Изобретение относится к новым терапевтически пригодным производным 2-(3Н)-оксазолона, способам их получения и к содержащим их фармацевтическим композициям.
Полагают, что механизм действия нестероидных противовоспалительных лекарственных средств состоит в ингибировании ферментной циклооксигеназы (СОХ) и в последовательном превращении арахидоновой кислоты в простагландины. Идентификация изоферментов циклооксигеназы-1 (СОХ-1) и циклооксигеназы-2 (СОХ-2) привела к гипотезе, состоящей в том, что ингибирование, в особенности селективное ингибирование, СОХ-2, будет снижать воспаление без побочных эффектов классических нестероидных противовоспалительных лекарственных средств, желудочную и почечную токсичность.
В соответствии с этой гипотезой, обнаружено, что некоторые производные 2-(3Н)-оксазолона ингибируют СОХ-2 и селективно ингибируют СОХ-2 в предпочтении к СОХ-1. Эти производные обладают эффективностью и толерантностью при лечении СОХ-2 опосредованных заболеваний, таких как воспаление, боль, жар и астма, и оказывают меньшие побочные действия, такие как ульцерогенная активность.
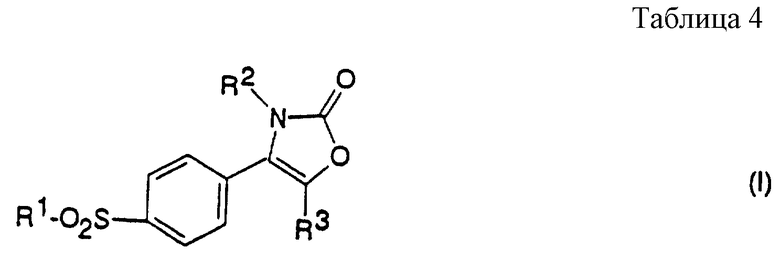
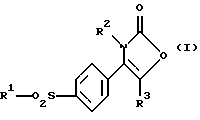
Соответственно настоящее изобретение предлагает соединение 2-(3Н)-оксазолона формулы (I):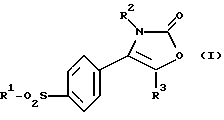
где R1 является алкильной группой или группой -NR4R5, где каждый R4 и R5 независимо являются водородом или алкильной или бензильной группой;
R2 является нафтильной (предпочтительно 2-нафтил), тетрагидронафтильной, незамещенной фенильной или фенильной группой, замещенной от 1 до 3 атомами галогена (предпочтительно хлором или фтором) или алкильной, гидрокси, алкокси или трифторметильной группой; и
R3 является водородом или алкильной группой.
Алкильные группы или фрагменты, как например в алкокси группах, упомянутые в отношении групп R1-R5, обычно представляют низший алкил, то есть содержащие до 6, и в особенности, до 4-х атомов углерода, при этом углеводородная цепь является разветвленной или неразветвленной. Предпочтительной алкильной группой или фрагментом является метил.
Заместители в фенильном кольце могут быть в любом положении. Так например, единственный заместитель может быть в 2-, 3- или 4-положении; или два заместителя могут быть в 2- и 4-положении или 3- и 4-положении.
Предпочтительными соединениями формулы (I) являются такие, где R1 является алкильной или аминогруппой, R2 является фенильной группой, замещенной одним или двумя атомами галогена (в особенности хлором или фтором), и R3 является водородом.
Заместители в фенильной группе, представленные R2, могут быть одинаковыми или различными.
Значительный интерес представляют следующие соединения:
3-(4-фторфенил)-4-(4-метилсульфонилфенил)-2-(3Н)- оксазолон, 3-(2-фторфенил)-4-(4-аминосульфонилфенил)-2-(3Н)-оксазолон, 3-(3,4-дихлорфенил)-4-(4-аминосульфонилфенил)-2-(3Н)-оксазолон и 3-(2, 4-дифторфенил)-4-(4-аминосульфонилфенил)-2-(3Н) -оксазолон.
Настоящее изобретение предлагает также способы получения соединения формулы (I), которые зависят от определения R1.
Настоящее изобретение предлагает способ получения соединения формулы (I), где R1 является алкильной группой или группой -NR4R5, в которой R4 и R5 отличны от водорода, а именно производного 2-(3Н)-оксазолона формулы (II):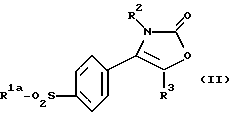
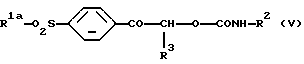
где R1a является алкильной группой -NR4aR5a, в которой каждый R4a и R5a независимо является алкильной или бензильной группой, и R2 и R3 являются такими, как они определены выше, который включает взаимодействие карбамата формулы (V):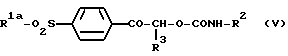
где R1, R2 и R3 являются такими, как они определены выше, с безводной уксусной кислотой.
Карбамат формулы (V) может быть получен, например, путем взаимодействия фенацильного спирта формулы (III):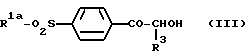
где R1a и R3 являются такими, как определено выше, с изоцианатом формулы (IV):
OCN-R2 (IV)
где R2 является таким, как определено выше.
Реакцию между фенацильным спиртом формулы (III) и изоцианатом формулы (IV) можно осуществить путем нагревания смеси этих двух исходных веществ, необязательно в присутствии органического растворителя, как например толуола или ксилола, при температуре от 80oС до 200oС.
Карбамат формулы (V) может быть также получен путем взаимодействия тиопроизводного формулы (VI):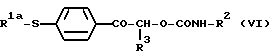
где R1, R2 и R3 являются такими, как определено выше, с окислителем, предпочтительно монопероксифталатом магния или 3-хлорпероксибензойной кислотой. Реакцию предпочтительно осуществляют в органическом растворителе, как например в смеси метиленхлорида с метанолом или этанолом, при температуре от 10oС до 40oС.
Карбамат формулы (V) после каждого процесса может быть выделен известными методами. Для получения соединения формулы (II) карбамат может быть нагрет до температуры от 80oС до 120oС с избытком безводной уксусной кислоты.
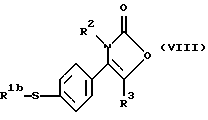
Настоящее изобретение также предлагает способ получения соединения формулы (I), где R1 является группой, а именно производного 2-(3Н)-оксазолона формулы (VII):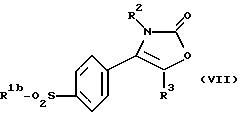
где R1b является алкильной группой, и R2 и R3 являются такими, как определено выше, путем взаимодействия меркаптопроизводного формулы (VIII):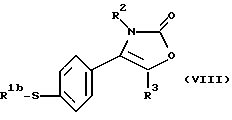
где R1b, R2 и R3 являются такими, как определено выше, с окислителем, предпочтительно с монопероксифталатом магния или 3-хлорпероксибензойной кислотой.
Реакцию между меркаптопроизводным формулы (VIII) и окислителем осуществляют предпочтительно, как описывалось ранее для соединения формулы (VI), в органическом растворителе, как например в смеси метиленхлорида с метанолом или этанолом, при температуре от 10oС до 40oС.
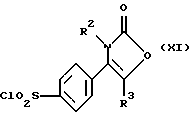
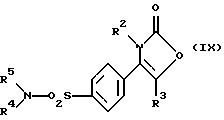
Настоящее изобретение дополнительно предлагает способ получения соединения формулы (I), где R1 является группой -NR4R5, а именно производного 2-(3Н)-оксазолона формулы (IX):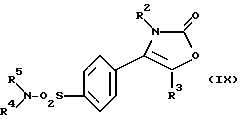
где R2, R3, R4 и R5 являются такими, как определено выше, путем взаимодействия производного хлорсульфонила формулы (XI):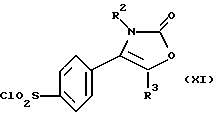
где R2, R3 являются такими, как определено выше, с амином формулы (XII):
R4-NH-R5 (XII)
где R4, R5 являются такими, как определено выше.
Данную реакцию предпочтительно осуществляют при температуре от 10oС до 40oС.
Производное хлорсульфонила формулы (XI) может быть получено, например, путем взаимодействия соединения формулы (X):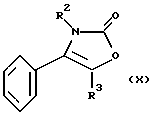
где R2, R3 являются такими, как определено выше, с хлорсульфоновой кислотой, предпочтительно при температуре от 80oС до 120oС.
Настоящее изобретение, кроме того, предлагает способ получения соединения формулы (I), где R1 является группой -NR4R5, где R4 и R5 являются водородом, а именно производного 2-(3Н)-оксазолона формулы (XIII):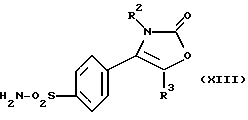
где R2, R3 являются такими, как определено выше, путем дебензилирования соответствующего соединения формулы (IX), где R4, R5 являются такими, как определено выше, при условии, что, по меньшей мере, один, предпочтительно оба, из R4, R5 является бензильной группой, например производного 2-(3Н)-оксазолона формулы (XIV):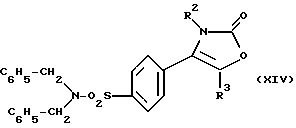
где R2, R3 являются такими, как определено выше.
Дебензилирование осуществляют предпочтительно с избытком трифторуксусной, серной и метансульфоновой кислоты при температуре от 0oС до 120oС.
Промежуточные соединения формул (III) и (VI), применяемые при получении соединений изобретения, могут быть получены способами, описанными в литературе, например, в: M.F. Saettone, J. Org. Chem. 31, р.1959 (1966).
Промежуточные соединения формул (XIII) и (X) могут быть получены тем же самым способом, раскрытым для получения соединений (II) с применением соответствующих исходных веществ.
Далее настоящее изобретение иллюстрирует следующие биологические испытания и данные.
Для анализа цельного клеточного СОХ-1 и СОХ-2 маточные растворы (10-3 М) лекарственных средств растворили в 50% диметилсульфоксиде и с помощью среды осуществили дополнительные разбавления. Наполнитель для лекарственного средства, при применяемых концентрациях, не оказывал влияния на ферментативные активности.
Ингибирование активности циклооксигеназы-1 (СОХ-1) в тромбоцитах человека
Из периферической крови человека, полученной от здоровых доноров, которые отказывались принимать какие-либо нестероидные противовосполительные лекарственные средства в течение, по меньшей мере, предшествующей недели, выделили тромбоциты. Для воспрепятствования свертывания крови ее обрабатывали 2 мг/мл натриевой соли EDTA и центрифугировали при 180 g в течение 10 минут при комнатной температуре с получением богатой тромбоцитами плазмы. Для получения тромбоцитного осадка обогащенную тромбоцитами плазму центрифугировали в течение 20 минут при 2000 g и 4oС. Клетки дважды промывали PBS без Са2+ и Мg2+ и повторно суспендировали сбалансированным солевым раствором Хенка (HBSS) до получения 5•107 клеток. Тромбоциты (107) предварительно инкубировали с лекарственными средствами в течение 15 минут при 37oС и затем инкубации продолжали в течение дополнительных 15 минут в присутствии 50μM арахидоновой кислоты. С применением твердофазного иммуноанализа (ELISA) измеряли в супернатантах получение тромбоксана B2 в ответ на арахидоновую кислоту. Результаты выражали в виде среднего от значений IC50, полученных в результате проведения трех независимых экспериментов.
Ингибирование активности циклооксигеназы-2 (СОХ-2) в клеточной линии HUV-EC-C
Эндотелиальная клеточная линия HUV-EC-C человека выражает селективность изофермента циклооксигеназы-2 после обработки форбол-12-миристат-13-ацетатом (РМА) (Miralpeix et al. , "Agents and Actions", 44: S274 (1995). HUV-EC-C клетки выращивали в F12K среде Хама (Ham), содержащей 10% эмбриональную бычью сыворотку, 100 μг/мл гепарина и 50 μг/мл добавки для роста эндотелиальных клеток (ECGS). Эксперименты осуществляли с применением 19-27 пассажей HUV-EC-C. Клетки (2•104) засевали на чашки Петри с 96-ю лунками и до начала экспериментов делали неактивными путем удаления в течение 48 часов фактора роста. Для индуцирования изофермента СОХ-2 активные HUV-EC-C клетки в течение 6 часов при 37oС обрабатывали 50 нМ ТРА. Затем культуральную среду изменили и клетки в течение 30 минут при 37oС инкубировали с лекарственными средствами. Затем добавили арахидоновую кислоту (50 μM) и клетки инкубировали в течение дополнительных 30 минут. С применением твердофазного иммуноанализа (ELISA) определили получение простагландина E2 в супернатантах в ответ на арахидоновую кислоту. Результаты выразили в виде среднего от значений IC50, полученных в результате проведения независимых экспериментов.
Ульцерогенная активность
Животные: Использовали крыс-самцов Wistar (Interfauna, U.K. Ltd), весящих около 120-150 г. Их выдерживали в цикле свет-темнота 12:12 часов (свет включали в 7.00 утра) при комнатной температуре (22±1oС). До осуществления эксперимента животные голодали в течение 18 часов, имея при этом свободный доступ к питьевой воде.
Методика: Эксперименты проводили с 9 до 17 часов. Соединения вводили оральным путем и через 6 часов после дозировки лекарственного средства животных исследовали. Желудок каждой крысы удаляли, открывали и осторожно промывали. Определяя количество и размер язв в гландулярном желудке, с применением параметрической шкалы (Cosen и Mazure), оценивали макроскопическую степень эрозий. Каждый желудок классифицировали, таким образом, с помощью изменения индекса и сравнивали с повреждением желудка, вызванным пероральным приемом 100 мг/кг кеторолака, применяемого в качестве положительного стандарта. Обработку в каждом эксперименте осуществляли произвольно.
Противовоспалительная активность (адъювантный артрит)
Использовали крыс самцов Wistar, весящих 175-200 г, имеющих свободный доступ к пище и воде. До начала эксперимента животные получали внутриплантарную инъекцию суспензии Mycobacterium tuberculosis в парафиновом масле (0,5 мг/крысу) в левую заднюю лапу. Группе из 8 контрольных крыс, не пораженных артритом, вводили парантерально только парафиновое масло. На 11 и 14 день после индуцирования артрита с применением водного плетизмографа измеряли объем задней лапы каждой крысы. Отбирали животных, объем лап которых за это время увеличился. Крыс распределяли на группы из 8 крыс, имеющих равные средние объемы лап и приблизительно равное стандартное отклонение.
Испытуемые соединения вводили перорально 1 раз ежедневно в течение 7 дней (14-20 день). Не пораженные и пораженные артритом контрольные крысы получали только наполнитель в течение 7 дней. Объемы задних лап измеряли через 20 часов после последней дозы (на 21 день). Массу тела определяли каждый второй день.
Результаты выражали в виде процента ингибирования воспаления (объем лапы) для каждой подверженной лечению группы, принимая во внимание как пораженных артритом, так и не пораженных артритом контрольных крыс с наполнителем. Для статистических исследований применяли испытание ANOVA.
Лекарственные средства
Для анализа цельного клеточного СОХ-1 и СОХ-2 маточные растворы (10-3 М) лекарственных средств растворяли в 50% диметилсульфоксиде и с помощью среды осуществляли дополнительные разбавления. Наполнители для лекарственного средства, при применяемых концентрациях, не оказывали влияния на ферментативную активность.
Для анализа in vitro все лекарственные средства вводили в наполнителе (0,1% Tween 80+0,5% метилцеллюлоза в дистиллированной воде) объемом 5 мл/кг.
Результаты
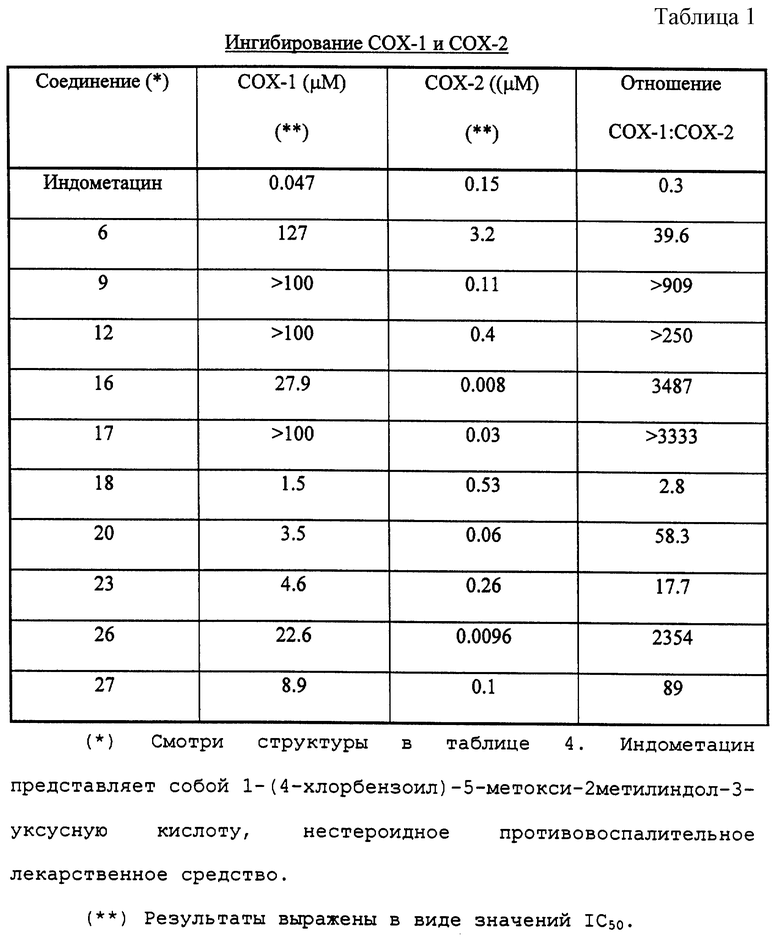
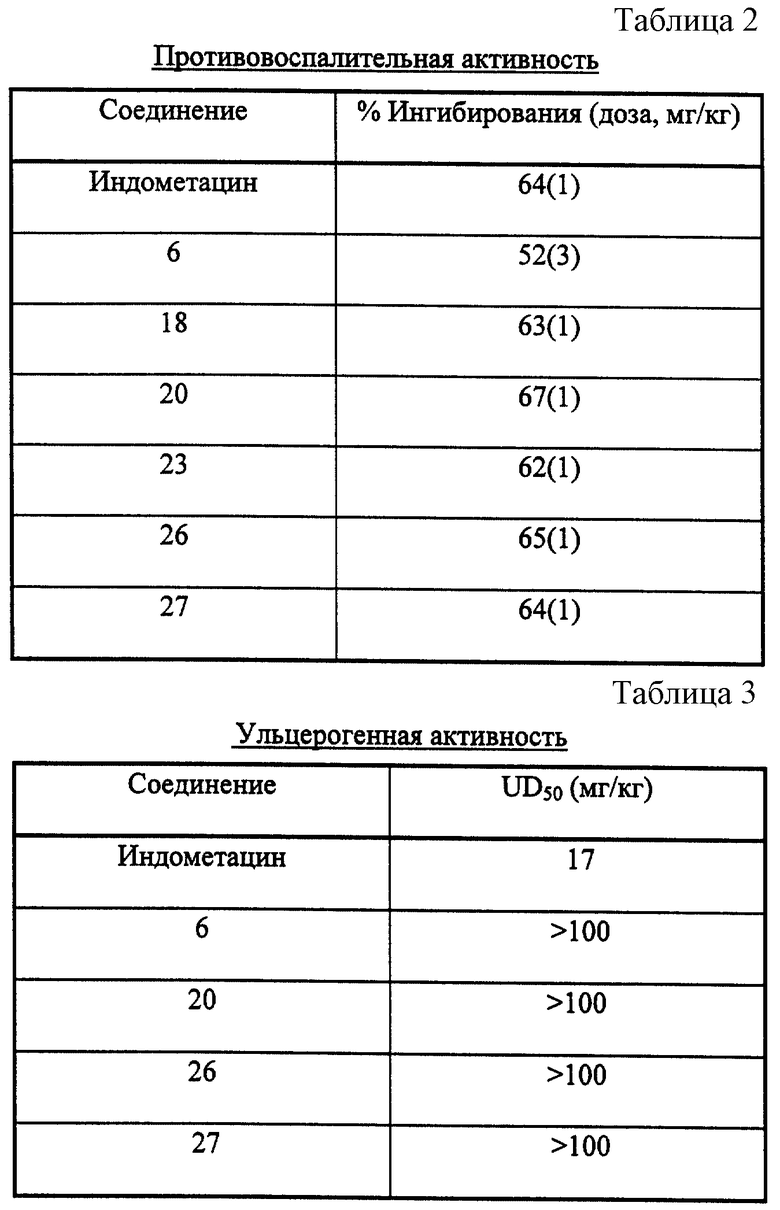
Результаты, полученные от проведения биологических анализов, показаны в табл. 1, 2 и 3.
Как показано в табл. 1, соединения формулы (I) являются селективными и сильнодействующими ингибиторами COX-2, Было обнаружено, что соединения примеров являются более эффективными при ингибировании активности СОХ-2, чем при ингибировании активности СОХ-1, тогда как эталонное соединение индометацин является сильнодействующим и эффективным ингибитором СОХ-1. Вследствие их низкой СОХ-1 активности соединения формулы (I) проявляют значительную противовоспалительную активность (см. табл. 2) и выгода от их применения состоит в том, что они оказывают значительно менее опасные побочные действия, чем обычно применяемые нестероидные противовоспалительные лекарственные средства (например, желудочно-кишечная токсичность (см. табл. 3), побочное действие на почки, ослабленное действие на время кровотечения и индуцирования астмы у субъектов, чувствительных к аспирину).
Настоящее изобретение предлагает соединение формулы (I) для применения в способе лечения человека или животного посредством терапии, в особенности для лечения боли, жара или воспаления, для ингибирования сокращения гладкой мышцы, вызванного простаноидом, или для профилактики колоректального рака.
Настоящее изобретение также предлагает применение соединения формулы (I) в производстве лекарственных препаратов для лечения боли, жара или воспаления для ингибирования сокращения гладкой мышцы, вызванного простаноидом, или для профилактики колоректального рака.
Соединения формулы (I) являются пригодными для ослабления боли, жара и воспаления при множестве состояний, включающих ревматическую атаку, симптомы, связанные с эпидемическим гриппом или другими вирусными инфекциями, насморк, задне-нижнюю и шейную боль, дисменорею, головную боль, зубную боль, растяжения и деформации, миозиты, невралгию, синовит, бурсит, тендинит, травмы, хирургические и стоматологические процедуры и артриты, включающие ревматоидные артриты, остеоартриты, подагрические артириты, спондилоартропатии, системную красную волчанку и болезнь Стилла-Шоффара. Они могут быть также использованы при лечении кожных воспалительных нарушений, как например псориаза, экземы, жжения и дерматитов. Такие соединения могут быть также использованы для профилактики колоректального рака.
Соединения формулы (I) будут ингибировать также вызванное простаноидом сокращение гладкой мышцы и поэтому могут быть использованы для лечения дисменореи, при преждевременных родах, для лечения астмы и бронхитов.
Соединения формулы (I) могут быть использованы в качестве альтернативы обычным нестероидным противовоспалительным лекарственным средствам, в частности, когда такие нестероидные противовоспалительные лекарственные средства могут быть противопоказаны, как например, для лечения больных, страдающих желудочно-кишечными расстройствами, включающими пептические язвы, гастриты, регионарный энтерит, неспецифический язвенный колит, дивертикулит, болезнь Крона, синдром воспалительного кишечника и синдром раздраженной толстой кишки, желудочно-кишечное кровотечение и нарушение коагуляции заболевания почек (например, нарушенной функцией почек), до хирургического вмешательства или приема антикоагулянтов, и астмой, вызванной чувствительностью к нестероидным противовоспалительным лекарственным средствам.
Соединения могут быть также использованы для лечения воспалений при заболеваниях таких, как сосудистые заболевания, мигрень, узелковый периартериит, тиреоидит, апластическая анемия, болезнь Ходжкина, склеродермия, диабет типа I, миастения, саркоидоз, нефротический синдром, синдром Бехчета, полимиозит, аллергия, конъюктивит, гингивит и миокардная ишемия.
Соединения настоящего изобретения являются ингибиторами фермента циклооксигеназы-2, и поэтому они являются пригодными для лечения перечисленных выше заболеваний, опосредованных циклооксигеназой-2.
Настоящее изобретение предлагает также фармацевтическую композицию, которая содержит в качестве активного ингредиента, по меньшей мере, одно производное 2-(3Н)-оксазолона формулы (I) и фармацевтически приемлемый носитель или разбавитель. Композиции предпочтительно находятся в форме, подходящей для перорального, местного, ректального, трансдермального, назального или парентерального введения, а также для ингаляции.
Фармацевтически приемлемые носители или разбавители, которые смешивают с активным соединением или соединениями для образования композиций данного изобретения, являются хорошо известными сами по себе, и действительные используемые эксципиенты зависят, между прочим, от предназначенного метода введения композиций. Композиции данного изобретения предпочтительно приспособлены для перорального введения.
В этом случае композиции для перорального введения могут иметь форму таблеток, капсул, лепешек или шипучих гранул или жидких препаратов, таких как эликсиры, сиропы или суспензии, всех содержащих одно или более соединений изобретения. Такие препараты могут быть получены способами, хорошо известными в данной области, например путем смешивания производного 2-(3Н)-оксазолона формулы (I) с фармацевтически приемлемым носителем или разбавителем.
Разбавители, которые могут быть использованы при получении композиций, включают те жидкие и твердые разбавители, которые совместимы с активным ингредиентом, вместе с красителями и вкусовыми добавками, если желательно. Таблетки или капсулы могут подходяще содержать активный ингредиент в количестве между 10 и 500 мг и предпочтительно от 15 до 100 мг. Соединения могут быть также введены в гранулы, покрытые соответствующими природными или синтетическими полимерами, известными в данной области, с получением свойств длительного выделения, или могут быть введены с полимерами в таблетированную форму с получением таких же свойств.
Жидкие композиции, приспособленные для перорального применения, могут быть в форме растворов, суспензий или аэрозолей. Растворы могут представлять собой водно-спиртовые растворы 2-(3Н)-оксазолона в сочетании, например, с сахарозой или сорбитом для образования сиропа. Суспензии могут содержать нерастворимую или микроинкапсулированную форму активного соединения изобретения в сочетании с водой и другими приемлемыми растворителями вместе с суспендирующим агентом или вкусовой добавкой.
Композиции для ингаляции могут быть в форме растворов, суспензий или микронизированного порошка, содержащегося в соответствующем ингаляторе.
Композиции для парентерального введения могут быть получены в форме микроэмульсий или микросуспензий в воде или соответствующей жидкости для парентерального введения.
При лечении человека дозы производных 2-(3Н)-оксазолона зависят от желательного эффекта и продолжительности лечения; дозы для взрослого человека составляют обычно между 15 мг и 500 мг в день. В общем, врач будет назначать дозу с учетом возраста и веса пациента, которому необходимо лечение.
Производные 2-(3Н)-оксазолона формулы (I) могут быть использованы в способе лечения любого из вышеперечисленных состояний, включающем введение субъекту, нуждающемуся в таком лечении, эффективного количества производного формулы (I).
Далее изобретение иллюстрируют следующие примеры.
Пример 1
a) Смесь 4-метилсульфонилфенацилового спирта (3 г; 0,014 моля), температура плавления 133-135oС, и 4-фторфенилизоцианата (5 мл; 0,044 моля) перемешивают в течение 1 часа при 100oС. После охлаждения полученное твердое обрабатывают диизопропиловым эфиром (30 мл), собирают фильтрацией и промывают 10% смесью метанола в диэтиловом эфире. Получают 4-метилсульфонилфенацил N-(4-фторфенил) карбамат (3,5 г) в виде белого твердого вещества, температура плавления: 198-200oС (разл.).
b) Раствор вышеназванного соединения (3 г; 0,0085 молей) в безводной уксусной кислоте (30 мл) кипятят с обратным холодильником в течение 8 часов. В вакууме удаляют растворитель, из смеси ацетонитрила (10 мл) и диизопропилового эфира (20 мл) выкристаллизовывают остаток и затем перекристаллизовывают из смеси этанола и метиленхлорида. Получают 3 -(4-фторфенил) - 4 - (4 -метилсульфонилфенил)-2-(3Н)-оксазолон (1,9 г), температура плавления: 170-172oС. Это соединение имеет другую кристаллическую форму с температурой плавления 152-153oС.
Пример 2
a) Раствор 4-метилтиофенацилового спирта (1 г; 5,5 ммоля) и 4-фенилизоцианата (1,08 г; 5,4 ммоля) в безводном ксилоле (10 мл) кипятят с обратным холодильником в течение 5 часов. Затем реакционную смесь охлаждают и твердое фильтруют и промывают диизопропиловым эфиром, получая 4-метилтиофенацил N-(4-бромфенил) карбамат в виде белого твердого вещества (1,8 г).
b) Раствор вышеуказанного карбамата (1,8 г; 4,7 ммоля) в безводной уксусной кислоте (18 мл) кипятят с обратным холодильником в течение 16 часов, в вакууме удаляют растворитель и остаток обрабатывают ацетоном. Образованное белое твердое отфильтровывают и получают 3-(4-бромфенил)-4-(4-метилтиофенил)-2-(3Н)-оксазолон (1 г).
c) К раствору вышеназванного соединения (1 г; 2,7 ммоля) в метаноле (3 мл) и метиленхлориде (17 мл) медленно добавляют гексагидрат монопероксифталата магния (2,13 г; 4,3 ммоля), и смесь перемешивают при комнатной температуре в течение 2 часов. Затем ее промывают 4М водным раствором бикарбамата натрия, сушат (Na2SO4) и при пониженном давлении удаляют растворитель. Остаток перекристаллизовывают из метиленхлорида/этанола с получением 3-(4-бромфенил)-4-(4-метилсульфонилфенил)-2-(3Н)-оксазолона (0,63 г), температура плавления: 217-219oС.
Пример 3
Раствор фенацил N-(4-фторфенил) карбамата (9,6 г; 35 ммолей) в безводной уксусной кислоте (96 мл) кипятят с обратным холодильником в течение 16 часов. При пониженном давлении удаляют растворитель и кристаллизуют твердое, которое собирают фильтрацией и промывают диэтиловым эфиром. Получают 3-(4-фторфенил)-4-фенил-2-(3Н)-оксазолон (7,8 г), температура плавления: 145-147oС.
b) Смесь вышеназванного соединения (4 г; 15,7 ммоля) и хлорсульфоновой кислоты (2,1 мл; 31,6 ммоля) нагревают при 100oС в течение 4 часов, охлаждают и затем вливают в смесь воды со льдом. Осажденное твердое экстрагируют этилацетатом, сушат (Na2SO4) и в вакууме удаляют растворитель. К остатку добавляют концентрированный гидроксид аммония (40 мл), перемешивают при комнатной температуре в течение получаса и экстрагируют метиленхлоридом. Органический раствор сушат (Na2SO4), при пониженном давлении удаляют растворитель, и остаток перекристаллизовывают из этанола. Получают 3-(4-фторфенил)-4-(4-аминосульфонилфенил)-2-(3Н)-оксазолон (0,89 г), температура плавления: 211-213oС.
Пример 4
a) Раствор 4-(N, N-дибензиламиносульфонил)фенацил N-(3,4-дихлорфенил)карбамата (2,6 г; 4,46 ммоля) в безводной уксусной кислоте (25 мм) кипятят с обратным холодильником в течение 6 часов. При пониженном давлении удаляют растворитель и полученное масло обрабатывают диэтиловым эфиром. Кристаллизуют 3- (3,4-дихлорфенил) -4- [ 4 (N, N-дибензиламиносульфонил) фенил] -2- (3Н) -оксазолон (2,0 г), температура плавления: 128-130oС.
b) Раствор вышеописанного соединения (2 г; 3,54 ммоля) в метансульфоновой кислоте (15 мл) перемешивают при 100oС в течение получаса. Реакционную смесь вливают в смесь воды со льдом, осажденное твердое собирают фильтрацией и затем обрабатывают этанолом. Отфильтровывают нерастворимое твердое и раствор пропускают через хроматографическую колонку, содержащую силикагель в качестве элюента метиленхлоридметанол (95: 5). Получают 3-(3,4-дихлорфенил)-4-(4-аминосульфонилфенил)-2-(3Н)-оксазолон (0,9 г): температура плавления: 158-161oС.
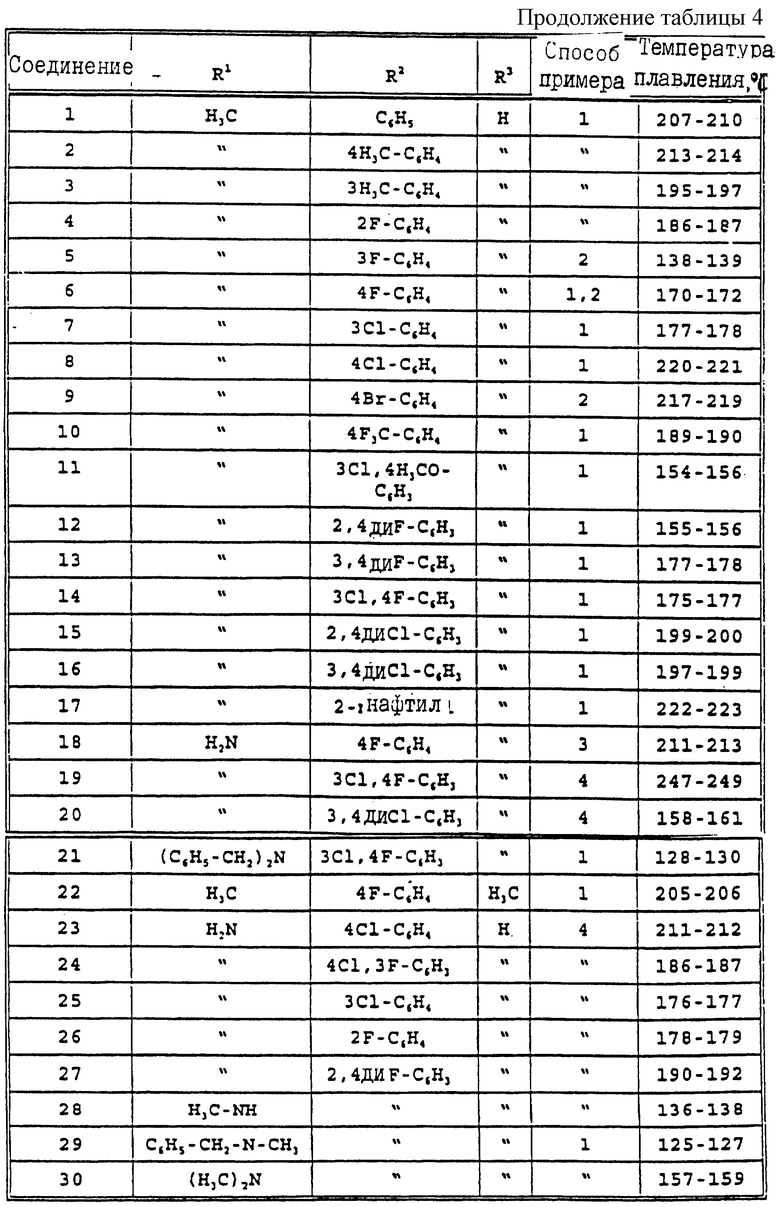
Другие производные 2-(3Н)-оксазолона формулы (I), представленные в табл. 4, получают в соответствии со способами, раскрытыми в этих примерах, но при соответствующих исходных веществах.
Последующие примеры иллюстрируют фармацевтические композиции в соответствии с настоящим изобретением и методикой их получения.
Пример 5
Из следующего состава получают 10000 таблеток, каждая из которых содержит 50 мг 3-(4-хлорфенил)-4-(4-метилсульфонилфенил)-2-(3Н)-оксазолона (активный ингредиент):
Активный ингредиент - 500 г
Микрокристаллическая целлюлоза - 390 г
Высушенная распылением лактоза - 1,990 г
Карбоксиметиловый крахмал - 80 г
Натрийстеарилфумарат - 20 г
Коллоидный диоксид кремния - 20 г
Методика
Все порошки пропускают через сито с размером ячейки 0,6 мм, затем в течение 20 минут перемешивают в соответствующем смесителе и прессуют в 300 мг таблетки с использованием 9 мм дисков и плоских скошенных пуансонов. Время расслоения таблеток составляет примерно 3 минуты.
Пример 6
Из следующего состава получают 100000 капсул, каждая из которых содержит 100 мг 3-(4-фторфенил)-4-(4-метилсульфонилфенил)-2-(3Н)-оксазолона (активный ингредиент):
Активный ингредиент - 10 кг
Моногидрат лактозы - 20 кг
Кукурузный крахмал - 2 кг
Стеарат магния - 0,4 кг
Коллоидный диоксид кремния - 0,2 кг
Методика
Вышеперечисленные ингредиенты просеивают через сито с размером ячеек 60 меш, загружают в подходящий смеситель и наполняют в 100000 желатиновых капсул.
Изобретение относится к новому производному 2-(3Н)-оксазолона формулы I, где R1 является алкильной группой или группой -NR4R5, где каждый R4 и R5 независимо являются водородом, алкильной или бензильной группой, R2 является нафтильной, незамещенной фенильной или фенильной группой, замещенной от 1 до 3 атомами галогена, алкильной группы, гидрокси, алкокси или трифторметильной группы, R3 является водородом или алкильной группой. Способ получения соединения формулы I: взаимодействием карбамата формулы V c безводной уксусной кислотой, или взаимодействием меркаптопроизводного формулы VIII с окислителем, или взаимодействием хлорсульфонила формулы XI с амином формулы R4-NH-R5, или дебензилирование соединения формулы IX. Фармацевтическая композиция, обладающая противовоспалительным действием, содержит соединение формулы I и фармацевтически приемлемый носитель или разбавитель. Способ ингибирования активности СОХ-2 у человека или животного включает введение эффективного количества соединения формулы I. Заявленные соединения могут использоваться для производства лекарственного средства. Технический результат - новые производные 2-(3Н)-оксазолона. 9 с. и 9 з.п. ф-лы, 4 табл.
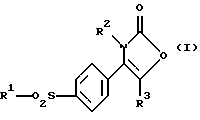
где R1 является алкильной группой или группой -NR4R5, где R4 и R5 каждый независимо является водородом или алкильной или бензильной группой;
R2 является нафтильной, тетрагидронафтильной, незамещенной фенильной или фенильной группой, замещенной от 1 до 3 атомами галогена или алкильной, гидрокси, алкокси или трифторметильной группой;
R3 является водородом или алкильной группой.
3-(4-фторфенил)-4-(4-метилсульфонилфенил)-2-(3Н)-оксазолон,
3-(2-фторфенил)-4-(4-аминосульфонилфенил)-2-(3Н)-оксазолон,
3-(3,4-дихлорфенил)-4-(4-аминосульфонилфенил)-2-(3Н)-оксазолон,
3-(2,4-дифторфенил)-4-(4-аминосульфонилфенил)-2-(3Н)-оксазолон.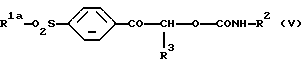
где R2 и R3 являются такими, как определено в п. 1;
R1a является алкильной группой или группой -NR4aR5a, где каждый R4a и R5a независимо является алкильной или бензильной группой,
с безводной уксусной кислотой.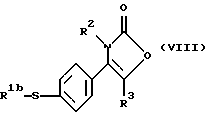
где R2 и R3 являются такими, как определено в п. 1;
R1b является алкильной группой,
с окислителем.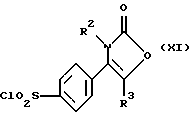
где R2 и R3 являются такими, как определено в п. 1,
с амином формулы ХII
R4-NH-R5 (ХII)
где R4 и R5 являются такими, как определено выше.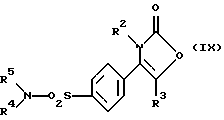
где R2, R3, R4 и R5 являются такими, как определено в п. 1, при условии, что по меньшей мере один из R4 и R5 является бензильной группой.
| WO 9427980 А1, 08.12.1994 | |||
| US 4632930 А, 30.12.1986 | |||
| US 3578671 А, 11.05.1971 | |||
| SU 1514245 A3, 07.10.1989. |

