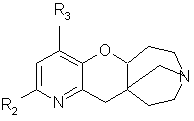
Настоящее изобретение относится к производным пиридипираноазепинов, в форме чистого геометрического или оптического изомера или смеси таких изомеров, общей формулы (I)

в которой R1 представляет собой атом водорода, (С1-С4)-алкильную группу или фуранилгидрокси(С1-С4)-алкильную группу;
R2 является атомом водорода или галогена, цианогруппой, фенильной или нафтильной группой, возможно замещенной атомом галогена или трифторметильной, трифторметокси, нитро, ацетильной, (С1-С4)-алкильной или метилендиоксигруппой, присоединенной по положениям 2 и 3 или 3 и 4 фенильного кольца, или фенилом, и
R3 представляет собой атом водорода или галогена или (С1-С4)-алкильную группу,
в форме основания или соли присоединения кислоты.
Соединения общей формулы (I) могут существовать в форме оснований или солей присоединения кислот. Кроме того, поскольку атомы в положениях 5а или 10а асимметричны, то соединение может существовать в форме чистых геометрических и оптических изомеров или смесей последних.
Одним из предпочтительных соединений по изобретению является (5aS,10aR)-5a,6,7,9,10,11-гексагидро-8,10a-метанопиридо[2',3':5,6]пирано[2,3-d]азепина гидрохлорид.
Согласно изобретению, можно получить соединения общей формулы (I) способом, проиллюстрированным на следующей схеме:
Cхема
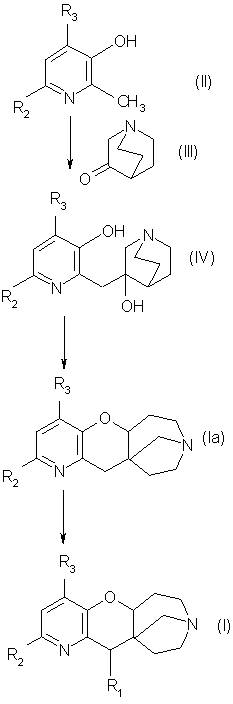
2-Метилпиридин-3-ол общей формулы (II), в которой R2 и R3 такие, как определено выше, подвергают взаимодействию с алкиллитием, затем полученное таким образом промежуточное соединение конденсируют с 1-азабицикло[2.2.2]октан-3-оном формулы (III) при низкой температуре и в апротонном растворителе, таком как тетрагидрофуран.
Получают соединение общей формулы (IV), в которое при желании можно ввести заместители R2 и R3 или модифицировать их любым из способов, известных специалистам в данной области техники.
Соединение общей формулы (IV) затем подвергают дегидратации, которая сопровождается перегруппировкой, в кислой среде, например метан-сульфоновой кислоте или серной кислоте, при высокой температуре.
Получают соединение общей формулы (Iа), в котором можно модифицировать R2 и R3 заместители и/или вводить R1 заместитель любым из способов, известных специалистам в данной области техники.
Исходные соединения общей формулы (II) и (III) имеются в продаже (R2=R3=H) или могут быть получены известными способами.
Таким образом, согласно еще одному аспекту данного изобретения предложен способ получения соединений формулы (I), отличающийся тем, что соединение общей формулы (IV)
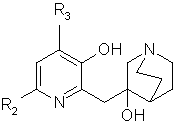
в которой R2 и R3 такие, как определено для формулы (I), подвергают дегидратации в кислой среде с последующей перегруппировкой при высокой температуре с получением соединения общей формулы (Iа)
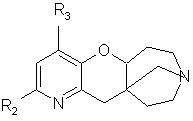
в которой, если желательно, модифицируют заместители R2 и R3 и/или вводят заместитель R1,такой, как определено выше для формулы (I).
Еще одним воплощением данного изобретения является лекарство против нарушений, связанных с дисфункцией никотиновых рецепторов, отличающееся тем, что оно состоит из соединения формулы (I).
Согласно изобретению, также предложена фармацевтическая композиция, обладающая действием против нарушений, связанных с дисфункцией никотиновых рецепторов, отличающаяся тем, что она содержит соединение формулы (I), объединенное с эксципиентом.
Следующие примеры иллюстрируют получение некоторых соединений по настоящему изобретению. Элементные микроанализы и ИК (инфракрасные) и ЯМР (ядерный магнитный резонанс) спектры, а также спектры дифракции рентгеновских лучей в конкретных случаях подтверждают структуры полученных соединений.
Цифры, указанные в скобках в названиях примеров, соответствуют цифрам 1-й колонки таблицы 1.
В названиях соединений черточка "-" является частью слова, а черточка "_" служит только для переноса в конце строки; ее следует убирать при отсутствии переноса и не нужно заменять ни нормальной черточкой, ни пробелом.
Пример 1 (Соединение №1)
(транс)-5а,6,7,9,10,11-гексагидро-8,10а-метанопиридо[2',3':5,6]пирано[2,3-d]азепина гидрохлорид (2:1).
1.1. 3-[(3-Гидроксипиридин-2-ил)метил]-1-азабицикло[2.2.2]октан-3-ол
52,9 г (484 ммоль) 2-метил-3-гидроксипиридина, растворенного в 1300 мл тетрагидрофурана, вносят в трехгорловую колбу на 2000 мл в атмосфере аргона. Раствор охлаждают до -56° C и по каплям в течение 3 часов добавляют 750 мл (975 ммоль) 1,3 М раствора 1-метилпропиллития в циклогексане, поддерживая температуру ниже -50° C. В конце добавления температуре позволяют подняться до -4° C в течение 45 мин и смесь затем снова охлаждают до -58° C для того, чтобы по каплям в течение 40 минут добавить 60,6 г (484 ммоль) 1-азабицикло[2.2.2]октан-3-она, растворенного в 250 мл тетрагидрофурана. Температуре позволяют подняться до температуры окружающей среды и продолжают перемешивание в течение 20 часов. Реакционную смесь охлаждают до 4° C и гидролизуют добавлением 110 мл водного раствора 36%-ной соляной кислоты. Добавляют 400 мл воды, двум фазам дают отстояться и органическую фазу экстрагируют водой. Водные фазы объединяют, смесь охлаждают до 4° C и добавляют концентрированный водный раствор гидроксида натрия до рН 8,4. Полученный осадок отфильтровывают и сушат под вакуумом при 80° C.
Таким образом получают 62,5 г продукта.
Точка плавления: 270-272° C.
1.2.(транс)-5а,6,7,9,10,11-Гексагидро-8,10а-метанопиридо[2',3':5,6]пирано[2,3-d]азепина гидрохлорид (2:1)
2,34 г (10 ммоль) 3-[(3-гидроксипиридин-2-ил)метил]-1-азабицикло[2.2.2]октан-3-ола, растворенного в 10 мл метансульфоновой кислоты, вносят в колбу на 50 мл и нагревают при 180°C в течение 48 часов.
Реакционную смесь охлаждают и выливают на лед. Ее подщелачивают добавлением концентрированного водного раствора гидроксида натрия и экстрагируют хлороформом. Органическую фазу сушат над сульфатом магния и концентрируют при пониженном давлении. Остаток очищают хроматографией на колонке с силикагелем, элюируя смесью 90/10/1 хлороформа, метанола и аммиака. Продукт получают в форме основания, из которого образуется соль при добавлении раствора соляной кислоты в этаноле. Выделяют 1,55 г гидрохлорида.
Точка плавления: выше 300° C.
Пример 2 (Соединение №2)
(5аS,10aR)-5а,6,7,9,10,11-гексагидро-8,10а-метанопиридо[2',3':5,6]пирано[2,3-d]азепина гидрохлорид (2:1)
2.1. (5аS, 10aR)-5а,6,7,9,10,11-Гексагидро-8,10а-метанопири-до[2',3':5,6]пирано[2,3-d]азепина (3R, 5R)-(-)-О,О'-дибензоил-L-тартрат (1:2)
15,335 г (0,0709 моль) (транс)-5а,6,7,9,10,11-гексагидро-8,10а-метанопиридо[2',3':5,6]пирано[2,3-d]азепина в 50 мл этилацетата вносят в колбу на 500 мл. Добавляют раствор 50,83 г (0,142 моль) (3R, 5R)-(-)-O,O'-дибензоил-L-винной кислоты в 50 мл этилацетата, растворитель выпаривают при пониженном давлении и остаток растворяют в 885 мл смеси 7/3 воды и этанола при кипячении с обратным холодильником. После охлаждения полученные кристаллы собирают фильтрацией и перекристаллизовывают в 50 мл горячего пропан-2-ола.
После охлаждения получают 13,7 г кристаллов.
Точка плавления: 145-148° C; [α ]
2.2. (5аS, 10aR)-5а,6,7,9,10,11-Гексагидро-8,10а-метанопиридо[2',3':5,6]пирано[2,3-d]азепина гидрохлорид (2:1)
Обработка предыдущего соединения водным раствором карбоната калия с последующей экстракцией дихлорметаном позволяет получить 3,1 г (0,0143 моль) соединения в форме основания.
Точка плавления: 69-71° C.
[α ]
Это основание растворяют в 10 мл этанола в колбе на 50 мл, добавляют 6 мл (0,030 моль) раствора 6 М соляной кислоты в пропан-2-оле, смесь концентрируют до сухости при пониженном давлении, остаток собирают снова в 40 мл пропан-2-ола, эту смесь нагревают до начала флегмообразования и добавляют 5 мл этанола. После охлаждения полученные кристаллы собирают фильтрацией и сушат при пониженном давлении.
Получают 3,4 г белых кристаллов.
Точка плавления: 330° C; [α ]
Пример 3 (Соединение №4)
(транс)-2-бром-5а,6,7,9,10,11-гексагидро-8,10а-метанопиридо[2',3':5,6]пирано[2,3-d]азепин
3.1 3-[(6-Бром-3-гидроксипиридин-2-ил)метил]-1-азабицикло[2.2.2]октан-3-ол
52,23 г (0,223 моль) 3-[(3-гидроксипиридин-2-ил)метил]-1-азабицикло-[2.2.2]октан-3-ола, суспендированного в 500 мл воды, при температуре окружающей среды вносят в колбу на 1000 мл. Добавляют 26,7 г (0,669 моль) гидроксида натрия, растворенного в 350 мл воды, и 26,5 г (0,223 моль) бромида калия, смесь перемешивают до полного растворения перед добавлением 11,5 мл (0,223 моль) брома по каплям в течение 2 часов.
Смесь перемешивают в течение 18 часов при температуре окружающей среды, затем реакционную смесь нейтрализуют добавлением 23 мл уксусной кислоты. Ее охлаждают на ледяной бане и полученный осадок отфильтровывают. Маточные растворы концентрируют, и полученный осадок растирают в пропан-2-оле, отфильтровывают и промывают.
Получают 27,9 г продукта.
Точка плавления: 215-221° C.
3.2.(транс)-2-бром-5а,6,7,9,10,11-Гексагидро-8,10а-метанопиридо[2',3':5,6]пирано[2,3-d]азепин
6,1 г 3-[(6-бром-3-гидроксипиридин-2-ил)метил]-1-азабицикло[2.2.2]октан-3-ола и 50 мл концентрированной серной кислоты вносят в колбу на 100 мл. Смесь нагревают при 130° C в течение 72 часов, затем охлаждают до температуры окружающей среды и выливают на лед. Водную фазу подщелачивают до рН 10 добавлением концентрированного водного раствора гидроксида натрия и экстрагируют хлороформом. Органические фазы сушат над сульфатом магния и концентрируют при пониженном давлении. Остаток очищают хроматографией на колонке с силикагелем, элюируя смесью 90/10/4 дихлорметана, метанола и аммиака.
Получают 1,2 г продукта.
Точка плавления: 157-159° C.
Пример 4 (Соединение №28)
(транс)-(-)-2-бром-5а,6,7,9,10,11-гексагидро-8,10а-метанопиридо[2',3':5,6]пирано[2,3-d]азепина гидробромид (1:1)
4.1. (5аS, 10aR)-2-Бром-5а,6,7,9,10,11-гексагидро-8,10а-метанопиридо[2',3':5,6]пирано[2,3-d]азепина (3R, 5R)-(-)-О,О'-дибензоил-L-тартрат (1:2)
0,3 г (1 ммоль) (транс)-2-бром-5а,6,7,9,10,11-гексагидро-8,10а-метанопиридо[2',3':5,6]пирано[2,3-d]азепина, растворенного в 10 мл этилацетата, вносят в колбу на 50 мл, добавляют 0,358 г (1 ммоль) О,О'-(-)-дибензоил-L-винной кислоты, растворенной в 3 мл этилацетата, растворитель выпаривают при пониженном давлении и остаток перекристаллизовывают в 5 мл горячего пропан-2-ола. После охлаждения полученные кристаллы собирают фильтрацией и сушат под вакуумом.
Получают 0,12 г кристаллов.
Точка плавления: 200° C.
[α ]
4.2.(транс)-(-)-2-бром-5а,6,7,9,10,11-гексагидро-8,10а-метанопиридо[2',3':5,6]пирано[2,3-d]азепина гидробромид (1:1)
Превращение основания проводят путем обработки предыдущего соединения водным раствором гидроксида натрия с последующей экстракцией дихлорметаном. 0,3 г (1 ммоль). Основания растворяют в 30 мл пропан-2-ола в колбе на 100 мл. Добавляют 0,36 мл (2 ммоль) раствора 33%-ной бромистоводородной кислоты в уксусной кислоте. После охлаждения до 4° C полученные кристаллы собирают фильтрацией и сушат под вакуумом.
Получают 0,25 г белых кристаллов.
Точка плавления: 350-352° C; [α ]
Пример 5 (Соединение №24)
(транс)-(-)-2-хлор-5а,6,7,9,10,11-гексагидро-8,10а-метанопиридо[2',3':5,6]пирано[2,3-d]азепина гидрохлорид (2:1)
0,2 г (0,68 ммоль) (транс)-(-)-2-бром-5а,6,7,9,10,11-гексагидро-8,10а-метанопиридо[2',3':5,6]пирано[2,3-d]азепина растворяют в 4 мл концентрированного водного раствора соляной кислоты и нагревают при 180° C в запечатанной пробирке в течение 48 часов. Водную фазу упаривают и остаток перекристаллизовывают в пропан-2-оле.
Получают 0,075 г кристаллов.
Точка плавления: 339-344° C; [α ]
Пример 6 (Соединение №27)
(транс)-2-циано-5а,6,7,9,10,11-гексагидро-8,10а-метанопиридо[2',3':5,6]пирано[2,3-d]азепина гидробромид (1:1)
0,45 г (1,52 ммоль) (транс)-2-бром-5а,6,7,9,10,11-гексагидро-8,10а-метанопиридо[2',3':5,6]пирано[2,3-d]азепина растворяют в 8 мл пиридина в колбе на 50 мл, добавляют 0,205 г (2,29 ммоль) цианида меди и смесь нагревают до начала дефлегмации в течение 30 часов. Добавляют 75 мл дихлорметана и органическую фазу промывают 45 мл насыщенного водного раствора хлорида аммония, затем 75 мл воды. После сушки и концентрирования органической фазы при пониженном давлении получают 0,22 г ожидаемого продукта. Его растворяют в пропан-2-оле и обрабатывают одним эквивалентом бромистоводородной кислоты, растворенной в концентрации 33% в уксусной кислоте. После охлаждения сбора кристаллов фильтрацией и сушки под вакуумом получают 0,21 г продукта.
Точка плавления: 329-332° C.
Пример 7 (Соединение №10)
(транс)-2-(4-метилфенил)-5а,6,7,9,10,11-гексагидро-8,10а-метанопиридо[2',3':5,6]пирано[2,3-d]азепина гидробромид (2:1)
0,3 г (1 ммоль) (транс)-2-бром-5а,6,7,9,10,11-гексагидро-8,10а-метанопиридо[2',3':5,6]пирано[2,3-d]азепина в 6 мл толуола, 0,193 г (1,4 ммоль) 4-метилфенилбороновой кислоты, 0,072 г (0,06 ммоль) тетракис(трифенил)фосфинпалладия, 1 мл (2 ммоль) карбоната натрия в виде 2 М водного раствора и 0,05 мл этанола вносят в реактор на 10 мл и реакционную смесь нагревают до начала флегмообразования в течение 72 часов. После отстаивания органическую фазу наносят на силикагель и элюируют смесью 97/3/0,3 дихлорметана, метанола и аммиака.
Получают 0,31 г продукта, который образует соль с двумя эквивалентами бромистоводородной кислоты, растворенной в уксусной кислоте.
Точка плавления: 355° C.
Пример 8 (Соединение №5)
(транс)-11-метил-5а,6,7,9,10,11-гексагидро-8,10а-метанопиридо[2',3':5,6]пирано[2,3-d]азепина гидрохлорид (2:1)
(транс)-5а,6,7,9,10,11-гексагидро-8,10а-метанопиридо[2',3':5,6]пирано[2,3-d]азепин в 20 мл безводного тетрагидрофурана вносят в трехгорлую колбу на 100 мл, реакционную смесь охлаждают до -78° C для добавления по каплям 1,2 мл (3 ммоль) 2,5 М бутиллития в гексане и перемешивание продолжают при -78° C в течение 30 мин.
Добавляют 0,19 мл (3 ммоль) иодметана и смесь оставляют медленно нагреваться до температуры окружающей среды перед добавлением 100 мл воды и экстракции дихлорметаном. Органическую фазу сушат над сульфатом магния, ее упаривают при пониженном давлении и остаток очищают хроматографией на колонке с силикагелем, элюируя смесью 90/10/1 дихлорметана, метанола и аммиака. Полученный продукт обрабатывают двумя эквивалентами соляной кислоты, растворенной в пропан-2-оле, и 0,15 г кристаллов выделяют фильтрацией.
Точка плавления: выше 330° C.
Пример 9 (Соединение №9)
(транс)-α -фуран-3-ил-5а,6,7,9,10,11-гексагидро-10аН-8,10а-метанопиридо[2',3':5,6]пирано[2,3-d]азепин-11-метанола гидробромид (2:1)
0,43 г (2 ммоль) (транс)-5а,6,7,9,10,11-гексагидро-8,10а-метанопиридо[2',3':5,6]пирано[2,3-d]азепина обрабатывают фуран-3-карбоксальдегидом в условиях, описанных в примере 8.
После образования соли с 2 эквивалентами бромистоводородной кислоты в уксусной кислоте получают 0,3 г соединения.
Точка плавления: 69-73° C с разложением.
Пример 10 (Соединение №26)
(транс)-2-4-дибром-5а,6,7,9,10,11-гексагидро-8,10а-метанопиридо[2',3':5,6]пирано[2,3-d]азепина гидробромид (1:1)
10.1. 3-[(4,6-Дибром-3-гидроксипиридин-2-ил)метил]-1-азаби-цикло[2.2.2]октан-3-ол
Раствор 24 г (0,426 моль) гидроксида калия в 600 мл воды вносят в колбу на 2000 мл, добавляют 50,0 г (0,213 моль) 3-[(3-гидроксипиридин-2-ил)метил]-1-азабицикло[2.2.2]октан-3-ола и затем по каплям в течение 40 минут добавляют раствор 10,93 мл (0,213 моль) брома и 152,4 г (1,280 моль) бромида калия в 600 мл воды, смесь перемешивают при температуре окружающей среды в течение 16 часов. рН Смеси доводят до 7,5 добавлением уксусной кислоты, и ее перемешивают в течение 1 часа. Ее фильтруют, полученное твердое вещество сушат, собирают его в 1000 мл этанола и полученную суспензию нагревают в течение 2 часов.
После охлаждения осадок собирают фильтрацией и сушат.
Получают 21,24 г твердого вещества.
Точка плавления: 260-265° C.
10.2 (транс)-2,4-Дибром-5а,6,7,9,10,11-гексагидро-8,10а-метанопири-до[2',3':5,6]пирано[2,3-d]азепина гидробромид (1:1)
10 г (25 ммоль) 3-[(4,6-дибром-3-гидроксипиридин-2-ил)метил]-1-азабицикло[2.2.2]октан-3-ола вносят в колбу на 500 мл, добавляют 150 мл концентрированной серной кислоты и 3,6 г (25 ммоль) пентаоксида фосфора, смесь нагревают при 150° C в течение 48 часов. Ее охлаждают, выливают на 300 г льда, рН доводят до 10 добавлением аммиака и эту смесь экстрагируют хлороформом. Органическую фазу сушат над сульфатом натрия и фильтруют, растворитель выпаривают при пониженном давлении и остаток очищают хроматографией на колонке с силикагелем, элюируя смесью 98/2/0,2 хлороформа, метанола и аммиака.
После образования соли твердого вещества, полученной с одним эквивалентом бромистоводородной кислоты в уксусной кислоте, получают 3,53 г гидробромида.
Точка плавления: 320° C с разложением.
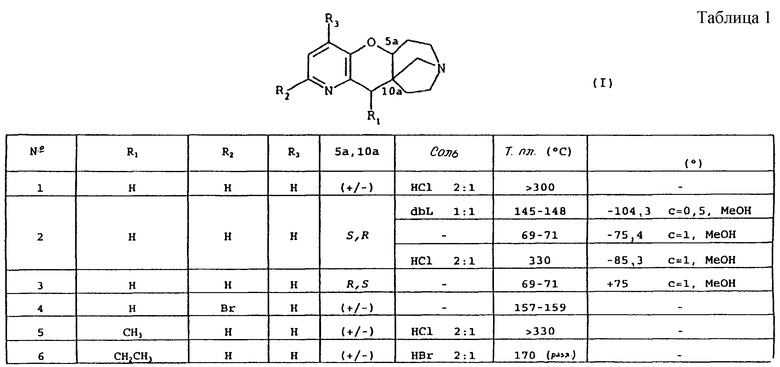
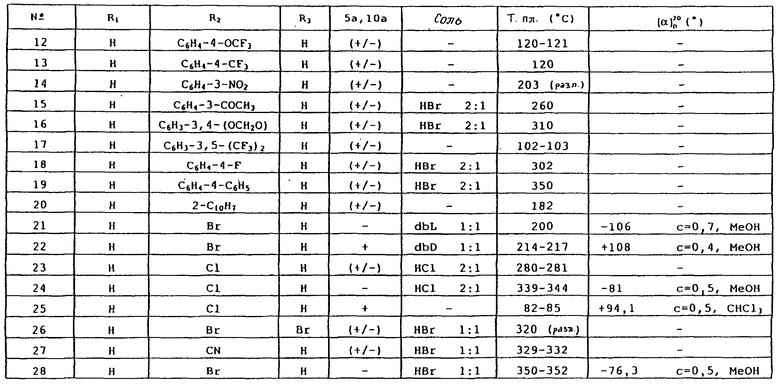
Таблица 1 иллюстрирует химические структуры и физические свойства некоторых соединений по изобретению. В колонках "R1" и "R2", "C6H5", "C6H4" и "C6H3" обозначают соответственно незамещенные, однозамещенные или двузамещенные фенильные группы. Указаны заместители и их положение. "C4H3О" обозначает фуран-3-ильную группу. "2-C10H7" обозначает нафталин-2-ильную группу. Колонка "5а, 10а" указывает конфигурацию хиральных центров 5а и 10а, и "+/-" обозначает рацемат.
В колонке "соль" символ "-" обозначает соединение в основной форме, "HCl" обозначает гидрохлорид, "HBr" обозначает гидробромид, "dbL" обозначает дибензоил-L-тартрат и "dbD" обозначает дибензоил-D-тартрат. Указаны молярные отношения кислота:основание.
В колонке "Т. пл." (° C), "(разл.)" указывает точку плавления с разложением.
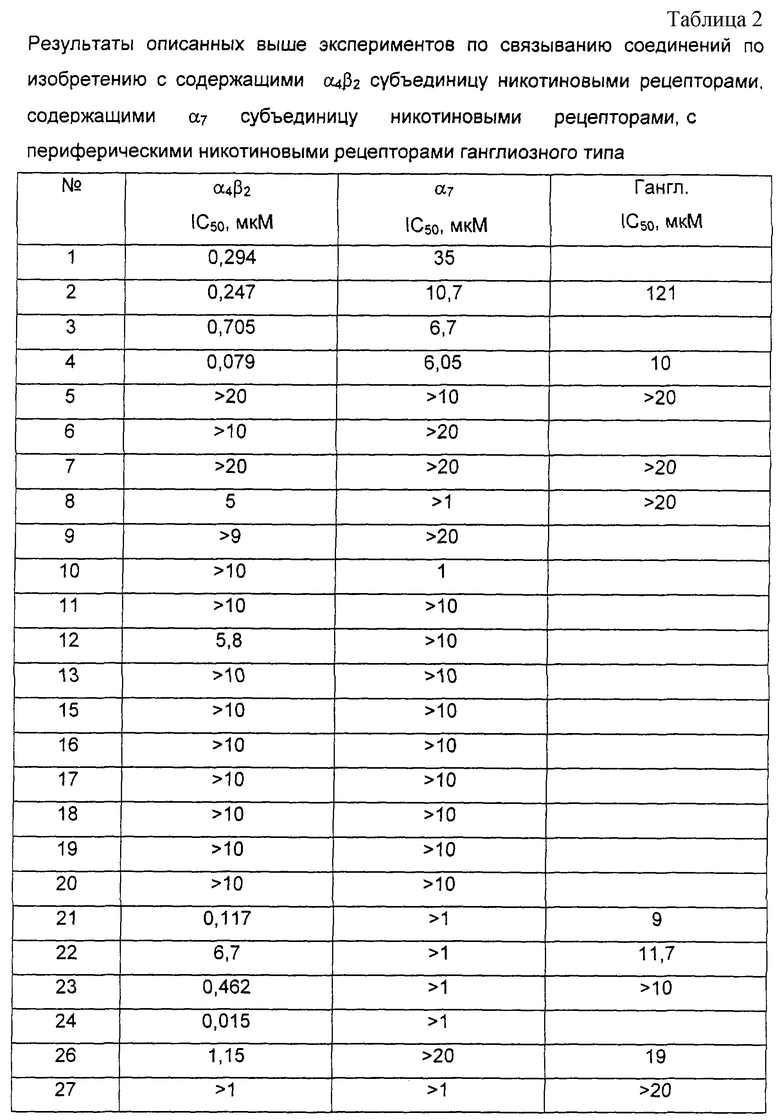
Соединения по настоящему изобретению являлись объектом экспериментов, которые продемонстрировали их терапевтические свойства (см. табл.2).
Так, их исследовали на их сродство по отношению к содержащим α 4β 2 субъединицу никотиновым рецепторам способами, описанными Anderson and Arneric, Eur. J. Pharmacol. (1994) 253 261, и Hall et al., Brain Res. (1993) 600 127. Самцов Sprague Dawley крыс массой от 150 до 200 г декапитируют и весь мозг быстро извлекают, гомогенизируют в 15 объемах 0,32 М раствора сахарозы при 4° C и затем центрифугируют при 1000 g в течение 10 минут. Осадок отбрасывают, а супернатант центрифугируют при 20000 g в течение 20 минут при 4° C. Осадок извлекают и гомогенизируют при помощи Polytron™ мельницы в 15 объемах дважды дистиллированной воды при 4° C, а затем центрифугируют при 8000 g в течение 20 минут. Осадок отбрасывают, а супернатант и светлый слой кровяного сгустка центрифугируют при 40000 g в течение 20 минут, осадок извлекают, ресуспендируют в 15 мл дважды дистиллированной воды при 4° C и центрифугируют еще раз при 40000 g перед хранением его при -80° C.
В день эксперимента ткань медленно оттаивают и суспендируют в 3 объемах буфера. 150 мкл этой мембранной суспензии инкубируют при 4°C в течение 120 мин в присутствии 100 мкл 1 нМ [3H]цитизина в конечном объеме 500 мкл буфера в присутствии или в отсутствии исследуемого соединения. Реакцию останавливают фильтрацией на Whatman GF/B™ фильтрах, предварительно обработанных полиэтиленимином, фильтры промывают дважды 5 мл буфера при 4° C и радиоактивность, оставшуюся на фильтре, измеряют при помощи жидкостной сцинтиграфии. Неспецифическое связывание определяют в присутствии 10 мкМ (-)-никотина; неспецифическое связывание составляет от 75 до 85% общего связывания, полученного на фильтре. Для каждой концентрации исследуемого соединения определяют процент ингибирования специфического связывания [3H]цитизина, затем рассчитывают IC50, концентрацию соединения, которая ингибирует 50% специфического связывания. IC50 значения наиболее активных соединений по изобретению находятся между 0,08 и 1 мкМ.
Соединения по настоящему изобретению также изучали с точки зрения их сродства по отношению к содержащим α 7 субъединицу никотиновым рецепторам способами, описанными Marks and Collins, J. Pharmacol. Exp. Ther. (1982) 22 554 и Marks et al., Mol. Pharmacol. (1986) 30 427. Самцов OFA крыс массой от 150 до 200 г декапитируют, весь мозг быстро удаляют, гомогенизируют при помощи Polytron™ мельницы в 15 объемах 0,32 М раствора сахарозы при 4° C, затем центрифугируют при 1000 g в течение 10 минут. Осадок отбрасывают, и супернатант центрифугируют при 8000 g в течение 20 минут при 4° C. Осадок извлекают и гомогенизируют при помощи Polytron™ мельницы в 15 объемах дважды дистиллированной воды при 4° C, затем центрифугируют при 8000 g в течение 20 минут. Осадок отбрасывают, а супернатант и светлый слой кровяного сгустка центрифугируют при 40000 g в течение 20 минут. Осадок извлекают, ресуспендируют в 15 мл дважды дистиллированной воды при 4° C и центрифугируют еще раз при 40000 g в течение 20 минут перед хранением при -80° C.
В день эксперимента ткань медленно оттаивают и суспендируют в 5 объемах буфера. 150 мкл этой мембранной суспензии предварительно инкубируют при 37° C в течение 30 мин в темноте в присутствии или в отсутствии исследуемого соединения. Мембраны затем инкубируют в течение 60 минут при 37° C в темноте в присутствии 50 мкл 1 нМ [3H]α -бунгаротоксина в конечном объеме 250 мкл 20 мМ HEPES (N-2-гидроксиэтилпиперазин-N'-2-этансульфоновая кислота) буфера с 0,05% полиэтиленимина. Реакцию останавливают фильтрацией на Whatman GF/С™ фильтрах, предварительно обработанных в течение 3 часов 0,5%-ным полиэтиленимином. Фильтры промывают дважды 5 мл буфера при 4° C и радиоактивность, оставшуюся на каждом фильтре, измеряют при помощи жидкостной сцинтиграфии. Неспецифическое связывание определяют в присутствии α -бунгаротоксина в конечной концентрации 1 мкМ; неспецифическое связывание составляет приблизительно 60% общего связывания, полученного на фильтре. Для каждой концентрации исследуемого соединения определяют процент ингибирования специфического связывания [3H]α -бунгаротоксина, затем рассчитывают IC50, концентрацию соединения, которая ингибирует 50% специфического связывания. IC50 Значения соединений по изобретению находятся между 1 и 20 мкМ.
Соединения по настоящему изобретению также изучали с точки зрения их сродства по отношению к периферическим никотиновым рецепторам ганглиозного типа способом, описанным Houghtling et al., Mol. Pharmacol. (1995) 48 280-287.
Способность соединения вытеснять [3H]α -эпибатидин из мембран бычьих надпочечников определяет его сродство к этому рецептору.
Бычьи надпочечники, хранящиеся при -80° C, оттаивают и гомогенизируют при помощи Polytron™ мельницы в 20 объемах 50 мМ трис-HCl буфера при рН 7,4 и 4° C, затем центрифугируют при 35000 g в течение 10 минут. Супернатант отбрасывают и осадок ресуспендируют в 30 объемах 50 мМ трис-HCl буфера при 4° C и снова гомогенизируют перед повторным центрифугированием при 35000 g в течение 10 минут. Конечный осадок переносят в 10 объемов трис-HCl буфера при 4° C. 100 мкл мембран или 10 мг свежей ткани инкубируют при 24° C в течение 3 часов в присутствии 50 мкл 0,66 нМ [3H]-эпибатидина в конечном объеме 250 мкл буфера, в присутствии или в отсутствии исследуемого соединения. Реакцию останавливают разбавлением образцов 50 мкМ трис-HCl буфера рН 7,4 при 4° С и затем все это фильтруют на Whatman GF/С™ фильтрах, предварительно обработанных в течение 3 часов 0,5%-ным полиэтиленимином. Фильтры промывают дважды 5 мл буфера и радиоактивность, оставшуюся на фильтре, измеряют при помощи жидкостной сцинтиграфии. Неспецифическое связывание определяют в присутствии (-)-никотина в конечной концентрации 2 мМ; неспецифическое связывание составляет от 30 до 40% общего связывания, полученного на фильтре. Для каждой концентрации исследуемого соединения определяют процент ингибирования специфического связывания [3H]-эпибатидина, затем рассчитывают IC50, концентрацию соединения, которая ингибирует 50% специфического связывания. IC50 Значения наиболее активных соединений по изобретению находятся между 9 и 20 мкМ.
Результаты предыдущих анализов показывают, что определенные соединения по настоящему изобретению являются селективными лигандами для α 4β 2 субъединиц никотинового рецептора.
И наконец, соединения по настоящему изобретению являлись предметом in vivo экспериментов, которые продемонстрировали их терапевтические свойства.
Так, например, их исследовали в модели с горячей пластиной известным способом (Eddy and Leimbach, J. Pharmacol. Exp. Ther. (1953) 107 385-393) с целью изучения и количественной оценки возможного анальгезирующего действия. Мышей массой от 20 до 30 г подвергали воздействию теплового раздражителя путем контакта лап с пластиной, поддерживаемой при постоянной температуре 57,5° C при помощи термостатированной водяной бани. Измеряли время реакции на боль, которая проявлялась лизанием лап или подпрыгиванием. Так, после периода предварительной обработки, проведенной подкожным или пероральным путем (каждая партия образована из восьми животных для одинаковой предварительной обработки), мышей помещали по отдельности на пластину и измеряли время реакции на боль. Животное удаляют с пластины немедленно после проявления боли. Максимальное время воздействия раздражителя составляет 30 секунд. Для каждой партии определяют среднее время реакции вместе со среднеквадратической ошибкой среднего (s. e. m.). Непараметрический дисперсионный анализ (Kruskal-Wallis) проводят для всей партии. Анализ Wilcoxon позволяет сравнить каждую исследуемую партию с контрольной партией. Различия считаются статистически значимыми при пределе 5%.
Это время реакции значительно увеличивается под действием анальгетиков, главным образом, с общим действием.
Соединения по настоящему изобретению проявляют активность в этом анализе в дозах между 3 и 30 мг/кг при внутрибрюшинном или пероральном пути введения.
Эти результаты подтверждают возможность применения соединений в лечении или предупреждении нарушений, связанных с дисфункцией никотиновых рецепторов, особенно на уровне центральной нервной системы или желудочно-кишечной системы.
На уровне центральной нервной системы эти нарушения включают в себя снижение познавательной способности, более конкретно ухудшения памяти, а также ослабление внимания, ассоциированные с болезнью Альцгеймера, с патологическим старением (возрастное ухудшение памяти (Age Associated memory Impairment, AAMI)), с болезнью Паркинсона, с монголизмом (синдромом Дауна), с алкогольным синдромом Корсакова (Korsakoff) и с сосудистым слабоумием (мультиинфарктное слабоумие (multi-infarct dementia, MID)). Соединения по настоящему изобретению также могут быть пригодны при лечении двигательных нарушений, наблюдаемых при болезни Паркинсона или других неврологических заболеваниях, таких как хорея Гентингтона, болезнь Туретта (Tourett), поздняя дискинезия или гиперкинезия.
Соединения по настоящему изобретению также могут служить основой терапевтического или симптоматического лечения инсультов и приступов церебральной гипоксии. Их можно применять в случае психиатрических патологий: шизофрении, депрессии, тревожности, приступов паники, компульсивного и обсессивного поведения.
Они могут предупреждать симптомы, связанные с отказом от табака, алкоголя и различных веществ, вызывающих зависимость, таких как кокаин, ЛСД, гашиш и бензодиазепины.
Наконец, их можно использовать в лечении боли.
На уровне желудочно-кишечной системы соединения по настоящему изобретению можно использовать в лечении болезни Крона, неспецифического язвенного колита, синдрома раздраженной толстой кишки и ожирения.
Для такого воздействия соединения по настоящему изобретению можно представить в любой форме композиции, подходящей для энтерального, парентерального или чрескожного введения, такой как таблетки, покрытые сахарной оболочкой, твердые и мягкие желатиновые капсулы, пригодные для питья, или инъекционные суспензии или растворы, такие как сиропы или ампулы, чрескожные пластыри и так далее, объединенными с подходящими эксципиентами и дозированными так, чтобы обеспечить суточное введение от 0,01 до 20 мг/кг.
Исследованные соединения являются нетоксичными или малотоксичны.
Результаты описанных выше экспериментов по связыванию соединений по изобретению с содержащими α 4β 2 субъединицу никотиновыми рецепторами, содержащими α 7 субъединицу никотиновыми рецепторами, и к периферическими никотиновыми рецепторами ганглиозного типа
Изобретение относится к производным пиридопираноазепинов, в форме чистого геометрического или оптического изомера или смеси таких изомеров, общей формулы (I)
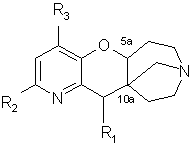 (I)
(I)
в которой R1 представляет собой атом водорода, (С1-С4)-алкильную группу или фуранилгидрокси(С1-С4)-алкильную группу; R2 является атомом водорода или галогена, цианогруппой, фенильной или нафтильной группой, возможно замещенной атомом галогена или трифторметильной, трифторметокси, нитро, ацетильной, (С1-С4)-алкильной или метилендиоксигруппой, присоединенной по положениям 2 и 3 или 3 и 4 фенильного кольца, или фенилом, и R3 представляет собой атом водорода или галогена или (С1-С4)-алкильную группу, в форме основания или соли присоединения кислоты, а также относится к лекарству против нарушений, связанных с дисфункцией никотиновых рецепторов, и фармацевтической композиции, обладающей действием против нарушений, связанных с дисфункцией никотиновых рецепторов, на основе этих соединений. Технический результат - получение новых производных пиридопираноазепинов, обладающих биологически активным действием. 4 н. и 1 з.п. ф-лы, 2 табл.
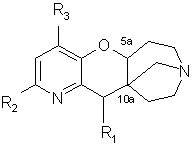 (I)
(I)
в которой R1 представляет собой атом водорода, (С1-С4)алкильную группу или фуранилгидрокси(С1-С4)алкильную группу;
R2 является атомом водорода или галогена, цианогруппой, фенильной или нафтильной группой, возможно замещенной атомом галогена или трифторметильной, трифторметокси, нитро, ацетильной, (С1-С4)алкильной или метилендиоксигруппой, присоединенной по положениям 2 и 3 или 3 и 4 фенильного кольца, или фенилом;
R3 представляет собой атом водорода или галогена или (С1-С4)алкильную группу,
в форме основания или соли присоединения кислоты.

в которой R2 и R3 такие, как определено в п.1,
подвергают дегидратации в кислой среде с последующей перегруппировкой при высокой температуре с получением соединения общей формулы (Iа)
в которой, если желательно, модифицируют заместители R2 и R3 и/или вводят заместитель R1,такой, как определено в п.1.
| ГЕКСАЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 1992 |
|
RU2071476C1 |

