Данное изобретение относится к композиции длительного высвобождения биологически активного вещества, способу получения этой композиции.
В не прошедшей экспертизу заявке на патент Японии №97334/1995 заявлен препарат длительного высвобождения, в состав которого входит биологически активный пептид или его соль и полимер, подвергающийся биологической деструкции и имеющий на одном конце свободную карбоксильную группу, и способ получения этого препарата.
В патентных публикациях GB 22009937, GB 2234169, GB 2234896, GB 2257909 и ЕР заявлены композиции на основе биодеградируемого полимера, содержащие отдельно приготовленную водонерастворимую соль, такую как памоат пептида или белка, и способ получения этих композиций.
В международной заявке WO 95/15767 заявлен эмбонат (памоат) цетрореликса (антагонист РГЛГ (LH-RH)) и способ его получения, и показано, что характер высвобождения пептида в том случае, когда памоат включен в биодеградируемый полимер, остается таким же, как при использовании только одного памоата.
Данное изобретение обеспечивает новую композицию с высоким содержанием биологически активного вещества, которая позволяет контролировать скорость высвобождения этого вещества.
После проведения всестороннего исследования, направленного на разрешение указанной выше проблемы, авторы данного изобретения обнаружили, что при высоком содержании биологически активного вещества, включенного в состав композиции путем одновременного введения биологически активного вещества и оксинафтойной кислоты в момент формирования композиции, и в том случае, когда оба эти компонента включены в биодеградируемый полимер, биологически активное вещество высвобождается со скоростью, отличной от скорости высвобождения биологически активного вещества из точно такой же композиции биологически активного вещества и оксинафтойной кислоты, приготовленной в отсутствие биодеградируемого полимера, причем скорость выхода биологически активного вещества контролируется выбором соответствующего вида биодеградируемого полимера. На основе этого наблюдения авторы изобретения провели дальнейшие исследования и разработали данное изобретение.
Таким образом, в настоящем изобретении представлены следующие объекты:
(1) композиция длительного высвобождения, содержащая биологически активное вещество или его соль, оксинафтойную кислоту или ее соль и биодеградируемый полимер или его соль,
(2) композиция длительного высвобождения по п.(1), указанному выше, в которой биологически активным веществом является биологически активный пептид,
(3) композиция длительного высвобождения по п.(2), указанному выше, в которой биологически активным пептидом является производное РГЛГ(LН-RН),
(4) композиция длительного высвобождения по п.(1), указанному выше, в которой оксинафтойной кислотой является 3-окси-2-нафтойная кислота,
(5) композиция длительного высвобождения по п.(1), указанному выше, в которой биодеградируемым полимером является полимер α-гидроксикарбоновой кислоты,
(6) композиция длительного высвобождения по п.(5), указанному выше, в которой полимером α-гидроксикарбоновой кислоты является полимер молочной кислоты - гликолевой кислоты,
(7) композиция длительного высвобождения по п.(6), указанному выше, в которой отношение содержания молочной кислоты к содержанию гликолевой кислоты составляет от 100/0 до 40/60 мол.%,
(8) композиция длительного высвобождения по п.(7), указанному выше, в которой отношение содержания молочной кислоты к содержанию гликолевой кислоты составляет 100/0 мол.%,
(9) композиция длительного высвобождения по п.(6), указанному выше, в которой среднемассовая молекулярная масса полимера составляет примерно от 3000 до 100000,
(10) композиция длительного высвобождения по п.(9), указанному выше, в которой среднемассовая молекулярная масса полимера составляет примерно от 20000 до 50000,
(11) композиция длительного высвобождения по п.(3), указанному выше, в которой производным РГЛГ является пептид, изображаемый следующей формулой:
5-оксо-Pro-His-Trp-Ser-Tyr-Y-Leu-Arg-Pro-Z,
в которой Y означает DLeu, DAla, DTrp, DSer(tBu), D2Nal или DHis(ImBzl); Z означает NH-C2H5 или Gly-NH2,
(12) композиция длительного высвобождения по п.(6), указанному выше, в которой содержание концевых карбоксильных групп полимера составляет 50-90 микромоль на единицу массы (грамм) полимера,
(13) композиция длительного высвобождения по п.(3), указанному выше, в которой молярное отношение оксинафтойной кислоты или ее соли к производному РГЛГ или его соли составляет от 3 : 4 до 4 : 3,
(14) композиция длительного высвобождения по п.(13), указанному выше, в которой концентрация производного РГЛГ или его соли составляет от 14% (вес/вес) до 24% (вес/вес),
(15) композиция длительного высвобождения по п.(1), указанному выше, в которой биоактивное вещество или его соль очень мало растворимы в воде или растворимы в воде,
(16) композиция длительного высвобождения по п.(1), указанному выше, которая предназначена для инъекций,
(17) способ получения композиции длительного высвобождения по п.(1), указанному выше, включающий в себя удаление растворителя из смеси биоактивного вещества или его соли, биодеградируемого полимера или его соли и оксинафтойной кислоты или ее соли,
(18) способ получения композиции длительного высвобождения по п.(17), указанному выше, включающий в себя смешивание и диспергирование биоактивного вещества или его соли в растворе, содержащем биодеградируемый полимер или его соль и оксинафтойную кислоту или ее соль в органическом растворителе, и последующее удаление органического растворителя,
(19) способ получения композиции длительного высвобождения по п.(18), указанному выше, в котором биоактивное вещество или его соль используется в виде водного раствора,
(20) способ получения по п.(17), указанному выше, в котором солью биоактивного вещества является соль, образованная свободным основанием или кислотой,
(21) фармацевтический препарат, содержащий композицию длительного высвобождения по п.(1), указанному выше,
(22) средство профилактики или лечения карциномы простаты, гипертрофии простаты, эндометриоза, миомы матки, фибромы матки, преждевременного полового созревания, дисменореи или рака молочной железы или средство контрацепции, содержащее композицию длительного высвобождения по п.3, указанному выше,
(23) композиция длительного высвобождения, содержащая оксинафтоат биоактивного вещества и биодеградируемый полимер или его соль,
(24) способ подавления бурного начального высвобождения биоактивного вещества из композиции длительного высвобождения, включающий в себя использование оксинафтойной кислоты или ее соли,
(25) способ повышения эффективности включения биоактивного вещества в композицию длительного высвобождения, включающий в себя использование оксинафтойной кислоты или ее соли,
(26) оксинафтоат биоактивного пептида,
(27) оксинафтоат биоактивного пептида по п.(26), указанному выше, который растворим в воде или очень мало растворим в воде, и
(28) композиция длительного высвобождения, содержащая оксинафтоат биоактивного пептида.
Далее в данном изобретении представлены следующие объекты:
(29) композиция длительного высвобождения по п.(28), указанному выше, в которой содержание оксинафтойной кислоты или ее соли составляет примерно от 1 до 7 моль, предпочтительно примерно от 1 до 2 моль, на 1 моль биоактивного пептида или его соли,
(30) способ получения композиции длительного высвобождения по п.(17), указанному выше, включающий в себя получение эмульсии вода/масло, в которой дисперсная водная фаза представлена раствором, содержащим биоактивное вещество или его соль, и масляная фаза представлена раствором, содержащим биодеградируемый полимер и оксинафтойную кислоту или ее соль, и последующее удаление растворителя,
(31) способ получения композиции длительного высвобождения по п.(17), указанному выше, включающий в себя получение эмульсии вода/масло, в которой дисперсная водная фаза представлена раствором, содержащим оксинафтойную кислоту или ее соль, и масляная фаза представлена раствором, содержащим биоактивное вещество или его соль и биодеградируемый полимер или его соль, и последующее удаление растворителя,
(32) способ получения композиции длительного высвобождения по п.(28), указанному выше, включающий в себя смешивание и растворение биоактивного пептида или его соли и оксинафтойной кислоты или ее соли, и последующее удаление растворителя, и
(33) способ получения композиции длительного высвобождения по любому из пп.30-32, в котором в качестве способа удаления растворителя используется способ сушки водосодержащих эмульсий.
В данном изобретении могут применяться все без ограничения биологически активные вещества при условии, что они допущены к фармакологическому применению, и это могут быть непептидные и пептидные вещества. К непептидным веществам относятся агонисты, антагонисты и вещества, обладающие ингибирующей ферменты активностью. К пептидным веществам относятся, например, биологически активные пептиды, в частности пептиды с молекулярной массой примерно от 300 до 40000, предпочтительно примерно от 400 до 30000 и более предпочтительно примерно от 500 до 20000.
К таким биологически активным пептидам относятся, например, рилизинг-гормон лютеинизирующего гормона (РГЛГ), инсулин, соматостатин, гормоны роста, рилизинг-фактор гормона роста (РФГР), пролактин, эритропоэтин, адренокортикотропный гормон, меланоцитстимулирующий гормон, рилизинг-фактор тиреоидного гормона, тиреотропный гормон, лютеинизирующий гормон, фолликулостимулирующий гормон, вазопрессин, окситоцин, кальцитонин, гастрин, секретин, панкреозимин, холецистокинин, ангиотензин, плацентарный лактоген человека, хорионический гонадотропин человека, энкефалин, эндорфин, киоторфин, туфтсин, тимопоэтин, тимозин, тимостимулин, тимический гуморальный фактор, сывороточный тимический фактор, фактор некроза опухолей, колониестимулирующий фактор, мотилин, дайнорфин, бомбезин, нейротензин, церулеин, брадикинин, атриальный натрийуретический фактор, фактор роста нервов, фактор роста клеток, нейротрофический фактор, пептиды - антагонисты эндотелина, их производные, фрагменты этих пептидов и производные этих фрагментов.
Биологически активный пептид может использоваться в данном изобретении либо как таковой, либо в виде соли, допущенной к фармакологическому применению.
К таким солям относятся соли неорганических кислот (их можно также назвать неорганическими свободными кислотами) (например, соли угольной кислоты (карбонаты и бикарбонаты), соляной кислоты, серной кислоты, азотной кислоты, борной кислоты), соли органических кислот (их можно также назвать органическими свободными кислотами) (например, янтарной кислоты, уксусной кислоты, пропионовой кислоты, трифторуксусной кислоты) и т.д., которые образуются в том случае, когда биологически активный пептид имеет основную группу, такую как аминогруппа.
Когда указанный биологически активный пептид содержит кислую группу, такую как карбоксильная группа, то такие соли образуются неорганическими основаниями (их можно также назвать неорганическими свободными основаниями) (например, щелочных металлов, таких как натрий и калий, щелочноземельных металлов, таких как кальций и магний), органическими основаниями (их можно также назвать органическими свободными основаниями) (например, органическими аминами, такими как триэтиламин, основными аминокислотами, такими как аргинин) и т.д. Биологически активный пептид может образовывать комплексные соединения с металлами (например, комплекс меди, комплекс цинка).
Наиболее предпочтительными примерами описанного выше биологически активного пептида являются производные РГЛГ или их соли, которые эффективны при лечении зависимых от половых гормонов заболеваний, таких как карцинома простаты, гипертрофия простаты, эндометриоз, миома матки, преждевременное половое созревание и рак молочной железы, и эффективны в качестве средств контрацепции.
Примерами производных РГЛГ или их солей могут служить, например, пептиды, описанные в "Treatment with GnRH Analogs: Controversies and Perspectives" (The Parthenon Publishing Group Ltd., публикация 1996 г.), в прошедшей экспертизу заявке на патент Японии №503165/1991, в не прошедших экспертизу заявках на патент Японии №№101695/1991, 97334/1995 и 259460/1996 и в других публикациях.
Производными РГЛГ могут быть агонисты РГЛГ или антагонисты РГЛГ; пригодными для данного изобретения антагонистами РГЛГ могут быть, например, биологически активные пептиды, представленные общей формулой [I]:
X-DD2Nal-D4ClPhe-D3Pal-Ser-A-B-Leu-C-Pro-DAlaNH2,
где X представляет собой N (4Н2 -фуроил)Gly или NAc; A представляет собой один из следующих остатков: NMeTyr, Туr, Aph(Atz) и NMeAph(Atz); В представляет собой один из следующих остатков: DLys(Nic), DCit, DLys(AzaglyNic), DLys(AzaglyFur), DhArg(Et2), DAph(Atz) и DhCi; С представляет собой Lys(Nisp), Arg или hArg(Et2), или их соли.
Пригодными для данного изобретения агонистами РГЛГ могут быть, например, биологически активные пептиды, представленные общей формулой [II]:
5-оксо-Pro-His-Trp-Ser-Tyr-Y-Leu-Arg-Pro-Z,
где Y представляет собой один из следующих остатков: DLeu, DAla, DTrp, DSer (tBu), D2Nal и Dhis (lmBzl); Z представляет собой NH2-C2H5, Gly-NH2, или их соли. В частности, наиболее предпочтительными пептидами являются пептиды, в которых Y является DLeu и Z является NH-C2H5, (то есть пептид, представленный формулой 5-оксо-Pro-His-Trp-Ser-Tyr-DLeu-Arg-Pro-NH-C2H5).
Эти пептиды могут быть получены способами, описанными в упоминаемых выше ссылках или заявках на патенты, или основанными на них способами.
Используемые здесь сокращения имеют следующие значения:
N (4Н2-фуроил)Gly: остаток N-тетрагидрофуроилглицина
NAc: N-ацетильная группа
D2Nal: остаток D-3-(2-нафтил)аланина
D4ClPhe: остаток D-3-(4-хлор)фенилаланина
D3Pal: остаток D-3-(3-пиридил)аланина
NMeTyr: остаток N-метилтирозина
Aph(Atz): остаток N-[5’-(3’-амино-1’Н-
1’,2’,4’-триазолил)]фенилаланина
NMeAph(Atz): остаток N-метил-[5’-(3’-амино-1’Н-
1’,2’,4’-триазолил)]фенилаланина
DLys(Nic): остаток D-(e-N-никотиноил)лизина
DCit: остаток D-цитруллина
DLys(AzaglyNic): остаток D-(азаглицилникотиноил)лизина
DLys(AzaglyFur): остаток D-(азаглицилфуранил)лизина
DhArg(Et2): остаток D-(N,N’-диэтил)гомоаргинина
DAph(Atz): остаток D-N-[5’-(3’-амино-1’Н-
1’, 2’, 4’-триазолил)]фенилаланина
DhCi: остаток D-гомоцитруллина
Lys(Nisp): остаток (e-N-изопропил)лизина
hArg(Etz): остаток (N,N’-диэтил)гомоаргинина
Сокращенные названия аминокислот соответствуют сокращениям, используемым в биохимической номенклатуре, принятой комиссией ИЮПАК-ИЮБ [European Journal of Biochemistry, Vol.138, pp.9-37 (1984)] или сокращениям, которые обычно используются в связанных с этой проблемой областях. В случае, когда присутствует оптический изомер аминокислоты, эта аминокислота находится в L-конфигурации, если только не оговорено особо.
Оксинафтойная кислота, используемая в данном изобретении, имеет в своем составе нафталиновое кольцо и имеет одну гидроксильную группу и 1 карбоксильную группу, обе группы могут связываться с различными атомами углерода нафталинового кольца. Поэтому существует всего 14 изомеров с различным положением гидроксильной группы относительно карбоксильной группы, локализованной в положениях 1 и 2 нафталинового кольца. Может быть использован любой из этих изомеров и может быть использована их смесь в различных соотношениях. Как описано далее, предпочтительным является высокое значение константы диссоциации кислоты, или чтобы значение рКа (рКа = -log10Ka, Ka представляет собой константу диссоциации кислоты) было низким. Предпочтение отдается также изомерам, которые очень слабо растворимы в воде.
Предпочтительными являются изомеры, которые растворимы в спиртах (например, этанол, метанол). Используемый здесь термин “растворимы в спиртах” означает, что растворимость составляет, например, не меньше чем 10 г/л метанола.
Что касается значений рКа описанных выше изомеров оксинафтойной кислоты, известно значение только для 3-окси-2-нафтойной кислоты (рКа=2,708, Kagaku Binran Kisohen II, опубликовано в Chemical Society of Japan, September 25,1969); однако полезная информация получена при сравнении значений рКа трех изомеров оксибензойной кислоты. В частности, значения рКа м-оксибензойной кислоты и п-оксибензойной кислоты составляют не менее 4, в то время как значение рКа о-оксибензойной кислоты (салициловая кислота) очень низкое (2,754). Поэтому предпочтительными из 14 приведенных выше изомеров являются изомеры, в состав которых входит нафталиновое кольцо, и карбоксильная и гидроксильная группы связаны с соседними атомами углеродного кольца, т.е. 3-окси-2-нафтойная кислота, 1-окси-2-нафтойная кислота и 2-окси-1-нафтойная кислота. Наиболее предпочтительна 3-окси-2-нафтойная кислота, в состав которой входит нафталиновое кольцо и гидроксильная группа связана с атомом углерода в 3 положении кольца и 1 карбоксильная группа связана с углеродом во 2 положении кольца.
Оксинафтойная кислота может использоваться в виде соли. К солям относятся, например, соли неорганических оснований (например, образованных щелочными металлами, такими как натрий и калий, щелочноземельными металлами, такими как кальций и магний), органических оснований (например, органических аминов, таких как триэтиламин, основных аминокислот, таких как аргинин), и соли и комплексные соли переходных металлов (например, цинка, железа, меди).
Пример способа получения соли оксинафтойной кислоты и биоактивного вещества, разработанного в данном изобретении, приводится ниже.
(1) Раствор оксинафтойной кислоты в гидратированном органическом растворителе пропускали через слабоосновную ионообменную колонку, чтобы адсорбировать кислоту и насытить колонку. Затем избыточную часть оксинафтойной кислоты удаляли с помощью гидратированного органического растворителя, после чего через колонку пропускали раствор биоактивного вещества в гидратированном органическом растворителе для осуществления ионного обмена; растворитель удаляли из полученного элюата. В качестве указанного гидратированного органического растворителя могут использоваться такие органические растворители как спирты (например, метанол, этанол), ацетонитрил, тетрагидрофуран и диметилформамид. Удаление растворителя для осаждения соли достигается использованием общеизвестного способа или способа, основанного на ранее известном методе. Примером такого способа может быть способ, в котором растворитель выпаривается в условиях вакуума с помощью роторного испарителя, и т.д.
(2) Через слабоосновную ионообменную колонку, после предварительно проведенного обмена ионов на гидроксильные ионы, пропускали раствор биоактивного вещества или его соли в гидратированном органическом растворителе для замещения основных групп гидроксильными группами. Оксинафтойную кислоту в количестве, не превышающем молярный эквивалент, добавляют к полученному элюату и растворяют с последующим концентрированием; преципитированную соль отмывают водой, сколько это необходимо, и высушивают.
Так как соль биоактивного вещества и оксинафтойной кислоты очень слабо растворима в воде, хотя растворимость зависит еще и от природы используемого биоактивного вещества, сама указанная соль биоактивного пептида как таковая, представляющая собой потенциально длительно высвобождающееся вещество, может использоваться в качестве препарата биоактивного вещества длительного высвобождения и так же может быть использована для получения композиции длительного высвобождения.
Биодеградируемыми полимерами, используемыми в данном изобретении, могут быть, например, полимеры и сополимеры, которые синтезируются на основе одного или более чем одного вида а - гидроксимонокарбоновых кислот (например, гликолевой кислоты, молочной кислоты), гидроксидикарбоновых кислот (например, яблочной кислоты), гидрокситрикарбоновых кислот (например, лимонной кислоты) и т.д., и на основе кислот, имеющих свободные карбоксильные группы, или их смесей; сложные полиэфиры α - цианакриловой кислоты; полиаминокислоты (например, поли-g-бензил-L-глутаминовая кислота); и сополимеры малеинового ангидрида (например, сополимеры стирола и малеиновой кислоты).
Связывание мономеров может быть в виде статистического полимера, блоксополимера или привитого сополимера. В том случае, когда в структуре молекул упоминаемых выше а - гидроксимонокарбоновых кислот, а - гидроксидикарбоновых кислот и а - гидрокситрикарбоновых кислот имеется оптически активный центр, они могут иметь D-, L- или DL-конфигурацию. Среди них полимеры молочной кислоты -гликолевой кислоты [упоминаемые в дальнейшем также как поли(лактид-со-гликолид), поли(молочная кислота-со-гликолевая кислота) или сополимер молочной кислоты - гликолевой кислоты, что в общем относится к гомополимерам и сополимерам молочной кислоты - гликолевой кислоты, если только не оговорено особо; гомополимеры молочной кислоты называются также полимерами молочной кислоты, полимолочными кислотами, полилактидами и т.д. и гомополимеры гликолевой кислоты называются полимерами гликолевой кислоты, полигликолевыми кислотами, полигликолидами и т.д.], при этом предпочтение отдается сложным поли(α-цианакриловым эфирам) и т.д. Большее предпочтение отдается полимерам молочной кислоты - гликолевой кислоты. Более предпочтительны для применения полимеры молочной кислоты - гликолевой кислоты, имеющие свободную карбоксильную группу на одном конце.
Биодеградируемый полимер может быть в виде соли. Примерами солей могут служить соли неорганических оснований (например, образованных щелочными металлами, такими как натрий и калий, щелочноземельными металлами, такими как кальций и магний), органических оснований (например, органических аминов, таких как триэтиленамин, основных аминокислот, таких как аргинин) и соли и комплексные соли переходных металлов (например, цинка, железа, меди).
В том случае, когда в качестве биодеградируемого полимера используется полимер молочной кислоты - гликолевой кислоты, предпочтительно соотношение их содержания (мол.%) примерно от 100/0 до 40/60, более предпочтительно примерно от 100/0 до 50/50. Также предпочтительными для использования являются гомополимеры молочной кислоты, в которых соотношение содержания кислот составляет 100/0.
Соотношение оптических изомеров молочной кислоты в одной минимальной повторяющейся единице указанного полимера молочной кислоты - гликолевой кислоты составляет предпочтительно от 75/25 до 25/75, речь идет об отношении D-конфигурации/L-конфигурации (моль/мол.%). Обычно используются полимеры молочной кислоты - гликолевой кислоты, в которых соотношение D-конфигурации/L-конфигурации (моль/мол.%) составляет примерно от 60/40 до 30/70.
Среднемассовая молекулярная масса указанного полимера молочной кислоты - гликолевой кислоты обычно составляет примерно от 3000 до 100000, преимущественно примерно от 3000 до 60000, более предпочтительно примерно от 3000 до 50000 и еще более предпочтительно примерно от 20000 до 50000.
Степень дисперсности (среднемассовая молекулярная масса/среднечисловая молекулярная масса) обычно примерно равна значению от 1,2 до 4,0, более предпочтительно от 1,5 до 3,5.
Содержание свободных карбоксильных групп в указанном полимере молочной кислоты - гликолевой кислоты составляет предпочтительно примерно от 20 до 1000 мкмоль, более предпочтительно от 40 до 1000 мкмоль на единицу массы (грамм) полимера.
Приведенные здесь среднемассовая молекулярная масса, среднечисловая молекулярная масса и степень дисперсности - это молекулярные массы и степень дисперсности, определенные на основе сравнения с полистиролом с помощью гель-проникающей хроматографии (ГПХ), при которой в качестве эталонов использовали 15 полистиролов со среднемассовой молекулярной массой 1110000, 707000, 455645, 354000, 189000, 156055, 98900, 66437, 37200, 17100, 9830, 5870, 2500, 1304 и 504 соответственно. Измерения проводили с помощью прибора для высокоскоростной ГПХ (производства Toso, HLC-8120GPC, определяемый показатель - показатель преломления) и колонки для ГПХ типа KF804Lx2 (производства Showa Denco), при этом в качестве подвижной фазы использовали хлороформ.
Термин “содержание свободных карбоксильных групп” используется здесь по отношению к содержанию, определяемому методом мечения (в дальнейшем упоминается в виде “содержания карбоксильных групп, определяемого методом мечения”). Детали методики определения содержания карбоксильных групп описаны ниже. Сначала навеску W (мг) полимолочной кислоты растворяют в 2 мл смеси 5 N соляной кислоты/ацетонитрила (объем/объем = 4/96); добавляют 2 мл 0,01 М раствора гидрохлорида о-нитрофенилгидразина (ОНФГ) (5 N соляная кислота/ацетонитрил/ этанол = 1,02/35/15) и 2 мл 0,15 М раствора 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорида (пиридин/этанол = 4/96 объем/объем), реакция продолжается в течение 30 минут при 40°С, после чего растворитель удаляется. После промывания водой (4 раза) остаток растворяют в 2 мл ацетонитрила; добавляют 1 мл 0,5 моль/л раствора гидроокиси калия в этаноле, после чего реакция продолжается 30 мин при 60°С. Реакционную смесь разбавляют 1,5 N водным раствором гидроокиси натрия до Y мл; определяется оптическая плотность А (см-1) при 544 нм с использованием в качестве контроля 1,5 N водного раствора гидроокиси натрия. Отдельно определяют содержание свободных карбоксильных групп С (моль/л) в стандартном водном растворе DL-молочной кислоты путем титрования щелочью. Определив значение оптической плотности В (см-1) при 544 нм гидразида DL-молочной кислоты, полученного методом мечения ОНФГ, можно рассчитать молярное содержание свободных карбоксильных групп на единицу массы (грамм) полимера с использованием уравнения:
[СООН] (моль/г)=(AYC)/(WB).
Хотя указанное содержание карбоксильных групп может быть также определено растворением биодеградируемого полимера в смеси растворителей толуол-ацетон-метанол и выявлением карбоксильных групп путем титрования этого раствора спиртовым раствором гидроокиси калия с использованием фенолфталеина в качестве индикатора (значение, полученное этим методом, в дальнейшем употребляется как “содержание карбоксильных групп, определяемое методом щелочного титрования”), желательно рассчитывать содержание на основе метода мечения, описанного выше, так как конечная точка титрования может быть определена не точно из-за конкуренции, обусловленной гидролитической реакцией главной цепи сложного полиэфира, во время титрования.
Скорость деструкции/элиминации биодеградируемого полимера варьирует в широких пределах, в зависимости от структуры сополимера, молекулярной массы или содержания свободных карбоксильных групп. Однако продолжительность выхода лекарственного средства может быть увеличена благодаря снижению относительной доли гликолевой кислоты или увеличению молекулярной массы и снижению содержания карбоксильных групп, так как в случае полимеров молочной кислоты - гликолевой кислоты деструкция/элиминация обычно замедляется при снижении относительной доли гликолевой кислоты. Однако поскольку содержание свободных карбоксильных групп влияет на эффективность включения биоактивного вещества в состав препарата, оно должно быть выше определенного уровня. По этой причине предпочтительно в биодеградируемом полимере, полученном для препарата длительного высвобождения пролонгированного типа (например, 6 месяцев и более), в данном случае для полимера молочной кислоты - гликолевой кислоты, использование полимолочной кислоты (например, D-молочной кислоты, L-молочной кислоты, DL-молочной кислоты, предпочтительно DL-молочной кислоты и т.д.), среднемассовая молекулярная масса и содержание карбоксильных групп в которой, определяемые, как описано выше, составляют примерно от 20000 до 50000 и примерно от 30 до 95 мкмоль/г, предпочтительно примерно от 40 до 95 мкмоль/г, наиболее предпочтительно примерно от 50 до 90 мкмоль/г.
Указанный “полимер молочной кислоты - гликолевой кислоты” может быть получен, например, способом некаталитической дегидратационной поликонденсации (не прошедшая экспертизу заявка на патент Японии №28521/1986) из молочной кислоты и гликолевой кислоты или полимеризацией с раскрытием цикла из лактида и соединения сложного циклического диэфира, такого как гликолид, с участием катализатора (Encyclopedic Handbook of Biomaterials and Bioengineering Part A: Materials, Volume 2, Marcel Dekker, Inc., 1995). Хотя полимер, полученный упомянутым выше известным способом полимеризации с раскрытием цикла, не всегда содержит свободную карбоксильную группу на одном конце, он также может использоваться после модификации, приводящей к образованию полимера, имеющего определенное количество карбоксильных групп на единицу массы благодаря проведению гидролитической реакции, описанной в ЕП-А-0839525.
Описанный выше “полимер молочной кислоты - гликолевой кислоты, имеющий свободную карбоксильную группу на одном конце”, может быть получен без проблем общеизвестным способом (например, некаталитической дегидратационной поликонденсацией, не прошедшая экспертизу заявка на патент Японии №28521/1986) или способом, описанным ниже.
(1) Сначала в присутствии производного гидроксимонокарбоновой кислоты (например, трет-бутил-D-лактата, бензил-L-лактата) с защищенной карбоксильной группой или производного гидроксидикарбоновой кислоты (например, дибензилтартроната, ди-трет-бутил-2-гидроксиэтилмалоната) с защищенной карбоксильной группой, соединение сложного циклического эфира подвергается реакции полимеризации с участием катализатора полимеризации.
Примерами описанных выше “производного гидроксимонокарбоновой кислоты с защищенной карбоксильной группой” или “производного гидроксидикарбоновой кислоты с защищенной карбоксильной группой” служат производные гидроксикарбоновых кислот с карбоксильной группой (-СООН), амидированной (-CONH2) или эстерифицированной (-COOR), причем предпочтение отдается производным гидроксикарбоновых кислот с карбоксильной группой (-СООН), которая эстерифицирована (-COOR) и т.д.
Примером R в сложных эфирах здесь могут служить C1-6 алкильные группы, такие как метил, этил, н-пропил, изопропил, н-бутил и трет-бутил, С3-8 циклоалкильные группы, такие как циклопентил и циклогексил, С6-12 арильные группы, такие как фенил и α-нафтил, и C7-14 аралкильные группы, такие как фенил-С1-2 алкильные группы, такие как бензил и фенетил, и α-нафтил-C1-2 алкильные группы, такие как α-нафтилметил. Среди этих групп предпочтительны трет-бутильные группы, бензильные группы и т.д.
Указанное “соединение сложного циклического эфира” относится к циклическим соединениям, имеющим, по меньшей мере, одну сложноэфирную связь в цикле. В частности, к таким соединениям относятся соединения сложных циклических моноэфиров (лактоны) или соединения сложных циклических диэфиров (лактиды).
Примером указанного “соединения сложного циклического моноэфира” могут служить лактоны с 4-членным кольцом (β-пропиолактон, β-бутиролактон, β-изовалеролактон, β-капролактон, β-изокапролактон, β-метил-β-валеролактон и т.д.), лактоны с 5-членным кольцом (γ-бутиролактон, γ-валеролактон и т.д.), лактоны с 6-членным кольцом (δ-валеролактон и т.д), лактоны с 7-членным кольцом (ε-капролактон и т.д.), п-диоксанон и 1,5-диоксепан-2-он.
Примером указанного “соединения сложного циклического диэфира” могут служить соединения, представленные формулой:
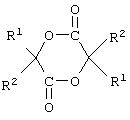
где R1 и R2, которые либо идентичны, либо не идентичны, представлены атомом водорода или C1-6 алкильной группой, такой как метил, этил, н-пропил, изопропил, н-бутил или трет-бутил, причем предпочтение отдается лактидам, имеющим в качестве R1 атом водорода и в качестве R2 метильную группу или имеющим атом водорода в качестве R1 и R2 и т.д.
В частности, к таким соединениям относятся гликолиды, L-лактиды, D-лактиды, DL-лактиды, мезо-лактиды и 3-метил-1,4-диоксан-2,5-дион (включая оптически активные конфигурации).
Примером указанного “катализатора полимеризации” являются оловоорганические катализаторы (например, октилат олова, ди-н-бутилоловодилаурилат, тетрафенилолово), алюминиевые катализаторы (например, триэтилалюминий) и цинковые катализаторы (например, диэтилцинк).
С точки зрения простоты удаления после реакции предпочтительны алюминиевые катализаторы и цинковые катализаторы; с точки зрения безопасности в случае неполного удаления предпочтительны цинковые катализаторы.
В качестве используемых растворителей катализаторов полимеризации могут быть бензол, гексан и толуол, причем предпочтение отдается гексану и толуолу.
Что касается “способа полимеризации”, то это может быть способ проведения полимеризации в массе, при котором продукт реакции находится в расплавленном состоянии, или способ проведения полимеризации в растворе, при котором продукт реакции растворен в соответствующем растворителе (например, бензол, толуол, ксилол, декалин, диметилформамид). Хотя нет ограничений для температуры полимеризации, при инициации реакции полимеризации в массе она не должна быть ниже, чем температура, при которой продукт реакции находится в расплавленном состоянии, обычно от 100 до 300°С, и для полимеризации в растворе это обычно от комнатной температуры до 150°С; если температура реакции превышает температуру кипения реакционного раствора, реакцию проводят с дефлегмацией с использованием конденсатора или в реакторе высокого давления. Принимая во внимание температуру полимеризации, определяют другие соответствующие условия реакции, желаемые физические свойства полимера и т.д., например время полимеризации может быть от 10 минут до 72 часов. После окончания реакции полимеризация останавливается добавлением кислоты (например, хлорной кислоты, уксусного ангидрида, трифторуксусной кислоты), с растворением реакционной смеси, если это необходимо, в соответствующем растворителе (например, ацетоне, дихлорметане, хлороформе), после чего смесь перемешивается с растворителем, который не растворяет нужный продукт (например, спирт, вода, эфир, изопропиловый эфир) или осаждается другим способом, после чего выделяется полимер, имеющий защищенную карбоксильную группу на ω-конце.
В способе полимеризации, представленном в данной заявке, вместо традиционных протонных агентов передачи цепи, таких как метанол, используются производные гидроксикарбоновых кислот (например, трет-бутил-D-лактат, бензил-L-лактат) с защищенной карбоксильной группой или производные гидроксидикарбоновых кислот (например, дибензилтартронат, ди-трет-бутил-L-2-гидроксиэтилмалонат) с защищенным карбоксилом.
Использование производных гидроксикарбоновых кислот (например, трет-бутил-D-лактата, бензил-L-лактата) с защищенной карбоксильной группой или производных гидроксидикарбоновых кислот (например, дибензилтартроната, ди-трет-бутил-L-2-гидроксиэтилмалоната) с защищенным карбоксилом в качестве протонных агентов передачи цепи делает возможным 1) контроль молекулярной массы за счет различной комбинации исходных материалов и 2) освобождение карбоксильной группы на ω-конце полученного биодеградируемого полимера благодаря реакции удаления защитных групп после полимеризации.
(2) Во-вторых, в результате того, что полимер, полученный в результате реакции полимеризации, описанной выше в параграфе (1), который имеет защищенную карбоксильную группу на ω-конце, подвергается реакции удаления защитных групп, может быть получен нужный биодеградируемый полимер, имеющий свободную карбоксильную группу на ω-конце.
Указанная защитная группа может быть удалена общеизвестными способами. К таким способам относятся все методы, которые дают возможность удалить защитную группу, не оказывая влияния на сложноэфирную связь поли(гидроксикарбоновой кислоты), в частности, в качестве примеров можно привести восстановление и кислотное расщепление.
К таким способам восстановления относится, например, каталитическое восстановление с использованием катализаторов (например, палладий на угольном носителе, палладиевая чернь, оксид платины), восстановление с использованием аммиачного раствора натрия и восстановление дитиотреитолом. В том случае, когда полимер, имеющий защищенную карбоксильную группу на ω-конце, подвергается каталитическому восстановлению, удаление защитных групп может достигаться, например, добавлением палладия на угольном носителе к раствору полимера в этилацетате, дихлорметане, хлороформе и других подобных растворителях и подачей водорода при комнатной температуре в течение примерно от 20 минут до 4 часов при энергичном встряхивании.
К таким способам кислотного расщепления относится, например, кислотное расщепление неорганическими кислотами (например, фтороводород, бромистый водород, хлористый водород), органическими кислотами (например, трифторуксусная кислота, метансульфоновая кислота, трифторметансульфоновая кислота) или их смесями. Также там, где это необходимо, добавляется соответственно акцептор протонов (например, анизол, фенол, тиоанизол). В том случае, когда полимер, имеющий на ω-конце карбоксильную группу, защищенную трет-бутильной группой, подвергается кислотному расщеплению, удаление защитных групп может достигаться, например, добавлением к раствору полимера в дихлорметане, ксилоле, толуоле или других подобных растворителях соответствующего количества трифторуксусной кислоты или растворением полимера в трифторуксусной кислоте и перемешиванием при комнатной температуре около 1 часа.
Предпочтительно указанное кислотное расщепление проводится сразу после реакции полимеризации; в этом случае оно может служить реакцией обрыва цепи полимеризации.
Более того, благодаря тому, что полимер, полученный в результате описанной выше реакции удаления защитных групп, подвергается, поскольку это необходимо, реакции кислотного гидролиза, можно регулировать, в зависимости от поставленной цели, среднемассовую молекулярную массу, среднечисловую молекулярную массу или содержание концевых карбоксильных групп указанного полимера. В частности, это может достигаться, например, способом, описанным в ЕП-А-0839525, или основанном на нем способом.
Биодеградируемый полимер, полученный, как описано выше, может использоваться как основа для получения препарата длительного высвобождения.
Кроме того, полимер, имеющий данную свободную карбоксильную группу на одном конце, может быть получен с помощью известных способов получения (например, смотри заявку на патент WO 94/15587).
Также в качестве полимера молочной кислоты - гликолевой кислоты, на одном конце которого в результате химической обработки после полимеризации с раскрытием цикла карбоксильная группа переходит в свободное состояние, может использоваться коммерческий продукт Boehringer Ingelheim KG.
Биодеградируемый полимер может быть в виде соли (к солям биодеградируемых полимеров относятся, например, соли, упоминавшиеся выше). К способам, используемым для их получения, относятся, например, следующие способы: (а) способ, при котором раствор описанного выше биодеградируемого полимера, имеющего карбоксильную группу, в органическом растворителе и водный раствор, содержащий ионы неорганических оснований (например, щелочных металлов, таких как натрий и калий, щелочноземельных металлов, таких как кальций и магний) или органических оснований (например, органических аминов, таких как триэтиламин, основных аминокислот, таких как аргинин) смешиваются вместе для ионообменной реакции, после которой выделяется полимер, находящийся теперь в виде соли, (b) способ, при котором соль слабой кислоты (например, ацетат, гликолат) и основания, одного из перечисленных выше в пункте (а), растворяется в растворе описанного выше биодеградируемого полимера, имеющего карбоксильную группу, в органическом растворителе, после чего выделяется полимер, находящийся теперь в виде соли, и (с) способ, при котором соль слабой кислоты (например, ацетат, гликолат) или оксид переходного металла (например, цинка, железа, меди) смешивается с раствором описанного выше биодеградируемого полимера, имеющего карбоксильную группу в органическом растворителе, после чего выделяется полимер, находящийся теперь в виде соли.
В качестве биодеградируемого полимера для препарата длительного высвобождения пролонгированного типа (например, 6 месяцев или более) предпочтителен “полимер молочной кислоты - гликолевой кислоты, имеющий свободную карбоксильную группу на одном конце”, полученный способом, описанным выше.
Массовая доля биоактивного вещества в композиции, представленной в данном изобретении, варьирует в зависимости от вида биоактивного вещества, желаемого фармакологического действия, продолжительности действия и других факторов. В случае композиции длительного высвобождения, содержащей три компонента (биоактивное вещество или его соль, оксинафтойная кислота или ее соль и биодеградируемый полимер или его соль) массовая доля биоактивного пептида или его соли составляет, например, приблизительно от 0,001 до 50% по массе, предпочтительно примерно от 0,02 до 40% по массе, более предпочтительно примерно от 0,1 до 30% по массе и наиболее предпочтительно примерно от 14 до 24% по массе, относительно суммарной массы трех компонентов. В случае непептидного биоактивного вещества или его соли массовая доля составляет примерно от 0,01 до 80% по массе, предпочтительно примерно от 0,1 до 50% по массе. Когда имеется соль, образованная оксинафтойной кислотой и биоактивным веществом, применимы сходные массовые доли. В случае композиции длительного высвобождения, содержащей соль, образованную биоактивным пептидом (обозначенным как (А)) и оксинафтойной кислотой (обозначенной как (В)), доля (А) по массе обычно составляет примерно от 5 до 90% по массе, предпочтительно примерно от 10 до 85% по массе, более предпочтительно примерно от 15 до 80% по массе и еще более предпочтительно примерно от 30 до 80% по массе, относительно суммарной массы соли, образованной (А) и (В).
В случае композиции длительного высвобождения, содержащей три компонента (биоактивное вещество или его соль, оксинафтойную кислоту или ее соль и биодеградируемый полимер или его соль), введенное в композицию количество оксинафтойной кислоты или ее соли составляет предпочтительно примерно от 1/2 до 2 моль, более предпочтительно примерно от 3/4 до 4/3 моль и еще более предпочтительно примерно от 4/5 до 6/5 моль на 1 моль биоактивного вещества или его соли.
Создание композиции, являющейся предметом данного изобретения, описывается в дальнейшем для композиции длительного высвобождения, состоящей из трех компонентов: основного биоактивного вещества, оксинафтойной кислоты и биодеградируемого полимера. В данном случае биоактивное вещество как основание и оксинафтойная кислота как кислота одновременно присутствуют в композиции; вводятся ли они в композицию в свободной конфигурации или в форме солей, для каждого компонента в гидратированном состоянии или в присутствии следовых количеств воды в момент образования композиции устанавливается равновесие реакции диссоциации. Поскольку соль, образованная оксинафтойной кислотой, которая очень слабо растворима в воде, и биоактивным веществом, предположительно тоже очень слабо растворима в воде, хотя растворимость зависит еще и от свойств указанного биоактивного вещества, равновесие диссоциации этой соли сдвигается в сторону образования соли, очень слабо растворимой в воде.
При создании композиции с высоким содержанием основного биоактивного вещества желательно, чтобы большая часть биоактивного вещества была протонирована, чтобы перевести его в форму очень слабо растворимой в воде соли, как описано выше, судя по описанному выше равновесию реакции диссоциации. С этой целью желательно, чтобы оксинафтойная кислота или ее соль были введены в композицию в количестве, по меньшей мере, близком к эквивалентному по отношению к количеству биоактивного вещества или его соли.
Далее описан механизм высвобождения биоактивного вещества, включенного в композицию. В описанной выше рецептуре композиции биоактивное вещество главным образом протонировано и присутствует вместе с противоионом. Противоионом является главным образом оксинафтойная кислота (предпочтительно оксинафтойная кислота). После введения композиции в живой организм в результате деструкции биодеградируемого полимера в течение определенного времени начинают образовываться олигомеры и мономеры. Когда указанным полимером является полимер молочной кислоты - гликолевой кислоты, образующийся олигомер (олигомер молочной кислоты - гликолевой кислоты) и мономер (молочная кислота или гликолевая кислота) всегда имеют одну карбоксильную группу, которая также может служить противоионом для биоактивного вещества. Биоактивное вещество высвобождается без переноса заряда или в форме соли с противоионом; к противоионам, способным к переносу заряда, относятся оксинафтойные кислоты, олигомеры молочной кислоты - гликолевой кислоты (такой молекулярной массы, при которой возможен перенос) и мономеры (молочная кислота или гликолевая кислота), как описано выше.
Когда одновременно присутствует множество кислот, обычно преимущественно образуются соли более сильных кислот, хотя исход также зависит от их относительного содержания. Что касается значений рКа оксинафтойных кислот, то известно, например, что 3-окси-2-нафтойная кислота имеет значение рКа 2,708 (Kagaku Binran Kisohen II, Chemical Society of Japan, опубликовано 25 сентября 1969). С другой стороны, значения рКа карбоксильных групп олигомеров молочной кислоты - гликолевой кислоты неизвестны, но могут быть рассчитаны на основе значения рКа молочной кислоты или гликолевой кислоты (3,86 или 3,83), в соответствии с теорией о том, что “изменение уровня свободной энергии вследствие введения заместителя может быть примерно рассчитано по правилу сложения”. Вклад заместителей в константы диссоциации уже определен и может быть использован для этой цели (таблица 4.1 в "рКа Prediction for Organic Acid and Bases", D.D.Perrin, B. Dempsey and E.P.Serjeant, 1981). Поскольку для гидроксильной группы и сложноэфирной связи характерны следующие значения:
ΔрКа (ОН) = -0,90,
ΔрКа (сложноэфирная связь) = -1,7,
значение рКа карбоксильной группы олигомеров молочной кислоты - гликолевой кислоты может быть определено при рассмотрении вклада ближайшей к диссоциирующей группе сложноэфирной связи следующим образом:
рКа = рКа (молочной кислоты или гликолевой кислоты) -ΔрКа(ОН) + ΔрКа (сложноэфирной связи) = 3,06 или 3,03.
Следовательно, поскольку оксинафтойные кислоты являются более сильными кислотами, чем молочная кислота (рКа=3,86), гликолевая кислота (рКа=3,83) и олигомеры молочной кислоты - гликолевой кислоты, можно предположить, что соль, образованная оксинафтойной кислотой и биоактивным веществом предпочтительна для получения описанной выше композиции, причем можно предположить, что природа длительного высвобождения биоактивного вещества из композиции определяется главным образом свойствами соли. Примерами указанного биоактивного вещества могут быть биоактивные вещества, описанные выше.
В данном случае тот факт, что соль, образованная оксинафтойной кислотой и биоактивным веществом, не совсем не растворима в воде, а все-таки, хотя и очень слабо, но растворима в воде, благоприятно сказывается на механизме длительного высвобождения. Другими словами, как показано в приведенном выше обсуждении, посвященном константе диссоциации кислоты, соль оксинафтойной кислоты, более сильной кислоты, чем описанные выше мономеры и олигомеры молочной кислоты - гликолевой кислоты, предпочтительна в начальной стадии высвобождения; характер начального высвобождения лекарственного средства может регулироваться относительным содержанием оксинафтойной кислоты, поскольку растворимость соли и характер ее распределения в тканях организма являются факторами, определяющими скорость высвобождения биоактивного вещества. Затем, по мере увеличения количества олигомеров и мономеров, в результате восстановления оксинафтойной кислоты и гидролиза биодеградируемого полимера постепенно преобладающее влияние на механизм высвобождения биоактивного вещества начинают оказывать вовлекаемые в него в качестве противоионов олигомеры и мономеры, даже если оксинафтойная кислота окончательно исчезает из указанной “композиции”, достигается стабильное высвобождение биоактивного вещества. Таким же образом может объясняться повышенная эффективность включения биоактивного вещества при образовании композиции длительного высвобождения и возможность подавления начального бурного высвобождения после введения в организм биоактивного вещества, включенного в состав композиции.
Роль оксинафтойной кислоты в композиции длительного высвобождения, содержащей соль оксинафтойной кислоты и биоактивного пептида, также может быть объяснена описанным выше механизмом.
Используемое здесь понятие “нерастворимо в воде” означает, что когда указанное вещество перемешивается в дистиллированной воде в течение 4 часов при температуре не выше 40°С, масса вещества, растворенного в 1 л раствора, не превышает 25 мг.
Используемое здесь понятие “очень слабо растворимо в воде” означает, что описанная выше масса вещества составляет не менее 25 мг, но не превышает 5 г. Когда указанное вещество представляет собой соль биоактивного вещества, приведенное выше определение применяется по отношению к массе биоактивного вещества, которое растворяется при описанной выше процедуре.
Хотя не существует ограничений в выборе формы композиции длительного высвобождения, представленной в данном изобретении, предпочтительны микрочастицы, причем большее предпочтение отдается микросферам (также называемым микрокапсулами в случае композиций длительного высвобождения, содержащих биодеградируемые полимеры). Используемый здесь термин “микросфера” означает пригодные для инъекций сферы, которые могут быть диспергированы в растворах. Их форма может быть подтверждена, например, с помощью сканирующей микроскопии.
Способы, используемые в разных вариантах изобретения
Ниже приведены примеры способов получения композиций длительного высвобождения, являющихся предметом данного изобретения, которые содержат биологически активное вещество или его соль, оксинафтойную кислоту или ее соль и биодеградируемый полимер или его соль, микросферы.
(I) Способ сушки водосодержащих эмульсий
(i) Способ на основе получения эмульсии М/В
При таком способе получают раствор оксинафтойной кислоты или ее соли и биодеградируемого полимера или его соли в органическом растворителе.
Примерами указанного органического растворителя могут являться галогенированные углеводороды (например, дихлорметан, хлороформ, дихлорэтан, трихлорэтан, четыреххлористый углерод), эфиры (например, этиловый эфир, изопропиловый эфир), сложные эфиры жирных кислот (например, этилацетат, бутилацетат), ароматические углеводороды (например, бензол, толуол, ксилол), спирты (например, этанол, метанол) и ацетонитрил. В качестве органического растворителя биодеградируемого полимера или его соли из названных растворителей предпочтителен дихлорметан. Спирты предпочтительны в качестве органического растворителя оксинафтойной кислоты или ее соли. Эти растворители могут использоваться в виде смесей в определенных соотношениях. Из таких растворителей предпочтительны смеси галогенированных углеводородов и спиртов, причем наибольшее предпочтение отдается смесям дихлорметана и этанола.
Когда в качестве органического растворителя используется смесь дихлорметана и этанола, соотношение их концентраций обычно выбирается в пределах примерно от 0,01 до 50% (объем/объем), предпочтительно примерно от 0,05 до 40% (объем/объем) и более предпочтительно примерно от 0,1 до 30% (объем/объем).
Концентрация биодеградируемого полимера в растворе в органическом растворителе варьирует в зависимости от молекулярной массы биодеградируемого полимера и вида органического растворителя. Например, когда в качестве органического растворителя используется дихлорметан, концентрация биодеградируемого полимера обычно выбирается в пределах примерно от 0,5 до 70% по массе, предпочтительно примерно от 1 до 60% по массе и более предпочтительно примерно от 2 до 50% по массе.
Концентрация оксинафтойной кислоты или ее соли в растворе в органическом растворителе обычно выбирается, например, в пределах примерно от 0,01 до 10% по массе, предпочтительно примерно от 0,1 до 5% по массе и более предпочтительно примерно от 0,5 до 3% по массе.
Биологически активное вещество или его соль добавляется к полученному таким образом раствору, содержащему оксинафтойную кислоту или ее соль и биодеградируемый полимер в органическом растворителе, и растворяется или диспергируется.
Полученный таким образом раствор, содержащий биологически активное вещество или его соль, оксинафтойную кислоту или ее соль и биодеградируемый полимер в органическом растворителе, добавляется затем к водной фазе для получения эмульсии М (масляная фаза)/В(водная фаза), после чего растворитель выпаривается из масляной фазы для получения микросфер. Для этой операции обычно выбирают объем водной фазы в пределах примерно от 1-кратного до 10000-кратного, предпочтительно примерно от 5-кратного до 50000-кратного и более предпочтительно примерно от 10-кратного до 2000-кратного, по сравнению с объемом масляной фазы.
К описанной выше водной фазе может быть добавлен эмульгатор. Указанным эмульгатором может быть любой эмульгатор при условии, что он способствует образованию стабильной эмульсии М/В. К таким эмульгаторам относятся, например, анионактивные поверхностно-активные вещества (например, олеат натрия, стеарат натрия, лаурилсульфат натрия), неионогенные поверхностно-активные вещества [например, полиоксиэтилированный сложный эфир сорбитана и жирной кислоты (твин 80, твин 60, Atlas Powder Company), производные полиоксиэтилированного касторового масла (например, НСО-60, НСО-50, Nikko Chemicals)], поливинилпирролидон, поливиниловый спирт, карбоксиметилцеллюлоза, лецитин, желатин и гиалуроновая кислота. Эти эмульгаторы могут использоваться по отдельности или в комбинации. Что касается концентрации, то они используются предпочтительно в пределах концентраций примерно от 0,01 до 10% по массе, более предпочтительно примерно от 0,05 до 5% по массе.
К описанной выше диспергирующей водной фазе может быть добавлен регулятор осмотического давления. Указанным регулятором осмотического давления может быть любой регулятор при условии, что он поддерживает осмотическое давление, если приготовлен в виде водного раствора.
Примерами указанного регулятора осмотического давления являются многоатомные спирты, одноатомные спирты, моносахариды, дисахариды, олигосахариды, аминокислоты и их производные.
К пригодным для использования многоатомным спиртам относятся, например, трехатомные спирты, такие как глицерин, пятиатомные спирты, такие как арабит, ксилит и адонит, и шестиатомные спирты, такие как маннит, сорбит и дульцит. Из этих спиртов предпочтительны шестивалентные спирты, причем наибольшее предпочтение отдается манниту.
К пригодньм для использования одноатомным спиртам относятся, например, метанол, этанол и изопропиловый спирт, причем предпочтение отдается этанолу.
К пригодным для использования моносахаридам относятся, например, пентозы, такие как арабиноза, ксилоза, рибоза и 2-дезоксирибоза, и гексозы, такие как глюкоза, фруктоза, галактоза, манноза, сорбоза, рамноза и фукоза, причем предпочтение отдается пентозам.
К пригодным для использования олигосахаридам относятся, например, трисахариды, такие как мальтотриоза и раффиноза, и тетрасахариды, такие как стахиоза, причем предпочтение отдается трисахаридам.
К пригодным для использования производным моносахаридов, дисахаридов и олигосахаридов относятся, например, глюкозамин, галактозамин, глюкуроновая кислота и галактуроновая кислота.
К пригодным для использования аминокислотам относятся, например, глицин, лейцин и аргинин, причем предпочтение отдается L-аргинину.
Эти регуляторы осмотического давления могут использоваться по отдельности или в комбинации.
Эти регуляторы осмотического давления обычно используются в таких концентрациях, при которых осмотическое давление диспергирующей водной фазы составляет примерно от 1/50- до 5-кратного, предпочтительно примерно от 1/25- до 3- кратного, по сравнению с осмотическим давлением физиологического раствора соли.
Удаление органического растворителя может достигаться общеизвестными способами или способами, основанными на известных методах. К таким способам относится, например, способ, при котором органический растворитель выпаривается при нормальном или постепенно снижающемся давлении при перемешивании с использованием пропеллерной мешалки, магнитной мешалки или подобным им мешалкам, и способ, при котором органический растворитель выпаривается в условиях вакуума с помощью роторного испарителя или подобного ему устройства.
Полученные таким образом микросферы выделяются путем центрифугирования или фильтрования, после чего они промываются несколько раз дистиллированной водой для удаления свободного биологически активного вещества, оксинафтойной кислоты, носителя лекарственного средства, эмульгатора и т.д., прилипших к поверхности микросфер, затем снова диспергируются в дистиллированной воде и т.д. и высушиваются вымораживанием.
Чтобы предотвратить взаимную агрегацию частиц в ходе процесса их получения, можно добавить антикоагулянт. Примерами указанного антикоагулянта могут быть растворимые в воде полисахариды, такие как маннит, лактоза, глюкоза и крахмал (например, кукурузный крахмал), аминокислоты, такие как глицин, и белки, такие как фибрин и коллаген. Из этих веществ предпочтительным является маннит.
В случае, когда это необходимо, для того чтобы удалить воду и органический растворитель из микросфер, после высушивания вымораживанием проводят нагревание в условиях разрежения, стараясь не вызвать взаимную агрегацию микросфер. Предпочтительно нагревание микросфер до температуры, лишь немного превышающей точку перехода биодеградируемого полимера в стеклообразное состояние, которая определяется с помощью дифференциального сканирующего калориметра при увеличении температуры со скоростью от 10 до 20°С в минуту. Более предпочтительно нагревание микросфер в пределах температур от точки перехода биодеградируемого полимера в стеклообразное состояние до температуры, превышающей температуру стеклования примерно на 30°С. В частности, когда в качестве биодеградируемого полимера используется полимер молочной кислоты - гликолевой кислоты, микросферы предпочтительно нагревают в пределах температур от точки перехода в стеклообразное состояние до температуры, превышающей температуру стеклования на 10°С, более предпочтительно в пределах температур от точки перехода в стеклообразное состояние до температуры, превышающей температуру стеклования на 5°С.
Хотя время нагревания варьирует в зависимости от количества микросфер и других факторов, обычно оно составляет приблизительно от 12 до 168 часов, предпочтительно примерно от 24 до 120 часов и более предпочтительно примерно от 48 до 96 часов, после того как температура микросфер достигнет заданной величины.
Могут быть использованы любые способы нагревания при условии, что нагревание агрегатов микросфер идет равномерно.
К пригодным для использования способам термической сушки относятся, например, способы, при которых термическая сушка проводится в термостатируемых камерах, в сушильных камерах с псевдоожиженным слоем, в сушильных камерах или печах с движущимся слоем, или с применением микроволновых печей для термической сушки. Из этих способов предпочтителен способ, при котором термическая сушка проводится в термостатируемых камерах.
(ii) Способ на основе получения эмульсии В/М/В (1)
Сначала готовится раствор, содержащий биодеградируемый полимер или его соль в органическом растворителе.
Концентрации органического растворителя и биодеградируемого полимера или его соли такие же, как описано выше в параграфе (I) (i).
В том случае, когда используется более двух видов растворителя, соотношение этих растворителей такое же, как соотношение растворителей, описанное выше в параграфе (I) (i). К полученному таким образом раствору, содержащему биодеградируемый полимер в органическом растворителе, добавляют биологически активное вещество или его соль, затем растворяют и диспергируют.
Затем к раствору биологически активного вещества и биодеградируемого полимера в органическом растворителе (масляная фаза) добавляют раствор оксинафтойной кислоты или ее соли [растворителем при этом могут быть, например, вода, спирты (например, метанол, этанол), раствор пиридина, раствор диметилацетамида и т.д.]. Эта смесь подвергается эмульгированию известными способами, такими как гомогенизация или ультразвуковое диспергирование, для образования эмульсии В/М.
Затем полученная таким образом эмульсия В/М, содержащая биологически активное вещество или его соль, оксинафтойную кислоту или ее соль и биодеградируемый полимер или его соль, добавляется к водной фазе для получения эмульсии В (дисперсная водная фаза)/М (масляная фаза)/В (диспергирующая водная фаза), после чего растворитель выпаривается из масляной фазы для получения микросфер. Для этой процедуры объем диспергирующей водной фазы обычно выбирается в пределах примерно от 1-кратного до 10000-кратного, предпочтительно примерно от 5-кратного до 50000-кратного и более предпочтительно от 10-кратного до 2000-кратного, по сравнению с объемом масляной фазы.
Описанные выше эмульгатор и регулятор осмотического давления, которые могут быть добавлены к диспергирующей водной фазе, и все последующие процедуры такие же, как описано выше в параграфе (I) (i).
(ii) Способ на основе получения эмульсии В/М/В (2)
Сначала готовится раствор, содержащий оксинафтойную кислоту и биодеградируемый полимер в органическом растворителе. Полученный таким образом раствор в органическом растворителе назван масляной фазой. Способ ее получения точно такой же, как способ, описанный выше в параграфе (I) (i).
Альтернативный способ состоит в том, что отдельно готовятся раствор, содержащий оксинафтойную кислоту в органическом растворителе, и раствор, содержащий биодеградируемый полимер в органическом растворителе, и они смешиваются вместе для получения масляной фазы.
Концентрация биодеградируемого полимера в растворе в органическом растворителе варьирует в зависимости от молекулярной массы биодеградируемого полимера и от вида органического растворителя. Например, когда в качестве органического растворителя используется дихлорметан, концентрация биодеградируемого полимера обычно выбирается в пределах примерно от 0,5 до 70% по массе, предпочтительно примерно от 1 до 60% по массе и более предпочтительно примерно от 2 до 50% по массе.
Затем готовится раствор биологически активного вещества или его соли [в качестве растворителя при этом используется, например, вода, спирты (например, метанол, этанол)]. Полученный таким образом раствор называется дисперсной водной фазой. Концентрация биологически активного вещества обычно составляет от 0,001 мг/мл до 10 г/мл, предпочтительно от 0,1 мг /мл до 5 г/мл, более предпочтительно от 10 мг/мл до 3 г/мл. Масляная фаза и дисперсная водная фаза подвергаются эмульгированию известными способами, такими как гомогенизация или ультразвуковое диспергирование для образования эмульсии В/М.
Для этой операции объем масляной фазы обычно выбирается в пределах примерно от 1-кратного до 1000-кратного, предпочтительно примерно от 2-кратного до 100-кратного и более предпочтительно примерно от 3-кратного до 10-кратного, по сравнению с объемом дисперсной водной фазы.
Вязкость эмульсии В/М обычно выбирается в пределах примерно от 10 до 10000 сП, предпочтительно примерно от 100 до 5000 сП, более предпочтительно примерно от 500 до 2000 сП.
Затем полученная таким образом эмульсия В/М, содержащая биологически активное вещество или его соль, оксинафтойную кислоту или ее соль и биодеградируемый полимер, добавляется к водной фазе для образования эмульсии В (дисперсная водная фаза)/М (масляная фаза)/В (диспергирующая водная фаза), после чего растворитель выпаривается из масляной фазы для получения микросфер. Для этой операции объем диспергирующей водной фазы обычно выбирается в пределах примерно от 1-кратного до 10000-кратного, предпочтительно примерно от 2-кратного до 100-кратного и более предпочтительно примерно от 3-кратного до 10-кратного, по сравнению с объемом дисперсной водной фазы.
Описанные выше эмульгатор и регулятор осмотического давления, которые могут быть добавлены к диспергирующей водной фазе, и последующие процедуры - такие же, как описано выше в параграфе (I) (i).
(II) Способ разделения фаз
Для получения микросфер этим способом к раствору веществ в органическом растворителе, описанном выше в параграфе (I), посвященном способу сушки водосодержащих эмульсий, который содержит композицию, состоящую из биологически активного вещества или его соли оксинафтойной кислоты или ее соли и биодеградируемого полимера или его соли, при перемешивании понемногу добавляют агент, вызывающий коацервацию, для того чтобы осадить и отвердить микросферы. Указанный агент, вызывающий коацервацию, добавляется в объеме, соответствующем примерно от 0,01- до 1000-кратного объема, предпочтительно примерно от 0,05- до 500-кратного объема и более предпочтительно от 0,1- до 200-кратного объема, по сравнению с объемом масляной фазы.
Указанным агентом, вызывающим коацервацию, может быть любой агент, при условии, что это полимерное соединение, соединение, относящееся к минеральным или растительным маслам, которое смешивается с органическим растворителем и которое не растворяет комплексную соль, образованную биологически активным веществом с оксинафтойной кислотой, и биосовместимый полимер. В частности, к пригодным для использования агентам, вызывающим коацервацию, относятся, например, силиконовое масло, кунжутное масло, соевое масло, кукурузное масло, хлопковое масло, кокосовое масло, льняное масло, минеральное масло, н-гексан и н-гептан. Они также могут использоваться и в комбинации.
Полученные таким образом микросферы собираются, после чего повторно промываются гептаном и т.д. для удаления вызывающего коацервацию агента и других примесей из композиции биологически активного вещества, оксинафтойной кислоты и биодеградируемого полимера, после чего высушиваются в условиях разрежения. Микросферы могут быть отмыты альтернативным способом, описанным выше в параграфе (I) (i), посвященном способу сушки водосодержащих эмульсий, затем высушены вымораживанием и высушены термически.
(III) Способ распылительной сушки
Для получения микросфер этим способом раствор, который содержит композицию, состоящую из биологически активного вещества или его соли, оксинафтойной кислоты или ее соли и биодеградируемого полимера или его соли в органическом растворителе, описанный выше в параграфе (I), посвященном способу сушки водосодержащих эмульсий, распыляют через форсунку в сушильную камеру распылительной сушилки для того, чтобы органический растворитель испарился из мелких капелек за очень короткое время для получения микросфер. Примерами указанных форсунок являются двухкомпонентная форсунка, сопло высокого давления, сопло с вращающимся диском. После промывания тем же способом, который описан выше в параграфе (I), посвященном способу сушки водосодержащих эмульсий, микросферы могут быть высушены вымораживанием и высушены термически.
Для получения лекарственных форм, отличных от описанных выше микросфер, раствор, который описан выше в параграфе (I), посвященном способу сушки водосодержащих эмульсий, который содержит композицию, состоящую из биологически активного вещества или его соли, оксинафтойной кислоты или ее соли и биодеградируемого полимера или его соли в органическом растворителе, может быть высушен выпариванием органического растворителя и воды в условиях вакуума с помощью роторного испарителя или подобного ему устройства с последующим измельчением в струйной мельнице или подобном ей устройстве для получения микрочастиц.
Измельченные микрочастицы после промывания тем же самым способом, который описан для случая получения микросфер в параграфе (I), посвященном способу сушки водосодержащих эмульсий, могут быть высушены вымораживанием и высушены термически.
Полученные таким образом микросферы или микрочастицы обеспечивают возможность высвобождения лекарственного средства со скоростью, соответствующей скорости деструкции биодеградируемого полимера или используемого полимера молочной кислоты - гликолевой кислоты.
(IV) Двухступенчатый способ
Биологически активное вещество или его соль добавляют к раствору оксинафтойной кислоты или ее соли в органическом растворителе в весовом отношении, соответствующем описанному выше относительному содержанию биологически активного вещества для получения раствора соли оксинафтойной кислоты и биологически активного вещества.
Указанным органическим растворителем является такой же растворитель, какой описан выше в параграфе (I) (i). В том случае, когда используются более двух видов органических растворителей в виде смеси растворителей, соотношение в смеси такое же, как описано выше в параграфе (I) (i).
Удаление органического растворителя для осаждения композиции, содержащей соль оксинафтойной кислоты и биологически активного вещества, достигается общеизвестными способами или способами, основанными на ранее известным методах. К таким способам относится, например, способ, при котором органический растворитель выпаривается в условиях вакуума с помощью роторного испарителя или подобных ему устройств.
Полученная таким образом композиция, содержащая соль оксинафтойной кислоты и биологически активного вещества, может быть снова растворена в органическом растворителе для получения композиции длительного высвобождения (микросфер или микрочастиц).
Примерами указанного органического растворителя могут быть галогенированные углеводороды (например, дихлорметан, хлороформ, дихлорэтан, трихлорэтан, четыреххлористый углерод), эфиры (например, этиловый эфир, изопропиловый эфир), сложные эфиры жирных кислот (например, этилацетат, бутилацетат) и ароматические углеводороды (например, бензол, толуол, ксилол). Эти растворители могут использоваться в виде смесей в соответствующих соотношениях. Из этих растворителей предпочтительны галогенированные углеводороды, причем наибольшее предпочтение отдается дихлорметану.
Затем раствор, содержащий соль оксинафтойной кислоты и биологически активного вещества в органическом растворителе, добавляют к водной фазе для получения эмульсии М (масляная фаза)/В (водная фаза), после чего растворитель выпаривается из масляной фазы для получения микросфер. Для этой операции объем водной фазы обычно выбирается в пределах примерно от 1-кратного до 10000-кратного, предпочтительно примерно от 5-кратного до 5000-кратного и более предпочтительно примерно от 10-кратного до 2000-кратного, по сравнению с объемом масляной фазы.
Эмульгатор, регулятор осмотического давления и последующие этапы - такие же, как описано выше в параграфе (I) (i).
Удаление органического растворителя может быть достигнуто общеизвестными способами или способами, основанными на известных методах. К таким способам относится, например, способ, при котором органический растворитель выпаривается при нормальном или постепенно снижающемся давлении при перемешивании с помощью пропеллерной мешалки, магнитной мешалки или подобных мешалок, и способ, при котором органический растворитель выпаривается в условиях вакуума с помощью роторного испарителя или подобных ему устройств.
Полученные таким образом микросферы выделяют путем центрифугирования или фильтрования, после чего они промываются несколько раз дистиллированной водой для удаления свободного биологически активного вещества, оксинафтойной кислоты, эмульгатора и т.д., прилипших к поверхности микросфер, затем снова диспергируются в дистиллированной воде и т.д. и сушатся вымораживанием.
Чтобы предотвратить взаимную агрегацию частиц во время процесса получения, может быть добавлен антикоагулянт. Примерами указанного антикоагулянта могут быть водорастворимые полисахариды, такие как маннит, лактоза, глюкоза и крахмал (например, кукурузный крахмал), аминокислоты, такие как глицин, и белки, такие как фибрин и коллаген. Из этих веществ предпочтителен маннит.
При необходимости после сушки вымораживанием может быть проведено нагревание при пониженном давлении, не вызывающее взаимного слипания микросфер, для того чтобы удалить воду и органический растворитель из микросфер.
Хотя время нагревания варьирует в зависимости от количества микросфер и других факторов, обычно это время составляет примерно от 12 до 168 часов, предпочтительно примерно от 24 до 120 часов и более предпочтительно примерно от 48 до 96 часов, после того как температура микросфер достигнет заданной величины.
Может быть использован любой способ нагревания при условии, что агрегаты микросфер нагреваются равномерно.
В качестве термических способов сушки пригодны, например, способы, при которых сушка проводится в термостатируемых камерах, в сушильных камерах с псевдоожиженным слоем, в сушильных камерах или печах с подвижным слоем, или способ с использованием микроволновых печей для термической сушки. Из этих способов предпочтителен способ, при котором термическая сушка проводится в термостатируемой камере. Полученные микросферы имеют относительно однородную сферическую форму и при инъекционном введении встречают незначительное сопротивление, так что засорение иглы маловероятно. К тому же возможно использование тонких игл для инъекций, уменьшающих у пациентов болевые ощущения при инъекции.
(V) Одноступенчатый способ
Биологически активное вещество или его соль добавляют к раствору оксинафтойной кислоты или ее соли в органическом растворителе в весовом отношении, соответствующем описанному выше относительному содержанию биологически активного вещества для получения соли оксинафтойной кислоты и биологически активного вещества, после чего готовится препарат длительного высвобождения (микросферы или микрочастицы).
Указанный органический растворитель - такой же, как растворитель, описанный выше в параграфе (I) (i). В том случае, когда используется больше двух органических растворителей в виде смеси растворителей, соотношение их в смеси такое же, как описано в (I) (i).
Затем раствор, содержащий соль оксинафтойной кислоты и биологически активного вещества в органическом растворителе, добавляют к водной фазе для образования эмульсии М (масляная фаза)/В (водная фаза), после чего растворитель выпаривается из масляной фазы для получения микросфер. Для этой операции объем водной фазы обычно выбирается в пределах приблизительно от 1-кратного до 10000-кратного, предпочтительно примерно от 5-кратного до 5000-кратного и более предпочтительно примерно от 10-кратного до 2000-кратного, по отношению к объему масляной фазы.
Описанные выше эмульгатор и регулятор осмотического давления, которые могут быть добавлены к диспергирующей водной фазе, и последующие процедуры - такие же, как описано выше в параграфе (IV).
Представленная в данном изобретении композиция длительного высвобождения может вводиться в организм как таковая либо в виде различных лекарственных форм, приготовленных с использованием этой композиции в качестве исходного материала, в частности в виде внутримышечных, подкожных, висцеральных или других инъекционных препаратов или имплантируемых препаратов, в виде назальных, ректальных, маточных и других трансдермальных препаратов, оральных препаратов [например, твердых препаратов, таких как капсулы (например, твердые капсулы, мягкие капсулы), гранул и порошков; жидких препаратов, таких как сиропы, эмульсии и суспензии] и т.д.
Например, представленная в данном изобретении композиция длительного высвобождения может быть приготовлена в виде инъекционных препаратов путем суспендирования в воде с диспергирующим агентом (например, поверхностно-активными веществами, такими как твин 80 и НСО-60, полисахаридами, такими как гиалуронат натрия, карбоксиметилцеллюлоза и альгинат натрия), консервантом (например, метилпарабеном, пропилпарабеном), агентом, обеспечивающим изотоничность раствора (например, хлоридом натрия, маннитом, сорбитом, глюкозой, пролином) и т.д., для получения водной суспензии или путем диспергирования в растительном масле, таком как кунжутное масло или кукурузное масло, для получения масляной суспензии, посредством чего получают инъекционный препарат длительного высвобождения для практического применения.
В том случае, когда представленная в данном изобретении композиция длительного высвобождения используется в виде инъекционной суспензии, диаметр частиц выбирается в таких пределах, которые удовлетворяют требованиям, предъявляемым степенью дисперсности и прохождением сквозь иглу. Например, значение диаметра частиц обычно находится в пределах примерно от 0,1 до 300 мкм, предпочтительно примерно от 0,5 до 150 мкм и более предпочтительно примерно от 1 до 100 мкм.
Представленная в данном изобретении композиция длительного высвобождения может быть приготовлена в виде стерильного препарата с помощью таких способов, при в которых весь процесс получения полностью проходит в асептических условиях, с помощью использования гамма-излучения для стерилизации и способом, при котором добавляется консервант, - это методы, которые не следует рассматривать, как ограничивающие.
Вследствие низкой токсичности представленная в данном изобретении композиция длительного высвобождения может быть использована в качестве безопасного фармацевтического препарата и т.д. для млекопитающих (например, человека, крупного рогатого скота, свиней, собак, кошек, мышей, крыс, кроликов).
Хотя доза композиции длительного высвобождения широко варьирует в зависимости от вида, количества и лекарственной формы биологически активного вещества, которое является активным компонентом, и продолжительности высвобождения биологически активного вещества, от типа заболевания, от вида заболевшего животного и других факторов, она может быть установлена на некотором уровне, в соответствии с продолжительностью эффективного действия биологически активного вещества. Доза активного компонента - биологически активного вещества - на одно введение может быть выбрана предпочтительно в пределах, соответствующих примерно интервалу от 0,01 до 10 мг/кг веса тела, более предпочтительно от 0,05 до 5 мг/кг веса тела для взрослого организма в случае использования препарата, продолжительность высвобождения которого составляет 1 месяц.
Доза композиции длительного высвобождения при однократном введении может быть выбрана предпочтительно в пределах, соответствующих интервалу примерно от 0,05 до 50 мг/кг веса тела, более предпочтительно примерно от 0,1 до 30 мг/кг веса тела взрослого организма.
Частота введения, в зависимости от вида, количества и лекарственной формы активного компонента – биологически активного вещества -, от продолжительности высвобождения биологически активного вещества, типа заболевания, вида заболевшего животного и других факторов, может быть соответственно выбрана, например, один раз каждые несколько недель, один раз каждый месяц или один раз каждые несколько месяцев (например, 3 месяца, 4 месяца, 6 месяцев).
В зависимости от биологически активного вещества, которое входит в состав композиции длительного высвобождения, представленная в данном изобретении композиция длительного высвобождения может использоваться как средство для лечения и профилактики различных видов заболеваний. Когда биологически активное вещество представлено производными РГЛГ, представленная в данном изобретении композиция длительного высвобождения пригодна для применения в качестве средства лечения или профилактики гормональных заболеваний, особенно заболеваний, зависимых от половых гормонов, таких как зависимый от половых гормонов рак (например, рак простаты, карцинома матки, рак молочной железы, гипофизома и т.д.), гипертрофия простаты, эндометриоз, миома матки, преждевременное половое созревание, дисменорея, аменорея, предменструальный синдром, синдром многокамерного яичника. Представленная в данном изобретении композиция длительного высвобождения также пригодна для применения в качестве средства контрацепции. В том случае, когда имеет место симптом отдачи после медикаментозного лечения, представленная в данном изобретении композиция длительного высвобождения пригодна для применения в качестве средства лечения или профилактики полиорганной недостаточности. Кроме того, представленная в данном изобретении композиция длительного высвобождения пригодна для применения в качестве средства лечения или профилактики независимых от половых гормонов, но чувствительных к РГЛГ доброкачественных или злокачественных опухолей.
Примеры
Данное изобретение в дальнейшем описывается более детально посредством следующих примеров, которые не следует рассматривать как ограничивающие изобретения.
Пример 1
3429,6 мг ацетата (производства ТРА) N-(S)-тeтpaгидpoфyp-2-oил-Gly-D2Nal-D4ClPhe-D3Pal-Ser-NMeTyr-DLys (Nic)-Leu-Lys(Nisp)-Pro-DAlaNH2 (в дальнейшем упоминаемого как пептид А), химическая формула которого
и 685,2 мг 3-окси-2-нафтойной кислоты растворяли в 15 мл этанола. Этот раствор постепенно перегоняли с помощью роторного испарителя, чтобы испарился органический растворитель. Полученный остаток снова растворяли в 5,5 мл дихлорметана и вливали в 400 мл 0,1% (вес/вес) водного раствора поливинилового спирта (EG-40, производства The Nippon Synthetic Chemical Industry), температура которого была предварительно доведена до 18°С; раствор перемешивали при 8000 об/мин, используя миксер-гомогенизатор турбинного типа, чтобы получить эмульсию типа М/В. Эту эмульсию М/В перемешивали при комнатной температуре в течение 3 часов, чтобы испарился дихлорметан и загустела масляная фаза, с последующим сбором микросфер при центрифугировании со скоростью 2000 об/мин на центрифуге (05PR-22, Hitachi, Ltd.). Микросферы снова диспергировали в дистиллированной воде, после чего проводили центрифугирование и вымывали свободное лекарственное средство и др. примеси. Собранные микросферы снова диспергировали в небольшом количестве дистиллированной воды, затем сушили вымораживанием для получения порошка. Выход составил 65%, содержание пептида А и молярное отношение 3-окси-2-нафтойная кислота/пептид А в микросферах составляли 75,4% и 1,94 соответственно.
Пример 2
1785,1 мг ацетата пептида А и 1370,4 мг 3-окси-2-нафтойной кислоты растворяли в 15 мл этанола. Этот раствор постепенно перегоняли с помощью роторного испарителя для того, чтобы испарился органический растворитель. Полученный остаток снова растворяли в 10 мл дихлорметана и вливали в 1000 мл 0,1% (вес/вес) водного раствора поливинилового спирта, температура которого предварительно была доведена до 18°С; затем следовали такие же процедуры для сбора микросфер, которые описаны в примере 1. Выход составил 58%, содержание пептида А и молярное отношение 3-окси-2-нафтойной кислота/пептид А в микросферах составляли 54,3% и 6,15 соответственно.
Пример 3
1800 мг ацетата пептида А, 173 мг 3-окси-2-нафтойной кислоты и 2 г сополимера молочной кислоты - гликолевой кислоты (молочная кислота - гликолевая кислота = 50/50 (мол.%), среднемассовая молекулярная масса 10100, среднечисловая молекулярная масса 5670, среднечисловая молекулярная масса 3720, как было найдено в результате количественного определения содержания концевых групп, производство Wako Pure Chemical Industries) были растворены в смеси 6 мл дихлорметана и 0,2 мл этанола. Этот раствор вливали в 900 мл 0,1% (вес/вес) водного раствора поливинилового спирта, содержащего 5% маннита, температура которого предварительно была доведена до 18°С, и перемешивали при 7000 об/мин с использованием миксера-гомогенизатора турбинного типа для получения эмульсии М/В. Эту эмульсию М/В перемешивали при комнатной температуре в течение 3 часов, для того чтобы испарился дихлорметан и этанол и загустела масляная фаза, после чего микросферы собирали центрифугированием со скоростью 2000 об/мин. Микросферы снова диспергировали в дистиллированной воде, после чего проводили центрифугирование и вымывали свободное лекарственное средство и др. примеси. Собранные микросферы снова диспергировали в небольшом количестве дистиллированной воды, содержащей 250 мг маннита, затем сушили вымораживанием для получения порошка. Выход составил 76%, эффективность включения пептида А в микросферы 84,6% и содержание пептида А и молярное отношение 3-окси-2-нафтойная кислота/пептид А в микросферах составили 34,7% и 1,19 соответственно.
Пример 4
1900 мг ацетата пептида А, 182 мг 3-окси-2-нафтойной кислоты и 1,9 г сополимера молочной кислоты - гликолевой кислоты (такой же как в примере 3) растворяли в смеси 6 мл дихлорметана и 0,2 мл этанола. Этот раствор вливали в 900 мл 0,1% (вес/вес) водного раствора поливинилового спирта, содержащего 5% маннита и 0,05% L-аргинина, температура которого была предварительно доведена до 18°С, после чего были проведены такие же процедуры для сбора микросфер, как процедуры, описанные в примере 3. Выход составил 85%, эффективность включения пептида А в микросферы составила 88,9% и содержание пептида А и молярное отношение 3-окси-2-нафтойная кислота/пептид А в микросферах составили 38,6% и 0,83 соответственно.
Пример 5
Микросферы были получены тем же способом, как в примере 4, за исключением того, что сополимер молочной кислоты -гликолевой кислоты, используемый в примере 4, был заменен сополимером молочной кислоты - гликолевой кислоты, в котором отношение содержания молочной кислоты/гликолевой кислоты было равно 75/25 (мол.%), среднемассовая молекулярная масса 10700, среднечисловая молекулярная масса 6100 и среднечисловая молекулярная масса 3770, как было найдено в результате количественного определения концевых групп, и количество дихлорметана было изменено и составило 6,5 мл. Выход составил 87%, эффективность включения пептида А в микросферы составила 88,3%, содержание пептида А и молярное отношение 3-окси-2-нафтойная кислота/пептид А в микросферах составили 38,3% и 0,92 соответственно.
Пример 6
Готовили раствор, содержащий 1800 мг ацетата пептида А и 1,8 г сополимера молочной кислоты - гликолевой кислоты (молочная кислота/гликолевая кислота = 50/50 (мол.%), среднемассовая молекулярная масса 12700, среднечисловая молекулярная масса 7090, среднечисловая молекулярная масса 4780, как было найдено путем количественного определения концевых групп, производства Wako Pure Chemical Industries) в 7,2 мл дихлорметана. К этому раствору был добавлен раствор 196 мг натриевой соли 3-окси-2-нафтойной кислоты в 2,3 мл воды, после чего проведено эмульгирование с помощью гомогенизатора для получения эмульсии В/М. Эту эмульсию вливали в 800 мл 0,1% (вес/вес) водного раствора поливинилового спирта, содержащего 5% маннита, температура которого была предварительно доведена до 18°С, и перемешивали при 7000 об/мин с помощью миксера-гомогенизатора турбинного типа для получения эмульсии В/М/В. После следовала такая же процедура для сбора микросфер, как процедура, описанная в примере 3. Выход составил 79%. Эффективность включения пептида А в микросферы составила 81,2% и содержание пептида А и молярное отношение 3-окси-2-нафтойная кислота/пептид А в микросферах составили 32,8% и 0,91 соответственно.
Экспериментальный пример 1
Около 40 мг микросфер, полученных в каждом из примеров 1 и 2, или около 60 мг микросфер, полученных в каждом из примеров с 3 по 5, были диспергированы в 0,5 мл диспергатора (дистиллированная вода с 0,25 мг карбоксиметилцеллюлозы, 0,5 мг полисорбата 80 и 25 мг маннита, все в растворенном виде) и введены подкожно в спину самцам крыс SD 8-10-недельного возраста, используя иглы для инъекций 22G. После введения каждую крысу забивали и отбирали микросферы, оставшиеся в месте введения, и исследовали на содержание в них пептида А. Результаты показаны в таблице 1.
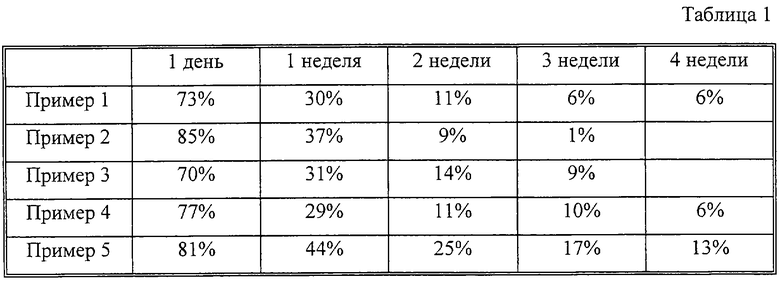
Экспериментальные результаты примеров 1 и 2 показывают, что скорость выхода пептида А из микросфер, состоящих из двух компонентов, т.е. пептида А и 3-окси-2-нафтойной кислоты, варьирует в зависимости от их соотношения; пептид А высвобождается быстрее при увеличении содержания 3-окси-2-нафтойной кислоты. Также экспериментальные результаты примеров 3, 4 и 5 показывают, что профиль высвобождения пептида А из микросфер, состоящих из трех компонентов, т.е. из указанных выше двух компонентов и сополимера молочной кислоты - гликолевой кислоты, отличается от профиля высвобождения из микросфер, состоящих из двух компонентов. Было также показано, что режим высвобождения из микросфер может контролироваться путем комбинирования различных составов сополимера молочной кислоты - гликолевой кислоты и среднемассовых молекулярных масс. Результаты примера 7 и приведенного для сравнения примера 1 показывают, что 3-окси-2-нафтойная кислота увеличивает содержание пептида В в микросферах.
Пример 7
Раствор, содержащий 0,8 г ацетата 5-оксо-Pro-His-Trp-Ser-Tyr-DLeu-Leu-Arg-Pro-NH-C2H5 (в дальнейшем упоминаемого как пептид В, производства Takeda Chemical) в 0,8 мл дистиллированной воды смешивали с раствором, содержащим 3,08 г полимера DL-молочной кислоты (среднемассовая молекулярная масса 36000, среднечисловая молекулярная масса 18000, содержание карбоксильных групп по результатам количественного определения методом мечения 70,4 мкмоль/г) и 0,12 г 3-окси-2-нафтойной кислоты в смеси органических растворителей, состоящей из 5 мл дихлорметана и 0,3 мл этанола, и эту смесь эмульгировали в гомогенизаторе для получения эмульсии В/М. Эту эмульсию В/М впрыскивали в 800 мл 0,1% (вес/вес) водного раствора поливинилового спирта (EG-40, производства Nippon Synthetic Chemical Industry), температура которого была предварительно доведена до 15°С, и перемешивали при 7000 об/мин с помощью миксера-гомогенизатора турбинного типа для получения эмульсии В/М/В. Эту эмульсию В/М/В перемешивали при комнатной температуре в течение 3 часов для испарения или диффузии в диспергирующую водную фазу дихлорметана и этанола, для загустения масляной фазы, после чего масляную фазу просеивали сквозь сито с величиной пор 75 мкм, после этого центрифугировали со скоростью 2000 об/мин в течение 5 минут на центрифуге (05PR-22, Hitachi, Ltd.), чтобы осадить микрокапсулы, которые были собраны. Микрокапсулы снова диспергировали в дистиллированной воде, затем центрифугировали, после чего вымывались свободное лекарственное средство и др. примеси и собирались микрокапсулы. Микрокапсулы еще раз диспергировали в небольшом количестве добавленной дистиллированной воды, после чего сушили вымораживанием для получения порошка. Выход микрокапсул по массе составил 46%, содержание пептида В и содержание 3-окси-2-нафтойной кислоты в микрокапсулах составили 21,3% и 2,96% соответственно. Эффективности включения, как было определено путем деления значений реального содержания этих компонентов на соответствующие значения количества израсходованного вещества, составили 106,6% для пептида В и 98,6% для 3-окси-2-нафтойной кислоты.
Пример 8
Раствор, содержащий 1,2 г ацетата пептида В в 1,2 мл дистиллированной воды, смешивали с раствором, содержащим 4,62 г полимера DL-молочной кислоты (среднемассовая молекулярная масса 25200, среднечисловая молекулярная масса 12800, содержание карбоксильных групп по результатам количественного определения методом мечения 62,5 мкмоль/г) и 0,18 г 3-окси-2-нафтойной кислоты в смеси органических растворителей, состоящей из 7,5 мл дихлорметана и 0,45 мл этанола, и эту смесь эмульгировали в гомогенизаторе для получения эмульсии В/М. Затем эту эмульсию В/М впрыскивали в 1200 мл 0,1% (вес/вес) водного раствора поливинилового спирта (EG-40, производства Nippon Synthetic Chemical Industry), температура которого была предварительно доведена до 15°С, и перемешивали при 7000 об/мин с помощью миксера-гомогенизатора турбинного типа для получения эмульсии В/М/В. Эту эмульсию В/М/В перемешивали при комнатной температуре в течение 3 часов для испарения или диффузии в диспергирующую водную фазу дихлорметана и этанола, для загустения масляной фазы, после чего масляную фазу просеивали сквозь сито с величиной пор 75 мкм, после этого центрифугировали со скоростью 2000 об/мин в течение 5 минут на центрифуге (05 PR-22, Hitachi, Ltd.), чтобы осадить микрокапсулы, которые были собраны. Микрокапсулы снова диспергировали в дистиллированной воде, затем центрифугировали, после чего вымывались свободное лекарственное средство и др. примеси и собирались микрокапсулы. Микрокапсулы еще раз диспергировали в небольшом количестве дистиллированной воды, добавляли и растворяли 0,3 г маннита, после чего раствор сушили вымораживанием для получения порошка. Выход микрокапсул по массе, как было определено после вычитания количества добавленного маннита, составил 55,2%, содержание пептида В и содержание 3-окси-2-нафтойной кислоты в микрокапсулах составили 21,3% и 2,96% соответственно. Эффективности включения, как было определено путем деления значений реального содержания этих компонентов на соответствующие значения количества израсходованного вещества, составили 99,7% для пептида В и 102,2% для 3-окси-2-нафтойной кислоты.
Пример 9
Микрокапсулы в виде порошка были получены тем же способом, который приведен в примере 8, за исключением того, что полимер DL-молочной кислоты, описанный в примере 8, был заменен на другой полимер DL-молочной кислоты (среднемассовая молекулярная масса 28800, среднечисловая молекулярная масса 14500, содержание карбоксильных групп по результатам количественного определения методом мечения 78,1 мкмоль/г). Выход микрокапсул по массе, как было определено после вычитания количества добавленного маннита, составил 50,2%, содержание пептида В и содержание 3-окси-2-нафтойной кислоты в микрокапсулах составили 20,8% и 2,78% соответственно. Эффективности включения, как было определено путем деления значений реального содержания этих компонентов на соответствующие значения количества израсходованного вещества, составили 103,4% для пептида В и 92,7% для З-окси-2-нафтойной кислоты.
Сравнительный пример 1
Раствор, содержащий 1,2 г ацетата пептида В в 1,2 мл дистиллированной воды смешивали с раствором, содержащим 4,8 г того же полимера DL-молочной кислоты, который описан в примере 9, в 7,8 мл дихлорметана, и эту смесь впрыскивали в 1200 мл 0,1% (вес/вес) водного раствора поливинилового спирта (EG-40, производства Nippon Synthetic Chemical Industry), температура которого была предварительно доведена до 15°С, и перемешивали при 7000 об/мин с помощью миксера-гомогенизатора турбинного типа для получения эмульсии В/М/В. Эту эмульсию В/М/В обрабатывали тем же способом, как в примере 8, для получения порошка микрокапсул. Выход микрокапсул по массе, как было определено после вычитания количества добавленного маннита, составил 53,6%, содержание пептида В в микрокапсулах составило 12,1%. Эффективность включения пептида В, как было определено путем деления значения его реального содержания на соответствующее значение количества израсходованного вещества, составила 60,6%, эта эффективность намного ниже, чем было получено в примере 9. Поэтому очевидно, что эффективность включения пептида В увеличивалась при добавлении 3-окси-2-нафтойной кислоты.
Пример 10
Раствор, содержащий 1,00 г ацетата пептида В в 1,00 мл дистиллированной воды, смешивали с раствором, содержащим 3,85 г полимера DL- молочной кислоты (среднемассовая молекулярная масса 49500, среднечисловая молекулярная масса 17500, содержание карбоксильных групп по результатам количественного определения методом мечения 45,9 мкмоль/г), и 0,15 г 3-окси-2-нафтойной кислоты в смеси органических растворителей, состоящей из 7,5 мл дихлорметана и 0,4 мл этанола, и эту смесь эмульгировали в гомогенизаторе для получения эмульсии В/М. Затем эту эмульсию В/М обрабатывали тем же способом, как в примере 8 для получения порошка микрокапсул, за исключением того, что количество 0,1% (вес/вес) водного раствора поливинилового спирта и количество добавленного маннита было изменено и составило 1000 мл и 0,257 г соответственно. Выход микрокапсул по массе, как было определено после вычитания количества добавленного маннита, составил 53,8%, содержание пептида В и содержание 3-окси-2-нафтойной кислоты в микрокапсулах составили 18,02% и 2,70% соответственно. Эффективности включения, как было определено путем деления значений реального содержания этих компонентов на соответствующие значения количества израсходованного вещества, составили 90,1% для пептида В и 90,1% для 3-окси-2-нафтойной кислоты.
Пример 11
Раствор, содержащий 1,202 г ацетата пептида В в 1,20 мл дистиллированной воды смешивали с раствором, содержащим 4,619 г полимера DL-молочной кислоты (среднемассовая молекулярная масса 19900, среднечисловая молекулярная масса 10700, содержание карбоксильных групп по результатам количественного определения методом мечения 104,6 мкмоль/г) и 0,179 г 3-окси-2-нафтойной кислоты в смеси органических растворителей, состоящей из 7,5 мл дихлорметана и 0,45 мл этанола, и эту смесь эмульгировали в гомогенизаторе для получения эмульсии В/М. Затем эту эмульсию В/М обрабатывали тем же способом, как в примере 8, для получения порошка микрокапсул, за исключением того, что количество добавленного маннита было 0,303 г. Выход микрокапсул по массе, как было определено после вычитания количества добавленного маннита, составил 61,4%, содержание пептида В и содержание 3-окси-2-нафтойной кислоты в микрокапсулах составили 15,88% и 2,23% соответственно. Эффективности включения, как было определено путем деления значений реального содержания этих компонентов на соответствующие значения количества израсходованного вещества, составили 77,75% для пептида В и 75,05% для 3-окси-2-нафтойной кислоты.
Пример 12
Раствор, содержащий 1,00 г ацетата пептида В в 1,00 мл дистиллированной воды, смешивали с раствором, содержащим 3,85 г полимера DL-молочной кислоты (среднемассовая молекулярная масса 25900, среднечисловая молекулярная масса 7100, содержание карбоксильных групп по результатам количественного определения методом мечения 98,2 мкмоль/г) и 0,15 г 3-окси-2-нафтойной кислоты в смеси органических растворителей, состоящей из 5,5 мл дихлорметана и 0,35 мл этанола, и эту смесь эмульгировали в гомогенизаторе для получения эмульсии В/М. Затем эту эмульсию В/М обрабатывали тем же способом, как в примере 7, для получения порошка микрокапсул. Выход микрокапсул по массе составил 48,8%, содержание пептида В и содержание 3-окси-2-нафтойной кислоты в микрокапсулах составили 21,31% и 2,96% соответственно. Эффективности включения, как было определено путем деления значений реального содержания этих компонентов на соответствующие значения количества израсходованного вещества, составили 106,5% для пептида В и 98,7% для 3-окси-2-нафтойной кислоты.
Сравнительный пример 1
Раствор, содержащий 1,00 г ацетата пептида В в 1,00 мл дистиллированной воды, смешивали с раствором, содержащим 4,00 г того же полимера DL-молочной кислоты, как в примере 12, в 5 мл дихлорметана, и эту смесь эмульгировали в гомогенизаторе для получения эмульсии В/М. Затем эту эмульсию В/М обрабатывали тем же способом, как в примере 7, для получения порошка микрокапсул. Выход микрокапсул по массе составил 48,7%, содержание пептида В в микрокапсулах составило 11,41%. Эффективность включения, как было определено путем деления значения реального содержания пептида на соответствующее значение количества израсходованного вещества, составила 57,1%, эта эффективность намного ниже, чем получено в примере 12. Поэтому очевидно, что эффективность включения пептида В увеличивалась при добавлении 3-окси-2-нафтойной кислоты.
Пример 13
Раствор, содержащий 89,2 г полимера DL-молочной кислоты (среднемассовая молекулярная масса 30600, среднечисловая молекулярная масса 14400, содержание карбоксильных групп по результатам количественного определения методом мечения 63,0 мкмоль/г) в 115,3 г дихлорметана, смешивали с раствором, содержащим 3,45 г 3-окси-2-нафтойной кислоты в смеси органических растворителей, состоящей из 38,8 г дихлорметана и 6,27 г этанола, и температуру этой смеси доводили до 28,5°С. Из этого раствора в органическом растворителе отвесили 224 г и смешали с водным раствором, содержащим 22,3 г ацетата пептида В в 20 мл дистиллированной воды, предварительно нагретым до 44,9°С, после этого перемешивали в течение 5 минут для грубого получения эмульсии, которую затем эмульгировали при 10000 об/мин в течение 5 минут в гомогенизаторе для получения эмульсии В/М. Эту эмульсию В/М охлаждали до 16,3°С и впрыскивали в течение 5-минутного периода в 20 литров 0,1% (вес/вес) водного раствора поливинилового спирта (EG-40, производства Nippon Synthetic Chemical Industry), температура которого была предварительно доведена до 15°С, и перемешивали при 7000 об/мин с помощью HOMOMIC LINE FLOW (производство Tokushu Kika) для получения эмульсии В/М/В. Эту эмульсию В/М/В перемешивали при температуре 15°С в течение 3 часов для испарения или диффузии в диспергирующую водную фазу дихлорметана и этанола, для загустения масляной фазы, после чего масляную фазу просеивали сквозь сито с размером пор 75 мкм, после этого центрифугировали со скоростью 2000 об/мин на центрифуге (H-600S, производства Kokusan Enshinki), чтобы сразу же осадить микрокапсулы, которые были собраны. Микрокапсулы снова диспергировали в небольшом количестве дистиллированной воды, затем просеивали сквозь сито с размером пор 90 мкм, после чего сушили вымораживанием для получения порошка. Выход микрокапсул по массе, как было определено после вычитания количества добавленного маннита, составил 66,5%, содержание пептида В и содержание 3-окси-2-нафтойной кислоты в микрокапсулах составили 22,3% и 2,99% соответственно. Эффективности включения, как было определено путем деления значений реального содержания этих компонентов на соответствующие значения количества израсходованного вещества, составили 104,5% для пептида В и 102,1% для 3-окси-2-нафтойной кислоты.
Экспериментальный пример 2
Дисперсию, содержащую 45 мг микрокапсул, полученных, как описано в примере 8, в 0,3 мл диспергатора (дистиллированная вода, содержащая 0,15 мг кабоксиметилцеллюлозы, 0,3 мг полисорбата 80 и 15 мг маннита, все в растворенном виде), вводили подкожно в спину самцам крыс SD 7-недельного возраста, используя иглы для инъекций 22G. Через определенные интервалы времени крыс забивали и отбирали микросферы, оставшиеся в месте введения, и проводили количественное исследование на содержание пептида В и 3-окси-2-нафтойной кислоты. Эффективности удерживания, как было определено путем деления значений, полученных при этом исследовании, на соответствующие значения исходного содержания компонентов, и краткие сведения о свойствах используемого полимера DL-молочной кислоты показаны в таблице 2.
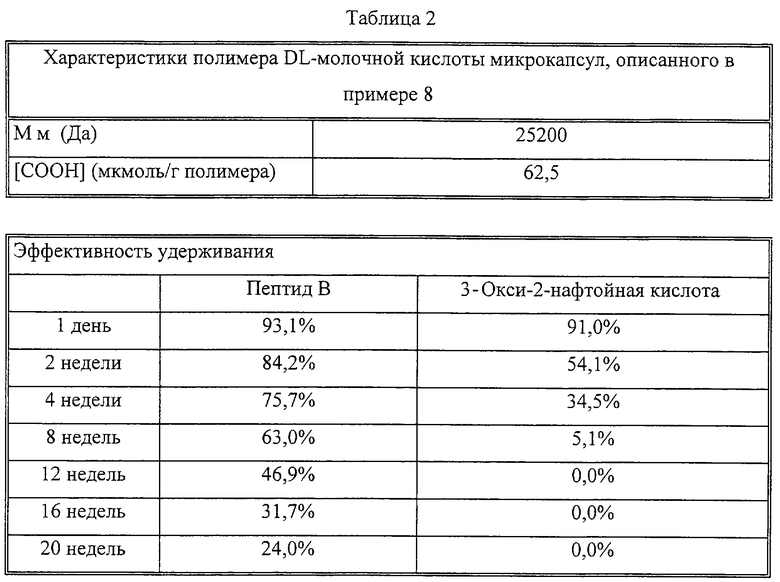
Как видно из таблицы 2, микрокапсулы, описанные в примере 8, обладают поразительно высокой эффективностью удерживания биоактивного вещества, не менее 90% в первый день после введения, несмотря на высокое содержание биоактивного вещества. Поэтому очевидно, что 3-окси-2-нафтойная кислота не только эффективна для обеспечения включения большого количества биоактивного вещества в препараты длительного высвобождения, но также очень хорошо подавляет бурное начальное высвобождение биоактивного вещества. Эти микрокапсулы обладают способностью высвобождать биоактивное вещество с постоянной скоростью в течение очень продолжительного периода времени. Кроме того, несмотря на то, что 3-окси-2-нафтойная кислота полностью элиминируется из микрокапсул через 12 недель, продолжает поддерживаться та же скорость высвобождения биоактивного вещества, что свидетельствует об эффективности данного препарата в качестве препарата длительного высвобождения.
Экспериментальный пример 3
После того как микрокапсулы, полученные, как описано в примерах 7, 9-12 и сравнительном примере 1, были введены животным и извлечены тем же способом, как описано в экспериментальном примере 2, было проведено количественное определение содержания пептида В. Эффективности удерживания, как было определено путем деления значений, полученных при этом исследовании, на соответствующие значения исходного содержания пептида, и краткие сведения о свойствах используемого полимера DL-молочной кислоты, показаны в таблице 3.
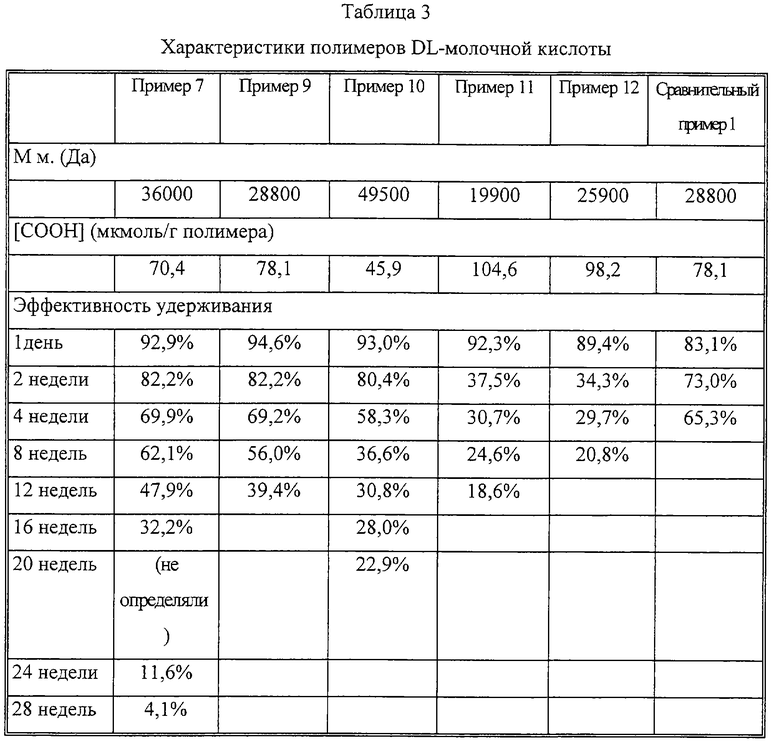
Как видно из таблиц 2 и 3, микрокапсулы, описанные в примерах 7 - 12, в отличие от сравнительного примера 1, обладают поразительно высокой эффективностью удерживания, примерно 90% и выше, в первый день после введения. Поэтому очевидно, что 3-окси-2-нафтойная кислота не только эффективна для обеспечения включения большого количества биоактивного вещества в препараты длительного высвобождения, но также очень хорошо подавляет бурное начальное высвобождение биоактивного вещества. В частности, эксперименты с использованием микрокапсул, описанных в примерах 7 - 9, показывают, что в случае использования DL-молочных кислот со среднемассовыми молекулярными массами примерно от 20000 до 50000 и содержанием карбоксильных групп f примерно от 50 до 90 мкмоль/г, как было найдено путем количественного определения методом мечения, возможно высвобождение биоактивного вещества с постоянной скоростью в течение очень продолжительного периода времени.
Экспериментальный пример 4
После того как микрокапсулы, полученные в примере 7, были введены подкожно крысам способом, описанным в экспериментальном примере 2, отбирали образцы крови и определяли концентрации пептида В и тестостерона в сыворотке. Результаты показаны в таблице 4.

Как видно из таблицы 4, концентрация биоактивного вещества в крови поддерживается на постоянном уровне вплоть до 28 недели после введения, это означает, что биоактивное вещество непрерывно высвобождалось из микрокапсул в течение 28-недельного периода. Было показано, что фармакологически активная концентрация тестостерона в течение этого периода постоянно удерживалась ниже нормального уровня и что биологически активное вещество стабильно присутствовало в микрокапсулах и высвобождалось из них в течение длительного периода времени, не теряя своей активности, даже тогда, когда 3-окси-2-нафтойная кислота присутствовала в препарате.
Пример 14
Смесь 0,5 N водного раствора гидроокиси натрия/метанола (объем/объем = 1/5) пропускали через сильноосновную ионообменную колонку (SeP-Pak Plus QMA Cartridge, производства WATERS) для удаления ионов хлора. После того как элюат переставал реагировать на добавление раствора нитрата серебра, подкисленного азотной кислотой, появлением белого помутнения, пропускали смесь вода/метанол (объем/объем = 1/5) для удаления избытка щелочи. После того как элюат становился нейтральным, 18,8 мг ацетата пептида В растворяли в 2 мл смеси вода/метанол (объем/объем = 1/5) и пропускали через колонку, предварительно обработанную, как описано выше. Этот элюат и другой элюат, полученный при прохождении только одной смеси объемом 6 мл, объединяли; эту смесь смешивали с раствором, содержащим 5,91 мг 3-окси-2-нафтойной кислоты в 1 мл смеси вода/метанол (объем/объем = 1/5), затем концентрировали с помощью роторного испарителя. Когда в смеси появлялось белое помутнение, добавляли 2 мл воды, после чего перемешивали. После центрифугирования (3000 об/мин, 20°С, 15 минут) супернатант удаляли, после этого проводили несколько циклов промывания водой, после чего осадок сушили в вакууме (40°С, в течение ночи) и получали 4,09 мг соли 3-окси-2-нафтойной кислоты и пептида В.
К этой соли добавляли 0,5 мл воды, затем перемешивали при комнатной температуре в течение 4 часов, после чего жидкость фильтровали через фильтр с размером пор 0,2 мкм и определяли количественно с помощью ВЭЖХ. Концентрации пептида В и 3-окси-2-нафтойной кислоты составляли 2,37 г/л и 0,751 г/л соответственно. Соль частично оставалась нерастворенной даже после перемешивания; приведенные выше значения позволяют считать, что растворимость в воде соли 3-окси-2-нафтойной кислоты и пептида В составляет не более 1/100 растворимости в воде ацетата пептида В, но не менее чем 1000 г/л. Это свидетельствует о том, что соль 3-окси-2-нафтойной кислоты и пептида В может использоваться в качестве препарата длительного высвобождения пептида В.
Результаты изобретения
Представленная в данном изобретении композиция длительного высвобождения характеризуется высоким содержанием биологически активного вещества и способна контролировать скорость его высвобождения.
Изобретение относится к области фармацевтической химии и касается композиции длительного высвобождения, содержащей физиологически активное вещество или его соль, оксинафтойную кислоту или ее соль и биодеградируемый полимер или его соль, обладающей улучшенной биодоступностью. 10 с. и 17 з.п. ф-лы, 4 табл.
5-оксо-Pro-His-Trp-Ser-Tyr-Y-Leu-Arg-Pro-Z,
где Y представляет собой DLeu, DAla, DTrp, Dser (tBu), D2Nal или Dhis (ImBzl);
Z представляет собой NH-C2H5 или Gly-NH2.
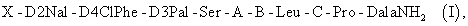
где Х представляет собой N (4Н2-фуроил) Gly или Nac;
А представляет собой один из следующих остатков: NMeTyr, Туr, Aph(Atz) и NMeAph(Atz);
В представляет собой один из следующих остатков:
DLys(Nic), DCit, DLys(AzaglyNic), DLys(AzaglyFur), DhArg(Et2), DAph(Atz) и DhCi;
С представляет собой Lys(Nisp), Arg или hArg(Et2);
или его соли,
и формулы II
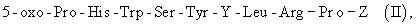
где Y представляет собой один из следующих остатков: DLeu, DAla, DTrp, DSer(tBu), D2Nal и DHis(lmBzl);
Z представляет собой NH2-C2H5, Gly-NH2;
или его соли.
| RU 94016167 А, 10.04.1994 | |||
| US 5478564 А, 26.12.1995 | |||
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |

