Область техники, к которой относится изобретение
Настоящее изобретение относится к способам сополимеризации олефинов с использованием замещенных гафноценовых каталитических соединений с некоординационными анионами.
Предпосылки создания изобретения
Олефиновые полимеры, включающие звенья этилена и по меньшей мере одного или нескольких α-олефинов, а также одного или нескольких необязательных диолефинов, составляют большой сектор полиолефиновых полимеров, который в настоящем описании упоминаются как "этиленовые сополимеры". Диапазон таких полимеров простирается от кристаллических полиэтиленовых сополимеров до в значительной степени аморфных эластомеров, между которыми находится новая область полукристаллических "пластомеров". Так, в частности, этиленовые сополимерные пластомеры являются хорошо зарекомендовавшим себя классом промышленных полимеров, которые находят самое разнообразное применение, связанное с их уникальными свойствами, такими, как эластомерные свойства и их термоокислительная устойчивость. Области применения пластомеров как обычных термопластичных полиолефинов включают получение пленок, покрытий для проводов и кабелей, модификацию полимеров (путем введения в смеси с другими полиолефинами), литье под давлением, изготовление вспененных материалов, обуви, листовых материалов, функционализованных полимеров (таких, как получаемые привитой свободнорадикальной сополимеризацией с полярными мономерами) и компонентов компаундов, клеев и герметиков.
Синтезируемые в промышленных условиях этиленовые сополимеры традиционно получают полимеризацией по Циглеру-Натту с использованием каталитических систем, которые в значительной мере основаны на ванадии или титане. Более новые металлоценовые каталитические соединения привлекли к себе внимание благодаря легкости введения с их помощью более существенных количеств мономерных звеньев и возможности повысить полимеризационную активность. В US 5324800 описаны металлоцены, содержащие замещенные и незамещенные циклопентадиенильные лиганды, которые приемлемы для получения высокомолекулярных олефиновых полимеров, включая линейные этиленовые сополимеры низкой плотности с небольшим количеством α-олефиновых звеньев.
Некоординационные анионы, которые могут быть использованы в качестве каталитических компонентов с такими металлоценами, известны. Понятие "некоординационный анион" в настоящее время принято в терминологии, относящейся к области полимеризации олефинов, как координационной полимеризации или полимеризации внедрением, так и карбокатионной полимеризации. Некоординационные анионы действуют как электронно-стабилизирующие сокатализаторы или противоионы в отношении катионоактивных металлоценов, которые активны при полимеризации олефинов. Понятие "некоординационный анион", используемое в настоящем описании и в литературных ссылках, применимо как к некоординационным анионам, так и к слабо координационным анионам, которые не так сильно координированы с катионоактивным комплексом, чтобы обладать подвижностью для замещения олефиново- или ацетиленово-ненасыщенными мономерами по месту внедрения. В US 5198401 в качестве предпочтительного некоординационного аниона описан тетра(перфторфенил)бор, [B(рfр)4]- или [B(C6F5)4]-, в котором перфторированные фенильные лиганды при атоме бора обуславливают подвижность и стойкость противоиона к потенциальным обратным взаимодействиям с комплексами с металлическим катионом.
Полезность ионогенных катализаторов на металлоценовой основе при высокотемпературной полимеризации олефинов описана в US 5408017 и 5408208, ЕР 0612768 и WO 96/33227. Каждая из этих публикаций посвящена металлоценовым катализаторам, приемлемым для проведения высокотемпературных процессов сополимеризации олефинов. Высокомолекулярные этилен/α-олефиновые сополимеры являются задачей по ЕР 0612768, для выполнения которой предназначены каталитические системы на основе бис(циклопентадиенил/инденил/флуоренил)гафноценов, которые совмещены с алюминийалкильным соединением и ионизирующим ионогенным соединением, обеспечивающими наличие некоординационного аниона.
Как сказано выше, общеизвестной проблемой в процессах высокотемпературной полимеризации, в частности в случаях, когда добиваются значительного содержания вводимых в этиленовые сополимеры сомономерных звеньев, является отмечаемое снижение молекулярной массы или повышение индекса расплава (ИР). Существует большая потребность в средствах сохранения высоких молекулярных масс или низкого ИР этиленовых сополимеров низкой плотности (с высоким содержанием сомономерных звеньев) в сочетании с одновременным ведением процесса при экономически предпочтительных высоких температурах реакции полимеризации и высокой производительности по полимеру.
Краткое описание изобретения
Таким образом, объектом изобретения являются замещенные, связанные мостиками каталитические комплексы, включающие некоординационные анионы, которые проявляют неожиданную стойкость в ходе проведения высокотемпературных процессов полимеризации олефинов, благодаря чему высокомолекулярные олефиновые сополимеры могут быть получены при относительно высокой производительности. Более конкретно объектом изобретения является способ полимеризации при получении этиленовых сополимеров, плотность которых составляет от примерно 0,850 до примерно 0,930, включающий введение в сверхкритических условиях или в условиях полимеризации в растворе при реакционной температуре, от равной или превышающей примерно 60 до 225°С, этилена и одного или нескольких сомономеров, способных к полимеризации внедрением, в контакт с гафноценовым каталитическим комплексом, дериватизированным из А) бициклопентадиенильного гафнийсодержащего металлорганического соединения, включающего I) по меньшей мере один незамещенный циклопентадиенильный лиганд или замещенный ароматическими конденсированными кольцами циклопентадиенильный лиганд, не имеющий дополнительных заместителей, II) один замещенный или не замещенный ароматическими конденсированными кольцами циклопентадиенильный лиганд и III) ковалентный мостик, связывающий оба циклопентадиенильных лиганда, причем такой мостик включает один атом углерода или кремния с двумя арильными группами, каждая из которых замещена С1-С20гидрокарбильной или гидрокарбилсилильной группой, по меньшей мере одна из которых представляет собой линейный заместитель С3- или с большим числом атомов углерода, и Б) активирующего со катализатора, предпочтительно ионогенного соединения-предшественника, включающего галоидированный тетраарилзамещенный анион с элементом 13-й группы.
Подробное описание изобретения
Гафниевые соединения с мостиковой связью по изобретению включают те, которые содержат по одному замещенному углеродному или кремниевому атому, связывающему мостиком два циклопентадиенилсодержащих (Ср) лиганда гафниевых металлических центров (III), замещенный ароматическими конденсированными кольцами циклопентадиенильный лиганд или лиганды, предпочтительно те, которые содержат гидрокарбильные или гидрокарбилсилильные С1-С30заместители при (II) нециклопентадиенильном ароматическом кольце. Предпочтительные мостиковые заместители включают линейный или разветвленный С1-С20алкил или С1-С20замещенный силил, замещенные фенильные группы, причем алкильные или замещенные силильные заместители находятся в пара- или мета-положениях арильных групп, предпочтительно у которых по меньшей мере один из алкильных заместителей представляет собой линейный н-алкильный заместитель С3- или с большим числом атомов углерода, предпочтительно С4- или с большим числом атомов углерода. Конкретные примеры включают метил, этил, н-пропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, неопентил и т.д. Заместители, находящиеся на нециклопентадиенильных ароматических кольцах замещенного ароматическим конденсированным кольцом циклопентадиенильного лиганда (II), такого, как содержащий инденильные и флуоренильные производные циклопентадиенильных групп, как правило, включают одну или несколько углеводородных или гидрокарбилсилильных C1-С30групп, выбранных из групп с линейным, разветвленным, циклическим, алифатическим, ароматическим или комбинированным строением, включая конфигурации с конденсированными кольцевыми или боковыми группами. Примеры включают метил, изопропил, н-пропил, н-бутил, изобутил, трет-бутил, неопентил, фенил, н-гексил, циклогексил и бензил. Принимая во внимание цели данной заявки на патент, понятие "углеводородный" или "гидрокарбильный" использовано применительно к тем соединениям или группам, которые обладают по существу углеводородными характеристиками, но включают не более примерно 10 мол.% неуглеродных атомов, таких, как атомы бора, кремния, кислорода, азота, серы и фосфора. Неограничивающими примерами "гидрокарбилсилила" служат диалкил и триалкилсилилы. Подобным же образом в настоящем описании понятия "циклопентадиенильный", "инденильный" и "флуоренильный" рассматриваются как охватывающие применение гетероатомсодержащих циклопентадиенильных колец или конденсированных колец, у которых один из циклических углеродных атомов в Ср кольце или в конденсированном с ним кольце замещен неуглеродным атомом элемента 14-й, 15-й или 16-й группы (см., например, содержание заявки WO 98/37106, имеющей общую дату приоритета с заявкой США серийный номер 08/999214, поданной 29.12.97, и заявки WO 98/41530, имеющей общую дату приоритета с заявкой США серийный номер 09/042378, поданной 13.3.98, включенные в настоящее описание в соответствии с существующей в США патентной практикой в качестве ссылок.
Конкретные гафниевые катализаторы с мостиковой связью включают те, которые дериватизированы из (1) комплексов на инденильной основе, таких, как изомеры и смеси (пара-н-бутилфенил)(пара-трет-диалкилбутилфенил)метилен-(флуоренил)(инденил)гафнийдиметила, (пара-н-пропилфенил)(пара-метил-фенил)метилен(флуоренил)(инденил)гафнийдиметила, ди(пара-н-бутилфенил)-метилен(2,7-ди-трет-бутилфлуоренил)(инденил)гафнийдиметила, (пара-н-бутилфенил)(пара-трет-бутилфенил)метилен(2,7-ди-трет-бутилфлуорен-ил)(инденил)гафнийдиметила, (пара-н-бутилфенил)(пара-трет-бутилфенил)-метилен(2,7-диметилфлуоренил)(инденил)гафнийдибензила и ди(пара-н-бут-илфенил)метилен(флуоренил)(инденил)гафнийдиметила; (2) комплексов на флуоренильной основе, таких, как (пара-н-пропилфенил)(пара-изопропилфен-ил)силил(флуоренил)(флуоренил)гафнийди-трет-бутил, ди(пара-н-пропилфен-ил)метилен(2,7-ди-трет-бутил-5-метилфлуоренил)(флуоренил)гафнийдиметил и (3) комплексов на циклопентадиенильной основе, таких, как изомеры и смеси (пара-н-пропилфенил)(пара-изопропилфенил)метилен(флуоренил)(инденил)-гафнийдиметила, (пара-н-бутилфенил)(пара-трет-бутилфенил)метилен(флуоренил)(циклопентадиенил)гафнийдиметила, ди(пара-н-бутилфенил)метилен-(2,7-ди-трет-бутилфлуоренил)(циклопентадиенил)гафнийдиметила, (пара-н-бутилфенил)(пара-трет-бутилфенил)метилен(2,7-ди-трет-бутилфлуоренил)(циклопентадиенил)гафнийдиметила и ди(пара-н-бутилфенил)метилен(2,7-диметилфлуоренил)(циклопентадиенил)гафнийдиметила и -дибензила. Было установлено, что замещенные соединения, содержащие мостиковые связи, такие, как те асимметричные соединения, которые перечислены выше, особенно эффективны для применения в соответствии с изобретением.
Так, в частности, что касается гафниевых соединений с мостиковой связью, то повышение степени замещения замещенного ароматическими конденсированными кольцами лиганда (II) обусловливает увеличение молекулярной массы, что можно сказать также об использовании по изобретению ковалентных мостиков (III) между циклопентадиенильными лигандами, как изложено выше. Замещенные арильные группы связывающего мостиком атома обусловливают неожиданное повышение активности или производительности катализатора в сравнении с более простыми диарильными замещенными аналогами без нежелательного влияния на молекулярную массу образующихся сополимеров. Предпочтительный вариант замещения флуоренильных или инденильных радикалов (II) в соединениях гафния обычно включает два или большее число гидрокарбильных или гидрокарбилсилильных C1-C30заместителей кольцевого водорода в по меньшей мере одном 6-членном конденсированном кольце, предпочтительнее в обоих в случае флуоренила.
По изобретению активирующие сокаталитические ионизирующие соединения-предшественники включают комплексы с элементом 13-й группы, содержащие по меньшей мере по два галоидированных арилзамещенных ароматических лиганда, такие, как галоидированные тетрафенилборные и -алюминиевые соединения, примеры которых приведены в описании известного уровня техники. Предпочтительные ароматические лиганды состоят из полициклических ароматических углеводородных остатков и ароматических кольцевых конгломератов, в которых два или большее число колец (или конденсированных кольцевых систем) связаны непосредственно друг с другом или находятся вместе. Эти лиганды, которые могут быть одинаковыми или различными, ковалентно связаны непосредственно с металлическим/металлоидным центром. В предпочтительном варианте арильные группы представляют собой галоидированные тетраарильные анионные комплексы с элементом 13-й группы, включающие по меньшей мере по одному конденсированному полициклическому ароматическому углеводородному остатку или боковому ароматическому кольцу. Примерами служат инденильные, нафтильные, антрацильные, гепталенильные и дифенильные лиганды. Так, например, приемлемые лиганды включают те, которые представлены ниже, причем незамкнутая связь обращена к атому элемента 13-й группы [для выбора дополнительных лигандов см. также примеры полициклических соединений в литературе, в частности Nomenclature of Organic Compounds, Chs. 4-5 (ACS, 1974)].
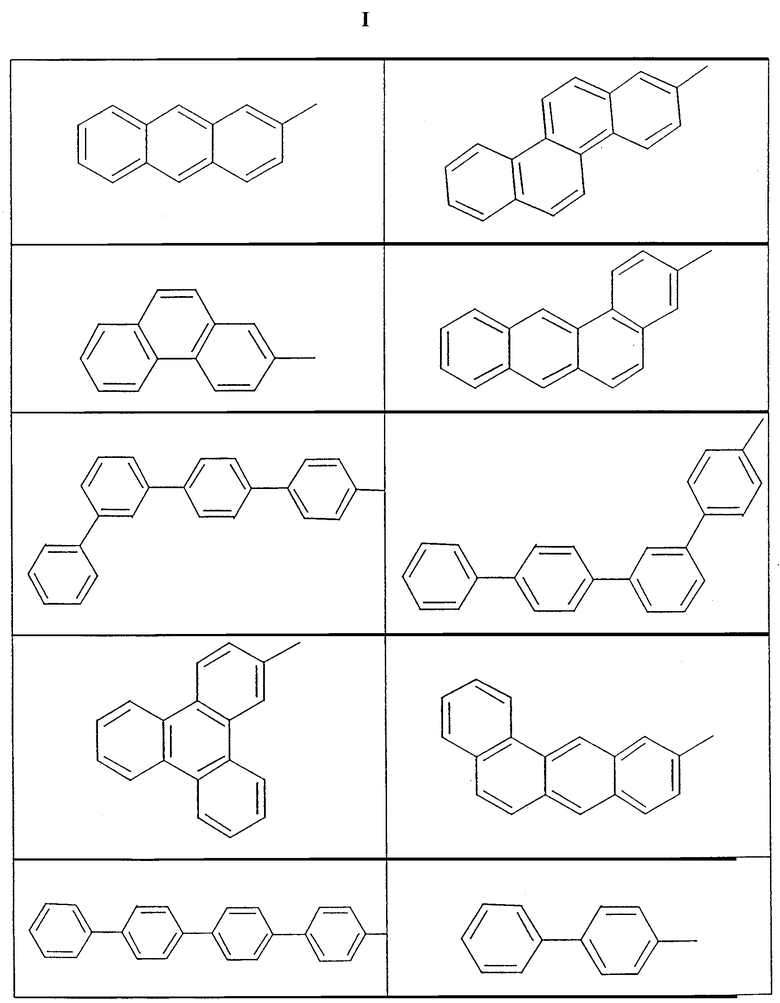
Эти предпочтительные ионизирующие соединения включают соли анионов, содержащих лиганды, способные к тетраэдрической ориентации. Таким образом, в предпочтительном варианте эти лиганды структурно совместимы с любым другим в смысле нахождения в связанном состоянии с металлом группы 13 в качестве центра и без пространственного затруднения для образования связи с ним дополнительных галоидированных арильных лигандов. Примеры охватывают те, которые содержат боковые арильные группы в пара- или мета-положении арильного кольца, ближайшего к металлическому/металлоидному центру, и те, которые содержат конденсированные арильные группы, связанные с арильным кольцом, ближайшим к металлическому/металлоидному центру, во 2-м, 3-м или 3-м, 4-м положениях (см. вышеприведенные формулы I). Приемлемы также анионы со смешанными лигандами. Примером комплекса является трис(перфторфенил)(перфторнафтил)борат. Таким образом, в общем комплексы с элементом 13-й группы, которые могут быть использованы в соответствии с изобретением, как правило, отвечают следующей формуле:
[M(A)4-n(Cn)]-
где М обозначает элемент 13-й группы, А обозначает незатрудненный лиганд, как он описан выше, С обозначает затрудненный лиганд, тот, который содержит объемистые заместители при ближайшем арильном кольце, связанном с металлическим/металлоидным центром, отличным от тех, которые описаны выше как приемлемые, а n обозначает 0, 1 или 2 (см. также совместно рассматриваемую заявку США серийный номер 60/087447, поданную 1 июня 1998 г., и ее аналог WO 99/45042, содержание которых в соответствии с существующей в США патентной практикой включено в настоящее описание в качестве ссылок.
Как для конденсированных ароматических колец, так и для ароматических кольцевых конгломератов весьма предпочтительно галоидирование с тем, чтобы позволить диспергироваться увеличенному заряду, что совместно с пространственным объемом в качестве независимых характеристик содействует снижению вероятности отщепления лиганда металлоценовым катионом со свойствами сильной кислоты Льюиса, образующимся при активировании катализатора. Кроме того, галоидирование подавляет взаимодействие гафниевого катиона со всеми оставшимися углерод-водородными связями ароматических колец, а пергалоидирование предотвращает возможность протекания таких потенциально нежелательных реакций. Таким образом, в предпочтительном варианте по меньшей мере одна треть водородных атомов при углеродных атомах арильных лигандов может быть замещена атомами галогена, а в более предпочтительном варианте арильные лиганды пергалоидированы. Наиболее предпочтительным галогеном является фтор, а наиболее предпочтительны пефторированные арильные лиганды.
Средства получения ионогенных каталитических систем, включающих каталитически активные катионы гафниевых соединений и подходящие некоординационные анионы, общеизвестны (см., например, US 5198401, WO 92/00333, WO 97/22639 и ЕР 0612768). Как правило, такие методы включают получение из технических источников или синтез соединений с выбранным переходным металлом, содержащих отщепляемые лиганды, например гидридную, галогенидную, алкильную, алкенильную или гидрокарбилсилильную группу, и их введение в контакт с источником некоординационного аниона или подходящими соединениями-предшественниками в приемлемом растворителе. Анионоактивное соединение-предшественник отщепляет одновалентный лиганд (или одну моноанионную связь бидентатных алкенильных лигандов), что удовлетворяет валентные потребности предпочтительных гафнийсодержащих металлоценовых соединений. Вследствие такого отщепления остаются гафноцены в практически катионоактивном состоянии, которое в соответствии с изобретением уравновешивается стабильными совместимыми и объемистыми некоординационными анионами. Содержание каждого из документов, упомянутых в этом абзаце, в соответствии с существующей в США патентной практикой включено в настоящее описание в качестве ссылки.
В предпочтительном варианте на стадии приготовления катализатора некоординационные анионы вводят в виде ионогенных соединений, включающих по существу катионоактивный комплекс, который отщепляет нециклопентадиенильный подвижный лиганд соединений переходного металла, которые при отщеплении нециклопентадиенильного лиганда в качестве побочного продукта оставляют некоординационный анионный фрагмент. Для ионогенных каталитических систем по настоящему изобретению исключительно предпочтительны гафниевые соединения, включающие при металлическом центре подвижные гидридные, алкильные или силильные лиганды, поскольку известно, что результатом протекающих in situ процессов алкилирования могут быть параллельные реакции и взаимодействия, которым свойственна тенденция снижать эффективность полимеризации в целом в высокотемпературных условиях, которые соответствуют предпочтительным вариантам осуществления способа по изобретению.
Подходящие катионы для соединений-предшественников, способных предоставлять для сокатализаторов по изобретению некоординационные анионы, включают те, которые в данной области техники известны. К ним относятся азотсодержащие катионы, такие, как те, которые представлены в US 5198401, карбениевые, оксониевые и сульфониевые катионы по US 5387568, металлические катионы, например Ag+ или Li+, силилиевые катионы по WO 96/08519 и гидратированные соли с металлами группы 1 или 2 в качестве катионов по WO 97/22635.
Примеры предпочтительных солей-предшественников с некоординационными анионами, способными к ионной катионизации металлоценовых соединений по изобретению и к последующей стабилизации образовавшимся некоординационным анионом, включают триалкилзамещенные аммониевые соли, такие, как триэтиламмонийтетракис(перфторнафтил)- и -тетра-кис(перфтор-4-дифенил)бор, три(н-бутил)аммонийтетракис(перфторнафтил)- и тетракис(перфтор-4-дифенил)бор, три(н-октил)аммонийтетракис(перфторнафтил)- и -тетракис(перфтор-4-дифенил)бор, триметиламмонийтетракис(перфторнафтил)- и -тетракис(перфтор-4-дифенил)бор, триметиламмонийтетратетракис(перфторнафтил)- и -тетракис(перфтор-4-дифенил)бор, трибутиламмонийтетракис(перфторнафтил)- и -тетракис(перфтор-4-дифенил)бор, трипропиламмонийтетракис(перфторнафтил)- и -тетракис(перфтор-4-дифенил)бор, три(н-бутил)аммонийтетракис(перфторнафтил)- и -тетракис(перфтор-4-дифенил)бор и т.п.;
N,N-диaлкилaнилиниeвыe соли, такие, как N,N-диметиланилинийтетракис(перфторнафтил)- и -тетракис(перфтор-4-дифенил)бор, N,N-ди(н-додецил)анилинийтетракис(перфторнафтил)- и тетракис(перфтор-4-дифенил)бор, N,N-2,4,6-пентаметиланилинийтетракис(перфторнафтил)- и -тетракис(перфтор-4-дифенил)бор и т.п.;
диалкиламмониевые соли, такие, как ди(н-додецил)аммониийтетракис(перфторнафтил)- и -тетракис(перфтор-4-дифенил)бор, дициклогексиламмониийтетракис(перфторнафтил)- и -тетракис(перфтор-4-дифенил)бор и т.п., и
триарилфосфониевые соли, такие, как трифенилфосфонийтетракис(перфторнафтил)- и -тетракис(перфтор-4-дифенил)бор, три(метилфенил)фосфонийтетракис(перфторнафтил)- и -тетракис(перфтор-4-дифенил)бор, три(диметилфенил)-фосфонийтетракис(перфторнафтил)- и -тетракис(перфтор-4-дифенил)бор и т.п. [см. также включающие длинноцепочечные группы комплексы азотсодержащих кислот Льюиса (например, протонированные аммониевые соли) по WO 97/35983, каталитические активаторы которых приемлемы для использования в соответствии с настоящим изобретением и описание которых в соответствии с существующей в США патентной практикой включено в настоящее описание в качестве ссылки].
Другие примеры приемлемых анионных предшественников включают те, которые содержат стабильный карбениевый ион и совместимый некоординационный анион. К ним относятся тропиллийтетракис(перфторнафтил)- и тетракис(перфтор-4-дифенил)борат, трифенилметилийтетракис(перфторнаф-тил)- и -тетракис(перфтор-4-дифенил)борат, бензол(диазоний)тетракис(перфторнафтил)- и -тетракис(перфтор-4-дифенил)борат. Точно так же приемлемы силилийборатные и алюминатные соли практически эквивалентной структуры.
В данной заявке понятие "очищающее средство" использовано в том смысле, который ему придают в данной области техники, для обозначения кислоты Льюиса, обладающей достаточной кислотностью для образования координационной связи с полярными загрязнителями и примесями, которые могут случайно попасть в полимеризационные исходные материалы или реакционную среду. Такие примеси могут быть неумышленно введены вместе с любыми компонентами реакции полимеризации, в частности с растворителем, мономерными и каталитическими исходными материалами, и оказывать нежелательное влияние на активность и стабильность катализатора. Так, в частности, для процессов, в ходе проведения которых применяют рецикловые потоки непревращенного мономера для повторного использования, потребность в применении полярных соединений в качестве дезактиваторов или "подавителей" катализатора, таких, как вода и низшие спирты, настоятельно требует применения очищающих средств, как это происходит в случае полярных примесей, естественно встречающихся в исходных мономерных материалах. Это может привести к снижению или даже полной потере каталитической активности, в особенности когда каталитической системой служит пара металлоценовый катион - некоординационный анион. Полярные примеси или каталитические яды включают воду, кислород, металлсодержащие примеси и т.д. В предпочтительном варианте перед их загрузкой в реакционный сосуд предпринимают некоторые меры, состоящие, например, в химической обработке или осуществлении методов осторожного разделения после или во время синтеза или получения различных компонентов, но для проведения собственно процесса полимеризации обычно все-таки требуются некоторые незначительные количества очищающего соединения.
Как правило, очищающим соединением служит металлорганическое соединение, такое, как металлорганические соединения с элементами 13-й группы, представленные в US 5241025, ЕР А-0426638, и те, которые представлены в US 5767208. Примеры соединений включают триэтилалюминий, триэтилборан, триизобутилалюминий, метилалюмоксан, изобутилалюмоксан, три-н-гексилалюминий и три-н-октилалюминий, причем для сведения к минимальному нежелательного взаимодействия с активным катализатором предпочтительны те соединения, у которых имеются объемистые заместители, ковалентно связанные с металлическим или металлоидным центром. Добавление избыточного количества очищающего средства приводит к уменьшенным производительности, молекулярной массы и степени введения сомономерных звеньев. Следовательно, молярное соотношение между алюминием и гафнием (Al:Hf) должно составлять меньше примерно 100:1, предпочтительно меньше примерно 75:1, более предпочтительно меньше примерно 50:1, а наиболее предпочтительно меньше примерно 30:1. Было отмечено, что для проведения непрерывных процессов, описанных в данной заявке, достаточны молярные соотношения меньше 20:1 и меньше 15:1.
Предпочтительным очищающим средством является линейное алюминийтриалкильное соединение с длинной цепью, причем средства с более длинными цепями предпочтительнее средств с более короткими цепями (для дальнейшего обсуждения см. WO 97/22635 и US 5767208; в соответствии с существующей в США практикой патентования изобретений этот документ включен в настоящее описание в качестве ссылки). Неограничивающие примеры эффективных длинноцепочечных линейных триалкиллигандсодержащих очищающих средств включают те, которые входят в ряд соединений, отвечающих формуле M’R’R’’R’’’, где М’ обозначает Аl, а каждая из групп R независимо друг от друга представляет собой C4- или более высокомолекулярную линейную, разветвленную или циклическую алкильную группу, предпочтительно С6- или более высокомолекулярную, наиболее предпочтительно С8- или более высокомолекулярную. Было установлено, что длинноцепочечные линейные алюминийалкилы, у которых длина каждого алкильного заместителя соответствует 8 или большему числу атомов С, предпочтительно 9 или большему числу атомов С, проявляют оптимальные рабочие свойства и расцениваются как оказывающие наименьшее нежелательное воздействие, когда их используют в количестве, превышающем оптимальное содержание, которое указано в следующем абзаце. Конкретно они включают алюминийтри-н-октил, алюминийтри-н-децил, алюминийтри-н-додецил, алюминийтри-н-гексадецил и их эквиваленты с более значительным числом углеродных атомов, например (C20)3Al, включая соединения со смешанными лигандами, а также смешанные очищающие соединения. Кроме того, обычно приемлемы гидролизованные производные этих алкиллигандсодержащих алюминийорганических соединений. Более того очевидно, что приемлемы также те очищающие соединения, которые включают как длинноцепочечные линейные, так и объемистые лиганды или смешанные линейные лиганды, каждый из которых описан выше, но они, вероятно, менее желательны вследствие более сложных или дорогостоящих синтезов.
Предпочтительным процессом полимеризации является тот, который планируют и проводят таким образом, чтобы сокаталитические компоненты, т.е. соединения переходного металла и анионные соединения-предшественники, содержались раздельно непосредственно до момента их использования или до введения в процесс полимеризации в выбранном реакторе или реакторах. Примером является применение параллельного введения каждого каталитического компонента прямо в реактор или применение смесительных камер Т-образного типа или с более значительным числом впускных патрубков непосредственно перед введением в реактор. Дополнительная оптимизация может быть достигнута, когда очищающее соединение вводят в реактор независимо от каталитической системы или соединений, предпочтительно после активации гафноценов анионными предшественниками сокатализаторов.
Способ по изобретению применим к гомогенной полимеризации под высоким давлением, предпочтительно с использованием меньше 30 мас.% растворителя в процессе, который является практически адиабатическим и в котором теплоту полимеризации расходуют на повышение температуры содержимого реактора, а не отводят за счет внутреннего или внешнего охлаждения. В этом случае содержимое состоит главным образом из непрореагировавшего мономера. Такой процесс можно вести в одно- или двухфазных гомогенных условиях под давлением 250-3000 бар, предпочтительно 500-2500 бар, с добавлением или без добавления нереакционноспособных разбавителей или растворителей при температуре, которая обычно превышает точку плавления получаемого полимера. Такие процессы в промышленности известны и могут включать применение очищающих соединений и стадий дезактивации или подавления катализатора (см., например, US 5408017, WO 95/07941 и WO 92/14766). Каждый из этих документов и их американские аналоги в соответствии с существующей в США практикой патентования изобретений включены в настоящее описание в качестве ссылок. Предпочтительные дезактиваторы или подавители катализаторов включают высокомолекулярные нереакционноспособные соединения, такие, как поливиниловый спирт, который проявляет функциональную способность к образованию с катализаторами комплексов, тем самым их дезактивируя без одновременного образования летучих полярных побочных продуктов или остаточных непрореагировавших соединений.
Предлагаемый по изобретению способ особенно эффективен также применительно к гомогенной полимеризации в растворе, которая является тоже по существу адиабатической, т.е., другими словами, теплота полимеризации расходуется на повышение температуры содержимого полимеризационного реактора, главным образом растворителя. В ходе проведения этого адиабатического процесса, как правило, не предусмотрено никакого внутреннего охлаждения и соответственно никакого внешнего охлаждения. Теплоту полимеризации отводит из реактора отходящий из реактора поток. Производительность такого адиабатического процесса можно повысить охлаждением поступающих растворителя и/или мономерного потока (потоков) перед вводом в реактор, что позволяет допустить более высокую экзотермию полимеризации. Таким образом, выбор катализатора, сокатализатора и очищающего средства, описанных в данной заявке, можно с успехом осуществить в ходе проведения непрерывного процесса в растворителе при температуре, от равной или превышающей 140°С, превышающей 150°С или превышающей 160°С до примерно 225°С. В наиболее предпочтительном варианте процесс полимеризации в растворе при получении полукристаллических полимеров проводят при температуре 140-220°С. Этот процесс, как правило, проводят в инертном линейном, циклическом или разветвленном алифатическом или ароматическом углеводородном растворителе под давлением 20-200 бар.
Способность этих катализаторов обеспечивать получение технически необходимого полимера при повышенных температурах содействует повышенной экзотермии, высокому содержанию полимера в реакторе благодаря пониженной вязкости и уменьшенному потреблению энергии на испарение и возврат в процесс растворителя, а также более высокой степени превращения мономера и сомономера.
Предпочтительными α-олефинами, приемлемыми для использования при получении этиленовых сополимеров или для полиэтиленовых сополимеров, являются С3-С20-α-олефины, но обычно к ним относятся олефины с более значительным числом углеродных атомов, такие, как способные полимеризоваться макромеры, включающие до пятисот или больше углеродных атомов. Иллюстрирующими, но неограничивающими примерами таких α-олефинов служат пропилен, 1-бутен, 1-пентен, 1-гексен, 1-октен и 1-децен. Принимая во внимание цели описания эффективно сополимеризующихся мономеров, понятие "олефины" охватывает циклические моноолефины с затрудненными кольцами, такие, как циклобутен, циклопентен, норборнен, алкилзамещенные норборнены, алкенилзамещенные норборнены и циклические олефины с более значительным числом углеродных атомов, известные в данной области техники (см., например, патент US 5635573, включенный в настоящее описание в качестве ссылки в соответствии с существующей в США практикой патентования изобретений), и известные сополимеризующиеся диолефины, например 1,4-гексадиен, этилиденнорборнен и винилнорборнен. Кроме того, приемлемы виниловые ароматические мономеры, например стирол и алкилзамещенные стирольные мономеры. Ряд полиэтиленовых сополимеров может охватывать от полукристаллических до практически аморфных, которые, как правило, характеризуются по существу неупорядоченным размещением звеньев по меньшей мере этилена и олефиновых сомономеров. Как очевидно для специалистов в данной области техники, применение асимметрично замещенных соединений гафния по изобретению дает возможность получать синдиотактичес-кие полимеры из прохиральных олефинов, например подобных пропилену. Дополнительные преимущества процессам такого рода придают увеличенные производительность и молекулярные массы, о которых говорилось в связи с этиленовыми сополимерами.
(Предпочтительные этиленовые сополимерные пластомеры по изобретению обычно проявляют полукристаллические характеристики, например точки плавления, находящиеся в интервале от примерно 85 до 115°С. Молекулярная масса (среднечисленная молекулярная масса) пластомеров по изобретению обычно находится в интервале от примерно 10000 до примерно 60000, предпочтительно от примерно 20000 до примерно 50000. Более часто молекулярную массу этиленовых сополимерных пластомеров отражают посредством значений их полиэтиленового индекса расплава (ИР) (определяют по стандарту ASTM D 1238, условие Е), которые, как правило, находятся в интервале 0,01-10,0, предпочтительно 0,005-6,0, а более предпочтительно от примерно 0,01 до меньше 3,0. Значения Мn этиленовых сополимерных эластомеров, как правило, составляют от ≥60000 до примерно 250000; в дополнение к звеньям этилена и одного или нескольких α-олефинов они могут включать необязательные звенья одного или нескольких несопряженных или циклических диолефинов, как правило, пропилена.
Что касается полимерной плотности, то у полимеров, которые могут быть получены в соответствии с изобретением, она может находиться в интервале от примерно 0,850 до примерно 0,930, предпочтительно 0,87-0,925, более предпочтительно 0,89-0,920. Пластомеры по изобретению обычно содержат от примерно 60 до 80 мас.% этиленовых звеньев, предпочтительно от примерно 60 до 75 мас.% этиленовых звеньев.
Каталитические комплексы по изобретению обеспечивают также введение значительных количеств сомономерных звеньев, например в случае этилена с С3-С8-альфа-олефинами и необязательными несопряженными С5-С20диолефинами или с любыми другими известными мономерами, способными сополимеризоваться с этиленом и способными проявлять высокую производительность по катализатору и обеспечивать получение высокомолекулярных сополимеров в промышленно эффективных условиях полимеризации в растворе. Такие условия, как правило, включают давление от нормального до умеренно высоких значений (т.е. ниже примерно 500 бар) и температуру в интервале от примерно 40 до 140°С, при которых способные полимеризоваться мономеры входят в контакт с каталитическими комплексами в практически жидкофазной полимеризационной среде, такой, как среда алифатического или ароматического растворителя или разбавителя. Такие катализаторы можно наносить на подложку в соответствии с известными методами нанесения на подложку металлоценовых катализаторов, в частности тех, которые используют в условиях суспензионной полимеризации. Условия проведения процесса как в растворе, так и в суспензии в данной области техники известны хорошо и могут быть легко адаптированы к применению катализаторов в соответствии с изобретением.
Примеры
Для иллюстрации вышеприведенного обсуждения приведены следующие примеры. Количества материалов во всех случаях, если не указано иное, выражены в массовых частях, пропорциях или процентах. Несмотря на то, что объектами этих примеров могут служить некоторые варианты выполнения настоящего изобретения, их не следует рассматривать как ограничивающие с какой-либо стороны объем изобретения. В таблицах 1 и 2 аббревиатурой "МЦН" обозначен металлоцен, в частности гафноцен по изобретению, а аббревиатурой "СК" обозначен сокатализатор.
Высокотемпературный полупериодический процесс полимеризации
Процессы сополимеризации этилена/1-октена проводили в 1-литровом реакторе периодического действия с хорошим перемешиванием, оборудованном с учетом возможности координационной полимеризации в среде инертного углеводородного растворителя (гексан) под давлением до 600 фунтов/кв. дюйм и при температурах до 150°С. В парожидкостной (ПЖ) полимеризационной системе полимеризация протекала в жидкой фазе, тогда как этилен вводили в реактор непрерывно для поддержания во время полимеризации постоянного давления в верхней части на уровне 265 фунтов/кв. дюйм. В ходе проведения этих экспериментов постоянную температуру (140°С) в реакторе поддерживали регулированием с помощью дросселя расхода водяного пара, вводимого в рубашку реактора, и регулированием количества катализатора, подаваемого в реактор насосом. В реактор, как правило, вводили 250 мл высушенного н-гексана, 18 мл высушенного 1-октена и 1,0 мл триизобутилалюминиевого раствора концентрацией 10 мас.% (толуол или гексан), очищающего средства, температуру которых затем доводили до 140°С. Далее введением этилена давление над содержимым реактора доводили до 265 фунтов/кв. дюйм и давление этилена во время всей полимеризации поддерживали постоянным. Полимеризацию начинали непрерывным введением во время полимеризации раствора (толуол или гексан) предварительно активированного катализатора. Предварительную активацию проводили введением катализатора и сокатализатора во взаимный контакт в толуоле перед подачей в реактор. Подачу потока катализатора прекращали, содержимому реактора давали остывать до комнатной температуры и сбрасывали давление. После выпадения продукта из раствора в осадок его в течение ночи сушили при комнатной температуре в вытяжном шкафу.
Пример 1А: получение 6-(п-трет-бутилфенил)-6’-(п-н-бутилфенил)-фульвена. В 1000-миллилитровой круглодонной колбе в 500 мл тетрагидрофурана растворяли 43,92 г соответствующего дизамещенного бензофенона. В этот раствор добавляли 90,0 мл циклопентадиенида натрия в тетрагидрофуране (2,0 М, фирма Aldrich). Реакционную смесь оставляли перемешиваться в течение 3 дней в инертной атмосфере (перчаточная камера). Далее реакционную смесь из камеры выносили и выливали в 300 мл воды. В эту смесь добавляли 400 мл диэтилового эфира. Органический слой отделяли. Водный слой один раз экстрагировали диэтиловым эфиром. Эфирные слои объединяли и сушили над сульфатом магния в течение 4 ч. Сульфат магния отделяли фильтрованием. После выпаривания растворителя в виде кирпично-красного масла получали продукт. Этот продукт очищали хроматографией в колонке (силикагель, гексан). В результате выделяли 39,11 г 6-(п-трет-бутилфенил)-6’-(п-н-бутилфенил)-фульвена. Строение этого продукта легко определяли по 1Н-ЯМР-спектрограмме, которую снимали в CDCl3 при комнатной температуре. С помощью спектрограммы отмечали наличие небольших количеств примесей, но влияния на последующую реакцию они не оказывали.
Пример 1Б: получение 6-(п-трет-бутилфенил)-6’-(п-метилфенил)фульвена. В 100-миллилитровой круглодонной колбе в 50 мл тетрагидрофурана растворяли 5,13 г соответствующего дизамещенного бензофенона. В этот раствор добавляли 10,0 мл циклопентадиенида натрия в тетрагидрофуране (2,0 М, фирма Aldrich). Реакционную смесь оставляли перемешиваться в течение 3 дней в инертной атмосфере (перчаточная камера). Далее реакционную смесь из камеры выносили и выливали в 30 мл воды. В эту смесь добавляли 100 мл диэтилового эфира. Органический слой отделяли. Водный слой один раз экстрагировали диэтиловым эфиром. Эфирные слои объединяли и сушили над сульфатом магния в течение 4 ч. Сульфат магния отделяли фильтрованием. После выпаривания растворителя в виде кирпично-красного масла, аналогичного соединению, описанному выше, получали продукт. Этот продукт очищали хроматографией в колонке (силикагель, метиленхлорид/гексан в соотношении 9/1). В результате выделяли 2,32 г 6-(п-трет-бутилфенил)-6’-(п-метилфенил)фульвена. Строение этого продукта легко определяли по 1Н-ЯМР-спектрограмме, которую снимали в CDCl3 при комнатной температуре. С помощью спектрограммы отмечали наличие небольших количеств примесей, но влияния на последующую реакцию они не оказывали.
Пример 2А: получение (п-тpeт-BuPh)(п-н-BuPh)C(Cp)(Flu)Li. В 40 мл толуола суспендировали 2,317 г флуоренила лития. В эту суспензию добавляли раствор, содержавший 4,608 г 6-(п-трет-бутилфенил)-6’-(п-н-бутилфенил)-фульвена, растворенного в приблизительно 80 мл толуола. Реакционную смесь оставляли перемешиваться в течение 30 мин. После удаления растворителя и растирания в пентане остатка в порошок твердый продукт собирали фильтрованием и промывали пентаном. В результате получали 6,17 г продукта. Продукт идентифицировали 1Н-ЯМР-спектроскопией при комнатной температуре в C6D6. В ароматическом диапазоне пики были широкими, но четкими и легко интерпретируемыми.
Пример 2Б: получение (п-Tpeт-BuPh)(п-MePh)C(Cp)(Flu)Li. В 40 мл толуола суспендировали 1,192 г флуоренила лития. В эту суспензию добавляли раствор, содержавший 2,081 г 6-(п-трет-бутилфенил)-6’-(п-метилфенил)фульвена, растворенного в приблизительно 80 мл толуола. Реакционную смесь оставляли перемешиваться в течение 1 ч. После удаления растворителя и растирания в пентане остатка в порошок твердый продукт собирали фильтрованием и промывали пентаном. В результате получали 2,960 г продукта. Продукт идентифицировали 1Н-ЯМР-спектроскопией при комнатной температуре в C6D6. В ароматическом диапазоне пики были широкими, но четкими и легко интерпретируемыми.
Пример 3А: получение (п-трет-ВuРh)(п-н-ВuРh)С(Ср)(Flu)НfСl2. В раствор в диэтиловом эфире, содержавшем 5,45 г (п-трет-ВuРh)(п-н-ВuРh)C(Cp)(Flu)Li, вводили 6,6 мл н-BuLi (1,6 М, фирма Aldrich). Реакционную смесь во время реакции литиирования оставляли перемешиваться в течение 2,5 ч. К дилитиевой соли в виде твердого вещества добавляли 3,45 г HfCl4. Реакционную смесь перемешивали в течение 14 ч. Фильтрованием отделяли хлорид лития. После выпаривания растворителя продукт экстрагировали дихлорметаном для удаления остаточного хлорида лития. Выпариванием удаляли растворитель. В результате получали остаток в виде темного масла. К этому маслу добавляли приблизительно 80 мл пентана и 10 мл диэтилового эфира. Это вызывало выпадение в осадок небольшого количества твердых частиц. Смесь оставляли стоять в холодильнике в течение 14 ч. Вследствие такого охлаждения происходило дальнейшее осаждение. Твердый продукт собирали фильтрованием и сушили под вакуумом с получением 2,532 г оранжевого твердого вещества. Выдержкой фильтрата при пониженной температуре в течение дальнейших 4 ч получали вторую порцию продукта (0,680 г) при совокупном выходе 3,212 г. Продукт идентифицировали 1Н-ЯМР-спектроскопией при комнатной температуре в C6D6.
Пример 3Б: получение (п-трет-ВuРh)(п-МеРh)С(Ср)(Flu)HfCl2. В раствор в диэтиловом эфире, содержавшем 2,96 г (п-тpeт-BuPh)(п-MePh)C(Cp)(Flu)Li, вводили 3,9 мл н-BuLi (1,6 М, фирма Aldrich). Реакционную смесь во время реакции литиирования оставляли перемешиваться в течение 4 ч. К дилитиевой соли добавляли 2,00 г HfCl4. Реакционную смесь перемешивали в течение 14 ч. После выпаривания растворителя продукт экстрагировали дихлорметаном для удаления остаточного хлорида лития. Выпариванием удаляли растворитель. В результате получали полутвердый продукт, который промывали пентаном. Продукт собирали фильтрованием и для удаления углеводородных примесей промывали небольшим количеством холодного пентана. В результате этого получали 3,733 г оранжевого твердого вещества. Продукт идентифицировали 1Н-ЯМР-спектроскопией при комнатной температуре в C6D6.
Пример 4А: метилирование (п-тpeт-BuPh)(п-н-BuPh)C(Cp)(Flu)HfCl2. Три эквивалента MeMgBr (3,0 М в диэтиловом эфире, фирма Aldrich) вводили в холодную (-35°С) суспензию, содержавшую 3,63 г (п-трет-ВuРh)(п-н-BuPh)C(Cp)(Flu)HfCl2 в толуоле. Реакционной смеси давали нагреться в течение 30 мин до комнатной температуры. Далее в течение двух часов реакционную смесь выдерживали при 80°С. Вследствие нагрева реакционная смесь становилась темно-коричневой. Реакционную смесь фильтровали с использованием броунмиллерита для удаления темного твердого вещества. В фильтрат добавляли избыток триметилхлорсилана и перемешивали в течение 2 ч. Осуществление этой последней стадии гарантировало нейтрализацию избытка MeLi. Растворитель заменяли метилендихлоридом и выпадавший в осадок LiCl отделяли фильтрованием. Объем уменьшали до минимума и для инициирования осаждения добавляли пентан. После выдержки при пониженной температуре в течение ночи фильтрованием собирали желтый продукт. В результате этого получали 1,70 г светло-желтого твердого вещества. Продукт идентифицировали 1Н-ЯМР-спектроскопией при комнатной температуре в C6D6.
Пример 4Б: метилирование (п-трет-ВuРh)(п-МеРh)С(Ср)(Flu)HfCl2. Три эквивалента MeMgBr (3,0 М в диэтиловом эфире, фирма Aldrich) вводили в холодную (-35°С) суспензию, содержавшую 3,70 г (п-трет-ВuРh)(п-МеРh)-C(Cp)(Flu)HfCl2 в толуоле. Реакционной смеси давали нагреться в течение 30 мин до комнатной температуры. Далее в течение двух часов реакционную смесь выдерживали при 80°С. Реакционную смесь фильтровали через броунмиллерит для удаления темного твердого вещества. В фильтрат добавляли избыток триметилхлорсилана и перемешивали в течение 3 ч. Осуществление этой последней стадии гарантировало нейтрализацию избытка MeLi. Растворитель заменяли метилендихлоридом и выпадавший в осадок LiCl отделяли фильтрованием. Объем уменьшали до минимума и для инициирования осаждения добавляли пентан. После выдержки при пониженной температуре в течение ночи фильтрованием собирали желтый продукт. Продукт идентифицировали 1Н-ЯМР-спектроскопией при комнатной температуре в C6D6.
Пример 5: синтез (п-н-ВиРh)(п-трет-ВuРh)С(Ср)(2,7-трет-ВuFlu)НfСl2. В раствор в диэтиловом эфире, содержавшем 1,35 г 6,6’-дифенилфульвена, вводили раствор, содержавший 1,12 г литий-2,7-ди-трет-бутилфлуорена. По прошествии 20 мин начинало выпадать в осадок бежевое твердое вещество. Реакционную смесь перемешивали в течение 6 ч. В реакционную смесь добавляли один эквивалент н-BuLi (7,38 мл, 1,6 М в диэтиловом эфире, фирма Aldrich). По прошествии 15 ч окраска реакционной смеси изменялась на бордово-красную и образовывался красный осадок. В красную смесь добавляли 1,26 г тетрахлорида гафния. Реакционную смесь оставляли перемешиваться в течение 3 ч. Смесь обладала оранжево-желтой окраской и обильным осадком. Растворитель заменяли дихлорметаном и смесь фильтровали. Для более полной экстракции продукта остаточные твердые частицы было необходимо промывать несколько раз. Под пониженным давлением удаляли растворитель. Продукт растирали в порошок с пентаном и собирали фильтрованием. В результате оставался оранжевый твердый продукт (1,045 г).
Синтез (п-н-BuPh)(п-тpeт-BuPh)C(Cp)(2,7-тpeт-BuFlu)HfMe2. В холодный (-35°С) раствор, содержавший 1,00 г (п-н-ВuРh)(п-трет-ВuРh)-С(Ср)(2,7-трет-ВuFlu)НfСl2 в толуоле вводили три эквивалента MeMgBr (3,0 М, фирма Aldrich). Реакционной смеси давали медленно нагреться до комнатной температуры, а затем в течение 3 ч реакционную смесь выдерживали при 80°С. В результате этого реакционная смесь становилась темно-коричневой. Реакционную смесь переносили в перчаточную камеру и пропускали через слой броунмиллерита. Это позволяло получить оранжевый раствор. Объем растворителя уменьшали и продукт растирали в порошок с пентаном. Фильтрованием собирали 0,300 г продукта.
Значения символов в приведенных ниже таблицах 1-3 приведены в таблице 1а
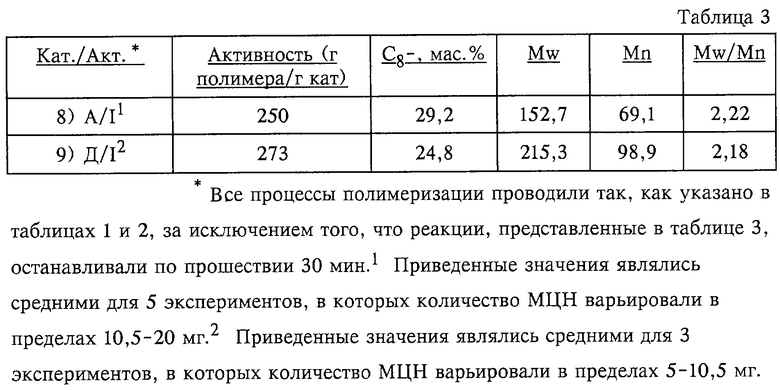
Как видно из данных таблицы 1, катализаторы по изобретению примеров 3)-5) демонстрируют значительное улучшение активности и в меньшей степени улучшение средневесовой и среднечисленной молекулярных масс, когда мостиковые арильные группы замещают в соответствии с изобретением в противоположность тем случаям, когда этого не производят. Данные определения Mw/Mn в примере 3) сомнительны, но отмеченное в этом случае противоречие непонятно. Полагают, что при повторе в создаваемых при этом условиях пришли бы к достижению значений, которые были бы соразмерны с полученными в примерах 4) и 5). Данные таблицы 2 создают возможность прямого сопоставления в МЦН "А" с незамещенными мостиковыми арильными группами и незамещенными флуоренильными группами с МНЦ "Г", обладавшим как замещенными мостиковыми арильными группами, так и замещенными флуоренильными группами в соответствии с изобретением. В таблице 3 проиллюстрирован МЦН "Д" с алкильными заместителями в мостиковых арильных группах, причем такие заместители не включают линейные н-алкилы С3- или с большим числом углеродных атомов по изобретению, и с заместителями во флуоренильных группах, как в случае МЦН "Г" по изобретению. Очевидно, что значения активности в примерах 8) и 9) сопоставимы, но эксперимент примера 9) демонстрирует улучшение молекулярных масс. Поскольку пример 8) иллюстрирует стандарт для сравнения с МЦН "А", от такого стандарта "Д" для таблиц 1 и 2 ожидается демонстрация значений активности, аналогичных тем, которые проявлял МЦН "А", и хуже, чем в случаях "В" и "Г" по изобретению.
| название | год | авторы | номер документа |
|---|---|---|---|
| СВЯЗАННЫЕ МОСТИКАМИ МЕТАЛЛОЦЕНЫ, СПОСОБ ПОЛИМЕРИЗАЦИИ | 1999 |
|
RU2232766C2 |
| МЕТАЛЛООРГАНИЧЕСКОЕ СОЕДИНЕНИЕ ПЕРЕХОДНОГО МЕТАЛЛА, БИСЦИКЛОПЕНТАДИЕНИЛЬНАЯ ЛИГАНДНАЯ СИСТЕМА, КАТАЛИТИЧЕСКАЯ СИСТЕМА И ПОЛУЧЕНИЕ ПОЛИОЛЕФИНОВ | 2004 |
|
RU2362779C2 |
| СПОСОБ РАЦЕМОСЕЛЕКТИВНОГО СИНТЕЗА АНСА-МЕТАЛЛОЦЕНОВ | 2005 |
|
RU2391350C2 |
| КОМПЛЕКСЫ ТИТАНА (II) ИЛИ ЦИРКОНИЯ (II), КАТАЛИТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПОЛИМЕРИЗАЦИИ ЭТИЛЕННЕНАСЫЩЕННЫХ ОЛЕФИНОВ | 1994 |
|
RU2135509C1 |
| БИСЦИКЛОПЕНТАДИЕНИЛДИЕНОВЫЕ КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ, СПОСОБ ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ | 1995 |
|
RU2135508C1 |
| СПОСОБ ПОЛУЧЕНИЯ КОМПОЗИЦИИ СОПОЛИМЕРОВ ЭТИЛЕНА И АЛЬФА-ОЛЕФИНОВ, КОМПОЗИЦИЯ СОПОЛИМЕРОВ ЭТИЛЕНА И АЛЬФА-ОЛЕФИНОВ | 1994 |
|
RU2113443C1 |
| СПОСОБ ПОЛУЧЕНИЯ СОПОЛИМЕРОВ И СОДЕРЖАЩИХ ИХ СМЕСЕВЫХ КОМПОЗИЦИЙ | 1997 |
|
RU2179558C2 |
| ПОЛИЭТИЛЕН ДЛЯ ЛИТЬЕВОГО ФОРМОВАНИЯ | 2005 |
|
RU2395527C2 |
| ПОЛИЭТИЛЕН И КАТАЛИТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЕГО ПОЛУЧЕНИЯ | 2005 |
|
RU2387681C2 |
| СПОСОБ РЕГУЛИРОВАНИЯ АКТИВНОСТИ БИМОДАЛЬНОГО КАТАЛИЗАТОРА В ПРОЦЕССЕ ПОЛИМЕРИЗАЦИИ | 2008 |
|
RU2479593C2 |
Изобретение относится к способам полимеризации олефинов с применением гафноценовых каталитических комплексов. Описан способ полимеризации при получении этиленовых сополимеров, плотность которых составляет от примерно 0,850 до примерно 0,930, включающий введение в гомогенных полимеризационных условиях при реакционной температуре, от равной или превышающей 60 до 225°С, этилена и одного или нескольких сомономеров, способных к полимеризации внедрением, в контакт с гафноценовым каталитическим комплексом с мостиковой связью, дериватизированным из А) бициклопентадиенильного гафнийсодержащего металлорганического соединения, включающего I) по меньшей мере один незамещенный циклопентадиенильный лиганд или замещенный ароматическими конденсированными кольцами циклопентадиенильный лиганд, II) один замещенный ароматическими конденсированными кольцами циклопентадиенильный лиганд и III) ковалентный мостик, связывающий оба циклопентадиенильных лиганда, причем такой мостик включает один атом углерода или кремния с двумя арильными группами, каждая из которых замещена С1-С20гидрокарбильной или гидрокарбилсилильной группой, по меньшей мере одна из которых представляет собой линейный заместитель С3 или с большим числом атомов углерода, и Б) активирующего сокатализатора. Технический результат: описан способ, позволяющий получать сополимеры с высокими молекулярными массами. 1 с. и 17 з.п.ф-лы, 4 табл.
А) бициклопентадиенильного гафнийсодержащего металлорганического соединения, включающего
I) по меньшей мере один незамещенный циклопентадиенильный лиганд или замещенный ароматическими конденсированными кольцами циклопентадиенильный лиганд,
II) один замещенный ароматическими конденсированными кольцами циклопентадиенильный лиганд и
III) ковалентный мостик, связывающий оба циклопентадиенильных лиганда, причем такой мостик включает один атом углерода или кремния с двумя арильными группами, каждая из которых замещена С1-С20 гидрокарбилом или гидрокарбилсилилом, по меньшей мере один из которых представляет собой линейный заместитель С3 или с большим числом атомов углерода, и
Б) активирующего сокаталитического соединения, включающего комплексы с элементом 13 группы, содержащие по меньшей мере два галоидированных ароматических лиганда.
(п-трет-бутилфенил) (п-н-бутилфенил)метилен(циклопентадиенил) (2,7-диметил-флуоренил)гафнийдиметил и (п-трет-бутилфенил) (п-н-бутилфенил) метилен(циклопентадиенил) (2,7-дитрет-бутил-флуоренил)гафнийдиметил.
триэтиламмонийтетракис(перфторнафтил)бор или -тетракис(перфтор4-дифенил)бор,
три(н-бутил)аммонийтетракис(перфторнафтил)бор или -тетракис(перфтор-4-дифенил)бор,
три(н-октил)аммонийтетракис(перфторнафтил)бор или -тетракис(перфтор-4-дифенил)бор,
триметиламмонийтетракис(перфторнафтил)бор или -тетракис(перфтор-4-дифенил)бор,
триметиламмонийтетратетракис(перфторнафтил)бор или -тетракис-(перфтор-4-дифенил)бор,
трибутиламмонийтетракис(перфторнафтил)бор или -тетракис (перфтор-4-дифенил)бор,
трипропиламмонийтетракис(перфторнафтил)бор или -тетракис(перфтор-4-дифенил)бор,
три(н-бутил) аммонийтетракис(перфторнафтил)бор или -тетракис(перфтор-4-дифенил)бор,
N,N-диметиланилинийтетракис(перфторнафтил)бор или -тетракис(перфтор-4-дифенил)бор,
N,N-ди(н-додецил)анилинийтетракис(перфторнафтил)бор или -тетракис (перфтор-4-дифенил)бор,
N,N-2,4,6-пентаметиланилинийтетракис (перфторнафтил)бор или -тетракис (перфтор-4-дифенил)бор,
ди(н-додецил)аммонийтетракис(перфторнафтил)бор или -тетракис(перфтор-4-дифенил)бор,
дициклогексиламмонийтетракис(перфторнафтил)бор или -тетракис(перфтор-4-дифенил)бор,
трифенилфосфонийтетракис(перфторнафтил)бор или -тетракис(перфтор-4-дифенил)бор,
три(метилфенил)фосфонийтетракис(перфторнафтил)бор или -тетракис(пер-фтор-4-дифенил)бор,
три(диметилфенил)-фосфонийтетракис(перфторнафтил)бор или -тетракис (перфтор-4-дифенил)бор,
тропиллийтетракис(перфторнафтил)борат или -тетракис(перфтор-4-дифе-нил)борат,
трифенилметилийтетракис(перфторнафтил)борат или -тетракис(перфтор-4-дифенил)борат,
бензол(диазоний)тетракис(перфторнафтил)борат или -тетракис(перфтор-4-дифенил)борат.
| RU 94033097 А1, 20.07.1996 | |||
| ЕР 0786466 А1, 30.07.1997 | |||
| JP 07247309 А1, 26.09.1995. |

