Изобретение относится к производным бензолсульфонамида, их получению и применению в терапии.
Известны [патент США N 4217305] производные бензолсульфонамида формулы
где R представляет собой атом водорода, атом галогена, гидроксильную группу, низшую алкильную, алкокси, алкилтио, амино, ациламино, алкилсульфонильную или алкилсульфониламиногруппу; R1, R2, R3 и R4 представляют собой водород или низшую алкильную группу; R5 представляет собой арильную группу, бензодиоксановую кольцевую группу, арилоксигруппу или арилтиогруппу, которые могут иметь заместитель; и n равно числу от 0 до 3. Данные соединения не являются селективными агонистами α1-адренорецепторов. Они обладают α- и β-адренергическим действием и используются в качестве антигипертензивных агентов.
Настоящее изобретение решает задачу создания новых производных бензолсульфонамида, обладающих активностью агониста α1-адренорецепторов, разработки способов их получения и создания новых фармацевтических композиций и лекарственных продуктов, обладающих активностью агониста α1 -адренорецепторов и пригодных для лечения недержания мочи.
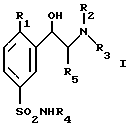
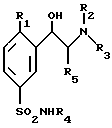
Соединения по изобретению соответствуют общей формуле (I)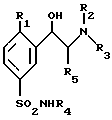
в которой:
R1 представляет собой атом водорода, атом галогена, такого как хлор или фтор, или линейную или разветвленную C1-4алкильную или C1-4алкоксильную группу,
R2, R3 и R4 представляют собой, независимо друг от друга, атомы водорода или линейные, разветвленные или циклические C1-4алкильные группы, и
R5 представляет собой атом водорода или C1-4алкильную группу.
Термин C1-4алкил включает в себя линейные радикалы, радикалы с разветвленной цепью или циклические радикалы, имеющие до 4-х углеродных атомов, в том числе метил, этил, пропил, изопропил, бутил, изобутил и трет-бутил, предпочтительно C1-2алкил, такой как метил и этил.
Термин C1-4алкоксил включает в себя линейные радикалы, имеющие до 4 углеродных атомов, присоединенные посредством атома кислорода, в том числе метокси, этокси, пропокси, изопропокси, бутокси, изобутокси и трет-бутокси, предпочтительно C1-2алкокси, метокси и этокси.
Соединения общей формулы (I) содержат один или более чем один асимметрический атом углерода. Поэтому они могут существовать в форме энантиомеров или диастереоизомеров. Такие энантиомеры и диастереоизомеры, а также их смеси, включающие в себя рацемические смеси, образуют часть изобретения.
Соединения общей формулы (I) могут принимать форму солей, полученных присоединением фармацевтически приемлемых кислот, которые также образуют часть изобретения. Согласно настоящему изобретению предпочтительными солями являются оксалаты и фумараты.
Соединения общей формулы (I), в которых R5 представляет собой C1-2алкильную группу, существуют в форме син- или анти-изомеров. Эти формы, а также их смеси, образуют часть изобретения.
Предпочтительными соединениями являются такие соединения, у которых R5 представляет собой атом водорода, метил или этил, предпочтительно водород или метил, в форме энантиомеров или диастереоизомеров, или смесей этих различных форм, включая рацемические смеси, а также их соли, полученные присоединением фармацевтически приемлемых кислот.
Другими предпочтительными соединениями являются соединения, у которых R1 представляет собой атом водорода, фтор, хлор или C1-4алкоксильную группу, предпочтительно метокси или этокси, в форме энантиомеров или диастереоизомеров, или смесей этих разных форм, включая рацемические смеси, а также их соли, полученные присоединением фармацевтически приемлемых кислот.
Другими соединениями по выбору являются соединения, у которых R2 и R3 представляют собой, независимо друг от друга, атом водорода, метил, этил или изопропил, предпочтительно водород, в форме энантиомеров или диастереоизомеров или смесей этих различных форм, включая рацемические смеси, а также их соли, полученные присоединением фармацевтически приемлемых кислот.
Среди них могут быть упомянуты соединения, у которых:
R1 представляет собой атом водорода, фтор, хлор или C1-4 алкоксильную группу, предпочтительно метокси или этокси,
R2 и R3 представляют собой, независимо друг от атом водорода, метил, этил или изопропил, предпочтительно водород,
R4 представляет собой водород или линейную, разветвленную или циклическую C1-4алкильную группу, и
R представляет собой атом водорода, метил или этил, предпочтительно атом водорода или метил,
в форме энантиомеров или диастереоизомеров или смесей этих различных форм, включая рацемические смеси, а также их соли, полученные присоединением фармацевтически приемлемых кислот,
и особое внимание может быть уделено
α-(аминометил)-2-метокси-5-сульфамоилбензолметанолу и его солям,
(+)-α-(аминометил)-2-метокси-5-сульфамоилбензолметанолу и его солям,
(-)-α-(аминометил)-2-метокси-5-сульфамоилбенэолметанолу и его солям,
α-(аминометил)-2-хлоро-5-сульфамоилбенэолметанолу и его солям,
α-(аминометил)-2-фторо-5-сульфамоилбенэолметанолу и его солям.
Соединения общей формулы (I), в которых R1 представляет собой алкоксильную группу, и R2 и R3 представляют собой атомы водорода, могут быть получены согласно способу, представленному в Приложении 1 (в конце описания), который состоит в том, что производное бензальдегида формулы (V), в котором R1 такой, как определено выше, обрабатывают этилортоформиатом в присутствии хлорида аммония, а затем хлорсульфоновой кислотой, производное 5-хлорсульфонилбензальдегида формулы (IV) обрабатывают амином формулы R4NH2, в котором R4 такой, как определено в общей формуле (I), а затем производное 5-сульфамоилбензальдегида формулы (III) обрабатывают триметилсилилцианидом (TMSCN) в присутствии иодида цинка, и в заключение восстанавливают полученное таким образом соединение формулы (II) боргидридом лития в присутствии триметилсилилхлорида (TNSCI).
Соединения общей формулы (I) могут также быть получены согласно способу, представленному в Приложении 2 (в конце описания), из производного сульфамоилацетофенона формулы (XII).
В случае, когда R1 такой, как определено в общей формуле (I), за исключением алкила, этот способ заключается в том, что производное 5-сульфамоилфенилкетона формулы (XII) обрабатывают бромом, затем соединение формулы (XI) подвергают взаимодействию либо с хлоридом лития с получением соединения формулы (X), которое затем восстанавливают бораном с получением соединения формулы (IX) и затем обрабатывают азидом натрия с получением соединения формулы (VIII), либо с азидом натрия, а затем боргидридом натрия с получением соединения формулы (VIII), либо с боргидридом натрия в присутствии карбоната калия с получением соединения формулы (VII), и наконец, обрабатывают соединение формулы (VIII) водородом в присутствии катализатора, такого как палладий на угле в случае, когда R1 не является атомом хлора, или трифенилфосфином, и затем водным аммиаком в случае, когда R1 является атомом хлора, с получением соединения общей формулы (I), в которой R2 и R3 представляют собой атомы водорода, или обрабатывают соединение формулы (VII) либо амином формулы R2(R3)NH, в которой R2 представляет собой атом водорода или C1-4алкильную группу, и R3 представляет собой C1-4алкильную группу, с получением соединения общей формулы (I), в которой R2 и R3 такие, как определено выше, либо амином формулы R2(Bn)NH, в которой R2 представляет собой C1-4алкильную группу, Bn представляет собой бензильную группу, с получением соединения формулы (VI), которое затем восстанавливают водородом в присутствии катализатора, такого как палладий на угле, с получением соединения общей формулы (I), в которой R2 представляет собой алкильную группу.
В случае, когда R1 представляет собой алкильную группу, этот способ заключается в том, что соединение формулы (XII) обрабатывают дихлориодатом бензилтриметиламмония с получением соединения формулы (X), в которой R1 представляет собой алкильную группу, которое затем обрабатывают, как описано выше, с получением соединения общей формулы (I), в которой R2 и R3 представляют собой атомы водорода, и R1 представляет собой алкильную группу, через соответствующие промежуточные соединения формул (IX) и (VIII).
Соединения формулы (XII)
в которой R1, R4 и R5 такие, как определено в общей формуле (I), могут быть получены путем взаимодействия производного фенилкетона формулы (XIV)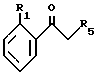
в которой R1 такой, как определено в общей формуле (I), с хлорсульфоновой кислотой с получением производного хлорсульфонилфенилкетона формулы (XIII)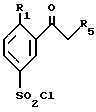
которое затем обрабатывают амином формулы R4NH2, в которой R4 такой, как определено в общей формуле (I).
Соединения формулы (XII), в которой R4 представляет собой атом водорода, также может быть получено путем взаимодействия соединения формулы (XVII)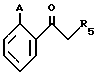
в которой A идентично R1, описанному в общей формуле (I), или альтернативно представляет собой гидроксильную группу с азотной кислотой с получением производного нитрофенилкетона формулы (XVI)
которое восстанавливают до производного аминофенилкетона с помощью водорода в присутствии палладия на угле или хлорида олова, если целесообразно, после обработки алкилиодидом в том случае, когда A представляет собой гидроксильную группу, с получением соответствующего производного 2-алкоксифенилкетона, и в заключение, обрабатывают соединение формулы (XV)
нитритом натрия, хлоридом меди и диоксидом серы.
Энантиомеры соединений общей формулы (I) получают из энантиомеров соединений формулы (VIII)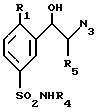
которые получают либо путем энантиоселективного синтеза; при котором соединение формулы (XI)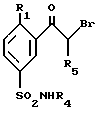
обрабатывают азидом натрия и полученное таким образом соединение формулы (XVIII)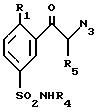
подвергают взаимодействию с (+)- или (-)-B- хлордиизопинокамфеилбораном (DIP-CI) с получением (+) и (-) энантиомеров, соответственно, соединения формулы (VIII),
либо путем ферментативного расщепления соединения (IX)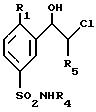
при котором рацемическое соединение формулы (IX) обрабатывают уксусной кислотой, проводят селективный ферментативный гидролиз липазой SP 523 (липаза, полученная техникой рекомбинантной ДНК из Aspergillus orysae) полученного таким образом соединения формулы (XIX)
приводящий к получению (+) энантиомера соединения формулы (IX) и (-) энантиомера негидролизованного соединения формулы (XIX), хроматографически разделяют (+) энантиомер соединения формулы (IX) и (-) энантиомер соединения формулы (XIX), и гидролизуют (-) энантиомер соединения формулы (XIX) с получением (-) энантиомера соединения формулы (IX), и в заключение осуществляют взаимодействие (+) и (-) энантиомеров соединения формулы (IX) с азидом натрия,
либо путем химического разделения, при котором соединение формулы (VIII) подвергают взаимодействию с N-карбобензилокси-L-аланином (N-CBZ-аланином), отделяют хроматографически и затем гидролизуют энантиомеры соединения формулы (XX)
Соли соединений общей формулы (I) получают путем взаимодействия соединений общей формулы (I) в форме оснований с фармацевтически приемлемыми кислотами.
Исходные материалы известны из литературы или непосредственно имеются в продаже.
Нижеследующие примеры иллюстрируют способ и технологии, приемлемые для осуществления данного изобретения, без ограничения объема изобретения. Структуры полученных соединений подтверждаются элементными микроанализами и ЯМР и ИК спектрами.
Пример 1: α-(аминометил)-2-метокси-5-сульфамоилбензолметанола метансульфонат
1.1.2-метокси-5-сульфамоилбензальдегид
Это соединение получают согласно известному (патент Франции FR 73/35277) способу путем пропускания потока аммиака в раствор 2-метокси-5-хлорсульфонилбензальдегида в хлороформе.
Согласно этому же способу путем обработки 2-метокси-5-хлорсульфонилбензальдегида 10-ю эквивалентами амина формулы R4NH2 в течение 3 часов при комнатной температуре, были получены следующие соединения:
- 2-метокси-5-метилсульфамоилбензальдегид.
Точка плавления: 118oC.
- 2-метокси-5-циклопропилсульфамоилбензальдегид.
Точка плавления: 162oC.
- 2-метокси-5-иэопропилсульфамоилбензальдегид.
Точка плавления: 125oC.
- 2-метокси-5-трет.-бутилсульфамоилбензальдегид.
Точка плавления: 99oC.
1.2. α-аминометил-2-метокси-5-сульфамоилбензолметанол
10,4 г (48,3 ммоль) 2-метокси-5-сульфамоилбензальдегида и 18,4 мл (96,6 ммоль) триметилсилилцианида вводят в 100-мл круглодонную колбу, затем добавляют 0,5 г (1,56 ммоль) иодида цинка и смесь перемешивают при комнатной температуре в течение 10 минут. Затем добавляют 20 мл безводного тетрагидрофурана и переносят раствор в капельную воронку.
Отдельно 100 мл безводного тетрагидрофурана и 2,6 г (119 ммоль) боргидрида лития вводят в 500-мл круглодонную колбу. Раствор перемешивают, затем по каплям добавляют 30 мл (236 ммоль) триметилсилилхлорида и смесь перемешивают при комнатной температуре в течение 10 минут.
Затем по каплям добавляют раствор приготовленного ранее триметилсилилцианогидрина. Смесь перемешивают в течение 16 часов, затем добавляют по каплям 20 мл этанола и раствор концентрируют. Затем по каплям добавляют 120 мл 20%-ного раствора гидроксида калия и раствор концентрируют. Остаток очищают посредством колоночной хроматографии с использованием смеси дихлорметана, метанола и водного аммиака в соотношении 90:9:1, затем его перекристаллизовывают в этаноле и сушат в эксикаторе под вакуумом над пентоксидом фосфора. Получают 0,30 г продукта.
Точка плавления: 217-220oC.
1.3. α-аминометил-2-метокси-5-сульфамоилбензолметанола метансульфонат
Один эквивалент метансульфоновой кислоты в 2М растворе в метаноле добавляют к продукту, полученному на стадии 1.2. После перекристаллизации в метаноле и диэтиловом эфире и сушки в эксикаторе под вакуумом над пентоксидом фосфора получают 0,370 г продукта.
Точка плавления: 210-212oC.
Пример 2: (-)-α-(аминометил)-2-метокси-5- сульфамоилбензолметанола метансульфонат
2.1. 2-Метокси-5-хлорсульфонилацетофенон
951 г (8,16 моль) хлорсульфоновой.кислоты вводят в 1-литровую круглодонную колбу. Смесь охлаждают до примерно -5oC и затем по каплям, добавляют 81,5 г (0,544 моль) 2-метоксиацетофенона так, чтобы температура не превышала 0oC. Затем смесь перемешивают при комнатной температуре в течение 16 часов, а затем медленно при перемешивании выливают в измельченный лед. Продукт отфильтровывают, промывают охлажденной на льду водой и затем сушат в эксикаторе под вакуумом над пентоксидом фосфора. Получают 87,5 г продукта.
Точка плавления: 85-86oC.
2.2. 2-метокси-5-сульфамоилацетофенон
86 г (0,344 моль) 2-метокси-5-хлорсульфонилацетофенона и 690 мл хлороформа вводят в 1-литровую круглодонную колбу. Смесь перемешивают до тех пор, пока не произойдет растворение, а затем охлаждают до 0oC на ледяной бане и пропускают через раствор поток аммиака в течение 1 часа. Затем смесь оставляют для нагревания до комнатной температуры, растворитель выпаривают и добавляют 250 мл 1М соляной кислоты. Полученную суспензию перемешивают в течение 3 часов, продукт отфильтровывают, промывают охлажденной на льду водой и сушат в эксикаторе под вакуумом над пентоксидом фосфора. Получают 72,8 г продукта.
Точка плавления: 161-162oC.
Согласно такому же процессу получают 2-метил-5-сульфамоилацетофенон.
Точка плавления: 215oC.
2.3. α-бром-2-метокси-5-сульфамоилацетофенон
60,36 г (0,262 моль) 2-метокси-5-сульфамоилацетофенона и 530 мл уксусной кислоты вводят в 1-литровую трехгорлую колбу. Смесь перемешивают и нагревают до 50oC. Затем по каплям добавляют 41,95 г (0,262 моль) брома и смесь перемешивают в течение 16 часов, пока она нагревается до комнатной температуры, и фильтруют. Осадок промывают минимальным количеством этанола и сушат в эксикаторе под вакуумом над пентоксидом фосфора. Получают 49 г продукта.
Точка плавления: 154-156oC.
2.4. α- азидо-2-метокси-5-сульфамоилацетофенон
7 г (0,023 моль) α-бром-2-метокси-5-сульфамоилацетофенона, 2,6 мл (0,045 моль) уксусной кислоты и 23 мл этанола вводят в трехгорлую колбу емкостью 100 мл. Суспензию нагревают до 50oC при перемешивании, а затем по каплям добавляют раствор 2,94 г (0,045 моль) азида натрия в 8 мл воды. Суспензию перемешивают при 50oC в течение 45 минут, а затем оставляют остывать до комнатной температуры. Осадок отфильтровывают, промывают минимальным количеством холодного этанола и затем сушат в эксикаторе под вакуумом над пентоксидом фосфора. Получают 5,46 г продукта.
Точка плавления: 155-160oC (с разложением).
2.5. (-)-α-азидо-2-метокси-5-сульфамоилбензолметанол
16,2 г (0,060 моль) α-азидо-2-метокси-5-сульфамоилацетофенона и 240 мл безводного тетрагидрофурана вводят в трехгорлую колбу емкостью 500 мл. Раствор охлаждают до -25oC и добавляют раствор 38,5 г (0,12 моль) (-)-DIP-CI в 30 мл безводного тетрагидрофурана со скоростью протока 1,5 мл/мин. Через 90 минут раствору позволяют нагреться до комнатной температуры и добавляют 10 мл метанола. Реакционную смесь затем концентрируют, а остаток очищают посредством колоночной хроматографии, используя смесь 40:60 петролейного эфира и этилацетата. После перекристаллизации из изопропанола и сушки в эксикаторе над пентоксидом фосфора получают 11,55 г продукта (эф. = 99,9%).
Точка плавления: 122-125oC.
[α]
2.6. (-)-α-аминометил-2-метокси-5-сульфамоилбензолметанол
11 г (0,040 моль) (-)-α-азидометил-2-метокси-5- сульфамоилбензолметанола, 500 мл этанола и 2,2 г 10%-ного палладия на угле вводят в 1-литровый реактор. Реактор закрывают и продувают азотом, смесь перемешивают под водородом с давлением 400 кПа при комнатной температуре в течение 2 часов. Затем реакционную смесь фильтруют через Whatman бумагу, восстановленный катализатор суспендируют в 200 мл метанола и смесь нагревают до кипения в течение 30 минут. Затем ее фильтруют через Whatman бумагу, фильтраты объединяют и концентрируют, а остаток сушат в эксикаторе под вакуумом над пентоксидом фосфора. Получают 9,4 г (-)-α-аминометил-2-метокси-5-сульфамоилбензолметанола.
После перекристаллизации продукта в 388 мл метанола получают 5,46 г продукта. Кроме того, путем концентрации маточных растворов и перекристаллизации остатка в 400 мл этанола получают 1,93 г продукта, поэтому общий выход продукта составляет 7,39 г.
Точка плавления: 217-220oC.
[α]
2.7. (-)-α-аминометил-2-метокси-5-сульфамоилбензолметанола метансульфонат
1 эквивалент метансульфоновой кислоты в виде 2М раствора в метаноле добавляют к продукту, полученному на предыдущей стадии. После перекристаллизации в метаноле и диэтиловом эфире и сушки продукта в эксикаторе под вакуумом над пентоксидом фосфора получают (-)-α-аминометил-2-метокси-5-сульфамоилбензолметанола метансульфонат.
Точка плавления: 232-233oC.
[α]
Пример 3: (+) и (-)-α-аминометил-2-метокси-5- сульфамоилбензолметанола метансульфонат
3.1. α-хлор-2-метокси-5-сульфамоилацетофенон
4,36 г (14,1 ммоль) α-бром-2-метокси-5-сульфамоилацетофенона, 200 мл безводного ацетона и 50 г хлорида лития вводят в круглодонную колбу емкостью 500 мл. Смесь нагревают до начала флегмообразования в течение 16 часов, затем раствор концентрируют, добавляют 200 мл воды и смесь экстрагируют три раза 80 мл этилацетата. Органические фазы объединяют, сушат над сульфатом магния и концентрируют. Получают 3,56 г продукта.
Точка плавления: 162oC.
3.2. α-хлорметил-2-метокси-5-сульфамоилбензолметанол
10 мл безводного тетрагидрофурана и 4 мл 1М раствора борана в тетрагидрофуране вводят в круглодонную колбу емкостью 100 мл. По каплям добавляют раствор. 1,0 г (3,8 ммоль) α-хлор-2-метокси- 5-сульфамоилацетофенона в 10 мл тетрагидрофурана. Смесь перемешивают в течение 10 часов при комнатной температуре и затем добавляют 10 мл метанола. Раствор концентрируют, добавляют 40 мл воды и смесь экстрагируют 3 раза 60 мл этилацетата. Органические фазы объединяют, сушат над сульфатом магния и концентрируют. Получают 1,0 г продукта.
Точка плавления: 112oC.
3.3. (±)-α-хлорметил-2-метокси-5-сульфамоилбензилацетат
2,64 г (9,9 ммоль) α-хлорметил-2-метокси-5-сульфамоилбензолметанола и 100 мл дихлорметана вводят в круглодонную колбу емкостью 250 мл. Затем при перемешивании добавляют 10 мл диметилформамида, затем 852 мкл уксусной кислоты, 2,56 г дициклогексилкарбодиимида и 121 мг диметиламинопиридина. Смесь оставляют для прохождения реакции на 1 час при комнатной температуре, в затем ее фильтруют и промывают 50 мл 5%-ного раствора гидрокарбоната натрия и 50 мл воды. Промывные воды экстрагируют 2 раза 20 мл уксусной кислоты, органические фазы объединяют, сушат и концентрируют. При хроматографировании остатка на колонке с силикагелем со смесью 25:75 этилацетата и циклогексана получают 1,9 г продукта.
Точка плавления: 131oC.
3.4. (+) и (-)-α-хлорметил-2-метокси-5-сульфамоилбензолметанол
2,86 г (9,3 ммоль) (±)-α-хлорметил-2-метокси-5- сульфамоилбензилацетата и 110 мл трет.-бутилметилового эфира вводят в трехгорлую колбу емкостью 500 мл. Смесь перемешивают в течение 15 минут, затем добавляют 170 мл фосфатного буфера и смесь интенсивно перемешивают до получения эмульсии. Затем добавляют 0,57 г (20%) липазы SP 523 и за ходом реакции следят при комнатной температуре, используя контроль pH (добавление 1М гидроксида натрия) и ЖХВР (HPLC) на хиральной колонке, степень конверсии сложного эфира и энантиомерный избыток сложного эфира и спирта определяют. После 45 часов прохождения реакции, когда энантиомерный избыток сложного эфира и спирта превышает 95%, реакционную среду разбавляют 800 мл этилацетата, органическую фазу отделяют, а водную фазу повторно экстрагируют 3 раза 500 мл этилацетата. Органические фазы объединяют, сушат и концентрируют, а остаток очищают, дважды последовательно подвергая его флэш- хроматографии на колонке с силикагелем, используя смесь 30:70 этилацетата и циклогексана. Получают 1,32 г (-)-α-хлорметил-2-метокси-5-сульфамоилбензилацетата (эф. = 99%) и 1,05г (+)-α-хлорметил-2-метокси-5-сульфамоилбензолметанола.
(+) энантиомер очищают путем растворения в 10 мл этилацетата и перекристаллизации при добавлении гексана (эф. = 98%).
Точка плавления: 117-118oC.
[α]
60 мкл ацетилхлорида добавляют к 100 мл метанола и смесь перемешивают в течение 15 минут. Затем добавляют 1,32 г (-)-α-хлорметил-2-метокси-5-сульфамоилбензилацетата и смесь нагревают до начала флегмообразования в течение 1 часа (степень конверсии составляет 97% по данным ЖХВР). Смесь затем упаривают, остаток переносят в 100 мл этилацетата и среду нейтрализуют 5 мл 2%-ного этилгидрокарбоната. Карбонатную фазу экстрагируют дважды 5 мл этилацетата, органические фазы объединяют, сушат и концентрируют до 30 мл, затем добавляют циклогексан. После ночи при комнатной температуре закристаллизовавшийся продукт отфильтровывают. Получают 1 г (-)-α-хлорметил-2-метокси-5-сульфамоилбензолметанола.
Точка плавления: 114-115oC.
[α]
3.5. (+)-α-азидометил-2-метокси-5-сульфамоилбензолметанол
1,58 г (5,9 ммоль) (+)-α-хлорметил-2- метокси-5- сульфамоилбензолметанола, 20 мл диметилформамида и 1,54 г азида натрия вводят в круглодонную колбу емкостью 250 мл. Реакционную смесь нагревают до 110oC в течение 16 часов, затем добавляют 200 мл воды и смесь экстрагируют 3 раза 80 мл этилацетата. Затем органические фазы объединяют, сушат над сульфатом магния и концентрируют. Получают 1,2 г продукта.
Точка плавления: 122oC.
[α]
Согласно такому же процессу, начиная с (-)-α-хлорметил-2-метокси-5-сульфамоилбензолметанола, получают (-)-α-азидометил-2-метокси-5- сульфамоилбензолметанол.
Точка плавления: 122oC.
[α]
3.6. (+)-α-аминометил-2-метокси-5-сульфамоилбензолметанол
Начиная с (+)-α-азидометил-2-метокси-5-сульфамоил6ензолметанола, обработанного при условиях, описанных на стадии 6 Примера 2, получают (+)-α-аминометил-2-метокси-5-сульфамоилбензолметанол.
Точка плавления: 217-220oC.
[α]
Согласно тому же процессу, начиная с (-)-α-азидометил-2-метокси-5-сульфамоилбензолметанола, получают (-)-α-аминометил-2-метокси-5-сульфамоилбензолметанол.
Точка плавления: 217-220oC.
[α]
3.7. (+)-α-аминометил-2-метокси-5-сульфамоилбензолметанола метансульфонат
Начиная с (+)-α-аминометил-2-метокси-5-сульфамоилбензолметанола, обработанного 1 эквивалентом метансульфоновой кислоты в виде 2М раствора в метаноле, после перекристаллизации в метаноле и диэтиловом эфире и сушки в эксикаторе под вакуумом над пентоксидом фосфора, получают (+)-α-аминометил-2-метокси-5-сульфамоилбензолметанола метансульфонат.
Точка плавления: 234oC.
[α]
Согласно тому же процессу, начиная с (-)-α-аминометил-2-метокси- 5-сульфамоилбензолметанола, получают (-)-α-аминометил-2- метокси-5-сульфамоилбензолметанола метансульфонат.
Точка плавления: 235oC.
[α]
Пример 4: (+) и (-)-α-аминометил-2-метокси-5- сульфамоилбензолметанола метансульфонат
4.1. (+)-α-азидометил-2-метокси-5-сульфамоилбензолметанол
5 г (18,5 ммоль) α-азидо-2-метокси-5-сульфамоилацетофенона и 150 мл метанола вводят в круглодонную колбу емкостью 500 мл. Раствор охлаждают до 0oC и затем добавляют 0,963 г (16,6 ммоль) борогидрида натрия. Раствор перемешивают в течение 10 минут и затем оставляют для нагревания до комнатной температуры, после чего добавляют 15 мл 5%-ного раствора соляной кислоты. Затем реакционную смесь концентрируют, а остаток затем очищают с помощью колоночной хроматографии со смесью 40:60 петролейного эфира и этилацетата и сушат в эксикаторе под вакуумом над пентоксидом фосфора.
Получают 3,85 г продукта.
Точка плавления: 123oC.
4.2. N-кapбoбeнзилoкcи-L-aлaнинoвый эфир (+) и (-)-α-азидометил-2-метокси-5-сульфамоилбензолметанола
4,66 г (20,9 ммоль) N-карбобензилокси-L-аланина, 25 мл дихлорметана и 3,58 г (17,4 ммоль) 1,3-дициклогексилкарбодиимида вводят в круглодонную колбу емкостью 250 мл. Смесь перемешивают в течение 20 минут при комнатной температуре и затем добавляют 3,8 г (13,9 ммоль) α-азидометил-2-метокси-5-сульфамоилбензолметанола и 0,17 г (0,14 ммоль) диметиламинопиридина, Реакционную среду перемешивают в течение 2 часов и затем концентрируют под вакуумом, остаток очищают путем неоднократного пропускания через хроматографическую колонку с силикагелем с использованием смеси 99:1 дихлорметана и ацетона. Получают 1,58 г N-карбобензилокси-L-аланинового эфира (+)-α-азидометил-2- метокси-5-сульфамоилбензолметанола и 2,92 г N-карбобензилокси-L-аланинового эфира (-)-α-азидометил-2-метокси-5-сульфамоилбензолметанола.
Точка плавления: 170oC (с разложением)
4.3. (+)-α-азидометил-2-метокси-5-сульфамоилбензолметанол
0,91 г (1,9 ммоль) N-карбобензилокси-L-аланинового эфира (+)-α-азидометил-2-метокси-5-сульфамоилбензолметанола, 20 мл этанола и 3 мл 1 М раствора гидроксида калия в смеси 1:1 этанола и воды вводят в круглодонную колбу. Реакционную смесь перемешивают в течение 25 минут при комнатной температуре и затем концентрируют под вакуумом, остаток очищают посредством колоночной хроматографии с использованием смеси 95:5 дихлорметана и метанола.
Получают 0,41 г продукта.
Точка плавления: 122oC.
Согласно тому же процессу, начиная с N-карбобензилокси-L-аланинового эфира (-)--α-азидометил-2-метокси-5-сульфамоил6ензолметанола, получают (-)α-азидометил-2-метокси-5-сульфамоилбензолметанол.
Точка плавления: 122oC.
4.4. (+)-α-аминометил-2-метокси-5-сульфамоилбензолметанола метансульфонат
Начиная с (+)-α-азидометил-2-метокси-5- сульфамоилбензолметанола, гидрогенизированного при условиях, описанных на стадии 6 Примера 2, и затем обработанного 1 эквивалентом метансульфоновой кислоты, получают (+)-α-аминометил-2-метокси-5-сульфамоилбензолметанол.
Точка плавления: 235oC.
[α]
Согласно тому же процессу, начиная с (-)-α-азидометил-2- метокси-5- сульфамоилбензолметанола, получают (-)-α-аминометил-2- метокси-6-сульфамоилбензолметанола метансульфонат.
Точка плавления: 233oC.
[α]
Пример 5: α-диэтиламинометил-2-метокси-5-сульфамоилбензолметанола метансульфонат
5.1. Оксид 2-метокси-5-сульфамоилстирола
2 г (6,5 ммоль) α-бром-2-метокси-5-сульфамоилацетофенона, 20 мл безводного этанола и 1,0 г (7,0 ммоль) карбоната калия вводят в круглодонную колбу емкостью 100 мл. Затем добавляют 0,41 г (10,8 ммоль) борогидрида натрия, смесь перемешивают при комнатной температуре в течение 20 минут, затем добавляют 0,1 М гидроксида натрия и смесь перемешивают в течение 30 минут. Раствор концентрируют, добавляют 30 мл воды и смесь экстрагируют 3 раза 30 мл этилацетата. Органические фазы объединяют, сушат над сульфатом магния и концентрируют. Получают 1,41 г продукта.
Точка плавления: 118oC.
5.2. α-диэтиламинометил-2-метокси-5-сульфамоилбензолметанола метансульфонат
1,41 г (6,1 ммоль) оксида 2-метокси-5-сульфамоилстирола, 10 мл безводного этанола и 17,8 г (244 ммоль) диэтиламина вводят в круглодонную колбу емкостью 100 мл. смесь нагревают до начала флегмообразования при перемешивании в течение 16 часов и затем раствор концентрируют. Остаток очищают посредством колоночной хроматографии, используя смесь 90:9:1 дихлорметана, метанола и водного аммиака, затем сушат в эксикаторе под вакуумом над пентоксидом фосфора. Получают 1,42 г продукта в виде масла, которое обрабатывают 1 эквивалентом метансульфоновой кислоты в виде 2М раствора в метаноле. После перекристаллизации в метаноле и диэтиловом эфире и сушки в эксикаторе под вакуумом над пентоксидом фосфора получают 0,875 г α-диэтиламинометил-2-метокси-5-сульфамоилбензолметанола метансульфоната.
Точка плавления: 90-92oC.
Пример 6: α-метиламинометил-2-метокси-5-сульфамоилбензолметанола метансульфонат
6.1. α-бензилметиламинометил-2-метокси-5-сульфамоилбензолметанол
Оксид 2-метокси-5-сульфамоилстирола, полученный на стадии 1 Примера 5, при обработке бензилметиламином при условиях, описанных на стадии 2 Примера 5, дает α-бензилметиламинометил-2-метокси-5- сульфамоилбензолметанол в форме масла.
6.2. α-метиламинометил-2-метокси-5-сульфамоилбензолметанола метансульфонат
При гидрогенизации 1,90 г (5,4 ммоль) α-бензилметиламинометил- 2-метокси-5-сульфамоилбензолметанола при условиях, описанных на стадии 6 Примера 2, получают α-метиламинометил-2-метокси-5- сульфамоилбензолметанол. После перекристаллизации в этилацетате и метаноле полученный продукт обрабатывают 1 эквивалентом метансульфоновой кислоты в виде 2М раствора в метаноле и соль перекристаллизовывают из метанола, дихлорметана и диэтилового эфира. Получают 0,396 г α- метиламинометил-2-метокси-5- сульфамоилбензолметанола метансульфоната.
Точка плавления: 194-196oC.
Пример 7: α-аминометил-2-фтор-5-сульфамоилбензолметанола метансульфонат
7.1. 2-фтор-5-нитроацетофенон
25 мл (180 ммоль) 2-фторацетофенона вводят по каплям в трехгорлую колбу емкостью 100 мл, содержащую 60 мл концентрированной серной кислоты, охлажденной до -5oC. Смесь 14 мл азотной кислоты (d=1,42) и 20 мл концентрированной серной кислоты затем по каплям добавляют так, чтобы температура не превысила 0oC. Смесь перемешивают при -5oC в течение 30 минут и затем выливают на крошеный лед. Полученную смесь затем экстрагируют три раза 60 мл этилацетата, органические фазы затем объединяют, сушат над сульфатом магния и концентрируют. Остаток очищают колоночной хроматографией с использованием смеси 70: 30 гексана и этилацетата, затем сушат в эксикаторе под вакуумом над пентоксидом фосфора. Получают 26 г продукта.
Точка плавления: 72oC.
Согласно тому же процессу получают следующие соединения:
- 2-хлор-5-нитроацетофенон.
Точка плавления: 65oC.
- 2-гидрокси-5-нитроацетофенон.
Точка плавления: 98oC,
которые в результате реакции с межфазным катализатором с изопропилиодидом превращают в 2-иэопропокси-5-нитроацетофенон,
Точка плавления: 78oC.
7.2. 5-амино-2-фторацетофенон
25,4 г (152 ммоль) 2-фтор-5-нитроацетофенона, 343 г (1,52 моль) дигидрата хлорида олова и 250 мл этилацетата вводят в литровую трехгорлую колбу. Реакционную смесь нагревают до 70oC в течение 30 минут и затем выливают в 1 литр крошеного льда, добавляют 30%-ный раствор гидроксида натрия и смесь экстрагируют 3 раза 350 мл этилацетата. Органические фазы объединяют, сушат над сульфатом магния и концентрируют. Получают 11,26 г продукта в форме масла.
Согласно тому же процессу получают следующие соединения:
- 5-амино-2-хлорацетофенон, в виде масла.
- 5-амино-2-изопропоксиацетофенон, в виде масла.
7.3. 2-фтор-5-сульфамоилацетофенон
15,3 г (100 ммоль) 5-амино-2-фторацетофенона и 50 мл уксусной кислоты вводят в трехгорлую колбу емкостью 250 мл, затем добавляют 50 мл концентрированной соляной кислоты. Реакционную смесь охлаждают до 0oC, затем по каплям добавляют раствор 10,3 г (150 ммоль) нитрита натрия в 25 мл воды и смесь оставляют при 0oC на 30 минут. Затем добавляют охлажденную до -15oC суспензию 5 г (29 ммоль) дигидрата хлорида меди и 30 г (470 ммоль) диоксида серы в 75 мл уксусной кислоты. Смесь выдерживают при 0oC в течение 48 часов, затем добавляют 20 мл воды, полученную смесь экстрагируют 3 раза 120 мл дихлорметана, органические фазы затем объединяют, сушат над сульфатом магния и концентрируют.
Остаток растворяют в 100 мл тетрагидрофурана, затем по каплям при 0oC добавляют 28%-ный водный раствор аммиака. Реакционную смесь перемешивают в течение 16 часов при комнатной температуре и затем концентрируют. Остаток очищают колоночной хроматографией с использованием смеси 60:40 гексана и этилацетата и сушат в эксикаторе под вакуумом над пентоксидом фосфора. Получают 11,23 г продукта.
Точка плавления: 112oC.
Согласно тому же процессу получают следующие соединения:
- 2-хлор-5-сульфамоилацетофенон.
Точка плавления: 106oC.
- 2-изопропокси-5-сульфамоилацетофенон.
Точка плавления: 85oC.
- 3-сульфамоилацетофенон.
Точка плавления: 144oC.
7.4. α-бром-2-фтор-5-сульфамоилацетофенон
Начиная с 2-фтор-5-сульфамоилацетофенона, обработанного при условиях, описанных на стадии 3 Примера 2, получают α- бром-2-фтор-5-сульфамоилацетофенон.
Точка плавления: 122oC.
Согласно тому же процессу получают следующие соединения:
-α-бром-2-хлор-5-сульфамоилацетофенон.
Точка плавления: 126oC.
-α-бром-2-изопропокси-5-сульфамоилацетофенон.
Точка плавления: 105oC.
-α-бром-3-сульфамоилацетофенон.
Точка плавления: 130oC.
7.5. α-хлор-2-фтор-5-сульфамоилацетофенон
Начиная с α-бром-2-фтор-5-сульфамоилацетофенона, обработанного при условиях, описанных на стадии 1 Примера 3, получают α-хлор-2-фтор-5-сульфамоилацетофенон.
Точка плавления: 114oC.
Согласно тому же процессу получают следующие соединения:
-α-хлор-2-хлор-5-сульфамоилацетофенон.
Точка плавления: 124oC.
-α-хлор-2-изопропокси-5-сульфамоилацетофенон.
Точка плавления: 98oC.
-α-хлор-3-сульфамоилацетофенон.
Точка плавления: 128oC.
7.6. α-хлорметил-2-фтор-5-сульфамоилбензолметанол
Начиная с α-хлор-2-фтор-5-сульфамоилацетофенона, обработанного при условиях, описанных на стадии 2 Примера 3, получают α-хлорметил-2-фтор-5-сульфамоилбензолметанол.
Точка плавления: 112oC.
Согласно тому же процессу получают следующие соединения:
-α-хлорметил-2-хлор-5-сульфамоилбензолметанол.
Точка плавления: 115oC.
-α-хлорметил-2-изопропокси-5-сульфамоилбензолметанол.
Точка плавления: 93oC.
-α-хлорметил-3-сульфамоилбензолметанол.
Точка плавления: 122oC.
7.7. α-азидометил-2-фтор-5-сульфамоилбензолметанол
Начиная с α-хлорметил-2-фтор-5-сульфамоилбензолметанола, обработанного при условиях, описанных на стадии 4 Примера 3, получают α-азидометил-2-фтор-5-сульфамоилбензолметанол.
Точка плавления: 86oC.
Согласно тому же процессу получают следующие соединения:
-α-азидометил-2-хлор-5-сульфамоилбензолметанол.
Точка плавления: 122oC.
-α-азидометил-2-изопропокси-5-сульфамоилбензолметанол.
Точка плавления: 95oC.
-α-азидометил-3-сульфамоилбензолметанол.
Точка плавления: 118oC.
7.8. α-аминометил-2-фтор-5-сульфамоилбензолметанола метансульфонат
Начиная с α-азидометил-2-фтор-5-сульфамоилбензолметанола, обработанного при условиях, описанных на стадии 6 Примера 2, получают α- аминометил-2-фтор-5-сульфамоилбензолметанола метансульфонат.
Точка плавления: 164oC.
Пример 8: α-аминометил-2-метил-5-сульфамоилбензолметанола метансульфонат
8.1. α- хлор-2-метил-5-сульфамоилацетофенон
26,4 г (76,0 ммоль) дихлориодата бензилтриметиламмония (полученного известным (Synthesis 7, (1988), 545) методом) 9,25 г (43,4 ммоль) 2-метил-5-сульфамоилацетофенона, 90 мл метанола и 220 мл 1,2-дихлорэтана вводят в круглодонную колбу емкостью 500 мл. Реакционную среду нагревают до начала флегмообразования в течение 16 часов и затем концентрируют, после чего добавляют 200 мл насыщенного раствора бикарбоната натрия. Смесь экстрагируют 3 раза 120 мл этилацетата, органические фазы объединяют, сушат над сульфатом магния и концентрируют. Остаток очищают посредством колоночной хроматографии с использованием смеси 60:40 гексана и этилацетата, сушат в эксикаторе под вакуумом над пентоксидом фосфора. Получают 1,7 г продукта.
Точка плавления: 114oC.
8.2. α-хлорметил-2-метил-5-сульфамоилбензолметанол
Начиная с α-хлор-2-метил-5-сульфамоилацетофенона, обработанного, при условиях, описанных на стадии 2 Примера 3, получают α-хлорметил-2- метил-5-сульфамоилбензолметанол.
Точка плавления: 126oC.
8.3. α-азидометил-2-метил-5-сульфамоилбензолметанол
Начиная с α-хлорметил-2-метил-5-сульфамоилбензолметанола, обработанного при условиях, описанных на стадии 4 Примера 3, получают α-азидометил-2-метил-5-сульфамоилбензолметанол.
Точка плавления: 98oC.
8.4. α-аминометил-2-метил-5-сульфамоилбензолметанола метансульфонат
Начиная с α-азидометил- 2-метил-5-сульфамоилбензолметанола, обработанного при условиях, описанных на стадии 6 Примера 6, получают α-аминометил- 2-метил-5-сульфамоилбензолметанола метансульфонат.
Точка плавления: 185oC.
Пример 9: α-аминометил-2-хлор-5-сульфамоилбензолметанола метансульфонат
1,35 г (4,9 ммоль) α-азидометил-2-хлор-5-сульфамоилбензолметанола, 90 мл безводного пиридина и 9,67 г (29,0 ммоль) трифенилфосфина в носителе вводят в круглодонную колбу емкостью 250 мл. Смесь перемешивают при комнатной температуре в течение 9 часов, затем добавляют 100 мл 28%-ного водного аммиака, суспензию перемешивают в течение 16 часов и фильтруют. Фильтрат концентрируют, а остаток перекристаллизовывают в метаноле. Получают 0,523 г α-аминометил-2-хлор-5-сульфамоилбензолметанола. Добавляют 1 эквивалент метансульфоновой кислоты в виде 2М раствора в метаноле. После перекристаллизации в метаноле и диэтиловом эфире и сушки в эксикаторе под вакуумом над пентоксидом фосфора получают 0,439 г α-аминометил-2-хлор-5-сульфамоилбензолметанола метансульфоната.
Точка плавления: 206-208oC.
Пример 10: син- и анти-(2'-метокси-5'-аминосульфонилфенил)- 2-амино-1-пропанол
10.1. 2-метокси-5-хлорсульфонилпропиофенон
Начиная с 2-метокси-пропиофенона, обработанного при условиях стадии 1 Примера 2, получают 2-метокси-5-хлорсульфонилпропиофенон.
Точка плавления: 86-89oC.
10.2. 2-метокси-5-сульфамоилпропиофенон
Начиная с 2-метокси-5-хлорсульфонилпропиофенона, обработанного при условиях стадии 2 Примера 2, получают 2-метокси-5-сульфамоилпропиофенон.
Точка плавления: 162-165oC.
10.3. 2-бром-2'-метокси-5'-сульфамоилпропиофенон
Начиная с 2-метокси-5-сульфамоилпропиофенона, обработанного при условиях стадии 3 Примера 2, получают 2-бром-2'-метокси-5'- сульфамоилпропиофенон.
Точка плавления: 108-110oC.
10.4. 2-азидо-2'-метокси-5'-сульфамоилпропиофенон
Начиная с α-бром-2-метокси-5-сульфамоилпропиофенона, обработанного при условиях стадии 4 Примера 2, получают 2-азидо-2'-метокси- 5'-сульфамоилпропиофенон.
Точка плавления: 113-114oC.
10.5. (2'-метокси-5'-аминосульфонилфенил)-2-азидо-1-пропанол
Начиная с 2-азидо-2'-метокси-5'-сульфамоилпропиофенона, обработанного при условиях стадии 1 Примера 4, получают (2'-метокси-5'-аминосульфонилфенил)-2-азидо-1-пропанол.
Точка плавления: 109-110oC.
10.6. Син- и анти-(2'-метокси-5'аминосульфонилфенил)-2- амино-1-пропанол
Начиная с (2'-метокси-5'-аминосульфонилфенил)-2-азидо-1-пропанола, обработанного при условиях стадии 6 Примера 2, получают смесь син- и анти- (2'-метокси-5'-аминосульфонилфенил)-2-амино-1-пропанола, эти продукты разделяют последовательным хроматографированием через колонку с силикагелем с использованием смеси растворителей для элюции 95:5:0,5 дихлорметан/метанол/аммиак с получением син- и анти-диастереоизомеров.
После перекристаллизации в изопропаноле и сушки в эксикаторе под вакуумом над пентоксидом фосфора получают син-(2'-метокси- 5'-аминосульфонилфенил)-2-амино-1-пропанол.
Точка плавления: 176-177oC,
и анти-(2'-метокси-5'-аминосульфонилфенил)-2-амино-1 -пропанол.
Точка плавления: 233-237oC.
Пример 11: (-)-син-(2'-метокси-5'-сульфамоилфенил)-2-амино- 1-пропанол
11.1. (-)-син-(2'-метокси-5'-сульфамоилфенил)-2-азидо-1-пропанол
Начиная с 2-азидо-2'-метокси-5'-сульфамоилпропиофенона, обработанного при условиях стадии 5 Примера 2, получают (-) -син-(2'-метокси-5'-сульфамоилфенил)-2-амино-1-пропанол после 2 перекристаллизаций в изопропаноле.
Точка плавления: 143-145oC.
[α]
11.2. (-)-син-(2'-метокси-5'-сульфамоилфенил)-2-амино-1-пропанол
Начиная с (-)-син-(2'-метокси-5'- аминосульфонилфенил)-2-азидо-1-пропанола, обработанного при условиях стадии 6 Примера 2, получают (-)-син-(2'-метокси-5'- сульфамоилфенил)-2-амино-1-пропанол после 2 перекристаллизаций в изопропаноле.
Точка плавления: 190-191oC.
[α]
Пример 12
К раствору 100 мг соединения Примера 3 в 15 мл теплого метанола добавляют 145,6 мг (+)-O-O'-дибензоил-D-винной кислоты в 5 мл этилацетата. Эту смесь перемешивают и упаривают при пониженном давлении. Твердое вещество переносят в 8 мл этанола и 4 мл метанола, фильтруют и кристаллизуют. Кристаллы выделяют и сушат с получением 130 мг (+)-O-O'-дибензоил-D-тартрата (-)-(R)-аминометил-2-метокси-5- сульфонамидо-бензолметанола.
Энантиомеры соединения Примера 3 могут быть выделены также на хиральной колонке DICEL с использованием смеси гексан:этанол:диэтиламин в соотношении 85:15:5 в качестве элюента (0,8 мл/мин).
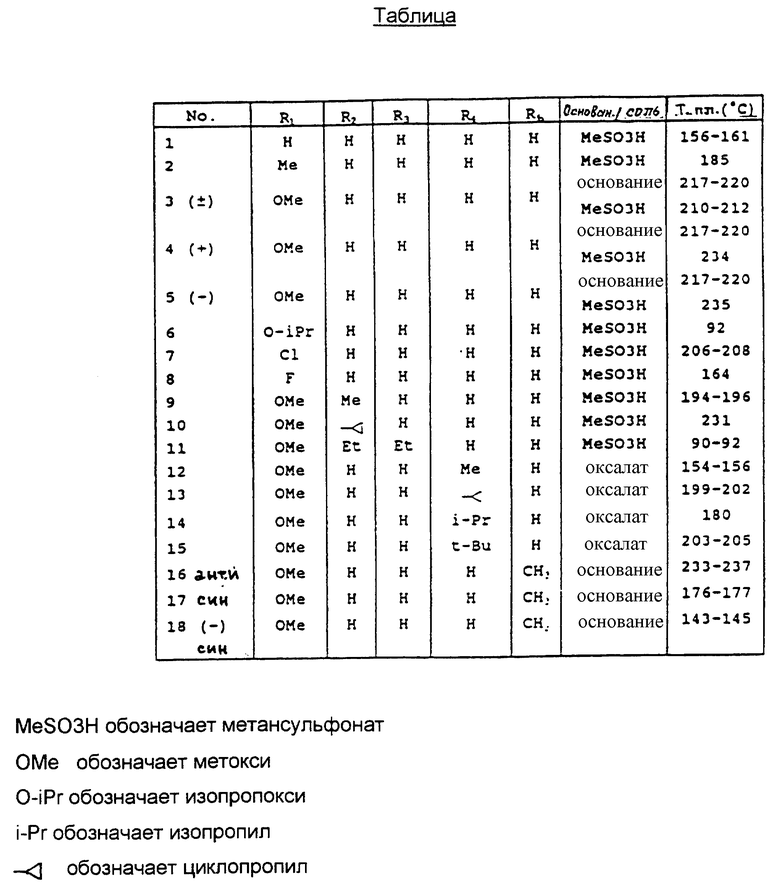
Соединения по изобретению приведены в таблице 1 вместе с их физическими свойствами.

Биологические тесты
Соединения по изобретению анализировали в биологических тестах для выявления их активности в качестве агонистов α1-адренорецепторов.
В частности, их подвергали тестированию на способность связываться с α1a, α1b и α1dсуб-рецепторами, которое проводили на ткани слюнной железы крыс, на ткани печени крыс и на трансфецированных CHO клетках.
Определяли сродство к каждому типу суб-рецепторов, выражаемое как ИК50 (концентрация, ингибирующая на 50% связывание с [3H]празозином), и рассчитывали относительные величины сродства к α1aрецептору относительно величин сродства к α1b и α1dрецепторам, выраженные как соотношения значений ИК50 [α1b/α1a] и [α1d/α1a].
Для соединений по изобретению эти соотношения варьируют от 9,3 до 21,6 и от 7,8 до 20,9, соответственно, свидетельствуя о существенной селективности в отношении α1aрецептора.
In vitro активность соединений по изобретению исследовали на уретральных и артериальных гладких мышцах.
Эти эксперименты проводили на самках новозеландских (New Zealand) кроликов весом от 3 до 3,5 кг. Животных умерщвляли путем позвоночного вывиха и затем извлекали тканевые кольца из мезентериальных артерий и из уретры. Кольца ткани погружали - в модифицированный раствор Кребса (Krebs) и насыщали кислородом с помощью смеси 95% O2 и 5% CO2. Каждый образец ткани подвергали давлению 1 г, затем вводили в кумулятивных дозах фенилэфрин и снимали кривую доза/ответ. После промывки образцов в кумулятивных дозах вводили тестируемое соединение и снимали кривую доза/ответ. α1-адренергическое действие каждого соединения оценивали путем расчета pD2 (отрицательный логарифм концентрации антагониста в присутствии которой действие дозы агониста делится на 2), а также максимальным действием, представляющим собой процент максимального сжатия, полученного с фенилфрином (% Emax).
Для соединений по изобретению уретральные и артериальные значения pD2 варьируют между 4,18 и 4,93 (pD2 фенилэфрина = 5,2- 5,5) и между 3,73 и 4,55 (pD2 фенилэфрина = 5,2-5,5), соответственно, а уретральные и артериальные значения % Emax варьируют между 58,4 и 76 и между 76 и 94,6, соответственно.
In vivo активность соединения по изобретению в отношении кровяного и уретрального давления исследовали на кроликах.
Эти эксперименты проводили на самках новозеландских (New Zealand) кроликов весом от 3 до 4 кг. После анестезии пентабарбиталом вводили катетеры в брюшную аорту через бедренную артерию, в яремную вену и в уретру (1 см ниже шейки мочевого пузыри).
Тестируемые соединения вводили через 5-15 дней после операции, либо внутривенно, либо перорально.
При внутривенном введении соединения вводили в течении 5 минут в виде разовой дозы, либо кумулятивным путем с интервалами 15 минут между каждой дозой, в дозах от 3 до 100 мкг/кг.
Кровяное давление (ВР) и уретральное давление (UP) измеряли непрерывно для каждой дозы.
Для соединений по изобретению увеличение ВР составляет приблизительно 5 мм рт. ст. в дозе 10 мкг/кг и 15 мм рт. ст. в дозе 100 мкг/кг, а увеличение UP составляет приблизительно 14 см водн. ст. в дозе 10 мкг/кг и 54 см водн. ст. в дозе 100 мкг/кг.
В различных тестировавшихся дозах соединения по изобретению обладают сильной уроселективностью, поскольку они очень существенно увеличивают уретральное давление без значительного изменения кровяного давления.
При пероральном введении соединения вводили путем зондового кормления в виде разовой дозы, составлявшей 300 и 1000 мкг/кг, в объеме 1 мл/кг. ВР и UP измеряли через 5, 10, 30, 45 и 60 минут после зондового введения.
Для соединений по изобретению изменения в ВР составляют приблизительно -0,2 и -0,9 мм рт. ст. в дозах 300 и 1000 мкг/кг, соответственно, через 30 минут, и приблизительно -5,3 и 1,1 мм рт.ст., соответственно, через 60 минут, а изменения в UP составляют приблизительно 1,6 и 7,8 см водн. ст. в дозах 300 и 1000 мкг/кг, соответственно, через 30 минут, и приблизительно 3,7 и 8,3 см водн. ст. соответственно, через 60 минут.
При пероральном введении соединения проявляют полную уроселективность, поскольку уретральное давление увеличивается значительно без изменения кровяного давления.
В совокупности полученные результаты свидетельствуют о том, что соединения по изобретению обладают сильным уретральным действием и слабым артериальным действием. Они являются агонистами α1-адренорецепторов, селективными в отношении α1a-рецепторов. Поэтому они могут быть использованы в лечении недержания мочи.
Для этой цели они могут быть представлены во всех формах, пригодных для энтерального и парентерального введения, в сочетании с фармацевтическими эксципиентами, например в форме таблеток, драже, капсул, в том числе твердых желатиновых капсул, растворов для перорального введения и для инъекций и суппозиториев, в дозах, обеспечивающих суточную дозу от 0,001 до 1000 мг активного вещества.
Пример фармацевтической композиции
Данный пример демонстрирует композицию и процедуру приготовления препарата в форме стандартной таблетки. В качестве активного ингредиента используют соединение Примера 1.
Таблетки, содержащие 1,0 мг активного ингредиента на таблетку, получают, используя композицию; представленную в табл. 2.
Смесь активного ингредиента с лактозой увлажняют гранулирующим раствором, содержащим воду и кукурузный крахмал, и гранулируют. Полученные гранулы сушат, затем смешивают с кристаллической целлюлозой, гидроксипропилцеллюлозой и стеаратом магния. Полученную смесь прессуют в таблетки.
Токсичность
Соединения по изобретению были испытаны на токсичность на крысах и собаках. Испытания показали, что в пределах эффективной дозы и выше заявленные соединения не токсичны.

Изобретение относится к бензолсульфонамидным соединениям общей формулы I, где R1 представляет собой атом водорода, галогена, такого как хлор или фтор, или линейную или разветвленную C1-4алкильную или C1-4алкоксильную группу, R2, R3 и R4 представляют собой, независимо друг от друга, атомы водорода или линейные, разветвленные или циклические C1-4алкильные группы, и R5 представляет собой атом водорода или C1-2алкильную группу в форме энантиомеров или диастереоизомеров или смесей этих различных форм, включая рацемические смеси, а также их соли, полученные присоединением фармацевтически приемлемых кислот. Соединения формулы I обладают активностью агониста α1-адренорецепторов и могут быть использованы в качестве лекарственного средства для лечения недержания мочи. Описаны разные способы получения соединения I. 8 с. и 10 з.п. ф-лы, 2 табл.
в которой R1 представляет собой атом водорода, атом галогена, такого, как хлор или фтор, или линейную или разветвленную C1-4алкильную или C1-4алкоксильную группу;
R2, R3 и R4 представляют собой, независимо друг от друга, атомы водорода или линейные, разветвленные или циклические, C1-4алкильные группы;
R5 представляет собой атом водорода или C1-2алкильную группу,
в форме энантиомеров, или диастереоизомеров, или смесей этих различных форм, включая рацемические смеси, а также их соли, полученные присоединением фармацевтически приемлемых кислот.
в которой R1 представляет собой линейную или разветвленную C1-4алкоксильную группу;
R2 и R3 представляют собой атомы водорода;
R4 представляет собой атом водорода или линейную, разветвленную или циклическую C1-4алкильную группу;
R5 представляет собой атом водорода,
в форме энантиомеров, или диастереоизомеров, или смесей этих различных форм, включая рацемические смеси, а также их солей, полученных присоединением фармацевтически приемлемых кислот, при котором
(а) соединение формулы III
в которой R1 и R4 такие, как определено выше, подвергают взаимодействию с триметилсилилцианидом в присутствии иодида цинка с получением производного формулы II
в которой R1 и R4 такие, как определено выше,
(б) восстанавливают полученное производное II боргидридом лития в присутствии триметилсилилхлорида с получением соединения формулы I,
(в) возможно превращают это полученное соединение I в его энантиомеры или диастереоизомеры, или в его фармацевтически приемлемые соли.
в которой R1 представляет собой атом водорода, атом галогена, такого, как фтор, или линейную или разветвленную C1-4алкильную или C1-4алкоксильную группу;
R2 и R3 представляют собой атомы водорода;
R4 представляет собой атом водорода или линейную, разветвленную или циклическую C1-4алкильную группу;
R5 представляет собой атом водорода или C1-2алкильную группу,
в форме энантиомеров, или диастереоизомеров, или смесей этих различных форм, включая рацемические смеси, а также их солей, полученных присоединением фармацевтически приемлемых кислот, при котором
(а) производное формулы VIII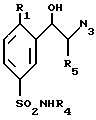
в которой R1, R4 и R5 такие, как определено выше,
в форме энантиомера, или диастереоизомеров, или смесей этих различных форм, включая рацемическую смесь, подвергают взаимодействию с водородом в присутствии катализатора, такого, как палладий на угле, с получением данного соединения общей формулы I; и
(б) возможно превращают это полученное соединение формулы I в его энантиомеры или диастереоизомеры, или его фармацевтически приемлемые соли.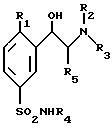
в которой R1 представляет собой атом хлора;
R2 и R3 представляют собой атомы водорода;
R4 представляет собой атом водорода или линейную, разветвленную или циклическую C1-4алкильную группу;
R5 представляет собой атом водорода или C1-2алкильную группу,
в форме энантиомеров, или диастереоизомеров, или смесей этих различных форм, включая рацемические смеси, а также их солей, полученных присоединением фармацевтически приемлемых кислот, при котором
(а) производное формулы VIII
в которой R1, R4 и R5 такие, как определено выше,
в форме энантиомера, или диастереоизомеров, или смесей этих различных форм, включая рацемическую смесь, подвергают взаимодействию с трифенилфосфином, а затем водным аммиаком с получением данного соединения общей формулы I;
(б) возможно превращают это полученное соединение формулы I в его энантиомеры, или диастереоизомеры, или его фармацевтически приемлемые соли.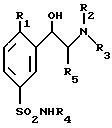
в которой R1 представляет собой атом водорода, атом галогена, такого, как хлор или фтор, или линейную или разветвленную C1-4алкильную или C1-4алкоксильную группу;
R2 представляет собой атом водорода или C1-4алкильную группу;
R3 представляет собой C1-4алкильную группу;
R4 представляет собой атом водорода или линейную, разветвленную или циклическую C1-4алкильную группу;
R5 представляет собой атом водорода или C1-2алкильную группу,
в форме энантиомеров, или диастереоизомеров, или смесей этих различных форм, включая рацемические смеси, а также их солей, полученных присоединением фармацевтически приемлемых кислот, при которой
(а) производное формулы VIII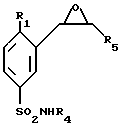
в которой R1, R4 и R5 такие, как определено выше,
подвергают взаимодействию с амином формулы R2(R3)NH, в которой R2 и R3 такие, как определено выше, с получением данного соединения общей формулы I;
(б) возможно превращают это полученное соединение формулы I в его энантиомеры, или диастереоизомеры, или его фармацевтически приемлемые соли.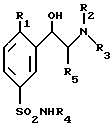
в которой R1 представляет собой атом водорода, атом галогена, такого, как хлор или фтор, или линейную или разветвленную C1-4алкильную или C1-4алкоксильную группу;
R2 представляет собой C1-4алкильную группу;
R3 представляет собой атом водорода;
R4 представляет собой атом водорода или линейную, разветвленную или циклическую C1-4алкильную группу;
R5 представляет собой атом водорода или C1-2алкильную группу,
в форме энантиомеров, или диастереоизомеров, или смесей этих различных форм, включая рацемические смеси, а также их солей, полученных присоединением фармацевтически приемлемых кислот, при которой
(а) производное формулы VIII
в которой R1, R4 и R5 такие, как определено выше,
подвергают взаимодействию с амином формулы R2(Bn)NH, в которой R2 такой, как определено выше, а Bu является бензильной группой, с получением производного формулы VI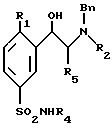
в которой R1, R4 и R5 такие, как определено выше,
которое затем восстанавливают водородом в присутствии катализатора, такого, как палладий на угле, с получением данного соединения общей формулы I;
возможно превращают это полученное соединение формулы I в его энантиомеры, или диастереоизомеры, или его фармацевтически приемлемые соли.
| СПОСОБ ИЗОЛЯЦИИ ПРИТОКА ПЛАСТОВЫХ ВОД В СКВАЖИНЕ СО СМЯТОЙ ЭКСПЛУАТАЦИОННОЙ КОЛОННОЙ В УСЛОВИЯХ АНОМАЛЬНО НИЗКИХ ПЛАСТОВЫХ ДАВЛЕНИЙ | 2009 |
|
RU2405931C1 |
| Пневмогидравлический прибор для испытания автосвечей на герметичность | 1932 |
|
SU34432A1 |
| US 3943254, 09.03.1976 | |||
| US 3860647, 14.01.1975 | |||
| Способ получения производных 2-окси-2-фенилэтиламина или их солей | 1979 |
|
SU932982A3 |
| М.Д.МАШКОВСКИЙ | |||
| Лекарственные средства | |||
| - М.: Медицина, ч.1, с.543, 1986, | |||
