Данное изобретение относится к новым терапевтически полезным производным 2-фенилпиран-4-она, способам их получения, а также содержащим их фармацевтическим композициям.
Известно, что неселективное ингибирование фермента циклооксигеназы (СОХ) предотвращает избыточное вырабатывание ассоциируемых с воспалением простагландинов, опосредуемых циклооксигеназой-2 (СОХ-2), но одновременно приводит к депривации (потере) в тканях базального уровня простагландинов, необходимых для здоровья определенных тканей, опосредуемых в основном циклооксигеназой-1 (СОХ-1). Нестероидные противовоспалительные лекарственные препараты являются неселективными ингибиторами циклооксигеназы и по этой причине оказывают побочное действие в виде снижения почечного кровотока и функции тромбоцитов, диспепсии и образования язв в желудке.
Обнаружено, что некоторые производные 2-фенилпиран-4-она селективно активнее ингибируют циклооксигеназу-2, чем циклооксигеназу-1, и могут быть использованы в лечении опосредуемых циклооксигеназой-2 заболеваний, таких как воспаление, боль, лихорадка и астма, с меньшими побочными эффектами.
Соответственно данное изобретение предлагает соединение 2-фенилпиран-4-она формулы (I)
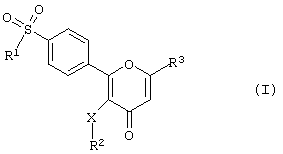
где R1 - алкил или - NR4R5-гpyппa, в которой каждый из R4 и R5 независимо друг от друга представляет собой атом водорода или алкильную группу;
R2 - алкил, С3-С7 циклоалкил, пиридил, тиенил, нафтил, тетрагидронафтил или инданил либо фенильная группа, которая может быть незамещенной или замещенной одним или несколькими атомами галогена или группами, включающими алкил, трифторметил, гидрокси, алкокси, метилтио, амино, моно- или диалкиламино, гидроксиалкильными или гидроксикарбонильными;
R3 - метил, гидроксиметил, алкоксиметил, С3-С7 циклоалкоксиметил, бензилоксиметил, гидроксикарбонил, нитрил, трифторметил или дифторметил либо CH2-R6-гpyппa, в которой R6 - алкильная группа, а
Х - простая связь, атом кислорода, атом серы или метиленовая группа,
или его фармацевтически приемлемую соль.
Алкильные группы и остатки, например, присутствующие в алкокси, оксиалкильных, моно- или диалкиламиногруппах, указанные в связи с группами R1-R6, обычно представляют собой "низший" алкил, содержащий от 1 до 6, особенно от 1 до 4, атомов углерода, при этом углеводородная цепь может быть разветвленной или прямой. Предпочтительные алкильные группы и группы с родственными алкильными остатками включают метил, этил, пропил, включая изо-пропил, а также бутил, включая н-бутил, трет-бутил и втор-бутил.
В фенильной группе, замещенной одним или несколькими атомами галогена или группами, включающими алкил, трифторалкил, гидрокси, алкокси, метилтио, амино, моно- или диалкиламино, гидроксиалкил или гидроксикарбонил, фенильное кольцо может быть замещено 1, 2, 3, 4 или 5 заместителями, предпочтительно 1, 2 или 3 заместителями, при этом каждый из них независимо выбирают из вышеуказанных возможных заместителей. Фенильная группа (присоединенная к Х или кольцу пиран-4-она через положение 1) может быть замещена в любом из оставшихся положений, т.е. 2, 3, 4, 5 или 6. Фенильная группа, имеющая более одного заместителя, может быть замещена в любом сочетании положений. Например, фенильная группа, имеющая два заместителя, может быть замещена в положениях 2 и 3, 2 и 4, 2 и 5, 2 и 6, 3 и 4 или 3 и 5.
В частности, R2 предпочтительно представляет собой разветвленный алкил, С3-C7 (предпочтительно С3, C5 или С6), циклоалкил, нафтил, тетрагидронафтил или инданил, незамещенную фенильную группу или фенильную группу, замещенную одним или несколькими атомами галогена, алкоксигруппами, предпочтительно метоксигруппами, и/или алкильными группами, предпочтительно метильными группами. Фенильная группа предпочтительно имеет 1, 2 или 3 заместителя, более предпочтительно 1 или 2 заместителя. Атомы галогена предпочтительно выбирают из атомов фтора, хлора и брома. Если R2 - фенильная группа, замещенная одним или несколькими атомами галогена, алкоксигруппами и/или алкильными группами, то один из заместителей предпочтительно находится в положении 4 фенильной группы. Если R2 - фенильная группа, замещенная одним или двумя атомами галогена, то по меньшей мере один из заместителей предпочтительно находится в положении 2 или 4.
R1 предпочтительно независимо представляет собой незамещенную алкильную группу, такую как метил, этил, пропил или бутил, предпочтительно метил, либо NН2-группу (т.е. в вышеуказанной формуле оба R4 и R5 независимо друг от друга представляют собой атом Н).
R3 также предпочтительно независимо представляет собой незамещенную алкильную группу, такую как метил, этил, пропил или бутил, предпочтительно метил, нитрил, гидроксиметил, метоксиметил, дифторметил или гидроксикарбонил.
Х предпочтительно независимо представляет собой простую связь, атом кислорода или метиленовую группу, более предпочтительно простую связь или атом кислорода.
Конкретные примеры производных 2-фенилпиран-4-она в соответствии с данным изобретением включают следующие соединения:
2-(4-метансульфонилфенил)-6-метил-3-фенилпиран-4-он,
3-(4-фторфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(3-фторфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(2-фторфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(4-хлорфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(3-хлорфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(2-хлорфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(4-бромфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
2-(4-метансульфонилфенил)-6-метил-3-п-толилпиран-4-он,
2-(4-метансульфонилфенил)-6-метил-3-м-толилпиран-4-он,
2-(4-метансульфонилфенил)-6-метил-3-о-толилпиран-4-он,
2-(4-метансульфонилфенил)-6-метил-3-(4-трифторметилфенил)пиран-4-он,
3-(2,4-дифторфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(3,4-дифторфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(3,5-дифторфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(2,5-дифторфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(2,6-дифторфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(2,4-дихлорфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(3,4-дихлорфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(3-фтор-4-метоксифенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(4-хлор-3-фторфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(2-хлор-4-фторфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(4-бромфенокси)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(4-фторфенокси)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(2,4-дифторфенокси)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-циклогексил-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
2-(4-метансульфонилфенил)-6-метил-3-нафтален-2-илпиран-4-он,
4-(6-метил-4-оксо-3-фенил-4Н-пиран-2-ил)бензолсульфонамид,
4-[3-(4-фторфенил)-6-метил-4-оксо-4Н-пиран-2-ил]бензолсульфонамид,
4-[3-(3,4-дихлорфенил)-6-метил-4-оксо-4Н-пиран-2-ил]бензолсульфонамид,
5-(2,4-дифторфенил)-6-(4-метансульфонилфенил)-4-оксо-4Н-пиран-2-карбонитрил,
3-(2-фторфенокси)-2-(метансульфонилфенил)-6-метилпиран-4-он,
3-(4-хлорфенокси)-2-(метансульфонилфенил)-6-метилпиран-4-он,
3-(2-хлорфенокси)-2-(метансульфонилфенил)-6-метилпиран-4-он,
3-(2,5-дифторфенокси)-2-(метансульфонилфенил)-6-метилпиран-4-он,
3-(3-хлор-4-метилфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
2-(4-метансульфонилфенил)-6-метил-3-феноксипиран-4-он,
3-(4-фторфенокси)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
2-(4-метансульфонилфенил)-6-метил-3-(4-метилфенокси)пиран-4-он,
3-(4-хлорфенил)-2-(4-метансульфонилфенил)-6-метоксиметилпиран-4-он,
3-(4-хлорфенил)-6-дифторметил-2-(4-метансульфонилфенил)пиран-4-он,
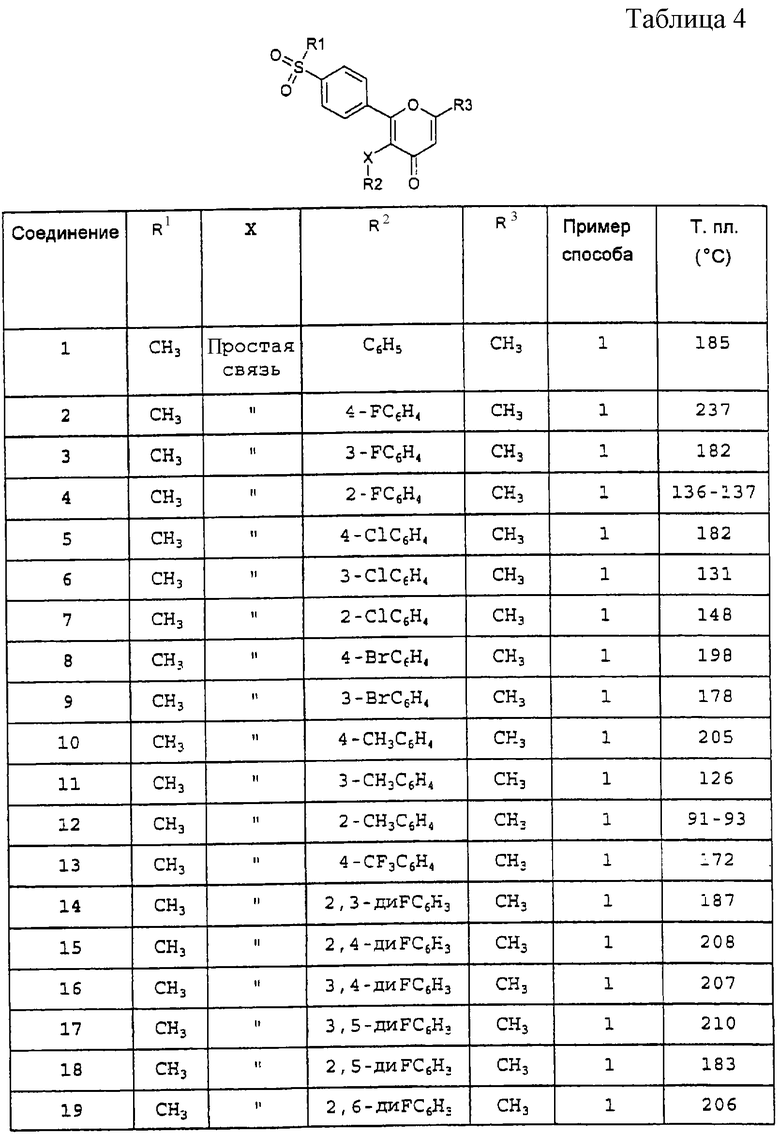
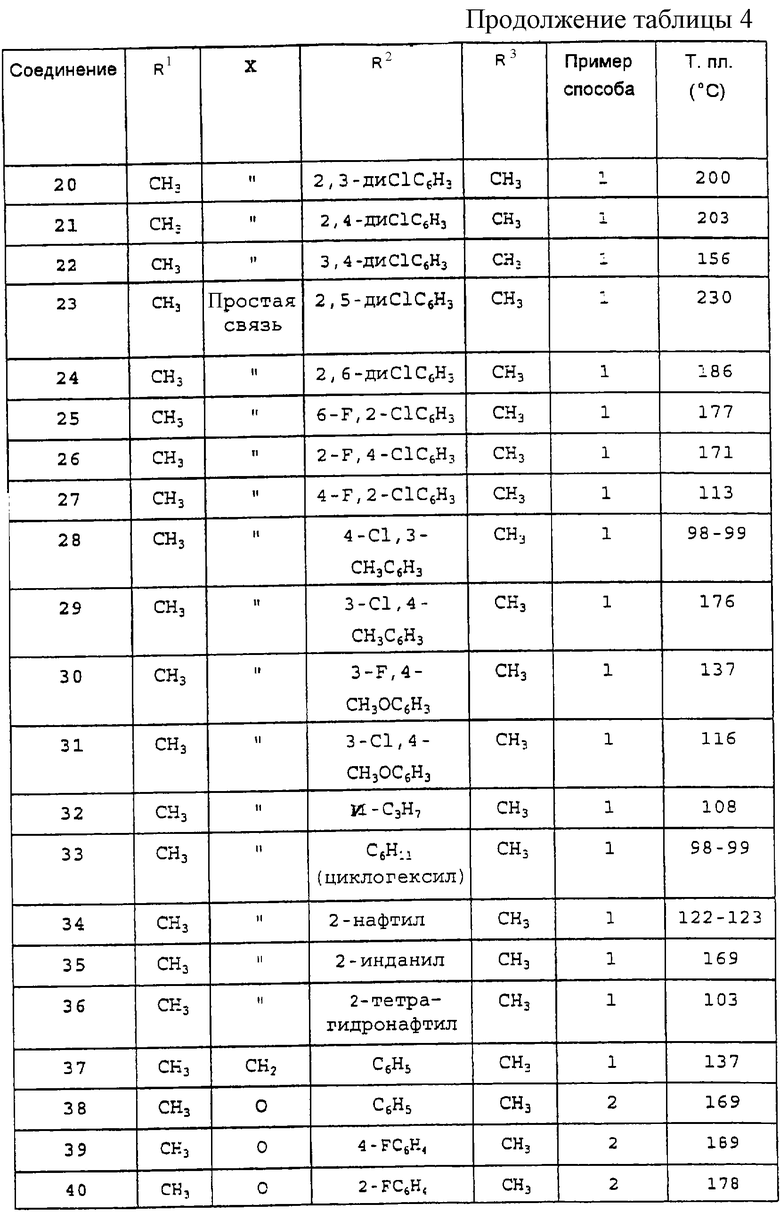
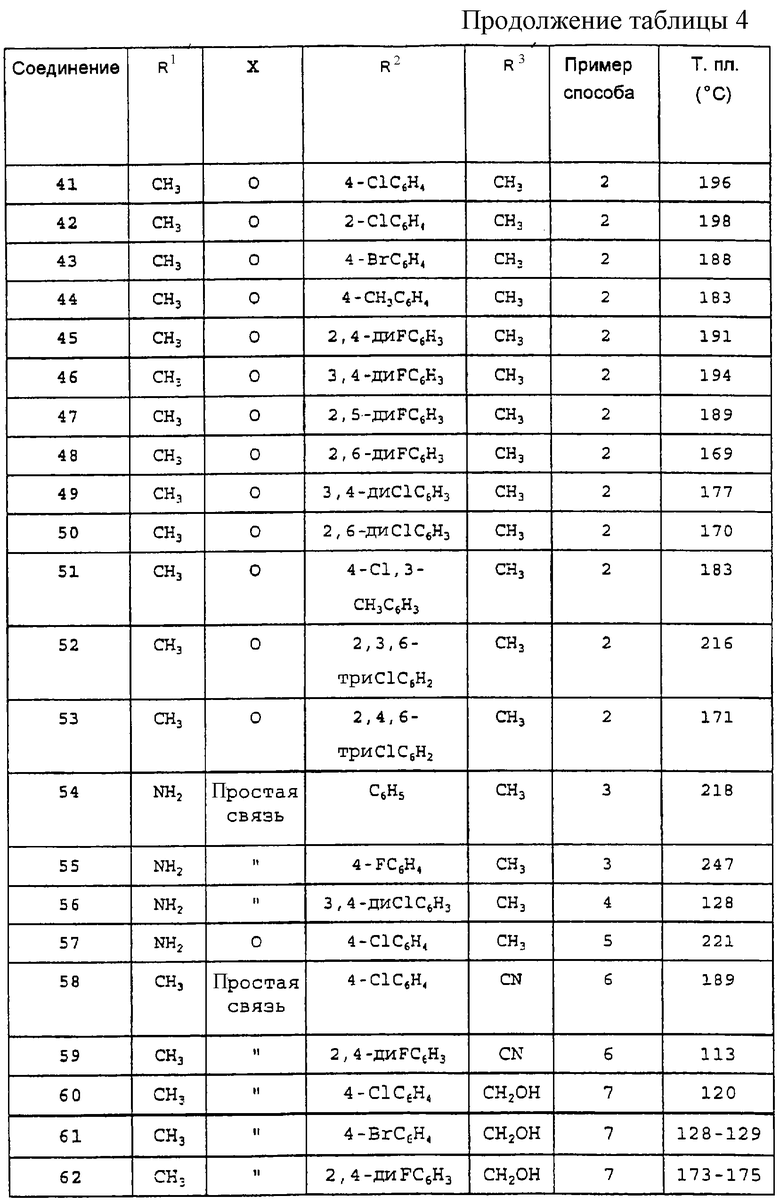
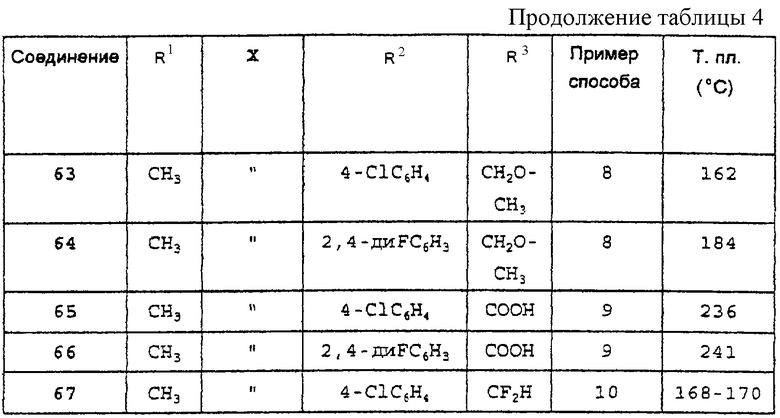
а также любое из соединений, конкретно указанных в Таблице 4, равно как и их фармацевтически приемлемые соли. Особый интерес представляют следующие соединения:
3-(4-фторфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(2-фторфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(4-хлорфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(4-бромфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(2,4-дифторфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(3,4-дихлорфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(3-хлор-4-метилфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
2-(4-метансульфонилфенил)-6-метил-3-феноксипиран-4-он,
3-(4-фторфенокси)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(2-фторфенокси)-2-(метансульфонилфенил)-6-метилпиран-4-он,
3-(4-хлорфенокси)-2-(метансульфонилфенил)-6-метилпиран-4-он,
3-(2-хлорфенокси)-2-(метансульфонилфенил)-6-метилпиран-4-он,
3-(4-бромфенокси)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
2-(4-метансульфонилфенил)-6-метил-3-(4-метилфенокси)пиран-4-он,
3-(2,4-дифторфенокси)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(2,5-дифторфенокси)-2-(метансульфонилфенил)-6-метилпиран-4-он,
3-(4-хлорфенил)-2-(4-метансульфонилфенил)-6-метоксиметилпиран-4-он,
3-(4-хлорфенил)-6-дифторфенил-2-(4-метансульфонилфенил)пиран-4-он,
а также их фармацевтически приемлемые соли.
Данное изобретение также предлагает способы получения соединения формулы (I), зависящие от значения R3. Если R3 - метильная группа, то соединения формулы (I) получают в соответствии со значением R1. Таким образом, соединение формулы (I), где R3 - метильная группа, а R1 - алкил или -NR4R5-гpyппa, в которой R4 и R5 - алкильные группы, т.е. производное 2-фенилпиран-4-она формулы (II)
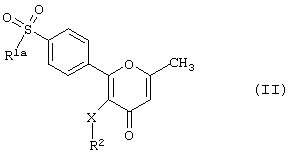
где R1a - алкил или -NR4aR5a- группа, в которой каждый из R4a и R5a независимо друг от друга представляет собой алкильные группы, a R2 и Х имеют вышеуказанные значения,
получают в результате реакции карбонильного производного формулы (III)
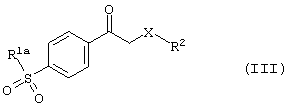
где R1a, R2 и Х имеют вышеуказанные значения,
с избытком безводной уксусной кислоты и полифосфорной кислоты при температуре от 100 до 150°С.
Производное карбонила формулы (III) может быть получено способами, хорошо известными в литературе (ЕР-А-714883, WО 96/06840, WО 96/31509 и DE-2064520), либо, когда Х - атом кислорода или серы, в результате реакции фенацильного производного формулы (IV)
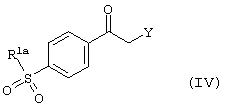
где R1a имеет вышеуказанные значения, a Y - атом хлора или брома,
с гидрокси или меркаптопроизводным формулы (V)
НХa-R2 (V)
где R2 имеет вышеуказанные значения, а X3 - атом кислорода или серы.
Реакция между производным фенацила формулы (IV) и промежуточным соединением формулы (V) может быть осуществлена путем нагревания смеси этих двух исходных материалов необязательно в смеси растворителя метиленхлорида, бензола или толуола и воды при температуре от 15 до 30°С и в присутствии катализатора межфазного переноса, такого как бензилтриэтиламмонийхлорид.
Производное карбонила формулы (III), в котором Х - не является атомом серы, может быть также получено в результате реакции тиопроизводного формулы (VI)
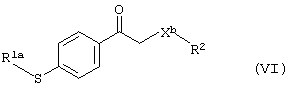
где R1 и R2 имеют вышеуказанные значения, а X1a - простая связь, атом кислорода или метиленовая группа,
с окислителем, предпочтительно монопероксифталатом магния или 3-хлорпероксибензойной кислотой. Реакцию предпочтительно проводят в органическом растворителе, таком как смесь метиленхлорида с метанолом или этанолом, при температуре от 10 до 40°С.
Данное изобретение также предлагает способ получения соединения формулы (I), в котором R3 - метильная группа, В1 - алкильная группа, а Х - отличен от атома серы, т.е. производного 2-фенилпиран-4-она формулы (VII)
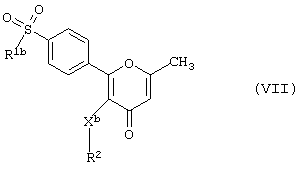
где R1b - алкил, а R2 и Xb имеют вышеуказанные значения,
в результате реакции меркаптопроизводного формулы (VIII)
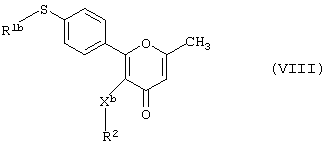
где R1b, R2 и Xb имеют вышеуказанные значения,
с окислителем, предпочтительно монопероксифталатом магния или 3-хлорпероксибензойной кислотой.
Реакцию между меркаптопроизводным формулы (VIII) и окислителем предпочтительно осуществляют, как описано выше для соединения формулы (VI), в органическом растворителе, таком как смесь метиленхлорида с метанолом или этанолом, при температуре от 10 до 40°С.
Данное изобретение также предусматривает способ получения соединения формулы (I), где R1-NR4R5-группа, а R3 - метил, т.е. производного 2-фенилпиран-4-она формулы (IX)
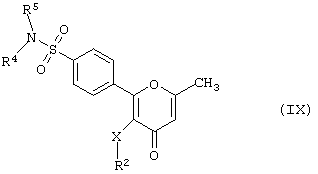
где R2, R4 R5 и Х имеют вышеуказанные значения,
в результате реакции производного хлорсульфонила формулы (X)
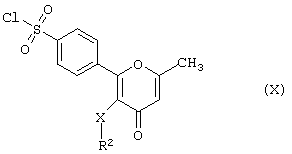
где R2 и Х имеют вышеуказанные значения,
с амином формулы (XI)
R4-NH-R5 (XI)
где R4 и R5 имеют вышеуказанные значения.
Эту реакцию предпочтительно проводят при температуре от 10 до 40°С.
Хлорсульфонильное производное формулы (X), например, может быть получено в результате реакции соединения формулы (XII)
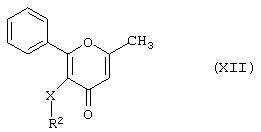
где R2 и X имеют вышеуказанные значения,
с хлорсульфоновой кислотой, предпочтительно при температуре от 80 до 120°С.
Данное изобретение далее предлагает способ получения соединения формулы (I), где R3 - метил, а R1 - -NR4R5-гpyппa, в которой R4 и R5 - водород, т.е. производного 2-фенилпиран-4-она формулы (XIII)
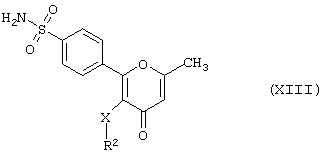
где R2 и Х имеют вышеуказанные значения,
в результате дебензилирования соответствующего N,N-дибензилпроизводного формулы (XIV)
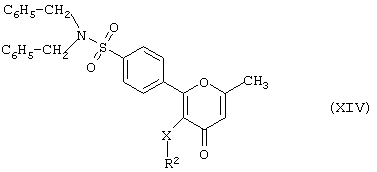
где R2 и X имеют вышеуказанные значения.
Дебензилирование предпочтительно проводят с избытком трифторуксусной, серной или метансульфокислоты при температуре от 0 до 120°С.
Промежуточное соединение формулы (XIV) может быть получено в соответствии с вышеприведенными способами с применением подходящих исходных материалов, в которых как R4, так и R5 (или R4a и R5a) представляют собой бензильные группы.
Промежуточные соединения формул (IV) и (VI), применяемые для получения соединений в соответствии с данным изобретением, могут быть получены описанными в литературе способами, например M.F. Saettone, J. Org. Chem., 31, р. 1959 (1966), и WO 9606840.
Промежуточные соединения формул (VIII) и (XII) могут быть получены способом, описанным для получения соединений формулы (II), с применением подходящих исходных материалов.
Производные 2-фенилпиран-4-она формулы (I), где R3 не означает метилгруппу, могут быть получены способами, представленными на следующей Схеме:
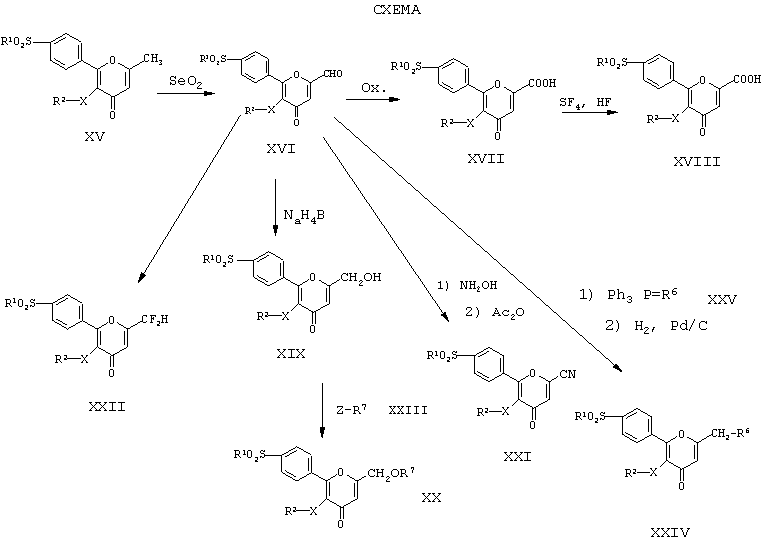
Как следует из Схемы, производные 2-фенилпиран-4-она формулы (I), где R3 не означает метил, т.е. соединения формул (XVII), (XVIII), (XIX), (XX), (XXI), (XXII) и (XXIV), получают из соединений формулы (I), в которой R3 - метил, т.е. соединения формулы (XV), способы получения которого описаны выше. На первой стадии соединения формулы (XV) обрабатывают окислителем, таким как диоксид селена, в органическом растворителе, таком как тетрагидрофуран или диоксан, в автоклаве и при температуре от 100 до 190°С. Получают соответствующий альдегид формулы (XVI), применяемый в качестве исходного материала для получения соединений формулы (I), в которой R3 не означает метил.
Соединения формулы (I), где R3 - оксикарбонил, т.е. соединение формулы (XVII), получают из соответствующего альдегида (XVI) в результате реакции с окислителем, таким как дихромат пиридиния или диоксид марганца, в органическом растворителе, таком как N,N-диметилформамид или этанол, при температуре от -5 до 35°С. Полученные соединения (XVII) применяют в качестве исходных материалов для получения соединений формулы (I), в которой R3 - трифторметил, т.е. соединение формулы (XVIII). Соединения (XVII) подвергают реакции со смесью тетрафторид серы - фтористый водород, необязательно в присутствии органического растворителя, такого как метиленхлорид, в автоклаве при температуре от 20 до 140°С.
Соединения формулы (I), где R3 - гидроксиметил, т.е. соединения формулы (XIX) получают, восстанавливая соединения (XVI) гидридом бора или алюминия, предпочтительно борогидридом натрия, в растворителе, таком как метанол или этанол, при температуре от 10 до 40°С. Дальнейшая реакция соединений (XIX) с подходящим галогенидом формулы (XXIII)
Z-R7 (ХХIII)
где Z - атом хлора, брома или иода, а R7 - алкил, С3-С7 циклоалкил или бензил,
приводит к получению соединений формулы (I), где R3 - алкоксиметил, С3-С7 циклоалкоксиметил или бензилоксиметил, т.е. соединений формулы (XX). Реакцию проводят в органическом растворителе, таком как ацетон, N,N-диметилформамид или тетрагидрофуран, в присутствии гидрида натрия или калия либо амида при температуре от 20 до 120°С.
Альдегиды формулы (XVI) также применяют в качестве исходного материала для получения соединений формулы (I), где R3 - нитрил, т.е. соединений формулы (XXI). Реакцию проводят на первой стадии путем обработки альдегидов (XVI) гидрохлоридом гидроксиламина и муравьиной кислотой при температуре от 80 до 120°С. Полученное производное оксима выделяют и подвергают нагреванию с избытком уксусного ангидрида при температуре от 100 до 180°С.
Соединение формулы (I), где R3 - дифторметилгруппа, т.е. соединения формулы (XII), получают из альдегидов формулы (XVI) в результате реакции с фторирующим реагентом, таким как трифторид диэтиламиносеры или смесь тетрафторид серы - фтористый водород, необязательно в присутствии органического растворителя, такого как метиленхлорид, бензол или толуол, при температуре от 0 до 130°С.
Производные 2-фенилпиран-4-она формулы (I), в которой R3 -CH2-R6-гpyппa, т.е. соединения формулы (XXIV), также получают из альдегидов формулы (XVI) в результате двустадийного процесса. На первой стадии альдегид (XVI) подвергают реакции с производным трифенилфосфина (XXV) в присутствии растворителя, такого как диоксан, диметоксиэтан или тетрагидрофуран, при температуре от 15°С до температуры кипения растворителя. Полученное соединение подвергают гидрированию на второй стадии процесса в присутствии катализатора, такого как палладий на активированном угле. Реакцию проводят в присутствии растворителя, такого как метанол, этанол или этилацетат при температуре от 15 до 40°С.
Производные 2-фенилпиран-4-она формулы (I), в которых присутствует основная группа, могут быть превращены способами, известными per se, в фармацевтически приемлемые соли, предпочтительно аддитивные соли кислот, в результате обработки органическими или неорганическими кислотами, такими как фумаровая, винная, янтарная или соляная кислота. Производные 2-фенилпиран-4-она формулы (I), в которой R3 - оксикарбонильная группа, также могут быть превращены в фармакологически приемлемые соли, например, со щелочными металлами, такими как натрий или калий, путем реакции с гидроксидом щелочного металла.
Следующие биологические тесты и данные дополнительно иллюстрируют данное изобретение.
Активность циклооксигеназы-1 и циклооксигеназы-2 в цельной крови человека
Свежую кровь от здоровых добровольцев, не принимавших никаких нестероидных противовоспалительных лекарственных препаратов в течение по меньшей мере 7 дней перед забором крови, собирают в промытые гепарином пробирки (20 ед. гепарина на 1 мл). Для определения активности циклооксигеназы-1 500 мкл аликвот крови инкубируют либо с 5 мкл наполнителя (диметилсульфоксид), либо с 5 мкл подвергаемого тесту соединения в течение 1 часа при 37°С. За 20 минут до прекращения инкубирования добавляют ионофор кальция А23187 (25 мкМ). Плазму отделяют центрифугированием (10 мин при 13000 об/мин) и хранят при -30°С до измерения уровня TXB2 с применением набора для иммунного анализа ферментов (ELISA). Действие соединений оценивают, инкубируя каждое соединение при 5-6 различных концентрациях и тройным определением. Величину IC50 получают в результате нелинейной регрессии, применяя математическое обеспечение InPLot, GraphPad на компьютере IBM.
Для определения активности циклокосигеназы-2 500 мкл аликвот крови инкубируют в присутствии LPS (10 мкг/мл) в течение 24 часов при 37°С, для того чтобы индуцировать экспрессию циклооксигеназы-2 (Patriagnani et al., J. Pharm. Exper. Ther., 271, 1705-1712 (1994)). Плазму отделяют центрифугированием (10 мин при 13000 об/мин) и хранят при -30°С до измерения уровня PGE2 с применением набора для иммунного анализа ферментов (ELISA). Действие ингибиторов оценивают, инкубируя каждое соединение (5-микролитровые аликвотные части) при 5-6 различных концентрациях и тройным определением в присутствии LPS в течение 24 час. Величины IC50 получают в результате нелинейной регрессии, применяя математическое обеспечение InPlot, GraphPad на компьютере IBM.
Противовоспалительная активность (стилизированный артрит)
Используют самцов крыс Wistar весом 175-200 г со свободным доступом к еде и воде. В 0 день животные получают внутриподошвенную инъекцию суспензии Mycobacterium tuberculosis в парафиновом масле (0,5 мг/крысу) в левую заднюю лапу. Группу контрольных крыс без артрита инъецируют только парафиновым маслом. На 11-й и 14-й день после возникновения артрита измеряют объем задней лапы каждой крысы, применяя водный плетизмограф. Отбирают животных, объем лап которых за это время увеличился. Крыс распределяют на группы по 8 особей, имеющих одинаковый средний объем лап и приблизительно одинаковое стандартное отклонение.
Тест-соединения вводят перорально один раз в сутки в течение 7 дней (дни 14-20). Контрольные крысы с артритом и без него получают только наполнитель в течение 7 дней. Объем задних лап измеряют через 20 часов после последней дозы (на 21-й день). Массу тела определяют через день.
Результаты выражают в виде процентной величины ингибирования воспаления (объем лапы) для каждой опытной группы, учитывая как артритные, так и неартритные контроли наполнителя. Для статистических исследований применяют тесты ANOVA.
Ульцерогенная активность
Животные. Используют самцов крыс Wistar (Interfauna, U.K., Ltd.) весом около 150-175 г. Животных содержат при режиме день-ночь (12:12 часов, свет включают в 7 часов утра) при комнатной температуре (22±1°С). Еду и воду предоставляют ad libitum.
Процедура. Соединения вводят перорально один раз в сутки 4 дня подряд. Каждый день перед введением лекарственного препарата определяют массу каждой, крысы. Животных подвергают анестезии через 24 часа после последней дозы и при помощи сердечной пункции забирают 1 мл крови, применяя гепарин (10 ед/мл) в качестве антикоагулянта. Измеряют процентную величину гематокрита. Кишечник удаляют, вскрывают горизонтально и осторожно промывают. Применяя параметрическую шкалу, оценивают макроскопическую тяжесть поражений кишечника, определяя количество перфоративных и неперфоративных язв кишечника с помощью индекса поражений от 0 до 3 (0: отсутствие язв, 1:<10 язв, 2:10-25 язв, 3:>25 язв). При использовании такого протокола исследований язв желудка не наблюдается.
Обработку в каждом эксперименте подвергают рандомизации. Результаты сравнивают с результатами, полученными в группе, получавшей носитель, с применением теста ANOVA.
Результаты
Результаты, полученные в ходе биологических анализов, представлены в Таблицах 1, 2 или 3.
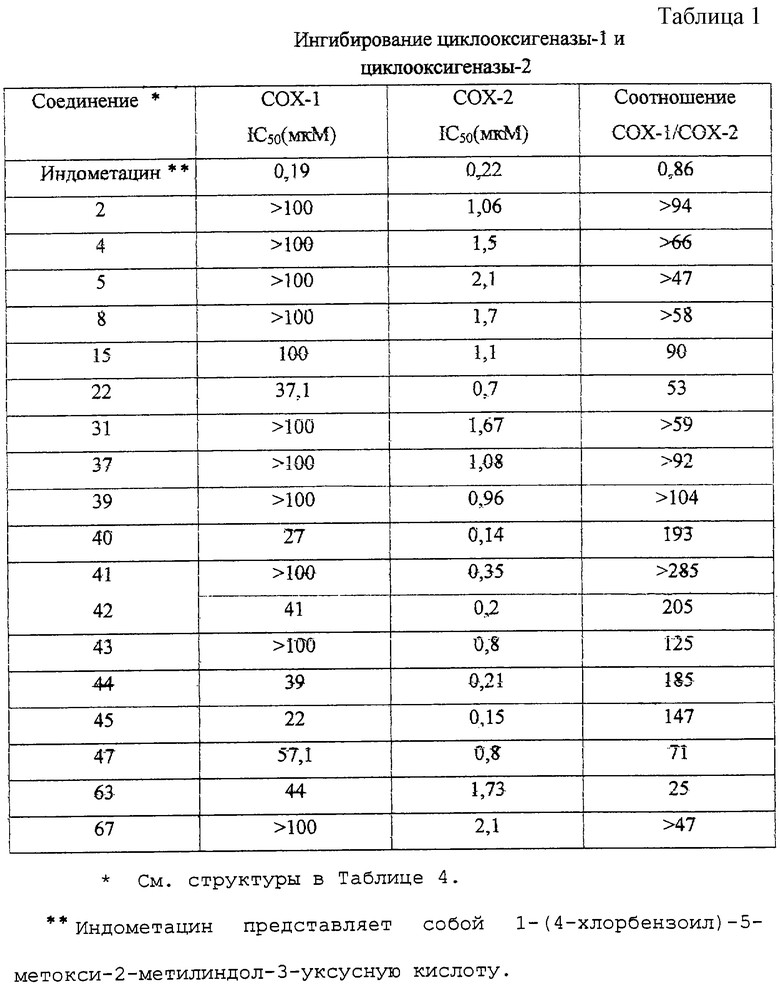
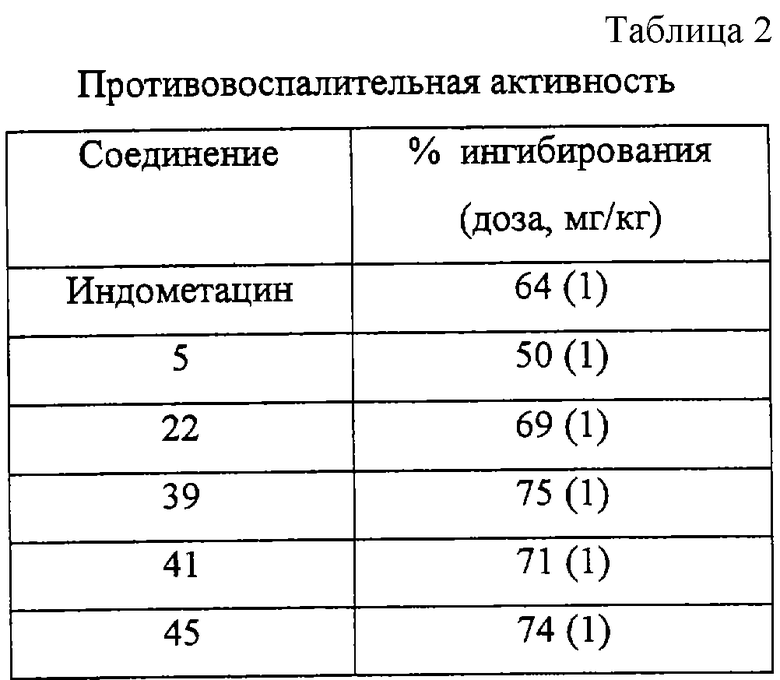
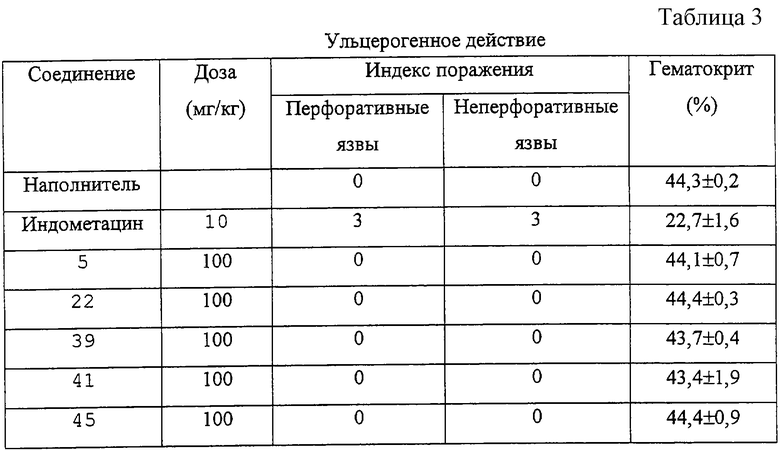
Как следует из Таблицы 1, производные 2-фенилпиран-4-она формулы (I) являются эффективными и селективными ингибиторами циклооксигеназы-2, в то время как соединение для сравнения - индометацин - эффективен как ингибитор циклооксигеназы-2, так и циклооксигеназы-1. Благодаря своей низкой ингибирующей активности по отношению к циклооксигеназе-1 соединения формулы (I) оказывают сильное противовоспалительное действие (см. Таблицу 2) и существенно более низкое побочное действие, чем обычно применяемые нестероидные противовоспалительные лекарственные препараты (например, желудочно-кишечная токсичность (см. Таблицу 3), побочное действие на почки, эффект снижения кровотечений и приступов астмы у людей, чувствительных к аспирину).
Таким образом, соединения в соответствии с данным изобретением предпочтительно представляют собой селективные ингибиторы циклооксигеназы-2 млекопитающих, например циклооксигеназы-2 человека. Соединения в соответствии с данным изобретением также предпочтительно проявляют низкую ингибирующую активность по отношению к циклооксигеназе-1 млекопитающих, например циклооксигеназе-1 человека. Обычно ингибирующая активность может быть установлена путем анализов in vitro, например, как описано выше.
Предпочтительные соединения в соответствии с данным изобретением имеют величину IC50 для циклооксигеназы-2, составляющую менее 5 мкМ, предпочтительно менее 3 и более предпочтительно менее 2,5 мкМ. Предпочтительные соединения в соответствии с данным изобретением также имеют величину IС50 для циклооксигеназы-1 свыше 10 мкМ, предпочтительно свыше 20 мкМ. В качестве индикатора селективности для ингибирования циклооксигеназы-2 в сравнении с циклооксигеназой-1, соотношение величин IC50 COX-1/COX-2 предпочтительно превышает 20, 30 или 50, более предпочтительно 80, 90 или 100.
Данное соединение также предлагает соединение формулы (I), применяемое в терапевтическом способе лечения человека или животного, в частности, для лечения боли, лихорадки или воспаления, для ингибирования индуцируемого простаноидами сокращения гладких мышц либо предупреждения колоректального рака или нейродегенеративных заболеваний.
Данное изобретение также предусматривает применение соединения формулы (I) при получении лекарственного препарата для лечения боли, лихорадки или воспаления, ингибирования индуцируемого простаноидами сокращения гладких мышц либо предупреждения колоректального рака или нейродегенеративных заболеваний.
Соединения формулы (I) могут быть использованы для облегчения боли, лихорадки и воспаления при различных состояниях, включая ревматическую лихорадку, симптомы, ассоциируемые с гриппом или другими вирусными инфекциями, насморк, боли в нижней части спины и шее, дисменорею, головную боль, зубную боль, растяжение и деформацию, миозит, невралгию, синовит, бурсит, тендинит, травмы после хирургических и стоматологических процедур, а также артрит, включая ревматоидный артрит, остеоартрит, подагрический артрит, спондилоартропатию, системную красную волчанку, ювенильный артрит и резорбцию костей. Они также могут быть использованы при лечении заболеваний, связанных с воспалением кожи, таких как псориаз, экзема, жжение и дерматит. Кроме того, эти соединения могут быть использованы для предупреждения колоректального рака.
Соединения формулы (I) также ингибируют индуцируемое простаноидами сокращение гладких мышц и поэтому могут быть использованы при лечении дисменореи, предупреждении преждевременных родов, астмы и бронхитов.
Соединения формулы (I) могут быть использованы в качестве альтернативы известным нестероидным противовоспалительным лекарственным средствам, особенно когда имеются противопоказания к применению таких нестероидных противовоспалительных лекарственных препаратов, например, при лечении пациентов с желудочно-кишечными нарушениями, включая пептические язвы, гастрит, региональный энтерит, неспецифический язвенный колит, дивертикулит, болезнь Крона, синдром воспаленного и раздраженного кишечника, желудочно-кишечное кровотечение и нарушения свертывания крови, заболевания почек (например, недостаточная функция почек), пациентов перед хирургической операцией или принимающих антикоагулянты, а также пациентов, подверженных астме, индуцируемой нестероидными противовоспалительными лекарственными препаратами.
Данные соединения также могут быть использованы для лечения воспалений при таких заболеваниях, как сосудистые заболевания, вызванные мигренью головные боли, узелковый периартрит, тиреоидит, апластическая анемия, лимфогранулематоз, склеродермия, диабет I типа, тяжелая миастения, саркоидоз, нефротический синдром, синдром Бехчета, полимиозит, повышенная чувствительность, конъюнктивит, гингивит, ишемия миокарда и удар.
Соединения в соответствии с данным изобретением являются ингибиторами фермента циклооксигеназы-2 и поэтому могут быть использованы для лечения вышеуказанных заболеваний, опосредуемых циклооксигеназой-2. Эти соединения могут быть также использованы для профилактики нейродегенеративных заболеваний, таких как болезнь Альцгеймера.
Соответственно производные 2-фенилпиран-4-она формулы (I) и их фармацевтически приемлемые соли, а также фармацевтические композиции, включающие такие соединения и/или их соли, могут быть использованы в способе лечения нарушений человеческого организма, включающем введение пациенту, нуждающемуся в таком лечении, эффективного количества производного 2-фенилпиран-4-она формулы (I) или его фармацевтически приемлемой соли.
Данное изобретение также предусматривает фармацевтические композиции, включающие в качестве активного ингредиента по меньшей мере производное 2-фенилпиран-4-она формулы (I) или его фармакологически приемлемую соль вместе с фармацевтически приемлемым наполнителем, таким как носитель или разбавитель. Активный ингредиент может включать от 0,001 до 99 маc.%, предпочтительно от 0,01 до 90 маc.% композиции в зависимости от природы состава и от того, требуется ли дальнейшее разбавление перед его применением.
Композиции предпочтительно имеют форму, подходящую для перорального, местного, назального, ингаляционного, ректального, чрескожного или инъецируемого введения.
Фармацевтически приемлемые наполнители, смешиваемые с активным соединением или солями такого соединения для получения композиций в соответствии с данным изобретением, хорошо известны per se, при этом применяемые наполнители зависят inter alia от предполагаемого способа введения композиций.
Композиции в соответствии с данным изобретением предпочтительно подвергают адаптации для инъецируемого или перорального введения. В этом случае композиции для перорального введения могут иметь форму таблеток, таблеток с отсроченным действием, сублингвальных таблеток, капсул или жидких препаратов, таких как смеси, эликсиры, сиропы или суспензии, содержащие соединение в соответствии с данным изобретением; такие препараты могут быть получены способами, хорошо известными в данной области.
Разбавители, которые могут быть использованы при получении композиций, включают такие жидкие и твердые разбавители, которые совместимы с активным ингредиентом, при желании вместе с красителями или отдушками. Таблетки или капсулы могут содержать от 2 до 500 мг активного ингредиента или эквивалентное количество его соли.
Жидкие композиции, адаптированные для перорального применения, могут иметь форму растворов или суспензий. Растворы могут представлять собой водные растворы растворимой соли или другого производного активного соединения, например, в сочетании с сахарозой, для получения сиропа. Суспензии могут включать нерастворимое активное соединение в соответствии с данным изобретением или его фармацевтически приемлемую соль в сочетании с водой вместе с суспендирующим агентом или отдушкой.
Композиции для парентеральных инъекций могут быть получены из растворимых солей, которые могут быть подвергнуты или не подвергнуты лиофилизации и которые могут быть растворены в апирогенной водной среде или иной подходящей для парентерального введения жидкости.
Эффективная доза обычно составляет 10-600 мг активного ингредиента в сутки. Суточная доза может быть введена за один или несколько приемов, предпочтительно за 1-4 раза в день.
Следующие примеры иллюстрируют данное изобретение, никоим образом не ограничивая его объем.
ПРИМЕР 1
а) К раствору 2-(4-фторфенил)-1-(4-метансульфонилфенил)этанона (1 г, 3,4 моль) в ледяной уксусной кислоте (15 мл) добавляют полифосфорную кислоту (10 г), а затем нагревают при 140°С в течение 16 часов. После охлаждения реакционную смесь выливают в ледяную воду, экстрагируют этилацетатом (2×50 мл), органический раствор подвергают сушке (Na2SO4), a растворитель удаляют при пониженном давлении. Остаточное масло очищают колоночной хроматографией на силикагеле и этилацетатом в качестве растворителя для элюирования, получая 3-(4-фторфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он (0,5 г), т.пл. 237°С (Соединение 2 в Таблице 4).
ПРИМЕР 2
a) К раствору 2,4-дифторфенола (3,71 г, 29 ммоль) и 2-бром-1-(4-метилсульфанилфенил)этанона (7,00 г, 29 ммоль) в метиленхлориде (50 мл) добавляют раствор карбоната калия (5,91 г, 43 ммоль) и кислого сернокислого тетрабутиламмония (0,48 г, 1,4 ммоль) в воде (20 мл). Смесь перемешивают при комнатной температуре в течение 16 часов. Добавляют воду (100 мл, органическую фазу сливают, а основную фазу экстрагируют метиленхлоридом (2×100 мл). Органический раствор сушат (Na2SO4), а растворитель удаляют при пониженном давлении. Полученное твердое вещество промывают этиловым эфиром, получая 2-(2,4-дифторфенокси)-1-(4-метилсульфанилфенил)этанон (6,60 г), т.пл. 70-71°С.
b) К раствору 2-(2,4-дифторфенокси)-1-(4-метилсульфанилфенил)этанона (6,60 г, 22 ммоль) в метиленхлориде (100 мл) добавляют воду (20 мл) и 80% гексагидрата монопероксифталата магния (15,26 г, 25 ммоль). Смесь перемешивают при комнатной температуре в течение 16 часов. Реакционную смесь выливают в насыщенный раствор бикарбоната натрия (200 мл) и экстрагируют метиленхлоридом (3×100 мл). Органическую фазу сушат (Na2SO4), а растворитель удаляют при пониженном давлении, получая 2-(2,4-дифторфенокси)-1-(4-метансульфонилфенил)этанона (4,97 г) в виде твердого вещества, т. пл. 161-163°С.
с) К раствору 2-(2,4-дифторфенокси)-1-(4-метансульфонилфенил)этанона (4,60 г, 14 ммоль) в уксусной кислоте (70 мл) добавляют полифосфорную кислоту (45 г), а затем нагревают при 140°С в течение 5 часов. После охлаждения реакционную смесь выливают в ледяную воду, экстрагируют этилацетатом (2×100 мл), органический раствор сушат (Nа2SO4), а растворитель удаляют при пониженном давлении. Остаточное масло очищают колоночной хроматографией на силикагеле и смесью этилацетат/н-гексан (1:2) в качестве растворителя для элюирования. В результате перекристаллизации из этанола получают 3-(2,4-дифторфенокси)-2-(4-метансульфонилфенил)-6-метилпиран-4-она (0,64 г), т.пл. 191°С (Соединение 45 в Таблице 4).
ПРИМЕР 3
a) Раствор 4-(дибензилсульфамоил)бензойной кислоты (24 г, 63 ммоль) в тионилхлориде (50 мл) подвергают кипячению с обратным холодильником в течение 2,5 часов, а избыток тионилхлорида удаляют при пониженном давлении, получая 4-(дибензилсульфамоил)бензоилхлорид (25 г) в виде масла, которое используют на следующей стадии без очистки.
b) К раствору N,О-диметилгидроксиламингидрохлорида (7,37 г, 75,6 ммоль) и триэтиламина (21,8 мл, 151 ммоль) в метиленхлориде (150 мл) медленно добавляют другой раствор 4-(дибензилсульфамоил)бензоилхлорида (25 г) в метиленхлориде (150 мл) и полученную смесь перемешивают при комнатной температуре в течение 16 часов. Твердое вещество отфильтровывают, растворитель удаляют при пониженном давлении, а остаточное масло очищают колоночной хроматографией на силикагеле и смесью н-гексан/этилацетат (1:1) в качестве растворителя для элюирования, получая N,O-диметиламид 4-(дибензилсульфамоил)бензойной кислоты (22 г), т. пл. 75°С.
с) К суспензии магния (2 г, 82,4 ммоль) в безводном тетрагидрофуране (20 мл) медленно добавляют другой раствор бензилхлорида (10,4 г, 82,4 ммоль) в безводном тетрагидрофуране (100 мл). По завершении реакции медленно добавляют раствор N,O-диметилформамида 4-(дибензилсульфамоил)бензойной кислоты (7 г, 16,5 ммоль) в безводном тетрагидрофуране (50 мл), поддерживая температуру на уровне 0°С. После перемешивания при этой температуре в течение получаса реакционную смесь выливают в насыщенный раствор хлорида аммония (100 мл), экстрагируют этиловым эфиром (3×75 мл), а органические экстракты сушат (Na2SO4). Растворитель удаляют при пониженном давлении, а остаточное масло очищают колоночной хроматографией на силикагеле и смесью н-гексан/этилацетат (1:3) в качестве растворителя для элюирования, получая N,N-дибензил-4-фенилацетилбензсульфонамид (9,4 г), т. пл. 143°С.
d) К раствору вышеуказанного соединения, получаемого на стадии с) (9,4 г, 20,7 ммоль), в ледяной уксусной кислоте (140 мл) добавляют полифосфорную кислоту (94 г) и полученную смесь нагревают до 140°С в течение 16 часов. После охлаждения реакционную смесь выливают в ледяную воду, экстрагируют этилацетатом (3×150 мл), а органический раствор сушат (Nа2SO4). Растворитель удаляют в вакууме, а к остаточному маслу добавляют концентрированную серную кислоту (38 мл), затем перемешивают при 0°С в течение 10 минут, еще в течение 60 минут при комнатной температуре и выливают в ледяную воду. Осажденные твердые вещества собирают с фильтрацией и очищают колоночной хроматографией на силикагеле и этилацетатом в качестве растворителя для элюирования, получая 4-(6-метил-4-оксо-3-фенил-4Н-пиран-2-ил)бензолсульфонамид (1,5 г), т. пл. 218°С (Соединение 54 в Таблице 4).
ПРИМЕР 4
а) К раствору 3,4-дихлорфенилацетофенона (5,3 г, 20 ммоль) в ледяной уксусной кислоте (90 мл) добавляют полифосфорную кислоту (64 г) и полученный раствор нагревают при 140°С в течение 24 часов. После охлаждения реакционную смесь выливают в ледяную воду, экстрагируют этилацетатом (3×75 мл), органический раствор сушат (Na2SO4), a растворитель удаляют при пониженном давлении. Полученный остаток очищают колоночной хроматографией на силикагеле и смесью н-гексан/этилацетат (3:2) в качестве растворителя для элюирования, получая 3-(3,4-дихлорфенил)-2-фенил-6-метилпиран-4-он (1,68 г), т.пл. 104°С.
b) Раствор вышеполученного соединения (1,4 г, 4,3 ммоль) в хлорсульфоновой кислоте (12 мл) нагревают при 70°С в течение 1,5 часов, после охлаждения реакционную смесь медленно выливают в ледяную воду и экстрагируют этилацетатом (2×50 мл). Органический раствор сушат (Na2SO4), растворитель удаляют при пониженном давлении и к остаточному маслу, предварительно растворенному в метаноле (10 мл), медленно добавляют насыщенный раствор аммиака в метаноле (40 мл). После перемешивания при комнатной температуре в течение 1 часа растворитель удаляют при пониженном давлении, остаток растворяют в ацетилацетате (100 мл), полученный раствор промывают водой (2×100 мл) и сушат (Na2SO4), a растворитель удаляют при пониженном давлении. Остаточное масло очищают колоночной хроматографией на силикагеле и смесью н-гексан/этилацетат (1:1) в качестве растворителя для элюирования, получая 4-[3-(3,4-дихлорфенил)-6-метил-4-оксо-4Н-пиран-2-ил]бензолсульфонамид (0,5 г), т. пл. 128°С (Соединение 56 в Таблице 4).
ПРИМЕР 5
а) К раствору N,N-дибензил-4-(2-бромацетил)бензолсульфонамида (10,5 г, 23 ммоль) и п-хлорфенола (2,94 г, 23 ммоль) в метиленхлориде (42 мл) добавляют карбонат калия (4,83 г, 34,7 ммоль) и тетрабутиламмонийбромид (0,42 г, 1,2 ммоль) в воде (140 мл). Реакционную смесь подвергают кипячению с обратным холодильником в течение 16 часов. После охлаждения смесь разбавляют метиленхлоридом (150 мл). Органический слой отделяют, промывают водой и сушат (Na2SO4). Растворитель удаляют при пониженном давлении, получая N,N-дибензил-4-[2-(4-хлорфенокси)ацетил]бензолсульфонамид (11,7 г) в виде полутвердого остатка, используемого на следующей стадии без дальнейшей очистки.
b) К раствору N,N-дибензил-4-[2-(4-хлорфенокси)ацетил]бензолсульфонамида (11,7 г, 23 ммоль) в уксусной кислоте (105 мл) добавляют полифосфорную кислоту (75 г) и полученный раствор нагревают при 140°С в течение 5 часов. После охлаждения реакционную смесь выливают в ледяную воду, экстрагируют этилацетатом (3×150 мл) и органический раствор сушат (Na2SO4). Растворитель удаляют при пониженном давлении и полученное масло растворяют в H2SO4 (33 мл). Смесь перемешивают при комнатной температуре в течение 15 минут и выливают в ледяную воду. Твердое вещество отфильтровывают и подвергают очистке колоночной хроматографией на силикагеле и смесью этилацетат/метиленхлорид/уксусная кислота (78:10:1) в качестве растворителя для элюирования, получая 4-[3-(4-хлорфенокси)-6-метил-4-оксо-4Н-пиран-2-ил]бензолсульфонамид (0,28 г), т. пл. 221°С (Соединение 57 в Таблице 4).
ПРИМЕР 6
а) К раствору 3-(2,4-дифторфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-она (1,7 г, 4,5 ммоль) (Соединение 13) в диоксане (45 мл) добавляют диоксид селена (2,2 г, 20 ммоль) и смесь нагревают в автоклаве при 180°С в течение 1 часа. После охлаждения реакционную смесь фильтруют, растворитель удаляют при пониженном давлении, а остаточное масло подвергают очистке колоночной хроматографией на силикагеле и этилацетатом в качестве растворителя для элюирования, получая 5-(2,4-дифторфенил)-6-(4-метансульфонилфенил)-4-оксо-4Н-пиран-2-карбальдегид (0,85 г).
b) К раствору вышеуказанного соединения (0,8 г, 2,1 ммоль) в муравьиной кислоте (6 мл) добавляют гидроксиламингидрохлорид (0,17 г, 2,7 ммоль) и смесь нагревают при 100°С в течение 2 часов. После охлаждения реакционную смесь выливают на лед, добавляют 2N гидроксид натрия до рН 7 и экстрагируют этилацетатом (2×50 мл). Органический раствор сушат (Na2SO4), растворитель удаляют в вакууме, остаток растворяют в уксусном ангидриде (15 мл) и нагревают при 150°С в течение 5 часов. Растворитель удаляют при пониженном давлении, остаток обрабатывают метиленхлоридом (50 мл), а полученный раствор промывают 2N гидроксидом натрия (2×25 мл). Органический раствор сушат (Na2SO4), растворитель удаляют в вакууме, а остаток подвергают очистке колоночной хроматографией на силикагеле и смесью н-гексан/этилацетат (1:1) в качестве растворителя для элюирования, получая 5-(2,4-дифторфенил)-6-(4-метансульфонилфенил)-4-оксо-4Н-пиран-2-карбонитрил (0,2 г), т.пл. 113°С (Соединение 59 в Таблице 4).
ПРИМЕР 7
a) К раствору 3-(4-хлорфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-она (4,0 г, 10,6 ммоль) в диоксане (50 мл) добавляют диоксид селена (5,9 г, 53 ммоль) и нагревают в запечатанной пробирке при 180°С в течение 30 минут. После охлаждения неочищенный материал фильтруют через целит, а растворитель удаляют при пониженном давлении. Полученное масло подвергают очистке колоночной хроматографией на силикагеле и смесью этилацетат/н-гексан (2:1) в качестве растворителя для элюирования, получая 5-(4-хлорфенил)-6-(4-метансульфонилфенил)-4-оксо-4Н-пиран-2-карбальдегид (1,80 г).
b) К раствору 5-(4-хлорфенил)-6-(4-метансульфонилфенил)-4-оксо-4H-пиран-2-карбальдегида (1,8 г, 4,6 ммоль) в метаноле (30 мл) при 0°С медленно добавляют борогидрид натрия (0,26 г, 6,9 ммоль). Полученную смесь перемешивают в течение 30 минут при комнатной температуре. Реакционную смесь концентрируют, а остаток растворяют в этилацетате. Органический слой промывают водой, сушат (Na2SO4), а растворитель удаляют при пониженном давлении. Полученный остаток подвергают очистке колоночной хроматографией на силикагеле и смесью этилацетат/н-гексан (1:1) в качестве растворителя для элюирования, получая 3-(4-хлорфенил)-6-гидроксиметил-2-(4-метансульфонилфенил)пиран-4-он (0,9 г), т. пл. 120°С (Соединение 60 в Таблице 4).
ПРИМЕР 8
а) К раствору 3-(4-хлорфенил)-6-гидроксиметил-2-(4-метансульфонилфенил)пиран-4-она (0,5 г, 1,3 ммоль) в метиленхлориде (10 мл) добавляют йодистый метил (0,24 мл, 3,86 ммоль), а также раствор гидроксида натрия (0,41 г, 10,3 ммоль) и тетрабутиламмонийхлорида (50 мл) в воде (0,8 мл). Реакционную смесь перемешивают при комнатной температуре в течение 18 часов. Органический слой разбавляют метиленхлоридом (20 мл), промывают водой и сушат (Na2SO4). Растворитель удаляют при пониженном давлении. Полученное твердое вещество подвергают очистке колоночной хроматографией на силикагеле и этилацетатом в качестве растворителя для элюирования, получая 3-(4-хлорфенил)-2-(4-метансульфонилфенил)-6-метоксиметилпиран-4-она (0,15 г), т.пл. 162°С (Соединение 63 в Таблице 4).
ПРИМЕР 9
а) К раствору нитрата серебра (0,88 г, 5,1 ммоль) в воде (4 мл) добавляют раствор гидроксида натрия (0,42 г, 6,2 ммоль) в воде (4 мл). Реакционную смесь перемешивают в течение 15 минут при комнатной температуре, после чего к ней добавляют раствор 5-(4-хлорфенил)-6-(4-метансульфонилфенил)-4-оксо-4Н-пиран-2-карбальдегида в тетрагидрофуране (10 мл). Реакционную смесь перемешивают в течение 3 часов при комнатной температуре и фильтруют через целит. Растворитель удаляют при пониженном давлении, а остаток растворяют в этилацетате. Органический слой промывают водой и сушат (Na2SO4). Растворитель удаляют при пониженном давлении. Полученное твердое вещество подвергают очистке колоночной хроматографией на силикагеле и смесью этилацетат/метиленхлорид/уксусная кислота (78:10:1) в качестве растворителя для элюирования, получая 5-(4-хлорфенил)-6-(4-метансульфонилфенил)-4-оксо-4Н-пиран-2-карбоновую кислоту (0,13 г), т. пл. 236°С (Соединение 65 в Таблице 4).
ПРИМЕР 10
а) К раствору 5-(4-хлорфенил)-6-(4-метансульфонилфенил)-4-оксо-4Н-пиран-2-карбальдегида (0,74 г, 1,9 ммоль) в метиленхлориде (10 мл) при 0°С медленно добавляют диэтиламиносульфид DAST (0,61 г, 3,8 ммоль). Реакционную смесь перемешивают при этой температуре в течение 1 часа и при комнатной температуре в течение 16 часов. Смесь разбавляют метиленхлоридом (10 мл). Органическую фазу промывают водой, сушат (Na2SO4), а растворитель удаляют при пониженном давлении. Полученный остаток подвергают очистке колоночной хроматографией на силикагеле и смесью этилацетат/ н-гексан (1:1) в качестве растворителя для элюирования, получая 3-(4-хлорфенил)-6-дифторметил-2-(4-метансульфонилфенил)пиран-4-она (0,1 г), т.пл. 168-170°С (Соединение 67 в Таблице 4).
Производные 2-фенилпиран-4-она общей формулы (I), указанные в Таблице 4, получают в соответствии со способами, описанными в вышеприведенных примерах, но с применением соответствующих исходных материалов.
Примеры 11 и 12 иллюстрируют фармацевтические композиции в соответствии с данным изобретением и процедуру их получения.
ПРИМЕР 11
25000 капсул, каждая из которых содержит по 100 мг 3-(4-хлорфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-она (активный ингредиент), получают в соответствии со следующей рецептурой, кг:
Активный ингредиент 2,5
Моногидрат лактозы 5
Коллоидный диоксид кремния 0,05
Кукурузный крахмал 0,5
Стеарат магния 0,1
Процедура
Вышеуказанные ингредиенты просеивают через сито размером 60 меш, загружают в подходящий смеситель и заполняют ими 25000 желатиновых капсул.
ПРИМЕР 12
100000 таблеток, каждая из которых содержит по 50 мг 3-(2,4-дифторфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-она (активный ингредиент), получают в соответствии со следующей рецептурой, кг:
Активный ингредиент 5
Лактоза, полученная распылительной сушкой 19,9
Микрокристаллическая целлюлоза 3,9
Натрия стеарилфумарат 0,2
Коллоидальный диоксид кремния 0,2
Карбоксиметилкрахмал 0,8
Процедура
Все порошки просеивают через сито с отверстиями 0,6 мм, затем перемешивают в подходящем смесителе в течение 20 минут и прессуют в 300-миллиграммовые таблетки, применяя 9-миллиметровые дисковые и плоские скошенные штампы. Время распада таблеток составляет около 3 минут.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 1,2,4-ТРИАЗОЛО[4,3-B]ПИРИДО[3,2-D]ПИРИДАЗИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ ФОСФОДИЭСТЕРАЗЫ-4 | 1998 |
|
RU2202552C2 |
| ДИАРИЛМЕТИЛИДЕНФУРАНОВЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1997 |
|
RU2189979C2 |
| ПРОИЗВОДНЫЕ ПИРИДОПИРИМИДИНОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2269527C2 |
| СОЕДИНЕНИЯ ФЕНИЛПИРИДАЗИНА И СОДЕРЖАЩИЕ ИХ ЛЕКАРСТВЕННЫЕ СРЕДСТВА | 2001 |
|
RU2269519C2 |
| ПРОИЗВОДНЫЕ 2-(3H)-ОКСАЗОЛОНА, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ И СПОСОБ ИНГИБИРОВАНИЯ АКТИВНОСТИ СОХ-2 | 1997 |
|
RU2194043C2 |
| СПОСОБ ПОЛУЧЕНИЯ 1,5-ДИАРИЛ-3-ЗАМЕЩЕННЫХ ПИРАЗОЛОВ | 2000 |
|
RU2243967C2 |
| АКТИВАТОРЫ ГЛЮКОКИНАЗЫ | 2000 |
|
RU2242469C2 |
| ПРОИЗВОДНЫЕ 2,3-ДИГИДРО-6-НИТРОИМИДАЗО [2, 1-b] ОКСАЗОЛА | 2003 |
|
RU2326121C2 |
| АЛКИЛАМИНОЗАМЕЩЕННЫЕ БИЦИКЛИЧЕСКИЕ АЗОТСОДЕРЖАЩИЕ ГЕТЕРОЦИКЛЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2000 |
|
RU2264404C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПИРИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2004 |
|
RU2366659C2 |
Изобретение относится к новым производным 2-фенил-4-она формулы I
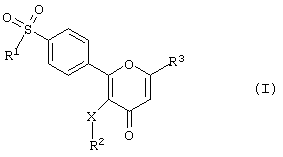
где R1 - алкил или - NR4R5 группа, в которой каждый из R4 и R5 независимо друг от друга представляет собой атом водорода или акильную группу;
R2 - алкил, С3-С7 циклоалкил, нафтил, тетрагидронафтил или инданил либо фенильная группа, которая может быть незамещенной или замещенной одним или несколькими атомами галогена либо группами алкил, или трифторметил, или алкокси;
R3 - группа метил, гидроксиметил, алкоксиметил, гидроксикарбонил, нитрил, трифторметил или дифторметил или группа CH2-R6, в которой R6 - алкильная группа, и
Х - простая связь, атом кислорода или метиленовая группа,
или к их фармацевтически приемлемым солям, а также к фармацевтической композиции на их основе, ингибирующей активность циклооксигеназы-2, и способу лечения. Технический результат - новые соединения, обладающие ценным фармакологическим действием. 3 н. и 5 з. п. ф-лы, 4 табл.
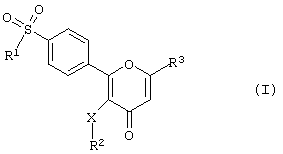
где R1 - алкил или -NR4R5 группа, в которой каждый из R4 и R5 независимо друг от друга представляет атом водорода или алкильную группу,
R2 - алкил, С3-С7 циклоалкил, нафтил, тетрагидронафтил или инданил, либо фенильная группа, которая может быть незамещенной или замещенной одним или несколькими атомами галогена либо группами алкила, трифторметила или алкокси;
R3 - группа метил, гидроксиметил, алкоксиметил, гидроксикарбонил, нитрил, трифторметил или дифторметил, или группа CH2-R6, в которой R6 - алкильная группа;
Х - простая связь, атом кислорода, атом серы или метиленовая группа,
или его фармацевтически приемлемая соль.
3-(4-фторфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(2-фторфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(4-хлорфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(4-бромфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(2,4-дифторфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(3,4-дихлорфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(3-хлор-4-метилфенил)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
2-(4-метансульфонилфенил)-6-метил-3-феноксипиран-4-он,
3-(4-фторфенокси)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(2-фторфенокси)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(4-хлорфенокси)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(2-хлорфенокси)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(4-бромфенокси)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
2-(4-метансульфонилфенил)-6-метил-3-(4-метилфенокси)пиран-4-он,
3-(2,4-дифторфенокси)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(2,5-дифторфенокси)-2-(4-метансульфонилфенил)-6-метилпиран-4-он,
3-(4-хлорфенил)-2-(4-метансульфонилфенил)-6-метоксиметилпиран-4-он,
3-(4-хлорфенил)-6-дифторметил-2-(4-метансульфонилфенил)-пиран-4-он,
или его фармацевтически приемлемая соль.
Приоритет по пунктам:
| RU 95112465 А1, 20.03.1997 | |||
| WO 9514014 А, 26.05.1995 | |||
| WO 9606840 А, 07.03.1996 | |||
| US 3901908 А, 26.08.1975. |

