Настоящее изобретение касается технологии рекомбинантных ДНК и, в частности, получения трансгенных растений, которые проявляют существенную резистентность (невосприимчивость) или существенную устойчивость к гербицидам по сравнению с подобными нетрансгенными растениями. Также изобретение, помимо прочего, касается нуклеотидных последовательностей (и продуктов их экспрессии), которые используются в получении указанных трансгенных растений или вырабатываются ими.
Растения, которые по существу “устойчивы” к гербициду тогда, когда они подвергаются его воздействию, формируют кривую зависимости ответа от дозы, смещенную вправо в случае сравнения с той кривой, которую получают на подобных неустойчивых растениях, обработанных сходным образом. При построении таких кривых зависимости ответа от дозы на ось абсцисс наносят “дозу”, а на ось ординат наносят “долю уничтоженных растений”, “эффект действия гербицида” и т.д. Для устойчивых растений обычно должно требоваться вдвое больше гербицида по сравнению с подобными, не обладающими устойчивостью растениями с целью достижения данного эффекта действия гербицида. Растения, которые по существу “резистентны” к гербициду, проявляют незначительные некротические, литические, хлоротические или иные повреждения или не проявляют их вовсе, когда подвергаются действию гербицида при концентрации и интенсивности, которые обычно используют в сельскохозяйственной практике для уничтожения сорняков в полевых условиях, где культурные растения должны быть выращены в промышленных целях.
Особенно предпочтительным является то, что растения по существу резистентны или по существу устойчивы к гербицидам (здесь и далее “глифосат”), мишенью действия которых является 5-енолпирувилшикимат-3-фосфатсинтаза (здесь и далее “EPSPS”) и прекрасным примером которых является N-фосфонометилглицин (и различные его соли).
Гербицид может быть нанесен как до, так и после появления всходов в соответствии с обычными методами применения гербицидов на полях, на которых выращивают культуры, которые резистентны к гербициду. Настоящее изобретение, помимо прочего, представляет нуклеотидные последовательности, применимые для получения указанных растений, устойчивых или резистентных к гербициду.
В соответствии с настоящим изобретением представляется выделенный полинуклеотид, включающий последовательность, приведенную в SEQ ID No 43. Также изобретение представляет полинуклеотид, исключая кДНК, кодирующую EPSPS риса и кукурузы, который кодирует EPSPS и который комплементарен полинуклеотиду, который в случае инкубации при температуре 65-70°С в 0,1 конц. цитратном солевом буфере, содержащем 0,1% ДСН, с последующей промывкой при той же температуре в 0,1 конц. цитратном солевом буфере, содержащем 0,1% ДСН, гибридизирует с последовательностью, приведенной в SEQ ID No 43. Полинуклеотид по настоящему изобретению, кодирующий EPSPS, может, однако, быть получен путем скрининга библиотек геномной ДНК растений нуклеотидом, составляющим интрон в составе последовательности SEQ ID No 43, и изобретение также охватывает такую последовательность, полученную в указанном скрининге.
Также настоящее изобретение охватывает выделенный полинуклеотид, включающий сегмент, кодирующий хлоропластный сигнальный (транзитный) пептид и глифосат устойчивую 5-енолпирувилшикимат-3-фосфатсинтазу (EPSPS), при этом последний расположен по ходу транскрипции за сегментом, кодирующим хлоропластный сигнальный пептид, причем экспрессия указанного сегмента находится под контролем функционального растительного промотора, при условии, что указанный промотор является негетерологичным по отношению к указанному сегменту и что хлоропластный сигнальный пептид является негетерологичным по отношению к указанной синтазе.
Термин “гетерологичный” подразумевает происхождение от отличающегося источника, и, соответственно, “негетерологичный” означает происхождение от одного и того же источника, но на генном уровне, а не на уровне организма или ткани. Например, промотор CaMV35S очевидно гетерологичен по отношению к кодирующей последовательности EPSPS петунии, поскольку промотор происходит от вируса, а последовательность, экспрессию которой он контролирует, от растения. Однако термин “гетерологичный” в соответствии с настоящим изобретением имеет еще более узкое значение. Например, “гетерологичный”, упоминаемый в связи с настоящим изобретением, означает, что кодирующая последовательность EPSPS петунии “гетерологична” по отношению, например, к промотору, также происходящему от петунии, но экспрессию гена EPSPS. В этом смысле промотор петунии, происходящий от гена EPSPS петунии и затем используемый для контроля экспрессии кодирующей последовательности EPSPS, также происходящей от петунии, является “негетерологичным” по отношению к указанной кодирующей последовательности. Однако термин “негетерологичный” не означает того, что промотор и кодирующая последовательность обязательно должны быть получены из состава одного и того же (исходного или предкового) полинуклеотида. То же самое касается сигнальных пептидов. Например, хлоропластный сигнальный пептид Rubisco, происходящий от подсолнечника, “гетерологичен” по отношению к кодирующей последовательности гена EPSPS, также происходящего от подсолнечника (того же растения, ткани или клетки). Последовательность, кодирующая сигнальный пептид Rubisco, происходящая от подсолнечника, “негетерологична” по отношению к последовательности, кодирующей фермент Rubisco, также происходящей от подсолнечника, даже если исходные для обеих последовательностей полинуклеотиды являются разными и могут присутствовать в разных клетках, тканях или растениях подсолнечника.
Предпочтительная форма полинуклеотида включает следующие компоненты, перечисленные в направлении транскрипции 5’→ 3’:
(i) по крайней мере один транскрипционный энхансер, являющийся энхансерным элементом, расположенным выше старт-сайта транскрипции в последовательности, от которой энхансер был получен, причем указанный энхансер сам по себе не функционирует в качестве промотора ни в последовательности, в которой он является эндогенным, ни в случае, когда он присутствует гетерологически как часть конструкции;
(ii) промотор гена EPSPS риса;
(iii) геномная последовательность риса, которая кодирует хлоропластный сигнальный пептид EPSPS риса;
(iv) геномная последовательность, которая кодирует EPSPS риса;
(v) терминатор транскрипции;
причем кодирующая последовательность EPSPS риса модифицирована таким образом, что первое положение мутировано так, что остатком в этом положении является Il вместо Thr, и второе положение мутировано так, что остатком в этом положении является Ser вместо Pro, причем мутации внесены в последовательности EPSPS, которые включают следующий консервативный сегмент GNAGTAMRPLTAAV в ферменте дикого типа, таким образом, что модифицированная последовательность читается как GNAGIAMRSLTAAV.
Энхансерный сегмент предпочтительно включает последовательность, 3’-конец которой находится по крайней мере на 40 нуклеотидов выше ближайшего старт-сайта транскрипции в последовательности, от которой энхансер получен. В следующем варианте полинуклеотида энхансерный сегмент включает сегмент, 3’-конец которого находится по крайней мере на 60 нуклеотидов выше указанного ближайшего старт-сайта, и еще в одном варианте полинуклеотида указанный энхансерный сегмент включает последовательность, 3’-конец которой находится по крайней мере на 10 нуклеотидов выше первого нуклеотида в консенсусном боксе ТАТА в последовательности, от которой энхансер получен.
Полинуклеотид в соответствии с настоящим изобретением может включать два или большее число транскрипционных энхансеров, которые в конкретном варианте полинуклеотида могут быть организованы тандемно (один за другим).
В заявляемом настоящим изобретением полинуклеотиде 3’-конец энхансера или первый энхансер, если их присутствует более одного, может находиться на расстоянии от примерно 100 до примерно 1000 нуклеотидов вверх от кодона, соответствующего старт-кодону трансляции сигнального пептида EPSPS, или первого нуклеотида интрона в 5’-нетранслируемом сегменте в случае, когда указанный сегмент включает интрон. В более предпочтительном варианте полинуклеотида 3’-конец энхансера или первый энхансер находится на расстоянии от примерно 150 до примерно 1000 нуклеотидов вверх от кодона, соответствующего старт-кодону трансляции сигнального пептида EPSPS, или первого нуклеотида интрона в 5’-нетранслируемом сегменте, и в еще более предпочтительном варианте 3’-конец энхансера или первый энхансер может находиться на расстоянии от примерно 300 до примерно 950 нуклеотидов вверх от кодона, соответствующего старт-кодону трансляции сигнального пептида EPSPS, или первого нуклеотида интрона в 5’-нетранслируемом сегменте. В еще более предпочтительном варианте 3’-конец энхансера или первый энхансер может находиться на расстоянии от примерно 770 до примерно 790 нуклеотидов вверх от кодона, соответствующего старт-кодону трансляции сигнального пептида EPSPS, или первого нуклеотида интрона в 5’-нетранслируемом сегменте.
В альтернативном заявляемом полинуклеотиде 3’-конец энхансера или первый энхансер может находиться на расстоянии от примерно 300 до примерно 380 нуклеотидов вверх от кодона, соответствующего старт-кодону трансляции сигнального пептида EPSPS, или от первого нуклеотида интрона в 5’-нетранслируемом сегменте, а в предпочтительном варианте указанного альтернативного полинуклеотида 3’-конец энхансера или первый энхансер расположен на расстоянии от примерно 320 до примерно 350 нуклеотидов вверх от кодона, соответствующего старт-кодону трансляции сигнального пептида EPSPS или первого нуклеотида интрона в 5’-нетранслируемом сегменте.
В полинуклеотиде по настоящему изобретению сегмент вверх от промотора гена EPSPS может включать по крайней мере один энхансер, происходящий из последовательности, которая находится выше старт-сайта транскрипции в промоторах CaMV35S или FMV35S.
Следовательно, полинуклеотид может включать в направлении 5’→ 3’ первый энхансер, включающий сегмент энхансера транскрипции, происходящий от последовательности, которая находится выше старт-сайта транскрипции промотора GOS-2, и второй энхансер, включающий сегмент энхансера транскрипции, происходящий от последовательности, которая расположена выше старт-сайта транскрипции промоторов CaMV35S или FMV35S.
Альтернативно, полинуклеотид может включать в направлении 5’→ 3’ первый энхансер, включающий сегмент энхансера транскрипции, происходящий от последовательности, которая находится выше старт-сайта транскрипции промотора гена актина риса, и второй энхансер, включающий сегмент энхансера транскрипции, происходящий от последовательности, которая расположена выше старт-сайта транскрипции промоторов CaMV35S или FMV35S.
Альтернативно, полинуклеотид может включать в направлении 5’→ 3’ первый энхансер, включающий сегмент энхансера транскрипции, происходящий от последовательности, которая находится выше старт-сайта транскрипции промотора гена пластоцианина ячменя, и второй энхансер, включающий сегмент энхансера транскрипции, происходящий от последовательности, которая расположена выше старт-сайта транскрипции промоторов CaMV35S или FMV35S.
Альтернативно, полинуклеотид может включать в направлении 5’→ 3’ первый энхансер, включающий сегмент энхансера транскрипции, происходящий от последовательности, которая находится выше старт-сайта транскрипции промотора гена полиубихитина кукурузы, и второй энхансер, включающий сегмент энхансера транскрипции, происходящий от последовательности, которая расположена выше старт-сайта транскрипции промоторов CaMV35S или FMV35S.
Альтернативно, полинуклеотид может включать в направлении 5’→ 3’ первый энхансер, включающий сегмент энхансера транскрипции, происходящий от последовательности, которая находится выше старт-сайта транскрипции промотора FMV35S, и второй энхансер, включающий сегмент энхансера транскрипции, происходящий от последовательности, которая расположена выше старт-сайта транскрипции промотора CaMV35S.
При любых идентичности и взаиморасположении различных энхансеров в составе полинуклеотида нуклеотиды с 5’-конца от кодона, который составляет старт-кодон трансляции хлоропластного сигнального пептида EPSPS риса, могут быть выбраны по Козаку. То, что это означает, хорошо известно специалистам в данной области техники и дополнительно станет понятным из приводимых далее примеров.
В частности, в предпочтительных вариантах настоящего изобретения полинуклеотид включает нетранслируемый сегмент, который включает последовательность, которая выполняет функции интрона, расположенного с 5’-конца от геномной последовательности риса, которая кодирует хлоропластный сигнальный пептид EPSPS риса. Нетранслируемый сегмент может включать последовательность, приведенную в SEQ ID No 55. В частности, нетранслируемый сегмент может включать интрон гена ADHI кукурузы, характеризующийся последовательностью SEQ ID No 55.
Полинуклеотид по настоящему изобретению может включать происходящий от вируса трансляционный энхансер, находящийся в пределах нетранслируемого сегмента 5’-конца от геномной последовательности риса, которая кодирует хлоропластный сигнальный пептид EPSPS риса. Специалистам в данной области техники известны индивидуальные свойства таких подходящих трансляционных энхансеров - таких как последовательности Omega и Omega-prime, происходящие от TMV, и те, которые происходят от вируса гравировки табака, а также известно, как такие трансляционные энхансеры могут быть внесены в полинуклеотид так, чтобы обеспечить желательный результат.
Полинуклеотид по настоящему изобретению может дополнительно включать сегменты, кодирующие белки, способные обеспечивать содержащему их растительному материалу по крайней мере один из следующих агрономически-полезных признаков: резистентность к насекомым, грибкам, вирусам, бактериям, нематодам, стрессам, обезвоживанию и гербицидам. При том, что такой полинуклеотид предусматривает иной ген, обеспечивающий резистентность к гербицидам, нежели ген EPSPS, такой как, например, ген глифосатоксидоредуктазы (GOX), гербицид может и не являться глифосатом, причем в этом случае гены, обеспечивающие резистентность, могут быть выбраны из группы генов, кодирующих следующие белки: фосфинотрицин-ацетилтрансфераза (PAT), гидроксифенилпируватдиоксигеназа (HPPD), глутатион-S-трансфераза (GST), цитохром Р-450, ацетил-КоА-карбоксилаза (АККаза), ацетолактатсинтаза (ALS), протопорфириногеноксидаза (РРО), дигидроптероатсинтаза, белки-переносчики полиаминов, супероксиддисмутаза (SOD), бромоксинилнитрилаза, фитоендесатураза (PDS), продукт гена tfdA, выделенного из Alcaligenes eutrophus, и известные мутированные или по-иному модифицированные варианты указанных белков. В случае, когда полинуклеотид обеспечивает множественную гербицидную резистентность, такие гербициды могут быть выбраны из группы, которая включает динитроанилиновый гербицид, триазолопиримидины, урацил, фенилмочевину, трикетон, изоксазол, ацетанилид, оксадиазол, триазинон, сульфонанилид, амид, анилид, RP201772, фторхлоридон, норфлуразон и гербицид триазолинонового типа, а гербициды, применяемые после появления всходов, выбирают из группы, которая включает глифосат и его соли, глуфозинат, асулам, бентазон, биалафос, бромацил, сетоксидим или иной циклогександион, дикамба, фозамин, флупоксам, феноксипропионат, квизалофоп или иной арилоксифеноксипропаноат, пиклорам, флуорметрон, атразин или иной триазин, метрибуцин, хлоримурон, хлорсульфурон, флуметсулам, галосульфурон, сульфометрон, имазахин, имазетапир, изоксабен, имазамокс, метосулам, пиритробак, римсульфурон, бенсульфурон, никосульфурон, фомесафен, флурогликофен, KIH9201, ЕТ751, карфентразон, ZA1296, сулькотрион, паракват, дикват, бромоксинил и феноксапроп.
В случае, когда полинуклеотид включает последовательности, кодирующие инсектицидные белки, такие белки могут быть выбраны из группы, которая включает кристаллические токсины, происходящие от Bacillus thuringiensis (Bt), включая секретируемые Bt-токсины; протеазные ингибиторы, лектины, токсины Xenorhabdus/Photorhabdus; гены, обеспечивающие резистентность к грибкам, могут быть выбраны из группы, которая включает гены, кодирующие известные AFP, дефензины, хитиназы, глюканазы, Avr-Cf9. В частности, предпочтительными инсектицидными белками являются cryIAc, cryIAb, сrу3А, Vip-1A, Vip-1B, ингибиторы цистеиновых протеаз и лектин подснежника. В случае, когда полинуклеотид включает гены, обеспечивающие резистентность к бактериям, такие гены могут быть выбраны из группы, которая включает гены, кодирующие цекропины и техиплезин и их аналоги. Гены, обеспечивающие резистентность к вирусам, могут быть выбраны из группы, которая включает гены, кодирующие вирусные капсидные белки, двигательные белки, вирусные репликазы и антисмысловые и рибозимные последовательности, для которых известно обеспечение резистентности к вирусам; кроме того, гены, обеспечивающие резистентность к стрессам, засолению и обезвоживанию, могут быть выбраны из генов, кодирующих глутатион-S-трансферазу и пероксидазу, последовательности которых включают известную регуляторную последовательность CBF1, и генов, для которых известно участие в накоплении трегалозы.
Полинуклеотид по настоящему изобретению может быть модифицирован с целью усиления экспрессии кодирующих белок последовательностей таким образом, что мотивы, приводящие к нестабильности мРНК, и/или случайные сайты сплайсинга могут быть удалены, или же предпочитаемые библиотекой кодонов данного растения кодоны могут быть использованы таким образом, что экспрессия модифицированного таким путем полинуклеотида в растении даст по существу такой же белок, обладающий по существу такими же активностью и функциями по сравнению с тем белком, который получают в результате экспрессии немодицифированного полинуклеотида в организме, для которого кодирующие белок сегменты немодифицированного полинуклеотида являются эндогенными. Степень идентичности между модифицированным полинуклеотидом и полинуклеотидом, являющимся эндогенным для указанного растения и кодирующего по существу такой же белок, может быть такой, чтобы предотвращать эффект взаимного подавления модифицированной и эндогенной последовательностей. В этом случае степень идентичности между последовательностями предпочтительно должна быть меньше, чем примерно 70%.
Далее настоящее изобретение охватывает биологический или трансформационный вектор, включающий заявляемый настоящим изобретением полинуклеотид. Следовательно, “вектор” означает, помимо прочего, одно из следующего: плазмиду, вирус, космиду или бактерию, трансформированную или трансфицированную таким образом, что она несет полинуклеотид.
Далее настоящее изобретение охватывает растительный материал, который был трансформирован указанным полинуклеотидом или вектором, а также такой трансформированный растительный материал, который был дополнительно трансформирован полинуклеотидом, включающим сегменты, которые кодируют белки, способные обеспечивать несущему их растительному материалу по крайней мере один из следующих агрономически-полезных признаков: резистентность к насекомым, грибкам, вирусам, бактериям, нематодам, стрессу, обезвоживанию и гербицидам.
Также настоящее изобретение охватывает морфологически нормальные фертильные цельные растения, которые были регенерированы из материала, представленного в предыдущем абзаце, образуемые на них семена и части потомства, которые несут полинуклеотид или вектор по настоящему изобретению, стабильно интегрированный в их геном и наследуемый по менделевскому типу.
Далее настоящее изобретение дополнительно охватывает морфологически нормальные фертильные цельные растения, несущие данный заявляемый полинуклеотид и которые получены в результате скрещивания растений, которые были регенерированы из материала, трансформированного данным заявляемым полинуклеотидом или вектором, а также растения, которые были трансформированы полинуклеотидом, включающим участки, которые кодируют белки, способные обеспечивать несущему их растительному материалу по крайней мере один из следующих агрономически-полезных признаков; резистентность к насекомым, грибкам, вирусам, бактериям, нематодам, стрессу, обезвоживанию и гербицидам, а также потомство получаемых в результате растений, их семена и части.
Растения по настоящему изобретению могут быть выбраны из группы, которая включает посевные культуры, фруктовые и овощные культуры, такие как брюква “canola”, подсолнечник, табак, сахарная свекла, хлопчатник, кукуруза, пшеница, ячмень, рис, сорго, помидор, манго, персик, яблоко, груша, земляника, банан, дыня, картофель, морковь, латук, капуста, лук, соя разных видов, сахарный тростник, горох, бобы, тополь, виноград, цитрусовые, люцерна, рожь, овес, дерновые и фуражные травы, лен и брюква, а также орехоносные растения, если только они уже не упоминались выше, а также их потомство, семена и части.
Особенно предпочтительными такими растениями являются кукуруза, соя, хлопчатник, сахарная свекла и брюква “canola”.
Далее настоящее изобретение представляет способ избирательной борьбы с сорняками в полевых условиях, причем на поле находятся сорняки и растения по настоящему изобретению или резистентное к гербициду их потомство, включающий нанесение на поле гербицида глифосатного типа в количестве, достаточном для борьбы с сорняками без существенного негативного влияния на культурные растения. В соответствии с данным способом один или большее число гербицидов, инсектицидов, фунгицидов, нематоцидов, бактериоцидов и антивирусных средств можно наносить на поле (и, соответственно, на растения, растущие на нем) как до, так и после применения глифосатного гербицида.
Далее изобретение предусматривает способ получения растений, которые по существу устойчивы или по существу резистентны к глифосатному гербициду, включающий следующие стадии:
(i) трансформацию растительного материала полинуклеогидом или вектором по настоящему изобретению;
(ii) отбор трансформированного таким путем материала; и
(iii) регенерацию отобранного таким путем материала в морфологически нормальные фертильные цельные растения.
Трансформация может включать внесение полинуклеотида в материал с помощью любого известного метода, в частности, с помощью: (i) биолистической бомбардировки материала частицами, покрытыми полинуклеотидом; (ii) прокалывания материала волокнами из карбида кремния, покрытыми раствором, содержащим полинуклеотид; или (iii) внесения полинуклеотида или вектора в Agrobacterium и совместного культивирования трансформированной таким образом Agrobacterium с растительным материалом, который тем самым трансформируется, с последующим регенерированием. Методы трансформации, отбора и регенерации растений, которые могут потребовать рутинной модификации по отношению к конкретным видам растений, хорошо известны специалистам. Трансформированный таким образом растительный материал может быть отобран по его резистентности к глифосату.
Далее настоящее изобретение представляет использование заявляемого здесь полинуклеотида или вектора в получении растительных тканей и/или морфологически нормальных фертильных цельных растений, которые по существу устойчивы или по существу резистентны к глифосатному гербициду.
Настоящее изобретение далее охватывает способ отбора биологического материала, трансформированного так, чтобы он экспрессировал интересующий ген, причем трансформированный материал несет полинуклеотид или вектор по настоящему изобретению и где данный отбор включает обработку трансформированного материала глифосатом или его солью с последующим отбором выживающего материала. Указанный материал может иметь растительное происхождение и может, в частности, происходить от однодольного растения, выбранного из группы, которая включает ячмень, пшеницу, кукурузу, рис, овес, рожь, сорго, ананас, сахарный тростник, банан, лук, спаржу и черемшу.
Кроме того, изобретение представляет способ регенерирования фертильного трансформированного растения, несущего чужеродную ДНК, включающий следующие стадии:
(a) получение способной к регенерации ткани от указанного растения, которое предстоит трансформировать;
(b) трансформацию указанной регенерируемой ткани указанной чужеродной ДНК, где указанная чужеродная ДНК включает селективную последовательность ДНК, где указанная последовательность функционирует в регенерируемой ткани в качестве селективного агента;
(c) по прошествии срока от одного дня до примерно 60 дней после стадии (b) помещение указанной регенерируемой ткани стадии (b) в среду, способную продуцировать побеги на указанной ткани, где указанная среда дополнительно содержит соединение, используемое для отбора регенерируемой ткани, несущей указанную селективную ДНК-последовательность, для обеспечения идентификации или отбора трансформированной регенерированной ткани;
(d) после того, как на отобранной ткани (с) сформировался по крайней мере один побег, перенос указанного побега во вторую среду, способную образовывать корни на указанном побеге с получением проростка, где вторая среда необязательно содержит указанное соединение; и
(e) выращивание указанного проростка в фертильное трансгенное растение, где чужеродная ДНК передается растениям-потомкам по менделевскому типу, характеризующееся тем, что чужеродная ДНК является или включающая селективную последовательность ДНК чужеродная ДНК включает полинуклеотид по настоящему изобретению, а указанное соединение является глифосатом или его солью. Растение может быть однодольным растением в соответствии с указанным выше: более предпочтительно выбранным из банана, пшеницы, риса, кукурузы и ячменя, а указанная регенерируемая ткань может включать эмбриогенные каллюсы, соматические зародыши, недоразвитые зародыши и т.п.
Далее настоящее изобретение дополнительно будет очевидно из нижеследующего описания, совместно с прилагающимися чертежами и списком последовательностей.
Список последовательностей
SEQ ID NO. 1-42 - праймеры для ПЦР.
SEQ ID NO. 43 - геномная последовательность EPSPS риса (от кодона ATG).
SEQ ID NO. 44 - геномная последовательность EPSPS риса, содержащая двойную мутацию.
SEQ ID NO. 45 - энхансер FMV.
SEQ ID NO. 46 - 1-й энхансер CaMV35S.
SEQ ID NO. 47 - 2-й энхансер CaMV35S.
SEQ ID NO. 48 - энхансер полиубихитина кукурузы.
SEQ ID NO. 49 - энхансер актина риса.
SEQ ID NO. 50 - энхансер GOS2 риса.
SEQ ID NO. 51 - энхансер пластоцианина ячменя.
SEQ ID NO. 52 - промотор EPSPS риса G1 + 5’ “верхний” конец.
SEQ ID NO. 53 - промотор EPSPS риса G3 + 5’ “верхний” конец.
SEQ ID NO. 54 - промотор EPSPS риса G2 + интрон Adhl в составе 5’ “верхнего” конца.
SEQ ID NO. 55 - интрон Adhl кукурузы.
Список чертежей
Фиг.1 - схематическая геномная карта EPSPS риса.
Фиг.2 - вектор pCR4-OSEPSPS (ген dmEPSPS риса в векторе pCR4-Blunt).
Фиг.3 - схематическое изображение стратегии, использованной для внесения двойной мутации.
Фиг.4 - вектор pTCV1001.
Фиг.5 - вектор pTCV1001OSEPSPS (включает ген dmEPSPS риса в векторе pTCV1001).
Фиг.6 - вектор pTCV1001EPSPSPAC (включает ген dmEPSPS риса в векторе pTCV1001).
Фиг.7 - вектор pBluSK + EPSPS (включает ген dmEPSPS риса в векторе pBluescript SK+).
Фиг.8 - вектор pPAC1.
Фиг.9 - вектор pTCVEPSPSPH.
Фиг.10 - вектор pTCVEPSPSADH.
Фиг.11 - вектор pBluSKEPSPSADH (включает ген dmEPSPS риса и интрон Adh1).
Фиг.12 - вектор pIGPD9.
Фиг.13 - вектор Zen-9.
Фиг.14 - вектор Zen-12.
Фиг.15 - вектор Zen-18.
Фиг.16 - внесение векторов Zen в супербифункциональные векторы.
Получение растений, устойчивых к обработке глифосатом за счет сверхэкспрессии мутантного EPSPS под контролем негетерологичного промотора
Термин “энхансер” в соответствии с использованием его в данной заявке обозначает те последовательности, расположенные выше промотора, которые сами не включают промотор, но действие которых проявляется в усилении или регуляции инициации транскрипции с промотора. Термин “делеция промотора EPSPS” по использованию в данной заявке относится к промотору EPSPS наряду с нуклеотидами, отделяющими его от энхансера, являющегося нативным для гена EPSPS, - т.е. нуклеотидами, расположенными выше (т.е. с 5’-конца) данного промотора.
В отношении трансформации растительного материала для специалиста в данной области техники должно быть понятно, что, хотя конкретные типы материала-мишени (например, суспензионная культура зародышевых клеток или дедифференцированные недоразвитые зародыши) и конкретные способы трансформации (например, с использованием Agrobacterium или бомбардировки частицами) описаны далее в примерах, при этом настоящее изобретение не ограничено данными конкретными вариантами, и указанные материалы-мишени и способы можно использовать в разных сочетаниях. Более того, термин “растительные клетки” по использованию в данном описании может относиться к выделенным клеткам, включая суспензионные культуры, а также к клеткам в интактной или частично интактной ткани, такой как зародыш, щиток, микроспора, производный от микроспоры зародыш или соматические клетки растительных органов. Сходным образом, хотя конкретные примеры ограничиваются кукурузой, пшеницей и рисом, настоящее изобретение в равной степени применимо к любому из широкого круга сельскохозяйственных культур и домашних растений, которые могут быть трансформированы с помощью подходящих методов трансформации растительных клеток.
Базовые молекулярно-биологические методы осуществляют, как описано Sambrook et al. (1989) Molecular Cloning: A Laboratory Manual, 2 nd Edn. Cold Spring Harbor Lab. Press.
Пример 1
Формирование кДНК-зонда для EPSPS риса
Неполную кДНК, кодирующую EPSPS риса, получают с использованием ПЦР с обратной транскриптазой (ОТ-ПЦР). Общую РНК выделяют из двухнедельных растений риса (Oryza sakiva L. indica, сорт Koshihikari) с использованием метода TRI-ZOL™ (Life Technologies). Синтез первой цепи кДНК осуществляют с использованием обратной транскриптазы Superscript II (Life Technologies) с 200 нг вырожденного обратного праймера 10 для EPSPS (SEQ ID NO. 1) и 2 мкг общей РНК в соответствии с прилагаемым протоколом. Синтез второй цепи и амплификацию кДНК с помощью ПЦР проводят с использованием вырожденных праймеров 10 и 4 для EPSPS (SEQ ID NO. 1 и SEQ ID NO. 2) и ПЦР-шариков (Pharmacia) в соответствии с инструкциями изготовителя. Все буквенные коды соответствуют стандартным аббревиатурам (Eur. J. Biochem. (1985) 150:15).
SEQ ID NO. 1
Вырожденный обратный праймер 10 для EPSPS

Вырожденный прямой праймер 4 для EPSPS
SEQ ID NO. 2

Полученные продукты клонируют в состав вектора pCR2.1 (Invitrogen) с использованием набора ТА Cloning kit™ в соответствии с рекомендациями поставщика. Плазмиду выделяют из отобранных колоний и последовательность анализируют с помощью способа, включающего компьютерный анализ гомологии (BLAST), для подтверждения того, что продукт ОТ-ПЦР проявляет высокий уровень гомологии с известными последовательностями EPSPS растений.
Пример 2
Выделение геномной последовательности EPSPS риса и клонирование гена EPSPS риса
Участок геномной ДНК, включающий полноразмерный ген EPSPS риса и 5’-верхний конец, выделяют из геномной библиотеки λ -EMBLSP6/T7, сконструированной на материале 5-дневных этиолированных побегов риса [Оrуzа sativa L. indica, сорт IR36) (Clontech). 1× 106 бляшкообразующих единиц (БОЕ) подвергают скринингу с использованием 32Р-меченого кДНК-зонда EPSPS риса (пример 1) с использованием протоколов, предоставляемых изготовителем. Позитивные бляшки подвергают последующим раундам гибридизационного скрининга до тех пор, как будет достигнута чистота на уровне перекрестно-гибридизующей бляшки. ДНК фага λ получают на материале маточного штамма фага в соответствии с методом, описанным у Sambrook et al., 1989. Полученную ДНК анализируют с помощью рестриктазного расщепления и Саузерн-блоттинга с использованием той же самой 32Р-меченой кДНК EPSPS риса в качестве зонда. У рестрикционных фрагментов, которые перекрестно гибридизуют, затупляют концы с использованием такого метода, как Perfectly Blunt™ (Novagen), и их клонируют в состав подходящего вектора, такого как pSTBlue (Novagen). Затем ДНК секвенируют с использованием автоматического ДНК-секвенатора ABI 377A PRISM. На фиг.1 схематически показан ген EPSPS риса с рядом отмеченных рестрикционных сайтов.
Фрагмент гена EPSPS риса длиной 3,86 тысячи пар нуклеотидов (т.п.н.), включающий кодирующий сегмент, промотор EPSPS, часть 5’-верхнего конца и терминатор, получают с помощью ПЦР. Олигонуклеотидный праймер OSGRA1 (SEQ ID NO. 3) используют в сочетании с праймером OSEPSPS3 (SEQ ID NO. 4) для амплификации желаемого сегмента. OSEPSPS3 включает дополнительные рестрикционные сайты рестриктаз SacI и SmaI, предназначенные для облегчения субклонирования гена в ходе последующих стадий конструирования вектора. Расположение указанных праймеров схематически показано на фиг.1.

Высокоточную полимеразу Pfu Turbo™ (Stratagene) используют для осуществления реакции ПЦР с ДНК, полученной из λ -препарата (описано выше), в качестве амплификационной матрицы. ПЦР-продукт ожидаемого размера клонируют в состав pCRBlunt 4-TOPO™ (Invitrogen) и секвенируют для проверки целостности.
Пример 3
Мутация Т→ 1 и Р→ S в EPSPS риса
Мутацию Т→ 1 и Р→ S получают путем внесения двух точковых мутаций. Указанные мутации вносят в геномный ген EPSPS риса с помощью ПЦР с использованием олигонуклеотидных праймеров, включающих желательную мутацию. Схематическая диаграмма, указывающая на сайты отжига использованных праймеров, показана на фиг.3. Осуществляют две раздельные реакции ПЦР (обе с использованием ДНК λ -фага в качестве матрицы).

Полученные в результате ПЦР-продукты объединяют с использованием эквимолярных концентраций каждого ПЦР-продукта, служащего матрицей, с двумя олигонуклеотидами SalIEnd и EcoRVEnd в новой реакции ПЦР. Аликвоту продукта реакции анализируют с помощью электрофореза в агарозном геле и клонируют в состав pCR-Blunt II™ (Invitrogen). Плазмидную ДНК выделяют и секвенируют с целью выявления успешного внесения двойной мутации.
ДНК-фрагмент, включающий двойную мутацию, вносят в геномный клон EPSPS риса (фиг.1) следующим образом. Клон, включающий двойную мутацию, расщепляют рестриктазами EcoRV и SalI. Плазмиду, включающую ДНК EPSPS риса, являющуюся ПЦР-продуктом, сходным образом расщепляют, и EсоRV/SаlI-фрагмент, включающий двойную мутацию, лигируют в ген EPSPS риса в составе pCR4Blunt-TOPO™ с использованием стандартных методов клонирования, описанных у Sarnbrook et al., 1989, и трансформируют в компетентные клетки Е.coli. Плазмиду выделяют из полученных в результате колоний и секвенируют с целью подтверждения присутствия двойной мутации при отсутствии других изменений. Данная плазмида, обозначенная pCR4-OSEPSPS, показана на фиг.2.
Геномный ген EPSPS риса, включающий двойную мутацию (фиг.2), вырезают из плазмиды pCR4-Blunt-TOPO™ с использованием рестриктаз PstI и NotI и лигируют в состав pTCV1001 (фиг.4) с получением pTCV1001OSEPSPS (фиг.5), которую трансформируют в Е. coli с целью амплификации. Далее рестрикционный РасI/EсоRV-фрагмент вырезают из ДНК фага λ , (фиг.1) и встраивают в pTCV1001OSEPSPS (фиг.5) с получением pTCV1001EPSPSPAC (фиг.6). Ген EPSPS риса, теперь включающий последовательность от РасI до SacI (фиг.6), вырезают из pTCV1001EPSPSPAC (фиг.6) в виде ЕаgI/SacI-фрагмента и лигируют в сходным образом расщепленную плазмиду pBluescript SK+ с получением pBluSK + EPSPS (фиг.7). Далее расположенные выше сегменты EPSPS риса и желательные энхансеры собирают (в соответствии с описанным ниже) и лигируют в вектор pBluescript SK+ с использованием рестриктаз XbaI/PacI.
Пример 4
Формирование химер отдельных энхансеров и промотора EPSPS риса
На фиг.1 показаны сайты связывания праймеров G1 и G2, использованных для получения серии делеций с 5’-конца гена EPSPS риса. Праймеры G1 и G2 используют в сочетании с праймером RQCR10, используя λ -ДНК-матрицу EPSPS риса и полимеразу Pfu Turbo™ (Stratagene) в соответствии с протоколами, предоставляемыми изготовителем.

Полученные продукты анализируют методом электрофореза в агарозном геле и клонируют в состав вектора pCR-Blunt II-ТОРО™ (Invitrogen). Последовательность полученных в результате продуктов определяют для того, чтобы убедиться в отсутствии изменений последовательности геномного клона EPSPS риса. Клоны для дальнейшего анализа выбирают на основе их ориентации в составе вектора, определяемой по тому, удаляет или нет расщепление рестриктазой XhoI только полилинкерную последовательность, а не полную вставку из вектора.
Последовательности генов CaMV35S и FMV35S и связанных с ними 5’-верхних концов опубликованы в базе данных EMBL (например, депозитарные №№ v00141 и х06166). Праймеры конструируют таким образом, чтобы амплифицировать только расположенные выше энхансерные сегменты указанных генов. Энхансер-1 CaMV35S (SEQ ID NO. 36) получают таким образом с помощью ПЦР с использованием праймеров SEQ ID NO. 12 и SEQ ID NO. 13 в сочетании с полимеразой Pfu Turbo™ и ДНК pMJB1 (А59870) в качестве матрицы. Альтернативно, отличающийся участок энхансера CaMV35S получают с использованием сходных методов (SEQ ID NO. 37). Энхансер FMV35S синтезируют химическим путем (SEQ ID NO. 35).
Используют следующие олигонуклеотидные праймеры.
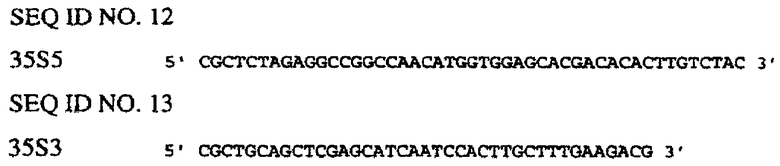
Последовательность амплифицированных и клонированных молекул подтверждают после клонирования в состав вектора pCR Blunt-II-TOPO (Invitrogen). Вектор pCR Blunt-II-TOPO, включающий делецию 5’UTR EPSPS, расщепляют рестриктазами XbaI /XhoI. Энхансер удаляют из соответствующего ему вектора pCR Blunt-II-TOPO также с использованием расщепления XbaI/XhoI и лигируют в первые векторы, включающие промоторные делеции EPSPS, сформированные с помощью ПЦР.
Пример 5
Формирование химер двойных энхансеров с промотором EPSPS риса
С целью дальнейшего усиления экспрессии с промотора EPSPS риса дополнительный энхансер может быть использован в сочетании с любым из энхансеров 35S или FMV. В одном примере стратегии клонирования с целью достижения указанного изначально формируют химеры энхансер/EPSPS (аналогично примеру 4), включающие единственный (первый) энхансер. Такие первые энхансеры выбирают из верхних 5’-концов от промоторов генов GOS2 риса, актина-1 риса, полиубихитина кукурузы, CaMV 35S, FMV 35S и пластоцианина ячменя. Нуклеотидные последовательности указанных сегментов опубликованы в базе данных EMBL (депозитарные №№ х51910, х15865, u29159, v00141, х06166 и z28347, соответственно). Праймеры конструируют таким образом, чтобы амплифицировать только желательные сегменты транскрипционных энхансеров из 5’-концов этих генов (SEQ ID NO. 15-22).

ДНК выделяют из растений (кукуруза, ячмень или рис), выращенных в теплице, с использованием протокола DNAeasy (Qiagen), и используют ее в качестве исходной матрицы для ПЦР-амплификации. Продукты ПЦР клонируют в состав pCR-Blunt-II-ТОРО и секвенируют с целью подтверждения аутентичности. Химеру энхансер/EPSPS получают, как описано в примере 4 (используя обмен по XbaI/XhoI-сайтам), за исключением случая с геном актина-1 риса, когда энхансер встраивают в виде XbaI/PstI-фрагмента (XhoI-сайт заменен PsfI-сайтом из-за присутствия XhoI-сайта в энхансере актина риса), и с геном полиубихитина кукурузы, когда энхансер встраивают в виде SреI/ХhоI-фрагмента (XbaI-сайт заменен SpeI-сайтом из-за присутствия XbaI-сайта в энхансере полиубихитина кукурузы). Следует отметить, что в связи с энхансерным сегментом полиубихитина кукурузы используют внутренний XhoI-сайт. Второй энхансер, который может быть либо энхансером CaMV35S, либо энхансером FMV, амплифицируют с использованием праймеров 35SXho и 35S3 (SEQ ID NO. 23 и SEQ ID NO. 13) (35S) или праймеров FMVXho и FMV3 (SEQ ID NO. 25 и SEQ ID NO. 26), соответственно. Указанные праймеры облегчают внесение XhoI-сайта (или PstI-сайта) по 5’- и 3’-концам энхансера. Альтернативно, PacI-сайт вносят по 3’-концу вместо XhoI-сайта с использованием праймеров 35SPac (SEQ ID NO. 24) или FMVPAC3 (SEQ ID NО. 27).

Будучи секвенированным, ПЦР-продукт в виде фрагмента XhoI:XhoI, PstI/PacI (когда первым энхансером является энхансер актина риса) или XhoI/PacI вносят в состав конструкции, которая включает химеру первого энхансера и гена EPSPS, либо по XhoI-сайту, либо между XhoI- и PacI-сайтами или между PstI- и PacI-сайтами, исходя из необходимости. Внесение по XhoI-сайту используют для конструирования химер с двумя энхансерами, включающими промоторные делеции EPSPS либо G1, либо G2. Как ХhоI/РасI-, так и PstI/PacI-фрагменты используют для конструирования химер с двумя энхансерами, включающими промоторную делецию EPSPS G3. Когда второй энхансер вносят в виде XhoI-фрагмента, ориентацию энхансера определяют с помощью ПЦР.
Пример 6
Внесение интрона Adhl в 5’-UTR гена EPSPS риса
Внесение 1-го интрона из гена Adh1 кукурузы в состав желательного делеционного промотора EPSPS риса (например, сформированного в соответствии с описанным в примере 4) осуществляют перед формированием химерной конструкции с желательным(ми) энхансером(ами). В данном конкретном примере интрон Adh1 вносят в делеционный промотор EPSPS G2. Для специалиста будет очевидно, что аналогичная методология может быть приспособлена для внесения интрона Adh1 в другие промоторы EPSPS с делециями. Интрон Adh1 кукурузы встраивают в состав конструкций с помощью ПЦР. Интрон Adh1 амплифицируют из подходящего источника, такого как геномная ДНК кукурузы или вектор, такой как рРАС1 (фиг.8), с использованием праймеров Adh5 (SEQ ID NO. 28) и Adh3 (SEQ ID NO. 29):
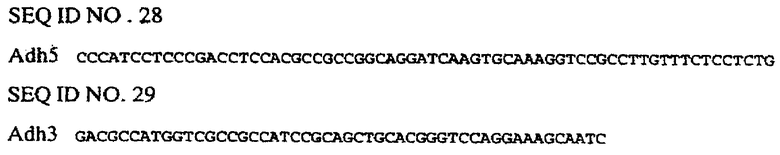
Полученный в результате ПЦР-продукт денатурируют и используют в качестве праймера в сочетании с праймером Adh5Рас (SEQ ID NO. 30) для амплификации желательного продукта с использованием вектора pTCV1001EPSPSPAC (фиг.6) в качестве матрицы.

Полученный ПЦР-продукт клонируют в состав pCR-Blunt-II (Invitrogen). PacI/HindIII-фрагмент вырезают из геномного клона риса (фиг.1) и встраивают в pTCV1001 с получением pTCVEPSPSPH (фиг.9). Далее PacI/NcoI-продукт, полученный выше с помощью ПЦР и включающий интрон Adh1, встраивают в состав pTCVEPSPS, как показано схематически (фиг.9). PacI/EcoRV-фрагмент, присутствующий в клонированном гене EPSPS, включающем двойную мутацию (фиг.10), вырезают и заменяют РасI/EсоRV-фрагментом из pTCVEPSPSPH, который включает последовательность интрона Adh1 (фиг.9). Наконец, полный ген EPSPS, который включает последовательность Adh1, вырезают из pTCVEPSPSADH (фиг.10) в виде EаgI/SасI-фрагмента и клонируют в вектор pBluescript SK+ с получением pBluSKEPSPSADH (фиг.11).
Пример 7
Внесение оптимизованной консенсусной последовательностью pre-ATG (по Козаку) с помощью направленного мутагенеза в конструкции, включающие интрон Adh1 кукурузы
Необязательно метод направленного (сайт-специфичного) мутагенеза осуществляют в отношении конструкций, включающих интрон Adh1, с использованием набора для мутагенеза QuickChange Site Directed Mutagenesis kit (Stratagene). Указанное проводят в отношении PacI/SacI-фрагмента EPSPS из pBluescript SK+ (фиг.11) перед лигированием на химеры энхансер/промотор EPSPS. Нижеследующие олигонуклеотиды используют в соответствии с протоколами изготовителя для оптимизации последовательности по Козаку.
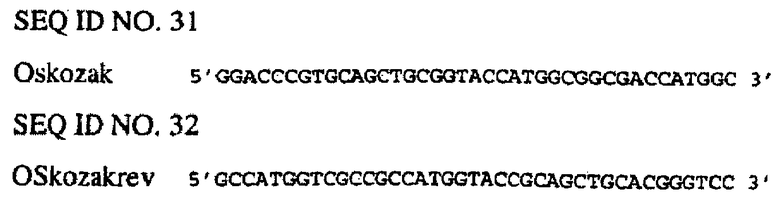
Клоны изучали с помощью рестрикционного анализа с использованием рестриктазы KpnI на материале выделенной плазмиды. Точно измененная ДНК характеризуется дополнительным рестрикционным KpnI-сайтом по сравнению с неизмененной ДНК. Затем последовательность подтверждают с помощью автоматического секвенирования ДНК. Измененная последовательность ДНК может быть перенесена в исходные конструкции с использованием уникальных рестрикционных сайтов для рестриктаз SphI или РасI на 5’-конце и AvrII или EcoRV на 3’-конце в зависимости от того, что подходит для каждого вектора.
Пример 8
Завершение экспрессионных кассет EPSPS, которые включают в направлении 5’→ 3’: энхансерный(ые) сегмент(ы), верхний участок промотора EPSPS риса, промотор EPSPS, 5’-UTR EPSPS + (необязательно) 1-й интрон Adh1 кукурузы, кодирующий участок пластидного сигнального пептида EPSPS риса, кодирующий участок зрелой EPSPS риса и терминаторный сегмент гена EPSPS риса
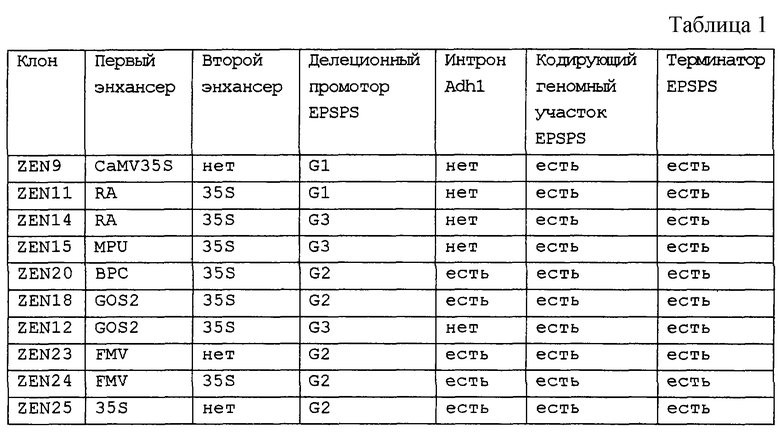
Химеры промотора EPSPS риса с одним или двумя энхансерами (примеры 4 и 5), входящие в состав векторов pCR-Blunt-II-TOPO, вырезают с использованием рестриктаз XbaI и РасI и встраивают в аналогичным образом расщепленный клон pBluescript SK+, включающий остальную часть последовательности EPSPS риса (фиг.7-11). Однако, если должна быть использована химера EPSPS с энхансером полиубихитина кукурузы, то ее сначала клонируют в pBluescript с использованием SpeI/PacI. Затем остальную часть последовательности EPSPS риса встраивают в качестве PacI/SacI-фрагмента с завершением экспрессионной кассеты. Данный заключительный этап клонирования приводит к получению требуемых генных конструкций. Примеры конструкций, получаемых с использованием указанных выше стратегий, приведены в таблице 1. Их схематические карты показаны на фиг.13-15.
Пример 9
Субклонирование экспрессионных кассет EPSPS из Bluescript в состав pIGPD9
Если желательно и особенно в случае трансформации растений с помощью методов прямого переноса ДНК (с вискерами, бомбардировка и протопласты), экспрессионные конструкции EPSPS вырезают из pBluescript с использованием рестриктазы XmaI и клонируют в pIGPD9 (фиг.12). Использование для трансформации данного вектора исключает перенос в растение генов резистентности к антибиотикам, потому что отбор осуществляется по комплементации ауксотрофного мутанта his-B E.coli геном, экспрессирующим IGPD (продукт his-B). Производные от pIGPD9 плазмиды, образованные в результате встраивания XmaI-фрагментов из pZEN9, 11, 12, 14, 15, 16, 18, 20, 23, 24 и 25, обозначают как pZEN9i, pZEN11i, pZEN12i... и т.п.
Препараты ДНК большого объема для использования в трансформации растений получают с использованием процедуры Maxi-prep (Qiagen) с использованием прописей, представляемых производителем.
Пример 10
Получение ДНК для трансформации растений
Приведенная выше процедура описывает сборку “экспрессионных кассет EPSPS”, включающих в направлении 5’→ 3’: последовательность(ти) энхансера, промотор EPSPS риса, участок кодирования сигнального пептида EPSPS риса, участок, кодирующий зрелый фермент EPSPS риса, который резистентен к глифосату за счет наличия замен Т на I и Р на S по конкретным положениям, а также терминатор гена EPSPS риса.
Необязательно желательные кассеты также дополнительно включают маркерный ген селективной резистентности к лекарственным средствам (например, резистентность к ампициллину, резистентность к канамицину и т.п.), левый или правый граничные сайты Т-ДНК и (необязательно) участок прикрепления к ядерному матриксу, присоединенный к 5’- и/или 3’-концу описанной выше конструкции. Для специалиста будет очевидно, что методы, аналогичные тем, которые были описаны выше, могут быть использованы для получения данных дополнительных компонентов и клонирования их в желательных положениях.
Пример 11
Трансформация линий кукурузы с использованием штамма Agrobacterium, несущего супербифункциональный вектор, который включает экспрессионную кассету EPSPS между правым и левым граничными сайтами Т-ДНК; отбор и регенерация растительных клеток и растений, которые резистентны к глифосату
Конструирование штамма Agrobacterium
Плазмидную ДНК Bluescript (например, ZEN9, ZEN11, ZEN14, ZEN15, ZEN18, ZEN20, ZEN12, ZEN23, ZEN24 или ZEN25) расщепляют рестриктазами XmaI или XbaI/SacI и полученный таким путем EPSPS-кодирующий фрагмент (примерно 5-6,5 т.п.н.) лигируют в положение в пределах сайта клонирования между правым и левым граничными сайтами Т-ДНК в аналогично обработанной рестриктазами pSB1. В случае использования, например, XmaI-фрагмента из ZEN18 такое лигирование создает плазмиду pZEN18SB11 (фиг.16). Конструирование плазмиды pSB11 и конструирование исходной для нее плазмиды pSB21 описано у Komari et al. (1996, Plant J., 10: 165-174). Участок Т-ДНК в pZEN18 встраивают в супербифункциональный вектор pSB1 (Saito et al., ЕР 672752 A1) по механизму гомологичной рекомбинации (фиг.16) с образованием плазмиды pSB1ZEN18. Для достижения этого плазмиду pZEN18SB11 трансформируют в штамм НВ101 E.coli, который затем в соответствии с методом тройного скрещивания Ditta et al. (1980, Proc. Natl. Acad. Sci. USA, 77: 7347-7351) скрещивают с Agrobacterium LBA4404, несущей pSB1, с образованием трансформированного штамма Agrobacterium, LBA4404 (pSB1ZEN18), в котором присутствие коинтегрированной плазмиды pSB1ZEN18 определяют на основе резистентности к спектиномицину. Также идентичность pSB1ZEN18 подтверждают на основе анализов рестрикции SalI (фиг.16). Штаммы LBA4404, несущие непосредственно аналогичные конструкции pSB1ZEN19, pSB1ZEN11, pSB1ZEN12, pSBZEN14 и т.д., аналогично конструируют, начиная с XmaI-фрагментов pZEN9, ZEN11, ZEN12, ZEN14 и т.д.
Альтернативно, используя методы, аналогично описанным выше, аналогичный фрагмент из pZEN9, ZEN11 и т.д. вносят по механизму гомологичной рекомбинации в положение между правым и левым граничными сайтами в супербифункциональном векторе рTOК162 (фиг.1 в US 5591616) с образованием аналогичного ряда коинтегрированных плазмид, отобранных в Agrobacterium по параметрам одновременной резистентности к канамицину и спектиномицину.
Штамм LBA4404 Agrobacterium, который несет хелперную плазмиду PAL4404 (включающую полный сегмент vir), доступен из Американской коллекции типовых культур (АТСС 37349). Альтернативным применимым штаммом Agrobacterium является штамм ЕНА101 (Hood et al., 1986, J. Bacteriol., 168(3), 1283-1290), который несет хелперную плазмиду, включающую сегмент vir от штамма Agrobacterium tumefaciens A281, характеризующегося высоким уровнем вирулентности.
Получение суспензий Agrobacterium
Каждый из штаммов Agrobacterium LBA4404 (pSB1ZEN9), LBA4404 (pSB1ZEN11) и т.д. высевают полосами на чашки, содержащие плотную культуральную среду PHI-L, и культивируют при 28°С в темноте в течение 3-10 дней.
Среда PHI-L соответствует описанному (пример 4) WO 98/32326. Среда PHI-L, приготовленная в дважды дистиллированной воде, содержит 25 мл/л маточного раствора А, 25 мл/л маточного раствора В, 450,9 мл/л маточного раствора С и 50 мг/л спектиномицина. Маточные растворы стерилизуют автоклавированием или фильтрованием. Маточный раствор А составлен 60 г/л К2НРO4 и 20 г/л NaH2PO4 с доведением до рН 7,0 с помощью КОН; маточный раствор В составлен 6 г/л MgSO4·7H2O, 3 г/л КСl, 20 г/л NH4Cl, 0,2 г/л CaCl2 и 50 мг/л FeSO4·7H2O; маточный раствор С составлен 5,56 г/л глюкозы и 16,67 г/л агара (А-7049: Sigma Chemicals, St.Louis, Mo, США).
Альтернативно, Agrobacterium культивируют в течение 3-10 дней на чашке, содержащей культуральную среду YP (5 г/л дрожжевого экстракта, 10 г/л пептона, 5 г/л NaCl, 15 г/л агара при рН 6,8), как описано Ishida et al. (1996, Nature Biotechnology, 14, 745-750) или, альтернативно, как описано Hei et al. в US 5591616 (культуральная среда АВ (Drlica & Kado, 1974; Proc. Natl. Acad. Sci. USA, 71: 3677-3681)), однако в каждом случае включая модификации для обеспечения соответствующего отбора с антибиотиками (например, включением 50 мг/мл спектиномицина в случае штамма Agrobacterium LBA4404 (pSB1ZEN9) и т.д. или включением 50 мг/мл спектиномицина и 50 мг/мл канамицина в случае, когда используют Agrobacterium, несущую происходящий от рТОК162 супербифункциональный вектор).
Полученные, как описано выше, чашки с Agrobacterium хранят при 4°С и используют в течение месяца с момента получения. Для получения суспензий отдельную колонию с основной чашки пересевают полосами на чашку, содержащую при рН 6,8 5 г/л дрожжевого экстракта (Difco), 10 г/л пептона (Difco), 5 г/л NaCl, 15 г/л агара (Difco) и 50 мг/л спектиномицина (или то, что соответствует конкретному штамму Agrobacterium). Чашки инкубируют при 28°С в темноте в течение 2 дней.
Суспензии Agrobacterium для трансформации растительного материала получают по аналогичной методике, как описано в US 5591616. (Используя соответствующую микробиологическую практику, чтобы избежать загрязнения стерильных культур). По три 5-мм петли Agrobacterium снимают с чашек, переносят и суспендируют в 5 мл стерильной жидкой среды АА в 14-мл пробирке Фалькона. Как используется в данном исследовании, жидкая культуральная среда ДА при рН 5,2 содержит основные минеральные соли, аминокислоты и витамины, определенные у Toriyama & Hinata, 1985, Plant Science, 41, 179-183, минорные минеральные соли культуральной среды по Мурасиге-Скоогу (Murashige & Skoog, 1962, Physiol. Plant, 15, 473-497), 0,5 г/л казаминовой кислоты (гидролизат казеина), 1 мг/л 2,4-дихлорфеноксиуксусной кислоты (2,4-D), 0,2 мг/л кинетина, 0,1 мг/л гиббереллина, 0,2 М глюкозы, 0,2 М сахарозы и 0,1 мМ ацетосирингона.
Альтернативно, суспензии Agrobacterium для трансформации растительного материала получают по аналогичной методике, как описано в WO 98/32326. По три 5-мм петли Agrobacterium снимают с чашек, переносят и суспендируют в 5 мл стерильной основной среды PHI-A, описанной в примере 4 WO 98/32326/ или, альтернативно, суспендируют в 5 мл комбинированной среды PHI-I, также описанной в примере 4 WO 98/32326. В любом случае также добавляют 5 мл 100 мМ 3’-5’-диметокси-4’-гидроксиацетофенона. Основная среда PHI-A при рН 5,2 содержит 4 г/л основных солей CHU (N6) (Sigma C-1416), 1,0 мл/л витаминной смеси Эрикссона (1000Х, Sigma E-1511), 0,5 мг/л гидрохлорида тиамина, 1,5 мг/мл 2,4-D, 0,69 г/л L-пролина, 68,5 г/л сахарозы и 68,5 г/л глюкозы. Комбинированная среда PHI-I, также доведенная до рН 5,2 с помощью КОН и стерилизованная фильтрованием, содержит 4,3 г/л солей MS (Gibco BRL), 0,5 мг/мл никотиновой кислоты, 0,5 мг/мл гидрохлорида пиридоксина, 1,0 мг/мл гидрохлорида тиамина, 100 мг/л миоинозитола, 1 г/л витаминного гидролизата казеина (Difco), 1,5 мг/мл 2,4-D, 0,69 г/л L-пролина, 68,5 г/л сахарозы и 36 г/л глюкозы.
Альтернативно, суспензии Agrobacterium для трансформации растительного материала получают по аналогичной методике, как описано Ishida et al (1996) Nature Biotechnology, 14, 745-750. По три 5-мм петли Agrobacterium снимают с чашек, переносят и суспендируют в 5 мл культуральной среды LS-inf. Среда LS-inf (Linsmaier & Skoog, 1965, Physiol. Plant, 18, 100-127), доведенная до рН 5,2 с помощью КОН, содержит основные и минорные минеральные соли LS, 0,5 мг/мл никотиновой кислоты, 0,5 мг/мл гидрохлорида пиридоксина, 1,0 мг/мл гидрохлорида тиамина, 100 мг/л миоинозитола, 1 г/л витаминного гидролизата казеина (Difco), 1,5 мг/мл 2,4-D, 68,5 г/л сахарозы и 36 г/л глюкозы.
После получения суспензию Agrobacterium перемешивают на вортексе с получением однородной суспензии и клеточную популяцию корректируют в диапазоне от 0,5× 109 до 2× 109 БОЕ/мл (предпочтительно ближе к нижнему значению). 1× 109 БОЕ/мл соответствует OD (1 см) примерно 0,72 при 550 нм.
Суспензии Agrobacterium разделяют на аликвоты по 1 мл в стерильные 2-мл микроцентрифужные пробирки и используют максимально быстро.
Линии кукурузы для трансформации
Подходящими для трансформации линиями кукурузы являются, но не ограничиваются ими, А188, F1 Р3732, F1 (А188 × B73Ht), F1 (B73Ht × А 188), F1 (А188 × BMS). Сорта А188, BMS (черная мексиканская кукуруза) и B73Ht получают из министерства сельского, лесного и рыбного хозяйства, которое хорошо известно специалистам. Р3732 получают от IWATA RAKUNOU KYODOKUMIAI. Подходящие линии кукурузы также включают различные кроссы А188 с инбредными линиями (например, PHJ90 × А188, PHN46 × А188, РНРР8 × А188 в таблице 8 WO 98/32326), а также элитные инбредные линии из различных гетерозисных групп (например, PHN46, РНР28 и PHJ90 в таблице 9 WO 98/32326).
Например, недоразвитые зародыши получают от кукурузы "HiII". "Hi-II" является гибридом между инбредными линиями (А188 × В73), образуемым путем реципрокных скрещиваний между родителем A Hi-II и родителем В Hi-II, доступными из Кооперативного центра по генетике кукурузы в университете Иллинойса в Шампейне (Urbana, IL). Семена, обозначенные как семена "Hi-II”, полученные в этих кроссах, высевают в теплице или в открытом грунте. Полученные в результате растения Hi-II воспроизводят с помощью самоопыления или перекрестного опыления с родственными растениями.
Получение недоразвитых зародышей, инфицирование и совместное культивирование
Трансформацию недоразвитых зародышей кукурузы осуществляют путем контактирования недоразвитых зародышей с подходящими рекомбинантными штаммами Agrobacterium, описанными выше. Недоразвитым зародышем является зародыш незрелого семени, которое находится на стадии созревания после опыления. Недоразвитые зародыши представляют собой интактную ткань, которая способна к клеточным делениям, приводящим к образованию каллюсных клеток, которые могут затем дифференцироваться с образованием тканей и органов цельного растения. Предпочтительным материалом для трансформации также является зародышевый щиток, который тоже способен индуцировать способные к дифференцировке каллюсы, обладающие способностью регенерировать нормальные фертильные растения, которые оказываются исходно трансформированными. Следовательно, предпочтительным материалом для трансформации также является каллюс, производный от такого дедифференцированного недоразвитого зиготического зародыша или щитка.
Недоразвитые зародыши кукурузы выделяют в стерильных условиях из развивающихся початков, как описано Green and Phillips (1976, Crop Sci., 15: 417-421) или, альтернативно, с помощью методов Neuffer et al. (1982, "Growing Maize for genetic purposes", in Maize for biological research, W.F. Sheridan ed., University Press, University of North Dakota, Grand Forks, North Dakota, США). Например, недоразвитые зародыши кукурузы длиной 1-2 мм (предпочтительно 1-1,2 мм) в стерильных условиях выделяют из женских соцветий через 9-12 (предпочтительно 11) дней после опыления с использованием стерильной лопатки. Обычно колосья стерилизуют по поверхности 2,63%-ным гипохлоритом натрия в течение 20 мин с последующей промывкой стерильной деионизованной водой и асептическим выделением недоразвитых зародышей. Недоразвитые зародыши (предпочтительно примерно по 100 штук) вносят непосредственно в 2-мл микроцентрифужную пробирку, содержащую примерно 2 мл той же самой культуральной среды, которую использовали для получения суспензии Agrobacterium (альтернативы которой были описаны выше). Пробирку закрывают пробкой и содержимое перемешивают на вортексе в течение нескольких секунд. Культуральную среду декантируют, добавляют 2 мл свежей среды и перемешивание на вортексе повторяют. После этого всю среду выливают, оставляя недоразвитые зародыши на дне пробирки.
После получения недоразвитых зародышей кукурузы следующей стадией процесса, стадией инфицирования, является контакт зародышей с трансформирующим штаммом Agrobacterium.
В одном примере указанного процесса стадию инфицирования осуществляют в жидкой культуральной среде, которая содержит основные минеральные соли и витамины среды N6 (1987, Chu С.С.Proc. Symp. Plant Tissue Culture, Science Press Peking, pp.43-50), как описано в примере 4 WO 98/32326. 1,0 мл суспензии Agrobacterium, полученной как описано выше в среде PHI-A, добавляют к зародышам в микроцентрифужной пробирке и перемешивают на вортексе в течение 30 с. Альтернативно, 1,0 мл суспензии Agrobacterium, полученной как описано выше, добавляют либо в среду PHI-I, либо в среду LS-inf.
После отстаивания в течение 5 мин суспензию Agrobacterium и зародыши переливают в чашку Петри, содержащую либо (1) среду PHI-B, либо (2) среду PHI-J, либо (3) среду LS-AS: это зависит от того, в какой среде была изначально получена суспензия Agrobacterium: в среде PHI-A, в среде PHI-I или в среде LS-inf, соответственно. Суспензию Agrobacterium отсасывают пастеркой, зародышами манипулируют таким образом, чтобы их осевая сторона оказывалась обращенной вниз к культуральной среде, чашки закрывают парафильмом и инкубируют в темноте при 23-25°С в течение 3 дней совместного культивирования. Среда PHI-B при pH 5,8 содержит 4 г/л основных солей CHU (N6) (Sigma C-1416), 1,0 мл/л витаминной смеси Эрикссона (1000Х, Sigma E-1511), 0,5 мг/л гидрохлорида тиамина, 1,5 мг/мл 2,4-D, 0,69 г/л L-пролина, 0,85 мг/л нитрата серебра, 30 г/л сахарозы, 100 мкМ ацетосирингона и 3 г/л гельрита (Sigma). Среда PHI-J, также доведенная до рН 5,8, содержит 4,3 г/л солей MS (Gibco BRL), 0,5 мг/мл никотиновой кислоты, 0,5 мг/мл гидрохлорида пиридоксина, 1,0 мг/мл гидрохлорида тиамина, 100 мг/л миоинозитола, 1,5 мг/мл 2,4-D, 0,69 г/л L-пролина, 20 г/л сахарозы, 10 г/л глюкозы, 0,5 г/л MES (Sigma), 100 мM ацетосирингона и 8 г/л очищенного агара (Sigma А-7049). Среда LS-AS (Linsmaier & Skoog, 1965, Physiol. Plant, 18, 100-127), доведенная до рН 5,8 с помощью КОН, содержит основные и минорные минеральные соли LS, 0,5 мг/мл никотиновой кислоты, 0,5 мг/мл гидрохлорида пиридоксина, 1,0 мг/мл гидрохлорида тиамина, 700 мг/л L-пролина, 100 мг/л миоинозитола, 1,5 мг/мл 2,4-D, 20 г/л сахарозы, 10 г/л глюкозы, 0,5 г/л MES, 100 мМ ацетосирингона и 8 г/л очищенного агара (Sigma А-7049).
После получения недоразвитых зародышей, как описано выше, альтернативным методом трансформации является инфицирование их в процессе и после периода дедифференцировки, как описано в US 5591616. Недоразвитые зародыши помещают на плотную культуральную среду LSD-1.5, содержащую минеральные соли и витамины LS наряду с 100 мг/мл гидролизата казеина, 700 мг/л L-пролина, 100 мг/л миоинозитола, 1,5 мг/мл 2,4-D, 20 г/л сахарозы и 2,3 г/л гельрита. Через 3 недели при 25°С образовавшиеся от щитков каллюсы собирают в 2-мл микроцентрифужную пробирку и погружают в 1 мл суспензии Agrobacterium, полученной, как описано выше, в среде АА. После 5-минутного отстаивания полученные в результате каллюсы переносят на плотную среду 2N6, содержащую 100 мМ ацетосирингона, и инкубируют в темноте при 25°С в течение 3 дней периода совместного культивирования. Плотная культуральная среда 2N6 содержит минеральные соли и витамины среды N6 (Chu С.С., 1978; Proc. Symp. Plant Tissue Culture, Science Press Peking, pp.43-50), содержащей 1 г/л гидролизата казеина, 2 мг/л 2,4-D, 30 г/л сахарозы и 2 г/л гельрита.
Хранение и отбор трансформантов
После совместного культивирования зародыши, необязательно, переносят на чашку, содержащую среду PHI-C, закрывают сверху парафильмом и инкубируют в темноте в течение 3 дней в качестве “стадии хранения” перед проведением отбора. Среда PHI-C при рН 5,8 содержит 4 г/л основных солей CHU(N6) (Sigma C-1416), 1,0 мл/л витаминной смеси Эрикссона (1000Х, Sigma E-1511), 0,5 мг/л гидрохлорида тиамина, 1,5 мг/мл 2,4-D, 0,69 г/л L-пролина, 0,85 мг/л нитрата серебра, 30 г/л сахарозы, 0,5 г/л MES, 100 мг/л карбенициллина и 8 г/л очищенного агара (Sigma A-7049). Как описано в WO 98/32326, желательность включения указанной стадии хранения в общий процесс получения трансформантов зависит от линии кукурузы и является объектом экспериментальной оценки.
Для стадии отбора примерно 20 зародышей переносят на каждую из ряда новых чашек, содержащих селективную среду PHI-D или селективную среду LSD-1,5, закрывают парафильмом и инкубируют в темноте при 28°С. Селективная среда PHI-D, доведенная до рН 5,8 с помощью КОН, содержит 4 г/л основных солей CHU(N6) (Sigma С-1416), 1,0 мл/л витаминной смеси Эрикссона (1000Х, Sigma Е-1511), 0,5 мг/л гидрохлорида тиамина, 1,5 мг/мл 2,4-D, 0,69 г/л L-пролина, 0,85 мг/л нитрата серебра, 30 г/л сахарозы, 0,5 г/л MES, 100 мг/л карбенициллина, 8 г/л очищенного агара (Sigma A-7049) и от 0,1 мМ до 20 мМ N-(фосфонометил) глицина чистоты для тканевых культур (Sigma Р-9556). Селективная среда LSD-1.5, доведенная до рН 5,8 с помощью КОН, содержит основные и минорные минеральные соли LS (Linsmaier & Skoog, 1965, Physiol. Plant, 18, 100-127), 0,5 мг/мл никотиновой кислоты, 0,5 мг/мл гидрохлорида пиридоксина, 1,0 мг/мл гидрохлорида тиамина, 700 мг/л L-пролина, 100 мг/л миоинозитола, 1,5 мг/мл 2,4-D, 20 г/л сахарозы, 0,5 г/л MES, 250 мг/л цефотаксима, 8 г/л очищенного агара (Sigma A-7049) и от 0,1 мМ до 20 мМ N-(фосфонометил)глицина чистоты для тканевых культур (Sigma Р-9556).
Альтернативно, в случае, когда исходным материалом для отбора являются каллюсы, происходящие от недоразвитых зародышей, как описано в US 5591616, такие каллюсы промывают стерилизованной водой, содержащей 250 мг/л цефотаксима, с последующим культивированием в селективной среде LSD-1,5.
Зародыши или клеточные кластеры, которые пролиферируют из недоразвитых зародышей, переносят (если необходимо, то с использованием стерильного скальпеля) на чашки, содержащие свежую селективную среду, с 2-недельными интервалами при общей продолжительности эксперимента примерно 2 месяца. Резистентные к гербициду каллюсы затем наращивают в объеме путем продолжающегося роста в той же культуральной среде до достижения диаметра отобранного каллюса свыше примерно 1,5 см.
Концентрацию N-(фосфонометил)глицина в селективной среде выбирают таким образом, чтобы отбирать желательное число истинных трансформантов, что предпочтительно составляет 0,3-5 мМ. Предпочтительно концентрация N-(фосфонометил)глицина, используемая в селективной среде, составляет примерно 1 мМ в первые две недели отбора и примерно 3 мМ после этого.
Регенерация трансформантов, размножение и анализ трансформированного растительного материала
Отобранные каллюсы регенерируют в нормальные фертильные растения в соответствии, например, с методами, описанными Duncan et al. (1985, Planta, 165, 322-332), Kamo et al. (1985, Bot. Gaz., 146(3), 327-334) и/или West et al. (1993, The Plant Cell, 5, 1361-1369), и/или Shillito et al. (1989, Bio/Technol, 7, 581-587).
Например, отобранные каллюсы диаметром 1,5-2 см переносят в среду для регенерации/созревания и инкубируют в темноте в течение примерно 1-3 недель, давая возможность созреть соматическим зародышам. Подходящую регенерационную среду, PHI-Е (WO 98/32326) доводят до рН 5,6 с помощью КОН, и она содержит 4,3 г/л солей MS (Gibco BRL), 0,5 мг/мл никотиновой кислоты, 0,5 мг/мл гидрохлорида пиридоксина, 0,1 мг/мл гидрохлорида тиамина, 100 мг/л миоинозитола, 2 мг/л глицина, 0,5 мг/л зеатина, 1,0 мг/мл индолилуксусной кислоты, 0,1 мМ абсцизовой кислоты, 100 мг/л карбенициллина, 60 г/л сахарозы, 8 г/л очищенного агара (Sigma А-7049) и, необязательно, от 0,02 мМ до 1 мМ N-(фосфонометил) глицина чистоты для тканевых культур (Sigma Р-9556).
Затем каллюсы переносят в регенерационную среду для образования корешков и выращивают при 25°С либо в условиях смены освещения (16 ч, 270 мЕ/м2/с) и темноты (8 ч), либо при постоянном освещении (примерно 250 мЕ/м2/с) до того момента, как образуются побеги и корешки. Подходящей регенерационной средой для образования корешков является либо среда LSZ, описанная в следующем абзаце (необязательно лишенная фосфонометилглицина), либо среда PHI-F при рН 5,6, которая содержит 4,3 г/л солей MS (Gibco BRL), 0,5 мг/мл никотиновой кислоты, 0,5 мг/мл гидрохлорида пиридоксина, 0,1 мг/мл гидрохлорида тиамина, 100 мг/л миоинозитола, 2 мг/л глицина, 40 г/л сахарозы и 1,5 г/л гельрита.
Альтернативно, отобранные каллюсы переносят непосредственно в регенерационную среду LSZ, доведенную до рН 5,8 с помощью КОН и содержащую основные и минорные минеральные соли LS (Linsmaier & Skoog, 1965, Physiol. Plant, 18, 100-127), 0,5 мг/мл никотиновой кислоты, 0,5 мг/мл гидрохлорида пиридоксина, 1,0 мг/мл гидрохлорида тиамина, 700 мг/л L-пролина, 100 мг/л миоинозитола, 5 мг/мл зеатина, 20 г/л сахарозы, 0,5 г/л MES, 250 мг/л цефотаксима, 8 г/л очищенного агара (Sigma A-7049) и, необязательно, от 0,02 мМ до 1 мМ N-(фосфонометил)глицина чистоты для тканевых культур (Sigma Р-9556). По прошествии периода инкубации в темноте культуру освещают (непрерывно или по типу “светового дня” в соответствии с указанным выше), в результате чего регенерируются проростки.
Небольшие проростки переносят в отдельные стеклянные пробирки, содержащие либо среду PHI-F, либо полуконцентрированную среду LSF при рН 5,8, содержащую основные соли LS (Linsmaier & Skoog, 1965, Physiol, Plant, 18, 100-127) в половинных концентрациях, минорные соли LS, 0,5 мг/мл никотиновой кислоты, 0,5 мг/мл гидрохлорида пиридоксина, 1,0 мг/мл гидрохлорида тиамина, 100 мг/л миоинозитола, 20 г/л сахарозы, 0,5 г/л MES, 8 г/л очищенного агара (Sigma A-7049), и выращивают в течение примерно еще недели. Затем проростки переносят в горшки с почвой, выставляют их в камеры для выращивания (относительная влажность 85%, 600 м.д. СО2 и освещение 250 мЕ/м2/с) и выращивают до созревания в почвенной смеси в условиях теплицы.
Первое поколение растений (Т0), полученное, как описано выше, самоопыляют с целью получения семян второго поколения (T1). Альтернативно (причем предпочтительно), первое поколение растений подвергают реципрокным скрещиваниям с другой нетрансгенной инбредной линией кукурузы с целью получения семян второго поколения. Потомство таких кроссов (T1) затем, как ожидается, расщепляется в соотношении 1:1 по признаку резистентности к гербициду. Семена T1 замачивают, выращивают в теплице или в открытом грунте, и уровень резистентности, наследование признака резистентности и расщепление по признаку резистентности к глифосатному гербициду в текущем и последующих поколениях оценивают по наблюдениям за параметрами выживаемости, фертильности растений и симптомов некроза тканей после обработки глифосатом (приготовленным подходящим образом и необязательно в виде соли) в диапазоне уровней от 25 до 2000 г/га и в диапазоне стадий роста между V2 и V8 включительно (или, альтернативно, на 7-21-й дни после проращивания). Данные оценки соотносят с параметрами восприимчивых сегрегантов и со сходными не трансформированными линиями кукурузы, которые не несут генов по настоящему изобретению или последовательностей, способных обеспечивать резистентность к глифосату. Трансгенные линии, которые проявляют резистентность к глифосату, отбирают и снова самоопыляют или бэккроссируют на нетрансгенные инбредные растения.
На всех стадиях описанных выше процессов образцы тканей трансформированных каллюсов, проростков, растительного материала Т0 и T1 необязательно берут и анализируют с помощью (1) Саузерн-блоттинга и ПЦР с целью определения присутствия, числа копий и целостности трансгенов, (2) Нозерн-блоттинга (или сходных методов) с целью измерения уровня экспрессии мРНК трансгенов, (3) количественного Вестерн-блоттинга ДСН-гелей с целью измерения уровней экспрессии EPSPS, и (4) измерения каталитической активности EPSPS в присутствии и в отсутствие глифосата с целью более точной оценки того, какая часть экспрессированной EPSPS происходит от трансгена.
Такие методы анализа хорошо известны в данной области техники. Подходящие методы для анализа присутствия, целостности и экспрессии трансгена с помощью ПЦР, для осуществления Саузерн-блоттинга, для клонирования и экспрессии зрелой EPSPS риса в клетках Е. coli, для очистки EPSPS риса, для формирования поликлональных антител к очищенной EPSPS риса, для Вестерн-блоттинга уровней EPSPS в каллюсе и в тканях растения и для измерения уровней активности EPSPS в производных от растений экстрактах при той концентрации глифосата, которая разграничивает эндогенную глифосат-восприимчивую EPSPS и резистентный к глифосату продукт, кодируемый трансгеном EPSPS, описаны более подробно далее в примерах 17-20.
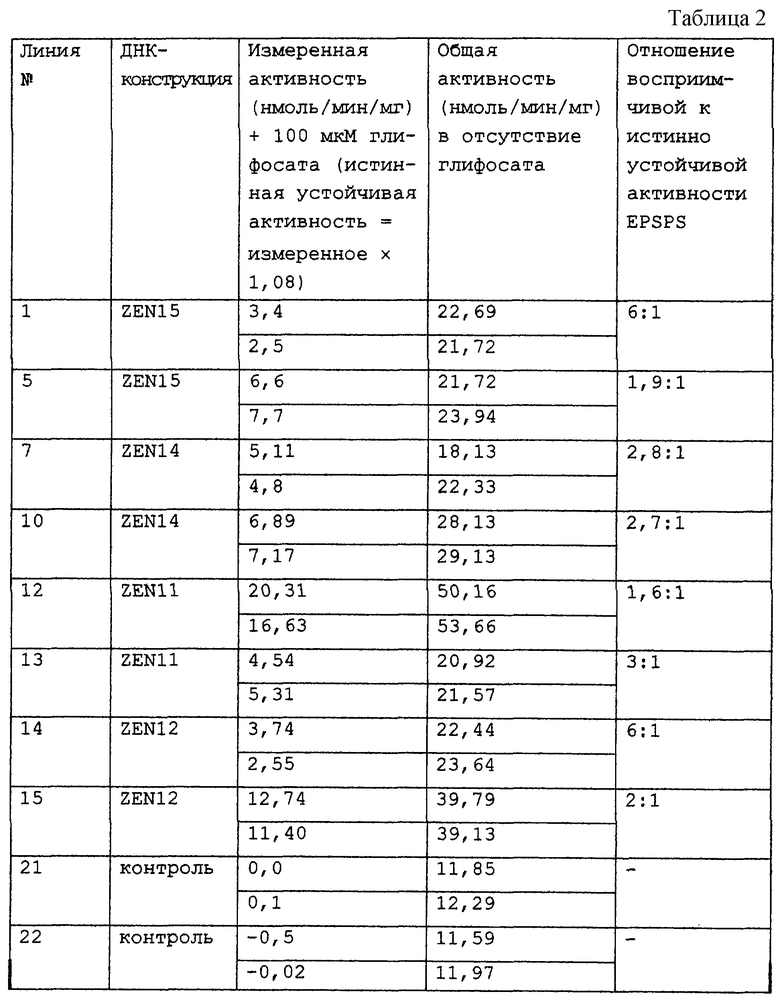
Экспрессия трансгена в каллюсах, образованных в результате трансформации недоразвитых зародышей кукурузы А188 × В73 (Hi-II) штаммом LBA4404 Agrobacterium, несущим супербифункциональный вектор, включающий экспрессионные кассеты EPSPS векторов Bluescript (pZEN11, 12, 14 или 15) в пределах граничных сайтов Т-ДНК показано в табл.2.
Таблица 2 показывает результаты ферментного теста для EPSPS (присутствие/отсутствие 100 мкМ глифосата при 100 мкМ PEP), полученные при ферментном тестировании экстрактов стабильно трансформированного каллюса способного к регенерации гибрида кукурузы А188 × В73, трансформированного с использованием Agrobacterium, несущей супербифункциональный вектор, включающий экспрессионные кассеты EPSPS плазмид pZEN11, ZEN12, ZEN14 или ZEN15 Bluescript. Каждая каллюсная линия представляет собой единственное событие, которое анализируют в двух повторностях. Подсчитывают отношение истинно устойчивой активности фермента, экспрессируемого трансгеном (допускается примерно 8% ингибирования), к активности эндогенного восприимчивого фермента (более 98% подавления в присутствии глифосата). Мутантный EPSPS экспрессируется относительно сильно в ряде линий, а именно в линиях 5 и 12, в которых, допуская снижение Vmax устойчивого фермента по отношению к дикому типу (примерно на треть), можно оценить, что устойчивый фермент экспрессируется в 2-6 раз выше нормального уровня эндогенной EPSPS (такой подсчет осложняется тем фактом, что в некоторых отобранных каллюсах экспрессия трансгена имеет место наряду с примерно 2-2,5-кратным усилением фоновой экспрессии восприимчивого эндогенного фермента).
Пример 12
Трансформация линий кукурузы с помощью бомбардировки частицами, покрытыми ДНК, которая включает экспрессионную кассету EPSPS; отбор и регенерация растительных клеток и растений, которые резистентны к глифосату
В следующем примере рыхлый эмбриогенный каллюс, производный от недоразвитых зародышей кукурузы, инициируют на плотной культуральной среде и трансформируют биолистическим путем. Затем, по аналогичной методике, как описано в примере 11, трансформированный каллюс отбирают на основании дифференцированного роста в среде, содержащей разные концентрации глифосата. Резистентный каллюс отбирают и регенерируют с получением проростков То, которые высаживают в горшки, выращивают до зрелости и самоопыляют или перекрестно опыляют в теплице. Семена-потомки (T1) затем выращивают для получения последующих поколений растений, которые оценивают по резистентности к глифосату и анализируют на присутствие, целостность и экспрессию трансгена в соответствии с описанным в примере 11.
Инициация каллюса из недоразвитых зародышей
Ломкий эмбриогенный каллюс II типа, пригодный для трансформации, извлекают из недоразвитых зародышей, например, кросса А188хВ73 кукурузы. Альтернативную инбредную линию, такую как производные от В73 и гибридные линии кукурузы, также можно использовать, включая, например, те, которые перечислены в примере 11. Недоразвитые зародыши кукурузы длиной 1-2 мм выделяют в стерильных условиях из женских соцветий обычно примерно на 11-й день после опыления с использованием методов, описанных в примере 11.
Недоразвитые зародыши высевают, например, в среду на основе N6 (Chu et al., 1975, Scientia Sinica, 18, 659-668), доведенную до рН 5,8 с помощью КОН и содержащую 1 мг/л 2,4-D, 2,9 г/л L-пролина, 2 мг/л L-глицина, 100 мг/л гидролизата казеина, основные соли N6, минорные соли N6, витамины N6, 2,5 г/л гельрита (или 2 г/л препарата Gelgro) и 20 г/л сахарозы. Альтернативно, культуральная среда включает, например, аналогичную среду, но содержащую соли MS (Murashige & Skoog, 1962, Physiol. Plant, 15, 473-497) вместо солей N6. Альтернативно, культуральная среда может содержать примерно 10 мг/л дикамбы вместо 2,4-D.
Недоразвитые зародыши инкубируют в темноте в указанной выше среде при примерно 25°С с целью инициации каллюса. Материал каллюса II типа отбирают путем визуального отбора быстрорастущих рыхлых эмбриогенных клеток с помощью методов, известных в данной области техники и описанных в WO 98/44140. Например, подходящие реципиентные клетки отбирают “вручную” путем выбора предпочтительных клеток, которые могут находиться на поверхности клеточного кластера, и далее идентифицируются по отсутствию у них дифференцировки, небольшому размеру и высокому соотношению объемов ядра и цитоплазмы. Суспензионную культуру инициируют из ткани в пределах каллюса, которая выглядит наименее дифференцированной, наиболее мягкой и наиболее рыхлой. Ткань, характеризующуюся такой морфологией, переносят на новые чашки со средой примерно через 8-16 дней после исходного высева недоразвитых зародышей. Затем ткань стандартным образом субкультивируют каждые 14-21 дней путем изъятия примерно 10% кусочков, которые достигают примерно грамма. На каждом этапе в субкультуру включают только тот материал, который удовлетворяет морфологии желательного типа II или типа III.
Получение суспензионных клеточных культур
Предпочтительно в течение 6 месяцев после описанной выше инициации каллюсов диспергированные суспензионные культуры инициируют в жидких культуральных средах, содержащих подходящие гормоны, такие как 2,4-D и NAA, необязательно представленные в форме капсулы с замедленным высвобождением гормона в соответствии с описанным в примерах 1 и 2 US 5550318. Необязательно, уровни гормонов в культурах поддерживают путем внесения свежих гормональных добавок по необходимости. Суспензионные культуры инициируют, например, добавлением приблизительно 0,5 г каллюсной ткани в 100-мл колбу, содержащую 10 мл среды для суспензионной культуры. Каждые 7 дней культуру дополнительно субкультивируют путем переноса с использованием стерильной пипетки с широким концом, 1 мл осевших клеток и 4 мл кондиционной среды в новую колбу, содержащую свежую среду. Крупные клеточные агрегации, которые не могут пройти через кончик пипетки, отбрасывают на каждом этапе субкультивирования. Необязательно, суспензионные культуры пропускают через подходящее сито (например, с ячейкой примерно 0,5-1,0 мм) на каждом этапе субкультивирования. Через 6-12 недель культура становится диспергированной. Подходящие культуральные среды для клеточных суспензий включают, например, среду, доведенную до рН 6,0 и содержащую основные и минорные соли Мурасиге-Скоога (Murashige & Skoog, 1962) (необязательно модифицированные так, чтобы содержать сниженный уровень нитрата аммония, 1,55 г/л), 30 г/л сахарозы, 0,25 мг/л тиамина, 10 мг/л дикамбы, 25 мМ L-пролина, 200 мг/л гидролизата казеина, 100 мг/л миоинозитола, 500 мг/л сульфата калия и 400 мг/л однозамещенного фосфата калия. Альтернативно, вместо дикамбы среда для клеточной суспензии содержит 2,4-D и/или NAA.
Криоконсервация суспензионных клеточных культур
Необязательно, суспензионные культуры, полученные в соответствии с описанным выше, криоконсервируют с использованием криоконсервантов и способов, описанных, например, в примере 2 US 5550318. Криоконсервация заключается в добавлении криоконсерванта при температуре льда к заранее охлажденным также до температуры льда клеткам постепенно в течение одного-двух часов. Полученную смесь выдерживают при температуре льда, в которой конечный объем криоконсерванта равен объему клеточной суспензии. Конечные концентрации криоконсервантов, например, составляют 10% диметилсульфоксида, 10% полиэтиленгликоля (молекулярная масса 6000), 0,23 М L-пролина и 0,23 М глюкозы. По прошествии 30-минутного периода уравновешивания при температуре льда смесь разделяют на аликвоты примерно по 0,5 мл, переносят в 2-мл микроцентрифужные пробирки и медленно охлаждают до температуры -8°С со скоростью 0,5°С/мин. После периода ледяной нуклеации образец далее медленно охлаждают до -35°С и затем погружают в жидкий азот. При необходимости использования замороженные образцы оттаивают сначала путем погружения их в своих контейнерах в воду при примерно 40°С на 2 мин и затем оставляют их медленно размораживаться полностью. Смесь клеток и криоконсервантов затем пипеткой наносят на фильтр, положенный сверху слоя фидер-клеток BMS при 25°С. После того, как размороженная ткань начинает расти, ее переносят обратно на свежую плотную культуральную среду, и после адаптации (в течение 1-2 недель) далее переносят в среду для суспензионной клеточной культуры. После повторного установления роста жидкой суспензионной культуры клетки используют для трансформации.
Трансформация с помощью частиц
ДНК, производную от плазмиды pIGPD9 (фиг.12), включающую XmaI-экспрессионные кассеты EPSPS (а именно pZEN9i, ZEN11i, ZEN12i, ZEN14i и т.д.), очищают, концентрируют (например, с помощью анионообменной хроматографии или денситометрического выделения плазмидной ДНК в градиенте хлорида цезия из клеток подходящего штамма-хозяина HisB-, RecA- E.coli (например, DH5α : hisB-) после роста до стационарной фазы культуры в минимальной среде 5хА (К2НРО4 52,5 г, KH2PO4 22,5 г, (NH4)2SO4 5 г и цитрат натрия· 2Н2O 2,5 г на литр) и представляют в виде концентрированного раствора (предпочтительно примерно 1 мг/мл) в стерильной воде. ДНК получают в виде кольцевой плазмидной ДНК или, альтернативно, в виде ДНК, расщепленной рестриктазой ХmаI с получением линейной экспрессионной кассеты, включающей EPSPS-фрагмент, и используют ее после очистки методом электрофореза в агарозном геле и электроэлюции.
Подходящим устройством для бомбардировки является, например, гелиевое ружье Biorad PDS1000. Чашку помещают на 5-6 см ниже блокирующего экрана, используемого для остановки каптонных макрочастиц. ДНК-конструкцию осаждают на частицы из вольфрама или золота, имеющие диаметр примерно 1,0 мкм, по аналогичной методике, как описано Klein et al., 1987, Nature, 327, 70-73. Например, 1,25-мг вольфрамовых или золотых частиц (предварительно промытых этанолом при 65°С в течение 12 ч) смешивают последовательно в следующем порядке: с примерно 20-30 мг ДНК, 1,1 М CaCl2 и 8,7 мМ спермидина до конечного объема примерно 0,6 мл. Смесь перемешивают в течение 10 мин при 0-4°С, подвергают низкоскоростному центрифугированию (примерно 500g) в течение 5 мин и концентрат супернатанта декантируют таким образом, чтобы оставить вольфрамовые частицы, суспендированные в конечном объеме примерно 30 мл. Аликвоты по 1-10 мкл пипеткой наносят на макрочастицы ружья.
Суспензионные культуры, производные от каллюса II-го типа и/или III-го типа, поддерживают в культуре в течение 3-5 месяцев (или, альтернативно, выделяют после криоконсервации), заново субкультивируют и затем просеивают через сито из нержавеющей стали с ячейкой примерно 0,5-1,0 мм. Затем приблизительно 0,5 мл объема собранных клеток, выделенных из фильтрата, пипеткой наносят на бумажные фильтры размером 5 см и высушивают в вакууме с последующим переносом в чашку Петри, содержащую “сэндвич” из трех 7-см бумажных фильтров, смоченных средой для суспензионной культуры. Каждую чашку с клеточной суспензией центруют на лотке для чашек с образцами, убирают крышку чашки Петри и подвергают двукратной бомбардировке в вакууме 28 дюймов рт.ст. Экраны 0,1 или 1,0 мм помещают примерно на 2,5 см ниже стоп-пластины для ослабления повреждаемости бомбардируемой ткани. После бомбардировки растительные клетки удаляют с фильтра, снова ресуспендируют в среде для клеточной суспензионной культуры и культивируют в течение 2-21 дней. Альтернативно, подвергнутый бомбардировке каллюс переносят с чашки на чашку в конечном счете на чашку, содержащую аналогичную плотную среду (например, содержащую 8 г/л очищенного агара) и аналогично культивируют при примерно 25°С в темноте.
Отбор трансформантов
После трансформации клетки, растущие без отбора в жидкой или плотной культуре, переносят на фильтры и переслаивают на плотную среду, содержащую диапазон (0,1-20 мМ) селективных концентраций N-(фосфонометил)глицина чистоты для тканевых культур (Sigma). Подходящими плотными селективными средами являются среды, доведенные до рН 5,8 или 6,0 с помощью КОН, содержащие соли либо MS, либо N6 (такие как те, которые описаны выше для инициации каллюсов, или, с соответствующим добавлением агара, те, которые описаны выше для роста клеток в жидкой суспензии) и N-(фосфонометил)глицин. Также подходящими селективными средами являются, например, селективные среды, описанные в примере 11, но в данном случае модифицированные так, чтобы не содержать антибиотики. Трансформированные каллюсы, экспрессирующие резистентный фермент ЕРSР-синтазу, отбирают на основе их роста при концентрациях, являющихся ингибиторными для аналогичных препаратов нетрансформированных клеток. Растущие скопления пересевают на свежую селективную среду. Предпочтительно концентрация N-(фосфонометил)глицина, используемого в селективной среде, составляет примерно 1 мМ в первые две недели отбора и примерно 3 мМ после этого. Через 6-18 недель предполагаемые резистентные каллюсы идентифицируют и отбирают.
Регенерация трансформантов, размножение и анализ трансформированного растительного материала
Отобранные каллюсы регенерируют в нормальные фертильные растения в соответствии, например, с методами, описанными Duncan et al. (1985, Planta, 165, 322-332), Kamo et al (1985, Bot. Gaz. 146(3), 327-334) и/или West et al. (1993, The Plant Cell, 5, 1361-1369), и/или Shillito et al. (1989) Bio/Technol. 7, 581-587).
Например, растения эффективно регенерируются путем переноса эмбриогенного каллюса в среду Мурасиге-Скоога, доведенную до рН 6,0, содержащую 0,25 мг/л 2,4-D, 10 мг/л 6-бензиламинопурина и, необязательно, от 0,02 до 1 мМ N-(фосфонометил)глицина. Спустя примерно 2 недели ткань переносят в аналогичную среду, но лишенную гормонов. Необязательно, содержание гормонов снижают постепенно с помощью большего числа переносов и за более длительный период времени вплоть до 6-8 недель. Побеги, которые развиваются через 2-4 недели, переносят в среду MS, содержащую 1% сахарозы, и уплотняют ее 2 г/л Gelgro, в которую затем прорастают корешки.
Альтернативные способы и среды, применяемые для регенерации, соответствуют описанным в примере 1, за исключением того, что используемые среды не содержат антибиотиков.
Способы выращивания растений до созревания, дальнейшего размножения растений в ряду поколений, анализа наследования резистентности к глифосату и анализа присутствия, целостности и экспрессии трансгена EPSPS соответствуют описанным в примере 11.
Пример 13
Трансформация линий кукурузы ДНК, включающей экспрессионную кассету EPSPS, нанесенной на вискеры из карбида кремния: отбор и регенерация растительных клеток и растений, которые резистентны к глифосату
В следующем примере линии кукурузы, включая, например, гибридные линии с генотипом А188× В73, получают в виде клеточных суспензий и трансформируют путем контактирования клеток с вискерами из карбида кремния, покрытыми ДНК, с использованием методов, сущность которых описана Frame et al. (1994, Plant J. 6, 941-948). Как было описано в предыдущих примерах, трансформированный каллюс, полученный таким путем, отбирают исходя из различного роста в среде, содержащей ряд концентраций глифосата, регенерируют в проростки (Т0), которые выращивают до созревания, и либо самоопыляют, либо перекрестно опыляют с получением семян потомства (T1) для дальнейшего воспроизведения. Растения и растительный материал оценивают на резистентность к глифосату и анализируют на присутствие, целостность и экспрессию трансгена в соответствии с описанным в предыдущих примерах.
Инициация каллюса из недоразвитых зародышей, получение суспензионных клеточных культур
Суспензии клеток кукурузы, пригодные для трансформации, необязательно подвергают криоконсервации и представляют таким же образом, как это описано в примере 12.
Трансформация
ДНК, производную от плазмиды pIGPD9 (фиг.12), включающую XmaI-экспрессионные кассеты EPSPS (а именно pZEN9i, ZEN11i, ZEN12i, ZEN14i и т.д.), очищают, концентрируют (например, с помощью анионообменной хроматографии или денситометрического выделения плазмидной ДНК в градиенте хлорида цезия из клеток подходящего штамма-хозяина HisB-, RecA- E.coli (например, DH5α : hisB-) после роста до стационарной фазы культуры в минимальной среде 5× А (К2НРO4 52,5 г, КН2РO4 22,5 г, (NH4)2SO4 5 г и цитрат натрия· 2Н2O 2,5 г на литр) и представляют в виде концентрированного раствора (предпочтительно примерно 1 мг/мл) в стерильной воде. ДНК получают в виде кольцевой плазмидной ДНК или, альтернативно, в виде ДНК, расщепленной рестриктазой XmaI с получением линейной экспрессионной кассеты, включающей EPSPS-фрагмент, и используют ее после очистки методом электрофореза в агарозном геле и электроэлюции.
Трансформацию проводят в точном соответствии с описанным у Frame et al. Альтернативно, процедуру в некоторой степени модифицируют, как описано ниже.
Клетки, выращенные в жидкой среде для клеточных суспензий, через день после субкультивирования оставляют отстаиваться в шейкерной колбе. Истощенную среду декантируют и отбрасывают, после чего 12 мл среды N6 (Сhu et al., 1975) при рН 6,0, модифицированной так, чтобы содержать 6 мМ L-пролина, 20 г/л сахарозы, 2 мг/л 2,4-D, 0,25 М сорбита и 0,25 М маннита, добавляют в расчете на объем 4 мл клеточного сгустка. Колбу возвращают в шейкер (роторное перемешивание при 125 об/мин, инкубация при 26-28°С) на 45 мин. По прошествии этого времени аликвоты по 1 мл клеточной суспензии переносят с использованием пипетки с широким отверстием в серию стерильных микроцентрифужных пробирок. После отстаивания клеток в каждой пробирке удаляют по 0,6 мл супернатанта истощенной среды так, чтобы остаток в основном состоял от отстоянных клеток. 50 мг вискеров из карбида кремния (вискеры Silar SC-9: Advanced Composite Materials Corp., Greer, SC, США) суспендируют на вортексе в 1 мл модифицированной среды N6, описанной выше. Затем 40 мл таких суспендированных вискеров и 25 мг плазмидной или линейной ДНК, включающей экспрессионную кассету EPSPS, добавляют в каждую пробирку с отстоявшимися клетками. Пробирки взбалтывают вручную 2-3 раза, взвихривают 1 с (с помощью стоматологического миксера Mixomat, (Degussa, Ontario, Канада)) и затем по 0,3 мл среды N6 (модифицированной в соответствии с описанным выше) добавляют в каждую микроцентрифужную пробирку. После этого суспендированные клетки высевают (200 мкл на чашку) на дисковый фильтр, наслоенный поверх среды N6 (та же модификация среды N6, которая описана выше, но при отсутствии сорбита и маннита и содержании 30 г/л сахарозы и 3 г/л гельрита). Затем каждую чашку оборачивают лентой Urgopore (Stelrico, Brussels) и оставляют инкубироваться в темноте в течение 1 недели при 26-28°С.
Отбор трансформантов
Трансформированный каллюс отбирают, как описано в примере 12 или, альтернативно, как описано Frame et al., 1994, за исключением того, что используют N-(фосфонометил)глицин в диапазоне концентраций от 1 до 5 мМ вместо биалафоса, указанного в публикации Frame et al.
Регенерация трансформантов, размножение и анализ трансформированного растительного материала
Растения регенерируют, воспроизводят и скрещивают, как описано в примере 12. Растения анализируют на резистентность к глифосату, а растительный материал тестируют на присутствие, целостность и экспрессию трансгена, как описано в примере 12.
Пример 14
Трансформация линий риса с использованием штамма Agrobacterium, несущего супербифункциональный вектор, который включает экспрессионную кассету EPSPS между правым и левым граничными сайтами Т-ДНК: отбор и регенерация растительных клеток и растений, которые резистентны к глифосату
В следующем примере зародышевые щитки выделяют из зрелых семян подходящих линий риса (например, включая сорта Koshihikari, Tsukinohikari и Asanohikari) и дедифференцируют, а полученные в результате каллюсы трансформируют инфицированием Agrobacterium. После отбора и регенерации получают трансгенные проростки (Т0), которые выращивают до созревания и либо самоопыляют, либо перекрестно опыляют с получением семян потомства (T1) для дальнейшего размножения. Растения и растительный материал оценивают по резистентности к глифосату и анализируют на присутствие, целостность и экспрессию трансгена, как описано в предыдущих примерах. В качестве альтернативы способам, описанным ниже, способы, описанные в примере 1 US 5591616, подходящим образом приспособленные для использования глифосата вместо гигромицина, используют для отбора.
Конструирование штамма Agrobacterium: получение суспензии Agrobaсterium
Штамм Agrobacterium, несущий супербифункциональный вектор, включающий желательную экспрессионную кассету EPSPS между первым и левым граничными сайтами, конструируют (трансформируя Agrobacterium с применением электропорации плазмидной ДНК), как описано в примере 1. Суспензии получают способами, описанными в примере 1. Альтернативно, трансформированный штамм Agrobacterium выращивают в течение 3 дней в среде АВ (Chilton et al., 1974, Proc. Natl. Acad. Sci. USA, 71, 3672-3676), содержащей подходящий селективный антибиотик (например, 50 мг/л спектиномицина в случае LBA4404 (pSB1ZEN18 и т.д.)), и снимают петлей с чашки для формирования суспензии в среде ААМ (Hiei et al., 1994, The Plant Journal, 6(2), 271-282) при плотности 1-5× 109 клеток/мл.
Сорта риса, получение каллюса из щитка
Сортами риса являются, например, сорта Tsukinohikari, Asanohikari и Koshihikari Oryza sativa L.
Зрелые семена отшелушивают, поверхность стерилизуют промывкой 70%-ным этанолом и затем замачивают в течение 30 мин в 1,5% NaOCl. После промывки в стерильной воде семена культивируют при 30°С в темноте в течение 3 недель при рН 5,8 в среде 2N6, содержащей основные соли, минорные соли и витамины среды N6 (Сhu, 1978, in Proc. Symp. Plant Tissue Culture, Peking: Science Press, pp. 43-50), 30 г/л сахарозы, 1 г/л гидролизата казеина, 2 мг/л 2,4-D и 2 г/л гельрита. Производные от семенного щитка пролиферированные клетки каллюса пересевают в течение 3-7 дней в свежую среду 2N6. Растущий каллюс (диаметром 1-2 мм) отбирают, суспендируют в жидкой среде 2N6 (без гельрита) и культивируют в колбах в темноте на роторном шейкере при 125 об/мин при 25°С. Среду заменяют каждые 7 дней. Для трансформации используют клетки, растущие в логарифмической фазе после 3-4 субкультивирования.
Инфицирование, трансформация и отбор
Суспендированные каллюсные клетки риса оставляют отстаиваться из суспензии, и затем их ресуспендируют в суспензии Agrobacterium, оставляя в контакте в течение нескольких минут, и после этого опять оставляют отстаиваться, без промывки высевают на среду 2N6-AS (среда 2N6, доведенная до рН 5,2 и содержащая 10 г/л D-глюкозы и 100 мМ ацетосирингона) и инкубируют в темноте при 25°С в течение 3-5 дней. Растущий материал тщательно промывают 250 мг/л цефотаксима в стерильной воде и затем переносят на среду 2N6-CH (среда 2N6, доведенная до рН 5,8 с помощью КОН, содержащая 250 мг/л цефотаксима и 0,5-5 мМ N-(фосфонометил)глицина чистоты для тканевых культур) или, альтернативно, среду 2N6K-CH (среда 2N6, модифицированная, как описано Hiei et al., 1994, но вместо гигромицина содержащая 0,5-5 мМ N-(фосфонометил)глицина чистоты для тканевых культур) и культивируют в течение 3 недель в темноте при 25°С. Пролиферировавшие колонии субкультивируют на вторую чашку с селективной средой на дополнительный срок 7-14 дней.
Регенерация и анализ растений
Растущие колонии высевают на регенерационную среду при рН 5,8, содержащую половину концентрации основных солей N6, минорные соли N6, аминокислоты N6, витамины среды АА (Chilton et al, 1974), 1 г/л гидролизата казеина, 20 г/л сахарозы, 0,2 мг/л нафталинуксусной кислоты, 1 мг/л кинетина, 3 г/л гельрита и, необязательно, 0,04-0,1 мМ N-(фосфонометил)глицина. Такие чашки инкубируют при 25°С и выдерживают в условиях постоянного освещения (примерно 2000 люкс). Как описано в примере 11, регенерированные растения в конечном счете переносят в горшки с почвой и выращивают до созревания в теплице.
Растения размножают и скрещивают (например, самоопыляют трансгенные растения), по существу как описано в примере 11. Растения анализируют на резистентность к глифосату и растительный материал тестируют на присутствие, целостность и экспрессию трансгена, по существу как описано в примере 11.
Пример 15
Трансформация линий пшеницы с использованием ДНК, которая включает экспрессионную кассету EPSPS, с помощью бомбардировки микрочастицами; отбор и регенерация растительных клеток и растений, которые резистентны к глифосату
В следующем примере недоразвитые зародыши выделяют на материале подходящих линий пшеницы (включая, например, яровую пшеницу сортов BobWhite или Jaggar), инкубируют в среде, содержащей гормон (2,4-D), в течение 2 дней и трансформируют путем бомбардировки частицами, покрытыми ДНК. После периода выделения и последующего роста каллюса каллюсные зародыши субкультивируют через серию сред, содержащих фиксированный уровень глифосата и (в серийном разведении) снижающиеся уровни 2,4-D таким образом, что индуцируется соматический эмбриогенез. Отобранный материал регенерируют с образованием побегов в среде, также содержащей глифосат, переносят в среду для развития корешков и, как и в предыдущих примерах с кукурузой, регенерируют в проростки (То), которые выращивают до созревания и либо самоопыляют, либо перекрестно опыляют с получением семян потомства (T1) для дальнейшего размножения. Растения и растительный материал оценивают по резистентности к глифосату и анализируют на присутствие, целостность и экспрессию трансгена, как описано в предыдущих примерах. В качестве альтернативы способам, описанным далее, используют способы, описанные в примере 1 US 5631152.
Получение недоразвитых зародышей
Линии пшеницы (например, обыкновенная пшеница Triticum aestivum ярового сорта BobWhite) выращивают до созревания в теплице, и зерновки выделяют на 11-15-й день после опыления. Зерновки стерилизуют по поверхности обработкой в течение 15 мин 5%-ным NaOCl и затем промывают несколько раз стерильной водой. Недоразвитые зародыши в стерильных условиях помещают на 3 см2 нейлоновой сети (размер ячейки 1,5 мм), расположенной поверх среды А2. Среда А2, доведенная до рН 5,8, содержит 4,32 г/л солей Мурасиге-Скоога, 20 г/л сахарозы, 0,5 г/л L-глутамина, 2 мг/л 2,4-D, 100 мг/л гидролизата казеина, 2 мг/л глицина, 100 мг/л миоинозитола, 0,5 мг/л никотиновой кислоты, 0,1 мг/л гидрохлорида тиамина и 2,5 г/л гельрита. Зародыши размещают на плотном диске 2,5 см в числе приблизительно 50. Чашки закрывают лейкопоровой лентой и инкубируют при 25°С в темноте в течение 2 дней. За 4 ч до проведения бомбардировки зародыши переносят на чашки, содержащие свежую среду А2, дополненную 36,44 г/л D-сорбита и 36,44 г/л D-маннита. Зародыши переносят с чашки на чашку с помощью нейлоновой сетки, на которой они размещены. Зародыши находятся на указанной среде с повышающейся осмотической силой в течение 4 ч при 25°С в темноте перед последующей бомбардировкой.
Трансформация с помощью частиц
ДНК, производную от плазмиды pIGPD9 (фиг.12), включающую XmaI-экспрессионные кассеты EPSPS (а именно pZEN9i, ZEN11i, ZEN12i, ZEN14i и т.д.), очищают, концентрируют (например, с помощью анионообменной хроматографии или денситометрического выделения плазмидной ДНК в градиенте хлорида цезия из клеток подходящего штамма-хозяина HisB-, RecA- E. coli (например, DH5α : hisB-) после роста до стационарной фазы культуры в минимальной среде 5хА (К2НРO4 52,5 г, КН2РО4 22,5 г, (NH4)2SO4 5 г и цитрат натрия· 2Н2O 2,5 г на литр) и представляют в виде концентрированного раствора (предпочтительно примерно 1 мг/мл) в стерильной воде. ДНК получают в виде кольцевой плазмидной ДНК или, альтернативно, в виде ДНК, расщепленной рестриктазой XmaI с получением линейной экспрессионной кассеты, включающей EPSPS-фрагмент, и используют ее после очистки методом электрофореза в агарозном геле и электроэлюции.
Частицы получают и покрывают ДНК по аналогичной методике, как описано Klein et al, 1987, Nature, 327, 70-73. Получение покрытых ДНК частиц и работа с “генным ружьем” соответствует описанному в примере 2. Альтернативно, некоторые подробности таковы. Например, 60 мг золотых или вольфрамовых частиц (примерно 1,0 мкм) в микроцентрифужной пробирке промывают несколько раз этанолом ВЭЖХ-чистоты и затем несколько раз в стерильной воде. Частицы ресуспендируют в 1 мл стерильной воды и аликвотами по 50 мкл распределяют по микроцентрифужным пробиркам. Золотые частицы хранят при 4°С, вольфрамовые частицы при -20°С. По 3 мг ДНК добавляют к каждой аликвоте (размороженных) частиц и содержимое пробирок перемешивают на вортексе с максимальной скоростью. Поддерживая практически непрерывное интенсивное перемешивание, добавляют 50 мкл 2,5 М CaCl2 и 20 мкл 0,1 М спермидина. Через 10 мин последующего встряхивания образцы центрифугируют в течение 5 с на эппендорфовской микроцентрифуге, супернатант удаляют и частицы промывают последовательно добавляемым этанолом ВЭЖХ-чистоты. Частицы тщательно ресуспендируют в 60 мкл этанола и затем распределяют аликвотами по 10 мкл на поверхности каждого макроносителя, которые должны быть использованы в генном ружье PDS1000.
Детали генного ружья PDS1000 стерилизуют по поверхностям погружением в 70%-ный этанол и высушивают на воздухе. Чашки-мишени, полученные как описано выше, несущие примерно по 50 зародышей, размещенных на диске ≈ 2,5 см, размещают в 6 см от стоп-экрана. Для бомбардировки затем используют пробойные диски при 1100 psi. Каждую чашку бомбардируют 1 или 2 раза.
Подвергнутые бомбардировке чашки закрывают пористой лентой и выдерживают при 25°С в темноте примерно в течение 16 ч. Зародыши, отделенные от поверхности среды ударной гелиевой волной, выделяют и также инкубируют в течение ночи на новых чашках в среде А2, таким же образом дополненной маннитом и сорбитом. Затем подвергнутые бомбардировке зародыши переносят на новые чашки со средой А2 и инкубируют в течение 1 недели при 25°С в темноте для последующего отбора.
Отбор и регенерация трансформантов
После указанного периода выделения каллюсные зародыши снимают с сеток и переносят в среду А2-2Р (среда А2, доведенная до рН 5,8, содержащая 2 мМ N-(фосфонометил)глицина) при плотности 20 эксплантатов на чашку. Через неделю культивирования в среде А2-2Р каллюсы переносят в среду А1-2Р (среда А2, содержащая только 1,0 мг/л 2,4-D и 2 мМ N-(фосфонометил)глицина) в течение 2 недель и затем в среду А0,5-2Р (среда А2, содержащая только 0,5 мг/л 2,4-D и 2 мМ N-(фосфонометил)глицина) в течение еще двух недель. Необязательно, 2-недельные периоды инкубации сокращают до 1 недели и/или средний этап инкубации в среде А1-2Р отменяют. Необязательно, селективная концентрация N-(фосфонометил) глицина составляет от 0,5 до 10 мМ, хотя предпочтительна концентрация 2 мМ. Общая продолжительность периода индукции каллюса при снижающейся концентрации 2,4-D в среде составляет 2-10 недель, предпочтительно 3-6 недель, а наиболее предпочтительно, примерно 4 недели.
После этого, для обеспечения максимального роста побегов и недопущения развития корешков, каллюсы переносят в среду Z. Среда Z является средой А2, содержащей 10 мг/л зеатина вместо 2,4-D и также содержащей 0,1 мМ N-(фосфонометил)глицина. Необязательно концентрация N-(фосфонометил)глицина находится в диапазоне 0,04-0,25 мМ. Регенерирующие каллюсы выдерживают в указанной среде в течение 3 недель перед субкультивированием, когда хорошо развитые побеги отрезают. Чтобы только одно событие приводило к образованию отдельного каллюса (что соответствует отдельному зародышу), весь каллюс переносят на новую чашку и выдерживают вместе с отрезанными побегами так, чтобы множественные клоны, возникающие на том же самом каллюсе, не воспринимались как отдельные события. Каллюсы, несущие лишь частично развитые побеги или не имеющие регенерирующих секторов, возвращают в среду Z еще на 3 недели. По окончании этого времени нерегенерированные каллюсы отбрасывают.
Побеги выдерживают в среде Z до тех пор, пока не образуются 4 или большее число хорошо развитых листьев (достигающих в длину примерно 2 см). Регенерированный растительный материал затем аккуратно переносят в пластиковые пробирки, содержащие среду 0,5MS. Среда 0,5MS при рН 5,8 содержит 2,16 г/л солей Мурасиге-Скоога, 15 г/л сахарозы, 2,5 г активированного угля, 2,5 г/л гельрита, 1 мг/л глицина, 50 мг/л миоинозитола, 0,25 мг/л никотиновой кислоты, 0,25 мг/л гидрохлорида пиридоксина, 0,05 мг/л гидрохлорида тиамина и 0,1 мМ N-(фосфонометил)глицина (необязательно 0,0-0,25 мМ).
После того, как у растений образуются корешки, их высаживают в почву и выращивают или переносят в отдельные стеклянные пробирки для кипячения, содержащие 0,5MS (без N-(фосфонометил)глицина) и 2,5 г/л угля. Предпочтительно включать уголь в среду для образования корешков с целью адсорбции любых оставшихся PGR или отбора химикатов, привнесенных с проростком, и с целью создания темной корневой среды, что тем самым исключает образование физиологически аномальных зеленых корней.
Индукция каллюсов и первая неделя регенерации проходят при 25°С в темноте. Вторая неделя регенерации проходит при слабом освещении при 25°С, а затем последующие недели проходят при приблизительно 2500 люкс при 16-часовом периоде освещенности.
Размножение, скрещивание и анализ трансформированного растительного материала.
Способы получения T1 и последующих потомств хорошо известны в данной области техники и по существу описаны в предыдущих примерах. Способы анализа наследования резистентности к глифосату и присутствия, целостности и экспрессии трансгенов соответствуют тому, что описано в предыдущих примерах.
Пример 16
Трансформация линий пшеницы ДНК, включающей экспрессионную кассету EPSPS, с помощью электропорации протопластов; отбор и регенерация растительных клеток и растений, которые резистентны к глифосату
В следующем примере плазмидную или линейную ДНК, включающую экспрессионную кассету EPSPS и идентичную той, которую использовали в примерах 12, 13 и 15, используют для прямой трансформации протопластов линии пшеницы, способной регенерировать в фертильные растения (см. US 5231019). Выделенные протопласты пшеницы, предпочтительно на материале ткани листьев или культивируемых клеток (см. Gamborg, O.L., and Wetter, L.R., Plant Tissue Culture Methods, 1975, 11-21), получают в концентрации примерно 2х106 протопластов на 1 мл в 0,4 М маннита при рН 5,8. К указанной суспензии добавляют сначала 0,5 мл 40%-ного (мас/об) полиэтиленгликоля (ПЭГ) молекулярной массы 6000 в модифицированной среде F (Nature (1982), 296, 72-74) при рН 5,8, и затем 65 мл воды, содержащей 15 мг желательной плазмидной или линейной ДНК и 50 мг ДНК вилочковой железы теленка. Смесь инкубируют вместе в течение 30 мин при 26°С, изредка помешивая, с последующим разведением средой F (Nature (1982), 296, 72-74). Протопласты выделяют с помощью низкоскоростного центрифугирования, отбирая их в 4 мл культуральной среды СС (Potrykus, Harms, Lorz, (1979) Theor. Appi. Genet., 54, 209-214), и инкубируют в темноте при 24°С.
Альтернативно, и в дополнение к обработке ПЭГ, трансформацию протопластов зерновых проводят с использованием дополнительных стадий теплового шока и/или электропорации (Neumann, E.et al. (1982), the EMBO J., 1, 841-845). Так, например, протопласты пшеницы инкубируют в водном растворе ДНК и маннита, нагревают до 45°С в течение 5 мин и затем охлаждают до 0°С в течение 10 с. Затем добавляют полиэтиленгликоль (молекулярная масса 3-8 кД) до конечной концентрации примерно 8% мас/об. После аккуратного, но тщательного перемешивания проводят обработку на электропораторе. Камеры поратора Dialog (Dialog, Dusseldorf, Германия) стерилизуют промывкой 70%-ным этанолом и затем высушивают в стерильном воздухе. Суспензии протопластов (примерно 2× 106 протопластов на 1 мл в 0,4 М маннита + ДНК) корректируют с помощью хлорида магния до измеренного электрического сопротивления примерно 1,4 кОм. Образцы объемом примерно 0,4 мл подвергают трем пульсам с 10-секундными интервалами при приложенном напряжении 1000-2000 В. Затем трансформированные таким путем протопласты собирают и вновь растворяют в культуральной среде СС.
Для специалистов в данной области техники будет понятно, что возможны разнообразные допущения и вариации указанных процедур трансформации, и что, например, трансформацию также можно оптимизировать увеличением рН до 9,5 и/или повышением концентрации ионов кальция в растворе, в котором осуществляют трансформацию.
Через 3-14 дней аликвоты культур развивающихся клеток переносят в среду, содержащую разные селективные концентрации N-(фосфонометил)глицина чистоты для тканевых культур (Sigma) от 1 до 5 мМ (предпочтительно 2 мМ). Колонии резистентных клеток, идентифицированных таким образом (проявляющие рост при концентрациях глифосата, которые по крайней мере вдвое выше, чем могут переносить нетрансформированные контроли), переносят в свежую агаровую среду, также содержащую диапазон селективных концентраций глифосата, и, как это было описано в примере 15, пересевают с чашки на чашку при постепенно снижающейся концентрации 2,4-D. Растущие резистентные колонии могут быть проанализированы (с помощью ПЦР и т.п.) на присутствие рекомбинантной ДНК. Проведение дополнительного отбора возможно или невозможно на стадии каллюса. В любом случае все растущие каллюсы должны быть отобраны в первую очередь.
Затем растущие каллюсы переносят в среду для регенерации побегов, содержащую зеатин и N-(фосфонометил)глицин, и из нее переносят в среду для образования корешков точно так же, как в примере 15. Фертильные трансгенные растения, экспрессирующие резистентную к глифосату ЕРSР-синтазу, затем регенерируют, отбирают и тестируют, как известно в данной области техники и как описано в примере 15, а также с использованием аналитических методов, описанных в примере 11.
Пример 17
Способ оценки активности EPSPS и определения кинетических констант. Способ оценки активностей EPSPS в неочищенных растительных материалах и определения доли, которая резистентна к глифосату
Ферментный анализ EPSPS
Анализ проводят по существу в соответствии с радиохимическим методом Padgette et al., 1987 (Archives of Biochemistry and Biophysics, 258 (2), 564-573) с ионами К+ как основным видом катионного контриона. Пробы общим объемом 50 мкл в 50 мМ HEPES (КОН) при рН 7,0 при 25°С содержат очищенный фермент или растительный экстракт (см. ниже), разведенный подходящим образом буфером HEPES при рН 7,0, содержащим 10% глицерина и 5 мМ ДТТ, 14С-РЕР либо в качестве варьирующегося субстрата (для кинетических определений), либо в фиксированной концентрации 100 или 250 мкМ, и шикимат-3-фосфат (калиевая соль) при 2 или 0,75 мМ, как указано. Необязательно, при анализе неочищенных растительных экстрактов пробы также содержат 5 мМ КF и/или 0,1 мМ молибдата аммония. Анализ начинают с добавления 14С-фосфоенолпирувата (циклогексиламмоний + соль) и останавливают через 2-10 мин (предпочтительно 2 мин) добавлением 50 мкл раствора 1 М уксусной кислоты и этанола (1:9). После остановки 20 мкл загружают на колонку Synchropak AX100 (25 см × 4,6 мм) и хроматографируют с использованием изократического элюирования 0,28 М фосфата калия при рН 6,5 при скорости протока подвижной фазы 0,5 мл/мин в течение 35 мин. При этих условиях время удерживания для PEP и EPSP составляет примерно 19 и 25 мин, соответственно. Сцинтилляционный счетчик СР 525TR подсоединяют к концу колонки AX100. Он заполняется 0,5 мл проточных клеток, и скорость протока сцинтиллянта (Ultima Flo АР) устанавливают на уровне 1 мл/мин. Относительные пиковые области PEP и EPSP интегрируют с целью определения процента превращения меченого PEP в EPSP. Значения Кm и Vmах определяют методом подбора с помощью наименьших квадратов к гиперболе при простом взвешивании с использованием программы Grafit 3.09b от Erithacus Software Ltd. Величины Кm обычно устанавливают с использованием 8-9 концентраций разного субстрата в диапазоне от Кm/2 до 10Km и тройных точек. За исключением специально оговариваемых случаев, данные включают в анализ только тогда, когда имеется менее чем 30%-ное превращение субстрата в EPSP.
Шикимат-3-Pi (S3P) получают следующим образом, К 7 мл 0,3 М TAPS при рН 8,5, содержащего 0,05 М шикимата, 0,0665 М АТФ (соль Na), 10 мМ KF, 5 мМ ДТТ и 0,05 М МgСl2·2Н2O, добавляют 75 мкл раствора 77 единиц (мкмоль/мин)/мл раствора шикиматкиназы. Через 24 ч при комнатной температуре реакцию останавливают кратковременным нагреванием до 95°С. Реакционный раствор разбавляют в 50 раз в 0,01 М Трис-НСl при рН 9 и хроматографируют на анионообменнике Dowex 1х8-400 с использованием градиента 0-0,34 М LiCl2. Фракции S3P объединяют, лиофилизуют и затем снова растворяют в 7 мл дистиллированной воды. Затем добавляют 28 мл 0,1 М Ва(СН3СООН)2 и 189 мл абсолютного этанола. Этот раствор оставляют перемешиваться в течение ночи при 4°С. Полученный преципитат трехбариевого S3P собирают и промывают 30 мл 67%-ного этанола. Затем промытый преципитат растворяют примерно в 30 мл дистиллированной воды. При необходимости добавлением K2SO4 получают либо К+, либо ТМА+ соль S3P. Необходима особая аккуратность при добавлении минимального избытка сульфата. Преципитат ВаSO4 удаляют и надосадочную фракцию, содержащую требуемую соль S3P, лиофилизуют. Каждую соль взвешивают и анализируют с помощью протонного ЯМР. Полученные таким путем препараты S3P характеризуются чистотой >90% по данным протонного ЯМР и (в соответствии с их массами и включением 31Р-ЯМР) содержат только следовые остатки сульфата калия.
Получение экстрактов растительного материала, пригодных для тестирования EPSPS
Материал каллюсов или проростков (0,5-1,0 г) измельчают в мелкий замороженный порошок с помощью охлаждаемых жидким азотом ступки и пестика. Указанный порошок отбирают в равный объем охлажденного экстракционного буфера (например, 50 мМ буфера HEPES/KOH при рН 7,5, содержащего 1 мМ ЭДТА, 3 мМ ДТТ, 1,7 мМ “pefabloc” (ингибитор сериновых протеаз), 1,5 мМ лейпептина, 1,5 мМ пепстатина-А, 10% глицерина (об/об) и 1% поливинилпирролидона), ресуспендируют, смешивают и центрифугируют в охлажденной центрифуге для осаждения клеточного дебриса. Супернатант обменивают в охлажденную колонку PD10 сефадекса G25 в 25 мМ буфера HEPES/KOH при рН 7,5, содержащего 1 мМ ЭДТА, 3 мМ ДТТ и 10% глицерина (об/об). Белок оценивают по Брэдфорду с нормализацией к бычьему сывороточному альбумину. Часть экстракта замораживают на жидком азоте, а часть анализируют сразу.
Тесты на EPSPS на материале растительных экстрактов проводят стандартным образом, как описано выше, с 0,1 мМ 14С-РЕР и 0,75 мМ шикимат-3-Pi либо в отсутствие, либо в присутствии 0,1 мМ N-(фосфонометил)глицина. При таких условиях тестирования резистентную форму EPSPS (см. далее) определяют, как ту, которая должна подавляться менее чем на 8,5%, в то время как восприимчивая форма дикого типа по существу подавляется полностью (>98%). Таким образом, уровень активности, выявленный в присутствии глифосата (А), принят примерно за 92% уровня резистентного фермента, образующегося в результате экспрессии трансгена, в то время как уровень восприимчивого EPSPS дикого типа принимается за общий уровень активности EPSPS, наблюдаемой в отсутствие глифосата за вычетом А: х≈ 1,08. Поскольку Vmах мутантного фермента оценивается на уровне всего одной трети от Vmах фермента дикого типа (и поскольку величины Кm для PEP и у мутантной формы, и у формы дикого типа были оценены на уровне примерно 20 мкМ или меньше), то уровень экспрессии полипептида мутантного фермента по отношению к уровню экспрессии эндогенного EPSPS дикого типа принимается как примерно в три раза более высокий по сравнению с отношением, подсчитанным на основе отношения их выявленных относительных активностей. Общий уровень экспрессии полипептида EPSPS (мутант + дикий тип) также оценивают с помощью Вестерн-блоттинга (см. далее).
Пример 18
Клонирование и экспрессия кДНК дикого типа и мутантной кДНК, кодирующих зрелый EPSPS риса, в Е.coli. Очистка и характеристика EPSPS дикого типа и мутантного EPSPS риса
Экспрессия, очистка и характеристика зрелого EPSPS риса дикого типа
кДНК EPSPS риса амплифицируют с применением ОТ-ПЦР на материале РНК, выделенной из клеток риса сорта Koshigari, с использованием обратной транскриптазы Superscript от BRL в соответствии с рекомендациями, предлагаемыми производителем. ПЦР проводят с использованием полимеразы Pfu turbo от Stratagene в соответствии с методами, предлагаемыми изготовителем. Приведенные далее олигонуклеотиды используют на этапах реакций амплификации и обратной транскрипции.
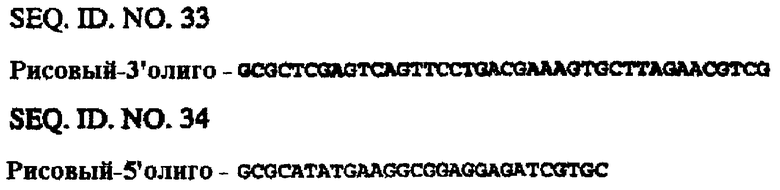
ПЦР-продукт клонируют в состав pCRBlunt II с использованием набора Zero Blunt TOPO kit от Invitrogen. Последовательность вставки подтверждают путем прямого секвенирования с подтверждением того, что предсказанная открытая рамка считывания соответствует той, которая предсказана для зрелого хлоропластного белка EPSPS риса, за исключением присутствия старт-кодона Met. Клонированную и подтвержденную последовательность EPSPS риса вырезают с использованием рестриктаз NdeI и XhoI, и очищенный фрагмент клонируют в плазмиду рЕТ24а (Novagen), расщепленную аналогичным образом. Рекомбинантные клоны вносят в BL21 (DE3), оптимизованный по библиотеке кодонов штамм RP Е.coli, предоставляемый Stratagene. Белок EPSPS экспрессируется указанным штаммом после добавления 1 мМ IPTG к питательной среде (среда LB, дополненная 100 мкг/мл канамицина). Рекомбинантный белок с точной предсказанной молекулярной массой идентифицируют: (i) на основе окрашивания по Кумасси ДСН-гелей клеточных экстрактов и параллельного сравнения с окрашенными по Кумасси гелями экстрактов аналогичных клеток Е. coli, трансформированных пустым вектором рЕТ24а, и (ii) с помощью Вестерн-блоттинга с использованием поликлонального антитела, сформированного к ранее очищенному растительному белку EPSPS. Зрелый белок EPSPS риса очищают при примерно 4°С следующим образом. Клетки (25 г сырого веса) промывают в 50 мл 0,1 М буфера HEPES/KOH при рН 7,5, содержащего 5 мМ ДТТ, 2 мМ ЭДТА и 20% глицерина (об/об). После низкоскоростного центрифугирования клеточный сгусток ресуспендируют в 50 мл того же буфера, но также содержащего 2 мМ “Pefabloc”, ингибитора сериновых протеаз. Клетки равномерно суспендируют с помощью стеклянного гомогенизатора и затем разрушают с помощью клеточного дезинтегратора Basic Z от Constant Systems (Budbrooke Road, Warwick, Великобритания) под давлением 10000 psi. Неочищенный экстракт центрифугируют при примерно 30000g в течение 1 ч и сгусток отбрасывают. Протаминсульфат (сальмин) добавляют в конечной концентрации 0,2%, смешивают и раствор оставляют отстаиваться в течение 30 мин. Осажденный материал удаляют центрифугированием в течение 30 мин при ≈ 30000g. Сульфат аммония от Aristar добавляют до конечной концентрации 40% насыщения, перемешивают в течение 30 мин и затем центрифугируют при ≈ 27000g в течение 30 мин. Сгусток ресуспендируют примерно в 10 мл тоже буфера, который использовался для разрушения клеток, после чего сульфат аммония добавляют с доведением раствора до примерно 70%-ного насыщения, раствор перемешивают в течение 30 мин и вновь центрифугируют с получением сгустка, который ресуспендируют примерно в 15 мл буфера S200 (10 мМ HEPES/KOH (рН 7,8), содержащего 1 мМ ДТТ, 1 мМ ЭДТА и 20% глицерина (об/об)). Полученный фильтрат (0,45 мкм) загружают и хроматографируют после загрузки на колонку К26/60, содержащую Superdex 200, уравновешенную буфером S200. Фракции, содержащие EPSPS, выявленные исходя из параметров ферментной активности EPSPS, объединяют и загружают на колонку xk16, содержащую 20 мл высокочистой Q-сефарозы, уравновешенную буфером S200. Колонку промывают буфером S200 и затем EPSPS элюируют линейным градиентом от 0,0 до 0,2 М КСl в том же буфере. EPSPS элюируется в пределах единственного пика, соответствующего концентрации соли 0,1 М или меньше. Фракции, содержащие EPSPS, выявленные исходя из параметров ферментной активности EPSPS, объединяют и загружают на колонку HiLoad xk26/60 супердекса-75, уравновешенную буфером Superdex 75 (25 мМ HEPES/KOH (рН 7,5), содержащим 2 мМ ДТТ, 1 мМ ЭДТА и 10% глицерина (об/об)). EPSPS элюируется с колонки позже, чем этого можно было ожидать исходя из молекулярной массы предполагаемого димера. Это может объясняться взаимодействием белка с гелем матрикса при низкой ионной силе раствора. Фракции, содержащие EPSPS, выявленные исходя из параметров ферментной активности EPSPS, объединяют и загружают на 1-мл колонку MonoQ, уравновешенную тем же буфером Superdex 75. Колонку промывают начальным буфером и EPSPS элюируется в виде единственного пика по ходу 15 мл линейного градиента, сформированного от 0,0 до 0,2 М КСl. На данной стадии очистки EPSPS получают с чистотой >90%. Необязательно, EPSPS дополнительно очищают путем обмена в буфер Superdex 75, содержащий 1,0 М сульфата аммония (Aristar) и загруженный в 10-мл фенилсефарозную колонку, уравновешенную тем же самым буфером. EPSPS элюируется в виде единственного пика в начале снижающегося линейного градиента сульфата аммония, сформированного от 1,0 до 0,0 М сульфата аммония.
Клонирование, экспрессия, очистка и характеристика резистентной к глифосату мутантной EPSPS риса
кДНК EPSPS риса в составе pCRBlunt используют в качестве матрицы в двух дополнительных ПЦР с использованием следующих пар праймеров, сконструированных так, чтобы внести конкретные изменения:

Полученные продукты очищают в геле и помещают в пробирку в эквимолярных концентрациях, чтобы они выполняли роль матрицы в следующем раунде ПЦР с олигомерами рисовый-5’ и рисовый-3’. Полученные продукты клонируют в pCRBlunt II с использованием набора Zero Blunt TOPO kit от Invitrogen. Подтверждено, что последовательность ДНК у вставки и ее предсказанная открытая кодирующая рамка соответствуют предсказанному для зрелого хлоропластного белка EPSPS риса (за исключением присутствия старт-кодона Met) и также тому, что кодируются желательные замены (конкретные мутации Т→ I и P→ S по конкретным положениям последовательности EPSPS). Клонированную таким путем и подтвержденную последовательность EPSPS риса вырезают с использованием рестриктаз NdeI и XhoI, и очищенный фрагмент клонируют в рЕТ24а (Novagen), расщепленную аналогичным образом. Рекомбинантные клоны вносят в BL21 (DЕ3), оптимизованный по библиотеке кодонов штамм RP Е.coli, предоставляемый Stratagene. Белок EPSPS экспрессируется указанным штаммом после добавления 1 мМ IPTG к питательной среде (среда LB, дополненная 100 мкг/мл канамицина). Рекомбинантный белок с точной предсказанной молекулярной массой идентифицируют: (i) на основе окрашивания по Кумасси ДСН-гелей клеточных экстрактов и параллельного сравнения с окрашенными по Кумасси гелями экстрактов аналогичных клеток Е.coli, трансформированных пустым вектором рЕТ24а, и (ii) с помощью Вестерн-блоттинга с использованием поликлонального антитела, сформированного к ранее очищенному растительному белку EPSPS. Данную мутантную форму EPSPS риса очищают и охарактеризовывают сходным образом со способом, описанным выше для EPSPS риса дикого типа.
Полученная таким образом мутантная форма EPSPS риса, протестированная, как описано выше, в присутствии 2 мМ шикимат-3-Pi, характеризуется Vmax примерно 10 мкмоль/мин/мг, а Кm для PEP 22 мкМ. При 40 мкМ PEP величина IC50 для калиевой соли глифосата составляет примерно 0,6 мМ. Оцененная величина Ki для глифосата калия составила у мутантной EPSPS примерно 0,2 мМ.
Пример 19
Получение антител к очищенной EPSPS риса и методы Вестерн-блоттинга
Используют стандартные методы получения поликлональных антисывороток у кроликов. Используют молодых крольчих новозеландской белой породы. Курс иммунизации составляет 4 дозы, каждая примерно по 100 мг, вводимые с месячным интервалом. Каждую дозу в фосфатно-солевом буфере вводят в виде эмульсии с полным адъювантом Фройнда (1-я доза) или неполным адъювантом Фройнда (2-4-я дозы), которые инъецируют в четыре разных подкожных сайта. Предварительную пробу крови берут до введения 1-й дозы. Тест-пробы крови берут через 14 дней после второй дозы для подтверждения иммунного ответа. Окончательное взятие крови проводят через 14 дней после 4-й дозы, которую используют в эксперименте. Пятую и последнюю дозы вводят по прошествии по крайней мере 6 недель после 4-й дозы, а заключительное взятие крови (также используемое в эксперименте) проводят через 14 дней после указанного. Антитела инициируют и к (i) очищенной нативной зрелой EPSPS риса дикого типа (пример 8), и также к (ii) ДСН-денатурированному полипептиду EPSPS риса, который элюирован из полосы, вырезанной из 12%-ного ДСН-геля (точное положение белка маркируется путем параллельного сопоставления бэндов, окрашенных по Кумасси).
Для электрофореза в ДСН-геле и Вестерн-блоттинга используют 12%-ный полиакриламидный гель. Электрофорез проводят с постоянным током при 100 В в течение 90 мин. Гели блотируют на нитроцеллюлозные пластины в течение ночи при 4°С при напряжении постоянного тока 30 В. Пластины блокируют 5% фосфатно-солевым буфером Marvel, содержащим 0,1% Твин (ФСБ-Твин) в течение 1-2 ч, трижды промывают ФСБ-Твином и инкубируют с первичным антителом к EPSPS-Rb1 риса из расчета примерно 1,3 мг IgG/мл (что в норме эквивалентно разведению окончательной пробы крови 1:4000-1:20000). Такие разведения антител проводят в ФСБ (фосфатно-солевой буфер), содержащем 1% Marvel и 0,05% Твин-20, в течение 2 ч. Вторичное антитело, козье антикроличье с пероксидазой хрена (Sigma А-6154), применяют в разведении 1:10000 или 1:20000 в ФСБ, содержащем 0,05% Твин и 1% Marvel. Инкубацию с вторичным антителом продолжают 1 ч, блот промывают 3 раза в ФСБ (0,05% Твин), субстрат для ECL вносят обычно в течение 1 мин и пленку экспонируют в течение 30-60 с. Негативными контрольными блотами являются блоты с (1) преиммунной сывороткой в таких разведениях, чтобы дать тот же выход IgG, что и в иммунных сыворотках (IgG стандартным образом очищают из аликвоты каждой сыворотки и количественно оценивают так, чтобы указанные разведения можно было подсчитывать напрямую), а также с (2) иммунной сывороткой только к адъюванту Фройнда. Концентрацию IgG в контрольных иммунных сыворотках корректируют таким образом, чтобы контроли имели соответствующую концентрацию IgG. С целью такого подсчета IgG очищают из неочищенной антисыворотки с помощью фильтрации через шприцевой фильтр 0,45 мкм и пропускают через 1-мл колонку Hi Trap с белком-G (Pharmacia: каталожный №17-0404-01). Связанный IgG элюируют из колонки 0,1 М глицин-HCl, рН 2,9 и диализуют против ФСБ в течение ночи. Концентрацию IgG определяют с помощью УФ-детектора (1-см пробег раствора 1 мг/мл чистого IgG характеризуется поглощением 1,35 при длине волны 280 нм). На основе выхода IgG может быть выполнен подсчет концентрации IgG в неочищенной антисыворотке с соответствующим точным подсчетом разведении для Вестерн-блоттинга.
Образцы растительных тканей получают, например, как описано в примере 17. Альтернативно, для Вестерн-блоттинга используют более простую процедуру. 100-200 мг растительной ткани, предназначенной для анализа, быстро гомогенизируют (например, с помощью диспергатора Ultra Turrax, шарикового измельчителя или стеклянного гомогенизатора) в равном объеме буфера (сходного с буфером примера 7), центрифугируют в течение 5 мин в охлажденной эппендорфовской центрифуге и (а) небольшую аликвоту надосадочной фракции анализируют на белок по Брэдфорду, или (b) большую часть надосадочной фракции смешивают 1:1 с “буфером для образца” Laemli-ДСН, нагревают и затем хранят готовым для загрузки в гель. Обычно ДСН-гели загружают десятью образцами белка в 10 лунках. Обычно используют 1-10 мкг неочищенных экстрактов растительного материала для анализа параллельно калибровочной кривой от 0 до 20 нг чистой EPSPS риса. В некоторых случаях Вестерн-блоты разгоняют с использованием антител, сформированных к очищенной EPSPS дикого типа от брюквы Brassica napus (экспрессированной и очищенной с использованием методов, сходных с описанными выше). В этом случае сила перекрестной реакции антител оказывается меньше по отношению к EPSPS риса (или с EPSPS, эндогенной для растений пшеницы или кукурузы) по сравнению с антителами, сформированными к EPSPS риса, но, несмотря на это, проявляет уровень реакции, достаточный для получения эффективной количественной информации по отношению к калибровочной кривой.
Пример 20
Выделение геномной ДНК из трансгенного растительного материала. ПЦР-анализ. Получение и гибридизация ДНК-зондов. Число копий и целостность трансгена
Геномную ДНК выделяют из материала растений, проростков и каллюсов с помощью, например, метода Dellporta et al. (1983) in Chromosome Structure and Function: Impact of New Concepts, 18’th Stadler Genetics Symposium. J.P.Gustafson and R.Appels, eds). New York: Plenum Press) pp. 263-283, или, альтернативно, с использованием набора DNAeasy kit (Qiagen). Трансгенные каллюсы и растения-сегреганты, которые несут мутантный трансген EPSPS риса, идентифицируют с использованием флуоресцентной ПЦР с олигонуклеотидными праймерами SEQ ID NO. 39 и 40, которые специфичны в отношении мутаций в пределах геномной последовательности EPSPS риса. Флуоресцентный краситель SYBR зеленки, который интеркалирует двухцепочечную ДНК, включают в ПЦР таким образом, что образцы, включающие мутантный ген EPSPS риса, идентифицируются по повышению флуоресценции образца, что выявляется на устройстве ABI 3377. Альтернативно, специалистам в данной области техники известно, что праймеры могут быть помечены флуоресцентным образом, причем доступны такие методы, как “молекулярный маяк” и “Scorpions”.

Обычная реакция ПЦР, приготовленная на 96-луночных планшетах Optical, закрытых лентой Optical (РЕ Biosystems), соответствует следующему:
5,0 мкл геномной ДНК-матрицы (препарат DNAeasy, Qiagen),
12,5 мкл 2х премикса SYBR зеленого;
2,5 мкл 5 пкмоль/мкл маточного прямого праймера;
2,5 мкл 5 пкмоль/мкл маточного обратного праймера;
2,5 мкл дважды дистиллированной воды;
общий объем - 25,0 мкл.
Параметры циклов следующие:
стадия 1:2 мин при 50°С,
стадия 2:10 мин при 95°С;
стадия 3: 50 циклов по 15 с при 95°С и по 45 с при 60°С.
Изменения флуоресценции в образцах регистрируют каждые 7 с со стадии 3 реакции.
Для Саузерн-блоттинга приблизительно 10 мкг ДНК используют для каждого расщепления рестриктазами. Геномную ДНК расщепляют подходящими рестриктазами (например, HindIII) в соответствии с инструкциями изготовителя (например, Promega). Выбирают рестрикционные ферменты, которые расщепляют ДНК и в пределах, и за пределами мутантной последовательности EPSPS. ДНК разделяют с использованием 0,8%-ных агарозных гелей с ТАЕ (0,04 М Трис-ацетат, 1 мМ ЭДТА). Саузерн-блоттинг осуществляют в соответствии с методами, предлагаемыми Sambrook et al., 1989, с использованием нитроцеллюлозной мембраны HyBond N+ для блоттинга (Amersham Pharmacia). ДНК перекрестно сшивают с указанной мембраной действием ультрафиолетового облучения.
Фрагменты ДНК, используемые для получения специфичных зондов, выделяют с помощью очистки в гелях рестрикционных фрагментов плазмидной ДНК или получают с помощью ПЦР. Например, фрагмент длиной 700 п.н., включающий 1-й интрон гена EPSPS риса, получают с помощью ПЦР с использованием показанных далее праймеров:
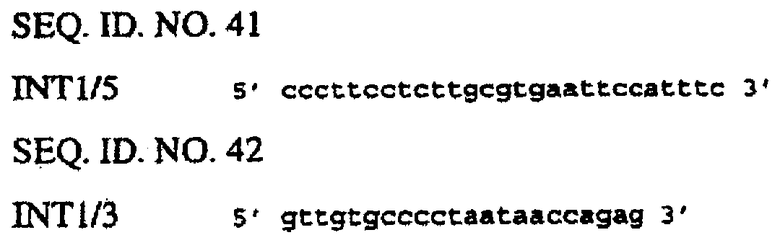
Такие зонды помечают 32P с использованием метода случайного мечения, например, с помеченными шариками Ready-To-Go NA labelling beads (Amersham Pharmacia), и очищают, например, на колонках MicroSpin G-25 (Amersham Pharmacia).
Блоты гелей с ДНК прегибридизуют при 65°С в 5× SSC, 0,5% ДСН, 2× раствор Денхардта, 0,15 мг/мл денатурированной ДНК спермы лосося по крайней мере в течение 1 ч. Затем блот гибридизуют с денатурированным зондом в течение 16-24 ч при 65°С в свежем прегибридизационном растворе. Мембраны блотируют досуха и проявляют с помощью авторадиографии.
Если Саузерн-блоттинг указывает на единственное событие встраивания трансгена по единственному локусу, о чем говорит гибридизация зонда с единственным рестрикционным фрагментом конкретного размера, то такой результат подтверждают путем повторной гибридизации блота с использованием альтернативного зонда. Для контроля используют нетрансформированный материал. Дополнительно блот может быть прозондирован еще с гибридизационными зондами, специфичными в отношении иных участков трансгенной конструкции (например, промотора, сегмента 5’UTR в интроне или расположенной выше энхансерной последовательности) с целью подтверждения целостности конструкции. Кроме того, в случае, когда применяют трансформацию бактерией Agrobacterium, конкретные зонды используют для указания на присутствие или отсутствие любой ДНК, расположенной вне пределов, ограниченных граничными сайтами RB и LB супербифункционального вектора.
SEQ ID NO. 43. Геномная последовательность EPSPS риса (от ATG - последовательность дикого типа)

SEQ ID NO. 44. Геномная последовательность EPSPS риса (от ATG - включая двойную мутацию: отмечены)




| название | год | авторы | номер документа |
|---|---|---|---|
| РАСТЕНИЕ КУКУРУЗЫ DBN9858, УСТОЙЧИВОЕ К ГЕРБИЦИДУ, И НУКЛЕОТИДНАЯ ПОСЛЕДОВАТЕЛЬНОСТЬ И СПОСОБ ДЛЯ ЕЕ ВЫЯВЛЕНИЯ | 2016 |
|
RU2712730C2 |
| УСТОЙЧИВЫЕ К ГЛИФОСАТУ РАСТЕНИЯ И СПОСОБЫ, СВЯЗАННЫЕ С НИМИ | 2013 |
|
RU2627169C2 |
| НОВЫЙ КЛАСС ГЕНОВ УСТОЙЧИВОСТИ К ГЛИФОСАТУ | 2013 |
|
RU2634411C2 |
| Белок устойчивости к гербицидам, кодирующий его ген и их применение | 2016 |
|
RU2692553C2 |
| Белок устойчивости к гербицидам, кодирующие его гены и их применение | 2016 |
|
RU2681162C1 |
| ГЕНЫ GRG23 И GRG51, ПРИДАЮЩИЕ УСТОЙЧИВОСТЬ К ГЕРБИЦИДАМ | 2006 |
|
RU2393225C2 |
| ТОЛЕРАНТНОЕ К СТРЕССУ ТРАНСГЕННОЕ РАСТЕНИЕ ПШЕНИЦЫ | 2005 |
|
RU2376377C2 |
| НОВЫЙ КЛАСС ГЕНОВ УСТОЙЧИВОСТИ К ГЛИФОСАТУ | 2013 |
|
RU2721122C2 |
| УСТОЙЧИВЫЕ К ГЛИФОСАТУ РАСТЕНИЯ И СПОСОБЫ, СВЯЗАННЫЕ С НИМИ | 2013 |
|
RU2636037C2 |
| ИЗОЛИРОВАННАЯ ПОСЛЕДОВАТЕЛЬНОСТЬ ДНК (ВАРИАНТЫ), РЕКОМБИНАНТНАЯ ДВУНИТЕВАЯ МОЛЕКУЛА ДНК И СПОСОБ ПОЛУЧЕНИЯ ГЕНЕТИЧЕСКИ ТРАНСФОРМИРОВАННЫХ РАСТЕНИЙ | 1991 |
|
RU2146290C1 |
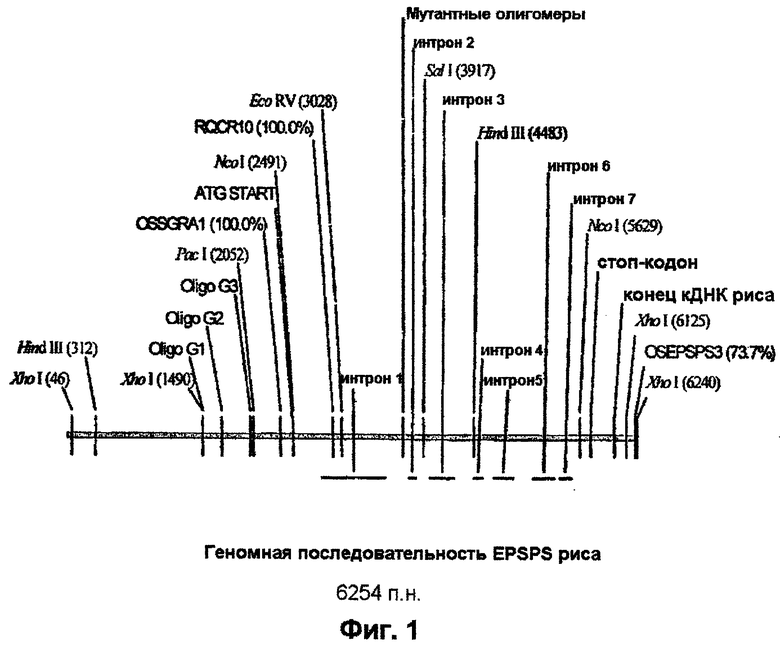























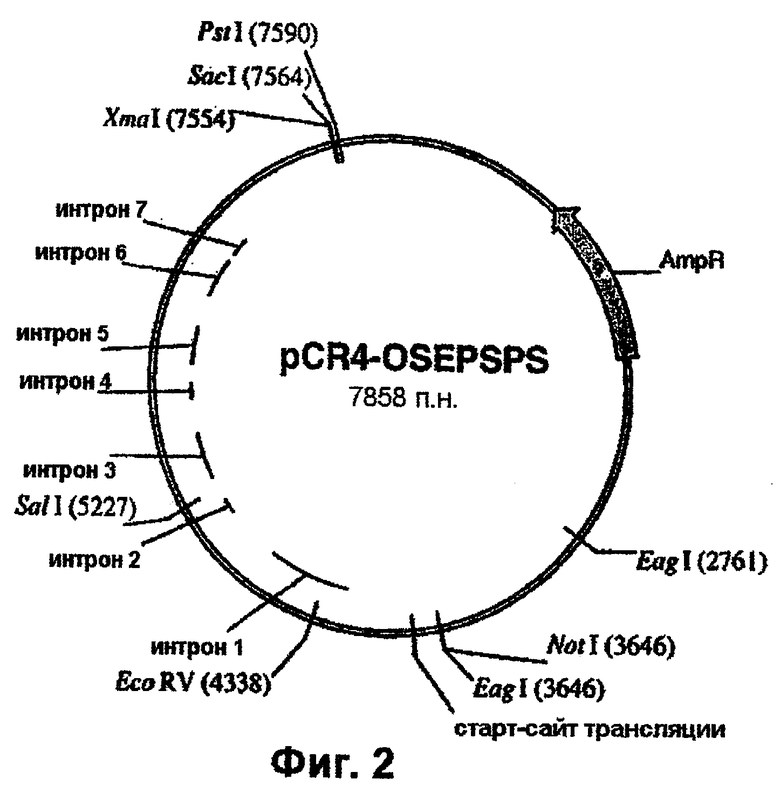
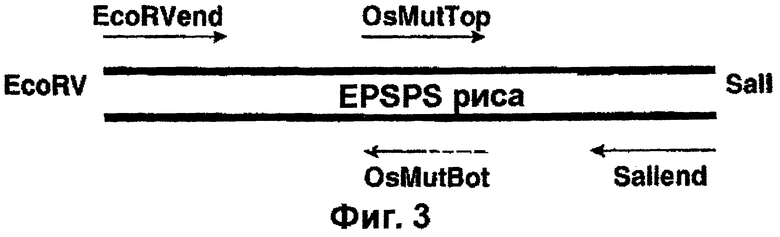
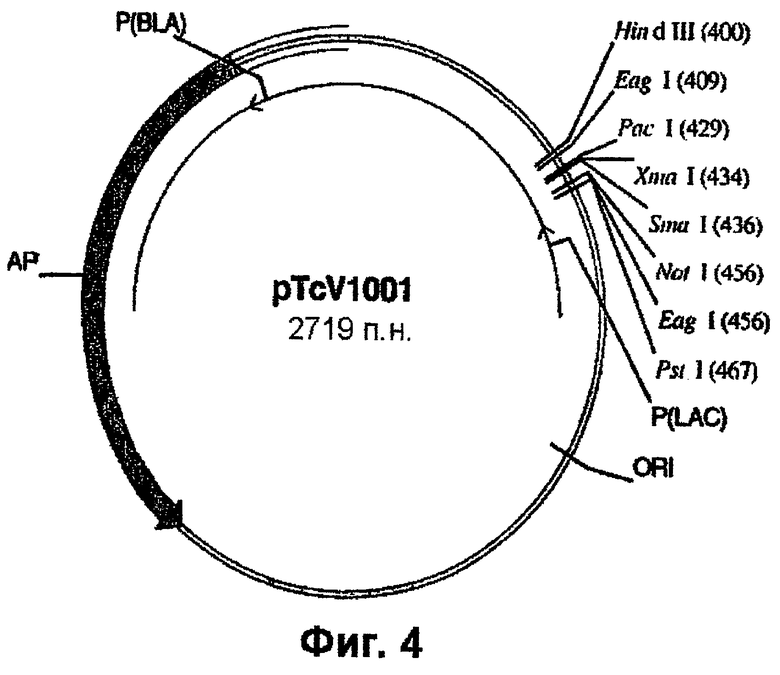
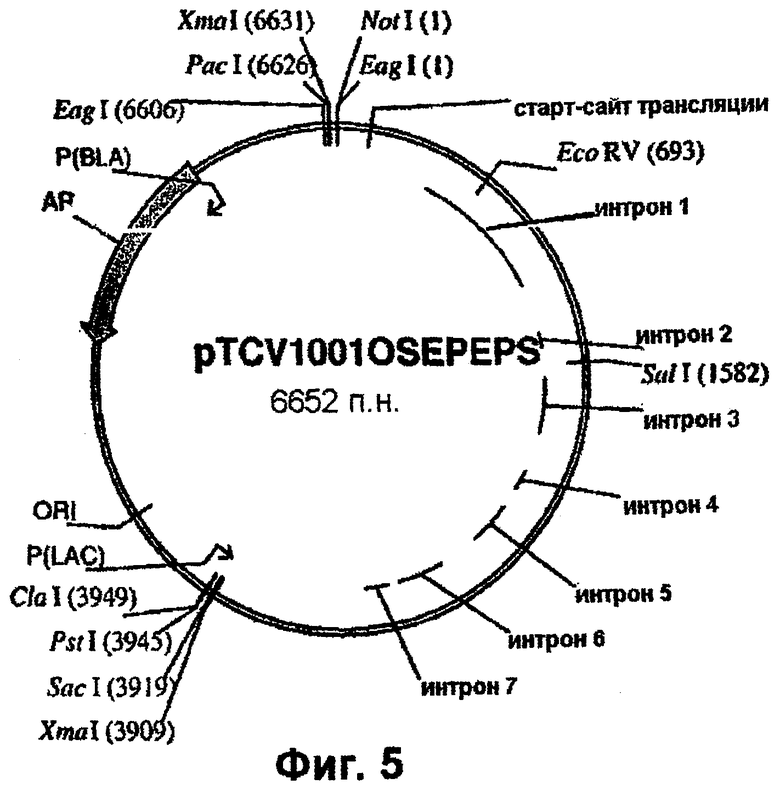
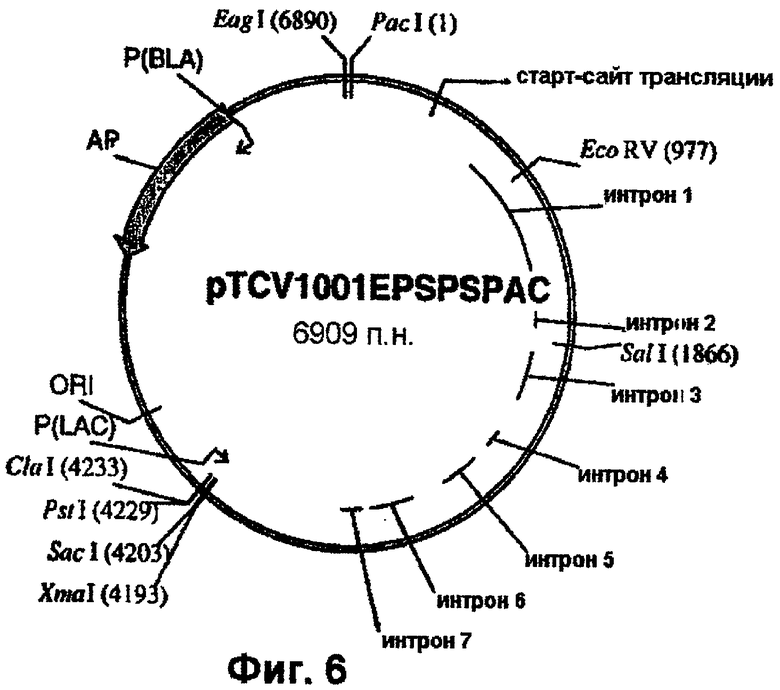
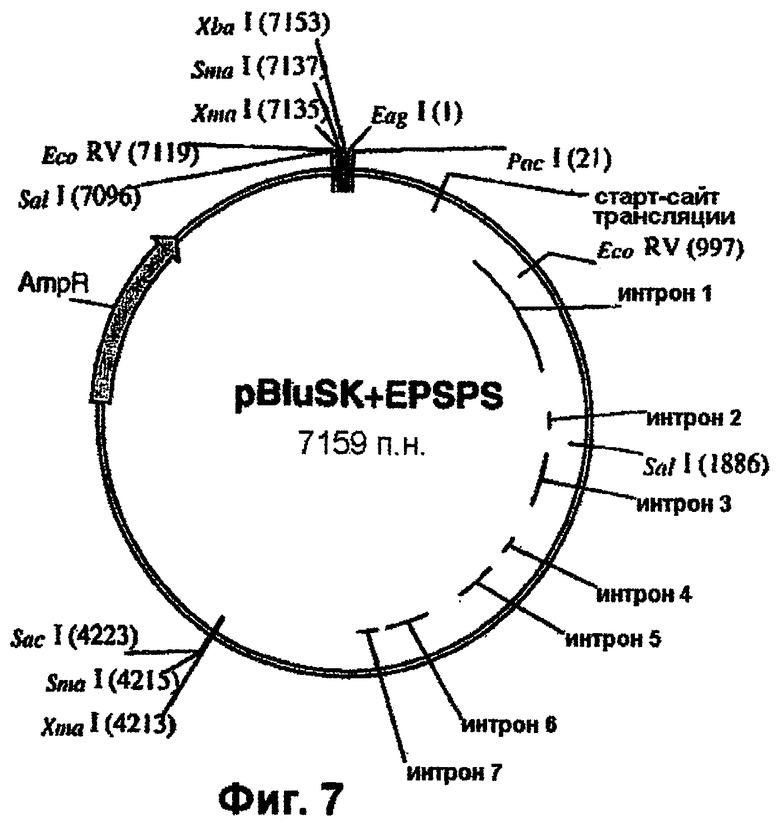
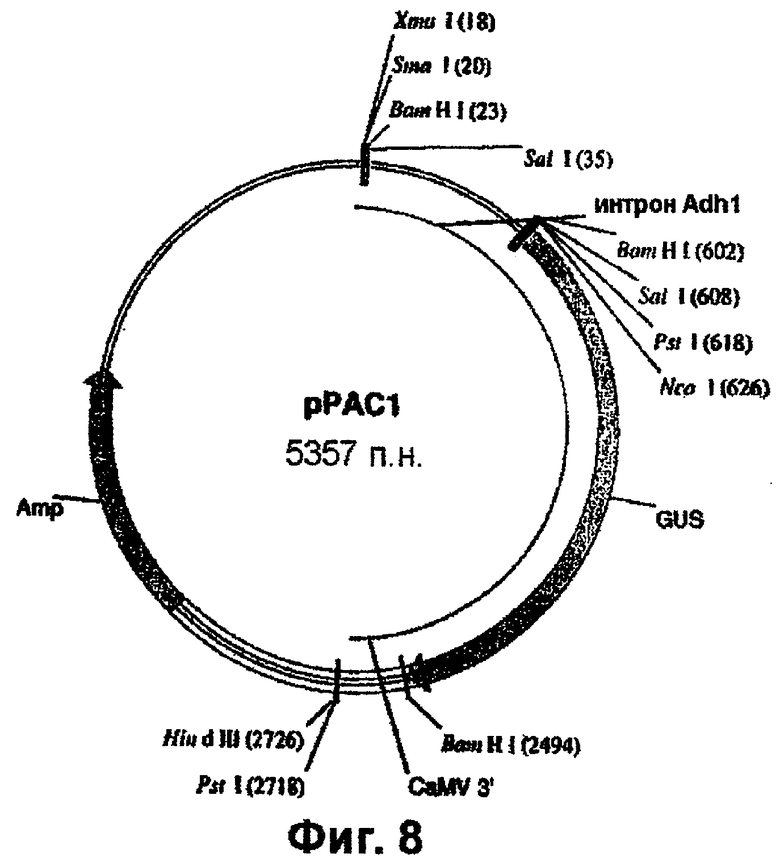
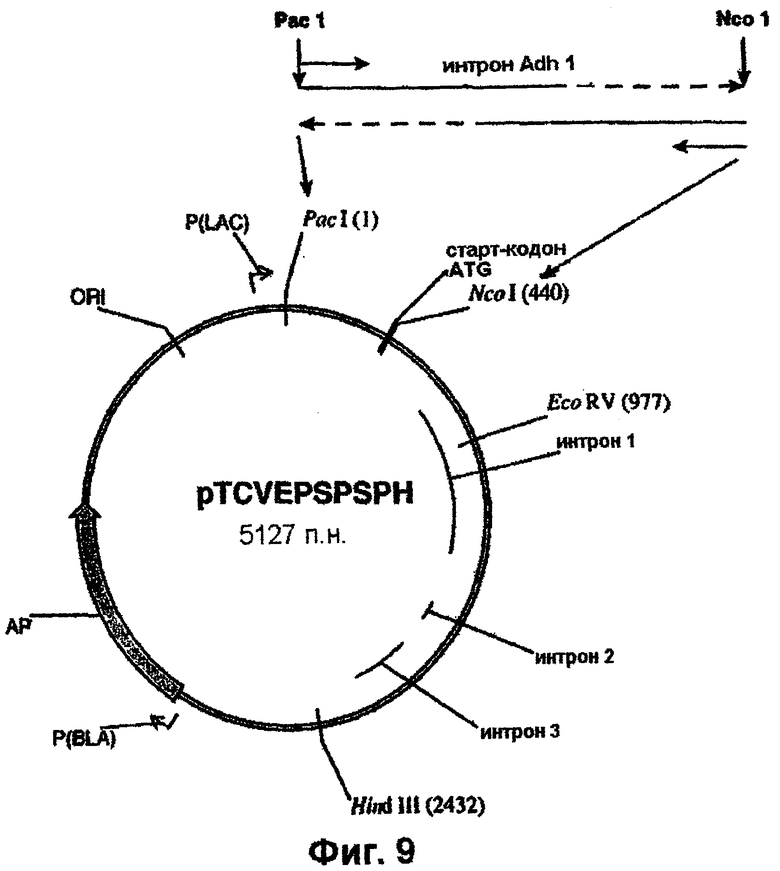
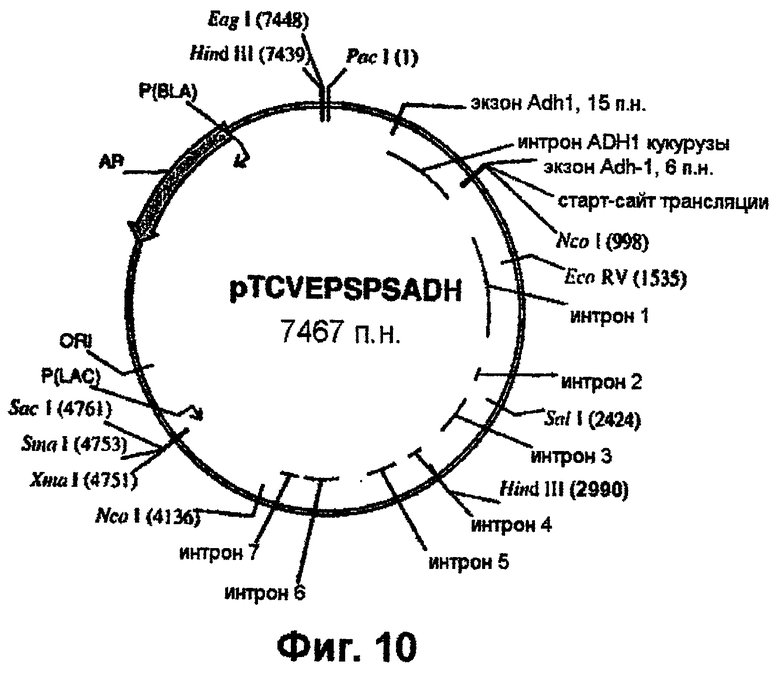
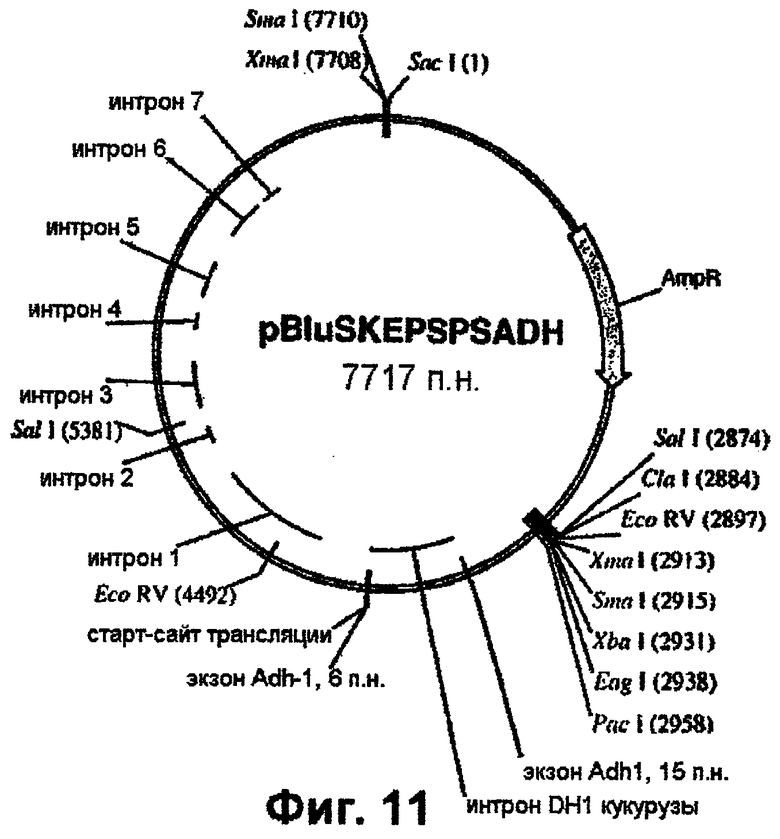
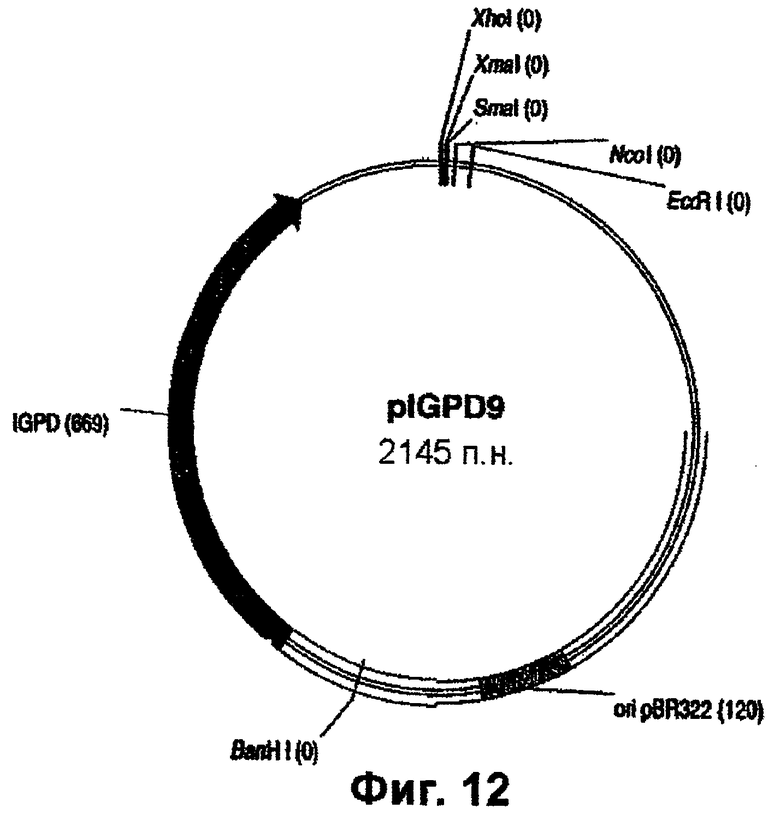
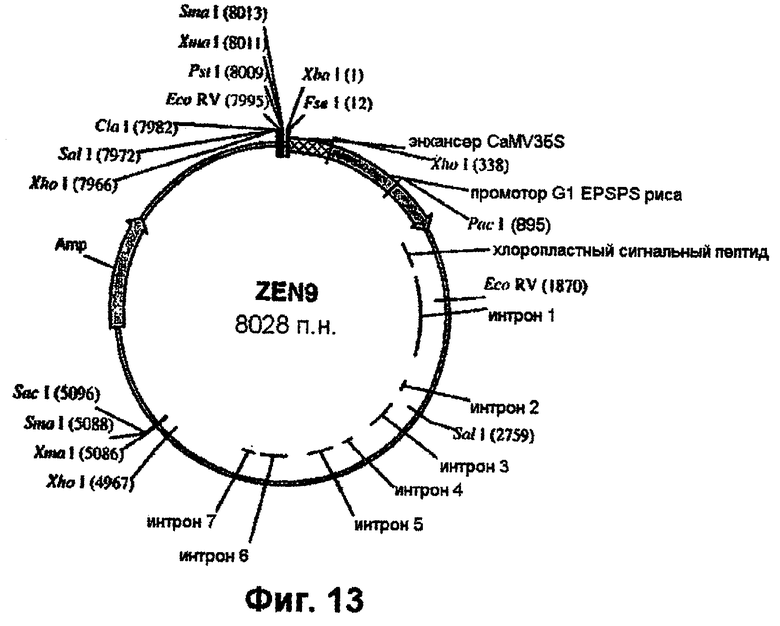
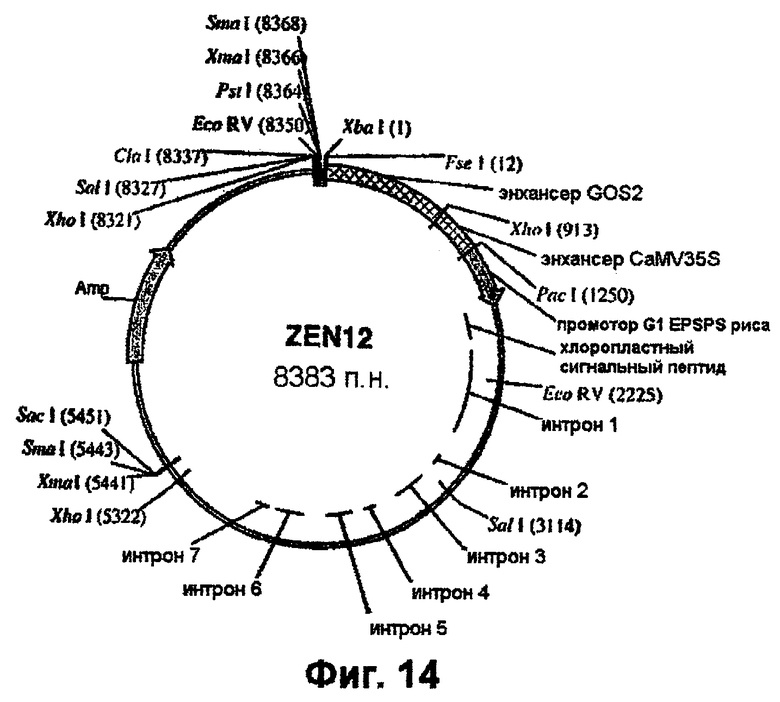
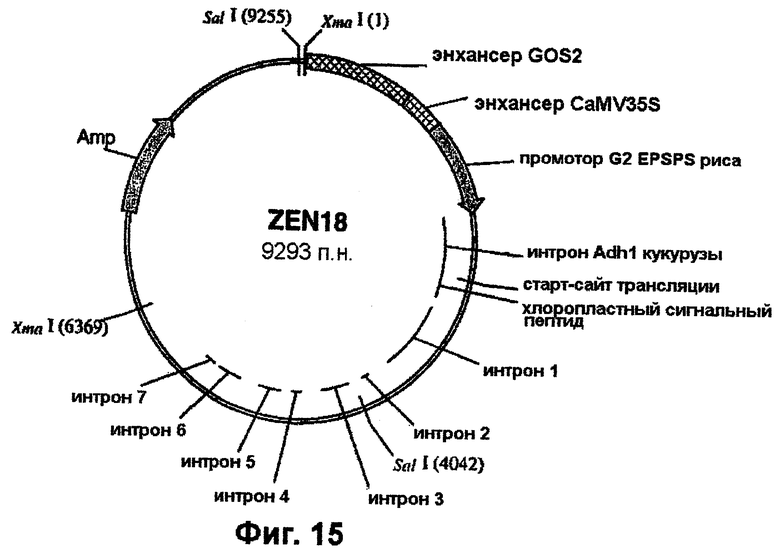
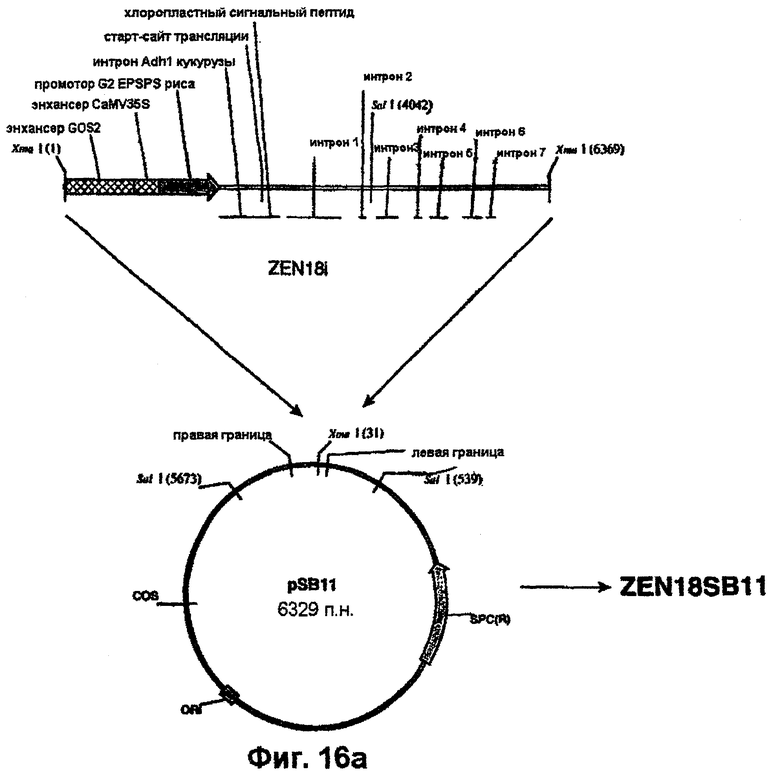
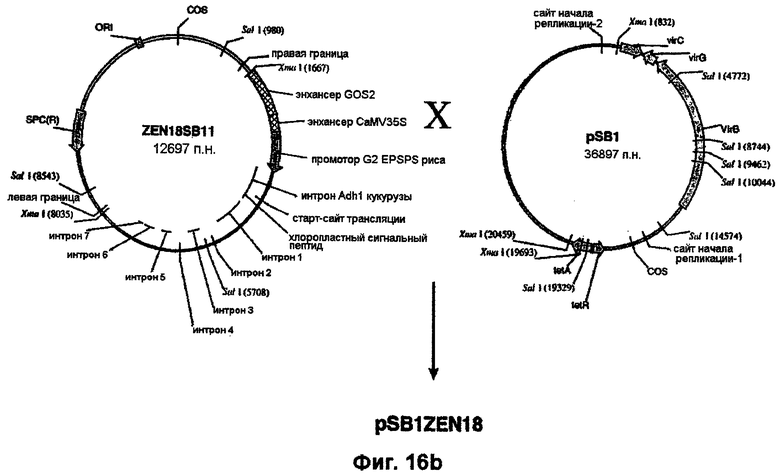
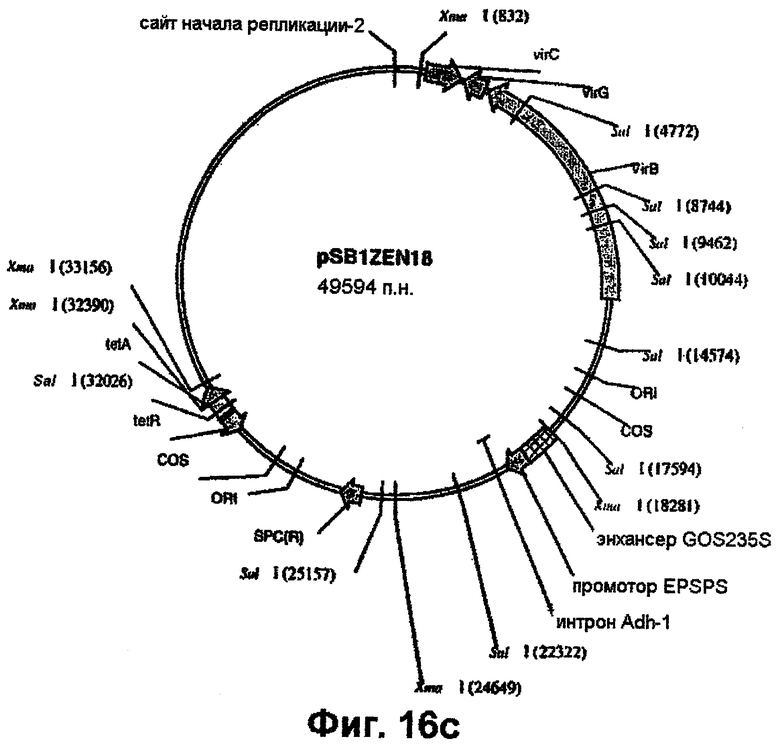
Изобретение относится к биотехнологии, в частности к технологии рекомбинантных ДНК, необходимых для получения трансгенных растений, устойчивых или проявляющих значительную толерантность к гербицидам. Выделяют полинуклеотид, кодирующий хлоропластный сигнальный пептид и глифосат-устойчивую 5-енолпирувилшикимат-3-фосфатсинтазу (EPSPS) риса, расположенную по ходу транскрипции за участком, кодирующим хлоропластный сигнальный пептид. Экспрессия указанного участка находится под контролем растительного функционального промотора, который не явлется гетерологичным по отношению к указанному участку, и хлоропластный сигнальный пептид не является гетерологичным по отношению к указанной синтазе. Указанные компоненты находятся в последовательности по направлению 5’→3’ транскрипции, с возможностью вариантов расположения энхансеров. Вместе с тем последовательность, кодирующая EPSPS риса, модифицирована таким образом, что по первому положению консервативного участка GNAGTAMRPLTAAV фермента дикого типа Thr заменен Ile, а по второму положению Pro заменен Ser, таким образом модифицированная последовательность представляет собой GNAGIAMRSLTAAV. Указанный полинуклеотид включают в векторную конструкцию, с помощью которой трансформируют растительный материал. Полученный трансформированный материал подвергают отбору и дальнейшей регенерации для получения целых растений, устойчивых или толерантных к глифосатному гербициду. Регенерация включает в себя получение ткани из растения, подлежащего трансформированию, помещение трансформированной указанным выше полинуклеотидом или вектором ткани на питательную среду, содержащую соединение для обеспечения возможности идентификации или отбора трансформированной регенерируемой ткани, перенос образовавшегося, по меньшей мере, одного побега во вторую среду, способствующую продуцированию корней, и выращивание указанного побега до стадии взрослого растения, способного к воспроизведению. Обрабатывают поле сельскохозяйственных трансгенных растений, засоренное сорняками, глифосатом в количестве, эффективном для борьбы с сорняками без существенного влияния на сельскохозяйственные растения. Изобретение позволяет снизить производственные затраты и повысить сбор сельскохозяйственной продукции. 5 н. и 9 з.п. ф-лы, 18 ил., 2 табл.
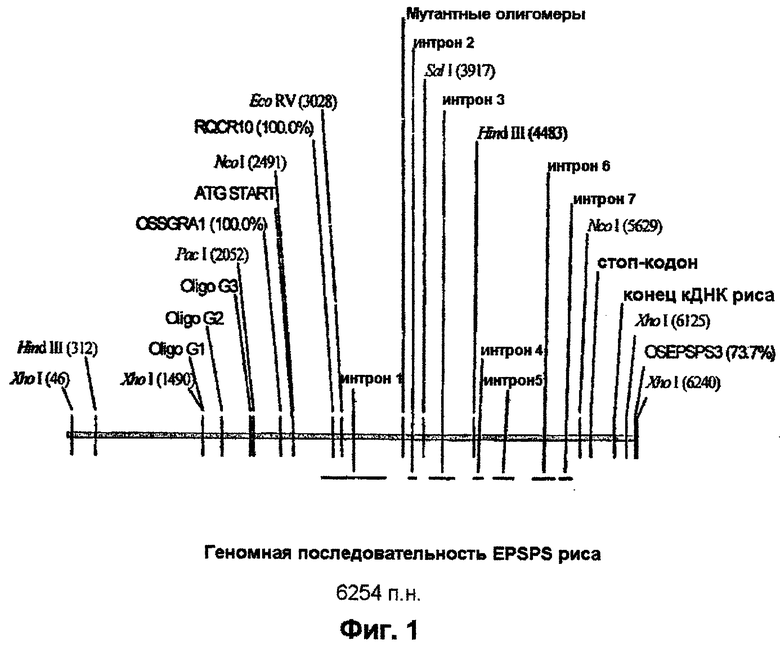
| WO 9104323, 04.04.1991 | |||
| GASSER С.S | |||
| ет al | |||
| Journal of biological chemistry | |||
| Механическая топочная решетка с наклонными частью подвижными, частью неподвижными колосниковыми элементами | 1917 |
|
SU1988A1 |
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| WO 9844140, 08.10.1998 | |||
| WO 9206201, 16.04.1992 | |||
| US 5188642, 23.02.1993 | |||
| RU 93038869, 10.03.1996. | |||

