Область техники, к которой относится изобретение.
Изобретение относится к фармацевтической промышленности, в частности к N-замещенным индол-3-глиоксиламидам, обладающим противоастматическим, противоаллергическим и иммунодепрессивным/иммуномодуляторным действием.
Уровень техники.
Индол-3-глиоксиламиды являются фармацевтически динамичными активными соединениями и широко используются в качестве исходных соединений для синтеза в фармацевтической химии.
В патентной заявке NL (Нидерланды) 6502481 описаны соединения, обладающие противовоспалительным и жаропонижающим свойствами и проявляющие обезболивающую активность.
В британской заявке GB-PS 1028812 производные индолил-3-глиоксиловой кислоты и вышеупомянутые амиды рассматриваются в качестве обезболивающих, противосудорожных и β-адренергических соединений.
В статье G. Domschke et al. (Ber., 94, 2353 (1961)) описаны 3-индолилглиоксиламиды, которые не охарактеризованы с точки зрения фармакологии.
Е. Walton et al. (J. Med. Chem., 11, 1252 (1968) сообщают о производных индолил-3-глиоксиловой кислоты, которые ингибируют глицерофосфатдегидрогеназу и лактатдегидрогеназу.
В европейской патентной заявке ЕР 0675110 А1 описаны амиды 1Н-индол-3-глиоксиловой кислоты, обладающие ингибирующим действием на SPLA2 и использующиеся при лечении септического шока, панкреатита, аллергического ринита и ревматического артрита.
Сущность изобретения.
Задачей изобретения является выбор новых соединений, обладающих противоастматическим, противоаллергическим и иммунодепрессивным/иммуномодуляторным действием, из ряда производных индолил-3-глиоксиловой кислоты.
Кроме того, задачей изобретения является разработка химического способа получения таких соединений, способов переработки новых соединений в галеновые препараты, а также способов получения их лекарственных форм.
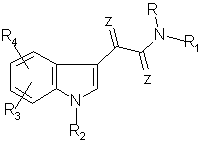
Предметом изобретения являются соединения общей формулы I

где R означает водород, (С1-С6)алкил, причем алкильная группа необязательно содержит один фенильный заместитель, который, в свою очередь, необязательно содержит по меньшей мере один заместитель, выбранный из группы, включающей галоген, метокси, этокси, (С1-С6)алкил;
R1 означает фенильный цикл, содержащий по меньшей мере один заместитель, выбранный из группы, включающей (С1-С6)алкокси, гидрокси, нитро, (С1-С6)алкоксикарбониламино или один фтор, или R1 представляет собой остаток пиридина формулы II
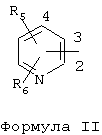
где углеродные атомы 2, 3 и 4 остатка пиридина необязательно имеют одинаковые или различные заместители R5 и R6, причем R5 и R6 обозначают (С1-С6)алкил или галоген, или R1 представлен аллиламинокарбонил-2-метилпроп-1-ильной группой, или R и R1 вместе с атомом азота, к которому они присоединены, образуют цикл пиперазина формулы III
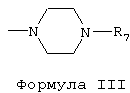
R7 обозначает фенил или пиридинил;
R2 означает (С1-С6)алкил, который необязательно содержит фенильный остаток, который, в свою очередь, необязательно замещен галогеном, метоксигруппой или этоксигруппой, или относящаяся к R2 (С1-С6)алкильная группа необязательно замещена 2-, 3- или 4-пиридильным остатком;
R3 и R4 являются одинаковыми или различными заместителями и обозначают водород, гидрокси, (С1-С6)алкокси, (С1-С3)алкоксикарбониламино- или (С1-С3)алкоксикарбониламино(С1-С3)-алкил или R3 представляет собой циклопентилоксикарбониламиногруппу;
Z означает О,
а также их фармацевтически приемлемые соли с кислотами.
Термины алкил-, алканол-, алкокси- или алкиламиногруппа относятся к остаткам R, R1, R2, R3, R4, R5, R6, R7 и означают упорядоченные как "неразветвленные", так и "разветвленные" алкильные группы, например, "неразветвленные группы" означают метил, этил, н-пропил, н-бутил, н-гексил, а в качестве "разветвленных" алкильных групп используют, например, изопропил или трет-бутильную группы.
Термин "галоген" означает фтор, хлор, бром или йод. "Алкоксигруппа" означает, например, метокси, пропокси, бутокси, изопропокси, изобутокси или пентоксигруппу.
Соединения согласно изобретению можно использовать в виде солей-аддуктов с кислотами, например солей минеральных кислот, таких как соляная кислота, серная кислота, фосфорная кислота; солей органических кислот, таких как уксусная кислота, молочная кислота, малоновая кислота, малеиновая кислота, фумаровая кислота, глюконовая кислота, глюкуроновая кислота, лимонная кислота, эмбоновая кислота (4,4’-метиленбис(3-гидрокси-2-нафтеновая кислота)), метансульфоновая кислота, трифторуксусная кислота и янтарная кислота.
Соединения формулы I, также, как и их соли, являются биологически активными. Соединения формулы I можно вводить в качестве лекарства как в свободной форме, так и в виде солей с физиологически переносимыми кислотами.
Соединения можно вводить перорально, парентерально, внутривенно, подкожно или через дыхательные пути.
Таким образом, изобретение относится также к лекарственным препаратам, обладающим противоастматическим, противоаллергическим и иммунодепрессивным/иммуномодуляторным действием и содержащим, по крайней мере, одно соединение формулы I или его соль с физиологически переносимыми неорганическими или органическими кислотами и в случае необходимости фармацевтически применимые вещества - носитель и/или разбавитель или вспомогательные вещества. Кроме этого, изобретение касается способа получения указанных лекарственных препаратов.
В качестве лекарственных форм используют, например, таблетки, драже, капсулы, растворы или соответственно ампулы, свечи, пластыри, ингаляционные композиции, суспензии, кремы и мази.
Изобретение также относится к способу получения N-замещенных индол-3-глиоксиламидов формулы I, где R, R1, R2, R3, R4 и Z имеют указанные выше значения, заключающемуся в том, что производное индола формулы IV

где R3 и R4 имеют вышеуказанные значения,
смешивают с основанием, суспендированным в протонном диполярном, апротонном или неполярном органическом растворителе, и с реакционноспособным соединением, содержащим остаток R2, где R2 имеет вышеуказанные значения, и получают производное 1-индола формулы V

где R2, R3 и R4 имеют вышеуказанные значения,
которое смешивают с апротонным или неполярным органическим растворителем и реакционноспособным соединением формулы VI
(C-Z-Hal)2
где Z является атомом кислорода и Hal обозначает галоген, выбранный из группы, содержащей фтор, хлор, бром или иод,
затем обрабатывают полученный продукт первичным или вторичным амином формулы VII
HNRR1
где R и R1 имеют вышеуказанные значения,
в апротонном или диполярном апротонном растворителе и выделяют целевое соединение формулы I.
Изобретение также касается способа получения N-замещенных индол-3-глиоксиламидов формулы I, где R, R1, R2, R3, R4 и Z имеют указанные выше значения, заключающемуся в том, что производное индола формулы IV

где R3 и R4 имеют вышеуказанные значения,
смешивают в апротонном или неполярном растворителе с реакционноспособным соединением формулы VI
(C-Z-Hal)2
где Z является атомом кислорода и Hal означает галоген, выбранный из группы, содержащей фтор, хлор, бром или иод,
обрабатывают полученный продукт в апротонном или диполярном апротонном растворителе первичным или вторичным амином формулы VII
HNRR1
где R и R1 имеют вышеуказанные значения,
затем полученное производное 3-индола формулы VIII

где R, R1, R3, R4 и Z имеют вышеуказанные значения,
смешивают в присутствии суспендированного основания в протонном диполярном, апротонном или неполярном органическом растворителе с реакционноспособным соединением, содержащим остаток R2, где R2 имеет вышеуказанные значения, и выделяют целевое соединение формулы I.
Соединения согласно изобретению обладают хорошим противоастматическим, противоаллергическим и иммунодепрессивным/иммуномодуляторным действием, например, при трансплантации и заболеваниях, таких как псориаз, ревматоидные заболевания и хронический полиартрит, которое было исследовано на следующих фармакологических моделях.
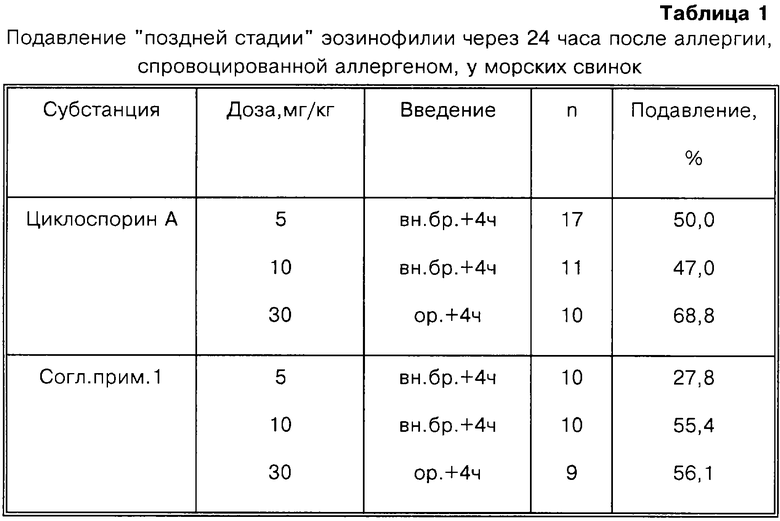
Подавление "поздней стадии" эозинофилии с помощью бронхоальвеолярного лаважа (BAL) в течение 24 часов после аллергии, спровоцированной у морских свинок.
Самцов морских свинок (200-250 г, Dunkin Hartley Shoe) активно сенсибилизируют овальбумином подкожно (10 мкг овальбумина + 1 мг Аl(ОН)3) и через 2 недели повторно иммунизируют. Через одну неделю после иммунизации овальбумином у животных вызывают аллергию овальбумином (0,5% раствором) ингаляционным способом в течение 20-30 секунд. Через 24 часа животных убивают сверхдозой уретана, обескровливают и проводят бронхоальвеолярный лаваж (BAL) 2×5 мл 0,9%-ного физиологического раствора. Лаважные смывы объединяют и центрифугируют в течение 10 минут при 400 g, полученный осадок суспендируют в 1 мл 0,9%-ного физиологического раствора. Число эозинофилов подсчитывают в микроскопе после окрашивания набором реактивов (Becton Dickinson Testkit Nr. 5877) с использованием камеры Нейбауэра. В этот набор входит флоксин В, который является реагентом, избирательно окрашивающим эозинофилы. При этом рассчитывают число эозинофилов в BAL для каждого животного и эозинофилию выражают величиной (миллионы/животное). Для каждой группы рассчитывают среднюю величину и стандартное отклонение. Процентное подавление эозинофилии для группы животных, обработанных тестируемым соединением, определяют по следующей формуле:
(А-В)-(В-С)/(А-С)x100 = % подавления,
где А - эозинофилия у группы необработанных животных со спровоцированной аллергией,
В - эозинофилия у группы обработанных животных и
С - эозинофилия у контрольной группы животных без спровоцированной аллергии.
Для предотвращения смерти за 2 часа до провокации аллергии животных обрабатывают Н1-антагонистами гистамина (азеластин, 0,01 мг/кг перорально). Тестируемые соединения или индифферентную основу лекарственного препарата вводят через 4 часа после провокации аллергии. Процентное подавление эозинофилии в BAL рассчитывают для группы из 6-10 животных. Полученные данные суммированы в таблице 1.
Метод определения активности пептидилпролилизомеразы (РРI) и подавления активности.
PPI-активность циклофилина определяли ферментативным методом согласно Fischer с соавт. (1984). Пептидилпролилизомераза (PPI) катализирует изомеризацию субстрата, после чего он становится доступным для химотрипсина и происходит отщепление хромофора (п-нитроанилина). Для определения подавления PPI-активности в присутствии субстанции используют рекомбинантный циклофилин В человека (Сур В). Взаимодействие Сур В с потенциальным ингибитором проводят следующим образом.
Раствор очищенного Сур В определенной концентрации инкубируют в присутствии 1 мкМ субстанции в течение 15 минут. PPI-реакцию инициируют добавлением в реакционную смесь раствора субстрата, HEPES-буфера, химотрипсина и либо тестируемой пробы, либо контрольной пробы. В этих условиях реакция подчиняется закону первого порядка с константой
Kнабл=Kо+Kферм,
где Кo - спонтанная изомеризация, Кферм - скорость изомеризации, катализируемой PPI.
Величины поглощения, соответствующие количеству отщепленного хромофора, измеряют при постоянной температуре реакции, равной 10°С, с использованием спектрофотометра Beckman DU 70.
Наблюдаемую остаточную активность различных субстанций сравнивают с циклофилинами, обработанными только растворителем. Результаты выражают в % остаточной активности. В качестве стандартного соединения используют циклоспорин A (CsA). Кроме того, подавление PPI-активности контролируют методом электрофореза в полиакриамидном геле в присутствии SDS-PAGE.
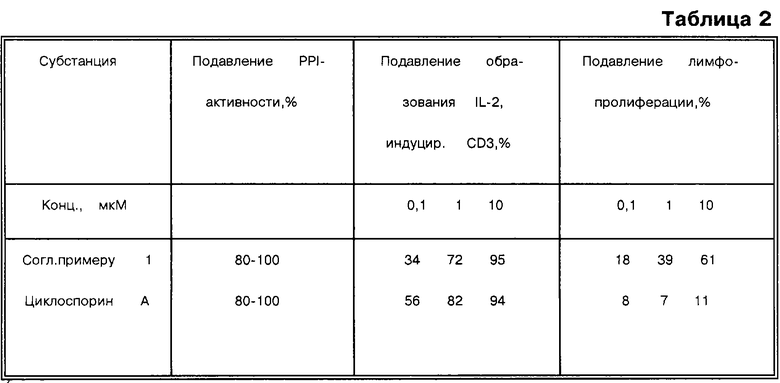
Колориметрический метод (основанный на использовании МТТ) для нерадиоактивного определения пролиферации клеток и их жизнеспособности.
МТТ используют для количественного определения пролиферации клеток и их активации, например, с помощью ростовых факторов и цитокинов, таких как IL-2 и IL-4, а также для количественной оценки антипролиферативного или токсического действия.
Метод основан на расщеплении желтой соли тетразолия МТТ до образования пурпурно-красных кристаллов формазана в присутствии метаболически активных клеток.
Клетки выращивают в 96-луночном планшете для клеточных культур и инкубируют с желтым раствором МТТ в течение приблизительно 4 часов. После указанного периода образуются пурпурно-красные кристаллы соли формазана, которые не растворяются в водном растворе, но которые можно растворить при добавлении реагента для растворения и после инкубирования в течение ночи. Количество растворенной соли формазана определяют спектрофотометрически с использованием ридера для иммуноферментного анализа (ELISA). Увеличение числа живых клеток приводит к повышению общей метаболической активности в пробе. Это увеличение напрямую коррелирует с количеством образующихся пурпурно-красных кристаллов формазана, которое измеряют по величине поглощения (таблица 2).
Способ получения соединений согласно изобретению описан в нижеследующих схемах 1 и 2, приведены также краткие описания синтеза. Все соединения могут быть получены по приведенным схемам или аналогичным способом.
Соединения общей формулы I синтезируют согласно следующей схеме 1. На схеме представлен синтез соединения согласно примеру 1.

Краткое описание получения соединений общей формулы I согласно схеме 1.
Стадия 1
Для синтеза можно использовать производное индола либо без заместителей, либо с заместителем в положении С-2, либо с одним или несколькими заместителями в фенильном цикле. Производное индола растворяют в протонном, диполярном апротонном или неполярном органическом растворителе, например изопропаноле, тетрагидрофуране, диметилсульфоксиде, диметилформамиде, диметилацетамиде, N-метилпирролидоне, диоксане, толуоле или хлористом метилене, и раствор медленно прикапывают в атмосфере азота к стехиометрическому количеству или к избытку от стехиометрического количества основания в виде суспензии, загруженного в трехгорлую колбу, например к гидриду натрия, порошкообразному гидроксиду калия, трет-бутилату калия, диметиламинопиридину или амиду натрия в подходящем растворителе. Затем к реакционной смеси добавляют, например, алкил-, аралкил- или гетероаралкилгалогенид, а также в подходящем случае добавляют катализатор, например медь, и реакционную смесь выдерживают, например, от 30 минут до 12 часов при температуре реакционной смеси от 0 до 120°С, предпочтительно от 30 до 80°С, более предпочтительно от 50 до 65°С. После завершения реакции реакционную смесь добавляют в воду. Раствор экстрагируют, например, диэтиловым эфиром, дихлорметаном, хлороформом, метил-трет-бутиловым эфиром или тетрагидрофураном и в каждом случае органическую фазу сушат безводным сульфатом натрия. Органическую фазу концентрируют в вакууме, остаток кристаллизуют затиранием или маслообразный остаток очищают перекристаллизацией, дистилляцией или колоночной хроматографией или тонкослойной хроматографией на силикагеле или окиси алюминия. В качестве подвижной фазы используют, например, смесь дихлорметана и диэтилового эфира в соотношении 8:2 (об./об.) или смесь дихлорэтана и этанола в соотношении 9:1 (об./об.).
Стадия 2
N-замещенный индол, полученный по указанной выше стадии 1, растворяют в апротонном или неполярном органическом растворителе, например диэтиловом эфире, метил-трет-бутиловом эфире, тетрагидрофуране, диоксане, толуоле, ксилоле, хлористом метилене или хлороформе, в атмосфере азота и реакционную смесь прибавляют к раствору, приготовленному в атмосфере азота и содержащему стехиометрическое количество или 60% избыток оксалилхлорида в апротонном или неполярном растворителе, например в диэтиловом эфире, метил-трет-бутиловом эфире, тетрагидрофуране, диоксане, толуоле, ксилоле, хлористом метилене или хлороформе, причем температуру реакционной смеси поддерживают в диапазоне от -5 до 20°С. Затем реакционную смесь нагревают в течение от 30 минут до 5 часов при температуре от 10 до 130°С, предпочтительно от 20 до 80°С, лучше всего от 30 до 50°С. После завершения реакции растворитель отгоняют. Остаток "хлорида индолил-3-глиоксиловой кислоты", полученный по этому методу, растворяют в апротонном растворителе, например в тетрагидрофуране (ТГФ), диоксане, диэтиловом эфире, толуоле, или также в диполярном апротонном растворителе, например диметилформамиде, диметилацетамиде или диметилсульфоксиде, охлаждают до температуры от 10 до -15°С, предпочтительно от -5 до 0°С, и к реакционной смеси добавляют раствор первичного или вторичного амина в разбавителе в присутствии акцептора кислоты. В качестве разбавителя используют растворители, применяемые для растворения хлорида индолил-3-глиоксиловой кислоты. В качестве акцептора кислоты используют триэтиламин, пиридин, диметиламинопиридин, основной ионообменник, карбонат натрия, карбонат калия, порошкообразный гидроксид калия, а также в реакционную смесь добавляют избыток первичного или вторичного амина. Реакцию проводят при температуре от 0 до 120°С, предпочтительно от 20 до 80°С, особенно предпочтительно от 40 до 60°С. Реакционную смесь выдерживают в течение 1-3 часов, 24 часа при комнатной температуре, затем гидрохлорид акцептора кислоты удаляют фильтрованием, фильтрат концентрируют в вакууме и остаток перекристаллизовывают из органического растворителя или очищают с помощью колоночной хроматографии на силикагеле или окиси алюминия. В качестве подвижной фазы используют, например, смесь, состоящую из дихлорметана и этанола (95:5, об./об.).
Сведения, подтверждающие возможность осуществления изобретения.
Пояснения к примерам.
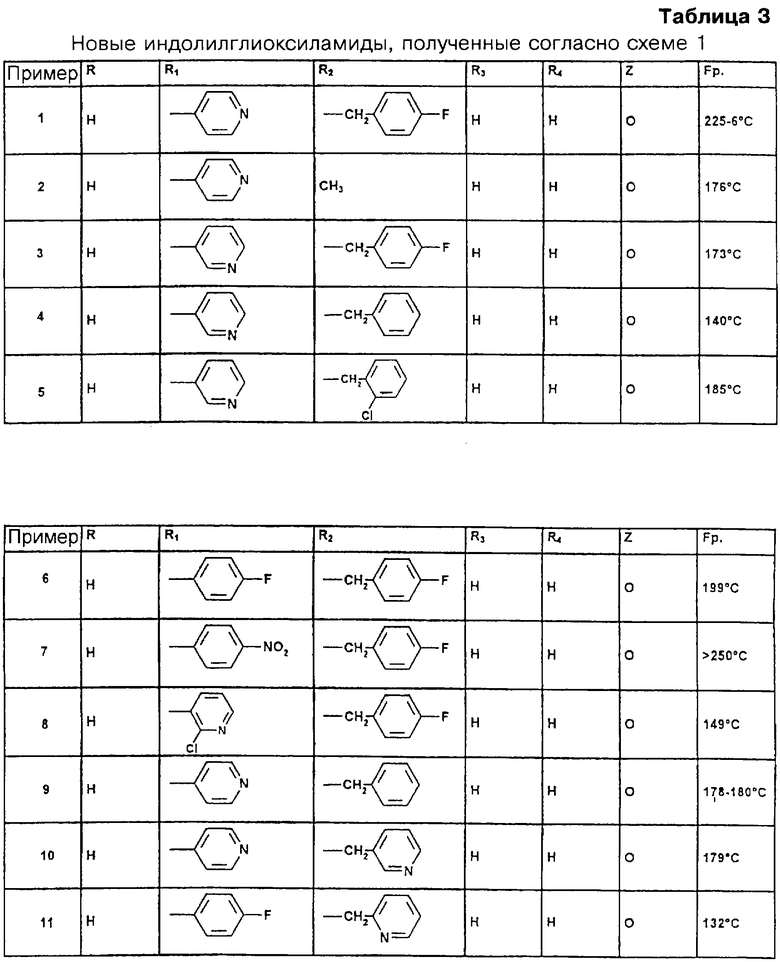
Следующие соединения были получены с использованием указанных общих принципов для стадий 1 и 2, которые являются основой схемы синтеза 1, соответствующие химические названия приведены ниже. В таблице 3 представлена структура заместителей R1-R4 и Z для соединений общей формулы I, а также температура плавления этих соединений.
Пример 1. N-(Пиридин-4-ил)-[1-(4-фторбензил)индол-3-ил]глиоксиламид.
Стадия 1. 1-(4-Фторбензил)индол.
Раствор 11,72 г (0,1 моль) индола в 50 мл диметилсульфоксида прибавляют к смеси 2,64 г гидрида натрия (0,11 моль, суспензия в минеральном масле) в 100 мл диметилсульфоксида. Реакционную смесь нагревают в течение 1,5 ч при 60°С, затем ее охлаждают и прикапывают 15,9 г (0,11 моль) 4-фторбензилхлорида. Раствор нагревают до 60°С, оставляют на ночь и затем при перемешивании вливают в 400 мл воды. Полученную смесь многократно экстрагируют хлористым метиленом (конечный объем 150 мл), органическую фазу отделяют и сушат над безводным сульфатом натрия, фильтруют и концентрируют в вакууме. Остаток перегоняют в высоком вакууме.
Выход 21,0 г (96% от теории). Т.кип. (0,5 мм) 140°С.
Стадия 2. N-(Пиридин-4-ил)-[1-(4-фторбензил)индол-3-ил]глиоксиламид.
Раствор 4,75 г (21,1 ммоль) 1-(4-фторбензил)индола в 25 мл эфира прикапывают к раствору 2,25 мл оксалилхлорида в 25 мл эфира при 0°С в атмосфере азота. Нагревают в течение 2 часов с обратным холодильником и отгоняют растворитель. Затем к остатку прибавляют 50 мл ТГФ, раствор охлаждают до -5°С и прикапывают раствор 4,66 г (49,5 ммоль) 4-аминопиридина в 200 мл ТГФ. Реакционную смесь нагревают в течение 3 часов с обратным холодильником и оставляют на ночь при комнатной температуре. 4-Аминопиридина гидрохлорид отфильтровывают, осадок на фильтре промывают ТГФ, фильтрат концентрируют в вакууме и остаток перекристаллизовывают из этилацетата.
Выход 7,09 г (90% от теории). Т.пл. 225-226°С.
Элементный анализ
Рассчитано, %: С 70,77; Н 4,32; N 11,25.
Найдено, %: С 71,09; Н 4,36; N 11,26.
Пример 2. N-(Пиридин-4-ил)-(1-метилиндол-3-ил)глиоксиламид.
Пример 3. N-(Пиридин-3-ил)-[1-(4-фторбензил)индол-3-ил]глиоксиламид.
Пример 4. N-(Пиридин-3-ил)-(1-бензилиндол-3-ил)глиоксиламид.
Пример 5. N-(Пиридин-3-ил)-[1-(2-хлорбензил)индол-3-ил]глиоксиламид.
Пример 6. N-(4-Фторфенил)-[1-(4-фторбензил)индол-3-ил]глиоксиламид.
Пример 7. N-(4-Нитрофенил)-[1-(4-фторбензил)индол-3-ил]глиоксиламид.
Пример 8. N-(2-Хлорпиридин-3-ил)-[1-(4-фторбензил)индол-3-ил]глиоксиламид.
Пример 9. N-(Пиридин-4-ил)-(1-бензилиндол-3-ил)глиоксиламид.
Пример 10. N-(Пиридин-4-ил)-[1-(3-пиридилметил)индол-3-ил]глиоксиламид.
Пример 11. N-(4-Фторфенил)-[1-(2-пиридилметил)индол-3-ил]глиоксиламид.
Пример 12. N-(4-Фторфенил)-[1-(3-пиридилметил)индол-3-ил]глиоксиламид.
Пример 13. N-(Пиридин-4-ил)-[1-(4-хлорбензил)индол-3-ил]глиоксиламид.
Пример 14. N-(Пиридин-4-ил)-[1-(2-хлорбензил)индол-3-ил]глиоксиламид.
Пример 15. N-(Пиридин-2-ил)-[1-(4-фторбензил)индол-3-ил]глиоксиламид.
Пример 16. N-(Пиридин-4-ил)-[1-(2-пиридилметил)индол-3-ил]глиоксиламид.
Пример 17. (4-Фенилпиперазин-1-ил)-[1-(4-фторбензил)индол-3-ил]глиоксиламид.
Пример 18. N-(Пиридин-2-ил)-(1-бензилиндол-3-ил)глиоксиламид.
Пример 19. N-(Пиридин-4-ил)-[1-(4-фторбензил)-6-этоксикарбониламиноиндол-3-ил]глиоксиламид.
Пример 20. N-(Пиридин-4-ил)-[1-(4-фторбензил)-5-этоксикарбониламиноиндол-3-ил]глиоксиламид.
Пример 21. N-(Пиридин-4-ил)-[1-(4-фторбензил)-6-циклопентилоксикарбониламиноиндол-3-ил]глиоксиламид.
Пример 22. [4-(Пиридин-4-ил)пиперазин-1-ил]-[1-(4-фторбензил)индол-3-ил]глиоксиламид.
Пример 23. N-(3,4,5-Триметоксибензил)-N-(аллиламинокарбонил-2-метилпроп-1-ил)-[1-(4-фторбензил)индол-3-ил]глиоксиламид.
Пример 24. N-(Пиридин-4-ил)-[1-(4-фторбензил)-5-метоксииндол-3-ил]глиоксиламид.
Пример 25. N-(Пиридин-4-ил)-[1-(4-фторбензил)-5-гидроксииндол-3-ил]глиоксиламид.
Пример 26. N-(Пиридин-4-ил)-[1-(4-фторбензил)-5-этоксикарбониламинометилиндол-3-ил]глиоксиламид.
Предварительные стадии синтеза соединений общей формулы I, полученных согласно схеме 1 (см. таблицу 3).
Все исходные соединения, используемые на конечных стадиях синтеза соединений согласно примерам 1-22 и 24-26, являются коммерческими препаратами.
Далее соединения общей формулы I были также получены по схеме 2, на которой представлен синтез соединения согласно примеру 27.

Краткое описание получения соединений формулы I согласно схеме 2.
Стадия 1. Раствор производного индола, содержащего замещенное или незамещенное в положении С-2 фенильное кольцо, медленно прикапывают к раствору оксалилхлорида в апротонном или неполярном растворителе, например в диэтиловом эфире, метил-трет-бутиловом эфире, ТГФ, диоксане или также дихлорметане в атмосфере азота при температуре от -5 до +5°С, причем раствор содержит оксалилхлорид в соотношении от эквимолярного до 60% избытка от стехиометрического количества. Затем реакционную смесь нагревают в течение от 1 до 5 часов при температуре от 10 до 120°С, предпочтительно от 20 до 80°С, особенно предпочтительно от 30 до 60°С, и растворитель отгоняют. Остаток, содержащий хлорид (индолил-3-ил)глиоксиловой кислоты, растворяют или суспендируют в апротонном растворителе, например ТГФ, диоксане, диэтиловом эфире, толуоле, или также в диполярном апротонном растворителе, например диметилформамиде, диметилацетамиде или диметилсульфоксиде, охлаждают до температуры от -10 до +10°С, предпочтительно от -5 до 0°С и прибавляют раствор первичного или вторичного амина в разбавителе в присутствии акцептора кислоты. В качестве разбавителя используют общепринятые для растворения "хлоридов индолил-3-глиоксиловой кислоты" растворители. В качестве акцептора кислоты используют триэтиламин, пиридин, диметиламинопиридин, основной ионообменник, карбонат натрия, карбонат калия, порошкообразный гидроксид калия, а также добавляют первичные и вторичные амины в избытке. Реакцию проводят при температуре от 0 до 120°С, предпочтительно от 20 до 80°С, особенно предпочтительно от 40 до 60°С. Реакционную смесь выдерживают при этой температуре в течение 1-4 часов, затем при комнатной температуре в течение 24 часов и фильтруют. Осадок многократно промывают водой и отфильтровывают.
Твердый остаток сушат в вакууме и проводят очистку соединения перекристаллизацией из органического растворителя или с помощью колоночной хроматографии на силикагеле или окиси алюминия. В качестве подвижной фазы используют, например, смесь дихлорметана и этанола (10:1, об./об.).
Стадия 2. "Индол-3-ил-глиоксиламид", полученный по указанному выше способу на стадии 1, растворяют в протонном, диполярном апротонном или органическом растворителе, например в изопропаноле, ТГФ, диметилсульфоксиде, диметилформамиде, диметилацетамиде, N-метилпирролидоне, диоксане, толуоле или дихлорметане, и медленно прикапывают к суспензии основания, например гидрида натрия, порошкообразного гидроксида калия, трет-бутилата калия, диметиламинопиридина или амида натрия, в соответствующем растворителе. Суспензию загружают в трехгорлую колбу в стехиометрическом соотношении или в избытке от стехиометрического соотношения. Все реакции проводят в атмосфере азота. Затем добавляют требуемый алкил-, аралкил- или соответственно гетероаралкилгалогенид без растворителя или в разбавителе, который, например, используют для растворения "индол-3-ил-глиоксиламида", в случае необходимости добавляют катализатор, например медь, реакционную смесь выдерживают в течение некоторого времени, например от 30 минут до 12 часов, и поддерживают температуру от 0 до 120°С, предпочтительно от 30 до 80°С, особенно предпочтительно от 50 до 70°С. После завершения реакции реакционную смесь выливают в воду, раствор экстрагируют, например, диэтиловым эфиром, дихлорметаном, хлороформом, метил-трет-бутиловым эфиром, ТГФ или н-бутанолом и органическую фазу сушат безводным сульфатом натрия.
Органическую фазу концентрируют в вакууме, полученный остаток кристаллизуют затиранием или маслообразный остаток очищают с помощью дистилляции, или колоночной, или тонкослойной хроматографии на силикагеле или окиси алюминия. В качестве подвижной фазы используют, например, смесь хлористого метилена и диэтилового эфира в соотношении 8:2 (об./об.) или смесь хлористого метилена и этанола в соотношении 9:1 (об./об.).
Пояснения к примеру.
Следующие соединения были получены с использованием указанных общих принципов для стадий 1 и 2, которые являются основой схемы синтеза 2. Эти соединения были также синтезированы согласно схеме 1 и представлены в таблице 3. Исходные соединения для получения указанных соединений следуют из таблицы 4.
Пример 27. N-(Пиридин-4-ил)-[1-(4-(фторбензил)индол-3-ил]глиоксиламид (конечная стадия аналогично примеру 1).
Стадия 1. N-(Пиридин-4-ил)-(индол-3-ил)глиоксиламид.
Раствор 10 г (85,3ммоль) индола в 100 мл эфира медленно прикапывают к раствору 9 мл оксалилхлорида в 100 мл безводного эфира. Реакционную смесь выдерживают в течение 3 часов при кипячении с обратным холодильником. Затем к реакционной смеси прикапывают при -5°С суспензию 12 г (127,9 ммоль) 4-аминопиридина в 500 мл ТГФ, реакционную смесь нагревают при перемешивании в течение 3 часов при кипячении с обратным холодильником и выдерживают при комнатной температуре в течение ночи. Реакционную смесь фильтруют, осадок обрабатывают водой и высушенное соединение очищают с помощью колоночной хроматографии на силикагеле (Кизельгель 60, выпускаемый фирмой Fa. Merck AG, Darmstadt). В качестве подвижной фазы используют смесь хлористого метилена и этанола (10:1, об./об.).
Выход 9,8 г (43,3% от теории). Т.пл. от 250°С.
Стадия 2. N-(Пиридин-4-ил)-1-[4-(фторбензил)индол-3-ил]глиоксиламид.
Полученный на стадии 1 N-(пиридин-4-ил)-(индол-3-ил)глиоксиламид обрабатывают согласно "стадии бензилирования" (стр. 8, стадия 1) 4-фторбензилхлоридом и выделяют полученное соединение.
Выход 41% от теории.
Т.пл. 224-225°С.
Элементный анализ
Рассчитано, %: С 70,77; Н 4,32; N 11,25.
Найдено, %: С 70,98; Н 4,40; N 11,49.
Пример 28. N-(4-Нитрофенил)-[1-(4-фторбензил)индол-3-ил]глиоксиламид (последняя стадия, аналогично примеру 7).
Пример 29. N-(4-Фторфенил)-[1-(4-фторбензил)индол-3-ил]глиоксиламид (последняя стадия, аналогично примеру 6).
Пример 30. N-(Пиридин-3-ил)-[1-(4-фторбензил)индол-3-ил]глиоксиламид (последняя стадия, аналогично примеру 3).
Следующие исходные соединения были получены по вышеприведенной схеме 2 (стадия 1 схемы 2, таблица 4).
Пример 31. N-(Пиридин-4-ил)-(индол-3-ил)глиоксиламид.
Пример 32. N-(4-Нитрофенил)-(индол-3-ил)глиоксиламид.
Пример 33. N-(4-Фторфенил)-(индол-3-ил)глиоксиламид.
Пример 34. N-(Пиридин-3-ил)-(индол-3-ил)глиоксиламид.
Данные исследования острой токсичности на моделях животных.
Исследование проводилось с использованием соединения D-24851, представляющего собой N-(пиридин-4-ил)-[1-(4-хлорбензил)индол-3-ил]глиоксиламид, полученный в примере 13, согласно изобретению.
Согласно протоколу изучения острой токсичности на мышах и крысах животным вводят перорально (п/о) единичную дозу соединения D-24851. Интервал дозы для мышей составляет от 100 до 2150 мг/кг п/о. Крысам мужского и женского пола вводят 100 мг/кг, 464 мг/кг, 681 мг/кг и 1000 мг/кг. Самцам дополнительно вводят 215 и 31,6 мг/кг. Соединение D-24851 для успешного введения суспендируют с тилозой (R).
Мыши. Доза 215 мг/кг п/о у самцов и 464 мг/кг п/о у самок не вызывала каких-либо проявлений системной токсичности. Летальные дозы LD10 и LD50, а также коэффициенты снижения приведены в таблице 5.
Крысы. Доза 215 мг/кг п/о у самцов и 31,6 мг/кг у самок не вызывала каких-либо проявлений системной токсичности. Летальные дозы LD10 и LD50, а также коэффициенты снижения приведены в таблице 5.



Изобретение относится к N-замещенным индол-3-глиоксиламидам общей формулы I, обладающим противоастматическим, противоаллергическим и иммунодепрессивным/иммуномодуляторным действием
где R означает водород, (С1-С6)алкил, причем алкильная группа необязательно содержит один фенильный заместитель, который, в свою очередь, необязательно содержит по меньшей мере один заместитель, выбранный из группы, включающей галоген, метокси, этокси, (С1-С6)алкил; R1 означает фенильный цикл, содержащий по меньшей мере один заместитель, выбранный из группы, включающей (С1-С6)алкокси, гидрокси, нитро, (С1-С6)алкоксикарбониламино или один фтор, или R1 представляет собой остаток пиридина формулы II

где углеродные атомы 2, 3 и 4 остатка пиридина необязательно имеют одинаковые или различные заместители R5 и R6, причем R5 и R6 обозначают (С1-С6)алкил или галоген, или R1 представлен аллиламинокарбонил-2-метилпроп-1-ильной группой, или R и R1 вместе с атомом азота, к которому они присоединены, образуют цикл пиперазина формулы III

где R7 обозначает фенил или пиридинил; R2 означает (С1-С6)алкил, который необязательно содержит фенильный остаток, который, в свою очередь, необязательно замещен галогеном, метоксигруппой или этоксигруппой, или относящаяся к R2 (С1-С6)алкильная группа необязательно замещена 2-, 3- или 4-пиридильным остатком; R3 и R4 являются одинаковыми или различными заместителями и обозначают водород, гидрокси, (С1-С6)алкокси, (С1-С3)алкоксикарбониламино- или (С1-С3)алкоксикарбониламино(С1-С3)алкил или R3 представляет собой циклопентилоксикарбониламиногруппу; Z означает О, причем алкил, алкокси или алкиламино означают как упорядоченные неразветвленные группы, такие как метил, этил, н-пропил, н-бутил, н-гексил, так и разветвленные алкильные группы, такие как изопропил или трет-бутильные группы; галоген означает фтор, хлор, бром или йод и алкоксигруппа означает метокси, пропокси, бутокси, изопропокси, изобутокси или пентоксигруппу, а также к их фармацевтически приемлемым солям с кислотами. Соединения с указанной активностью могут быть использованы для получения лекарственного препарата. 4 н. и 3 з.п. ф-лы, 5 табл.

где R означает водород, (С1-С6)алкил, причем алкильная группа необязательно содержит один фенильный заместитель, который, в свою очередь, необязательно содержит по меньшей мере один заместитель, выбранный из группы, включающей галоген, метокси, этокси, (С1-С6)алкил;
R1 означает фенильный цикл, содержащий по меньшей мере один заместитель, выбранный из группы, включающей (С1-С6)алкокси, гидрокси, нитро, (С1-С6)алкоксикарбониламино или один фтор, или R1 представляет собой остаток пиридина формулы II

где углеродные атомы 2, 3 и 4 остатка пиридина необязательно имеют одинаковые или различные заместители R5 и R6, причем R5 и R6 обозначают (С1-С6)алкил или галоген; или
R1 представлен аллиламинокарбонил-2-метил-проп-1-ильной группой; или
R и R1 вместе с атомом азота, к которому они присоединены, образуют цикл пиперазина формулы III

где R7 обозначает фенил или пиридинил;
R2 означает (С1-С6)алкил, который необязательно содержит фенильный остаток, который, в свою очередь, необязательно замещен галогеном, метоксигруппой или этоксигруппой, или относящаяся к R2 (С1-С6)алкильная группа необязательно замещена 2-, 3- или 4-пиридильным остатком;
R3 и R4 являются одинаковыми или различными заместителями и обозначают водород, гидрокси, (С1-С6)алкокси, (С1-С3)алкоксикарбониламино- или (С1-С3)алкоксикарбониламино(С1-С3)-алкил или R3 представляет собой циклопентилоксикарбониламиногруппу;
Z означает О,
причем алкил, алкокси или алкиламино означают как упорядоченные неразветвленные группы, такие, как метил, этил, н-пропил, н-бутил, н-гексил, так и разветвленные алкильные группы, такие, как изопропил или трет. бутильные группы; галоген означает фтор, хлор, бром или йод и алкоксигруппа означает метокси, пропокси, бутокси, изопропокси, изобутокси или пентоксигруппу, а также их фармацевтически приемлемые соли с кислотами.
N-(пиридин-4-ил)-[1-(4-фторбензил)-индол-3-ил]-глиоксиламид,
N-(пиридин-4-ил)-(1-метил-индол-3-ил)-глиоксиламид,
N-(пиридин-3-ил)-[1-(4-фторбензил)-индол-3-ил]-глиоксиламид,
N-(пиридин-3-ил)-(1-бензилиндол-3-ил)-глиоксиламид,
N-(пиридин-3-ил)-[1-(2-хлорбензил)-индол-3-ил]-глиоксиламид,
N-(4-фторфенил)-[1-(4-фторбензил)-индол-3-ил]-глиоксиламид,
N-(4-нитрофенил)-[1-(4-фторбензил)-индол-3-ил]-глиоксиламид,
N-(2-хлорпиридин-3-ил)-[1-(4-фторбензил)-индол-3-ил]-глиоксиламид,
N-(пиридин-4-ил)-(1-бензилиндол-3-ил]-глиоксиламид,
N-(пиридин-4-ил)-[1-(3-пиридилметил)-индол-3-ил]-глиоксиламид,
N-(4-фторфенил)-[1-(2-пиридилметил)-индол-3-ил]-глиоксиламид,
N-(4-фторфенил)-[1-(3-пиридилметил)-индол-3-ил]-глиоксиламид,
N-(пиридин-4-ил)-[1-(4-хлорбензил)-индол-3-ил]-глиоксиламид,
N-(пиридин-4-ил)-[1-(2-хлорбензил)-индол-3-ил]-глиоксиламид,
N-(пиридин-2-ил)-[1-(4-фторбензил)-индол-3-ил]-глиоксиламид,
N-(пиридин-4-ил)-[1-(2-пиридилметил)-индол-3-ил]-глиоксиламид,
(4-фенил-пиперазин-1-ил)-[1-(4-фторбензил)-индол-3-ил]-глиоксиламид,
N-(пиридин-2-ил)-(1-бензилиндол-3-ил)-глиоксиламид,
[4-(пиридин-4-ил)-пиперазин-1-ил]-[1-(4-фторбензил)-индол-3-ил]-глиоксиламид,
N-(пиридин-4-ил)-[1-(4-фторбензил)-6-этоксикарбониламино-индол-3-ил]-глиоксиламид,
N-(пиридин-4-ил)-[1-(4-фторбензил)-5-этоксикарбониламино-индол-3-ил]-глиоксиламид,
N-(пиридин-4-ил)-[1-(4-фторбензил)-6-циклопентилоксикарбониламино-индол-3-ил]-глиоксиламид,
N-(3,4,5-триметоксибензил)-N-(аллиламинокарбонил-2-метилпроп-1-ил)-[1-(4-фторбензил)-индол-3-ил]-глиоксиламид,
N-(пиридин-4-ил)-[1-(4-фторбензил)-5-метокси-индол-3-ил]-глиоксиламид,
N-(пиридин-4-ил)-[1-(4-фторбензил)-5-гидрокси-индол-3-ил]-глиоксиламид или
N-(пиридин-4-ил)-[1-(4-фторбензил)-5-этоксикарбониламинометил-индол-3-ил]-глиоксиламид.
где R3 и R4 имеют указанные значения,
смешивают с основанием, суспендированным в протонном диполярном, апротонном или неполярном органическом растворителе, и с реакционноспособным соединением, содержащим остаток R2, где R2 имеет указанные значения, и получают производное 1-индола формулы V

где R2, R3 и R4 имеют указанные значения,
которое смешивают с апротонным или неполярным органическим растворителем и реакционноспособным соединением формулы VI
(C-Z-Hal)2,
где Z является атомом кислорода;
Hal обозначает галоген, выбранный из группы, содержащей фтор, хлор, бром или иод,
затем обрабатывают полученный продукт первичным или вторичным амином формулы VII
HNRR1,
где R и R1 имеют указанные значения,
в апротонном или диполярном апротонном растворителе и выделяют целевое соединение формулы I.
где R3 и R4 имеют указанные значения,
смешивают в апротонном или неполярном растворителе с реакционноспособным соединением формулы VI
(C-Z-Hal)2,
где Z является атомом кислорода;
Hal означает галоген, выбранный из группы, содержащей фтор, хлор, бром или иод,
обрабатывают полученный продукт в апротонном или диполярном апротонном растворителе первичным или вторичным амином формулы VII
HNRR1,
где R и R1 имеют указанные значения,
затем полученное производное 3-индола формулы VIII
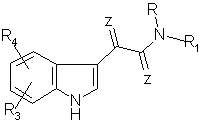
где R, R1, R3, R4 и Z имеют указанные значения,
смешивают в присутствии суспендированного основания в протонном диполярном апротонном или неполярном органическом растворителе с реакционноспособным соединением, содержащим остаток R2, где R2 имеет указанные значения, и выделяют целевое соединение формулы I.
| Способ автоматического контроля и регулирования процесса периодической варки растительного сырья | 1978 |
|
SU675110A1 |

