Настоящее изобретение относится к производным хинолин-2(1H)-она, их получению и их использованию в терапии.
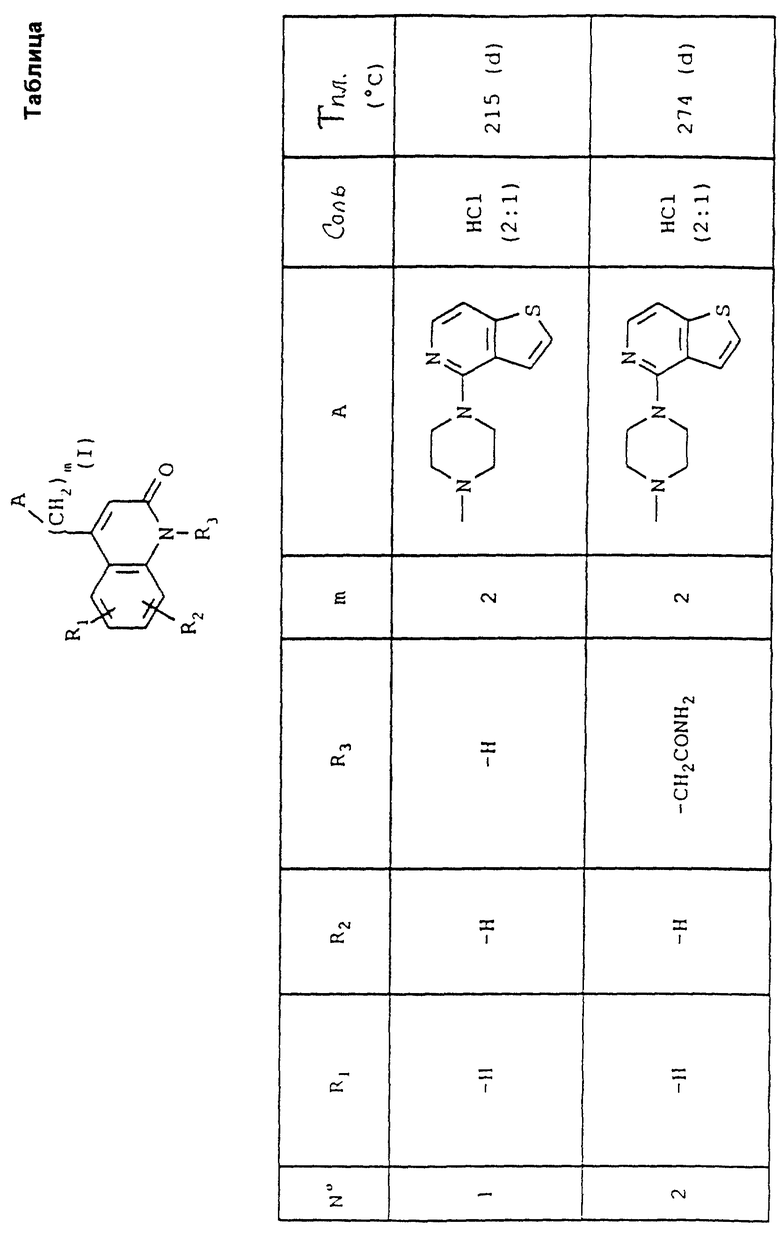
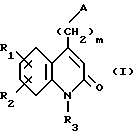
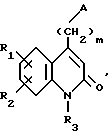
Соединения по изобретению соответствуют формуле (I),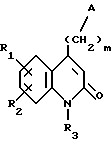
в которой A представляет собой либо группу 4-(тиено[3,2-с]пиридин-4-ил)пиперазин-1-ил, либо группу 4-(4-фторбензоил)пиперидин-1-ил,
R1 и R2 каждый представляет собой, независимо друг от друга, либо атом водорода, либо атом галогена, либо амино-группу, либо гидроксильную группу, либо нитро-группу, либо циано-группу, либо группу (C1-C6)алкил, либо группу (C1- C6)алкокси, либо трифторметильную группу, либо трифторметокси-группу, либо группу -COOH, либо группу -COOR4, либо группу -CONH2, либо группу -CONHR4, либо группу -CONR4R5, либо группу -SR4, либо группу -SO2R4, либо группу -NHCOR4, либо группу -NHSO2R4, либо группу -N(R4)2, где R4 и R5 каждый является группой (C1-C4)алкил,
R3 представляет собой либо атом водорода, либо группу (C1-C4)алкил, либо группу -(CH2)pОН, либо группу -(CH2)pNH2, либо группу -(CH2)nCOOH, либо группу -(CH2)nCOOR4, либо группу -(CH2)nCONH2, либо группу -(CH2)nCONHOH,
либо группу -(CH2)pSH, либо группу -(CH2)nSO3H, либо группу -(CH2)nSO2NH2, либо группу - (CH2)nSO2NHR4, либо группу -(CH2)nSO2NR4R5, либо группу - (CH2)nCONHR4, либо группу -(CH2)nCONR4R5, либо группу - (CH2)pNHSO2R4, либо группу -(CH2)pNHCOR4, либо группу -(CH2)pOCOR4, где R4 и R5 каждый является группой (C1-C4)алкил, n равно 1, 2, 3 или 4, p равно 2, 3 или 4, и
m равно 2, 3 или 4,
так же как их соли присоединения с фармацевтически приемлемыми кислотами или основаниями.
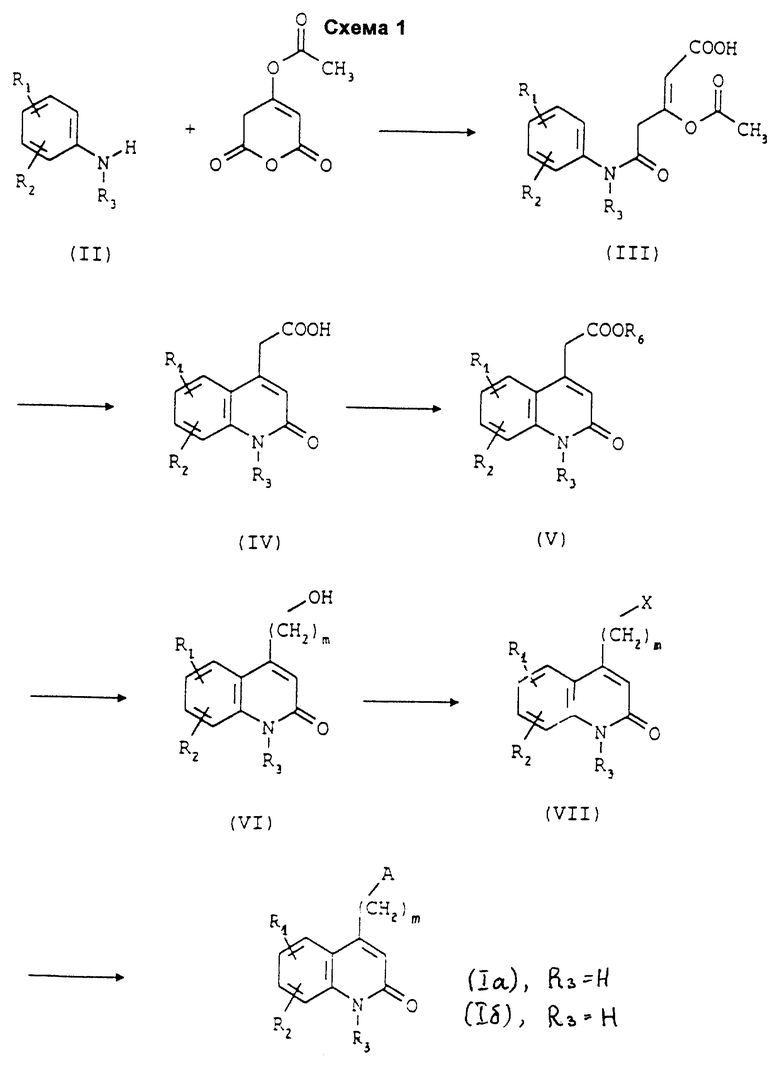
Согласно данному изобретению соединения формулы (I) могут быть синтезированы в соответствии со Схемой 1.
4-(Ацетилокси)-2H, 3H-пиран-2,6-дион подвергают взаимодействию с соединением формулы (II), в которой R1 и R2 такие, как определено выше, и R3 представляет собой атом водорода или группу (C1-C4)алкил, при комнатной температуре в полярном растворителе, таком как уксусная кислота. После сушки полученное таким образом соединение формулы (III) циклизуют в присутствии минеральной или органической кислоты, предпочтительно безводной, такой как концентрированная серная кислота, полифосфорная кислота или трифторметансульфоновая кислота, при температуре между 10 и 150oC, и получают замещенную или незамещенную 2-оксо-1,2- дигидрохинолин-4-уксусную кислоту формулы (IV), которую этерифицируют спиртом формулы R6OH, где R6 представляет собой группу (C1-C4)алкил, любым способом этерификации, предпочтительно действием тионилхлорида. Затем полученный таким образом сложный эфир формулы (V) восстанавливают гидридом в апротонном растворителе, например литийалюмогидридом в диоксане или избытком боргидрида натрия в тетрагидрофуране в условиях образования флегмы, или боргидридом лития в тетрагидрофуране при комнатной температуре, с получением спирта формулы (VI), в которой m равно 2; соединения формулы (VI), в которой m равно 3 или 4, получают из тех соединений, где m равно 2, с использованием известных специалистам способов получения гомологов. Затем соединения формулы (VI), в которой m равно 2, 3 или 4, активируют в соединения формулы (VII), в которой X представляет собой уходящую группу, такую как атом хлора или брома, например взаимодействием с тионилхлоридом в хлороформе в условиях образования флегмы или с дибромтрифенилфосфораном при комнатной температуре в дихлорметане, или в соединения формулы (VII), в которой Х представляет собой уходящую группу, такую как группа метансульфонилокси, трифторметансульфонилокси или паратолуолсульфонилокси, например взаимодействием с сульфоновым ангидридом или хлоридом сульфоновой кислоты в присутствии основания, такого как пиридин или триэтиламин. Наконец, соединения формулы (VII) подвергают взаимодействию с 4-(пиперазин-1-ил)тиено[3,2-с]пиридином или с 4- (4-трифторбензоил)пиперидином с или без апротонного или протонного растворителя в присутствии неорганического основания при температуре между 20 и 150oC, предпочтительно в ацетонитриле или диметилформамиде в контакте с бикарбонатом натрия, и получают соединение формулы (I).
Для получения соединения формулы (Iб), в которой R3 другой чем атом водорода, можно провести алкилирование соответствующего соединения формулы (Iа), в которой R3 представляет собой атом водорода, с использованием электрофильного агента типа R3Br или R3l, такого как, например, трет-бутиловый эфир бромуксусной кислоты, бромметансульфонамид, N-метилбромметансульфонамид, бромацетамид, N-метилбромацетамид, N,N-диметилбромацетамид или 2-бромэтилацетат, в присутствии основания, такого как гидрид натрия или гидрид калия, в апротонном растворителе, таком как тетрагидрофуран или диметилформамид, в присутствии или в отсутствии катализатора фазового переноса, такого как тетрабутиламмонийбромид. Затем, если желательно получить соединения формулы (Iб), в которой R3 представляет собой группу -(CH2)mCOOH, проводят омыление соответствующих соединений формулы (Ib), в которой R3 представляет собой группу -(CH2)nCOOR4. Если желательно получить соединения формулы (Iб), в которой R3 представляет собой группу -(CH2)pOH, проводят деацетилирование соответствующих соединений формулы (Iб), в которой R3 представляет собой группу -(CH2)pOCOR4.
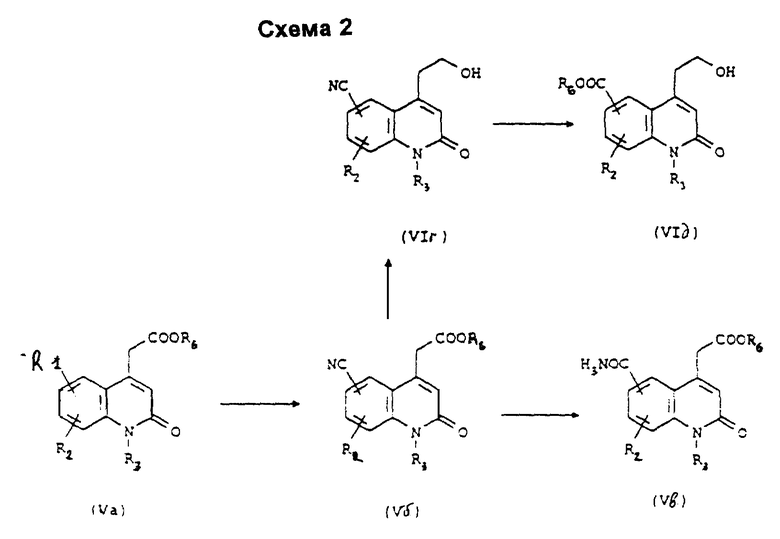
Для получения соединения формулы (I), в которой R1 и/или R2 представляет собой группу циано, -CONH2, -COOH, -COOR4, -SR4 или -SO2R4, где R4 представляет собой группу (C1-C4)алкил, причем циклизация соединения формулы (III) в хинолинон формулы (IV) является нежелательной, проводят синтез соответствующих соединений формул (V) и (VI) согласно Схемам 2 и 3.
Согласно Схеме 2 соединение формулы (Va), соответствующее соединению формулы (V), в которой R1 представляет собой атом иода, R2 и R6 такие, как определено выше, и R3 представляет собой атом водорода или группу (C1-C4)алкил, подвергают взаимодействию с солью цианистоводородной кислоты в присутствии соли меди в полярном растворителе, таком как диметилформамид или N-метилпирролидон, или с триметилсилилцианидом в присутствии палладиевого катализатора, предпочтительно тетракис(трифенилфосфин)палладия (0) в триэтиламине в условиях образования флегмы с получением соединения формулы (Vб), которое можно либо превратить в соединение формулы (VIг), а затем в соединение формулы (VIд), в которой R7 представляет собой атом водорода или группу (C1-C4)алкил, либо превратить в карбоксамидное производное формулы (Vв) с помощью способов, известных специалистам.
Согласно схеме 3 соединение формулы (VIa), соответствующее соединению формулы (VI), в которой R1 представляет собой атом иода, R2 такой, как определено выше, R3 представляет собой атом водорода или группу (C1-C4)алкил и m равно 2, подвергают взаимодействию с тиолатом, таким как тиометоксид натрия, в присутствии тетракис(трифенилфосфин)палладия (0) в спирте, таком как этанол, пропанол или н-бутанол, с получением соединения формулы (VIб), в которой R4 представляет собой группу (C1-C4)алкил, которое можно превратить окислением в соединение формулы (VIв).
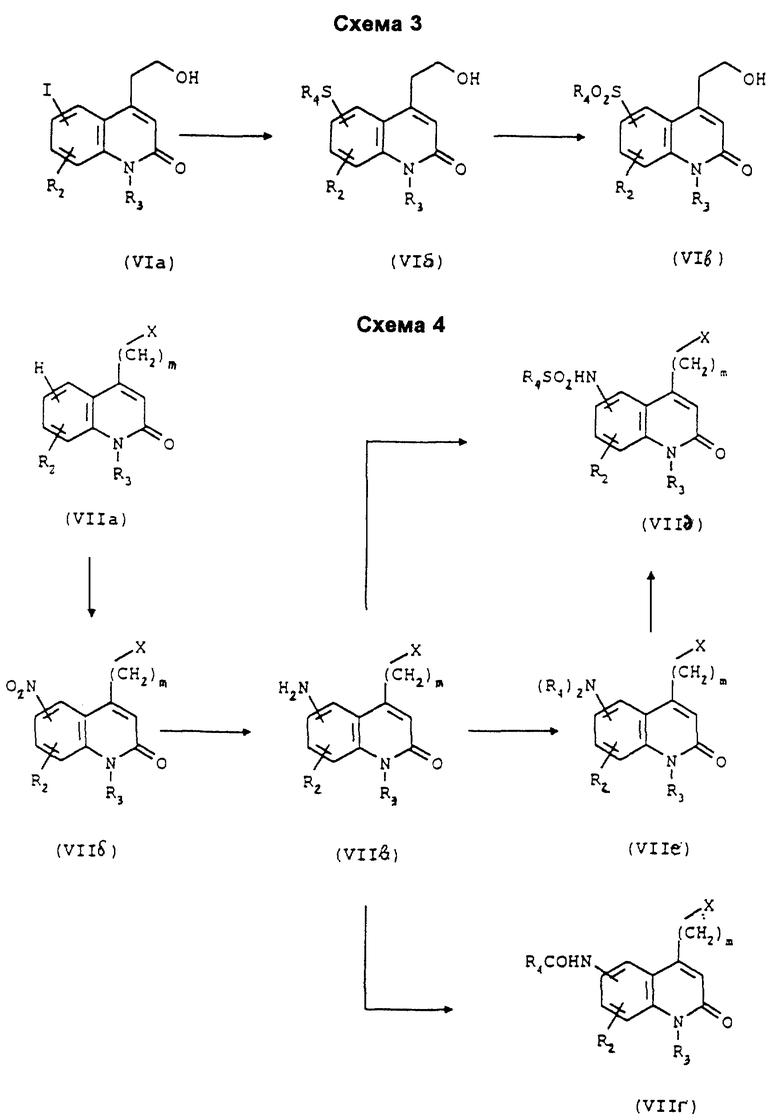
Для получения соединений формулы (I), в которой R1 и/или R2 представляет собой группу нитро, амино, -NHCOR4, -NHSO2R4 или -N(R4)2, причем R4 является группой (C1-C4)алкил, проводят синтез соответствующих соединений формулы (VII) согласно Схеме 4.
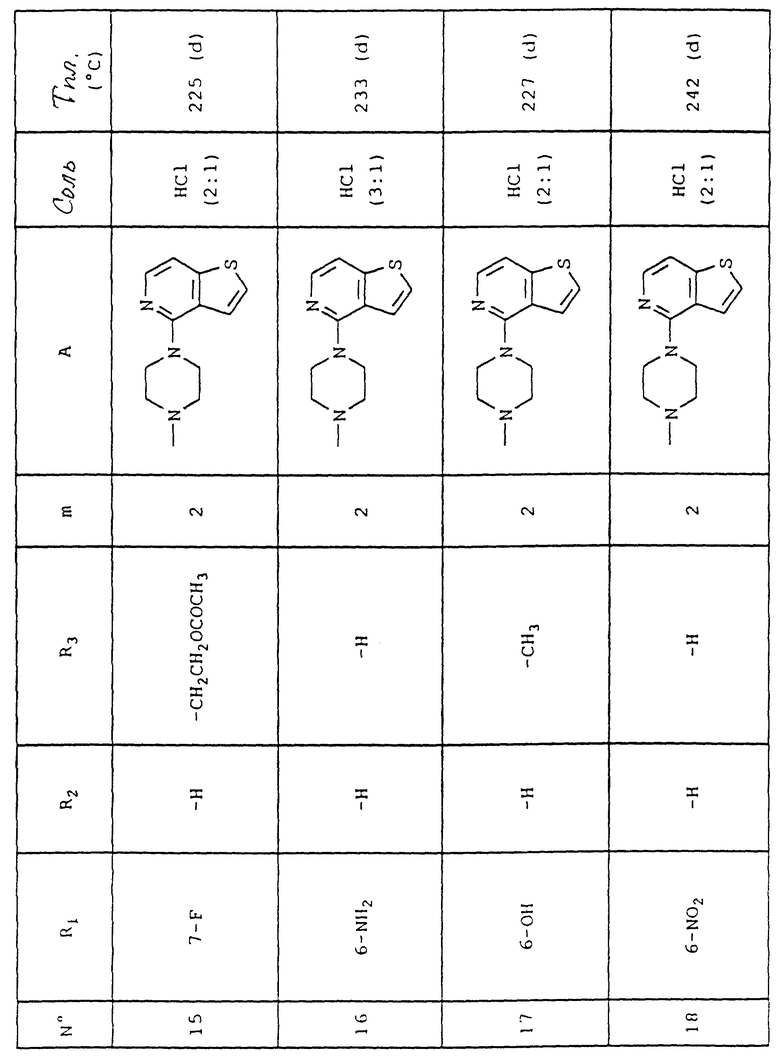
Проводят нитрование соединения формулы (VIIa), соответствующее соединению формулы (VII), в которой R1 является атомом водорода, X является атомом галогена и R3 представляет собой атом водорода или группу (C1-C4)алкил, с получением соединения формулы (VIIб), которое восстановлением водородом превращают в соединение формулы (VIIa), которое превращают либо в соединение формулы (VIIг) взаимодействием с хлоридом карбоновой кислоты формулы R4COCl, либо в соединение формулы (VIIд) взаимодействием хлоридом сульфоновой кислоты формулы R4SO2Cl, либо в соединение формулы (VIIe) реакцией N-диалкилирования. Затем эти соединения подвергают взаимодействию с 4-(пиперазин-1-ил)тиено[3,2-с] пиридином или с 4-(4-фторбензоил)пиперидином согласно Схеме 1.
Для получения соединений формулы (I), в которой R1 и/или R2 представляет(ют) собой гидроксильную группу, можно провести деалкилирование соответствующего алкоксилированного соединения формулы (I), в которой R1 и/или R2 представляет(ют) собой алкоксильную группу в стандартных условиях известных специалисту, такое как, например, обработка 48%-ой бромоводородной кислотой.
Исходные соединения коммерчески доступны или описаны в литературе, либо могут быть получены способами, которые описаны или которые известны специалисту.
Так, 4-(ацетилокси)-2H, 3H-пиран-2,6-дион получают из 3-оксоглутаровой кислоты согласно E.G. Frandsen и N. Jacobsen, J. Chem. Soc. Perkin I, 933-6 (1978).
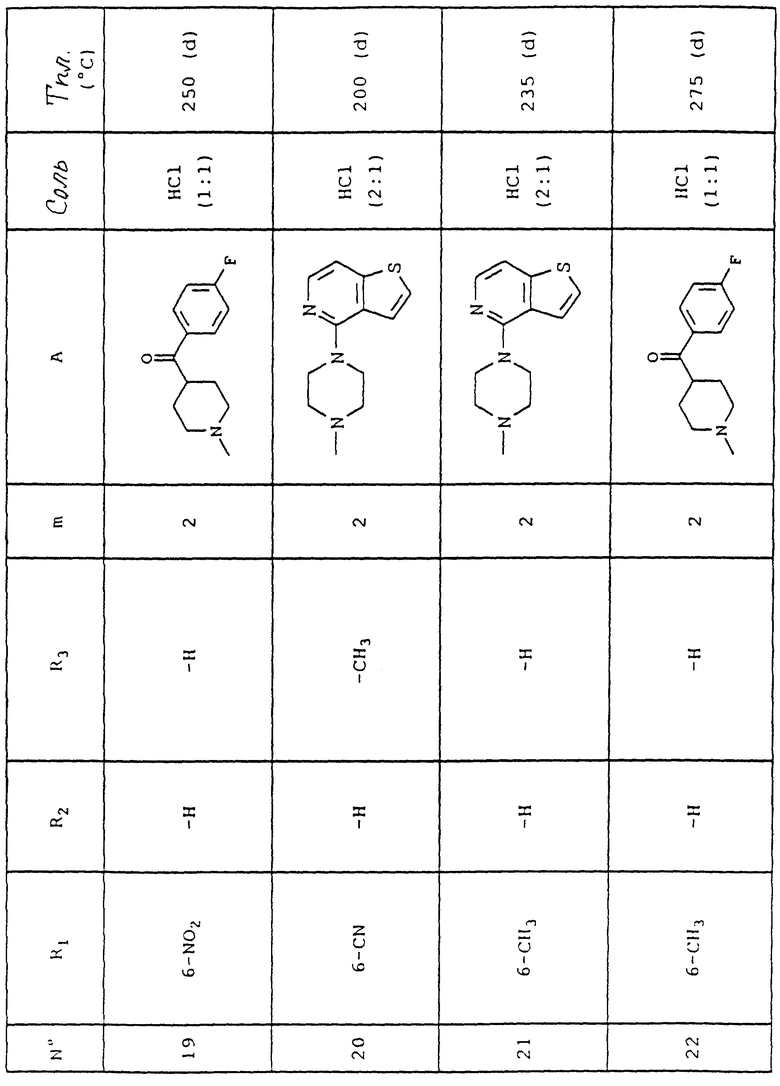
Способ циклизации описан в заявках на Европейский патент EP 0364327 и EP 0577325.
Введение нитрила в соединения формулы (V) проводят согласно методике, описанной N. Chantani и T. Hanafusa, J. Org. Chem. 51, 4714-4716 (1986).
Ароматическое нуклеофильное замещение иодированных арилов тиолатами основано на способе T. Migital и др. , Bull. Chem. Soc. Japan, 53, 1385 (1980).
4-(Пиперазин-1-ил)тиено[3,2-с] пиридин синтезируют согласно J.S. New и др., J. Med. Chem. 32, N 6, 1147-56 (1989).
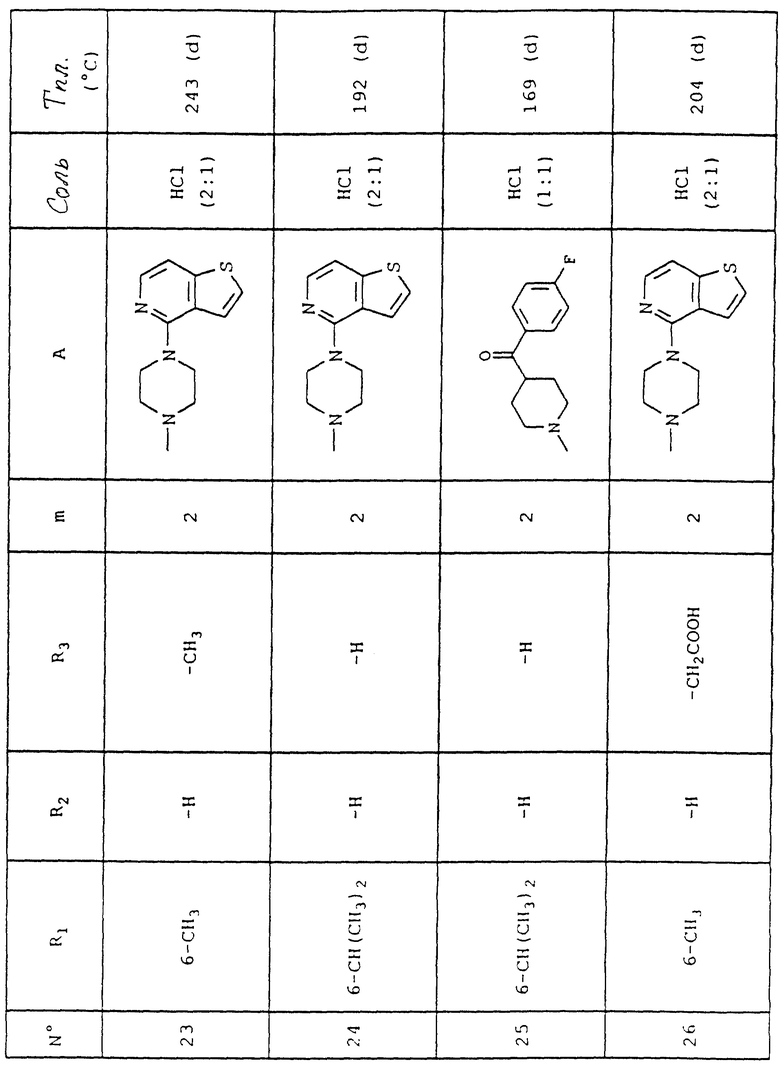
Следующие далее примеры иллюстрируют изобретение, не ограничивая его. Микро-анализы и ИК, ЯМР и масс-спектры подтверждают структуру полученных соединений.
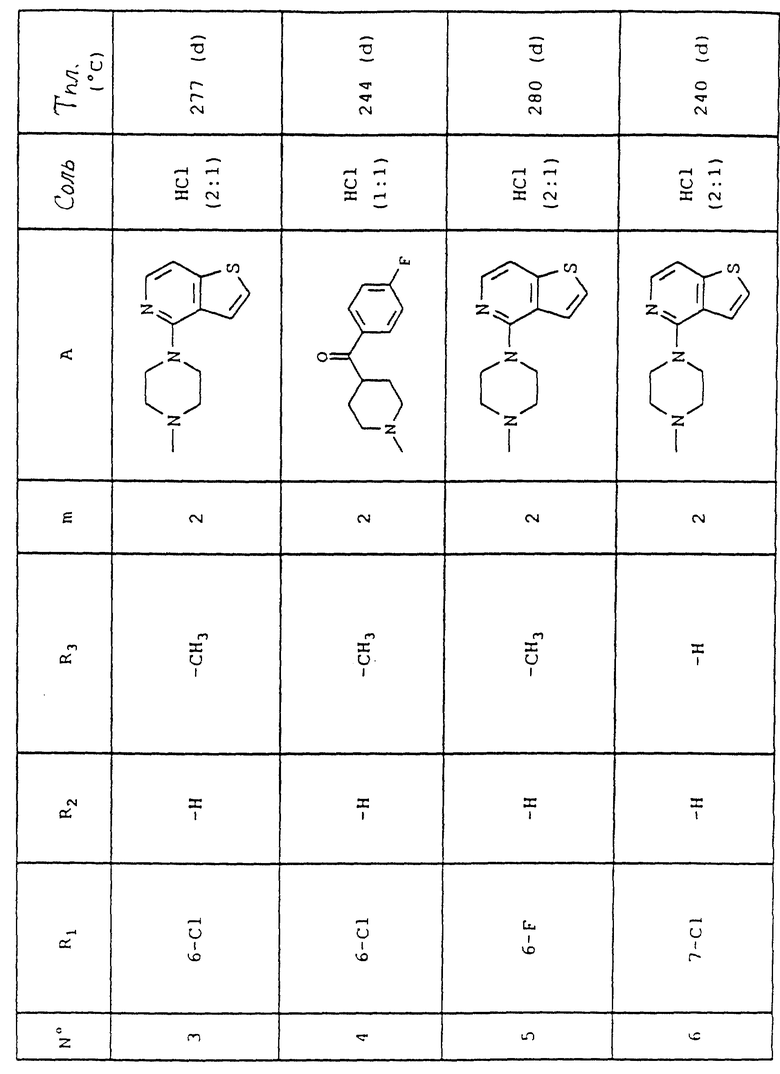
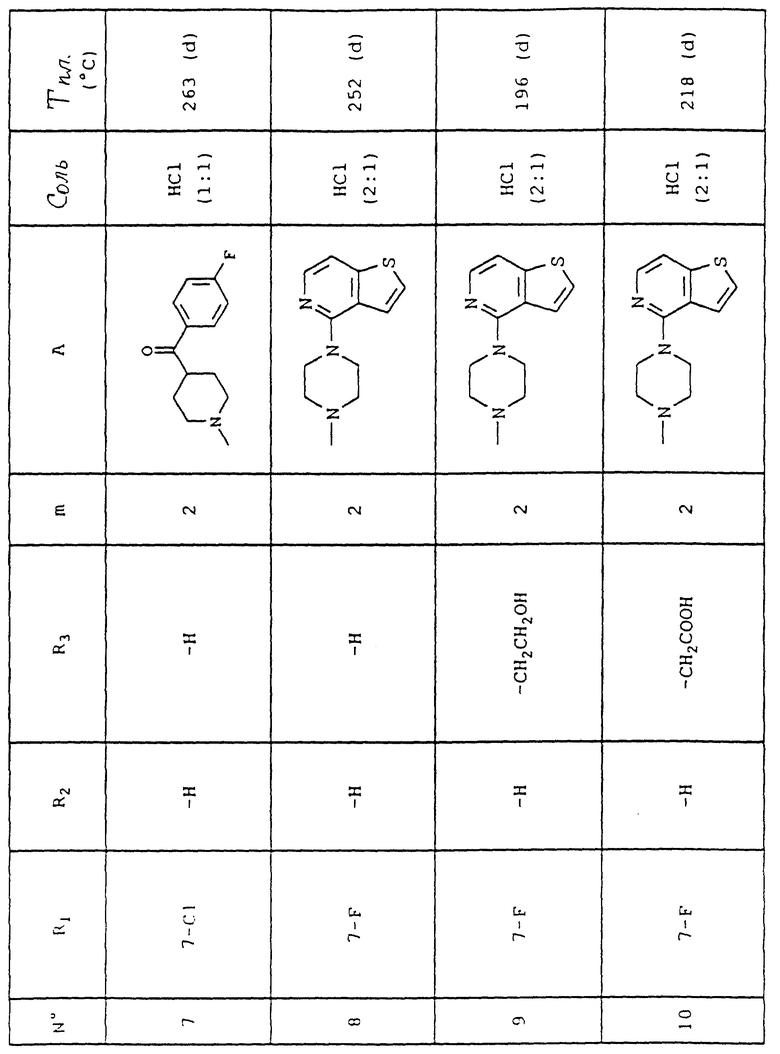
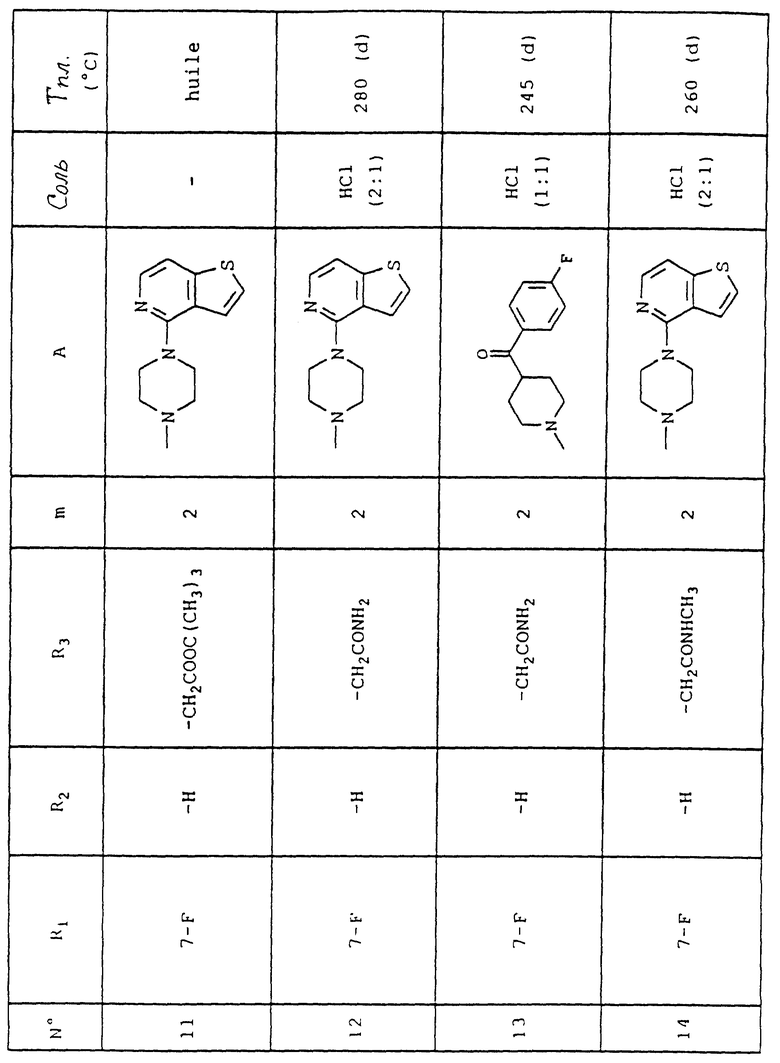
Номера приведенных в примерах соединений соответствуют номерам приведенной таблицы, которая иллюстрирует химические структуры и физические свойства некоторых соединений согласно данному изобретению.
Соотношения (x:y) соответствуют соотношению (кислота:основание).
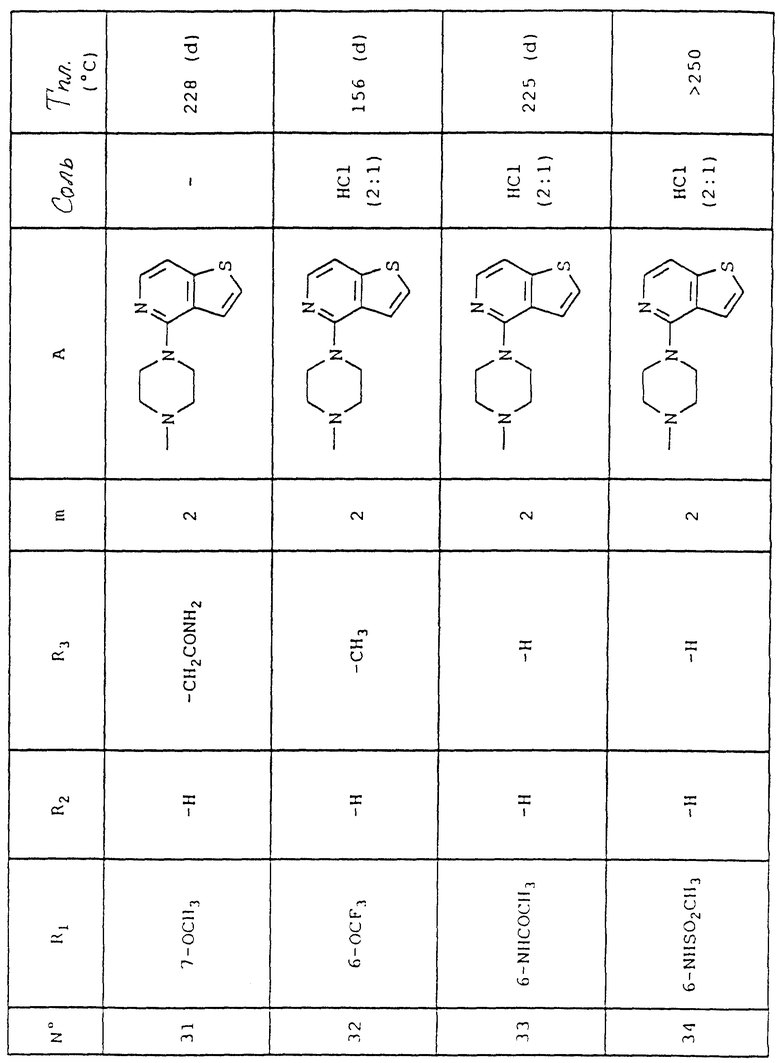
Пример 1 (соединение N 27)
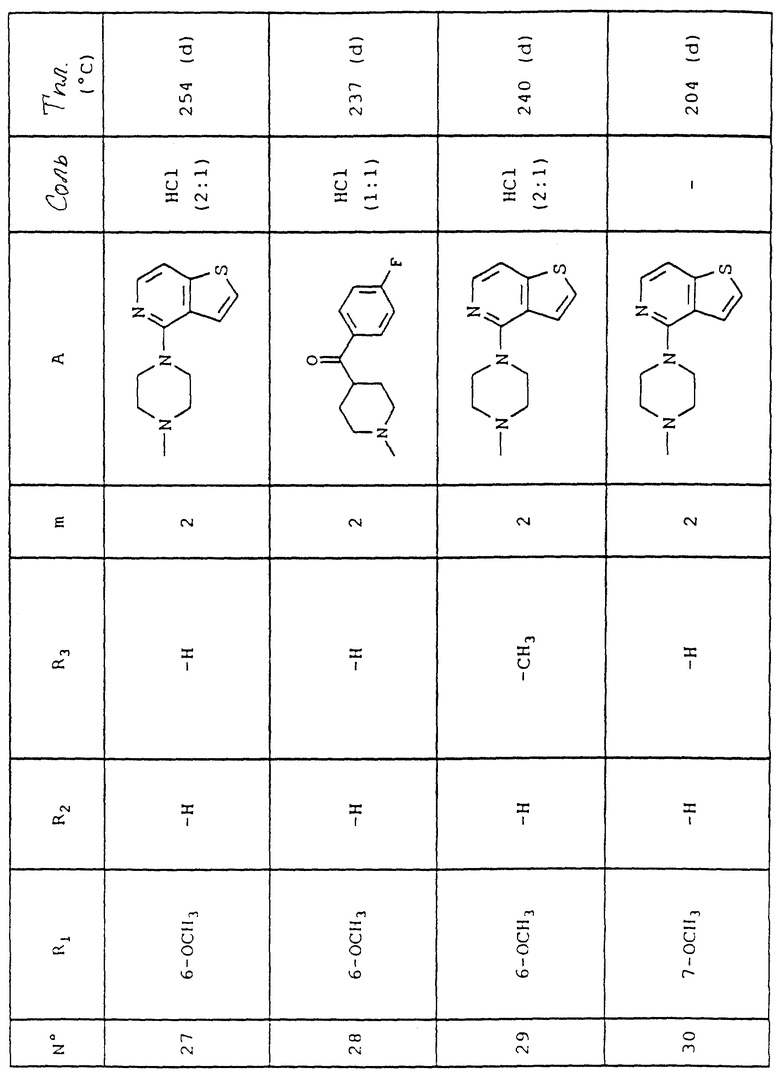
6-Метокси-4-[2-[4-(тиено[3,2-с] пиридин-4-ил)пиперазин-1-ил] этил] хинолин-2(1H)он гидрохлорид (2:1)
1.1. 3-(Ацетилокси)-5-[(4-метоксифенил)метиламино] -5-оксопент-2-еновая кислота
К раствору 20,0 г (146 ммоль) N-метил-4-метоксианилина в 100 мл уксусной кислоты добавляют при сильном перемешивании при комнатной температуре 27 г (158 ммоль) 4-(ацетилокси)-2H, 3H-пиран-2,6-диона. После 5 ч перемешивания при комнатной температуре добавляют 700 мл ледяной воды и перемешивают еще в течение 30 мин. Получают твердое вещество бежевого цвета, которое отжимают, промывают водой, растирают в диэтиловом эфире и сушат над пентоксидом фосфора при 40oC в течение 24 ч.
Получают 28,1 г продукта в виде твердого вещества.
Температура плавления = 85-88oC.
Выход = 76%.
1.2. 6-Метокси-2-оксо-1, 2-дигидрохинолин-4-уксусная кислота
К 70 мл серной кислоты (96-97%) при комнатной температуре добавляют небольшими порциями 41 г (133 ммоль) 3- (ацетокси)-5[(4-метоксифенил)-метиламино] -5-оксопент-2-еновой кислоты, затем смесь нагревают при перемешивании до 80oC в течение 1 ч 30 мин. После охлаждения реакционную среду вливают в 100 г льда и 100 мл воды, смесь перемешивают в течение 15 мин, и твердое вещество отжимают и обильно промывают водой, затем сушат при 50oC в течение 48 ч. Собирают 14,9 г смеси 6-метокси-1-метил-2-оксо-1,2-дигидрохинолин-4-уксусной кислоты и 6-метокси-2-оксо-1,2-дигидрохинолин-4-уксусной кислоты.
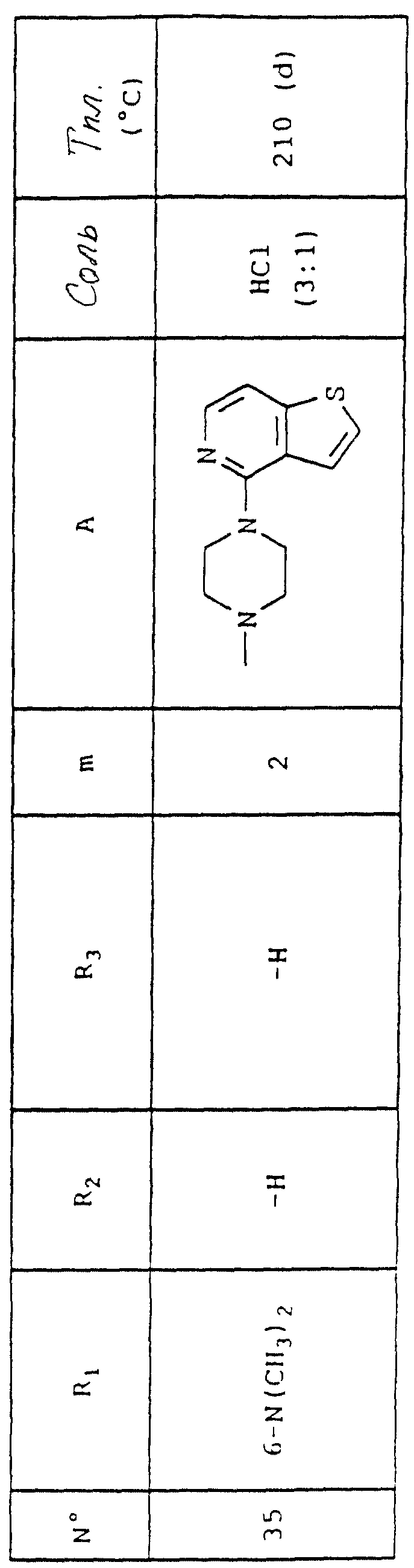
Выход = 45%.
1.3. Метиловый эфир 6-метокси-2-оксо-1,2-дигидрохинолин-4-уксусной кислоты
16 мл (219 ммоль) тионилхлорида добавляют по каплям к перемешиваемой суспензии 16,8 г (68 ммоль) смеси 6- метокси-1-метил-2-оксо-1,2-дигидрохинолин-4-уксусной кислоты и 6-метокси-2-оксо-1,2-дигидрохинолин-4-уксусной кислоты в 250 мл метанола при комнатной температуре, перемешивание затем продолжают в течение 16 ч. Растворитель отгоняют в вакууме, и остаток переносят в 400 мл дихлорметана. Смесь промывают насыщенным раствором гидрокарбоната натрия, затем водой, и органическую фазу сушат над сульфатом натрия. После фильтрации и концентрирования получают 12,6 г смеси двух сложных эфиров (71%). Разделяют эти два эфира посредством флэш-хроматографии на диоксиде кремния, элюируя смесью метанол/дихлорметан (3:97).
Получают 4,0 метилового эфира 6-метокси-1-метил-2-оксо-1,2-дигидрохинолин-4-уксусной кислоты,
Температура плавления = 129-130oC,
и 7,8 г метилового эфира 6-метокси-2-оксо-1,2-дигидрохинолин-4-уксусной кислоты.
Температура плавления = 223-224oC.
1.4. 4-(2-гидроксиэтил)-6-метоксихинолин-2(1H)-он
К суспензии 3,1 г (12,5 ммол) метилового эфира 6-метокси-2-оксо-1,2-дигидрохинолинуксусной кислоты в 100 мл сухого тетрагидрофурана и 1 мл метанола при комнатной температуре добавляют 1,4 г боргидрида натрия (37 ммоль), и реакционную среду нагревают с обратным холодильником в течение 16 ч. После охлаждения до 5oC добавляют по каплям 1 мл метанола, затем через 30 мин 0,5 г боргидрида натрия, и реакционную смесь нагревают в течение следующих 8 ч. После охлаждения и обработки 5 мл метанола растворители отгоняют, а остаток переносят в 200 мл дихлорметана и 100 мл 1 н. соляной кислоты. Органическую фазу отделяют, промывают водой и сушат над сульфатом натрия. После фильтрации и концентрирования в вакууме получают 1,95 г ожидаемого спирта.
Выход = 72%
1.5. 4-(2-хлорэтил)-6-метоксихинолин-2(1Н)-он
К суспензии 3,11 г (14,2 ммоль) 4-(2-гидроксиэтил)-6-метоксихинолин-2-(1H)-она в 50 мл хлороформа и 3 каплях диметилформамида добавляют 3,4 мл (46,6 ммоль) тионилхлорида при перемешивании при комнатной температуре. Суспензию нагревают с обратным холодильником в течение 14 ч (полное растворение). После охлаждения до комнатной температуры к реакционной среде добавляют по каплям 50 мл воды, и смесь перемешивают в течение 30 мин. Органическую фазу выделяют, промывают водой, сушат над сульфатом магния и фильтруют. Фильтрат концентрируют в вакууме.
Получают 3,2 г твердого светло-желтого вещества.
Температура плавления = 231-232oC.
Выход = 94%.
1.6. 6-Метокси-4-[2-[4-(тиено-[3,2-с]пиридин-4-ил)пиперазин-1-ил]этил] хинолин-2(1H)-она гидрохлорид (2:1)
1,2 г (5 ммоль) (2-Хлорэтил)-6-метоксихинолин-2(1H)-она добавляют к суспензии 1,2 г (5,5 ммоль) 4-(пиперазин-1-ил)тиено [3,2- с]пиридина и 0,44 г (5,25 ммоль) гидрокарбоната натрия в 15 мл ацетонитрила, затем реакционную смесь нагревают с обратным холодильником в течение 10 ч. После выпаривания растворителя в вакууме остаток переносят в 100 мл дихлорметана и промывают последовательно насыщенным водным раствором бикарбоната натрия, затем водой. После сушки над сульфатом натрия, фильтрации и концентрирования фильтрата, неочищенный продукт очищают посредством флэш-хроматографии на диоксиде кремния, элюируя смесью метанол/дихлорметан (5:95), содержащей следы водного аммиака.
Получают 0,50 г продукта в форме основания.
Выход = 24%.
Дигидрохлорид получают в смеси метанол/хлороводородная кислота/эфир.
Температура плавления = 254oC (разложение).
Пример 2 (соединение N 28)
4-[2-[4-(4-Фторбензоил)пиперидин-1-ил] этил] -6-метоксихинолин-2(1H)-он гидрохлорид (1:1)
Смесь 1,1 г (4,6 ммоль) 4-(2-хлорэтил)-6-метоксихинолин-2(1H)-она, 1,0 г (5,5 ммоль) 4-(4-фторбензоил)пиперидина и 0,38 г (4,6 ммоль) гидрокарбоната натрия в 20 мл ацетонитрила нагревают с обратным холодильником в течение 8,5 ч. Затем реакционную среду выпаривают до сухости, и неочищенный продукт очищают посредством флэш-хроматографии на диоксиде кремния, элюируя смесью метанол/дихлорметан (5:95), содержащей следы водного аммиака.
Получают 0,53 г ожидаемого продукта в форме основания.
Выход = 30%.
Гидрохлорид получают в смеси метанол/хлороводородная кислота.
Температура плавления = 237oC (разложение).
Пример 3 (соединение N 4)
6-Хлор-4-[2-[4-(4-фторбензоил)пиперидин-1-ил] этил] 1-метилхинолин-2(1H)-он гидрохлорид (1:1)
3.1. 3-(Ацетокси)-5-[(4-хлорфенил)метиламино]-5-оксопент-2-еновая кислота
К перемешиваемому раствору 15,0 г (106 ммоль) 4-хлор-N- метилбензоламина в 40 мл чистой уксусной кислоты добавляют небольшими порциями 19,8 г (116 ммоль) 4-(ацетилокси)-2H,3H-пиран-2,6-диона. Реакционную среду перемешивают в течение 3 ч при 35oC. Ей дают возможность охладиться до комнатной температуры и разбавляют в 10 мл ледяной воды. Твердое вещество отжимают, обильно промывают водой и сушат при 40oC в течение 48 ч.
Получают 25,5 г ожидаемого соединения в виде аморфного твердого вещества, которое без дальнейшей обработки используют на следующей стадии.
Выход = 77%.
3.2. 6-Хлор-1-метил-2-оксо-1,2-дигидрохинолин-4-уксусная кислота
25,5 г (81,8 ммоль) 3-(ацетилокси)-5-[(4-хлорфенил)метиламино]5-оксопент- 2-еновой кислоты вносят небольшими порциями в 40 мл концентрированной серной кислоты при комнатной температуре при интенсивном перемешивании, и реакционную среду нагревают до 85oC в течение 60 мин. После охлаждения этот раствор вливают в смесь 500 г льда и 500 мл воды. Полученное таким образом твердое вещество серого цвета отжимают, промывают водой, затем растирают в эфире и сушат в течение 24 ч при 40oC.
Получают 9,47 г ожидаемого продукта, который без дальнейшей обработки используют на следующей стадии.
Выход = 46%.
3.3. Метиловый эфир 6-хлор-1-метил-1,2-дигидрохинолин-4-уксусной кислоты
11 мл (147 ммоль) тионилхлорида добавляют по каплям к перемешиваемой суспензии 12,5 г (49 ммоль) 6-хлор-1-метил-2-оксо-1,2-дигидрохинолин-4-уксусной кислоты в 150 мл метанола. Смесь перемешивают при комнатной температуре в течение 17 ч, и растворитель удаляют в вакууме. Остаток растворяют в 400 мл дихлорметана, а затем промывают насыщенным водным раствором бикарбоната натрия, затем водой. После сушки над сульфатом натрия органическую фазу фильтруют, и фильтрат конденсируют.
Получают 11,16 г ожидаемого продукта.
Выход = 85%.
Температура плавления = 99-101oC.
3.4. 6-Хлор-4-(2-гидроксиэтил)-1-метилхинолин-2(1H)-он
К суспензии 5,9 г (23,4 ммоль) метилового эфира 6-хлор-1-метил- 1,2-дигидрохинолин-4-уксусной кислоты в 10 мл метанола и 100 мл сухого тетрагидрофурана, добавляют 3,0 г (79 ммоль) боргидрида натрия, и смесь затем нагревают с обратным холодильником в течение 9 ч. После охлаждения растворители в вакууме отгоняют, и остаток переносят в 400 мл дихлорметана и 100 мл 3 н. хлороводородной кислоты. Органическую фазу промывают водой, сушат над сульфатом натрия, фильтруют, и фильтрат конденсируют. Неочищенный продукт очищают посредством флэш-хроматографии на диоксиде кремния, элюируя смесью метанол/дихлорметан (5:95).
Получают 5,9 г ожидаемого спирта.
Выход = 92%.
Температура плавления = 169-170oC.
3.5. 6-Хлор-4-(2-хлорэтил)-1-метилхинолин-2(1H)-он
К суспензии 5,9 г (24,8 ммоль) 6-хлор-4-(2-гидроксиэтил)-1-метилхинолин-2(1H)-она в 120 мл хлороформа, двух каплях пиридина и двух каплях диметилформамида добавляют по каплям 5,5 мл (75 ммоль) тионилхлорида. Реакционную среду нагревают с обратным холодильником при несильном кипении в течение 2,5 ч, а затем обрабатывают, как описано в примере 1.5.
Получают 5,4 г ожидаемого продукта.
Выход = 86%.
Температура плавления = 120-122oC.
3.6. 6-Хлор-4-[2-[4-(4-фторбензоил)пиперидин-1-ил] этил] -1-метилхинолин-2(1H)-она гидрохлорид (1:1)
Смесь 0,90 г (3,5 ммоль) 6-хлор-4-(2-хлорэтил)-1-метилхинолин-2(1H)-она, 0,71 г (4,0 ммоль) 4-(4-фторбензоил)пиперидина и 0,60 г (7,0 ммоль) бикарбоната натрия в 15 мл ацетонитрила нагревают до образования флегмы в течение 11 ч. Затем реакционную среду выпаривают до сухости, и неочищенный продукт очищают посредством флэш-хроматографии на диоксиде кремния, элюируя смесью метанол/дихлорметан (4:96), содержащей следы водного аммиака.
Получают 0,86 г ожидаемого продукта в форме основания.
Выход = 62%.
Гидрохлорид получают в смеси метанол/хлороводородная кислота/эфир.
Температура плавления = 244oC (разложение).
Пример 4 (соединение N 5)
6-Фтор-1-метил-4-[4-(тиено)-[3,2-с]пиридин-4-ил)пиперазин-1-ил]этил]-хинолин-2(1H)-он гидрохлорид (2:1)
4.1. 3-(Ацетокси)-5-[(4-фторфенил)метиламино]-5-оксопент-2-еновая кислота
К перемешиваемому раствору 6,64 г (53,1 ммоль) N-метил-4- фторанилина в 25 мл чистой уксусной кислоты добавляют небольшими порциями 9,93 г (58,4 ммоль) 4-(ацетилокси)-2H,3H-пиран-2,6-дион. Реакционную среду перемешивают в течение 2 ч при 35oC, дают возможность охладиться до комнатной температуры и разбавляют в 500 мл ледяной воды. Полученное твердое вещество выделяют и отжимают, обильно промывают водой и сушат в термостате (40oC) в течение 48 ч.
Получают 12,05 г ожидаемого соединения в виде аморфного твердого вещества, которое плавится ниже 50oC.
Выход = 76%.
4.2. 6-Фтор-1-метил-2-оксо-1,2-дигидрохинолин-4-уксусная кислота
31,8 г (107 ммоль) 3-(ацетилокси)-5-[(4-фторфенил)метиламино]-5-оксопент-2-еновой кислоты вносят небольшими порциями в 130 мл концентрированной серной кислоты при комнатной температуре при интенсивном перемешивании, и затем реакционную среду нагревают до 90oC в течение 90 мин. После охлаждения этот раствор вливают в смесь 500 г льда и 500 мл воды. Полученное таким образом твердое вещество серого цвета отжимают. Промывают его водой, затем растирают в эфире и сушат при 40oC в течение 24 ч.
Получают 11,37 г продукта.
Температура плавления = 230oC.
Выход = 45%.
4.3. Метиловый эфир 6-фтор-1-метил-2-оксо-1,2-дигидрохинолин-4-уксусной кислоты
16 мл (219 ммоль) тионилхлорида добавляют по каплям в течение приблизительно 30 мин к перемешиваемой суспензии 11,37 г (49,38 ммоль) смеси 6-фтор-1-метил-2-оксо-1,2-дигидрохинолин-4-уксусной кислоты в 120 мл метанола. Смесь перемешивают при комнатной температуре в течение ночи (13 ч), и растворитель удаляют в вакууме. Остаток растворяют в 400 мл дихлорметана, а затем промывают насыщенным водным раствором бикарбоната натрия, затем водой. После сушки над сульфатом натрия, фильтрации и концентрирования фильтрата получают 9,6 г ожидаемого продукта.
Выход = 78%.
Температура плавления = 134-135oC.
4.4. 6-Фтор-4-(2-гидроксиэтил)-1-метилхинолин-2(1H)-он
К суспензии 8,0 г (32 ммоль) метилового эфира 6-фтор-1-метил-2-оксо-1,2-дигидрохинолин-4-уксусной кислоты в 100 мл сухого тетрагидрофурана добавляют 3,78 г (100 ммоль) боргидрида натрия, и смесь нагревают с обратным холодильником в течение 20 ч. После охлаждения до 5oC по каплям добавляют 2 мл метанола, затем добавляют 3 г боргидрида натрия, и смесь нагревают с обратным холодильником в течение 12 ч. Растворители выпаривают в вакууме, и остаток переносят в 400 мл дихлорметана и 150 мл 2 н. соляной кислоты, органическую фазу промывают водой, затем сушат над сульфатом натрия и фильтруют, и фильтрат концентрируют.
Получают 4,7 г ожидаемого спирта.
Выход = 66%.
Температура плавления = 153-154oC.
4.5. 4-(2-Хлорэтил)-6-фтор-1-метилхинолин-2(1H)-он
К суспензии 2,2 г (9,95 ммоль) 6-фтор-4-(2-гидроксиэтил)- 1-метилхинолин-2(1H)-она в 100 мл хлороформа, двух каплях пиридина и двух каплях диметилформамида по каплям добавляют 3 мл (41 ммоль) тионилхлорида. Реакционную среду нагревают при несильном кипении с обратным холодильником в течение 4,5 ч. После охлаждения до комнатной температуры в реакционную среду добавляют по каплям 50 мл воды, и смесь перемешивают еще 30 мин. Органическую фазу выделяют, промывают водой, сушат над сульфатом магния и фильтруют. Фильтрат концентрируют в вакууме.
Получают 2,36 г ожидаемого хлорида.
Выход = 98%.
Температура плавления = 141-142oC.
4.6. 6-Фтор-1-метил-4-[2-[4-(тиено)-[3,2-с]пиридин-4-ил)пиперазин-1 -ил] этил]-хинолин-2(1H)-он гидрохлорид (2:1)
1,4 г (5,8 ммоль) 4-(2-хлорэтил)-6-фтор-1-метил-хинолин-2(1H)-она добавляют к смеси 1,3 г (5,9 ммоль) 4-(пиперазин-1-ил)тиено[3,2-с]пиридина и 0,50 г (5,95 ммоль) гидрокарбоната натрия в 20 мл ацетонитрила, и реакционную среду нагревают при 55-60oC в течение 18 ч. Растворитель выпаривают, и остаток переносят в 100 мл дихлорметана. Его промывают насыщенным водным раствором бикарбоната натрия, затем водой. Органическую фазу сушат над сульфатом натрия, фильтруют, и фильтрат конденсируют. Неочищенный продукт очищают флэш-хроматографией на диоксиде кремния, элюируя смесью метанол/дихлорметан (5: 95), содержащей следы водного аммиака. Получают 0,70 г ожидаемого продукта в форме основания.
Выход = 27%.
Основание растворяют в 10 мл метанола и переводят в соль с избытком 2 н. раствора хлороводородной кислоты в эфире. Полученный осадок отжимают, перекристаллизовывают из метанола и сушат в вакууме.
Получают 0,38 г дигидрохлорида.
Температура плавления = 280oC (разложение).
Пример 5 (соединение N 10)
7-Фтор-2-оксо-4-[2-[4-(тиено[3,2-с] пиридин-4-ил)пиперазин-1-ил] этил]-1,2-дигидрохинолин-1-уксусной кислоты гидрохлорид (2:1)
2,9 мл 0,5 М раствора трет-бутилового эфира бромуксусной кислоты в тетрагидрофуране добавляют по каплям к смеси 0,50 г (1,23 ммоль) 7-фтор-4-[2-[4-(тиено[3,2-с] пиридин-4-ил)пиперазин-1-ил] этил] хинолин-2(1H)-она (полученного из 3-фторанилина согласно способу, описанному в примере 4), 0,10 г (1,79 ммоль) свежеразмолотого гидроксида калия и 0,12 г (0,37 ммоль) тетрабутиламмонийбромида в 20 мл тетрагидрофурана при 0-5oC. После 30 мин при 0-5oC дают возможность температуре подняться до комнатной температуры, и продолжают перемешивание в течение 6 ч. Растворитель выпаривают в вакууме, и остаток переносят в 100 мл дихлорметана, промывают водой, и органическую фазу сушат над сульфатом натрия и конденсируют. Неочищенный продукт очищают флэш-хроматографией на диоксиде кремния, элюируя смесью метанол/дихлорметан (5: 95), содержащей следы водного аммиака, и получают 0,48 г трет-бутил-N-ацетата в виде густого бесцветного масла.
Выход = 75%.
К этому маслу добавляют 50 мл 3 н. раствора хлороводородной кислоты в этилацетате, и смесь перемешивают при комнатной температуре в течение 4 ч. Ее выпаривают до сухости, и полученное белое твердое вещество растирают с эфиром и сушат в вакууме.
Получают 0,47 г ожидаемой кислоты в форме дигидрохлорида.
Выход = 87%.
Температура плавления = 218-220oC (разложение).
Пример 6 (соединение N 12)
7-Фтор-2-оксо-4-[2-[4-(тиено[3,2-с] пиридин-4-ил)пиперазин-1-ил] этил]-1,2-дигидрохинолин-1-ацетамид гидрохлорид (2:1)
3,9 мл 0,5 М раствора бромацетамида в тетрагидрофуране добавляют по каплям к перемешиваемой смеси 0,53 г (1,3 ммоль) 7-фтор-4-[2-[4-(тиено[3,2-с] пиридин-4-ил)этил] хинолин-2(1H)-она, 0,1 г (1,79 ммоль) измельченного гидроксида калия и 0,13 г (0,4 ммоль) трет-бутиламмонийбромида в 25 мл тетрагидрофурана при 0-5oC. Через 30 мин дают возможность температуре подняться до комнатной температуры, и смесь перемешивают при этой температуре в течение 20 ч. Реакционную среду выпаривают до сухости в вакууме, и остаток переносят в 100 мл дихлорметана. Этот раствор промывают водой. Органическую фазу сушат над сульфатом магния и концентрируют. Неочищенный продукт растирают в смеси диэтиловый эфир/дихлорметан (1:3), затем твердое вещество отжимают и очищают хроматографией на диоксиде кремния, элюируя смесью метанол/этилацетат (10:90), а затем смесью метанол/дихлорметан (10: 90), содержащей следы водного аммиака.
Получают 0,303 г белого твердого вещества, которое превращают в дигидрохлорид в смеси 2 М хлороводородная кислота/диэтиловый эфир/метанол.
Получают 0,32 г дигидрохлорида.
Температура плавления = 280oC (разложение).
Пример 7 (соединение N 20)
1-Метил-2-оксо-4-[2-[4-(тиено[3,2-с] пиридин-4-ил)пиперазин-1-ил]этил]-1,2-дигидрохинолин-6-карбонитрил гидрохлорид (2:1)
7.1. Метиловый эфир 6-циано-1-метил-2-оксо-1, 2-дигидрохинолин-4-уксусной кислоты
К раствору 0,50 г (1,4 ммоль) метилового эфира 6-иод-1-метил-2-оксо-1,2-дигидрохинолин-4-уксусной кислоты (полученной из N-метил-4-иоданилина согласно способу, описанному в примере 1) в 6 мл безводного триэтиламина добавляют 1,1 мл триметилсилилцианида (8,4 ммоль), затем 0,15 г (0,13 ммоль) тетракистрифенилфосфинпалладия. Затем реакционную среду нагревают с обратным холодильником в течение 4 ч в атмосфере азота. После охлаждения до комнатной температуры эту среду вливают в 60 мл толуола и 60 мл воды. Органическую фазу промывают водой, и начальную водную фазу повторно экстрагируют дихлорметаном. Органические фазы объединяют, сушат над сульфатом натрия и концентрируют в вакууме. Остаток очищают флэш-хроматографией на диоксиде кремния, элюируя смесью метанол/дихлорметан (5:95). Получают 0,313 г ожидаемого нитрила.
Выход = 87%.
Температура плавления = 202-203oC.
7.2. 6-Циан-4-(2-гидроксиэтил)-1-метилхинолин-2(1H)-он
7.2.1. 6-Циан-1-метил-2-оксо-1,2-дигидрохинолин-4-уксусная кислота
К 1,21 г (4,7 ммоль) метилового эфира 6-циан-1-метил-2-оксо-1,2-дигидрохинолин-4-уксусной кислоты в 10 мл метанола при 0-5oC добавляют по каплям 10,4 мл 0,5 н. раствора гидроксида лития (5,2 ммоль). Температуре дают возможность подняться до комнатной температуры, и реакционную среду перемешивают в течение 2 ч. Реакционную среду вливают в 250 мл ледяной воды и подкисляют до pH 2-3 4 н. соляной кислотой. Образовавшийся белый осадок отжимают, промывают водой, затем сушат в вакууме при 40oC.
Получают 0,85 г ожидаемого продукта.
Выход = 75%.
Температура плавления = 238oC.
7.2.2 6-Циан-4-(2-гидроксиэтил)-1-метилхинолин-2(1H)-он
К суспензии 0,365 г (1,51 ммоль) 6-циан-1-метил-2-оксо-1,2-дигидрохинолин-4-уксусной кислоты в 10 мл тетрагидрофурана при -10oC добавляют 0,22 мл (1,58 ммоль) триэтиламина, затем по каплям добавляют 0,16 мл (1,6 ммоль) этилхлорформиата. После перемешивания при -10oC в течение 45 мин реакционную среду фильтруют, и твердые вещества промывают 3 х 8 мл тетрагидрофурана. К фильтрату при 5-10oC добавляют 0,25 г (6,61 ммоль) боргидрида натрия, а затем 0,94 мл метанола. После перемешивания при 5-10oC в течение 2 ч добавляют 13 мл 1 н. раствора соляной кислоты. Смесь экстрагируют дихлорметаном, а затем этилацетатом. Органические фазы сушат над сульфатом натрия, затем концентрируют в вакууме.
Получают 0,315 г продукта.
Выход = 92%.
Температура плавления = 231-233oC.
7.3. 4-(2-Бромэтил)-6-циан-метилхинолин-2(1H)-он
К 0,48 г (1,14 ммоль) дибромтрифенилфосфорана в 14 мл дихлорметана при комнатной температуре небольшими порциями добавляют 0,24 г (1,05 ммоль) 6-циан-4-(2-гидроксиэтил)-1-метилхинолин-2(1H)-она. После 75 мин перемешивания при комнатной температуре реакционную среду вливают в 200 мл дихлорметана, и смесь промывают водой. Органическую фазу сушат над сульфатом натрия, фильтруют и конденсируют в вакууме. Белый остаток растирают в диэтиловом эфире. Полученное твердое вещество переносят в минимальное количество дихлорметана, смесь быстро фильтруют через слой диоксида кремния, элюируя эфиром, и фильтрат выпаривают.
Получают 0,20 г продукта, который используют без дальнейшей обработки.
Выход = 65%.
7.4. 1-Метил-2-оксо-4-[2-[4-(тиено[3,2-с] пиридин-4-ил)пиперазин-1-ил] этил]хинолин-2(1H)-он гидрохлорид (2:1)
Смесь 0,19 г (0,65 ммоль) 4-(2-бромэтил)-6-циан-1-метилхинолин-2(1H)-она, 0,15 г (0,65 ммоль) 4-(пиперазин-1-ил)тиено[3,2-с]пиридина и 0,09 г (0,11 ммоль) бикарбоната натрия в 10 мл ацетонитрила нагревают до 55oC в течение 36 ч. Реакционную среду выпаривают до сухости, остаток переносят в 100 мл хлороформа и промывают водой. Его сушат над сульфатом натрия и концентрируют, и неочищенный продукт очищают флэш-хроматографией на диоксиде кремния, элюируя смесью метанол/дихлорметан (1:9), содержащей следы водного аммиака.
Получают 0,211 г основания в виде бесцветного масла.
Выход = 48%.
Дигидрохлорид получают в смеси метанол/эфир/ 1 н. хлороводородная кислота.
Получают 0,182 г продукта в форме дигидрохлорида.
Температура плавления = 200oC (разложение).
Пример 8 (соединение N 17)
6-Гидрокси-1-метил-4-[2-[4-(-тиено[3,2-с] пиридин-4-ил)пиперазин-1-ил] этил]-хинолин-2(1H)-он гидрохлорид (2:1)
0,47 г (1,08 ммоль) 6-метокси-1-метил-4-[2-[4-(тиено[3,2-с]пиридин-4-ил)пиперазин-1-ил] этил] хинолин-2(1H)-она (полученного из метилового эфира 6-метокси-1-метил-2-оксо-1,2-дигидрохинолин-4-уксусной кислоты согласно Примеру 1) добавляют к 25 мл 48%-ной бромоводородной кислоты и доводят до образования флегмы в течение 3 ч. После охлаждения серый осадок отфильтровывают, промывают холодной водой и сушат в вакууме при 40oC. Получают 0,444 г продукта в форме дигидробромида.
Выход = 71%.
0,14 г (0,24 ммоль) этого продукта переносят в 22 мл 3,7 н. хлороводородной кислоты в безводном метаноле, и смесь перемешивают при комнатной температуре в течение 3 ч. Осадок отжимают, промывают диэтиловым эфиром и сушат в термостате. Получают 0,112 г ожидаемого продукта.
Выход = 95%.
Температура плавления = 227oC (разложение).
Пример 9 (соединение N 18)
6-Нитро-4-[2-[4-(тиено[3,2-с] пиридин-4-ил)пиперазин-1-ил]этил]хинолин-2(1H)-он гидрохлорид (2:1)
9.1. 4-(2-Хлорэтил)-6-нитрохинолин-2(1H)-он
К смеси 120 мл 65%-ной азотной кислоты и 80 мл концентрированной серной кислоты, охлажденной до 5oC, добавляют небольшими порциями 20,0 г (96,4 ммоль) 4-(2-хлорэтил)хинолин-2(1H)-она, и смесь нагревают при 45oC в течение 2 ч. Реакционную среду вливают в 600 мл ледяной воды, отжимают светло-желтый осадок, промывают его водой и сушат в вакууме.
Получают 22,5 г ожидаемого продукта.
Выход = 92%.
Температура плавления = 239-237oC.
9.2. 6-Нитро-4-[2-[4-(тиено[3,2-с]пиридин-4-ил)пиперазин-1-ил]этил]хинолин-2(1H)-он гидрохлорид (2:1)
Смесь 1 г (3,96 ммоль) 4-(2-хлорэтил)-6-нитрохинолин-2(1H)-она, 0,87 г (4 ммоль) 4-(пиперазин-1-ил)тиено[3,2-с]пиридина и 0,5 г (5,95 ммоль) бикарбоната натрия в 10 мл диметилформамида нагревают до 50oC в течение 20 ч. Затем остаток отфильтровывают и промывают водой, к фильтрату добавляют 200 мл воды, отжимают образовавшийся осадок и сушат его в вакууме.
Получают 1,28 г ожидаемого продукта в форме основания.
Выход = 74%.
Гидрохлорид получают в смеси метанол/диэтиловый эфир/хлороводородная кислота.
Температура плавления = 242oC (разложение).
Пример 10 (соединение N 16)
6-Амино-4-[2-[4-(тиено[3,2-с] пиридин-4-ил)пиперазин-1-ил]этил]хинолин-2(1H)-он гидрохлорид (3:1)
10.1. 6-Амино-4-(2-хлорэтил)хинолин-2(1H)-он гидрохлорид (1:1)
К суспензии 3,5 г (13,8 ммоль) 4-(2-хлорэтил)-6- нитрохинолин-2(1H)-она в 300 мл метанола при комнатной температуре добавляют 0,70 г палладия на угле (5% Pd), и смесь перемешивают при давлении водорода 8 psi (0,06 МПа) в течение 3 ч. Катализатор отфильтровывают, и фильтрат конденсируют.
Получают 2,97 г продукта в форме основания.
Гидрохлорид получают в смеси метанол/эфир/хлороводородная кислота.
Температура плавления выше 290oC.
10.2. 6-Амино-4-[2-[4-(тиено[3,2-с]пиридин-4-ил)пиперазин-1-ил]этил]хинолин-2(1H)-он гидрохлорид (3:1)
Смесь 0,35 г (1,35 ммоль) 6-амино-4-(2-хлорэтил)хинолин-2(1H)-он гидрохлорида, 0,33 г (1,5 ммоль) 4-(пиперазин-1-ил)тиено[3,2- с]пиридина и 0,17 г (2 ммоль) бикарбоната натрия в 10 мл диметилформамида нагревают до 60oC в течение 24 ч. После охлаждения до комнатной температуры реакционную среду разбавляют в 50 мл воды, и неочищенный продукт экстрагируют хлороформом. Органическую фазу сушат над сульфатом натрия и концентрируют, неочищенный продукт очищают посредством флэш-хроматографии на диоксиде кремния, элюируя сначала смесью метанол/этилацетат (6,5:93,5), содержащей следы триэтиламина, затем смесью метанол/дихлорметан (6,5:93,5), содержащей следы водного аммиака.
Получают 0,14 г продукта в форме основания.
Выход = 26%.
Затем получают тригидрохлорид в стандартных условиях.
Температура плавления = 233oC (разложение).
Пример 11 (соединение N 33)
6-Ацетиламино-4-[2-[4-(тиено[3,2-с] пиридин-4-ил)пиперазин-1-ил]этил]хинолин-2(1H)-он гидрохлорид (2:1)
11.1. 6-Ацетиламино-4-(2-хлорэтил)хинолин-2(1H)он гидрохлорид (1:1)
К суспензии 1,0 г (4,49 ммоль) 6-амино-4-(2-хлорэтил)хинолин-2(1H)-она в 50 мл хлороформа при комнатной температуре добавляют 0,75 мл (5,39 ммоль) триэтиламина, а затем 0,35 мл (4,9 ммоль) ацетилхлорида. Смесь перемешивают в течение 16 ч, затем разбавляют в 200 мл хлороформа. Суспензию промывают 1 н. раствором соляной кислоты и отжимают осадок.
Получают 0,72 г ожидаемого продукта.
Выход = 60%.
11.2. 6-Ацетиламино-4-[2-[4-(тиено[3,2-с]пиридин-4-ил)пиперазин-1-ил]этил]-хинолин-2(1H)-он гидрохлорид (2:1)
Смесь 0,35 г (1,32 ммоль) 6-ацетиламино-4-(2-хлорэтил)хинолин-2(1H)-он гидрохлорида, 0,38 г (1,75 ммоль) 4- (пиперазин-1-ил)тиено[3,2-с]пиридина и 0,17 г (2 ммоль) бикарбоната натрия в 10 мл диметилформамида. После охлаждения до комнатной температуры реакционную среду разбавляют в 100 мл воды и оставляют стоять в течение ночи при 5oC. Образовавшееся твердое вещество отжимают и сушат в вакууме. Неочищенный продукт очищают флэш-хроматографией на диоксиде кремния, элюируя сначала смесью метанол/этилацетат (5:95), затем смесью метанол/дихлорметан (10:90), содержащей следы водного аммиака.
Получают 0,20 г продукта в форме основания.
Выход = 34%.
Затем получают дигидрохлорид в стандартных условиях.
Температура плавления = 225oC (разложение).
Объяснения к таблице:
- в графе "Соль":
HCl представляет собой гидрохлорид,
соотношение (x:y) соответствует соотношению (кислота:основание),
отсутствие любого примечания означает, что данное соединение находится в форме основания;
- в графе "Температура плавления":
"(d)" соответствует плавлению с разложением.
Соединения по изобретению были объектом фармакологических исследований, которые продемонстрировали их свойства антагонистов серотонина и их значение как веществ, обладающих терапевтической активностью.
Так, соединения по изобретению были подвергнуты испытанию на ингибирование сосудосжимающего действия серотонина. Используют самцов крыс (Spragut-Dawley, Charles River France) весом от 250 до 300 г, которых анестезируют пентобарбитоном натрия (60 мг/кг/i.p.) и поддерживают под искусственным дыханием (респиратор HarvardTM - частота дыхания 70 мл в мин, объем воздуха 1 мл на 100 г веса тела). Животных забивают металлическим стержнем, введенным через орбиту правого глаза вдоль вертебрального столба. Правый и левый блуждающие нервы рассекают (биваготомия), и правую сонную артерию зашивают, причем в левую сонную артерию вставляют катетер, чтобы измерять кровяное давление с использованием датчика давления (StathamTM тип P23Db). В бедренную вену вставляют катетер с целью введения различных соединений. Увеличения среднего артериального кровяного давления, вызываемые серотонином, введенным внутривенно в дозе 30 мг/кг, измеряют. Соединения по изобретению или разбавитель вводят за 5 мин (для опытов при внутривенном введении) или за 75 мин (для опытов при пероральном введении) до введения серотонина. Соединения по изобретению вводят в дозах от 0,001 до 10 мг/кг. Процент ингибирования контрольной ответной реакции на серотонин используют для оценки потенциала соединений по изобретению как антагонистов серотонина.
Соединения по изобретению также были испытаны на модели сужения сосудов суматриптаном на изолированной бедренной вене собаки (антагонистическая активность по 5-НТ1-подобному рецептору согласно Humphrey и др. в Br.J.Pharmacol. 1988, 94, 1123).
Бедренные вены гончих или собак породы Англо-Пуатье удаляют под анестезией посредством внутривенной инъекции. Сосуд нарезают в спирали 0,4 см шириной, а затем делят на сегменты по 0,5 см длиной. Каждый фрагмент, закрепленный между двумя зажимами, помещают в ванну для изолированных органов, содержащую 20 мл физиологического раствора Кребса следующего состава (мМ): NaCl 118; KCl 4,7; MgCl2 1,2; CaCl2 2,6; NaHCO3 25; глюкоза 11,1; аскорбиновая кислота 0,11. Орган, выдерживаемый при 37oC в потоке карбогена (95% O2/5% CO2) при pH 7,4, связывают с изометрическим счетчиком типа Hugo Sachs 351 при базовом натяжении 2 г и подсоединяют к полиграфу Gould 2400S, дающему возможность регистрировать изменения натяжения. Получение данных осуществляется автоматически посредством системы микроЭвм. После 90 мин отдыха, перемежаемого частыми полосканиями, во время которого базовое натяжение настраивают заново, орган стимулируют 3 мкМ норадреналина, чтобы проверить его жизнеспособность. Затем строят кривую концентрация против сокращательной реакции на суматриптан по кумулятивной модели между 10 нМ и 10 мкМ. Когда получают максимальное сокращение (плато эффекта при последовательных концентрациях суматриптана), препарат обильно ополаскивают, перемежая периоды отдыха, чтобы дать органу возможность вернуться к начальному натяжению. Затем исследуемое соединение добавляют в ванну для органов за 15 мин до того, как строить вторую кривую концентрация-сокращательная реакция на суматриптан. Сокращательные реакции, полученные в присутствии соединения, выражают в процентах от максимального сокращения, наблюдаемого на первой суматриптановой кривой. Кривые анализируют с помощью нелинейной регрессии так, чтобы определить Emax (максимальную ответную реакцию) и EC50 (концентрацию, продуцирующую 50% максимальной реакции). Антагонистический потенциал соединений оценивают путем расчета константы диссоциации Кв согласно уравнению Кв = [концентрация соединения в M]/(CR - 1), где CR представляет собой соотношение значений EC50 суматриптана, в присутствии и в отсутствии данного соединения. Результат выражают как рА2 = -log Кв. Значения рА2 соединений по изобретению выше 6.
Соединения по изобретению также были подвергнуты испытанию на ингибирование связывания [3H]спироперидола с 5-НТ2 серотонинергическими рецепторами коры головного мозга крысы. Для этого испытания мозг крысы удаляют, отсекают кору и гомогенизируют при 0oC в 20 объемах смеси, содержащей, на литр, 50 ммол буфера Трис/HCl при pH 7,4, 120 ммол NaCl и 5 ммол KCl. Гомогенную смесь центрифугируют при 4000 х g в течение 10 мин, затем, в два приема, выделяют осадок, промывают его, суспендируя в той же самой буферной смеси, гомогенизируют снова и центрифугируют. В завершение, последний осадок разбавляют в той же самой буферной смеси в пропорции 500 мг влажной ткани на 10 мл буфера. Затем ткань предварительно инкубируют в течение 10 мин при 37oC в присутствии 10 мкм/л паргилина, затем инкубируют при 37oC в течение 20 мин в присутствии [3H]спироперидола (специфическая активность: 19 Ки на ммол) при концентрации 0,3 нМ и при концентрации исследуемого соединения от 0,0001 до 100 мкМ.
Отбирают аликвоты по 1 мл, которые фильтруют в вакууме, фильтры дважды промывают 5 мл холодного буфера, сушат и измеряют их радиоактивность.
Для оценки активности соединений строят кривую процент ингибирования специфического связывания [3H]спироперидола как функция концентрации вытесняемого соединения. Графически определяют IC50, концентрацию, которая ингибирует 50% специфического связывания. Специфическое связывание определяют как связывание, вытесняемое 100 мкМ 5-НТ.
IC50 соединений по изобретению менее чем 1 мкМ.
Результаты этих испытаний показывают, что соединения по изобретению проявляют антагонистические по отношению к серотонину свойства.
На этом основании они могут быть использованы при лечении и предупреждении различных форм патологий, связанных с серотонином, таких как артериальная, венозная, легочная, воротной вены, почечная или глазная гипертония, сердечная, почечная, глазная, церебральная ишемия или ишемия нижних конечностей, сердечная недостаточность, инфаркт миокарда, стенокардия, спазм коронарных или периферических сосудов, тромбоз (соединения сами по себе или как адъюванты при тромболизе), артериит, перемежающаяся хромота, рестеноз после ангиопластики и различные патологические состояния, связанные с атеросклерозом, нарушениями микроциркуляции или легочной дисфункцией. Их также можно использовать, отдельно или в сочетании с другими веществами, при операциях по пересадке сосудов.
Соединения по изобретению могут быть использованы в сочетании с другими веществами, обладающими кардиососудистой или кардиолегочной активностью, такими как противотромботики, тромболитики, β-блокаторы, антагонисты кальция, антагонисты тромбоксана, ингибиторы тромбоксан синтетазы.
С этой целью эти соединения могут быть представлены в любой форме, походящей для перорального или парентерального введения, такой как таблетки, драже, капсулы, включая твердые желатиновые капсулы, местные глазные составы в сочетании с подходящими эксципиентами. Дозы, присутствующие в этих формах таковы, чтобы обеспечить введение от 0,1 мг до 1 г от одного до нескольких раз в течение дня.
Их также можно представить во всех формах, подходящих для введения через кожу.
Настоящее изобретение относится к производным хинолин-2(1Н)-она общей формулы I, где R1, R2, R3, A и m имеют указанные в формуле изобретения значения и к способам получения соединений формулы I. Эти соединения обладают антагонистической активностью по отношению к серотонину и они полезны для применения в терапии. 5 с. и 2 з.п. ф-лы, 1 табл.
в которой А представляет собой группу 4-(тиено[3,2-с] пиридин-4-ил)пиперазин-1-ил либо группу 4-(4-фторбензоил)пиперидин-1-ил;
R1 и R2 каждый представляет собой, независимо один от другого, атом водорода, атом галогена, аминогруппу, гидроксильную группу, нитрогруппу, цианогруппу, группу (C1-C6)алкил, группу (C1-C6)алкокси, группу трифторметокси, группу -NHCOR4, или группу -NHSO2R4, или группу -N(R4)2, где R4 является группой (C1-C4) алкил;
R3 представляет собой атом водорода, группу (C1-C4)алкил, группу -(СН2)рОН, группу -(СН2)nСООН, группу -(СН2)nСООR4, группу -(СН2)nСОNH2, группу -(СН2)nСОNHR4 или группу -(СН2)рOCOR4, где R4 является группой (C1-C4)алкил;
n = 1, p = 2, m = 2,
также как их соли присоединения с фармацевтически приемлемыми кислотами или основаниями.
6-фтор-1-метил-4-[2-[4-(тиено[3,2-с] пиридин-4-ил)-1-пиперазинил]этил]-хинолин-2(1Н)-она,
7-фтор-2-оксо-4-[2-[4-(тиено[3,2-с] пиридин-4-ил)-1-пиперазинил] этил]-1,2-дигидро-1-хинолинацетамида,
7-фтор-N-метил-2-оксо-4-[2-[4-(тиено[3,2-с]пиридин-4-ил)-1 -пиперазинил] этил]-1,2-дигидро-1-хинолинацетамида,
6-хлор-1-метил-4-[2-[4-(тиено[3,2-с] пиридин-4-ил)-1-пиперазинил]этил]-хинолин-2(1Н)-она,
6-хлоро-4-[2-[4-(4-фторбензоил)-1-пиперидил] этил] -1-метилхинолин-2(1H)-она,
7-фтор-4-[2-[4-(тиено[3,2-с] пиридин-4-ил)-1-пиперазинил] этил]хинолин-2(1Н)-она,
4-[2-[4-(4-фторбензоил)-1-пиперидил]этил]-6-метоксихинолин-2(1Н)-она,
6-гидрокси-1-метил-4-[2-[4-(тиено[3,2-с] пиридин-4-ил)-1-пиперазинил] этилхинолин-2(1Н)-она,
2-[7-фтор-2-оксо-4-[2-[4-(тиено[3,2-с] пиридин-4-ил)-1-пиперазинил]этил] -1,2-дигидро-1-хинолинил]этилацетата и
6-метил-4-[2-[4-(тиено[3,2-с] пиридин-4-ил)-1-пиперазинил]этил]хинолин-2(1Н)-она,
также как их соли присоединения с фармацевтически приемлемыми кислотами или основаниями.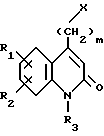
в которой R1, R2, R3 и m такие, как определено в п.1, а Х представляет собой уходящую группу.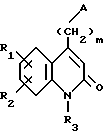
в которой A, R1, R2 и m такие, как определено в п.1, а R3 иной чем атом водорода, отличающийся тем, что соединение формулы Ia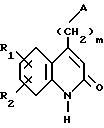
подвергают взаимодействию с электрофильным агентом.
Приоритет по пунктам:
15.09.1995 - по пп.1 - 2 и 4 - 7, касающийся соединения 4-(тиено[3,2-с] пиридин-4-ил)пиперазин-1-ил;
21.09.1995 - по пп.1 - 2 и 4 - 7, касающийся соединения 4-(4-фторбензоил)пиперидин-1-ил;
15.09.1995 - по п.3, касающийся соединений с 1 - 4, 6, 8 - 10;
21.09.1995 - по п.3, касающийся соединений 5, 7;
15.09.1995 - остальные признаки по пп.1 - 2 и 4 - 7.
| Способ получения производных изохинолина или их солей | 1974 |
|
SU528035A3 |
| Способ получения производных пиперидинилалкилхиназолина или их солей с фармацевтически приемлемыми кислотами | 1980 |
|
SU1041034A3 |
| ДИЭТИЛАМИНОЭТИЛАМИДА 1-ПРОПИЛ-2-ОКСО-4-ГИДРОКСИХИНОЛИН-3-КАРБОНОВОЙ КИСЛОТЫ ГИДРОХЛОРИД, ПРОЯВЛЯЮЩИЙ АНЕСТЕЗИРУЮЩУЮ, ПРОТИВОАРИТМИЧЕСКУЮ, АНТИОКСИДАНТНУЮ, АНТИМИКРОБНУЮ И ФУНГИЦИДНУЮ АКТИВНОСТЬ | 1990 |
|
RU1774624C |
| Электрогидравлический привод с управлением по ускорению | 1975 |
|
SU577325A1 |
| Машина для очистки наружной поверхности труб | 1951 |
|
SU98499A1 |
| Способ и устройство для коагуляции | 1932 |
|
SU35821A1 |
| 0 |
|
SU389352A1 |

