Данное изобретение относится к замещенным фенилалкеноилгуанидинам и их фармацевтически приемлемым солям и физиологически функциональным производным.
Образование желчных конкрементов (камней) определяется, наряду с рядом факторов, по существу составом желчи, в частности концентрацией и соотношением холестерина, фосфолипидов и солей желчных кислот. Предпосылкой для образования холестериновых желчных камней является наличие перенасыщенной холестерином желчи (Carey, M.C. and Small, D.M. (1978). The physical chemistry of cholesterol in bile. Relationship to gallstone formation and dissolution in man. J. Clin. Invest., 61:998-1026).
Желчные камни до сих пор преимущественно удаляются хирургически, так что существует большая терапевтическая потребность в медикаментозном растворении желчных камней и в предупреждении образования желчных камней.
Задачей изобретения являются соединения, которые способны предотвращать образование желчных камней, препятствуя перенасыщению желчи холестерином или замедляя образование кристаллов холестерина из перенасыщенной желчи.
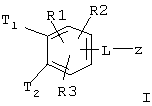
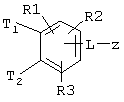
Таким образом, изобретение относится к соединениям формулы I
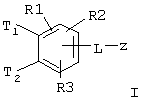
где Т1 и Т2 независимо обозначают
или водород, причем Т1 и Т2 не могут одновременно быть водородом;
z обозначает
R(A), R(B), R(C), R(D) независимо обозначают водород, F, Cl, Br, I, CN, OH, NH2, -(C1-C8) алкил, -О-(C1-C8) алкил, где алкильные остатки могут быть одно или многократно замещены F, (С3-C8)циклоалкил, фенил, бензил, NHR(7), NR(7)R(8), О-(С3-С6)алкенил, O-(С3-С6) циклоалкил, O-фенил, O-бензил, причем фенильное ядро может быть до трех раз замещено F, Cl, СF3, метилом, метокси, NR(9)R(10);
R(7), R(8) независимо друг от друга обозначают водород, -(C1-С8) алкил, причем алкильный остаток может быть одно или многократно замещен F, (С3-С8) циклоалкил, (С3-С6) алкенил, (С3-С8) циклоалкил, фенил, бензил, причем фенильное ядро может быть до трех раз замещено F, Cl, СF3, метилом, метокси, NR(9)R(10), или
R(7), R(8) образуют вместе цепь из 4 или 5 метиленовых групп, из которых одна СН2-группа может быть заменена кислородом, серой, NH, N-СН3 или N-бензилом;
R(9), R(10) независимо друг от друга обозначают водород, (C1-C4) алкил, (C1-C4) перфторалкил;
х равно нулю, 1 или 2;
y равно нулю, 1 или 2;
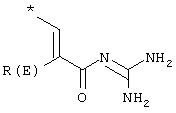
R(E), R(F) независимо друг от друга обозначают водород, F, Cl, Br, I, CN, (C1-C8) алкил, О-(C1-С8) алкил, где алкильный остаток может быть одно или многократно замещен F, (С3-С8) циклоалкил, О-(С3-С6) алкенил, О-(С3-С8) циклоалкил, O-фенил, O-бензил, причем фенильное ядро может быть до трех раз замещено F, Cl, СF3, метилом, метокси, NR(9)R(10);
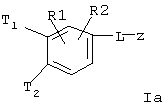
R(1), R(2), R(3) независимо друг от друга обозначают водород, F, Cl, Br, I, CN, -(С1-С8) алкил, -О-(С1-С8) алкил, где алкильные остатки могут быть одно- или многократно замещены F, -(С=O)-N=C(NH2)2, -(SO0-2)-(C1-C8) алкил, -(SO2)-NR(7)R(8), -О-(С0-С8)алкиленфенил, -(С0-С8) алкиленфенил, причем фенильные ядра могут быть до трех раз замещены F, Cl, СF3, метилом, метокси, -(C0-C8) алкилен-NR(9)R(10);
L обозначает -О-, -NR(47)-, -(C1-C8) алкилен-, -(C1-С8)алкенилен-, -(C1-C8)алкинилен-, -COO-, -CO-NR(47)-, -SO2-NR(47)-, -O-(CH2)n-O-, -NR (47)-(CH2) n-O-, -NR(48)-CO-(CH2)n-O-, -CO-NR(48)-(CH2)n-O-, -O-CO-(CH2)n-O-, -SO2-NR (48) -(CH2)n-O-, -NR (48)-CO-CH2-CH2-CO-NR (48) -(CH2)n-O-, -NR (48) -CO-CH=CH-CO-NR (48)-(CH2)n-О-, -NR (48)-SO2-(CH2)n-O-;
R(47) обозначает водород, (C1-C8) алкил, R(48)-CO-, фенил, бензил;
R(48) обозначает водород, (C1-C8) алкил, фенил и бензил, причем фенильное ядро может быть до трех раз замещено F, Сl, СF3, метилом, метокси;
n равно 1-8;
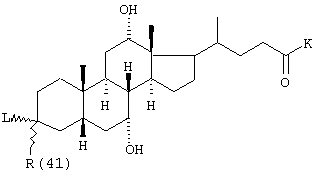
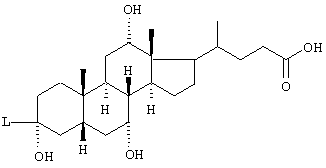
R(40)-R(45) независимо друг от друга обозначают водород, -OR(50), -SR(50), NHR(50), -NR(50)2 -O-(CO)-R(50), -S-(CO)-R(50), -NH-(CO)-R(50), -O-PO-(OR(50))-OR(50), -O-(SO2)-OR(50), -R(50), связь с L или
R(40) и R(41), R(42) и R(43), R(44) и R(45) образуют в каждом случае вместе с кислородом карбонильную группу, причем всегда точно один из остатков R(40)-R(45) обозначает связь с L;
К обозначает -OR(50), -NHR(50), -NR(50)2, -NH-CH2-CH2-CO2H, -НN-СН2-СН2-SО3Н, -NH-CH2-COOH, -N(СН3)СН2СO2Н, -HN-CH(R46)CO2H, -OKa, причем Ка обозначает катион, как, например, ион щелочного или щелочно-земельного металла или ион четвертичного аммония;
R(46) обозначает водород, С1-С4-алкил, бензил, -СН2-ОН, Н3СSСН2СН2-, НO2ССН2-, НO2ССН2СН2-;
R(50) обозначает водород, (C1-C4) алкил, фенил или бензил, причем фенильное ядро может быть до трех раз замещено F, Сl, СF3, метилом, метокси;
а также к их фармацевтически приемлемым солям и физиологически функциональным производным.
Предпочтительными являются соединения формулы I
где Т1 и Т2 независимо обозначают
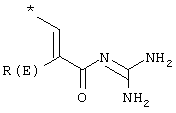
или водород, причем Т1 и Т2 не могут одновременно быть водородом;
L-z обозначает
R(E) обозначает водород, F, Cl, CN, (C1-C4) алкил, -O-(C1-C4) алкил, причем алкильные остатки могут быть одно- или многократно замещены F, (С3-С6) циклоалкил, (С3-С6)алкенил, О-(С3-С6) циклоалкил, O-фенил, O-бензил, причем фенильное ядро может быть до трех раз замещено F, Cl, СF3, метилом, метокси, NR(9)R(10);
R(9), R(10) независимо друг от друга обозначают водород, СН3, СF3;
R(1), R(2), R(3) независимо друг от друга обозначают водород, F, Cl, CN, -SО2-(C1-C4) алкил, -SO2-N ((C1-C4)алкил)2, -SO2-NH(C1-C4) алкил, -SO2-NH2, -SO2-(C1-C4) алкил, -(C1-C4) алкил, -О-(C1-C4) алкил, причем алкильные остатки могут быть одно- или многократно замещены F, -О-(С0-С4) алкиленфенил, -(С0-С4) алкиленфенил, причем фенильные ядра могут быть до трех раз замещены F, Cl, СF3, метилом, метокси;
L обозначает -О-, -NR(47)-, -(C1-C4) алкилен-, -(C1-C4)алкенилен-,
-(C1-C4)алкинилен-, -COO-, -CO-NR(47)-, -SO2-NR(47)-, -O-(CH2)n-O-, -NR (47)-(CH2)n-O-, -NR(48)-CO-(CH2)n-O-, -CO-NR(48)-(CH2)n-O-, -SO2-NR(48)-(CH2)n-O-;
R(47) обозначает водород, (C1-C4) алкил, R(48)-CO-, фенил, бензил;
R(48) обозначает водород, (C1-C4) алкил, фенил и бензил, причем фенильное ядро может быть до трех раз замещено F, Сl, СF3, метилом, метокси;
n равно 1-4;
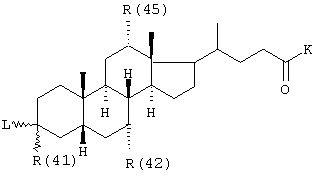
R(41), R(42), R(45) независимо друг от друга обозначают водород, -OR(50), NHR(50), -NR(50)2, -О-(СО)-R(50), -NH-(CO)-R(50);
R(50) обозначает водород, (C1-C4) алкил, фенил или бензил, причем фенильное ядро может быть до трех раз замещено F, С1, СF3, метилом, метокси;
К обозначает -OR(50), -NHR(50), -NR(50)2, -NH-CH2-CH2-CO2H, -NH-СН2-СН2-SО3Н, -NH-CH2-COOH, -N(СН3)СН2СO2Н, -ОКа, причем Ка обозначает катион, как, например, ион щелочного или щелочно-земельного металла или ион четвертичного аммония;
а также их фармацевтически приемлемые соли и физиологически функциональные производные.
Особенно предпочтительными являются соединения формулы I
где Т1 и Т2 независимо обозначают
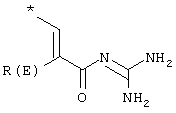
или водород, причем Т1 и Т2 не могут одновременно быть водородом;
L-z обозначает
R(E) обозначает водород, F, Cl, CN, (C1-C4) алкил, -О-(C1-C4) алкил, СF3, -ОСF3;
R(1), R(2) независимо друг от друга обозначают водород, F, Сl, CN, -SО2-СН3, SO2NH2, -(C1-C4) алкил, -О-(C1-C4) алкил, причем алкильные остатки могут быть одно- или многократно замещены F, -О-(С0-С4) алкиленфенил, -(С0-С4) алкиленфенил, причем фенильные ядра могут быть до трех раз замещены F, С1, СF3, метилом, метокси;
R(3) обозначает водород;
L обозначает -О-, NR(47), -СН2-СН2-, СН=СН-, -(С≡С)-, -COO, -CO-NR(47), -SO2-NR(47)-, -О-(СН2)n-O-, -NR(47)-(CH2)n-O-, -NR(48)-CO-(CH2)n-O-, -CO-NR (48) -(CH2)n-O-, -SO2-NR(48)-(CH2)n-O-;
R(47) обозначает водород, (C1-C4) алкил, R(48)-CO-, фенил, бензил;
R(48) обозначает водород, (C1-C4) алкил, фенил и бензил, причем фенильное ядро может быть до трех раз замещено F, С1, СF3, метилом, метокси;
n равно 1-4;
R(41) обозначает водород, -ОН;
К обозначает-OR(50), -NHR(50), -NR(50)2, -NH-CH2-CH2-CO2H, -NH-СН2-СН2-SО3Н, -NH-CH2-COOH, -N(СН3)СН2СO2Н, -ОКа, причем Ка обозначает катион, как, например, ион щелочного или щелочно-земельного металла или ион четвертичного аммония;
R(50) обозначает водород, (C1-C4) алкил, фенил или бензил, причем фенильное ядро может быть до трех раз замещено F, Сl, СF3, метилом, метокси;
а также их фармацевтически приемлемые соли.
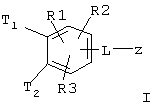
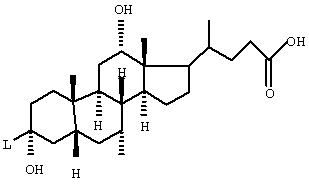
Особенно предпочтительными являются соединения формулы I со структурой Iа
где T1 и Т2 независимо обозначают
или водород, причем T1 и Т2 не могут одновременно быть водородом;
L-z обозначает
L обозначает -С≡С-, -NH-CH2-CH2-O-;
R(E) обозначает водород, (C1-C4) алкил;
R(1), R(2) независимо друг от друга обозначают водород, F, С1, CN, -SO2-СН3, -(C1-C4) алкил, -О-(C1-C4) алкил, причем алкильные остатки могут быть одно- или многократно замещены F;
а также их фармацевтически приемлемые соли.
* Отмечает в приведенных выше формулах точку присоединения T1 или Т2 к фенильному кольцу формулы I.
Если соединения формулы I содержат один или несколько центров асимметрии, то они могут иметь конфигурацию как S, так и R. Эти соединения могут существовать в виде оптических изомеров, в виде диастереомеров, в виде рацематов или в виде их смесей.
Геометрия двойных связей соединений данного изобретения может быть как Е, так и Z. Соединения могут существовать в смеси в виде изомеров по двойной связи.
Выражение “причем алкильный остаток может быть одно- или многократно замещен F”, относится также к перфорированным алкильным остаткам.
Указанные алкильные остатки могут быть остатками как с линейной, так и с разветвленной цепью.
Фармацевтически приемлемые соли на основе их более высокой растворимости по сравнению с исходными соединениями или основными соединениями особенно пригодны для медицинских целей. Эти соли должны иметь фармацевтически приемлемый анион или катион. Пригодными фармацевтически приемлемыми кислотно-аддитивными солями соединений данного изобретения являются соли неорганических кислот, таких как соляная кислота, бромистоводородная кислота, фосфорная, метафосфорная, азотная, сульфоновая и серная кислоты, а также органических кислот, таких как, например, уксусная кислота, бензолсульфоновая кислота, бензойная, лимонная, этансульфоновая, фумаровая, глюконовая, гликолевая, изэтионовая, молочная, лактобионовая, малеиновая, яблочная, метансульфоновая, янтарная, п-толуолсульфоновая, винная и трифторуксусная кислоты. Для медицинских целей особенно предпочтительной является хлорсодержащая соль. Пригодными фармацевтически приемлемыми основно-аддитивными солями являются соли аммония, соли щелочных металлов (такие как соли натрия и калия) и соли щелочно-земельных металлов (такие как соли магния и кальция).
Используемое здесь понятие “физиологически функциональное производное” обозначает любое физиологически переносимое производное соединения данного изобретения формулы I, например сложный эфир, которое при введении млекопитающему, такому как, например, человек, способно (прямо или опосредованно) образовывать соединение формулы I или один из его активных метаболитов.
К физиологически функциональным производным относят также пролекарства рассматриваемых соединений. Такие пролекарства могут метаболизироваться in vivo в соединение данного изобретения. Эти пролекарства сами могут быть или не быть активными.
Соединения данного изобретения могут также существовать в различных полиморфных формах, например в аморфной или кристаллической полиморфных формах. Все полиморфные формы соединений данного изобретения входят в объем изобретения и составляют объект данного изобретения.
В дальнейшем описании все ссылки на "соединение (соединения) в соответствии с формулой (I)" относятся к соединению (соединениям) формулы (I), описанным выше, а также к их солям, сольватам и физиологически функциональным производным.
Количество соединения формулы (I), которое требуется для достижения желаемого биологического эффекта, зависит от ряда факторов, например от выбранного конкретного соединения, предполагаемого применения, способа введения и клинического состояния пациентов.
В общем, суточная доза находится в диапазоне от 0,1 до 100 мг (обычно от 0,1 до 50 мг) в сутки на килограмм массы тела, например 0,1-10 мг/кг в сутки. Таблетки или капсулы могут содержать, например, от 0,01 до 100 мг, обычно от 0,02 до 50 мг. В случае фармацевтически приемлемых солей приведенные весовые данные относятся к весу производимых из соли ионов аминопропанола. Для профилактики или лечения вышеназванных состояний соединения формулы (I) могут применяться индивидуально в виде соединения, но предпочтительно они находятся вместе с приемлемым носителем в виде фармацевтической композиции. Носитель, конечно, должен быть приемлемым, в том смысле, что он является совместимым с другими компонентами композиции и не вреден для здоровья пациентов. Носитель может быть твердым веществом или жидкостью (или и тем и другим) и готовится с соединением в виде разовой дозы, например, в виде таблетки, которая может содержать от 0,05 до 95 мас.% активного вещества. Могут присутствовать также другие фармацевтически активные вещества, в том числе дополнительные соединения формулы (I). Фармацевтические композиции данного изобретения могут быть приготовлены согласно известным фармацевтическим способам, которые по существу состоят в том, что компоненты смешивают с фармакологически приемлемыми носителями и вспомогательными веществами (добавками).
Фармацевтически приемлемые композиции данного изобретения представляют собой композиции, которые пригодны для введения внутрь через рот или перорального (например, сублингвального) введения, хотя самые подходящие способы введения в каждом отдельном случае зависят от типа и тяжести подлежащего лечению состояния и от вида применяемого в каждом случае соединения формулы (I). Дражированные композиции и дражированные формы пролонгированного действия также входят в рамки данного изобретения. Предпочтительными являются кислотоустойчивые и устойчивые к желудочному соку композиции. Подходящие устойчивые к желудочному соку покрытия включают в себя ацетат-фталат целлюлозы, поливинилацетат-фталат, гидроксипропилметилцеллюлозофталат и анионные полимеры метакриловой кислоты и метилметакрилата.
Подходящие фармацевтические соединения для введения внутрь могут существовать в виде отдельных единиц, таких как, например, капсулы, капсулы с облатками, сосательные таблетки или таблетки, которые в каждом отдельном случае содержат определенное количество соединения формулы (I); в виде порошков или гранулятов; в виде растворов или суспензий в водной или неводной жидкости или в виде эмульсий типа "масло в воде" или типа "вода в масле". Эти композиции могут быть приготовлены, как уже упоминалось, согласно любому из подходящих фармацевтических способов, которые включают в себя стадию, на которой активное вещество и носитель (который может состоять из одного или нескольких дополнительных компонентов) приводят в контакт. В общем виде композиции готовят однородным и гомогенным смешиванием активного вещества с жидким и/или мелкоизмельченным твердым носителем, после чего продукт, если требуется, формуют. Так, например, таблетка может быть приготовлена прессованием или формованием порошка или гранулята, в случае необходимости с одним или несколькими дополнительными компонентами. Прессованные таблетки могут быть изготовлены таблетированием соединения в свободно текучей форме, такой как, например, порошок или гранулят, в случае необходимости смешанные со связывающим веществом, мягчителем, инертным разбавителем и/или одним (несколькими) поверхностно-активными веществами, в пригодном для таблетирования аппарате. Формованные таблетки могут быть приготовлены формованием порошкообразного, увлажненного инертными жидкими разбавителями соединения в пригодной для этого машине.
Фармацевтические композиции, которые пригодны для перорального (подъязычного) введения, включают в себя сосательные таблетки, которые содержат соединение формулы (I) со вкусовым веществом, обычно сахарозой и аравийской камедью или трагантом, и пастилки, которые содержат в себе соединение в инертной основе, такой как желатин и глицерин или сахароза и аравийская камедь.
Данное изобретение относится, далее, к способу получения соединения формулы I, отличающемуся тем, что соединение формулы II
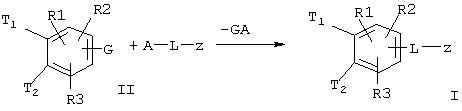
где T1, T2, R(1), R(2) и R(3) имеют приведенные выше значения и G обозначает обмениваемую с L-z функциональную группу, известным для специалиста образом подвергают реакции с соединением A-L-z, при этом GA отщепляется и образуется соединение формулы (I).
Функциональная группа G соединения формулы (II) может, например, быть бромом или иодом. Посредством Pd(0)-катализа может быть затем достигнуто известным образом желательное связывание С-С-связи.
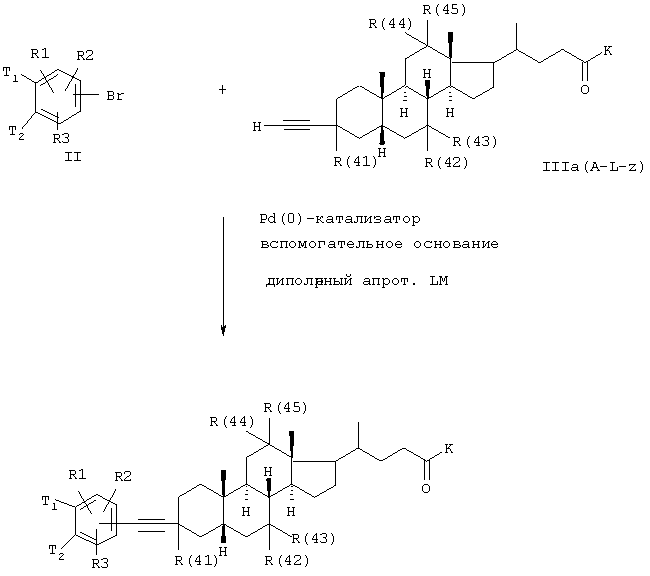
Ацетиленпроизводные желчных кислот формулы (III) получают из подходящих кетонов желчных кислот. Для этого ацетилид лития добавляют аналогично известному способу (патент США 5641767) к кетожелчным кислотам.
Соединения формулы (I) и их фармацевтически приемлемые соли и физиологически функциональные производные отличаются благоприятным действием на состав желчи и препятствуют образованию желчных камней, препятствуя перенасыщению желчи холестерином или замедляя образование кристаллов холестерина из перенасыщенной желчи. Эти соединения могут применяться индивидуально или в комбинации с активными веществами, понижающими содержание липидов в крови. Эти соединения пригодны, в частности, для профилактики, а также лечения желчных камней.
Соединения данного изобретения формулы (I) попадают в гепатобилиарную систему и поэтому действуют на эти ткани. Таким образом ингибируется поглощение воды из желчного пузыря ингибированием апикального NHE-антипорта подтипа 3 эпителия желчного пузыря, следствием чего является разбавленная жидкость желчи.
Биологическое испытание соединений данного изобретения проводили определением ингибирования натрий/водородного антипортера подтипа 3.
1. Описание теста
Для определения величины IC50 для ингибирования NHE-3-белка человека (экспрессируемого клеточной линией LAP1) определяли восстановление рН (pHi) после подкисления, которое наступает в случае функционально способного NHE также в условиях отсутствия бикарбоната. Для этого pHi определяют с помощью рН-чувствительного флуоресцентного красителя BCECF (Calbiochem, применяли предшественник BCECF-AM). Клетки сначала нагружали BCECF. Флуоресценцию BCECF определяли спектрометром ″Ratio Fluorescence Spectrometr" (Proton Technology International, South Brunswick, N.J., USA) при возбуждающих длинах волн 505 и 440 нм и длине волны испускания 535 нм и при помощи калибровочных кривых преобразовывали в pHi. Клетки инкубировали уже при загрузке BCECF в NН4С1-буфере (рН 7,4) (NH4Cl-буфер: 115 мМ NaCl, 20 мМ NH4Cl, 5 мМ КСl, 1 мМ CaCl2, 1 мМ MgSO4, 20 мМ Hepes, 5 мМ глюкоза, 1 мг/мл БСА; рН доводят до 7,4 с помощью 1 М NaOH). Внутриклеточное подкисление индуцировали добавлением 975 мкл не содержащего NH4Cl буфера к аликвотам 25 мкл инкубируемых в NН4Сl-буфере клеток. Последующую скорость восстановления рН регистрировали в течение 3 минут. Для расчета ингибирующей способности проверяемых веществ клетки сначала исследовали в буферах, при которых имело место полное восстановление или не было вообще никакого восстановления рН. Для полного восстановления рН (100%) клетки инкубировали в Nа+-содержащем буфере (133,8 мМ NaCl, 4,7 мМ КСl, 1,25 мМ CaCl2, 1,25 мМ MgCl2, 0,97 мМ Na2HPO4, 0,23 мМ NaH2PO4, 5 мМ Нереs, 5 мМ глюкоза, рН доводили до 7,0 с помощью 1 М NaOH). Для определения 0%-показателя клетки инкубировали в буфере, свободном от Nа+ (133,8 мМ холинхлорид, 4,7 мМ КСl, 1,25 мМ CaCl2, 1,25 мМ MgCl2, 0,97 мМ K2HPO4, 0,23 мМ КН2РO4, 5 мМ Нереs, 5 мМ глюкоза, рН доводили до 7,0 с помощью 1 М NaOH). Исследуемые вещества тестировали в Nа+-содержащем буфере.
Восстановление внутриклеточных рН при каждой тестируемой концентрации вещества выражали в процентах максимального восстановления. Из процентных показателей восстановления рН с использованием программы SigmaPlot (Version 3.0, Jandel Scientific, USA) рассчитывали IC50 каждого тестируемого вещества.
Результаты
Пример 1: IС50 = 1,7 мкМ/л.
Нижеследующие примеры служат для более подробного объяснения изобретения, не ограничивая изобретение описанными в примерах продуктами и формами осуществления.
Список сокращений:
МеОН - метанол
LAH - литийалюминийгидрид
ДМФ - N,N-диметилформамид
EI - электронный удар
CI - химическая ионизация
КТ - комнатная температура
ЕЕ - этилацетат (EtOAc)
т.пл. - точка плавления
НЕР - н-гептан
ДМЭ - диметоксиэтан
ES - электрораспыление
FAB - бомбардировка ускоренными атомами
CH2Cl2 - дихлорметан
ТГФ -тетрагидрофуран
экв. - эквивалент
Общий способ сочетания арилгалогенидов и замещенных конечных ацетиленов.
Арилгалогенид (1 экв.) помещают вместе с вспомогательным основанием (4 экв.), таким как, например, триэтиламин, и Pd-катализвтором, таким как, например, палладийбистрифосфинодихлорид (3 мол.%) в ДМФ. В течение 0,5-3 часов медленно добавляют производное ацетилена и в случае необходимости добавляют еще раз указанное выше количество катализатора. Температура реакции может при этом превышать КТ и достигать приблизительно 100°С, обычно она равна приблизительно 60°С. Неочищенный продукт может быть осажден этилацетатом и отфильтрован. Последующее образование соли достигается добавлением кислоты в ацетоне.
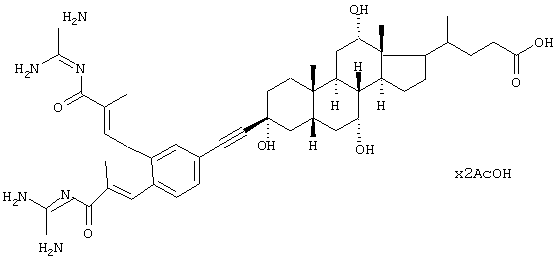
Пример 1.
Диацетат 4-{3β-[3,4-бис-(3-гуанидино-2-метил-3-оксопропенил)фенилэтинил]-3α,7α,12α-тригидрокси-10β,13β-диметилгексадекагидроциклопента[а]фенантрен-17-ил}пентановой кислоты, желтоватое твердое вещество, т.пл. 250°С (разложение).
MS:
М++Н (FAB)=880.
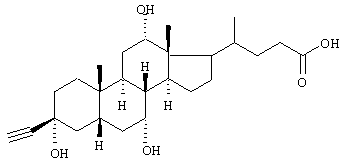
Получение промежуточных продуктов 1 и 2.
Промежуточный продукт 1: 3β-ацетиленхолевая кислота
Проведение синтеза.
а) Метиловый эфир 3,7,12-триацетилхолевой кислоты
90 г метилового эфира холевой кислоты и 3,0 г диметиламинопиридина растворяли в 500 мл пиридина, смешивали с 500 мл уксусного ангидрида и перемешивали в течение ночи при комнатной температуре. Смесь выливали на ледяную воду и экстрагировали этилацетатом (3х). Сушкой (МgSO4) и выпариванием органической фазы выделяют 92 г метилового эфира 3,7,12-триацетилхолевой кислоты.
MS: М++Н (FAB)=555.
b) Метиловый эфир 7,12-диацетилхолевой кислоты
При 5°С 150 мл уксусного ангидрида медленно добавляли по каплям в 1,5 л метанола. Спустя 15 мин добавляли 92 г метилового эфира 3,7,12-триацетилхолевой кислоты и перемешивали 1 час при комнатной температуре. Смесь выливали на ледяную воду и экстрагировали этилацетатом (3х). Органическую фазу промывали 1 н. раствором Nа2СО3, сушили MgSO4 и упаривали. Получали 85 г неочищенного продукта.
MS: M++Li (FAB)=513.
c) Метиловый эфир 3-кето-7,12-диацетилхолевой кислоты
85 г (168 ммоль) метилового эфира 1,12-диацетилхолевой кислоты, 183,7 г хлорформиата пиридиния и 175 г молекулярного сита перемешивали в 2,5 л дихлорметана 2 час при комнатной температуре. Смесь выливали на 7 л диэтилового эфира, твердые вещества отфильтровывали. Растворитель выпаривали и остаток растворяли в этилацетате. После хроматографии через Florisil-колонку получали 59,6 г продукта.
MS: M++Li (FAB)=511.
d) Метиловый эфир 3β-ацетилен-7,12-диацетилхолевой кислоты
В 750 мл абсолютного тетрагидрофурана при -55°С в атмосфере аргона в течение 25 мин вводили ацетилен. К этому раствору добавляли по каплям 145 мл 15% н-бутиллития в гексане и перемешивали 10 мин. Затем добавляли 45 г (89 ммоль) метилового эфира 3-кето-7,12-диацетилхолевой кислоты и перемешивали 1,5 час при -40°С. Для обработки добавляли 500 мл насыщенного водного раствора хлорида аммония и экстрагировали этилацетатом (3х), органическую фазу сушили над MgSO4 и упаривали. Остаток хроматографировали на силикагеле (н-гептан/этилацетат, 1:1). Получали 35,3 г продукта, MS: M++Li (FAB) =537.
e) 3β-Ацетиленхолевая кислота
35,2 г (66 ммоль) продукта из d) растворяли в 1 л метанола, соединяли с 300 мл 2 н. раствора гидроксида натрия и кипятили с обратным холодильником 25 час. Растворитель выпаривали, остаток растворяли в воде и подкисляли 2 н. соляной кислотой до рН 2. Осадок отфильтровывали и промывали водой до нейтральной реакции. Высушивание остатка давало 14,6 г продукта, MS: M++Li (FAB)=439.
Промежуточный продукт 2: дигидрохлорид 1,2-бис-[3-(гуанидид Е-2-метилпропеновой кислоты)]-4-бромбензола
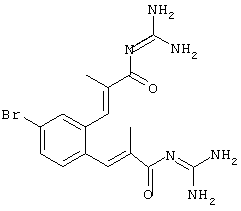
Проведение синтеза:
a) 4-бром-1,2-фталевый диол получали из диметилового эфира 4-бромфталевой кислоты согласно стандартным способам (например, восстановлением LAH), бесцветное масло, MS: M++H (FAB)=217;
b) 4-бром-1,2-фталдиальдегид получали из 2а) посредством, например, окисления по Swern при стандартных условиях, аморфное твердое вещество, MS: M++H (FAB)=213;
c) 4-бром-1,2-ди-[3-(этиловый эфир Е-2-метилпропеновой кислоты)]бензол получали депротонированием 1 экв. триэтилового эфира 2-фосфонопропионовой кислоты с 1 экв. н-бутиллития в гексане при 0°С и последующей реакцией при комнатной температуре с 0,5 экв. 4-бром-1,2-фталдиальдегида 2b). После полного использования диальдегида проводили обработку водой и трижды экстрагировали встряхиванием с толуолом. После сушки объединенных органических фаз над сульфатом магния растворитель удаляли в вакууме и оставшийся неочищенный продукт хроматографически разделяли на силикагеле со смесями ЕЕ/НЕР (этилацетат/н-гептан) в качестве элюента, бесцветное масло, MS: М++Н (FAB)=381;
d) 4-бром-1,2-ди-[3-(Е-2-метилпропеновая кислота)]бензол получали из 2с) омылением согласно стандартному способу (гидроксид натрия в метаноле), бесцветное аморфное твердое вещество, MS: M++H (FAB)=325;
e) дигидрохлорид 1,2-бис-[3-(гуанидид Е-2-метилпропеновой кислоты)]-4-бромбензола получали из 2d) согласно общему варианту, бесцветное твердое вещество, т.пл. 240°С, MS: М++Н (FAB)=407;
f) диацетат 4-{3β-[3,4-бис-(3-гуанидино-2-метил-3-оксопропенил)фенилэтинил]-3α,7α,12α-тригидрокси-10β,13β-диметилгексадекагидроциклопента[а]фенантрен-17-ил}пентановой кислоты получали из 2е) и 3β-ацетиленхолевой кислоты посредством Pd(0)-сочетания согласно общему способу в ДМФ при 60°С в течение 2 час.
Пример 2.
Бензиловый эфир 4-{3β-[3,4-бис-(3-гуанидино-2-метил-3-оксопропенил)фенилэтинил]-3α,7α,12α-тригидрокси-10β,13β-диметилгексадекагидроциклопента[а]фенантрен-17-ил}пентановой кислоты, желтоватое твердое вещество, т.пл. 155°С, MS: M++H (ES)=849.
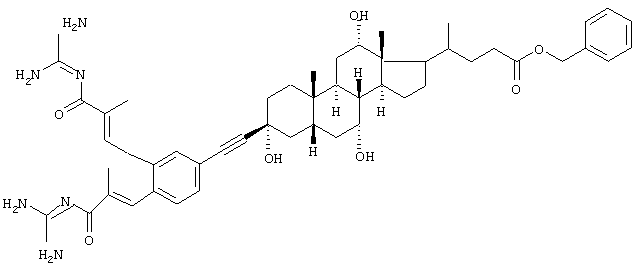
Синтез проводили аналогично примеру 1 с использованием бензилового эфира 3β-ацетиленхолевой кислоты.
Пример 3.
Бензиловый эфир 4-{3β-[4-(3-гуанидино-3-оксопропенил)фенилэтинил]-3α,7α,12α-тригидрокси-10β,13β-диметилгексадекагидроциклопента[а]фенантрен-17-ил}пентановой кислоты
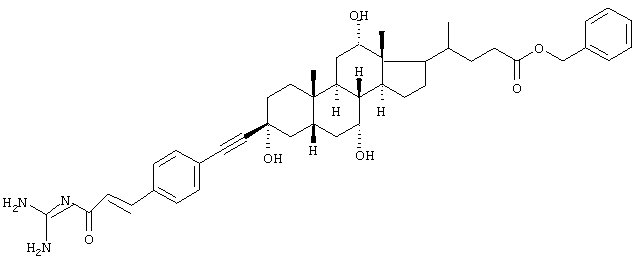
желтоватое твердое вещество, т.пл. 189°С, MS: M++H (FAB)=710.
Синтез проводили аналогично общему способу с использованием бензилового эфира гуанидида 4-бромкоричной кислоты и бензилового эфира 3β-ацетиленхолевой кислоты.
Пример 4.
Метиловый эфир 4-{3β-[4-(3-гуанидино-3-оксопропенил)фенилэтинил]-3α,7α,12α-тригидрокси-10β,13β-диметилгексадекагидроцик-лопента[а]фенантрен-17-ил}пентановой кислоты, желтоватое твердое вещество, т.пл. 60°С, MS: M++H (FAB)=718.
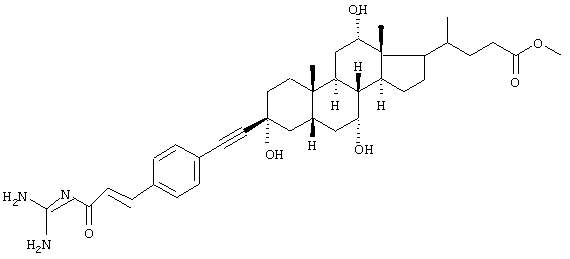
Синтез проводили аналогично общему способу реакцией гуанидида 4-бромкоричной кислоты и бензилового эфира 3β-ацетиленхолевой кислоты.
Пример 5.
(4-{3β-[3,4-бис-(3-Гуанидино-2-метил-3-оксопропенил)фенилэтинил]-3α,7α,12α-тригидрокси-10β,13β-диметилгексадекагидроциклопента[а]фенантрен-17-ил}пентаноиламино)уксусная кислота
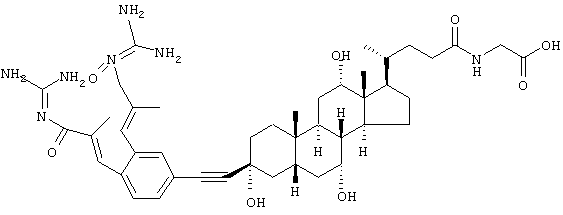
а) Метиловый эфир [4-(3β-этинил-3α,7α,12α-тригидрокси-10β,13β-диметилгексадекагидроциклопента[а]фенантрен-17-ил}пентаноиламино)уксусной кислоты
530 мг 3β-ацетиленхолевой кислоты (промежуточного продукта 1е), а также 510 мкл триэтиламина растворяют в 30 мл ТГФ и добавляют по каплям при 0°С 175 мкл этилового эфира хлормуравьиной кислоты. Перемешивают в течение 15 мин при 0°С, затем добавляют по каплям раствор 340 мг гидрохлорида метилового эфира глицина в 10 мл ДМФ и перемешивают в течение 4 ч при комнатной температуре. Разбавляют 200 мл этилацетата и дважды промывают по 50 мл в каждом случае 5% водным раствором NaHSO4. Сушат над МgSO4 и растворитель удаляют в вакууме. Остаток помещают в 100 мл этилацетата и трижды промывают по 50 мл в каждом случае насыщенного водного раствора Na2CO3. Сушат над MgSO4 и растворитель удаляют в вакууме. Хроматографией на силикагеле со смесью ЕЕ/МеОН, 10:1, и затем второй раз с ЕЕ получают 280 мг бесцветной пены.
Rf (EE) = 0,37, MS (FAB) =518 (М+Н)+.
b) [4-(3β-Этинил-3α,7α,12α-тригидрокси-10β,13β-диметилгексадекагидроциклопента [а]фенантрен-17-ил}пентаноиламино)уксусная кислота
270 мг метилового эфира [4-(3β-этинил-3α,7α,12α-тригидрокси-10β,13β-диметилгексадекагидроциклопента[а]фенантрен-17-ил}пентаноиламино)уксусной кислоты и 630 мкл 1 н. водного раствора NaOH растворяют в 5 мл этанола и выдерживают 16 час при комнатной температуре. Растворитель удаляют в вакууме, остаток помещают в 50 мл насыщенного водного раствора NaH2PO4 и трижды экстрагируют по 50 мл этилацетата в каждом случае. Сушат над МgSO4 и растворитель удаляют в вакууме. Получают 230 мг аморфного твердого вещества.
Rf (ацетон/вода, 10:1) = 0,25, MS(FAB): 502 (М+2Li)+.
с) (4-{3β-[3,4-бис-(3-Гуанидино-2-метил-3-оксопропенил)фенилэтинил]-3α,7α,12α-тригидрокси-10β,13β-диметилгексадекагидроциклопента[а]фенантрен-17-ил}пентаноиламино)уксусная кислота
230 мг [4-(3β-этинил-3α,7α,12α-тригидрокси-10β,13β-диметилгексадекагидроциклопента[а]фенантрен-17-ил}пентаноиламино)уксусной кислоты, а также 183 мг N-{3-[4-бром-2-(3-гуанидино-2-метил-3-оксопропенил)фенил]-2-метилакрилоил}гуанидина подвергают взаимодействию посредством Pd(0)-сочетания согласно общему способу в ДМФ при 60°С в течение 3 час. После препаративной ВЭЖХ через С18 LiChrosorb со смесью ацетонитрил/вода, 2:4, + 0,1% уксусная кислота + 0,1% ацетат аммония получают 70 мг аморфного твердого вещества.
Rf (н-бутанол/уксусная кислота/вода, 3:1:1) = 0,33, MS (ES): 816 (M+H)+.
Пример 6.
4-{3-[2-Фтор-4-(3-гуанидино-2-метил-3-оксопропенил)фенилэтинил]-3,7,12-тригидрокси-10,13-диметилгексадекагидроциклопента[а]фенантрен-17-ил}пентановая кислота
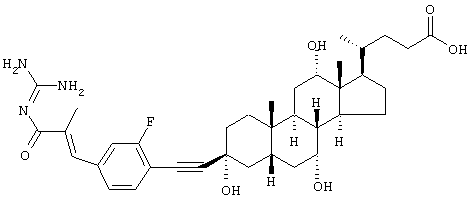
а) Бутиловый эфир 3-(4-бром-3-фторфенил)-2-метилакриловой кислоты
2 г 1-бром-2-фтор-4-иодбензола и 1,1 мл диизопропилэтиламина растворяют в 20 мл диметилацетамида (безводного) и 5 мин барботируют легкий ток аргона через этот раствор. Затем добавляют 1,4 мл бутилового эфира акриловой кислоты и 10 мг 2,6-ди-трет-бутил-4-метилфенола и нагревают до 100°С. Наконец дегазируют следующие 4 мл диметилацетамида при помощи тока аргона и суспендируют в них 80 мг транс-бис(β-ацетато)бис[о-толилфосфино)бензил]дипалладия (Tetrahedron Lett., 1996, 37(36), 6535-6538). Эту суспензию добавляют к смеси остальных компонентов реакции и перемешивают 90 мин при 140°С. Затем смесь разбавляют 200 мл ЕЕ, промывают 2 раза по 100 мл воды и 1 раз 100 мл насыщенного водного раствора NaCl. Сушат над МgSО4 и растворитель удаляют в вакууме. Хроматографией на силикагеле получают 230 мг бесцветного масла.
Rf (EE/HEP) = 0,27, MS (DCl): 315 (М+Н)+.
b) Бутиловый эфир 3-{4-[17-(3-карбокси-1-метилпропил)-3,7,12-тригидрокси-10,13-диметилгексадекагидроциклопента[а]фенантрен-3-илэтинил]-3-фторфенил}-2-метилакриловой кислоты
64 мг бис(трифенилфосфин)палладий(II)хлорида, 17 мг CuI, 0,5 мл триэтиламина, а также 230 мг бутилового эфира 3-(4-бром-3-фторфенил)-2-метилакриловой кислоты растворяют в 10 мл безводного ДМФ и добавляют по каплям при 60°С на протяжении одного часа раствор 395 мг 3β-ацетиленхолевой кислоты в 10 мл безводного ДМФ. Перемешивают в течение 1 час при 60°С и затем снова медленно добавляют по каплям раствор 395 мг 3β-ацетиленхолевой кислоты в 10 мл безводного ДМФ при 60°С. Перемешивают еще в течение 2 час при 60°С, затем еще раз добавляют 64 мг бис(трифенилфосфин)палладий(II)хлорида и 17 мг Cul и снова перемешивают 2 час при 60°С. Наконец добавляют еще 80 мг 3β-ацетиленхолевой кислоты и перемешивают 2 час при 60°С. Растворитель удаляют в вакууме, остаток помещают в 100 мл 5% водного раствора NaHSO4 и 3 раза экстрагируют 100 мл ЕЕ в каждом случае. Сушат над Na2SO4 и растворитель удаляют в вакууме. Хроматографией на силикагеле со смесью ЕЕ/МеОН, 5:1, получают 90 мг воскообразного вещества.
Rf (EE/MeOH, 5:1) = 0,56, MS (FAB): 667 (М+Н)+.
c) 4-{3-[2-фтор-4-(3-Гуанидино-2-метил-3-оксопропенил)фенилэтинил]-3,7,12-тригидрокси-10,13-диметилгексадекагидроциклопента[а]фенантрен-17-ил}пентановая кислота
73 мг гидрохлорида гуанидина и 71 мг трет-бутилата калия растворяют в 2 мл безводного ДМФ и перемешивают 30 мин при комнатной температуре. Эту суспензию наносят распылением на 85 мг бутилового эфира 3-{4-[17-(3-карбокси-1-метилпропил)-3,7,12-тригидрокси-10,13-диметилгексадекагидроциклопента[а]фенантрен-3-илэтинил]-3-фторфенил}-2-метилакриловой кислоты и 5 час перемешивают при 100°С. После охлаждения добавляют 10 мл воды, доводят рН до 4 водным раствором HCl и три раза экстрагируют 10 мл ЕЕ каждый раз. Сушат над МgSO4 и растворитель удаляют в вакууме. Хроматографией на силикагеле смесью ацетон/вода, 10:1, получают 15,5 мг аморфного твердого вещества.
Rf (ацетон/вода, 10:1) = 0,19, MS (ES): 652 (М+Н)+.
Пример 7.
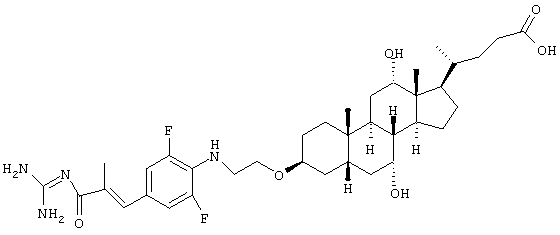
4-(3-{2-[2,6-Дифтор-4-(3-гуанидино-2-метил-3-оксопропенил)фениламино]этокси)-7,12-дигидрокси-10,13-диметилгексадекагидроциклопента[а]фенантрен-17-ил}пентановая кислота
a) 4-(7,12-Дигидрокси-3-метансульфонилокси-10,13-диметилгексадекагидроциклопента[а]фенантрен-17-ил)пентановая кислота
100 г холевой кислоты растворяют в 500 мл пиридина и при 0°С добавляют по каплям на протяжении 30 мин 23,1 мл мезилхлорида. Перемешивают в течение 3 час при комнатной температуре, затем выливают на раствор 400 мл H2SO4 в 3 л воды и 4 раза экстрагируют 750 мл ЕЕ каждый раз. Сушат над Na2SO4 и растворитель удаляют в вакууме. Остаток кристаллизуют с диизопропиловым эфиром и получают 117,1 г, т.пл. 121°С (разложение).
Rf (ЕЕ/НЕР/уксусная кислота, 5:5:1)=0,31, MS (FAB): 487 (M+H)+.
b) Метиловый эфир 4-[7,12-дигидрокси-3-(2-гидроксиэтокси)-10,13-диметилгексадекагидроциклопента[а]фенантрен-17-ил]пентановой кислоты
116 г 4-(7,12-дигидрокси-3-метансульфонилокси-10,13-диметилгексадекагидроциклопента[а]фенантрен-17-ил)пентановой кислоты, а также 130 мл триэтиламина растворяют в 650 мл гликоля и перемешивают 3 час при 100°С, а также 7,5 часов при 115°С. Реакционную смесь выливают при 0°С на раствор 400 мл Н2SO4 в 3 л воды и 7 раз экстрагируют 750 мл ЕЕ каждый раз. Сушат над Na2SO4 и растворитель удаляют в вакууме. Получают промежуточный продукт ZWP.
Добавляют при 0°С 130 мл ацетилхлорида к 900 мл метанола. Затем добавляют раствор ZWP в 400 мл и перемешивают 6 час при комнатной температуре. Выдерживают 60 час при комнатной температуре, затем выливают на 2,6 л воды и 8 раз экстрагируют по 500 мл диизопропилового эфира (DIP). Затем органическую фазу еще 6 раз промывают по 600 мл каждый раз полунасыщенным водным раствором NаНСО3. Сушат над Na2SO4 и растворитель удаляют в вакууме. Хроматографией на силикагеле с этилацетатом получают 32 г смолообразного твердого вещества.
Rf (EE) = 0,19, MS (FAB): 467 (М+Н)+.
c) Метиловый эфир 4-{3-[2-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)этокси]-7,12-дигидрокси-10,13-диметилгексадекагидроциклопента[а]фенантрен-17-ил}пентановой кислоты
1,5 г метилового эфира 4-[7,12-дигидрокси-3-(2-гидроксиэтокси)-10,13-диметилгексадекагидроциклопента[а]-фенантрен-17-ил]пентановой кислоты, 950 мг трифенилфосфина и 550 мг фталимида нагревают в 26 мл ТГФ до 45°С и при этой температуре добавляют по каплям 1,14 мл диэтилового эфира азодикарбоновой кислоты. Перемешивают 2 час при 45°С, затем реакционную смесь выливают в 200 мл полуконцентрированного водного раствора NaHCO3 и трижды экстрагируют по 200 мл ЕЕ. Сушат над Na2SO4 и растворитель удаляют в вакууме. Хроматографией на силикагеле с трет-бутилметиловым эфиром (МТВ) получают 1,76 г вязкого масла.
Rf (EE) = 0,60, MS (FAB): 602 (M+Li)+.
d) Метиловый эфир 4-[3-(2-аминоэтокси)-7,12-дигидрокси-10,13-диметилгексадекагидроциклопента[а]фенантрен-17-ил]пентановой кислоты
1,7 г метилового эфира 4-{3-[2-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)этокси]-7,12-дигидрокси-10,13-диметилгексадекагидроциклопента[а]-фенантрен-17-ил]пентановой кислоты, а также 0,52 мл гидразингидрата (80%) растворяют в 14 мл метанола и 3 час кипятят с обратным холодильником. Затем охлаждают до 40°С и реакционную смесь объединяют с 8,7 мл 2 н. водного раствора HCl. Перемешивают далее при 40°С, затем удаляют летучие компоненты в вакууме. Хроматографией на силикагеле со смесью ацетон/вода, 10:1, получают 540 мг смолообразного твердого вещества.
Rf (ацетон/вода, 10:1) = 0,06, MS (FAB): 466 (М+H)+.
e) 4-[3-(2-Аминоэтокси)-7,12-дигидрокси-10,13-диметилгексадекагидроциклопента[а]фенантрен-17-ил]пентановая кислота
3 г метилового эфира 4-[3-(2-аминоэтокси)-7,12-дигидрокси-10,13-диметилгексадекагидроциклопента[а]фенантрен-17-ил]пентановой кислоты и 310 мг NaOH перемешивают в 5 мл воды, а также 30 мл метанола 24 час при комнатной температуре. Растворители удаляют в вакууме, остаток помещают в 200 мл воды и доводят рН до 7-7,5 водным раствором HCl. Перемешивают 1 час и затем продукт отфильтровывают. Получают 1,6 г бледно-желтого кристаллического твердого вещества, т.пл. 185-195°С.
Rf (СН2С12/МеОН/уксусная кислота/вода, 32:8:1:1) = 0,18, MS (ES): 452 (M+H)+.
f) Этиловый эфир 2-метил-3-(3,4,5-трифторфенил)акриловой кислоты
4,3 мл триэтилового эфира 2-фосфонопропионовой кислоты растворяют в 30 мл безводного ТГФ и добавляют по каплям при 0°С 12,5 мл 1,6 н. раствора н-бутиллития в гексане. 15 мин перемешивают при комнатной температуре и затем добавляют по каплям раствор 3,2 г 3,4,5-трифторбензилового альдегида в 8 мл безводного ТГФ. Один час перемешивают при комнатной температуре и оставляют стоять при комнатной температуре 16 час. Реакционную смесь разбавляют 300 мл воды, добавляют 30 мл насыщенного водного раствора Na2СО3 и 3 раза экстрагируют по 100 мл ЕЕ. Сушат над Na2SO4 и растворитель удаляют. Хроматографией на силикагеле со смесью ЕЕ/НЕР, 1:8, получают 3,8 г бесцветных кристаллов, т.пл. 54°С.
Rf (ЕЕ/НЕР, 1:8) = 0,35, MS (DCI): 245 (М+Н)+.
д) Этиловый эфир 3-(4-{2-[17-(3-карбокси-1-метилпропил)-7,12-дигидрокси-10,13-диметилгексадекагидроциклопента[а]фенантрен-3-илокси]этиламино}-3,5-дифторфенил)-2-метилакриловой кислоты
600 мг 4-[3-(2-аминоэтокси)-7,12-дигидрокси-10,13-диметилгексадекагидроциклопента[а]фенантрен-17-ил]пентановой кислоты, 390 мг этилового эфира 2-метил-3-(3,4,5-трифторфенил)акриловой кислоты и 828 мг К2СО3 перемешивают в 10 мл диметилацетамида 2,5 час при 130°С. Реакционную смесь после охлаждения разбавляют 400 мл СН2С12 и промывают 400 мл 5% раствора NaHSO4. Сушат над MgSO4 и растворитель удаляют в вакууме. Хроматографией на силикагеле со смесью CH2Cl2/MeOH, 10:1, получают 155 мг бесцветного масла.
Rf (CH2Cl2/MeOH, 10:1) = 0,27, MS (ES): 676 (М+Н)+.
i) 4-(3-{2-[2,6-Дифтор-4-(3-гуанидино-2-метил-3-оксопропенил)фениламино]этокси)-7,12-дигидрокси-10,13-диметилгексадекагидроциклопента[а]фенантрен-17-ил)пентановая кислота
130 г гидрохлорида гуанидина и 125 г калий-трет-бутилата перемешивают в 1 мл безводного ДМФ 30 мин при комнатной температуре. Затем добавляют раствор 150 мг этилового эфира 3-(4-{2-[17-(3-карбокси-1-метилпропил)-7,12-дигидрокси-10,13-диметилгексадекагидроциклопента[а]фенантрен-3-илокси]этиламино}-3,5-дифторфенил)-2-метилакриловой кислоты в 1 мл безводного ДМФ и перемешивают 6 час при 110-115°С. Реакционную смесь выливают затем на 100 мл воды, доводят рН до 6 водным раствором HCl и продукт отфильтровывают. В небольшом вакууме сушат и получают 8,0 мг аморфного твердого вещества.
Rf (СН2С12/МеОН/уксусная кислота/вода, 32:8:1:1) = 0,21, MS (ES): 689 (М+Н)+.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОНЪЮГАТЫ 4-БЕНЗИЛАМИНОХИНОЛИНОВ С ЖЕЛЧНОЙ КИСЛОТОЙ И ИХ ГЕТЕРОАНАЛОГИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ЛЕКАРСТВЕННЫЕ СРЕДСТВА НА ОСНОВЕ ЭТИХ СОЕДИНЕНИЙ | 2000 |
|
RU2247125C2 |
| ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ С БИФЕНИЛСУЛЬФОНИЛЬНЫМ ЗАМЕЩЕНИЕМ, ЛЕКАРСТВЕННОЕ СРЕДСТВО | 1998 |
|
RU2242466C2 |
| ЗАМЕЩЕННЫЕ АМИНОКИСЛОТОЙ БЕНЗОИЛГУАНИДИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, СПОСОБ ИНГИБИРОВАНИЯ ЦЕЛЛЮЛЯРНОГО NA/Н-ИОНООБМЕНА И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 1995 |
|
RU2154055C2 |
| СЛОЖНЫЙ 17-ДЕЗОКСИ-КОРТИКОИД-21-ЭФИР КАРБОНОВОЙ КИСЛОТЫ, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ, ЛЕКАРСТВЕННОЕ СРЕДСТВО | 1995 |
|
RU2161624C2 |
| ИМИДАЗО-АННЕЛИРОВАННЫЕ ИЗО- И ГЕТЕРОЦИКЛЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2076105C1 |
| БИФЕНИЛСУЛЬФОНИЛЦИАНАМИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 1999 |
|
RU2247111C2 |
| ЗАМЕЩЕННЫЕ БЕНЗИЛОКСИКАРБОНИЛГУАНИДИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СРЕДСТВО, ИНГИБИРУЮЩЕЕ NA/H-ОБМЕН | 1996 |
|
RU2188191C2 |
| ИНДАНИЛЗАМЕЩЕННЫЕ БЕНЗОЛКАРБОНАМИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, А ТАКЖЕ СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2000 |
|
RU2238934C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛИДИНА, СПОСОБ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ | 1998 |
|
RU2239641C2 |
| ФЕНИЛЗАМЕЩЕННЫЕ ГУАНИДИДЫ АЛКЕНИЛКАРБОНОВОЙ КИСЛОТЫ И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 1997 |
|
RU2193026C2 |
Изобретение относится к замещенным фенилалкеноилгуанидинам, их фармацевтически приемлемым солям и физиологически функциональным производным. Описаны соединения формулы Iа
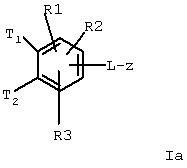
где Т1 и Т2 независимо
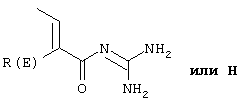
и Т1 и Т2 одновременно не могут быть Н,
L-z означает
L – –C≡C–, –NH–CH2–CH2–O–; R(E) – H, (C1-C4)алкил, R(1), R(2) – H, F, Cl, CN, –SO2–CH3, (C1–C4)алкил, –О–(С1–С4)алкил, а также их физиологически приемлемые соли, физиологически функциональные производные и способ их получения. Эти соединения пригодны, например, в качестве лекарственных средств для профилактики или лечения желчных камней. 2 с. и 2 з.п. ф-лы.
где Т1 и Т2 независимо друг от друга обозначают
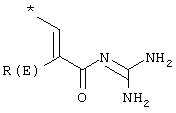
или водород, причем Т1 и Т2 не могут одновременно быть водородом;
L-z обозначает
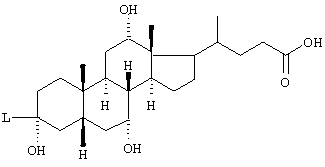
L обозначает -C≡С-, -NH-CH2-CH2-O-;
R(E) обозначает водород, (C1-C4)алкил;
R1, R2 независимо друг от друга обозначают водород, F, Cl, CN, -SО2-СН3, -(C1-C4)алкил, -О-(C1-C4)алкил, причем алкильные остатки могут быть одно или многократно замещены F,
а также их фармацевтически приемлемые соли.
| Устройство для сортирования плодов | 1977 |
|
SU624594A1 |
| Способ получения производных транс-триолов стеринов | 1975 |
|
SU537085A1 |
| 3,6-Диформиат ситостан-3 ,5 ,6 ,триола, проявляющий гиполипидемическое действие | 1976 |
|
SU574450A1 |

