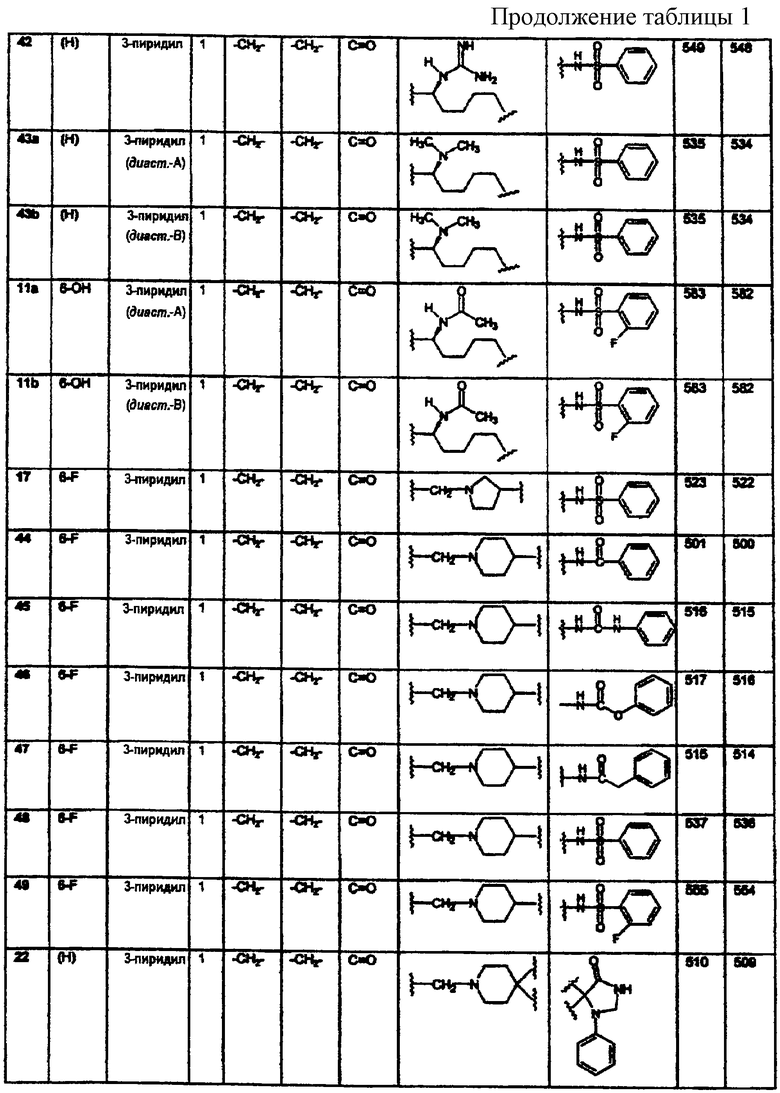
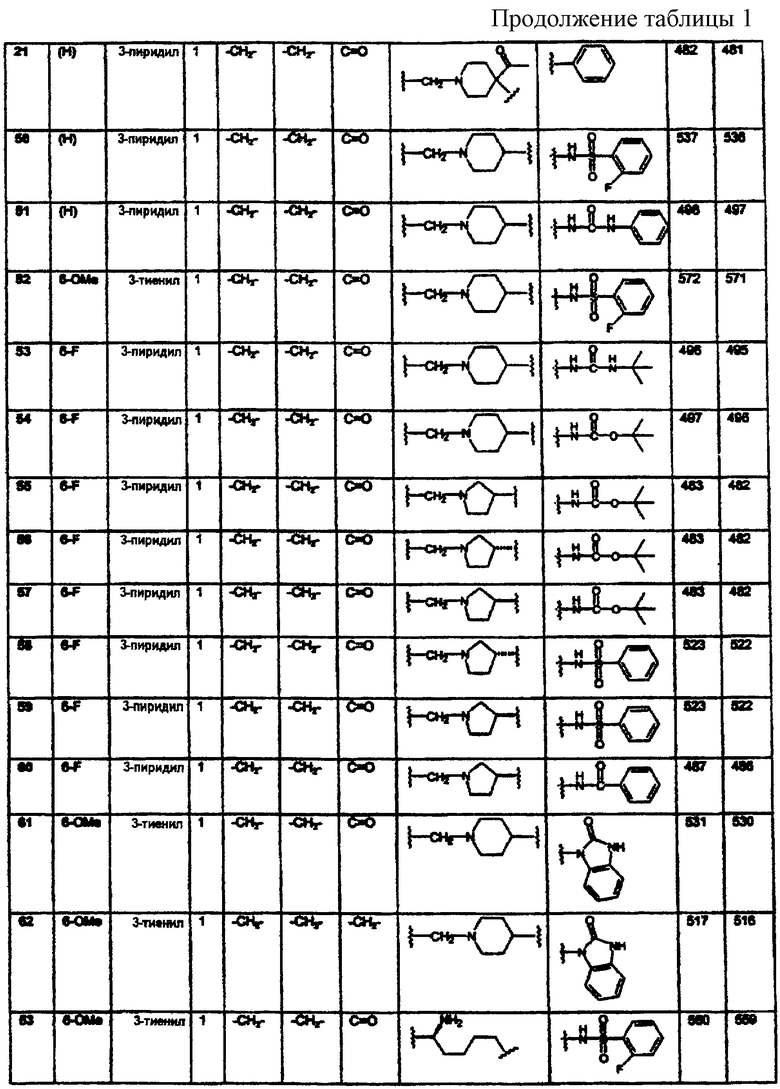
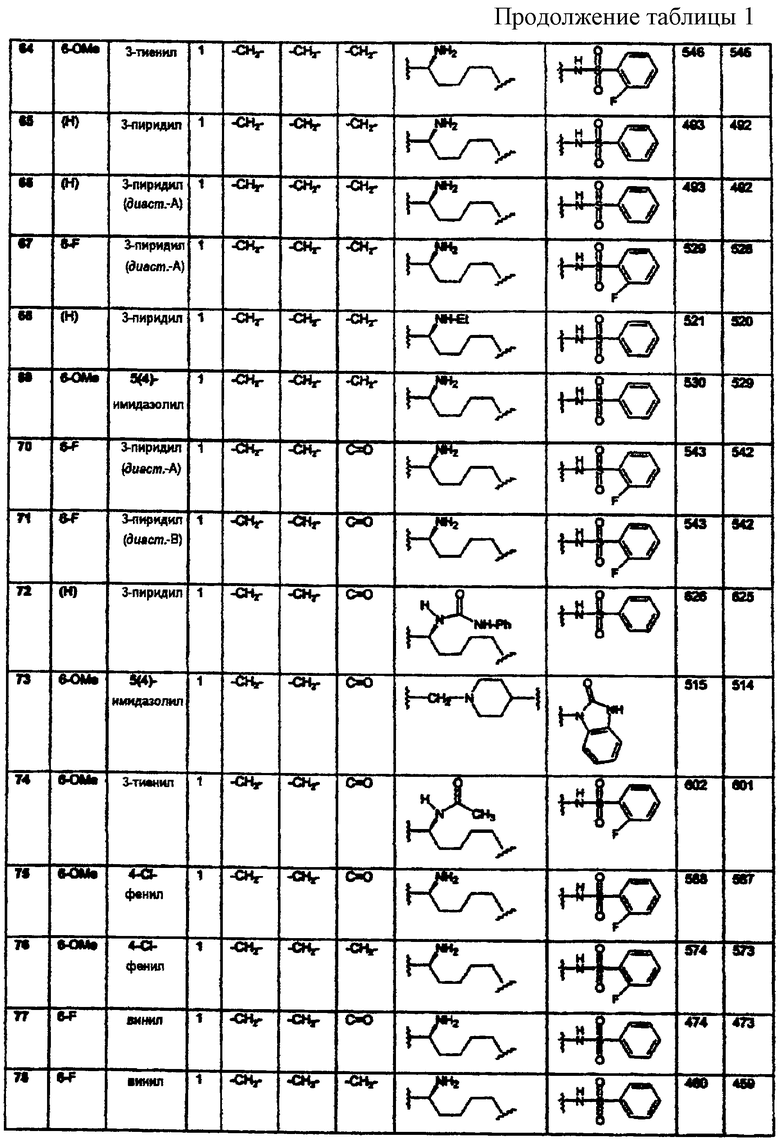
Анализы in vitro
Анализ путем центрифугирования NPY5 HTS
Описанные соединения настоящего изобретения были оценены на связывание с рецептором Y5 нейропептида человека.
Стабильная трансфекция
кДНК рецептора NPY5 человека (Genbank, номер доступа U66275) встраивали в вектор pCLneo (Invitrogen) и трансфецировали в эмбриональные клетки почек человека (НЕК-293) кальцийфосфатным методом (Cullen 1987). Стабильно трансфецированные клетки отбирали с использованием G-418 (600 мкг/мл). Эти стабильно трансфецированные клетки служили источником мембран для анализа на связывание с рецептором NPY5.
Мембранный препарат
NPY5-трансфецированные клетки НЕК-293 культивировали до конфлюэнтности в 150 см2 чашках для культивирования. Клетки промывали один раз забуференным фосфатом физиологическим раствором (Gibco Cat# 14040-133). Затем клетки инкубировали в забуференном фосфатом физиологическом растворе, который не содержал кальция и магния и в который было добавлено 2 мМ EDTA. Клетки инкубировали в течение 10 минут при комнатной температуре и эти клетки собирали путем повторного пипетирования. Клетки собирали в виде осадка, а затем замораживали при -80°С вплоть до их использования. Замороженный осадок гомогенизировали с использованием политрона на полной скорости в течение 12 секунд в буфере для гомогенизации (20 мМ Трис-HCl, 5 мМ EDTA, рН 7,4). Гомогенаты центрифугировали в течение 5 минут при 4°С и 200g. Супернатанты переносили в пробирки из корекса и центрифурировали в течение 25 минут при 28000g. Осадки ресуспендировали в буфере для связывания (20 мМ HEPES, 10 мМ NaCl, 0,22 мМ КН2РO4, 1,3 мМ CaCl2, 0,8 мМ MgSO4, pH 7,4). Мембраны выдерживали на льду вплоть до их использования.
Был проведен анализ на конкурентное связывание, известный специалистам, в котором соединения формулы А конкурируют с 125I-PYY за связывание с клеточными мембранами. Проще говоря, чем меньше 125I-PYY связывается с мембранами, тем лучшим ингибитором (конкурентом) является данное соединение. Связывание 125I-PYY определяли путем центрифугирования мембран, отсасывания супернатантов, отмывки остаточного 125I-PYY и последующего подсчета образца для связывания в гамма-счетчике.
Процедура проведения анализа на связывание с радиолигандом
Соединения, предназначенные для тестирования, получали в виде 10× маточных растворов в буфере для связывания и добавляли в первую аналитическую пробирку (РИА-сосуды, Sarstedt). 20 мкл каждого маточного 10× раствора соединения пипетировали в сосуды и 80 мкл 125I-PYY (NEN, каталожный номер NEX240), который был разведен до концентрации 200 пМ в 0,25% BSA в буфере для связывания, добавляли в указанные сосуды с соединениями (конечная концентрация 125I-PYY составляла 80 пМ). В каждую пробирку добавляли 100 мкл мембран и смесь интенсивно перемешивали путем 2-кратного пипетирования. Образцы инкубировали в течение 1 часа при комнатной температуре. Затем алюминиевые формы (Sarstedt), содержащие указанные сосуды, центрифугировали в течение 10 минут при 3200 об/мин в центрифуге Sorvall RT6000. После этого супернатанты отсасывали. В каждый сосуд добавляли 400 мкл PBS и супернатанты снова отсасывали. Затем сосуды помещали в полиэтиленовую направляющую трубку 12×75 и подсчитывали на гамма-счетчике (Packard). Неспецифическое связывание определяли в присутствии 300 нМ NPY. Процент ингибирования связывания 125I-PYY вычисляли путем вычитания значения для неспецифического связывания из величины, полученной для тестируемых образцов (соединение (I)), и полученные величины делили на величину общего связывания и умножали на 100. Значения ингибирующей концентрации (IC50) для соединений, обнаруживающих заметное ингибирование связывания 125I-PYY, вычисляли путем определения процента ингибирования величин связывания 125I-PYY при различных концентрациях тестируемого соединения с использованием графической программы, такой как GraphPad Prism (San Diego, CA), для вычисления концентрации тестируемого соединения, которое на 50% ингибирует связывание 125I-PYY (см. таблицу 2). Эти операции хорошо известны специалистам.
Аффиности связывания соединений А с рецептором Y5 NPY человека (выражено как % ингибирования связывания 125I-PYY)
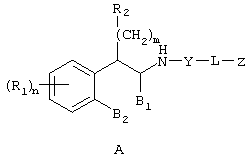
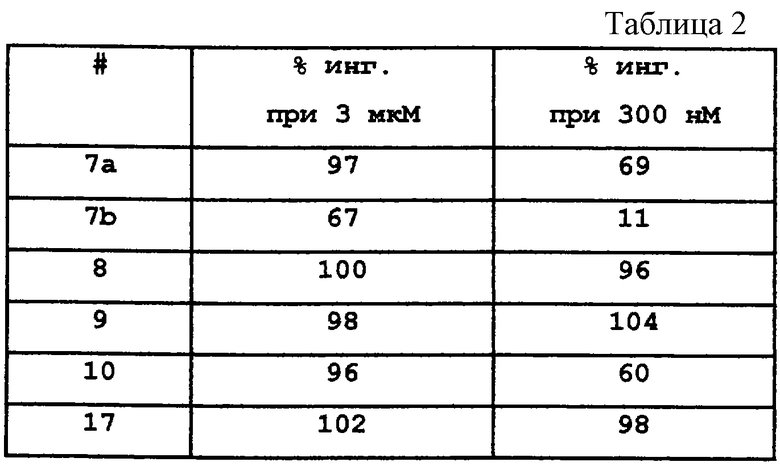
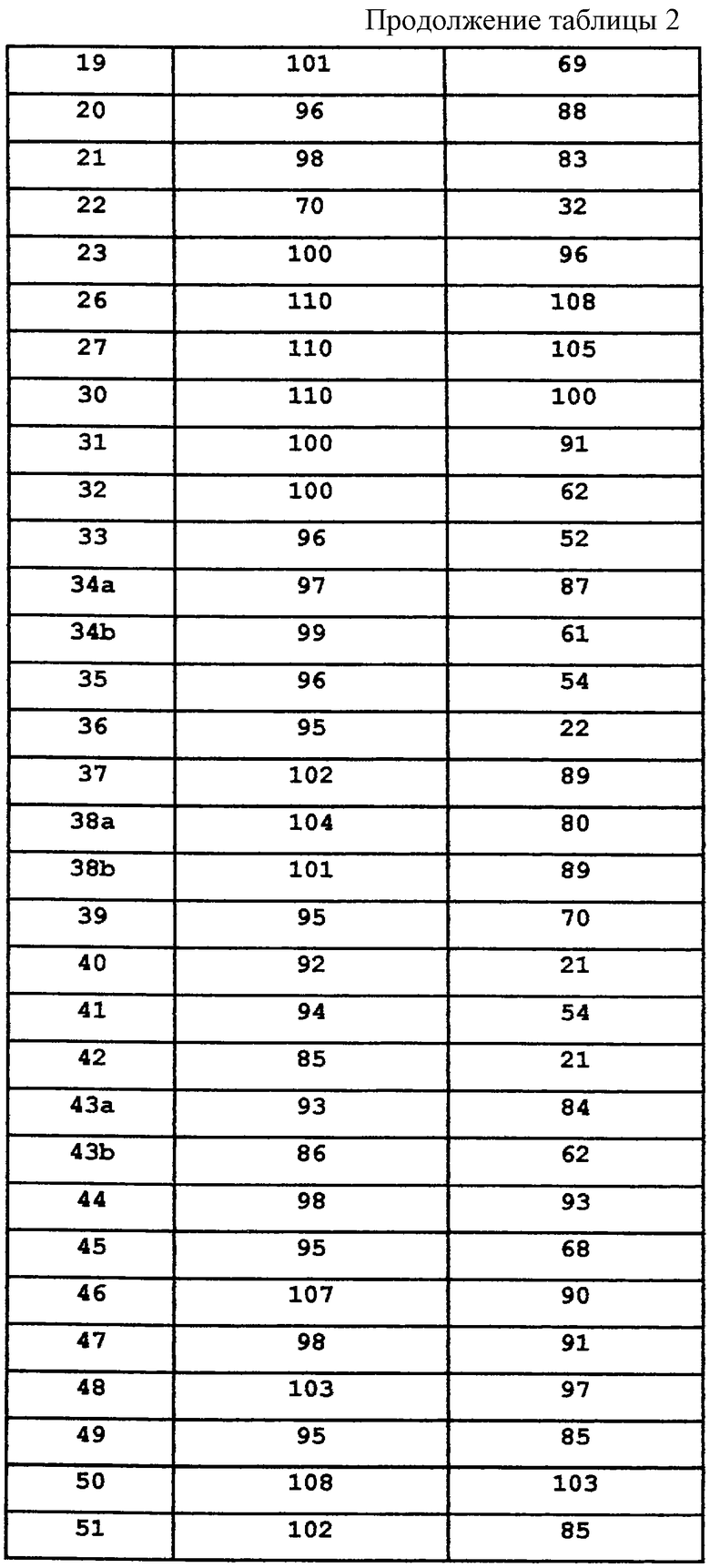
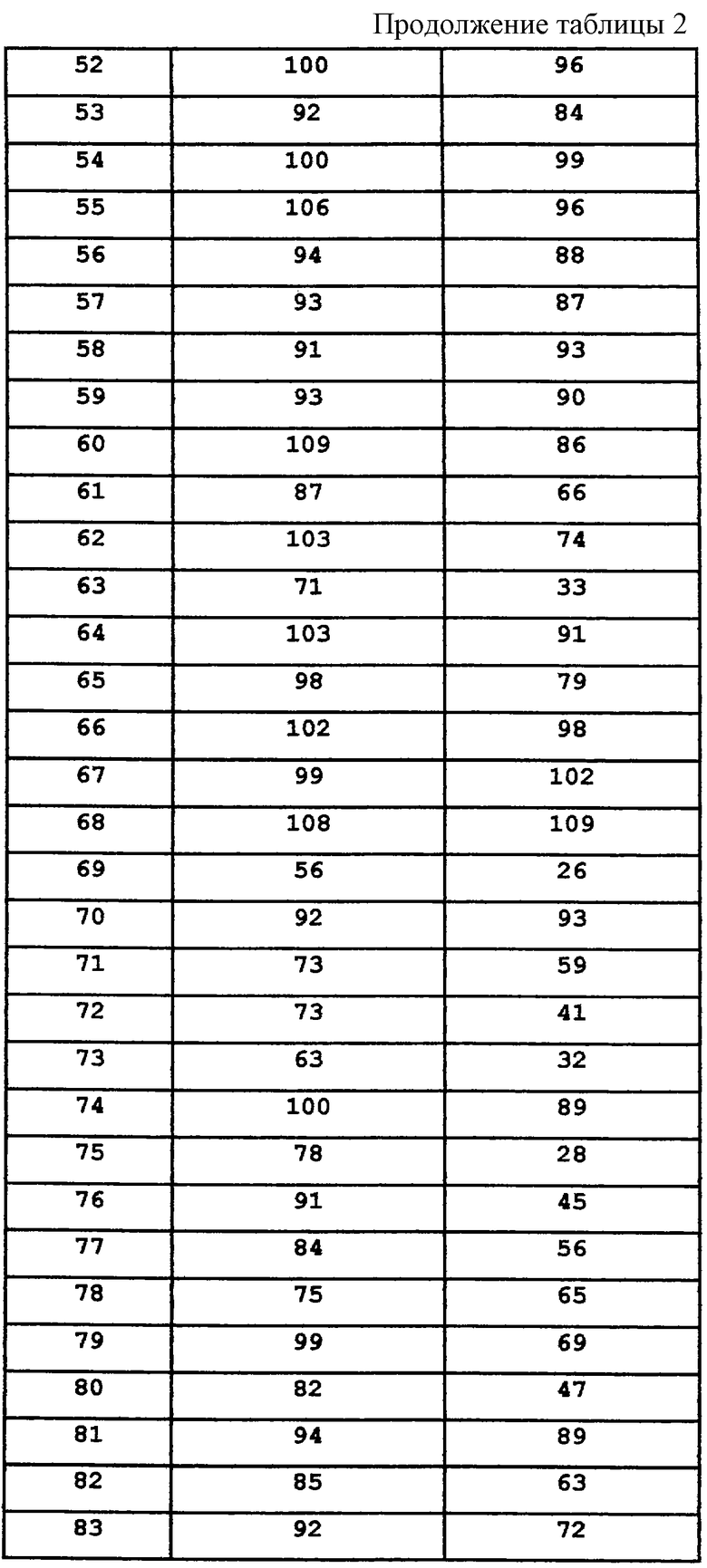
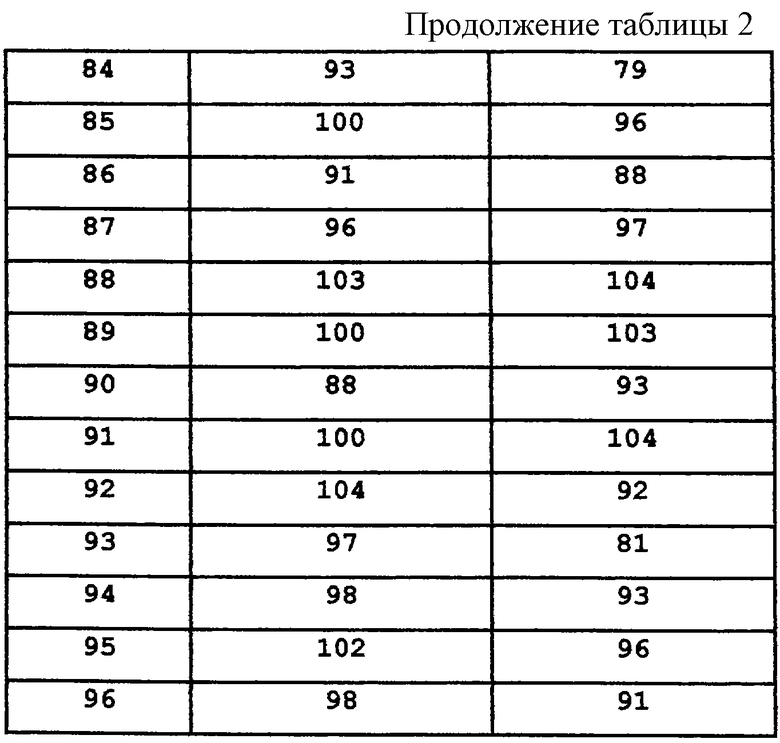
Анализы in vivo
Модель питания с использованием грызунов: измерение потребления пищи крысами в условиях голодания
Самцов крыс Long-Evans (180-200 граммов) помещали в отдельные клетки и, после привыкания животных к питанию порошкообразным кормом (сертифицированный корм для грызунов PMI #5002) в течение определенного промежутка времени, их поддерживали в режиме кормления 1 раз в день (то есть в период с 10 a.m. до 4 p.m.) в течение пяти дней. Корм помещали в открытый сосуд, прикрепленный к клетке проволокой, причем, для минимизации потери корма, этот корм покрывали подвижной металлической пластиной. Воду давали по желанию.
За 18 часов до испытаний животным не давали корма. По окончании периода голодной выдержки, животным вводили либо соединения настоящего изобретения, либо наполнитель. Наполнитель и тестируемые соединения вводили либо перорально (5 мл/кг) за 60 минут до начала эксперимента, либо подкожно (1 мл/кг) или внутрибрюшинно (1 мл/кг) за 30 минут до эксперимента. Соединения настоящего изобретения вводили перорально в виде суспензии в водной смеси 0,5% метилцеллюлозы - 0,4% Твин 80 или внутрибрюшинно в виде раствора или суспензии в ПЭГ 200; при этом концентрации соединений обычно составляли от 1 до 100 мг/кг, а предпочтительно от 10 до 30 мг/кг. Потребление пищи измеряли через 2, 4 и 6 часов после введения путем взвешивания специального сосуда, содержащего пищу, до начала эксперимента и через определенные промежутки времени. После завершения эксперимента, перед повторным тестированием, всем животным осуществляли промывку ЖКТ в течение недели.
Процент снижения потребления пищи вычисляли путем вычитания количества (в граммах) пищи, поглощенной животными группы обработки, из количества (в граммах) пищи, поглощенной животными контрольной группы, деления полученного значения на количество (в граммах) пищи, поглощенной животными контрольной группы, и умножения на 100.
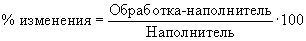
Отрицательное значение указывает на снижение потребления пищи, а положительное значение указывает на увеличение потребления пищи (см. таблицу 3).
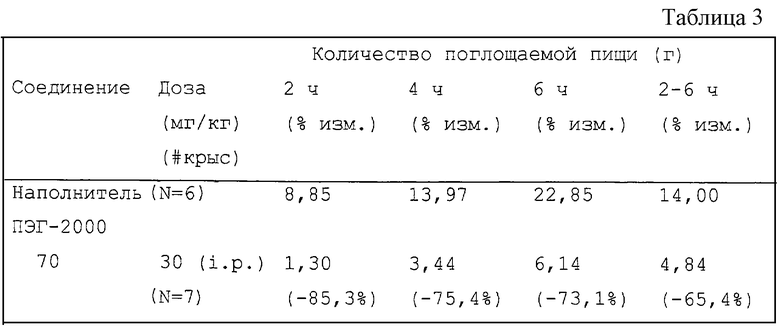
| название | год | авторы | номер документа |
|---|---|---|---|
| N-ЗАМЕЩЕННЫЕ АМИНОТЕТРАЛИНЫ КАК ЛИГАНДЫ ДЛЯ РЕЦЕПТОРА Y5 НЕЙРОПЕПТИДА Y, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ ОЖИРЕНИЯ И ДРУГИХ РАССТРОЙСТВ | 1999 |
|
RU2219167C2 |
| АЛКИЛЗАМЕЩЕННЫЕ ЦИКЛИЧЕСКИЕ АМИНЫ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ЛИГАНДОВ D3-ДОПАМИНА | 1997 |
|
RU2185372C2 |
| СОЕДИНЕНИЯ | 2003 |
|
RU2327690C2 |
| ПРОИЗВОДНЫЕ 1,2,3,4-ТЕТРАГИДРО-2-НАФТИЛАМИНА | 1990 |
|
RU2086535C1 |
| НОВЫЕ СОЛИ МОРФОЛИНОБЕНЗАМИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 2000 |
|
RU2237668C2 |
| АРИЛПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1997 |
|
RU2189976C2 |
| СОЕДИНЕНИЯ ТИЕНОПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ | 2004 |
|
RU2331648C2 |
| ПРОИЗВОДНЫЕ ГУАНИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРА НЕЙРОПЕПТИДА FF | 2004 |
|
RU2337911C2 |
| ЗАМЕЩЕННЫЕ 4-ИМИДАЗОЛЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2007 |
|
RU2456281C2 |
| СПОСОБ СИНТЕЗА ПРОИЗВОДНЫХ АМИНО-МЕТИЛТЕТРАЛИНА | 2009 |
|
RU2512285C2 |
Изобретение относится к новым производным аминов или амидов общей формулы А
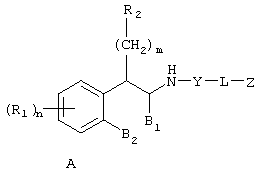
где R1 – водород, гидрокси, галоген, С1-8алкил, С1-8алкокси, трифторалкил, С1-8алкилтио и другие; n равно 1-2; В1 и В2 - водород либо В1 и В2 представляют метилен и, взятые вместе, они образуют шестичленное кольцо, или один из В1 и В2 представляет водород, тогда как второй представляет метилен и, взятые вместе, они образуют пятичленное кольцо; m равно 0-3; R2 – водород, гидрокси, С1-6алкил, С2-6алкенил, галоген, С3-7циклоалкил, фенил и другие; L – С1-8алкилен, С2-10алкенилен, С2-10алкинилен, С3-7циклоалкилен и другие; Y - метилен или карбонил; Z – арил, N-сульфонамидо, ариламидо, арилуреидо и другие; R3 -С1-6алкил, циклоалкил, нафтил, гетероарил и другие; R4 – водород, С1-8алкил, С1-8алкокси, гидрокси, галоген, циано и другие; R5 – водород, С1-8алкил, С1-8алкилкарбонил, ароил, карбамоил и другие; R6 - водород или С1-8алкил; р и q равны 1-3. Соединения формулы А представляют собой лиганды для рецептора Y5 нейропептида NPY и могут быть использованы в медицине для лечения расстройств центральной нервной системы. 3 с. и 12 з.п. ф-лы, 3 табл.
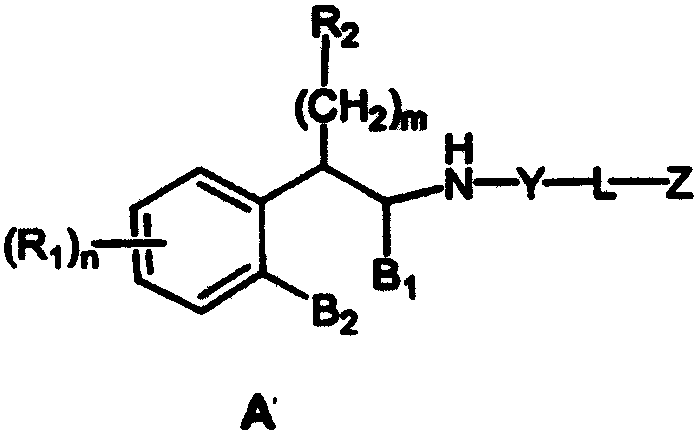
где R1 независимо выбран из группы, состоящей из водорода; гидрокси; галогена; С1-8алкила; замещенного С1-8алкила; С1-8алкокси; замещенного С1-8алкокси; трифторалкила; С1-8алкилтио; замещенного С1-8алкилтио; С3-6циклоалкила; С3-8циклоалкокси; нитро; амино; С1-6алкиламино; С1-8диалкиламино; С4-8циклоалкиламино; циано; карбокси; С1-5алкоксикарбонила; С1-5алкил-карбонилокси; формила; карбамоила; фенила и замещенного фенила;
n = 1-2;
В1 - водород;
В2 - водород; либо В1 и В2 представляют метилен и, взятые вместе, образуют шестичленное кольцо, или один из В1 и В2 представляет водород, тогда как второй представляет метилен, и, взятые вместе, они образуют пятичленное кольцо;
m = 0-3;
R2 независимо выбран из группы, состоящей из водорода; гидрокси; С1-6алкила; С2-6алкенила; галогена; С3-7циклоалкила; фенила; замещенного фенила; нафтила; замещенного нафтила; фенокси; замещенного фенокси; гетероарила; замещенного гетероариала; и гетероциклоалкила;
L выбран из группы, состоящей из С1-8алкилена; С2-10алкенилена; С2-10алкинилена; С3-7циклоалкилена; С3-7циклоалкилС1-4алкилена; арилС1-4алкилена; α-аминоС4-7алкилена;
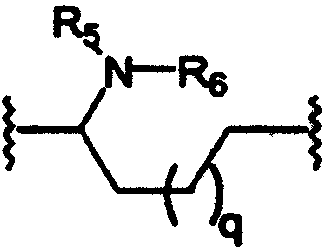
(N-метилен)пиперидин-4-ила;
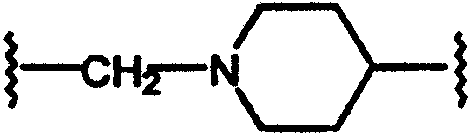
(N-метилен)пиперазин-4-ила;
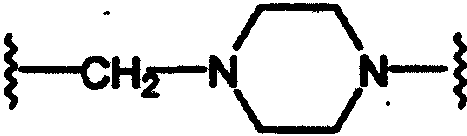
(N-метилен)пирролидин-3-ила;
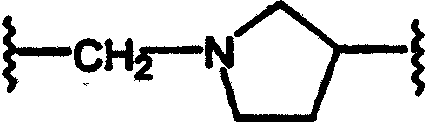
(N-метилен)-4-ацетилпиперидин-4-ила;
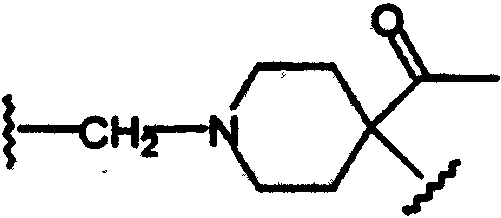
и (N-метилен)пиперидин-4,4-диила;
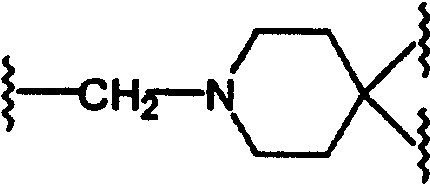
Y представляет метилен или карбонил;
Z выбран из группы, состоящей из арила;
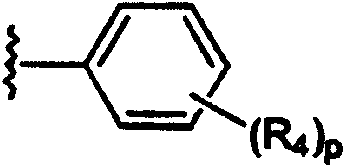
N-сульфонамидо;
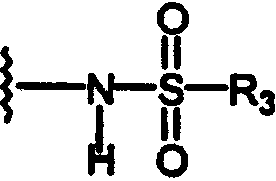
N-(арил)сульфонамидо;
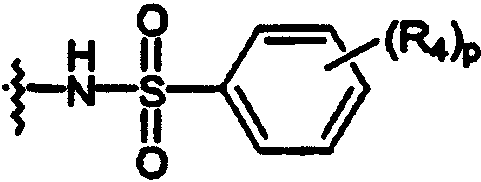
ариламидо;
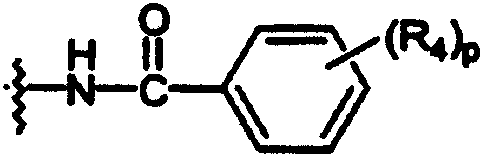
арилуреидо;
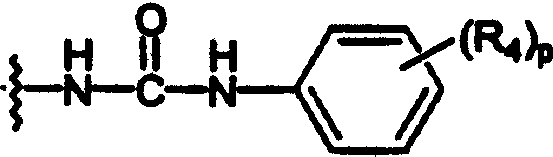
арилацетамидо;
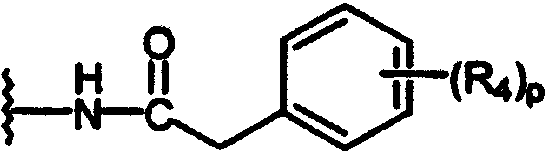
(арилокси)карбониламино;
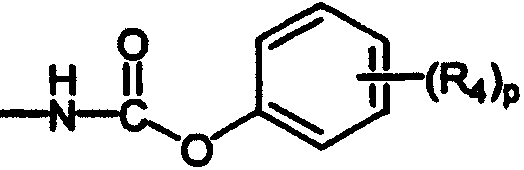
2,3-дигидро-2-оксо-1Н-бензимидазол-1-ила;
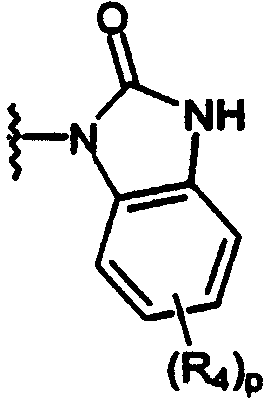
и 1-арил-2,3-дигидро-4-оксо-имидазол-5,5-диила;
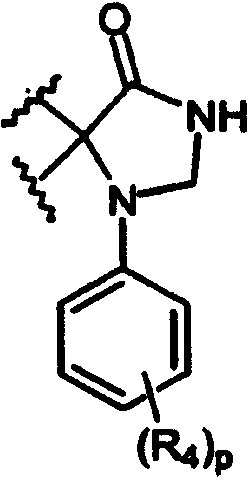
R3 независимо выбран из группы, состоящей из С1-6алкила; замещенного С1-8алкила; циклоалкила; замещенного циклоалкила; нафтила; замещенного нафтила; гетероарила и замещенного гетероарила;
R4 независимо выбран из группы, состоящей из водорода; С1-8алкила; С1-8алкокси; замещенного С1-8алкокси; гидрокси; галогена; циано; нитро; амино; С1-8алкиламино и С1-8диалкиламино;
R5 независимо выбран из группы, состоящей из водорода; С1-8алкила; С1-8алкилкарбонила; ароила; карбамоила; амидино; С1-8алкиламинокарбонила; (ариламино)карбонила и арилС1-8алкилкарбонила;
R6 независимо выбран из водорода и С1-8алкила;
р = 1-3;
q = 1-3;
и его энантиомеры, диастереомеры и фармацевтически приемлемые соли при условии, что если L представляет С1-6алкилен; С2-10алкенилен; С2-10алкинилен; С3-7циклоалкилен; С3-7циклоалкилС1-4алкилен; арилС1-4алкилен или α-аминоС4-7алкилен, то Z представляет фенил, N-сульфонамидо или N-(арил)сульфонамидо; если L представляет (N-метилен)пиперазин-4-ил, то Z представляет фенил или нафтил; если L представляет (N-метилен)пирролидин-3-ил или (N-метилен)пиперидин-4-ил, то Z представляет N-сульфонамидо, N-(арил)сульфонамидо, 2,3-дигидро-2-оксо-1Н-бензимидазол-1-ил; бензамидо, фенилуреидо, фенилацетамидо или (фенокси)-карбониламино; если L представляет (N-метилен)-4-ацетилпиперидин-4-ил; то Z представляет фенил или нафтил, а Y представляет карбонил; если L представляет (N-метилен)пиперидин-4,4-диил; то Z представляет 1-арил-2,3-дигидро-4-оксоимидазол-5,5-диил, а Y представляет карбонил; и если В1 и В2 оба представляют метилен, образуя, таким образом шестичленное кольцо, и если L представляет С1-10алкилен, С2-10алкенилен, С2-10алкинилен или арилС1-4алкилен, то Z не является N-сульфонамидо, N-(арил)сульфонамидо или фенилом.
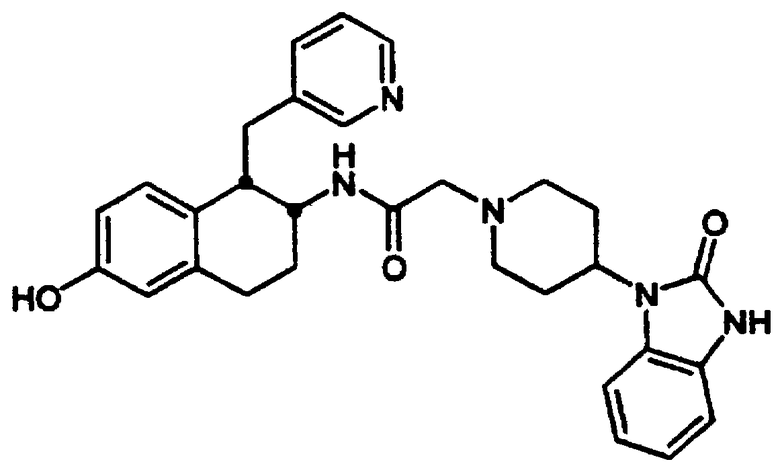
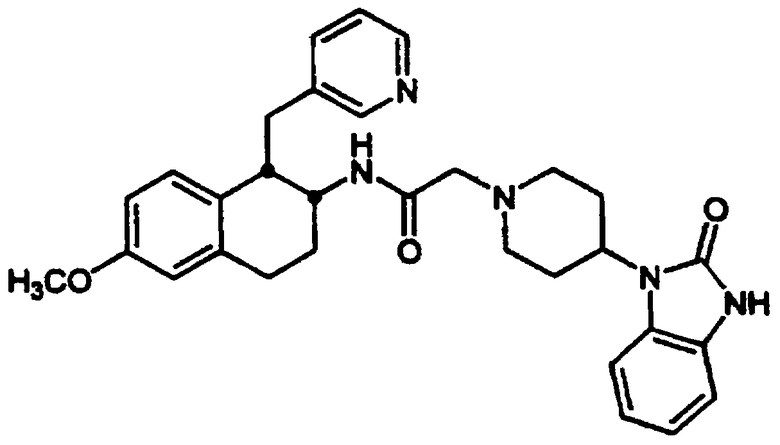
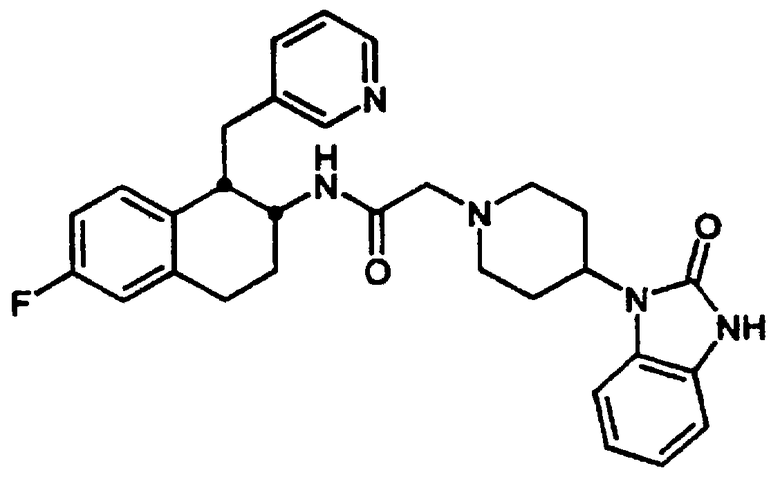
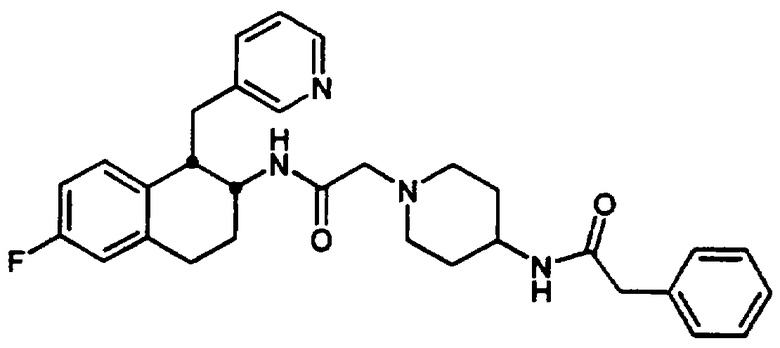
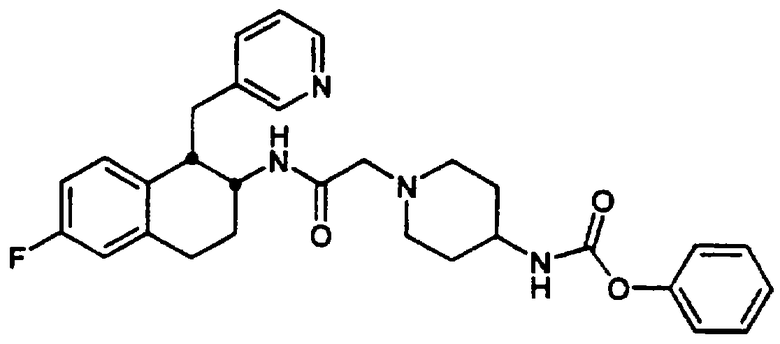
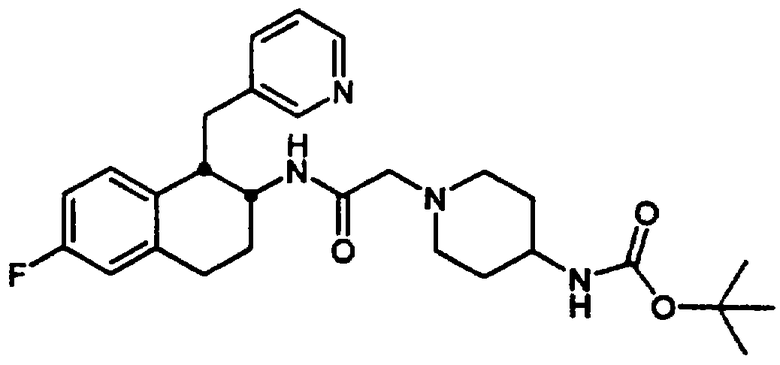
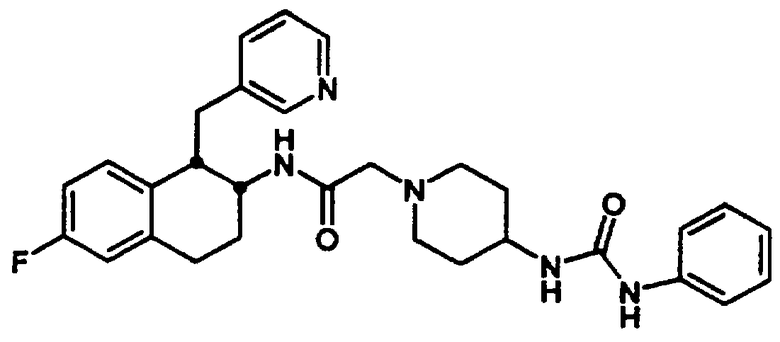
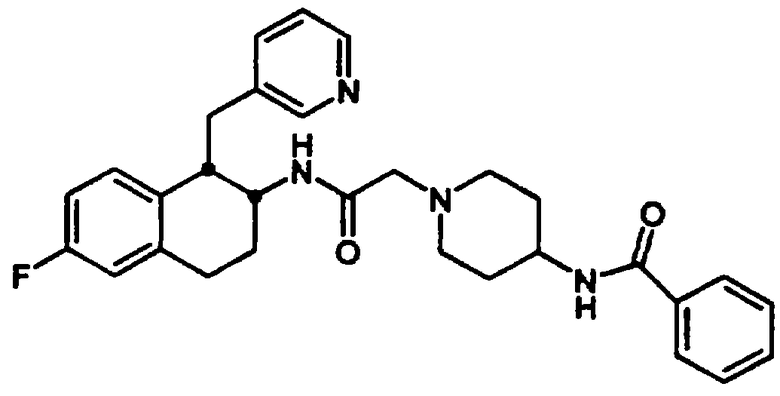
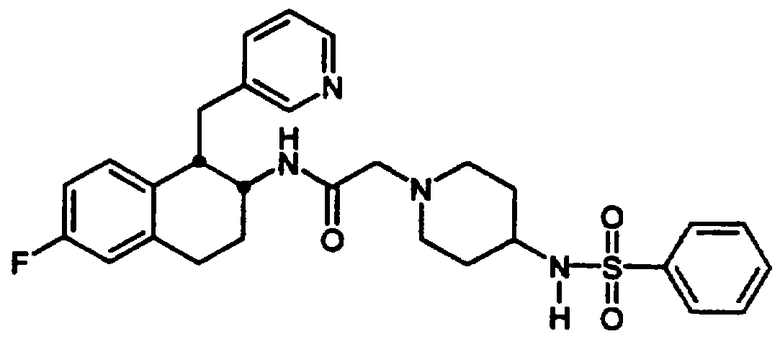
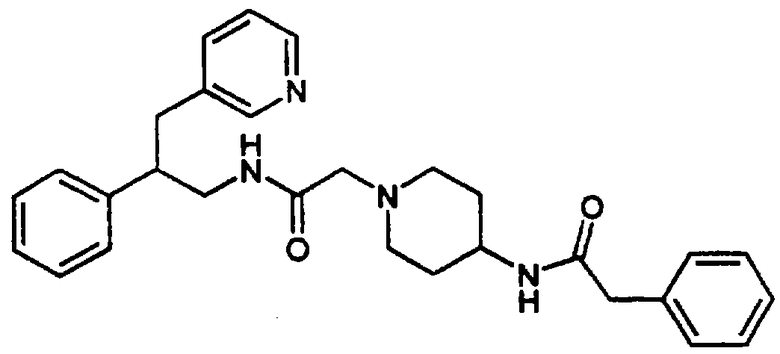
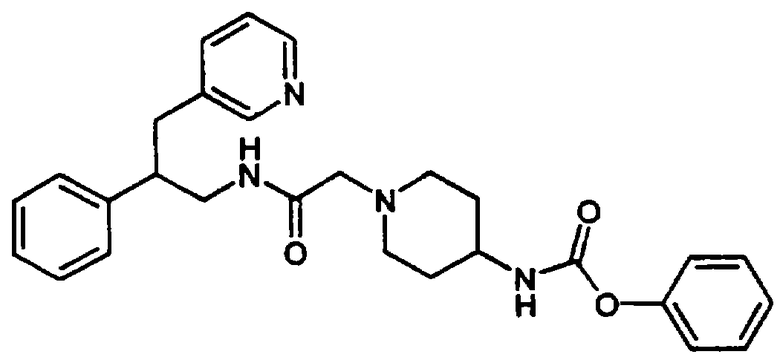
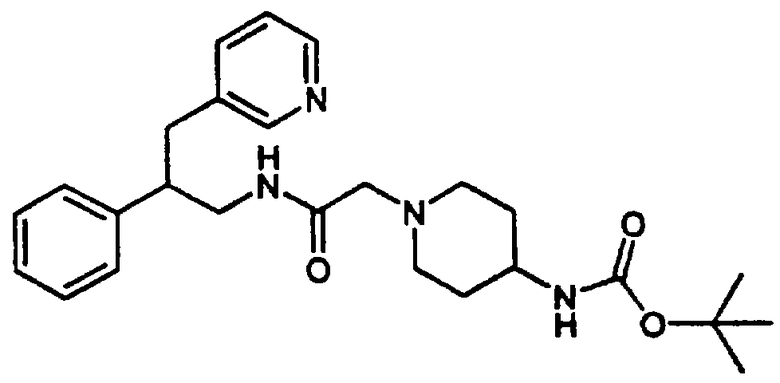
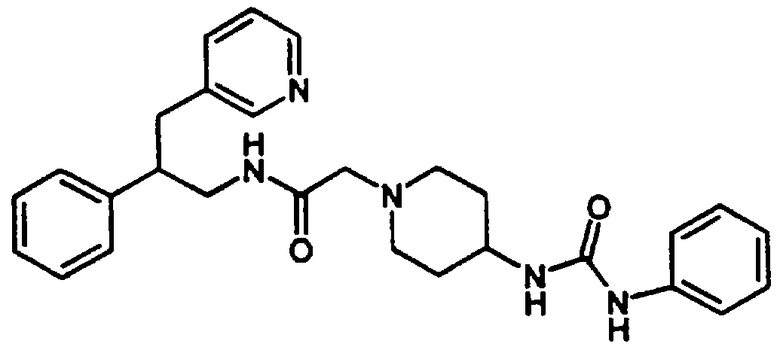
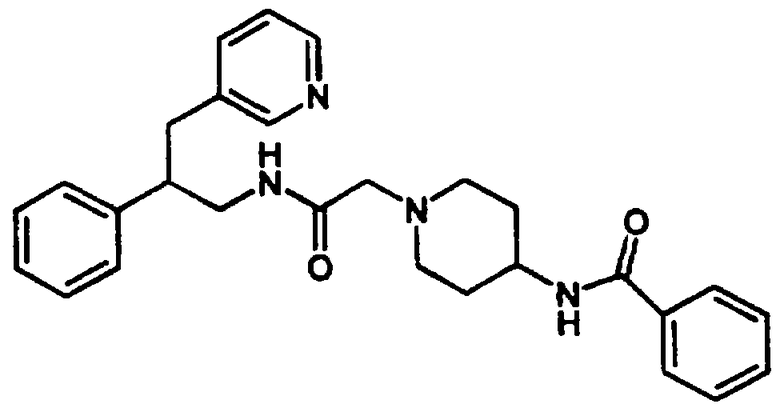
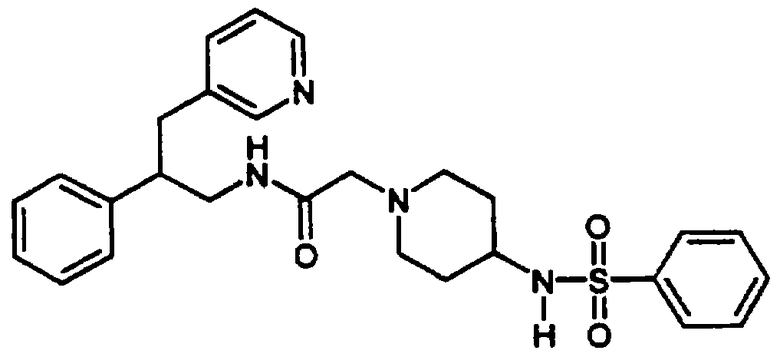
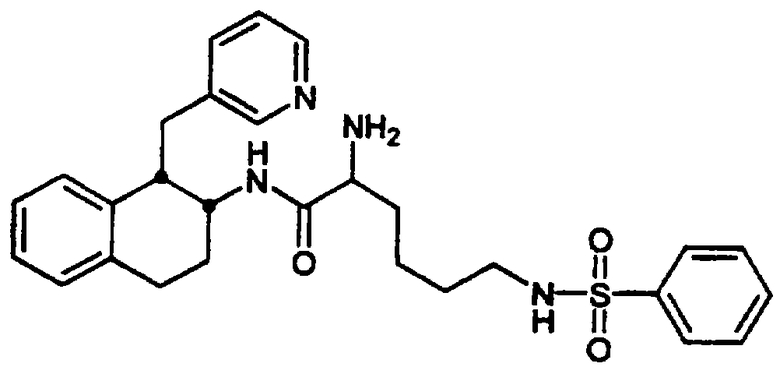
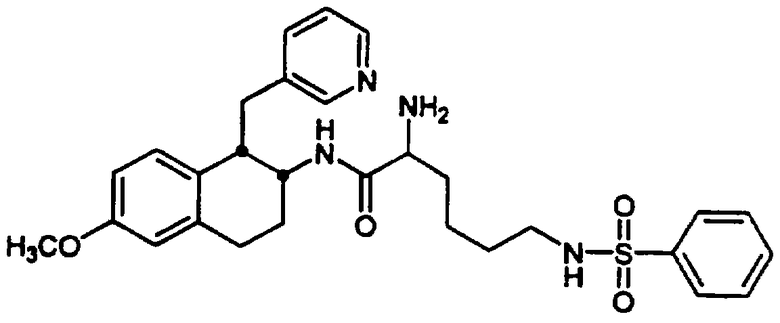
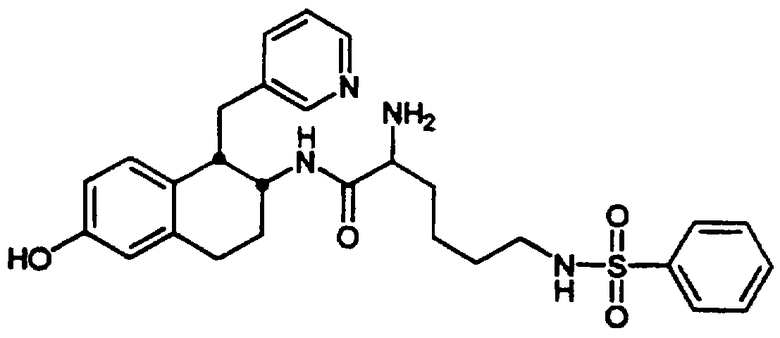
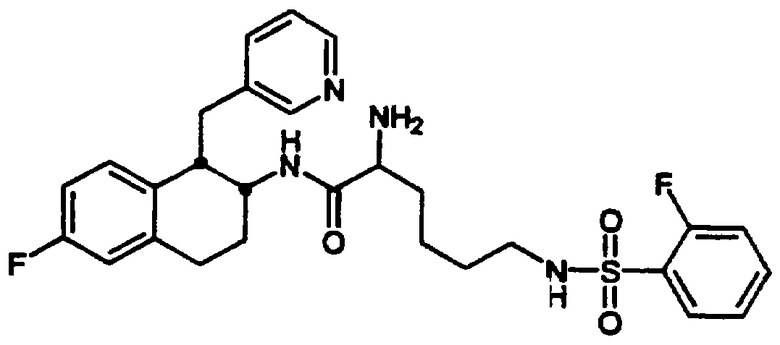
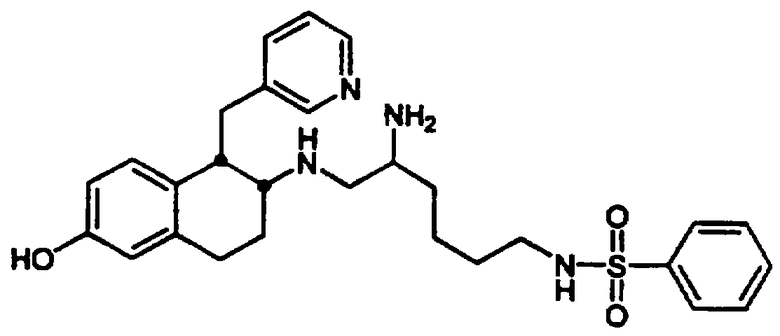
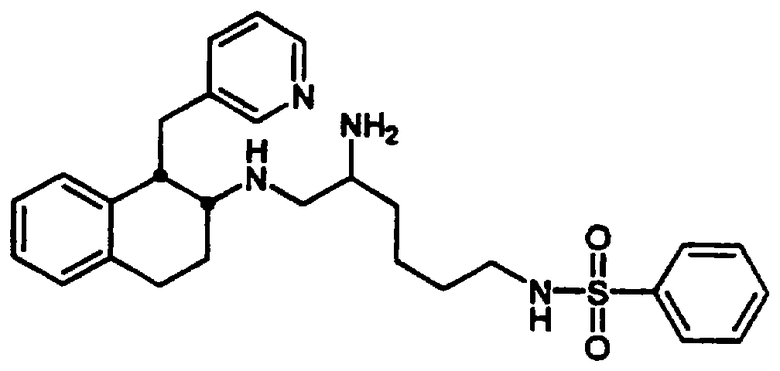
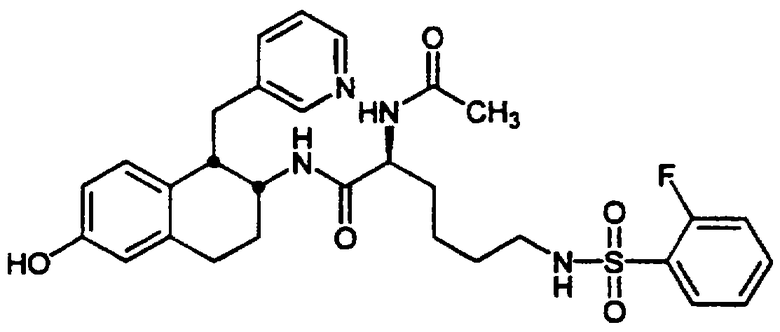
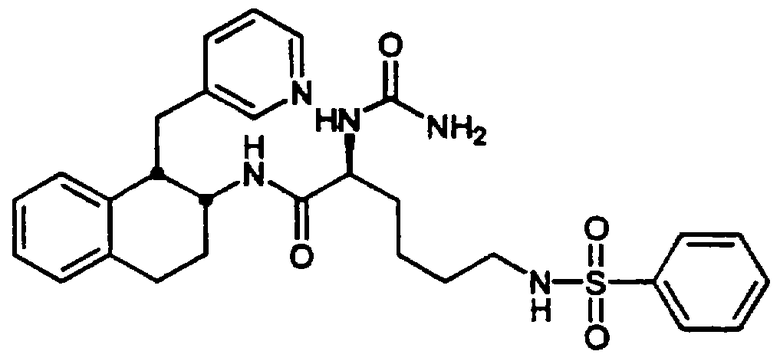
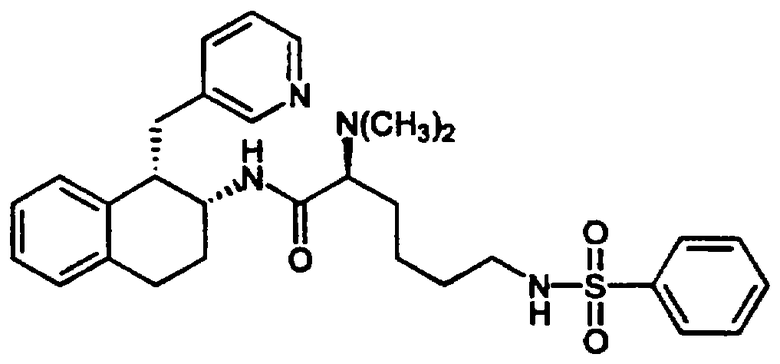
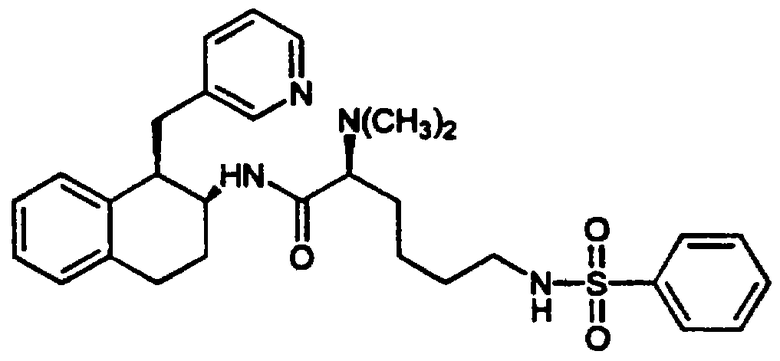
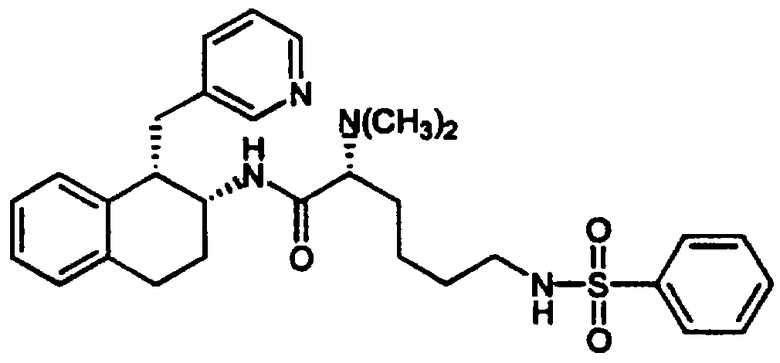
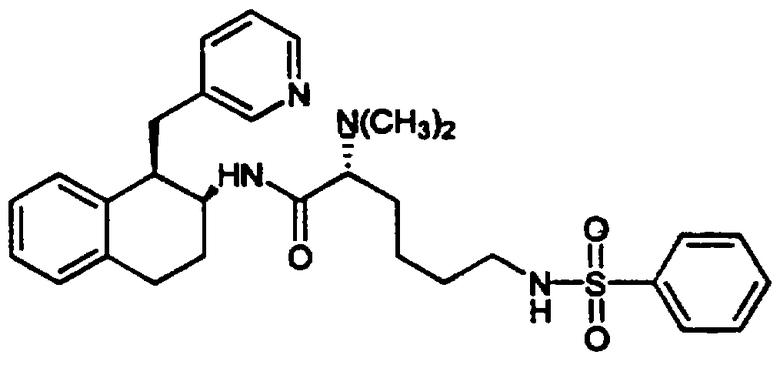
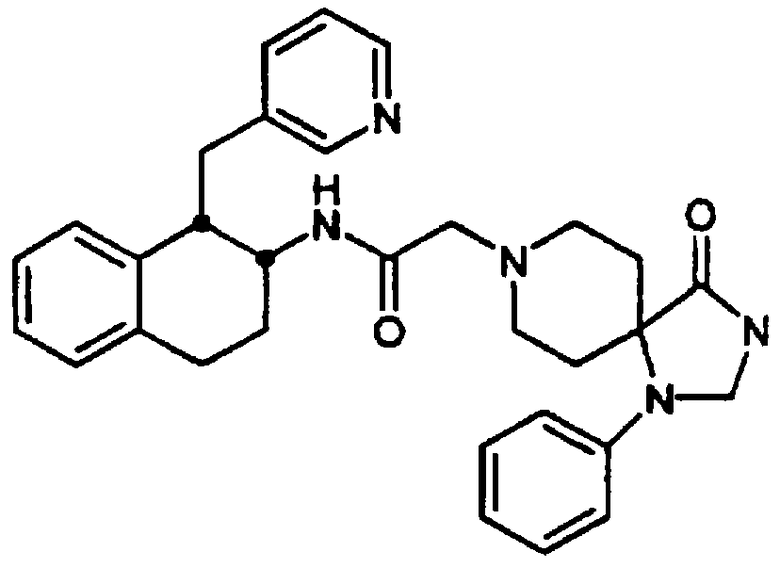
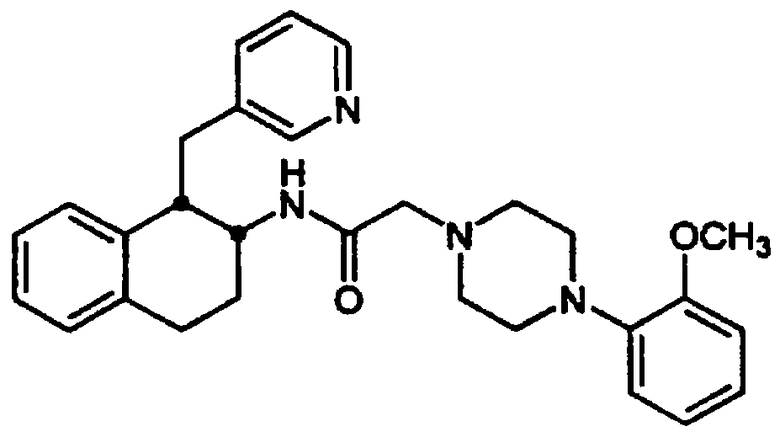
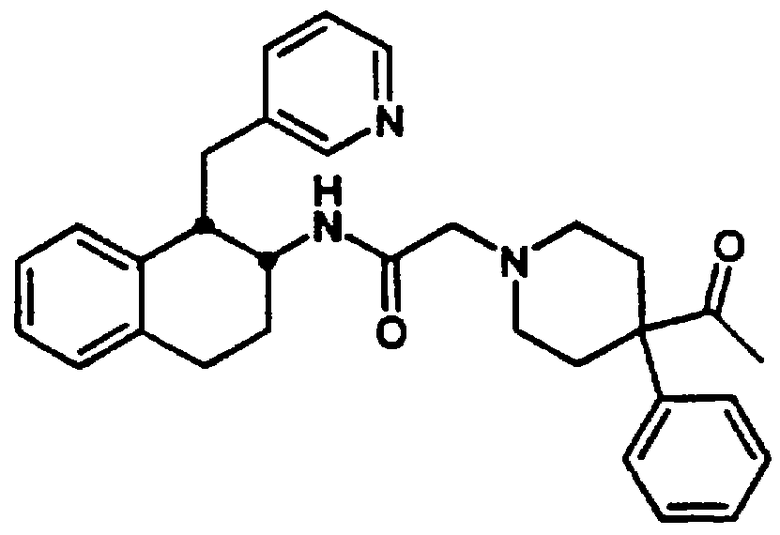
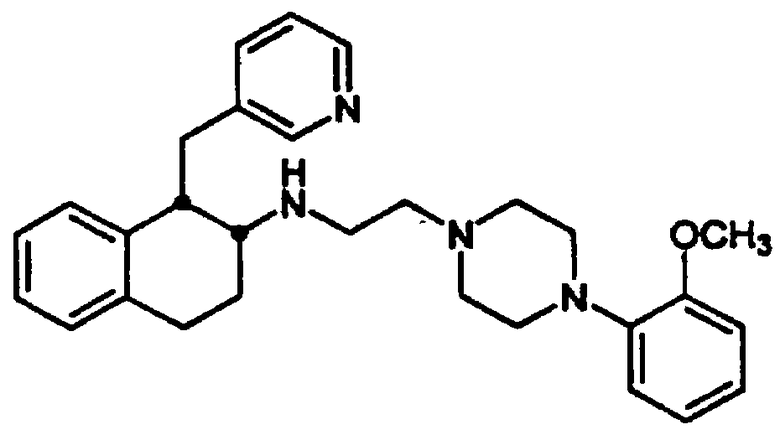
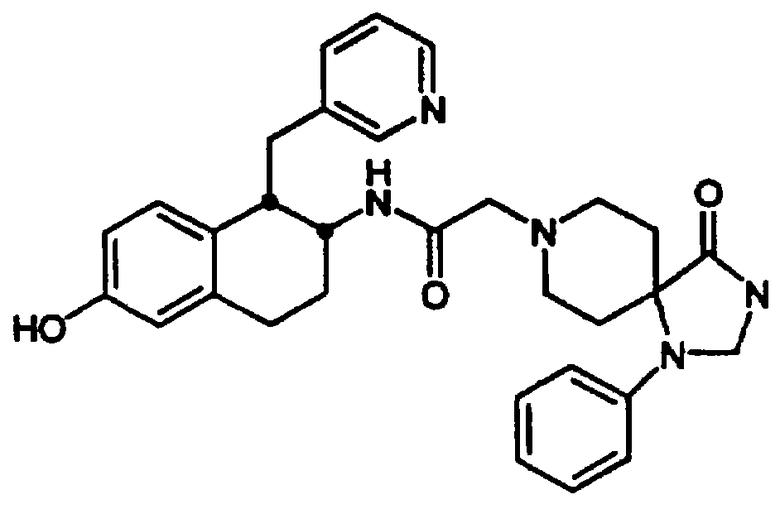
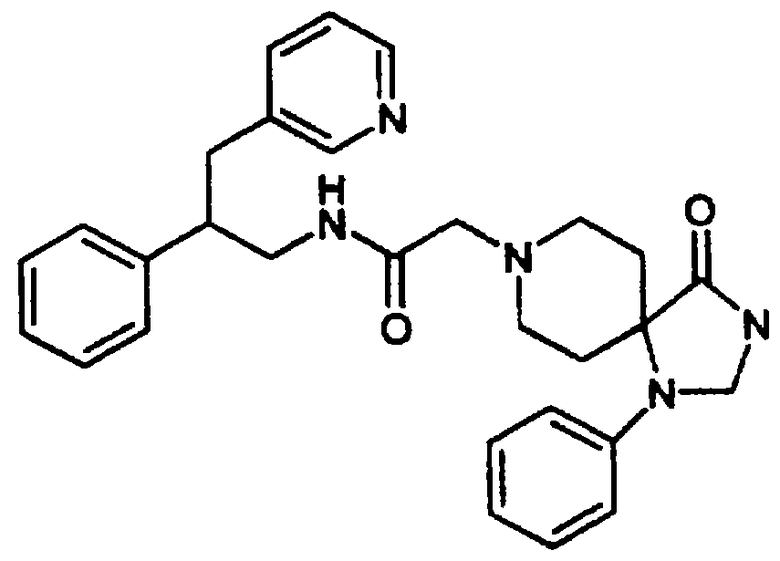
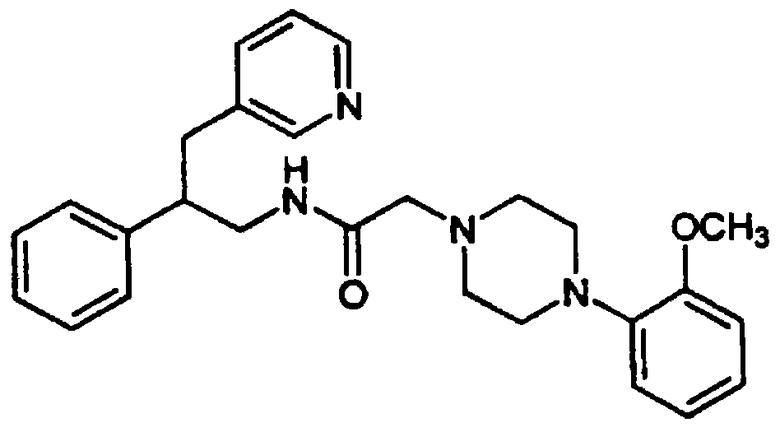
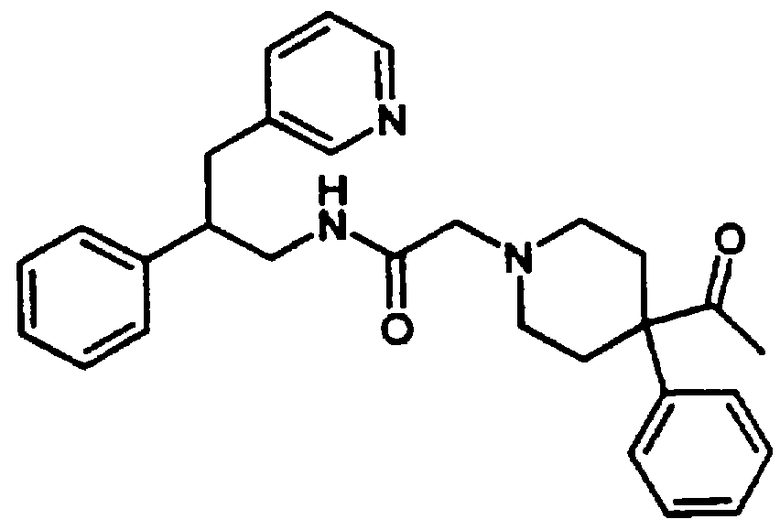
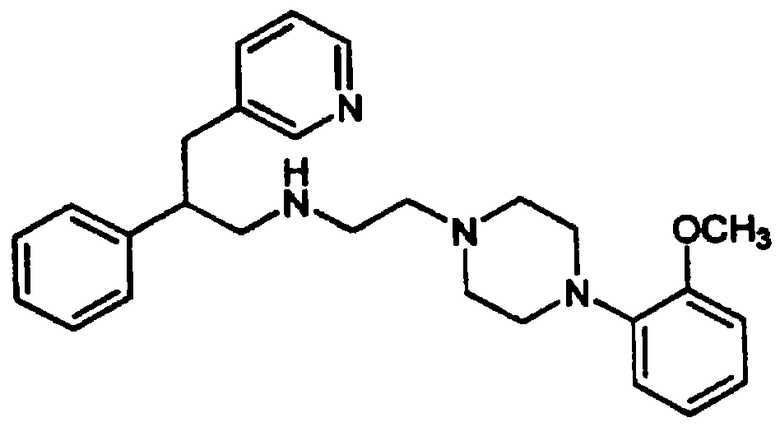
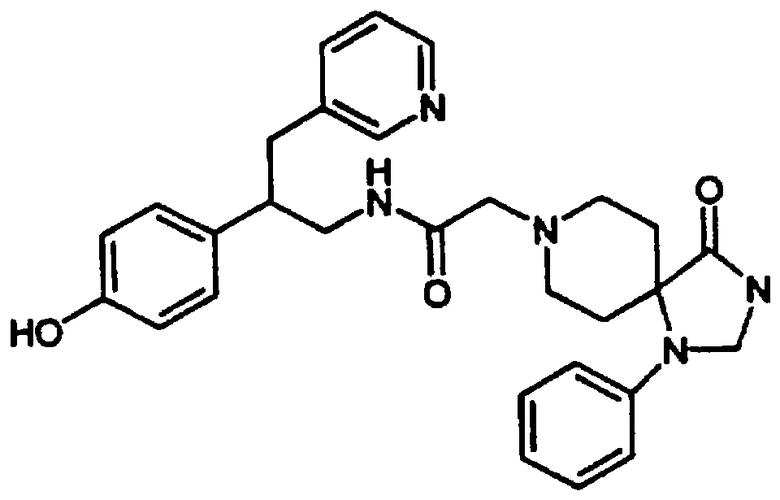
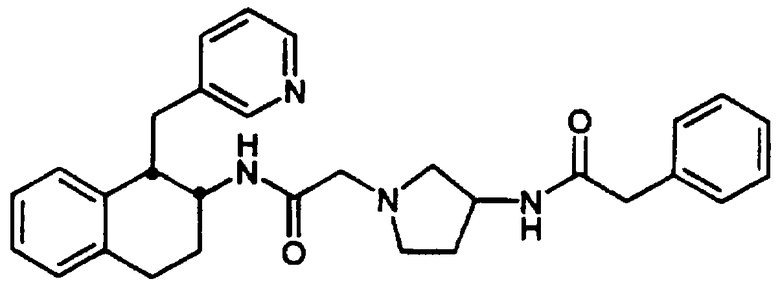
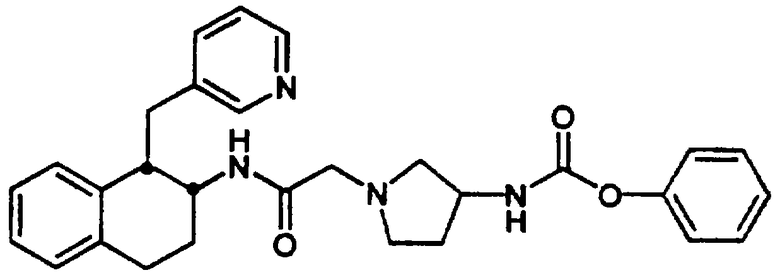
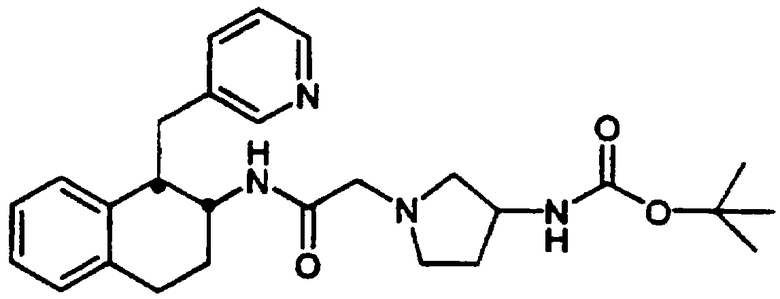
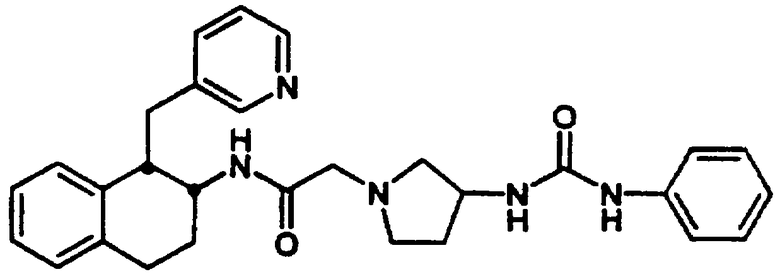
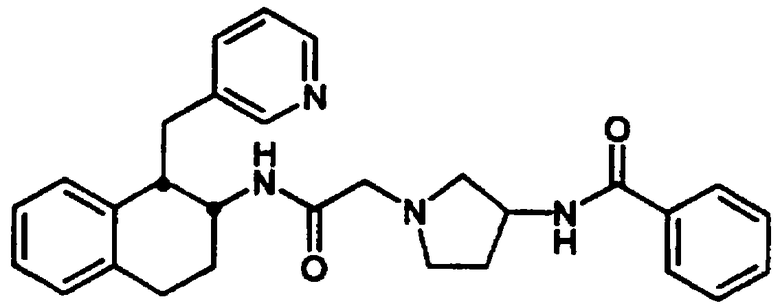
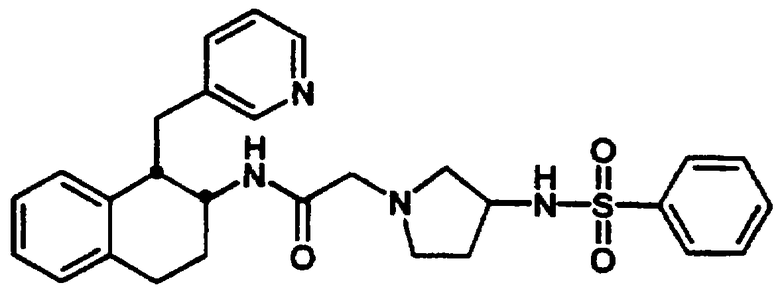
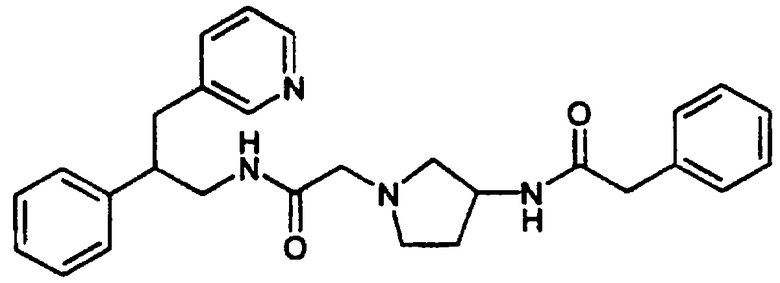
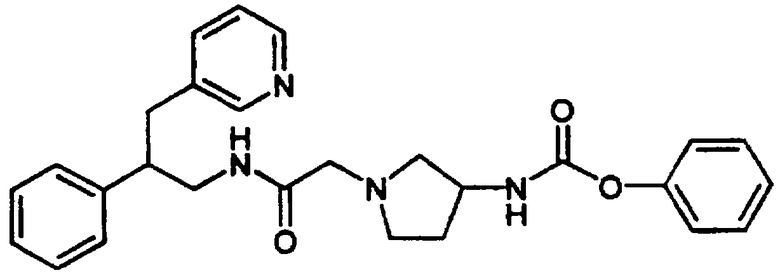
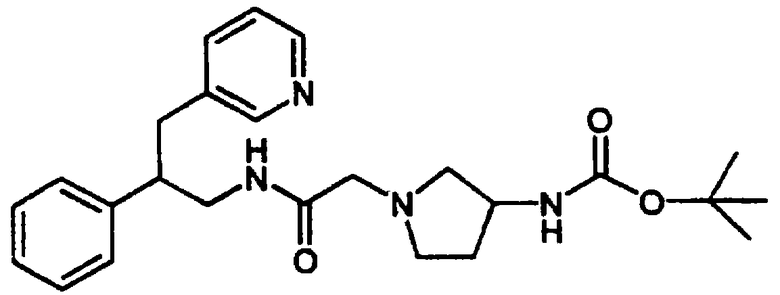
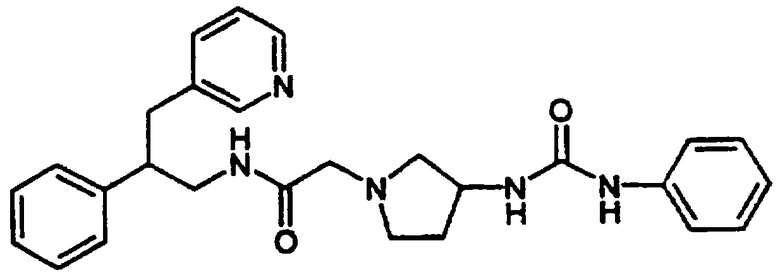
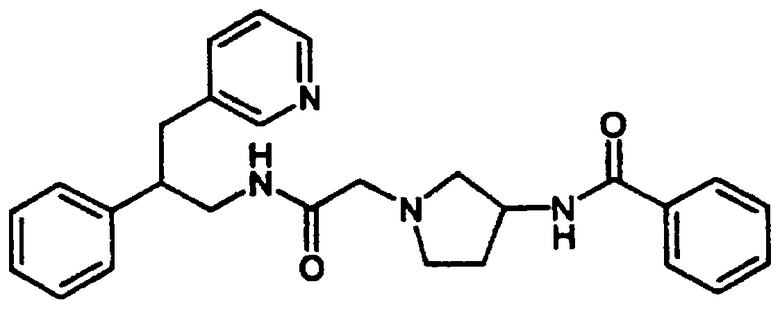
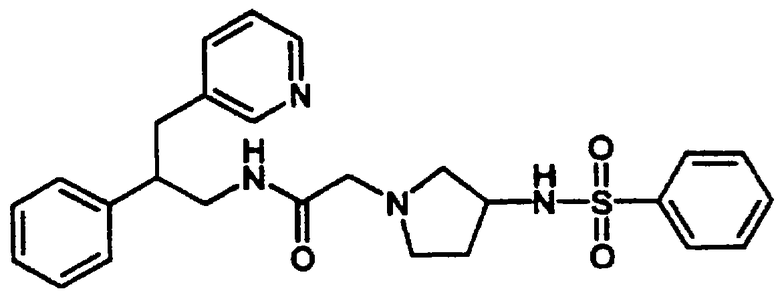
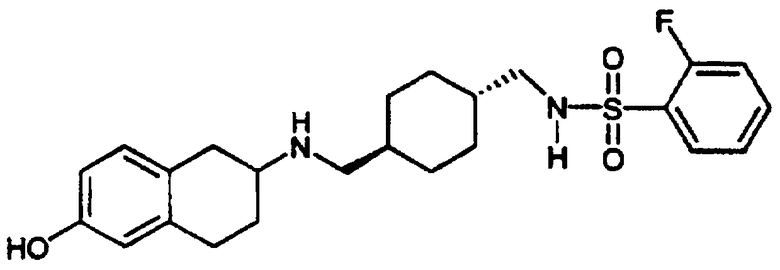
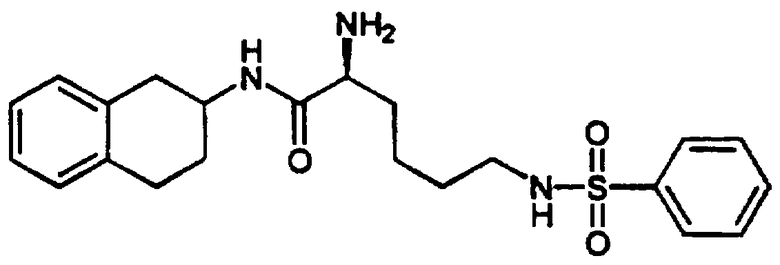
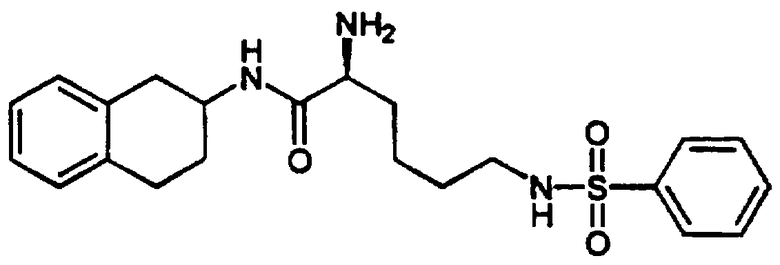
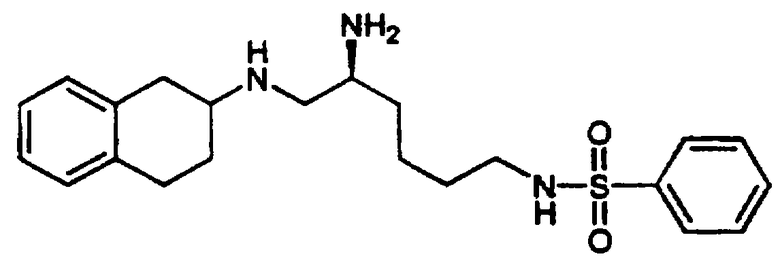
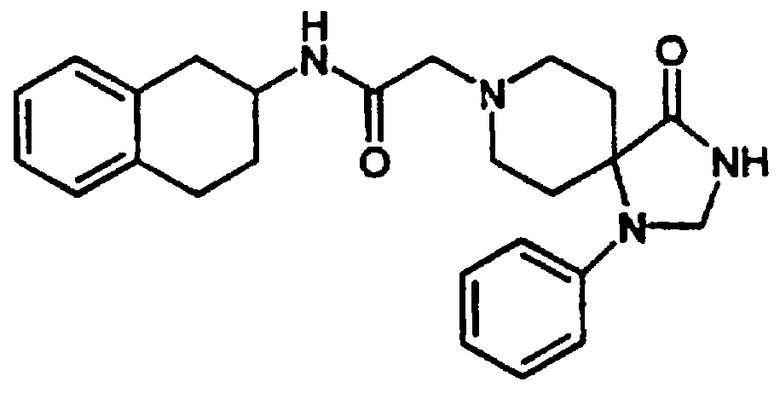
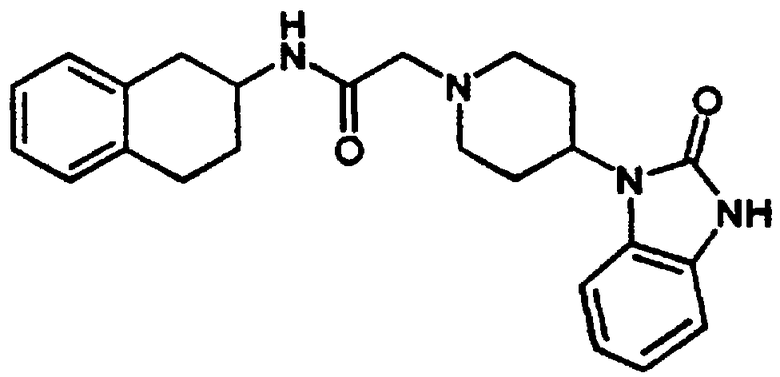
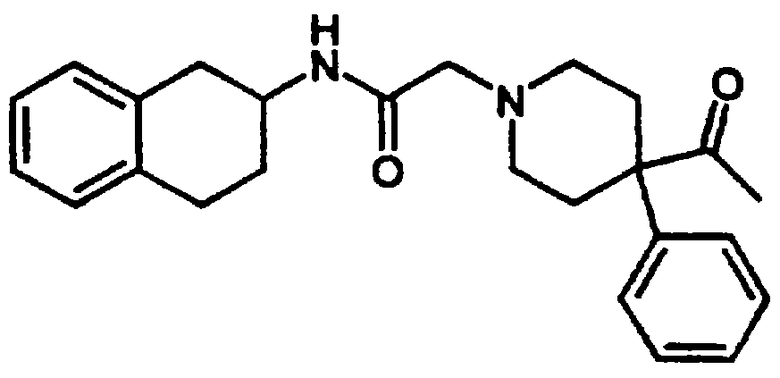
2-амино-6-[(2-фторфенилсульфонил)амино]-N-[цис-1,2,3,4-тетрагидро-6-метокси-1-(3-пиридинилметил)-2-нафтенил-(2S)-гексанамида бис-гидрохлорида;
N-[5-aмино-6-[[цис-1,2,3,4-тетрагидро-6-метокси-1-(3-пиридинилметил)-2-нафтил]амино]гексил-2-фторбензолсульфонамида трис-гидрохлорида;
N-[5-амино-6-[[цис-1,2,3,4-тетрагидро-6-гидрокси-1-(3-пиридинилметил)-2-нафтил]амино]гексил-2-фторбензол-сульфонамида трис-гидрохлорида;
(2S)-2-(ацетиламино)-6-[(2-фторфенилсульфонил)-амино]-N-[цис-1,2,3,4-тетрагидро-6-метокси-1-(3-пиридинилметил)-2-нафтенил]гексанамида бис-гидрохлорида;
(2S)-2-(ацетиламино)-6-[(2-фторфенилсульфонил)-амино]-N-[цис-1,2,3,4-тетрагидро-6-гидрокси-1-(3-пиридинилметил)-2-нафтенил]гексанамида бис-гидрохлорида;
3-[(фенилсульфонил)амино)-N-[цис-1,2,3,4-тетра-гидро-6-фтор-1-(3-пиридинилметил)-2-нафтил]-1-пирролидинацетамида бис-трифторацетат;
4-(2,3-дигидро-2-оксо-1Н-бензимидазол-1-ил)-N-[цис-1,2,3,4-тетрагидро-6-метокси-1-(3-пиридинилметил)-2-нафтил]-1-пиперидинацетамида бис-гидрохлорида;
4-(2,3-дигидро-2-оксо-1Н-бензимидазол-1-ил)-N-[транс-1,2,3,4-тетрагидро-6-метокси-1-(3-пиридинил-метил)-2-нафтил]-1-пиперидинацетамида бис-гидрохлорида;
4-ацетил-4-фенил-N-[цис-1,2,3,4-тетрагидро-1-(3-пиридинилметил)-2-нафтил]-1-пиперидинацетамида бис-гидрохлорида;
4-оксо-1-фенил-N-[цис-1,2,3,4-тетрагидро-1-(3-пиридинилметил)-2-нафтил]-1,3,8-триазаспиро [4.5]декан-8-ацетамида бис-гидрохлорида;
4-(2,3-дигидро-2-оксо-1Н-бензимидазол-1-ил)-N-[цис-1,2,3,4-тетрагидро-6-гидрокси-1-(3-пиридинилметил)-2-нафтил]-1-пиперидинацетамида бис-гидрохлорида;
транс-N-[2-(4-фторфенил)-3-(3-пиридинил)пропил]-4-[((2-фторфенилсульфонил)амино)метил]-1-циклогексанамида гидрохлорида;
транс-N-[[[2-(4-фторфенил)3-(3-пиридинил)пропил]-амино]метил]-4-циклогексил]метил]-2-фторбензол-сульфонамида бис-гидрохлорида;
N-[2-(4-фторфенил)-3-(3-пиридинил)пропил]-4-[(2-фторфенилсульфонил)амино]-1-пиперидинацетамида бис-трифторацетата.
| (S)-α-ФЕНИЛ-2-ПИРИДИНЭТАНАМИН И ЕГО СОЛЬ С НЕОРГАНИЧЕСКИМИ КИСЛОТАМИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ЛЕКАРСТВЕННЫЙ ПРЕПАРАТ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ СУДОРОГ | 1993 |
|
RU2128650C1 |
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |

