Изобретение относится к производным 1Н-пиридо[3,4-b]индол-4-карбоксамида, их получению и применению в терапии.
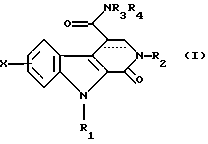
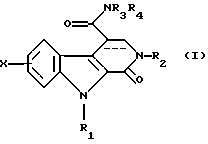
Соединения по изобретению соответствуют общей формуле (I)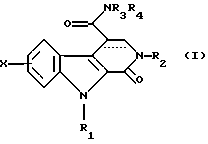
в которой Х представляет собой атом водорода или галогена или (С1-С3)алкильную, (С1-С3)алкоксильную, трифторметильную или фенилметокси группу,
R1 представляет собой атом водорода или (С1-С3)алкильную, циклопропильную или фенилметильную группу,
R2 представляет собой либо (С1-С3)алкильную группу, возможно замещенную метоксигруппой, либо фенил(С1-С3)алкильную группу, возможно замещенную по фенильному кольцу атомом галогена или метильной или метоксигруппой, либо циклогексилметильную группу, либо тиенилметильную группу, либо пиридинилметильную группу, либо фенильную группу, возможно замещенную одним или более чем одним атомом галогена или (С1-С3)алкильной или (С1-С3)алкоксильной группой, либо пиридинильную группу, либо 5-метил-1,2-оксазолильную группу, либо 5-метил-1,3,4-тиадиазолильную группу, либо нафтильную группу,
R3 и R4, независимо один от другого, каждый представляет собой атом водорода, (С1-С3)алкильную группу, 2-метоксиэтильную группу, гидрокси(С2-С4)алкильную группу, карбокси(С1-С3)алкильную группу, (С1-С3)алкоксикарбонил(С1-С3)алкильную группу или фенил(С1-С3)алкильную группу, или же совместно с атомом азота, который их несет, образуют либо пирролидинильную группу, возможно замещенную гидроксильной, этоксильной, метоксикарбонильной или метоксиметильной группой, либо пиперидинильную группу, либо морфолинильную группу, либо 4-метилпиперазинильную группу, либо азетидинильную группу, либо тиазолидинильную группу, а связь между атомами углерода в положениях 3 и 4 является простой или двойной.
В зависимости от природы этой связи, соединение по изобретению возможно может существовать в форме чистого оптического изомера или смеси таких изомеров.
Предпочтительными соединениями являются те, в общей формуле которых Х находится в положении 6 и представляет собой атом фтора, R1 представляет собой метильную группу, R2 представляет собой фенильную группу, R3 представляет собой метильную группу и R4 представляет собой этильную группу, или же R3 и R4 вместе с атомом азота, который их несет, образуют пирролидинильное кольцо.
Соединения общей формулы (I) могут быть получены способами, поясняемыми следующими схемами.
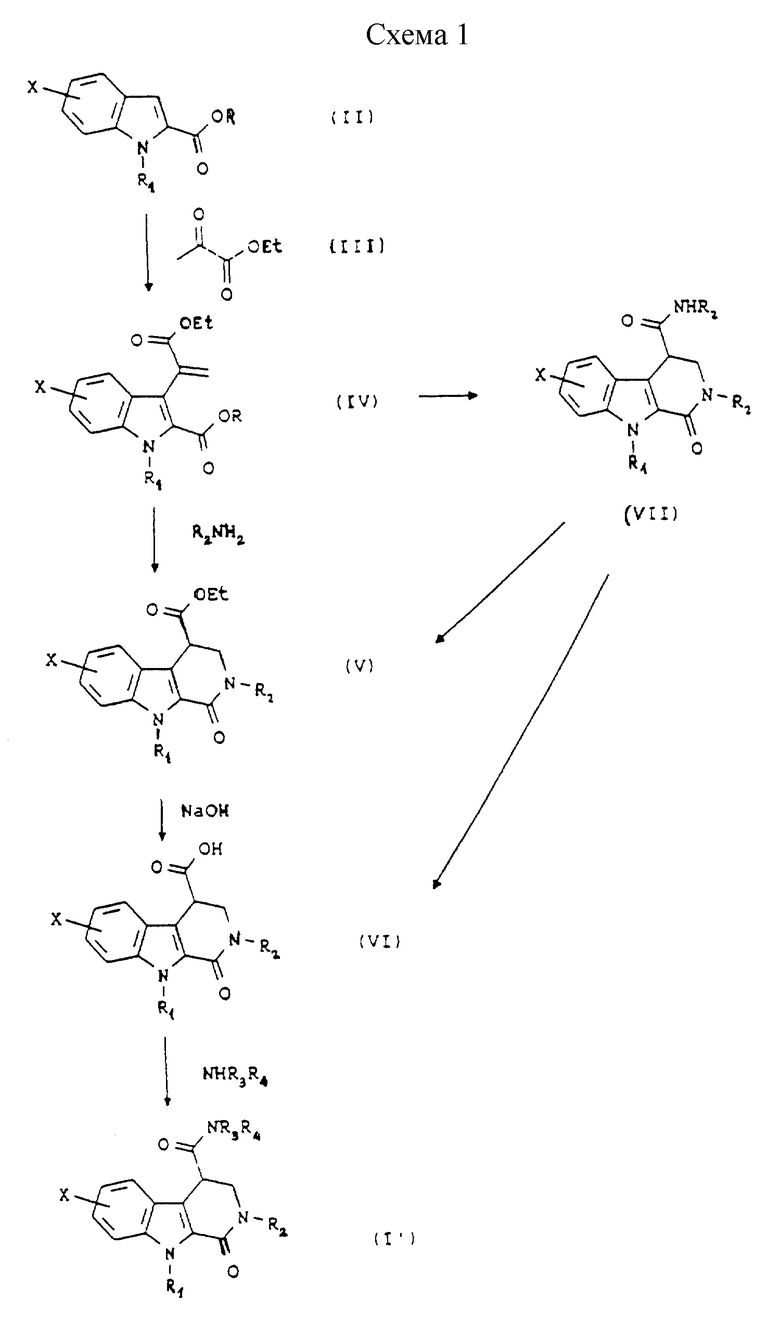
Согласно Схеме 1 исходное соединение соответствует общей формуле (II), в которой Х такой, как определено выше, и R1 такой, как определено выше; когда R1 представляет собой водород, возможно, по желанию, осуществлять алкилирование для получения соединения общей формулы (II), в которой R1 представляет собой (С1-С3)алкильную группу. Соединение общей формулы (II), таким образом, подвергают взаимодействию с этилпируватом формулы (III) в кислой среде, например в присутствии газообразной соляной кислоты в этаноле или в присутствии серной кислоты или эфирата трифторида бора в уксусной кислоте, при температуре между комнатной и температурой дефлегмации, для получения диэфира общей формулы (IV).
Диэфир общей формулы (IV) затем обрабатывают в этаноле при температуре дефлегмации амином общей формулы R2NH2, в которой R2 такой, как определено выше. Получают сложный эфир формулы (V), который превращают в соответствующую кислоту общей формулы (VI) путем гидролиза в щелочной среде.
Эту кислоту затем превращают в первичный, вторичный или третичный амид общей формулы (I') путем взаимодействия с амином общей формулы НNR3R4, в которой R3 и R4 такие, как определено выше, либо через имидазолид, полученный путем взаимодействия с N,N'-карбонилдиимидазолом или через хлорангидрид.
В соединении общей формулы (I'), полученном таким образом, связь между положениями 3 и 4 является простой. В тех случаях, когда требуется приготовить соединение, в котором эта связь является двойной, соединение общей формулы (I') окисляют при помощи 2,3-дихлор-5,6-дицианоциклогекса-2,5-диен-1,4-диона или 3,4,5,6-тетрахлорциклогекса-3,5-диен-1,2-диона в растворителе, таком как толуол или дихлорметан, при температуре между комнатной и температурой дефлегмации, для получения соответствующего соединения, в структуре которого связь между атомами углерода в положениях 3 и 4 является двойной, которое соответственно имеет общую формулу (I"):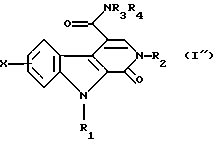
Наконец, и если это имеет место, энантиомеры могут быть получены из рацематов любым известным способом; таким образом, например, кислота общей формулы (VI) может быть подвергнута взаимодействию с оптически чистым хиральным амином, таким как α-метилбензиламин, и диастереоизомеры могут быть отделены фракционированной кристализацией, чтобы добиться оптически чистой кислоты и затем сложных эфиров и амидов, получаемых из нее.
В случае оптически чистой кислоты общей формулы (VI) нерацемизирующая реакция сочетания может быть выполнена любым известным способом, например с использованием (бензотриазол-1-илокси)трис-(пирролидин-1-ил)фосфониум гексафторфосфата.
В случае, когда R2 представляет собой ароматическое кольцо, возможно, по желанию, превратить диэфир (IV) в амид (VII) путем нагревания реакционной смеси при температуре от 100oС до 200oС, в инертном растворителе или без растворителя, например, при нагревании с обратным холодильником соответствующего амина общей формулы R2NH2. Далее можно превратить соединение общей формулы (VII) либо в сложный эфир общей формулы (V) в нагреваемом с обратным холодильником этаноле в кислой среде, например в присутствии концентрированной соляной кислоты, или в кислоту общей формулы (VI) путем гидролиза в щелочной среде.
Исходное соединение общей формулы (II), главным образом с R1=H, описано в литературе; пируват формулы (III) является коммерчески доступным.
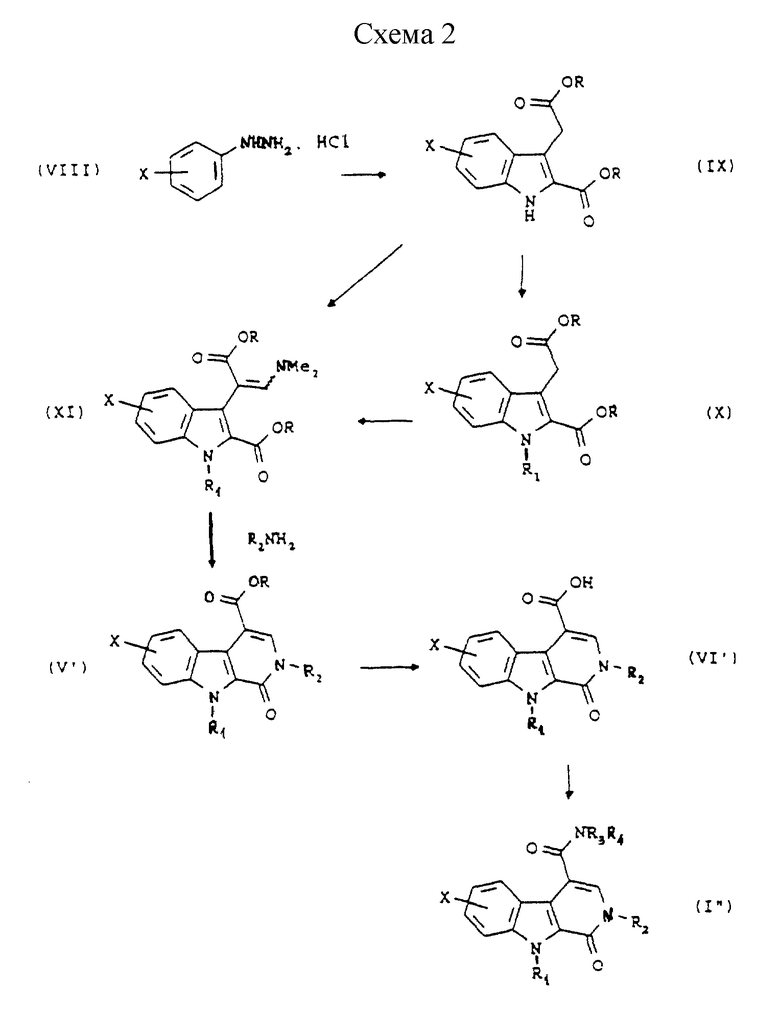
Согласно Схеме 2 исходное соединение соответствует общей формуле (VIII), в которой Х такой, как определено выше. Это соединение подвергают взаимодействию с 2-кетоглутаровой кислотой и затем обрабатывают в кислой спиртовой среде, например в этаноле, насыщенной газообразной соляной кислотой, при температуре дефлегмации, для получения диэфира формулы (IX), в которой R представляет собой (С1-С3)алкильную группу. По желанию, далее осуществляют реакцию алкилирования этого соединения для получения соединения общей формулы (X), в которой R1 представляет собой (С1-С3)алкильную группу, и затем его подвергают превращению в протонном растворителе, например в N,N-диметилформамиде, в присутствии диметилацеталя диметилформамида, при температуре дефлегмации, для получения соединения формулы (XI). По желанию, соединение общей формулы (IX) может быть непосредственно превращено в соединение общей формулы (XI), в которой R1 представляет собой метильную группу, в условиях, описанных выше.
Соединение общей формулы (XI) затем обрабатывают амином общей формулы H2NR2,в которой R2 такой, как определено выше, в протонном растворителе, например в N,N-диметилформамиде, возможно в присутствии кислоты, например 4-метилбензолсульфоновой кислоты, при температуре дефлегмации, для получения сложного эфира общей формулы (V').
Сложный эфир общей формулы (V') превращают в соответствующую кислоту общей формулы (VI') путем гидролиза в щелочной среде.
Наконец эту кислоту превращают в первичный, вторичный или третичный амид общей формулы (I") либо через имидазолид, полученный путем взаимодействия с N,N'-карбонилдиимидазолом, или через хлорангидрид.
Исходные соединения общей формулы (VIII) являются коммерчески доступными. Некоторые соединения общих формул (IX) и (X) описаны в литературе.
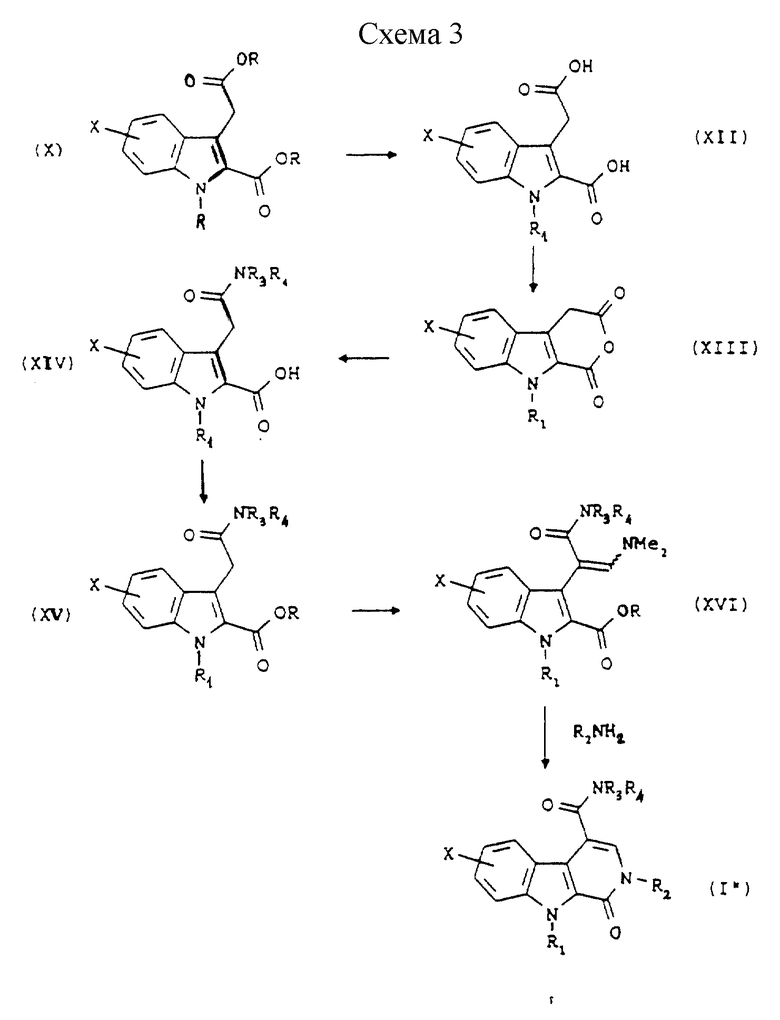
Согласно Схеме 3 исходным веществом является диэфир общей формулы (X), описанный в отношении способа по Схеме 2. Этот диэфир гидролизуют в кислой среде для получения двухосновной кислоты общей формулы (XII), которую превращают в ангидрид, например, путем использования ацетилхлорида при температуре дефлегмации, для получения соединения общей формулы (XIII). Это соединение превращают путем взаимодействия формулы HNR3R4, в которой R3 и R4 такие, как определено выше, в хлорированном растворителе, например дихлорметане, для получения соединения общей формулы (XIV), которое превращают в сложный эфир общей формулы (XV), этот сложный эфир затем подвергают обработке в протонном растворителе, например в N,N-диметилформамиде, в присутствии диметилацеталя диметилформамида при температуре дефлегмации для получения соединения общей формулы (XVI) и, наконец, это соединение подвергают взаимодействию с амином общей формулы R2NH2, в которой R2 такой, как определено выше, в протонном растворителе, например N,N-диметилформамиде, возможно в присутствии кислоты, например 4-метилбензолсульфоновой кислоты, при температуре дефлегмации, для получения общей формулы (I").
Наконец, по желанию, вторичный амид общей формулы (I"), в которой R3 или R4 представляет собой водород, может быть превращен в третичный амид при помощи реакции алкилирования, известной специалистам в данной области, при помощи алкилирующего агента, например алкилгалогенида. Таким же образом соединение общей формулы (I') или (I"), в которой R1 представляет собой атом водорода, может быть превращено в соединение, в формуле которого R1 представляет собой алкильную группу, при помощи реакции алкилирования известного типа. Соединения с химическими структурами, аналогичными структурам соединений по изобретениям, описаны в (СА 83(13) 114712с, СА 94(9) 64698g и СА 96(9) 68779у).
Некоторые соединения общих формул (XII), (XIII), (XIV) и (XV) описаны в литературе. Соединение общей формулы (I), в которой R3 и/или R4 представляют собой гидрокси(С2-С4)алкильную группу, может быть получено при помощи взаимодействия соответствующей кислоты общей формулы (VI) или (VI') со спиртом, защищенным обычной защитной группой, с последующим снятием защиты.
Соединение общей формулы (I), в котором R3 и/или R4 представляет собой карбокси(С2-С4)алкильную группу, может быть получено при помощи гидролиза соответствующего сложного эфира. Соединение общей формулы (I), в которой Х представляет собой фенилметоксильную группу, может быть получено в две стадии, известные специалистам в данной области, из соединения общей формулы (I), в которой Х представляют собой метоксильную группу.
Нижеследующие примеры поясняют получение нескольких соединений согласно изобретению. Приводятся данные по элементному микроанализу, данные по инфракрасной спектроскопии и ЯМР-спектроскопии, подтверждающие структуры полученных соединений.
Номера соединений, указанные в скобках в заголовках, соответствуют тем, которые указаны в таблице 1.
Пример 1 (соединение 20)
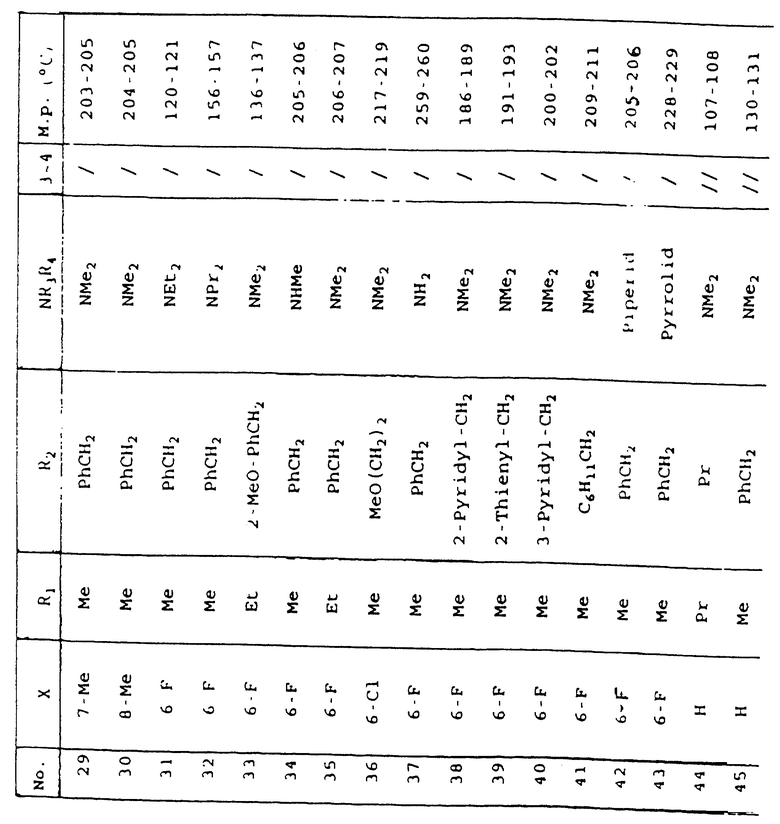
(±)-6-(фтор-N, N, 9-триметил-1-оксо-2-(фенилметил)-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-4-карбоксамид.
1.1. Этил 5-фтор-1-метил-1Н-индол-2-карбоксилат.
1.1.1. Этил 5-фтор-1Н-индол-2-карбоксилат.
150 г (0,92 моль) 4-фторфенилгидразингидрохлорида добавляют до тех пор, пока смесь охлаждается, к приготовленному раствору 23 г (1 моль) натрия в 1,5 л метанола и смесь перемешивают при комнатной температуре в течение 30 минут.
Раствор концентрируют при пониженном давлении, остаток растворяют в дихлорметане и хлорид натрия отделяют фильтрованием. Растворитель выпаривают при пониженном давлении, остаток растворяют в 830 мл этанола, содержащего 4,4 мл уксусной кислоты в 102 мл (0,91 моль) этилпирувата, и смесь нагревают с обратным холодильником в течение двух часов.
Реакционную смесь концентрируют при пониженном давлении, остаток растворяют в этилацетате, раствор промывают водой и сушат над сульфатом натрия и растворитель выпаривают при пониженном давлении. Получают 181,6 г (0,83 моль) гидразона.
214 г (1,12 моль) моногидрата 4-метилбензолсульфоновой кислоты в растворе 2,5 л толуола дегидратируют нагреванием реакционной смеси в течение двух часов с обратным холодильником, используя ловушку Дина-Старка. 181,6 г (0,83 моль) гидразона, полученного выше, добавляют, пока он холодный, и смесь нагревают с обратным холодильником в течение трех часов. Смесь охлаждают, добавляют этилацетат и воду, органическую фазу отделяют и сушат, и растворитель выпаривают при пониженном давлении. Остаток перекристаллизовывают из пропан-2-ола, и маточный раствор очищают хроматографией на колонке с силикагелем, осуществляя элюцию дихлорметаном. Получают 144 г (0,7 моль) продукта, который используют на следующей стадии.
1.1.2. Этил 5-фтор-1-метил-1Н-индол-2-карбоксилат.
11,7 г (0,39 моль) гидрида натрия в виде 80%-ной суспензии в масле промывают петролейным эфиром и затем добавляют раствор 62,1 г (0,3 моль) этил 5-фтор-1H-индол-2-карбоксилата в 600 мл диметилформамида. Смесь перемешивают в течение двух часов при комнатной температуре и затем добавляют 24,3 мл (0,39 моль) йодистого метила в растворе 50 мл диметилформамида. Смесь перемешивают в течение 20 часов при комнатной температуре и затем выливают на ледяную воду. Реакционную смесь экстрагируют этилацетатом, органическую фазу промывают и сушат над сульфатом натрия, растворитель выпаривают при пониженном давлении и получают 62,5 г (0,28 моль) твердого продукта, который используют на следующей стадии.
1.2. Этил 2-(этоксикарбонил)-5-фтор-1-метил-α-метилен-1Н-индол-3-ацетат.
Раствор 10,5 г (48 ммоль) этил 5-фтор-1-метил-1H-индол-2-карбоксилата, 18 г (155 ммоль) этилпирувата и 7,8 мл концентрированной серной кислоты в 100 мл уксусной кислоты перемешивают при комнатной температуре в течение 1,5 часа. Смесь концентрируют при пониженном давлении, гидролизуют ледяной водой, добавляют нашатырный спирт до щелочного рН и осуществляют экстракцию дихлорметаном. Органическую фазу промывают водой и сушат над сульфатом натрия, растворитель выпаривают при пониженном давлении и остаток перекристаллизовывают из смеси пентана и диэтилового эфира. Получают 13 г (42 ммоль) твердого вещества.
Температура плавления: 86-88oС.
1.3. Этил (±)-6-фтор-9-метил-1-оксо-2-(фенилметил)-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-4-карбоксилат.
Раствор 7 г (23 ммоль) этил 2-(этоксикарбонил)-5-фтор-1-метил-α-метилен-1H-индол-3-ацетата и 15 мл (140 ммоль) бензиламина в 200 мл этанола нагревают с обратным холодильником в течение 8 часов. Растворитель выпаривают при пониженном давлении и остаток растворяют в дихлорметане и 1 н. соляной кислоте. Органическую фазу промывают водой и сушат над сульфатом натрия. Растворитель выпаривают при пониженном давлении и остаток очищают хроматографией на колонке с силикагелем, осуществляя элюцию смесью дихлорметана и этилацетата. Получают 7 г (18 ммоль) твердого продукта, который используют на следующей стадии.
1.4. (±)-6-фтор-9-метил-1 -оксо-2-(фенилметил)-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-4-карбоновая кислота.
6 г (16 ммоль) этил (±)-6-фтор-9-метил-1-оксо-2-(фенилметил)-2,3,4,9-тетрагидро-1H-пиридо[3,4-b]индол-4-карбоксилата гидролизуют 2,5 г гидроксида натрия в смеси воды и этанола, эту смесь концентрируют при пониженном давлении, добавляют воду и уксусную кислоту, осуществляют экстракцию этилацетатом, органическую фазу промывают водой и сушат над сульфатом натрия, и растворитель выпаривают при пониженном давлении. Получают 4 г (11 ммоль) твердого вещества, которое используют на следующей стадии.
Температура плавления: 264-265oС.
1.5. (±)-6-фтор-N,N,9-триметил-1-оксо-2-(фенилметил)-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-4-карбоксамид.
Раствор 4 г (11 ммоль) (±)-6-фтор-9-метил-1-оксо-2-(фенилметил)-2,3,4,9-тетрагидро-1H-пиридо[3,4-b] индол-4-карбоновой кислоты и 2,2 г (30 ммоль) 1,1'-карбонилдиимидазола в 200 мл тетрагидрофурана нагревают при 40oС в течение двух часов. Реакционную смесь охлаждают и добавляют избыток разжиженного диметиламина и смесь перемешивают в течение нескольких часов.
Растворитель выпаривают при пониженном давлении, остаток растворяют в дихлорметане и воде, органическую фазу отделяют, промывают водой и сушат над сульфатом натрия, растворитель выпаривают при пониженном давлении и остаток очищают хроматографией на колонке с силикагелем, осуществляя элюцию смесью дихлорметана и метанола. Продукт перекристализовывают из этилацетата. Получают 1,2 г (3 ммоль) продукта.
Температура плавления: 185-186oС.
Пример 2 (соединение 59)
6-фтор-N, N, 9-триметил-1 -оксо-2-(фенилметил)-2,9-дигидро-1Н-пиридо[3,4-b]индол-4-карбоксамид.
Раствор 2 г (5 ммоль) (±)-6-фтор-N,N,9-триметил-1-оксо-2-(фенилметил)-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b] индол-4-карбоксамида и 1,6 г (7 ммоль) 2,3-дихлоро-5,6-дициано-1,4-бензохинона в 250 мл дихлорметана перемешивают в течение одного часа. Органическую фазу промывают и сушат сульфатом натрия. Растворитель выпаривают при пониженном давлении и остаток очищают хроматографией на колонке с силикагелем, осуществляя элюцию смесью дихлорметана и этилацетата. Продукт перекристаллизовывают из диэтилового эфира. Получают 1 г (2,6 ммоль) продукта.
Температура плавления: 192-193oС.
Пример 3 (соединение 36)
6-хлор-2-(2-метоксиэтил)-N, N, 9-триметил-1-оксо-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-4-карбоксамид.
3.1. Этил 5-хлор-1-метил-1Н-индол-2-карбоксилат.
Получение осуществляют так, как в Примере 1.1.2., из 8,95 г (40 ммоль) этил 5-хлор-1H-индол-2-карбоксилата, 1,6 г (52 ммоль) гидрида натрия в виде 80%-ной суспензии в масле и 11,35 г (80 ммоль) йодистого метила. Получают 9,5 г (40 ммоль) твердого продукта, который используют на следующей стадии.
3.2. Этил 5-хлор-2-(этоксикарбонил)-1-метил-α-метилен-1H-индол-3-ацетат.
Раствор, насыщенный соляной кислотой и содержащий 9,5 г (40 ммоль) этил 5-хлор-1H-индол-2-карбоксилата и 8,8 мл (80 ммоль) этилпирувата, нагревают с обратным холодильником в течение 4 часов. Растворитель выпаривают при пониженном давлении, остаток растворяют в этилацетате, который промывают до нейтральности, органическую фазу высушивают над сульфатом натрия и растворитель выпаривают при пониженном давлении. Продукт очищают хроматографией на колонке с силикагелем, осуществляя элюцию дихлорметаном. Получают 9,6 г (32 ммоль) твердого продукта, который используют на следующей стадии.
3.3. Этил (±)-6-хлор-2-(2-метоксиэтил)-9-метил-1-оксо-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-4-карбоксилат.
Раствор 9,5 г (31 ммоль) этил 5-хлор-2-(этоксикарбонил)-1-метил-α-метилен-1H-индол-3-ацетата и 8,1 г (93 ммоль) 2-метоксиэтиламина в 20 мл этанола нагревают с обратным холодильником в течение трех часов.
Растворитель выпаривают при пониженном давлении и остаток растворяют этила цетатом, который промывают водой и сушат над сульфатом натрия. Получают 9,7 г (27 ммоль) твердого продукта, который используют на следующей стадии.
3.4. (±)-6-хлор-2-(2-метоксиэтил)-9-метил-1-оксо-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-4-карбоновая кислота.
9,6 г (26 ммоль) этил (±)-6-хлор-2-(2-метоксиэтил)-9-метил-1-оксо-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b] индол-4-карбоксилата гидролизуют раствором 3,1 г (80 ммоль) гидроксида натрия в 260 мл этанола и 50 мл воды. Реакционную смесь концентрируют, остаток растворяют в воде и водную фазу промывают этилацетатом и подкисляют до рН 1 концентрированной соляной кислотой. Экстракцию осуществляют этилацетатом. Органическую фазу промывают водой и сушат над сульфатом натрия. Растворитель выпаривают при пониженном давлении и получают 8,3 г (26 ммоль) твердого продукта, который используют на следующей стадии.
3.5. (±)-6-хлор-2-(2-метоксиэтил)-N, N,9-триметил-1-оксо-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-4-карбоксамид.
Получение осуществляют так, как в примере 1.5., из 8,3 г (26 ммоль) (±)-6-хлоро-2-(2-метоксиэтил)-9-метил-1-оксо-2,3,4,9-тетрагидро-1H-пиридо[3,4-b] индол-4-карбоновой кислоты и из диметиламина. Выделяют 8,6 г продукта, который перекристаллизовывают из пропан-2-ола. Получают 6,9 г (19 ммоль) продукта.
Температура плавления: 217-219oС.
Пример 4 (соединение 77)
(+)-6-фтop-N, N,9-триметил-1-оксо-2-(фенилметил)-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-4-карбоксамид.
4.1. (+)-6-фтор-9-метил-1-оксо-2-(фенилметил)-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-4-карбоновая кислота.
Раствор 20,5 г (58 ммоль) (±)-6-фтор-9-метил-1-оксо-2-(фенилметил)-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b] индол-4-карбоновой кислоты и 7,5 мл (58 ммоль) (R)-(+)-α-метилбензиламина в 1000 мл метанола перемешивают. Смесь концентрируют при пониженном давлении, остаток разводят в 50 мл этилацетата и 400 мл диэтилового эфира, и осадок отделяют фильтрованием и перекристаллизовывают 3 раза из пропан-2-ола.
Выделяют 7,3 г (15 ммоль) диастереоизомерной соли, которую перерастворяют в 100 мл метанола, добавляют 16 мл 1 н. соляной кислоты и 200 мл воды, и осадок отделяют фильтрованием и сушат при пониженном давлении при комнатной температуре. Получают 5,1 г (15 ммоль) правовращающей кислоты.
Температура плавления: 264-269oС.
[α]
4.2. (+)-6-(фтор-N, N, 9-триметил-1-оксо-2-(фенилметил)-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-4-карбоксамид.
Раствор 0,5 г (1,3 ммоль) (+)-6-фтор-9-метил-1-оксо-2-(фенилметил)-2,3,4,9-тетрагидро-1H-пиридо[3,4-b] индол-4-карбоновой кислоты, 0,11 г (1,3 ммоль) диметиламин гидрохлорида, высушенный заранее при пониженном давлении, и 0,68 г (13 ммоль) (бензотриазол-1-илокси)трис(пирролидино)фосфониумгексафторфосфата в 10 мл дихлорметана, пропущенного заранее через колонку с окисью алюминия, охлаждают до -30oС и добавляют по каплям 0,68 мл (3,9 ммоль) N,N-ди(1-метилэтил)этиламина в растворе 5 мл дихлорметана. Смесь перемешивают в течение 6 часов при температуре между -30oС и -20oС, осуществляют гидролиз при помощи 10 мл 5%-ного водного раствора бисульфата калия, смесь экстрагируют дихлорметаном, органическую фазу промывают и сушат, растворитель выпаривают при пониженном давлении, остаток очищают хроматографией на колонке с силикагелем, осуществляя элюцию смесью дихлорметана и этилацетата, полученный продукт перекристаллизовывают из пропан-2-ола. Получают 0,37 г (1 ммоль) правовращающего амида.
Температура плавления: 199-202oС.
[α]
Пример 5 (соединение 76)
(-)-6-фтор-N, N,9-триметил-1-оксо-2-(фенилметил)-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-4-карбоксамид.
5.1. (-)-6-фтор-9-метпил-1-оксо-2-(фенилметил)-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-4-карбоновая кислота.
Растворитель от различных маточных растворов перекристаллизации диастереоизомера соли из примера 4.1. выпаривают при пониженном давлении, добавляют воду и концентрированную соляную кислоту, и осадок отделяют фильтрованием и сушат при пониженном давлении при комнатной температуре. Получают 13,6 г (38 ммоль) неочищенной левовращающей кислоты, к которой добавляют 250 мл метанола и 4,97 мл (38 ммоль) (S)-(-)-α-метилбензиламина.
Смесь перемешивают и концентрируют при пониженном давлении и осадок собирают фильтрованием и перекристаллизовывают три раза из пропан-2-ола. Получают 7 г (15 ммоль) диастереоизомера соли, которую растворяют в небольшом количестве метанола, добавляют 15 мл 1 н. соляной кислоты и объем удваивают водой. Осадок отделяют фильтрованием, промывают водой, поверхностно высушивают и сушат при комнатной температуре при пониженном давлении. Получают 5 г (14 ммоль) левовращающей кислоты.
Температура плавления: 264-269oС.
[α]
5.2. (-)-6-фтор-N,N,9-триметил-1-оксо-2-(фенилметил)-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-4-карбоксамид.
Получение осуществляют так, как в Примере 4.2., из (-)-6-фтор-9-метил-1-оксо-2-(фенилметил)-2,3,4,9-тетрагидро-1H-пиридо[3,4-b] индол-4-карбоновой кислоты и из диметиламина, получают 0,3 г (0,8 ммоль) левовращающего амина после окончательной перекристаллизации из этилацетата.
Температура плавления: 204-205oС.
[α]
Пример 6 (Соединение 101)
6-фтор-N, N, 9-триметил-1-оксо-2-фенил-2,9-дегидро-1Н-пиридо[3,4-b] индол-4-карбоксамид.
6.1. Этил 2-(этоксикарбонил)-5-фтор-α-метилен-1Н-индол-3-ацетат.
Раствор 37,2 г (180 ммоль) этил 5-фтор-1H-индол-2-карбоксилата, 25,8 г (222 ммоль) этилпирувата и 31 мл концентрированной серной кислоты в 400 мл уксусной кислоты перемешивают в течение 20 часов.
Растворитель выпаривают при пониженном давлении, остаток разводят в воде и этилацетате, органическую фазу отделяют, промывают разбавленным нашатырным спиртом, а затем насыщенным водным раствором хлористого натрия и сушат над сульфатом натрия, и растворитель выпаривают при пониженном давлении.
Получают 37,1 г (122 ммоль) твердого продукта, который используют на следующей стадии.
6.2. 6-фтор-1-оксо-2-фенил-2,3,4,9-тетрагидро-1Н-пиридо [3,4-b]индол-4-карбоновая кислота.
Смесь 25 г (82 ммоль) этил 2-(этоксикарбонил)-5-фтор-α-метилен-1H-индол-3-ацетата и 31,8 г (342 ммоль) анилина нагревают с обратным холодильником в течение 17 часов. Добавляют раствор разбавленной кислоты и этилацетат, органическую фазу отделяют, промывают водой и сушат над сульфатом натрия, и растворитель выпаривают при пониженном давлении. Получают 30 г остатка, который гидролизуют раствором 43 мл 30%-ного гидрида натрия в 400 мл этанола с обратным холодильником в течение 1 часа. Смесь концентрируют при пониженном давлении, добавляют воду, смесь промывают этилацетатом и дихлорметаном, водную фазу подкисляют концентрированной соляной кислотой и осадок собирают фильтрованием и сушат при пониженном давлении.
Получают 18,5 г (57 ммоль) твердого вещества, которое используют на следующей стадии.
6.3. б-фтор-N,N,9-триметил-1-оксо-2-фенил-2,9-дигидро-1Н-пиридо[3,4-b] индол-4-карбоксамид.
5 г (15 ммоль) 6-фтор-1-оксо-2-фенил-2,3,4,9-тетрагидро-1H-пиридо[3,4-b] индол-4-карбоновой кислоты в 40 мл тионилхлориде нагревают с обратным холодильником в течение 1 часа. Растворитель выпаривают при пониженном давлении, остаток разводят в дихлорметане, добавляют избыток разжиженного диметиламина, смесь перемешивают в течение нескольких часов, добавляют воду и осадок отделяют фильтрованием и сушат при пониженном давлении.
Получают 3,4 г (9 ммоль) соединения.
2,5 г его растворяют в 50 мл диметилсульфоксида, добавляют 0,6 г порошкообразного гидроксида калия и 1,2 мл йодистого этила, смесь перемешивают в течение 5 часов при 50oС и затем в течение 20 часов при комнатной температуре.
Добавляют разбавленную соляную кислоту, осуществляют экстракцию этилацетатом, органическую фазу сушат над сульфатом натрия, растворитель выпаривают при пониженном давлении и остаток очищают хроматографией на колонке с силикагелем, осуществляя элюцию смесью циклогексана и этилацетата.
Получают 2,2 г смеси, содержащей 6-фтор-N,N,9-триметил-1-оксо-2-фенил-2,9-дигидро-1H-пиридо[3,4-b] индол-4-карбоксамида и 6-фтор-N,N,9-триметил-1-оксо-2-фенил-2,3,4,9-тетрагидро-1H-пиридо[3,4-b]индол-4-карбоксамида.
Эту смесь перемешивают с 1 г 2,3-дихлора-5,6-дициано-1,4-бензохинона в течение 20 часов и промывают насыщенным водным раствором бикарбоната натрия, органическую фазу отделяют и сушат над сульфатом натрия, растворитель выпаривают при пониженном давлении, остаток очищают хроматографией на колонке с силикагелем, осуществляя элюцию смесью цилкогексана и этилацетата, и продукт перекристаллизовывают из этилацетата.
Получают 0,5 г (1,5 ммоль) соединения.
Температура плавления: 195-197oС.
Пример 7 (Соединение 97)
N, N, 9-триметил-1-оксо-2-фенил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b] индол-4-карбоксамид.
7.1. Этил 2-(этоксикарбонил)-α-метилен-1Н-индол-3-ацетат.
Раствор этанола, насыщенный газообразной соляной кислотой, содержащий 39,1 г (207 ммоль) этил 1H-индол-2-карбоксилата и 45,3 мл (410 ммоль) этилпирувата, доводят до приблизительно 60oС в течение 3 часов. Смесь концентрируют при пониженном давлении и остаток разводят в диэтиловом эфире. Органическую фазу промывают водой и сушат над сульфатом натрия. Растворитель выпаривают при пониженном давлении и остаток кристаллизуют из циклогексана. Получают 45,4 г (158 ммоль) твердого продукта, который используют на следующей стадии.
7.2. 1-оксо-N, 2-дифенил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b] индол-4-карбоксамид.
Смесь 32 г (111 ммоль) этил 2-(этоксикарбонил)-α-метилен-1Н-индол-3-ацетата и 47,5 г (511 ммоль) анилина нагревают с обратным холодильником в течение 13 часов. Добавляют дихлорметан и органическую фазу промывают 1 н. соляной кислотой. Ее сушат над сульфатом натрия и выпаривают растворитель при пониженном давлении. Получают 37,8 г неочищенного продукта, который используют на следующей стадии.
7.3. Этил 1-оксо-2-фенил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b] индол-4-карбоксилат.
Раствор 38,8 г неочищенного 1-оксо-N,2-дифенил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b] индол-4-карбоксамида в смеси этанола, воды и 37%-ной соляной кислоты нагревают с обратным холодильником в течение 8 часов. Реакционную смесь нейтрализуют концентрированным гидроксидом натрия и осуществляют экстракцию этилацетатом. Органическую фазу сушат над сульфатом натрия и выпаривают при пониженном давлении. Остаток очищают хроматографией на колонке с силикагелем, осуществляя элюцию смесью дихлорметана и этилацетата. Получают 22,6 г (68 ммоль) продукта, который используют на следующей стадии.
7.4. 1-оксо-2-фенил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b] индол-4-карбоновая кислота.
22,6 г (68 ммоль) этил 1-оксо-2-фенил-2,3,4,9-тетрагидро-1H-пиридо[3,4-b] индол-4-карбоксилата гидролизуют 200 мл 1 н. гидроксида натрия в 500 мл метанола. Реакционную смесь подкисляют 1 н. соляной кислотой и осадок отфильтровывают. Его сушат при пониженном давлении. Получают 18,1 г (59 ммоль) твердого продукта, который используют на следующей стадии.
7.5. N,N-диметил-1-оксо-2-фенил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-4-карбоксамид.
Раствор 10 г (33 ммоль) 1-оксо-2-фенил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b] индол-4-карбоновой кислоты в 30 мл тионилхлорида нагревают с обратным холодильником в течение 4 часов. Растворитель выпаривают при пониженном давлении, остаток разводят в дихлорметане и добавляют избыток разжиженного диметиламина. Смесь перемешивают в течение нескольких часов и растворитель выпаривают при пониженном давлении. Добавляют воду и этилацетат. Осадок отфильтровывают и сушат при пониженном давлении. Получают 8,2 г неочищенного продукта, который используют на следующей стадии.
7.6. N,N,9-триметил-1-оксо-2-фенил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b] индол-4-карбоксамид.
Смесь 1,3 г (23 ммоль) порошкообразного гидроксида калия и 6 г неочищенного N, N-диметил-1-оксо-2-фенил-2,3,4,9-тетрагидро-1H-пиридо[3,4-b] индол-4-карбоксамида в 60 мл диметилсульфоксида нагревают при 40oС в течение 30 минут. Добавляют 2,5 мл (40 ммоль) йодистого метила и реакционную смесь перемешивают в течение 5 часов при комнатной температуре.
Затем добавляют воду и дихлорметан. Органическую фазу сушат над сульфатом натрия и выпаривают при пониженном давлении. Остаток очищают хроматографией на колонке с силикагелем, осуществляя элюцию смесью дихлорметана и этилацетата. Продукт перекристаллизовывают из этилацетата. Получают 1,9 г (5,5 ммоль) продукта.
Температура плавления: 196-197oС.
Пример 8 (Соединение 123)
N-этил-6-фтор-N, 9-диметил-1-оксо-2-фенил-2,9-дигидро-1Н-пиридо[3,4-b] индол-4-карбоксамид.
8.1. Этил 2-(этоксикарбонил)-5-фтор-1Н-индол-3-ацетат.
Раствор 7,4 г (185 ммоль) гидроксида натрия в 75 мл воды добавляют к раствору 30 г (185 ммоль) 4-фторфенилгидрозингидрохлорида в 300 мл воды, смесь перемешивают в течение 15 минут и затем добавляют раствор 29 г (198 ммоль) кетоглутаровой кислоты в 60 мл воды. Реакционную смесь перемешивают при комнатной температуре в течение 3 часов и экстрагируют этилацетатом, и органическую фазу промывают водой, сушат над сульфатом натрия и выпаривают при пониженном давлении. Получают 42 г (144 ммоль) продукта, который растворяют в 420 мл этанола, насыщенного газообразной соляной кислотой, и нагревают с обратным холодильником в течение 4 часов.
Реакционную смесь концентрируют при пониженном давлении, остаток разводят в этилацетате и органическую фазу промывают нормальным гидроксидом натрия и затем водой, сушат над сульфатом магния и выпаривают при пониженном давлении. Получают 42 г (143 ммоль) твердого продукта, который используют на следующей стадии.
8.2. Этил 2-(этоксикарбонил)-5-фтор-1-метил-1Н-индол-3-ацетат.
Раствор 7,36 г (184 ммоль) 60%-ного гидрида натрия, промытого заранее петролейным эфиром, и 45 г (153 ммоль) этил 2-(этоксикарбонил)-5-фтор-1H-индол-3-ацетата в 450 мл N,N-диметилформамида перемешивают в течение 2 часов при комнатной температуре и затем добавляют раствор 19 мл (306 ммоль) йодистого этила в 100 мл N,N-диметилформамиде. После перемешивания в течение 20 часов реакционную смесь выливают на ледяную воду, осуществляют экстракцию диэтиловым эфиром, органическую фазу промывают водой и сушат над сульфатом магния, и растворитель выпаривают при пониженном давлении. Получают 44,3 г (144 ммоль) продукта, который используют на следующей стадии.
8.3. Этил α-(диметиламинометилиден)-2-(этоксикарбонил)-5-фтор-1-метил-1H-индол-3-ацетат.
Раствор 44,3 г (144 ммоль) этил 2-(этоксикарбонил)-5-фтор-1-метил-1H-индол-3-ацетата и 57,4 мл диметилацеталя диметилформамида в 450 мл N,N-диметилформамида нагревают с обратным холодильником в течение 50 часов. Растворитель выпаривают при пониженном давлении и остаток разводят в диэтиловом эфире. Нерастворимый материал удаляют фильтрованием и растворитель концентрируют при пониженном давлении. Получают 49,3 г (136 ммоль) твердого продукта, который используют на следующей стадии.
8.4. Этил 6-фтор-9-метил-1-оксо-2-фенил-2,9-дигидро-1Н-пиридо[3,4-b]индол-4-карбоксилат.
Раствор 16,3 г (45 ммоль) этил α-(диметиламинометилиден)-2-(этоксикарбонил)-5-фтор-1-метил-1H-индол-3-ацетата, 4,64 мл (50 ммоль) анилина и 1,6 г (8 ммоль) моногидрат 4-метилбензолсульфоновой кислоты в 160 мл N, N-диметилформамида нагревают с обратным холодильником в течение 24 часов. 3,3 г (31 ммоль) карбоната натрия добавляют небольшими порциями и продолжают нагревать с обратным холодильником в течение 2 часов. Раствор охлаждают и выливают на ледяную воду. Осуществляют экстракцию этилацетатом и органическую фазу промывают водой, сушат над сульфатом магния и выпаривают при пониженном давлении. Остаток очищают хроматографией на колонке с силикагелем, осуществляя элюцию смесью дихлорметана и этилацетата. Получают 10,9 г (30 ммоль) продукта, который используют на следующей стадии.
8.5. 6-фтор-9-метил-1-оксо-2-фенил-2,9-дигидро- 1Н-пиридо[3,4-b]индол-4-карбоновая кислота.
Раствор 22,8 г (65 ммоль) этил 6-фтор-9-метил-1-оксо-2-фенил-2,9-дигидро-1H-пиридо[3,4-b] индол-4-карбоксилата и 7,46 г (186 ммоль) гидроксида натрия в смеси 1 л этанола и 100 мл воды нагревают с обратным холодильником в течение 3 часов. Смесь концентрируют при пониженном давлении, остаток разводят водой и водную фазу промывают этилацетатом.
Осуществляют подкисление концентрированной соляной кислотой до рН 1 и отфильтровывают продукт, который промывают несколько раз водой. Его сушат при пониженном давлении. Получают 20,4 г (64 ммоль) твердого вещества, которое используют на следующей стадии.
8.6. 6-фтор-N,9-диметил-1-оксо-2-фенил-2,9-дигидро-1Н-пиридо[3,4-b]индол-4-карбоксамид.
Раствор 5 г (15,6 ммоль) 6-фтор-9-метил-1-оксо-2-фенил-2,9-дигидро-1H-пиридо[3,4-b] индол-4-карбоновой кислоты и 4,8 г (30 ммоль) N,N'-карбонилдиимидазола в 100 мл N,N-диметилформамида перемешивают при 60oС в течение 4 часов. Добавляют избыток разжиженного метиламина при комнатной температуре и реакционную смесь перемешивают в течение 20 часов. Раствор выливают на ледяную воду и осадок собирают фильтрованием, промывают насыщенным раствором бикарбоната натрия, водой и затем этилацетатом и сушат при пониженном давлении. Выделяют 3,5 г неочищенного продукта. Фильтраты объединяют и экстрагируют дихлорметаном. Органическую фазу промывают раствором бикарбоната натрия и затем водой и сушат над сульфатом натрия, растворитель выпаривают при пониженном давлении. Выделяют 1,5 г дополнительного продукта. Две партии объединяют и очищают хроматографией на колонке с силикагелем, осуществляя элюцию смесью дихлорметана и этилацетата. Получают 4,7 г (13,5 ммоль) твердого продукта.
8.7. N-этил-6-фтоp-N, 9-диметил-1-оксо-2-фенил-2,9-дигидро-1Н-пиридо[3,4-Ь]индол-4-карбоксамид.
Раствор 2,5 г (7,1 ммоль) 6-фтор-N,9-диметил-1-оксо-2-фенил-2,9-дигидро-1Н-пиридо[3,4-b] индол-4-карбоксамида и 0,36 г (9 ммоль) 60%-ного гидрида натрия, заранее промытого петролейным эфиром, перемешивают в течение 3 часов при 50oС. Добавляют 1,67 мл (21 ммоль) йодистого этила, перемешивание поддерживают в течение 20 часов, и раствор вливают в ледяную воду и экстрагируют этилацетатом. Органическую фазу промывают водой, сушат над сульфатом натрия и выпаривают при пониженном давлении. Остаток очищают хроматографией на кремнеземе, осуществляя элюцию смесью дихлорметана и этилацетата. Продукт перекристаллизовывают из пропан-2-ола.
Получают 2,3 г твердого вещества.
Температура плавления: 181-182oС.
Пример 9 (соединение 98)
6-фтор-9-метил-2-фенил-4-(пирролидин-1-илкарбонил)-2,9-дигидро-1Н-пиридо[3,4-b]индол-1-он.
Раствор 3,2 г (10 ммоль) 6-фтор-9-метил-1-оксо-2-фенил-2,9-дигидро-1H-пиридо[3,4-b] индол-4-карбоновой кислоты и 3,2 г (20 ммоль) N,N'-карбонилдиимидозола в 65 мл N,N-диметилформамида перемешивают в течение 4-х часов при 60oС. 2,5 мл (30 ммоль) пирролидина добавляют при комнатной температуре и реакционную смесь перемешивают в течение 20 часов.
Раствор выливают на ледяную воду и экстрагируют этилацетатом. Органическую фазу промывают водой и сушат над сульфатом магния, и растворитель выпаривают при пониженном давлении. Остаток очищают хроматографией на колонке с силикагелем, осуществляя элюцию смесью дихлорметана и этилацетата. Продукт перекристаллизовывают из пропан-2-ола.
Получают 3 г (7,7 ммоль) твердого вещества.
Температура плавления: 203-205oС.
Пример 10 (соединение 122)
6-фтор-N, N, 9-триметил-1-оксо-2-(пиридин-2-ил)-2,9-дигидро-1Н-пиридо[3,4-b]индол-4-карбоксамид.
10.1. Метил 6-фтор-9-метил-1-оксо-2-(пиридин-2-ил)-2,9-дигидро-1Н-пиридо[3,4-b]индол-4-карбоксилат.
Смесь 4,1 г (12,2 ммоль) метил α-(диметиламинометилиден)-5-фтор-2-(метоксикарбонил)-1-метил-1H-индол-3-ацетата и 1,73 г (18,4 ммоль) 2-аминопиридина нагревают при 180oС в течение 30 минут. Добавляют 7 мл N,N-диметилформамида и нагревание продолжают в течение 4-х часов.
Реакционную смесь охлаждают и вливают в смесь воды и этилацетата, и нерастворимый материал собирают фильтрованием и сушат при пониженном давлении. Получают 1,8 г (5,1 ммоль) твердого вещества, которое используют на следующей стадии.
10.2. 6-фтор-9-метил-1-оксо-2-(пиридин-2-ил)-2,9-дигидро-1Н-пиридо[3,4-b]индол-4-карбоновая кислота.
Раствор 3,3 г (9,4 ммоль) метил 6-фтор-9-метил-1-оксо-2-(пиридин-2-ил)-2,9-дигидро-1H-пиридо[3,4-b] индол-4-карбоксилата в смеси с 100 мл этанола и 28 мл 1 н. гидроксида натрия нагревают с обратным холодильником в течение 4-х часов. Смесь концентрируют при пониженном давлении, добавляют воду, подкисление осуществляют концентрированной соляной кислотой и осадок собирают фильтрованием. Его промывают водой и сушат при пониженном давлении. Получают 2,8 г (8,3 ммоль) твердого продукта, который используют на следующей стадии.
10.3. 6-фтор-N,N,9-триметил-1-оксо-2-(пиридин-2-ил)-2,9-дигидро-1Н-пиридо[3,4-b]индол-4-карбоксамид.
Раствор 2,5 г (7,4 ммоль) 6-фтор-9-метил-1-оксо-2-(пиридин-2-ил)-2,9-дигидро-1H-пиридо[3,4-b] индол-4-карбоновой кислоты и 2,4 г (14,8 ммоль) N, N'-карбонилдиимидозола в 65 мл N,N-диметилформамида перемешивают в течение 4-х часов. Реакционную смесь охлаждают и добавляют избыток разжиженного диметиламина. Смесь перемешивают в течение 48 часов при комнатной температуре, выливают на воду и экстрагируют этилацетатом. Органическую фазу промывают водой и сушат над сульфатом магния, и растворитель выпаривают при пониженном давлении. Остаток очищают хроматографией на колонке с силикагелем, осуществляя элюцию смесью дихлорметана и этилацетата. Продукт перекристаллизовывают из этилацетата.
Получают 1,6 г (4,4 ммоль) твердого вещества.
Температура плавления: 220-221oС.
Пример 11 (соединение 125)
6-фтор-9-метил-2-(5-метил-1,3,4-тиадиазол-2-ил)-4-(пирролидин-1-илкарбонил)-2,9-дигидро-1Н-пиридо[3,4-b]индол-1-он.
11.1. 2-карбокси-5-фтор-1Н-индол-3-уксусная кислота.
Раствор 26,5 г (103 ммоль) этил 2-(этоксикарбонил)-5-фтор-1H-индол-3-ацетата и 24 г гидроксида натрия в смеси 530 мл этанола и 100 мл воды нагревают с обратным холодильником. Смесь концентрируют при пониженном давлении, добавляют воду, и водную фазу промывают этилацетатом и подкисляют концентрированной соляной кислотой. Осадок отфильтровывают, промывают водой и сушат при пониженном давлении. Получают 23,4 г (98,7 ммоль) продукта, который используют на следующей стадии.
11.2. 6-фтор-1,3,4,9-тетрагидропирано[3,4-b]-индол-1,3-дион.
Раствор 4,7 г (19,8 ммоль) 2-карбокси-5-фтор-1H-индол-3-уксусной кислоты в 94 мл ацетилхлориде нагревают с обратным холодильником в течение 5 часов. Смесь концентрируют при пониженном давлении, добавляют толуол и растворитель выпаривают при пониженном давлении. Получают 4,5 г твердого продукта, который используют на следующей стадии.
11.3. 5-фтop-3-(2-oкco-2-(пиppoлидин-1-ил)этил)-1H-индoл-2-карбоновая кислота.
Раствор 4,4 г (20 ммоль) 6-фтор-1,3,4,9-тетрагидропирано[3,4-b]-индол-1,3-диона и 8,3 мл (100 ммоль) пирролидина в 100 мл дихлорметана перемешивают в течение 24 часов при комнатной температуре. Смесь концентрируют при пониженном давлении, добавляют воду и водную фазу промывают этилацетатом. Подкисление осуществляют концентрированной соляной кислотой и добавляют этилацетат. Осадок собирают фильтрованием, промытый водой и высушенный при пониженном давлении. Получают 5 г (17,2 ммоль) твердого продукта, который используют на следующей стадии.
11.4. Метил 5-фтор-3-(2-оксо-2-(пирролидин-1-ил)этил)-1Н-индол-2-карбоксилат.
3,8 мл (51 ммоль) тионилхлорида добавляют по каплям к раствору 5 г (17,2 ммоль) 5-фтор-3-(2-оксо-2-(пирролидин-1-ил)этил)-1H-индол-2-карбоновой кислоты в 50 мл метанола, охлажденной на ледяной бане и затем смесь нагревают с обратным холодильником в течение 3-х часов. Смесь концентрируют при пониженном давлении, добавляют воду и дихлорметан, и органическую фазу промывают водой, сушат над сульфатом магния и выпаривают при пониженном давлении. Получают 4,5 г (14 ммоль) продукта, который используют на следующей стадии.
11.5. Метил 3-(1-диметиламинометилиден-2-оксо-2-(пирролидин-1-ил)этил)-5-фтор-1-метил-1Н-индол-2-карбоксилат.
Раствор 3,4 г метил 5-фтор-3-(2-оксо-2-(пирролидин-1-ил)этил)-1H-индол-2-карбоксилата и 4,66 мл (35 ммоль) диметилацеталя диметилформамида в 34 мл N,N-диметилформамида нагревают с обратным холодильником в течение 30 часов. Смесь концентрируют при пониженном давлении, добавляют ксилол и растворитель выпаривают при пониженном давлении. Получают 4 г остатка, который содержит приблизительно 50% требуемого продукта (согласно спектру протонного магнитного резонанса) и который используют на следующей стадии.
11.6. 6-фтор-9-метил-2-(5-метил-1,3,4-тиадиазол-2-ил)-4-(пирролидин-1-илкарбонил)-2,9-дигидро-1Н-пиридо[3,4-b] индол-1-он.
Раствор 4 г остатка, полученного на предыдущей стадии, и 1,04 г (5,5 ммоль) моногидрата 4-метилбензолсульфоновой кислоты в 40 мл N,N-диметилформамида перемешивают в течение 15 минут. Добавляют 0,65 г (6,4 ммоль) 2-амино-5-метил-1,3,4-тиадиазола и смесь нагревают с обратным холодильником в течение 24 часов. Смесь выливают на воду и этилацетат.Осадок собирают фильтрованием, промытый водой, высушенный при пониженном давлении и перекристаллизованный из N,N-диметилформамида.
Получают 1 г (2,4 ммоль) продукта.
Температура плавления: 299-301oС.
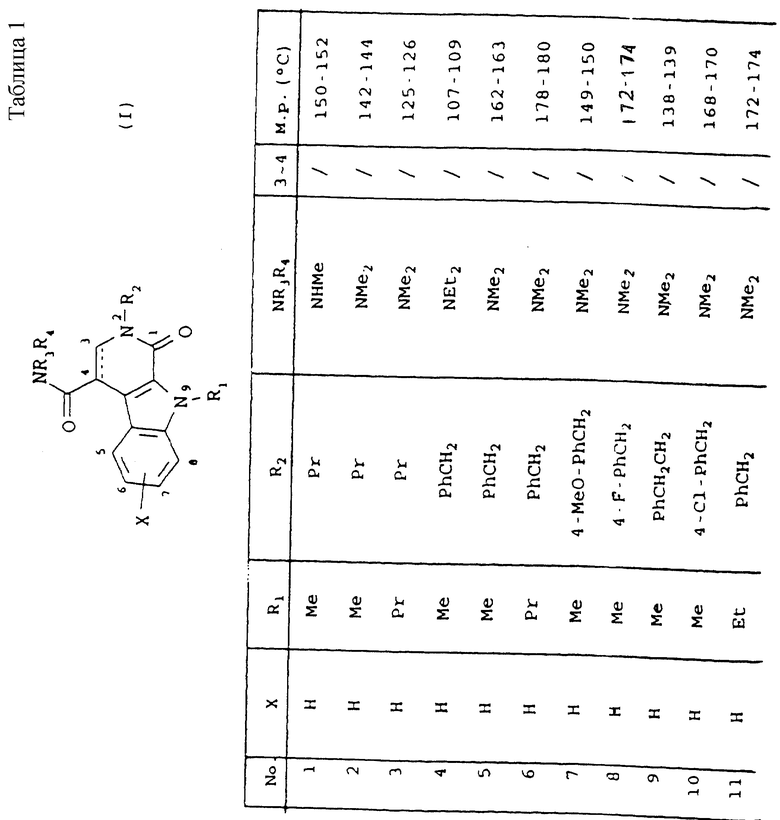
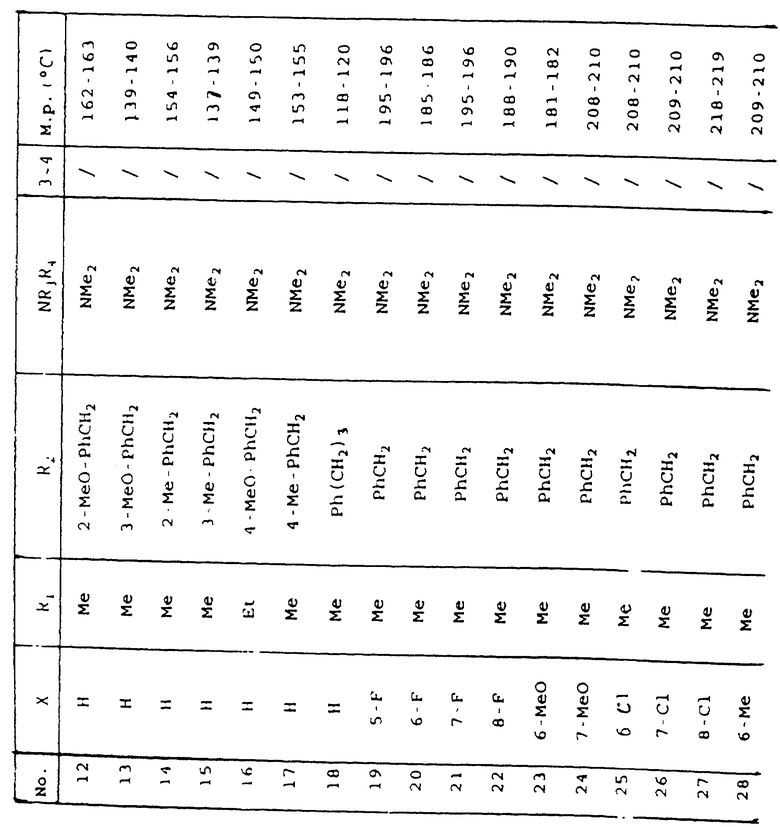
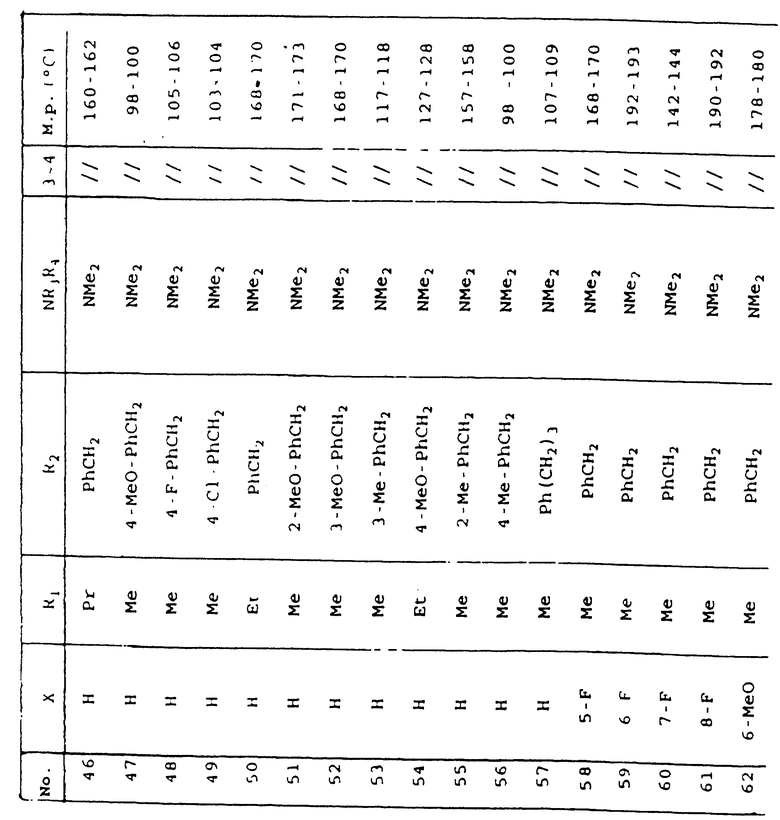
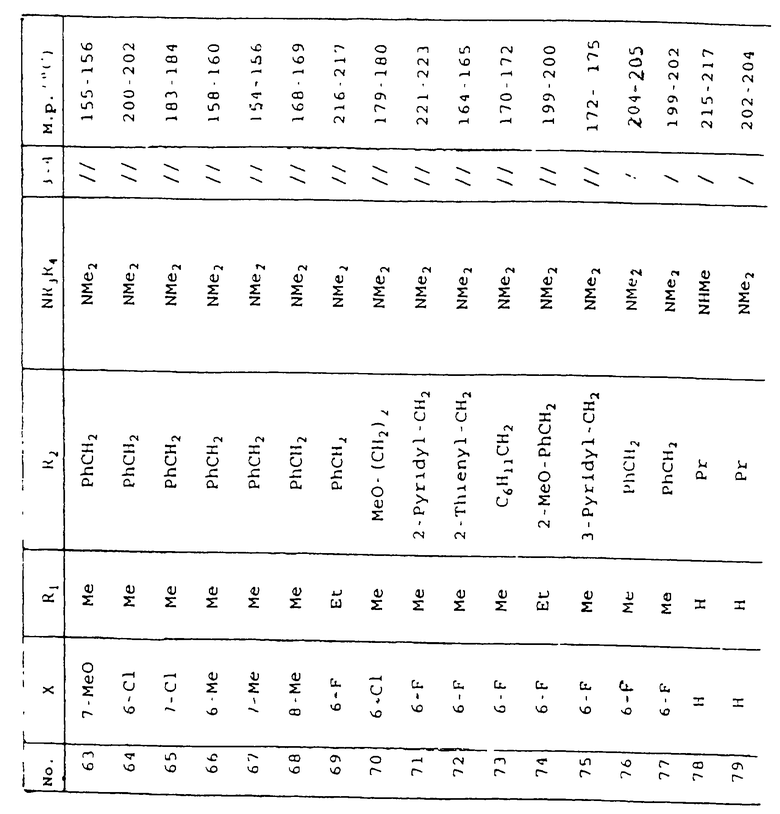
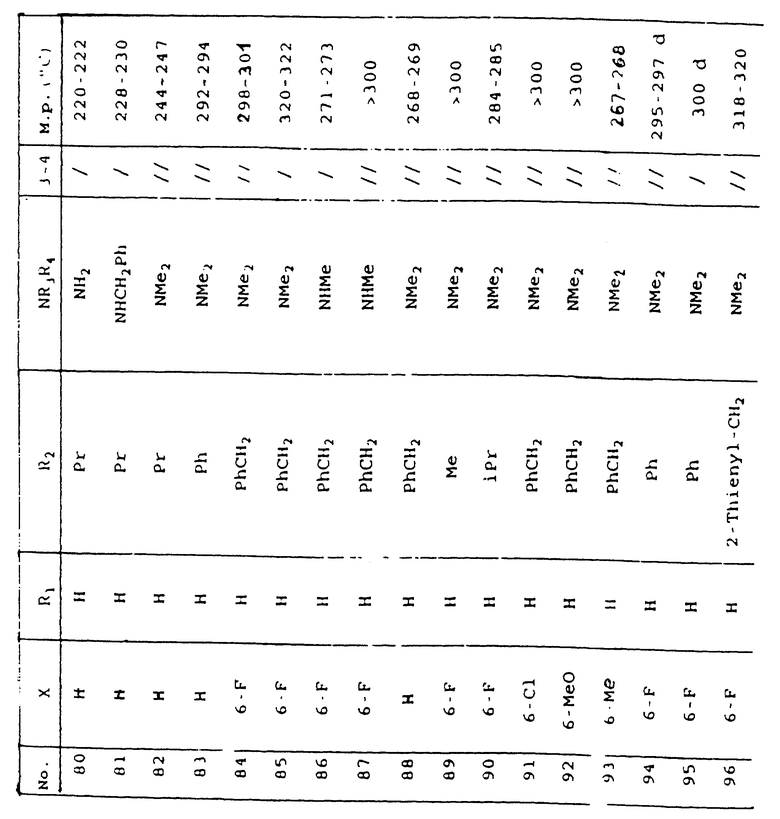
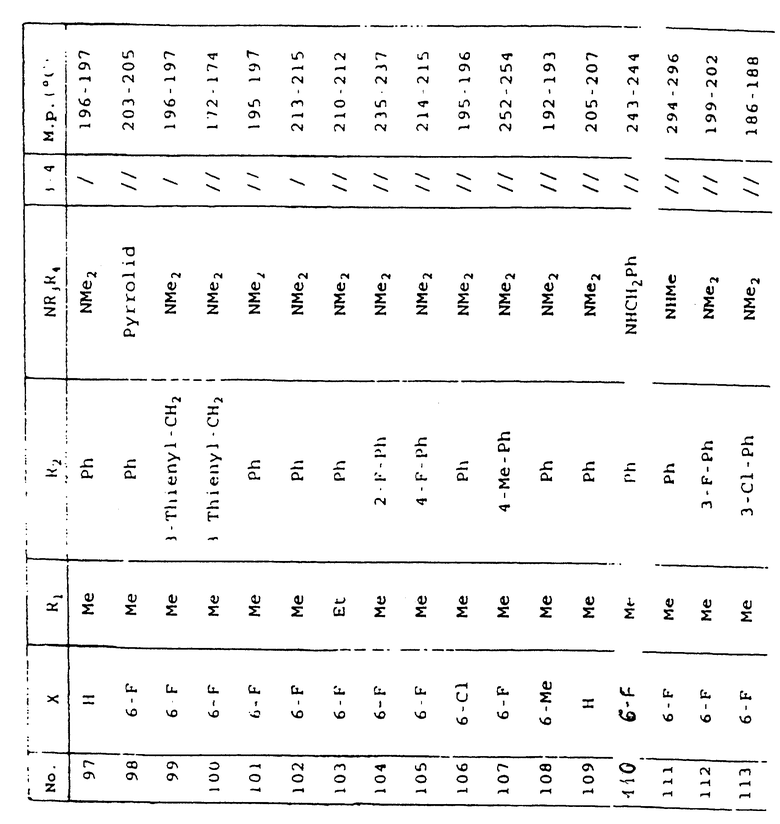
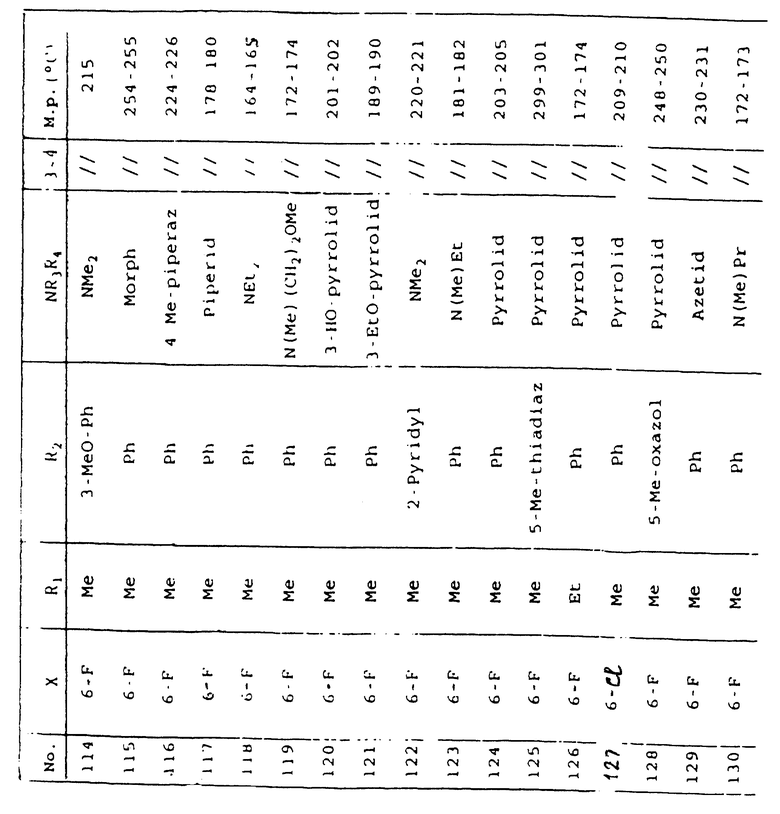
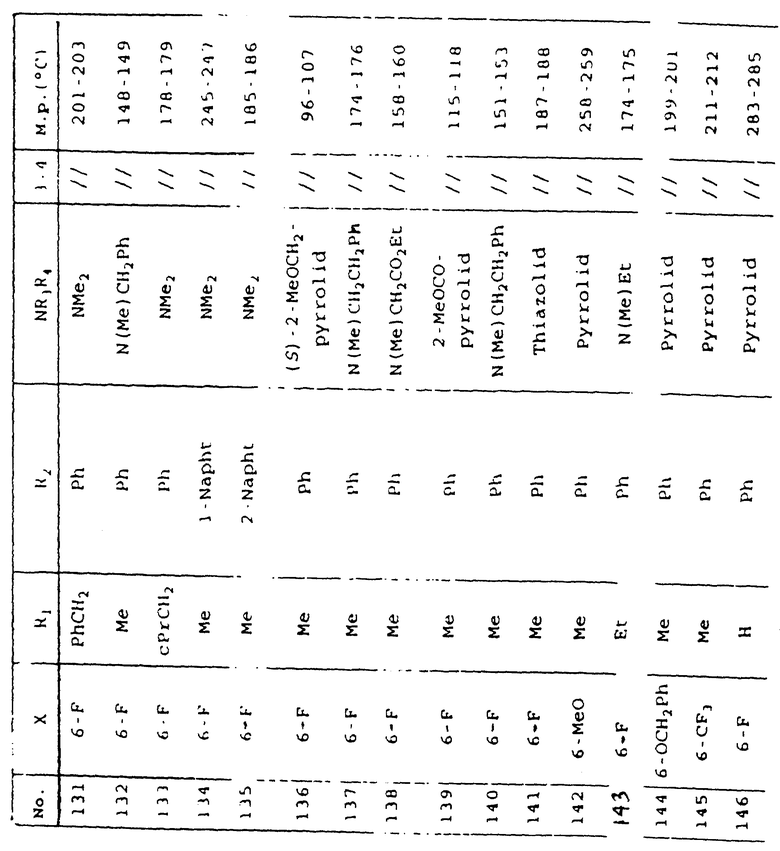
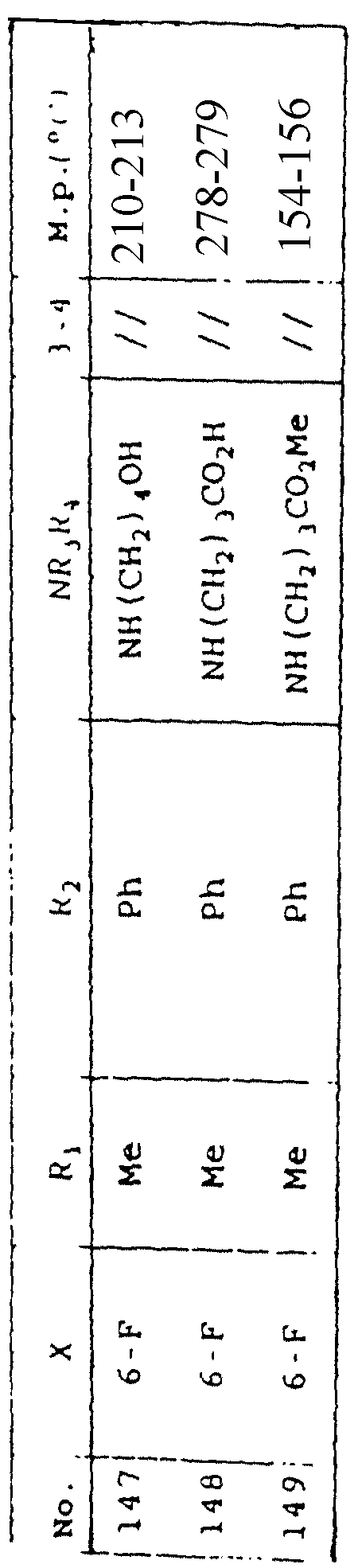
Таблица 1 иллюстрирует химические структуры и физические свойства нескольких соединений по изобретению.
Условные обозначения
Me означает метильную группу, Et означает этильную группу, Рr означает пропильную группу, iPr означает изопропильную группу, сРr означает циклопропильную группу, Рh означает фенильную группу, 1-Napht и 2-Napht соответственно означают нафт-1-ильную группу и нафт-2-ильную группу, x-Pyridyl означает перидин-х-ильную группу, x-Thienyl означает тиен-х-ильную группу, Piperid означает пиперидинильную группу, Pyrrolid означает пирролидинильную группу, Моrрh означает морфолин-4-ильную группу, Azetid означает азетидин-1-ильную группу, 4-Me-piperaz означает 4-метилпиперазин-1-ильную группу, 5-Me-thiadiaz означает 5-метил-1,3,4-тиадиазол-2-ильную группу, 5-Me-oxazol означает 5-метил-1,2-оксазол-2-ильную группу и Thiazolid означает тиазолидинильную группу.
В колонке "3~4", "/" означает углерод-углерод простую связь и "//" означает углерод-углерод двойную связь между 3 и 4 атомами в молекуле.
В колонке "М.р. (oС)", "d" означает температура плавления с разложением.
Соединения по изобретению подвергали фармакологическим тестам, которые демонстрировали их преимущества в качестве веществ, имеющих терапевтические активности.
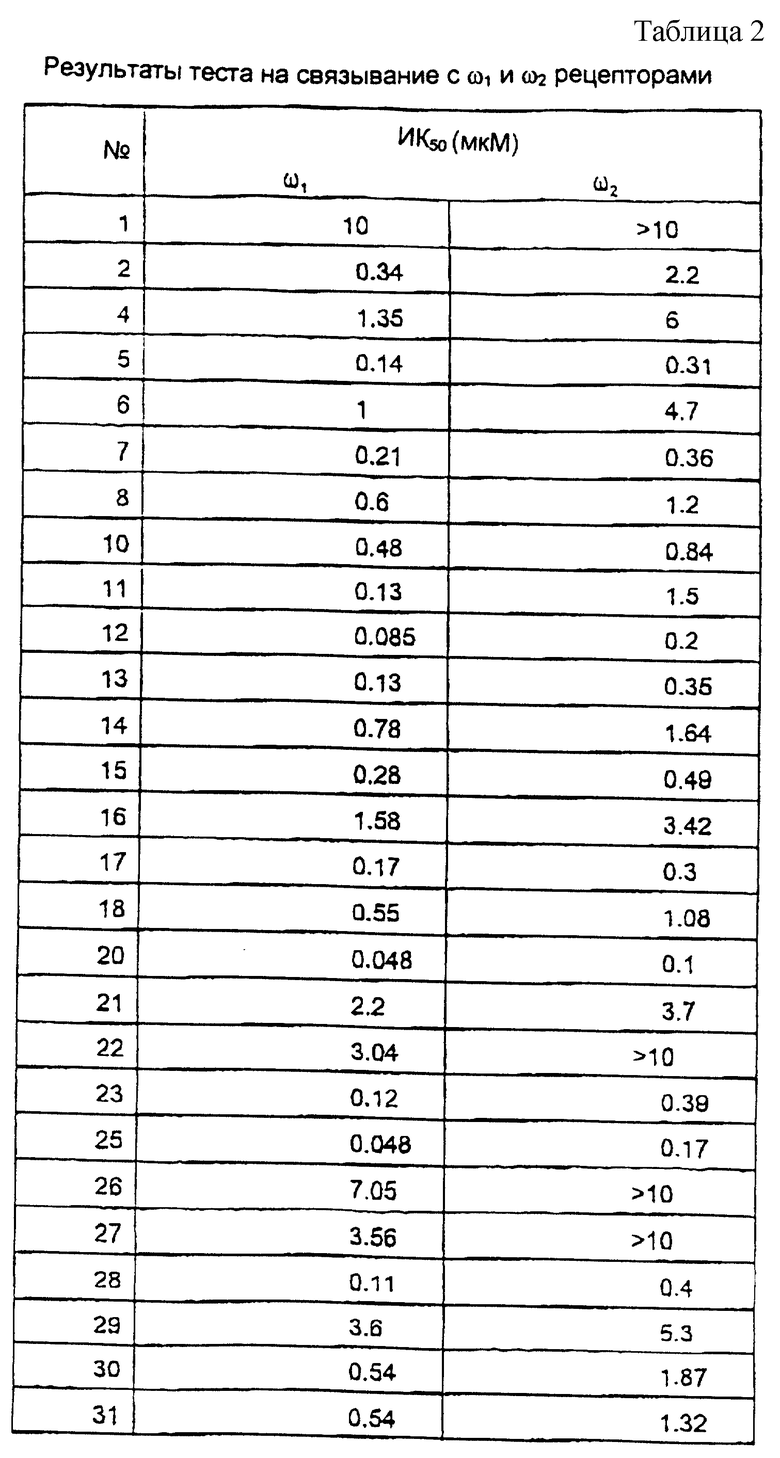
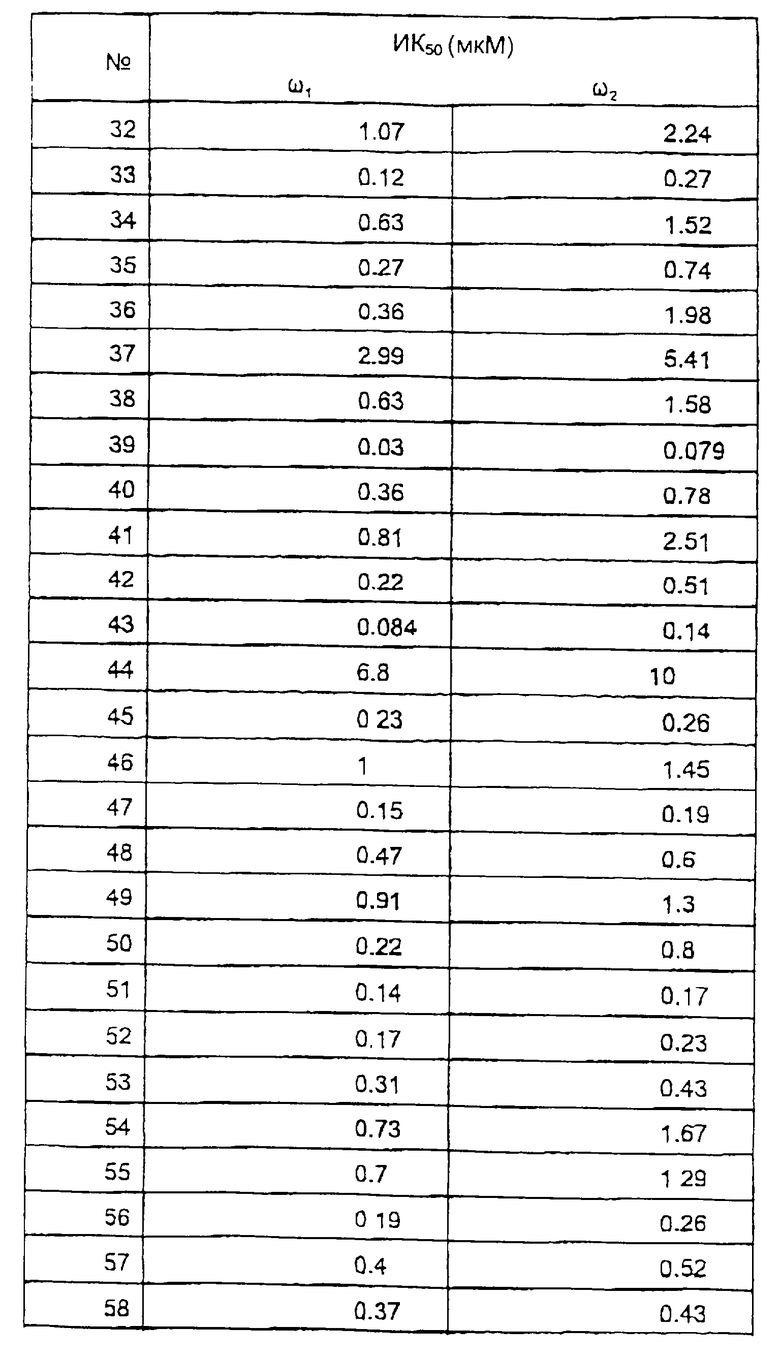
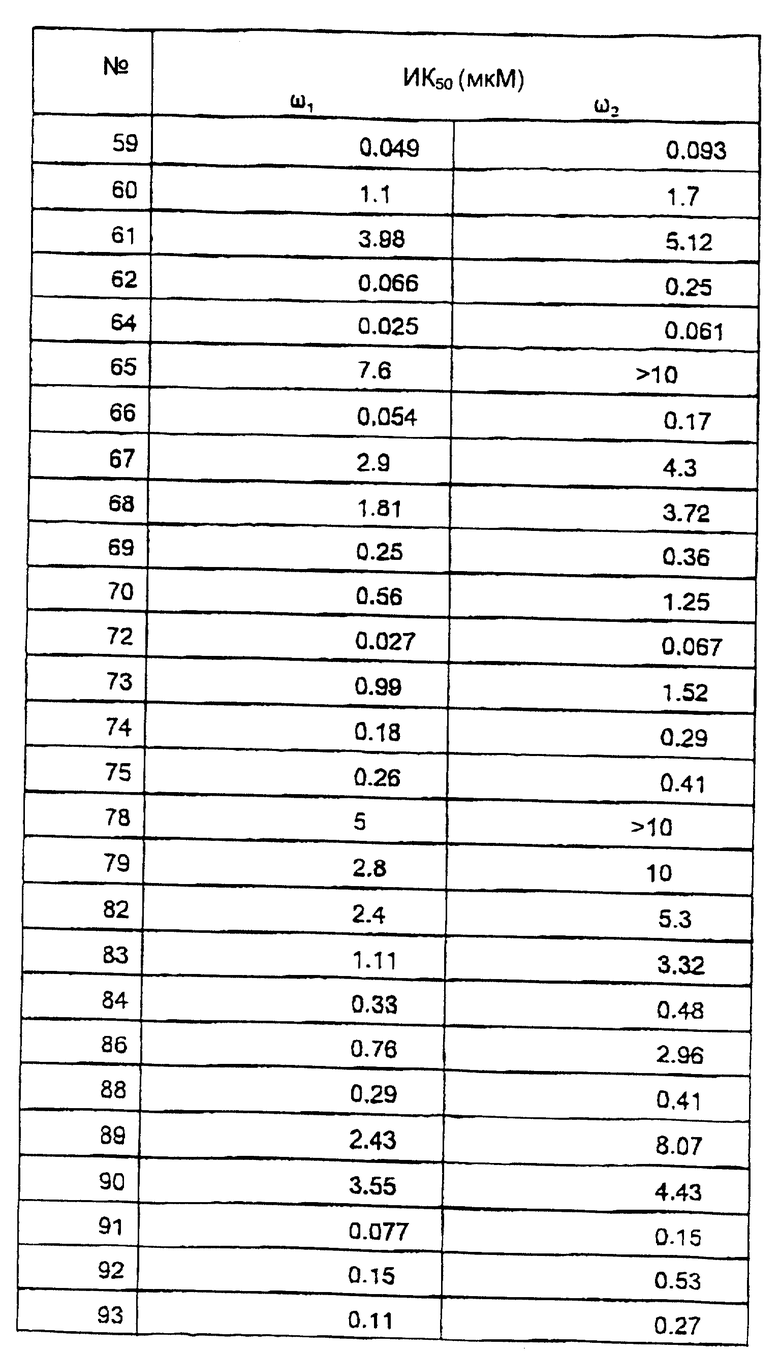
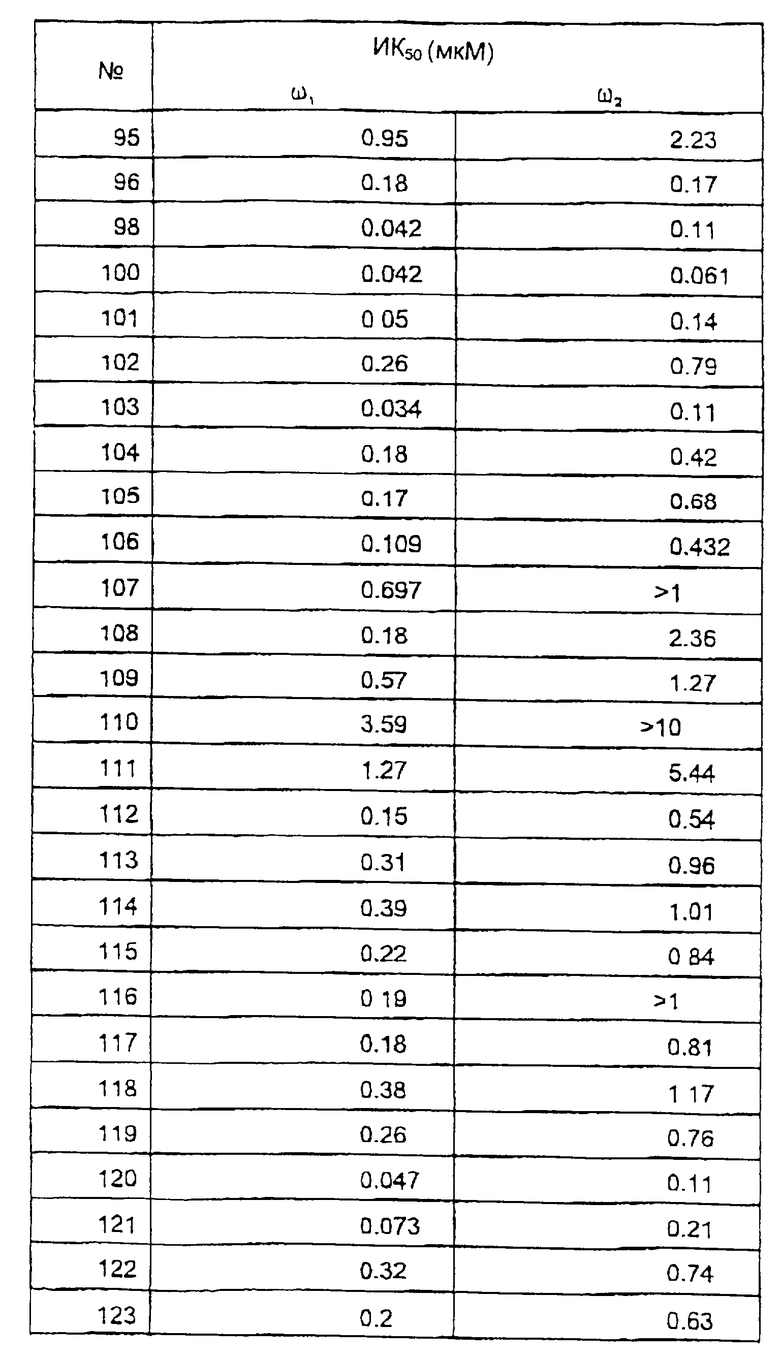
Исследование мембранного связывания в отношении ω1 (бензодиазепин типа 1) и ω2 (бензодиазепин типа 2) рецепторов.
Сродство соединений к ω1 рецепторам мозжечка и ω2 рецепторам спинного мозга определяли согласно варианту способа, описанного С.З. Лангер и С. Арбилла (Fund. Clin. Pharmacol. , 2, 159-170 (1988)), с использованием [3Н] флумазенила вместо [3Н] диазепама в качестве радиоактивно-меченного лиганда.
Ткань мозжечка или спинного мозга гомогенизируют в течение 60 секунд в 120 или в 30 объемах соответственно ледяного буферного раствора (50 мМ трис-HCl, рН 7,4, 120 мМ NaCl и 5 мМ KCl) и затем, после разведения 1/3 суспензию инкубируют с [3H] флумазенилом (удельная активность 78 Ки/ммоль, New England Nuclear) при концентрации 1 нМ и с соединениями по изобретению в различных концентрациях, в конечном объеме 525 мкл. После 30 минут инкубирования при 0oС, образцы фильтруют при пониженном давлении через фильтры (Whatman GF/B®) и немедленно промывают ледяным буферным раствором. Удельное связывание [3Н] флумазенила определяли в присутствии 1 мкМ немеченного диазепама. Данные анализируют стандартным способом и рассчитывают концентрацию ИК50, концентрацию, которая ингибирует на 50% связывание [3Н] флумазенила. В этом тесте величины ИK50 наиболее активных соединений по изобретению находятся между 10 и 1000 нМ.
Исследование анксиолитической активности: конфликтный тест на прием воды.
Анксиолитическую активность оценивают у крыс в конфликтном тесте на прием воды согласно способу, описанному Д. Р. Вогель, Б.Бир и Д.Е. Клоди (Psychopharmacologia (Berl.), 21, 1-7 (1971)).
Крысу, лишенную в течение 48 часов воды, помещали в звуконепроницаемую камеру, оборудованную пипеткой для воды, связанной с анксиометром, который наносит легкий электрический удар каждые 20 лаканий (глотков). Число полученных ударов автоматически подсчитывают в теченние 3 минут, что позволяет оценить анксиолитическую активность испытуемых соединений. Результаты выражают как минимальную эффективную дозу (МЭД), дозу, которая вызывает значительное увеличение числа получаемых ударов по отношению к числу ударов, наблюдаемому у контрольных животных.
В этом тесте величины МЭД наиболее активных соединений находятся между 5 и 50 мг/кг при внутрибрюшинном способе введения.
Исследование анксиолитической активности: тест в лабиринте с повышенным количеством перекрестов.
Условия проведения этого теста являются модификацией тех, которые описаны С. Пеллоу и С. Файл (Pharmacol. Biochem. Behav. (1986), 24, 525-529).
После периода привыкания к экспериментальной комнате, продолжающегося приблизительно 24 часа, крыс помещают по одной на центральную платформу мордой в направлении одного из закрытых ходов и наблюдают в течение 4 минут с использованием видеокамеры. Регистрируют время, проведенное животным в открытом ходе, число входов в закрытые и открытые ходы, число попыток войти в открытые ходы и следуемые за этим реакции избегания и обследования на концах открытых ходов. Результаты, полученные для каждого животного, выражают: 1) в виде процента вхождений в открытые ходы по отношению к общему числу входов в четырех ходах аппарата, 2) в виде процента времени, проведенного в открытом ходе по отношению к общему времени теста (4 минуты), 3) в виде общего числа абортивных попыток, сделанных животным, 4) в виде общего числа обследований.
Испытуемые продукты вводят внутрибрюшинным или пероральным способом в увеличивающихся дозах. Результаты выражают как минимальную эффективную дозу (МЭД), которая вызывает либо значительное увеличение (активность в открытых ходах) либо значительное уменьшение (попыток) по отношению к эффекту, наблюдаемому у контрольных животных.
В этом тесте величины МЭД наиболее активных соединений находятся между 3 и 50 мг/кг при внутрибрюшинном или пероральном способе введения.
Исследования снотворной активности.
Седативную или снотворную активность соединений определяют, наблюдая их действие на электрокортикограмму крыс согласно способу, описанному X. Депортере (Rev. E.E.G. Neurophysuol., 10, 3, 207-214 (1980)) и X. Депортере и М. Декоберт (J. Pharmacol., (Paris), 14, 2, 195-265 (1983)).
Испытуемые продукты вводили внутрибрюшинно в увеличивающихся дозах. Они вызывали сон подопытных при величине дозы, варьирующей от 1 до 30 мг/кг.
Исследование противосудорожной активности: активность в отношении клонических судорог, вызываемых у крыс инъекцией пентетразола.
Условия проведения этого теста являются модификацией теста, описанного Е. А. Свиниярд и Дж.Х. Вудхед (Antiepileptic Drugs, Raven Press, New York, 111-126(1982)).
Испытуемые продукты вводят животным внутрибрюшинно за 30 минут до внутривенной инъекции 20 мг/кг пентетразола. Немедленно после инъекции в течение 5 минут регистрируют число животных, проявляющих клонические судороги.
Результаты выражают в виде АД50, дозы, которая защищает 50% животных, вычисляемой согласно способу Дж.Т. Лихтфилд и Ф. Вилкоксон (J. Pharm. Exp. Ther. (1949), 96, 99-113)) на основе 3 или 4 доз, вводимых группе крыс численностью от 8 до 10.
Значения АД50 наиболее активных соединений находятся в пределах от 0.3 до 30 мг/кг при внутрибрюшинном или пероральном способе введения.
Исследование противосудорожной активности: активность в отношении судорог у мышей, вызываемых изониазидом.
Активность, свойственную соединениям, определяют при помощи латентного времени начала судорог, вызванных подкожным введением изониазида (800 мг/кг) одновременно с внутрибрюшинным введением испытуемого соединения согласно условиям проведения теста, описанных Г. Перро, Е. Море, Д. Санже и Б. Зивкович (Eur. J. Pharmacol., 156, 189-196 (1988)). Результаты выражают в виде АД50, дозы, которая дает 50% максимального эффекта по отношению к контрольным животным, определяемой на основе 3 или 4 доз, вводимых группе мышей численностью от 8 до 10. В этом тесте величины АД50 для соединений по изобретению находятся в пределах от 1 до 50 мг/кг при внутрибрюшинном способе введения и в зависимости от соединения максимальный эффект может достигать не менее 300%. Результаты тестов, проводимых для соединений по изобретению, показывают, что in vitro они вытесняют [3H] флумазенил с его специфических сайтов связывания в мозжечке и спинном мозге; они проявляют смешанное сродство к ω1 и ω2 (бензодиазепины типа 1 и типа 2) сайтам, расположенным в макромолекулярном комплексе  сайты - хлорный канал.
сайты - хлорный канал.
In vivo они ведут себя как полные или частичные агонисты в отношении этих рецепторов.
Они обладают анксиолитическими, снотворными и противосудорожными свойствами и могут, следовательно, быть применены для лечения заболеваний, связанных с нарушениями GABA-эргической передачи, таких как тревога, нарушения сна, эпилепсия, мышечная спастичность, мышечная контрактура, расстройство познавательной способности, синдромы отмены, связанные с алкоголизмом, табаком или наркотиками, и тому подобное.
Они также могут быть использованы для лечения болезни Паркинсона и всех типов экстрапирамидальных синдромов.
Наконец, они могут быть использованы для премедикации и как общие анестетики при осуществлении и/или поддержании анестезии, или как местные анестетики, возможно в комбинации с другими анестетиками и/или мышечными релаксантами и/или анальгетиками.
С этой целью они могут быть представлены в любой фармацевтической лекарственной форме, в комбинации с подходящими эксципиентами, для энтерального или парентерального введения, например в форме таблеток, драже, капсул, включая твердые желатиновые капсулы, растворов или суспензий для глотания, или инъекций, суппозиториев, и тому подобное, содержащих дозу, допускающую ежедневное введение от 1 до 1000 мг активной субстанции.
Результаты исследований представлены в таблице 2.
| название | год | авторы | номер документа |
|---|---|---|---|
| МОДУЛЯТОРЫ ЭСТРОГЕНОВЫХ РЕЦЕПТОРОВ | 2017 |
|
RU2738646C2 |
| ПРОИЗВОДНЫЕ N-(3-АМИНОПРОПИЛ)-N-ФЕНИЛ-5,6,7,8-ТЕТРАГИДРОНАФТАЛИН-2-КАРБОКСАМИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2138479C1 |
| ПРОИЗВОДНЫЕ 4-ОКСО-3,5-ДИГИДРО-4Н-ПИРИДАЗИНО[4,5-B]ИНДОЛ-1-АЦЕТАМИДА, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ В ТЕРАПИИ | 1998 |
|
RU2197490C2 |
| ПРОИЗВОДНЫЕ 3-(2-АМИНОЭТИЛ)-4-[3-(ТРИФТОРМЕТИЛ)БЕНЗОИЛ]-3,4-ДИГИДРО-2H-1,4- БЕНЗОКСАЗИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЙ ИХ ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ | 1995 |
|
RU2136670C1 |
| ПРОИЗВОДНЫЕ 5-(ГИДРОКСИМЕТИЛ)-ОКСАЗОЛИДИН-2-ОНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ЛЕКАРСТВО И ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ | 1996 |
|
RU2164226C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИН-2(1Н)-ОНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ЛЕКАРСТВЕННОЕ СРЕДСТВО И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1996 |
|
RU2167874C2 |
| ПРОИЗВОДНЫЕ 5-АРИЛ-3-(8-АЗАБИЦИКЛО[3.2.1]ОКТ-3-ИЛ)-1,3,4-ОКСАДИАЗОЛ-2(3H)-ОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ЛЕКАРСТВЕННОЕ СРЕДСТВО И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2186066C2 |
| ПРОИЗВОДНЫЕ ГЕТЕРОАРИЛАЛКИЛКАРБАМАТОВ, ИХ ПОЛУЧЕНИЕ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ФЕРМЕНТА FAAH | 2005 |
|
RU2364586C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОПИРИДО[3,4-b]ИНДОЛА, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ РАКА | 2014 |
|
RU2664109C2 |
| ПРОИЗВОДНЫЕ 5-ФЕНИЛ-3-(ПИПЕРИДИН-4-ИЛ)-1,3,4-ОКСАДИАЗОЛ-2(3Н)-ОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 1996 |
|
RU2167160C2 |
Изобретение относится к соединениям общей формулы (I), в которой Х представляет собой водород или галоген или алкильную, алкоксильную, трифторметильную или фенилметоксигруппу, R1 представляет собой водород или алкильную, циклопропильную или фенилметильную группу, R2 представляет собой либо алкильную группу, возможно замещенную, либо фенилалкильную группу, возможно замещенную, либо циклогексилметильную группу, либо тиенилметильную группу, либо пиридинилметильную группу, либо фенильную группу, возможно замещенную, либо пиридинильную группу, либо 5-метил-1,2-оксазолильную группу, либо 5-метил-1,3,4-тиадиазолильную группу, либо нафталинильную группу, R3 и R4 каждый представляет собой водород или алкильную, 2-метоксиэтильную, гидроксиалкильную, карбоксиалкильную, алкоксикарбонилалкильную или фенилалкильную группу, или совместно с атомом азота, который их несет, образуют либо пирролидинильную группу, возможно замещенную, либо пиперидинильную группу, либо морфолинильную группу, либо 4-метилпиперазинильную группу, либо азетидинильную группу, либо тиазолидинильную группу, а связь между атомами углерода в положениях 3 и 4 является простой или двойной. Изобретение применимо в терапии. 3 с. и 8 з.п. ф-лы, 2 табл.
в которой Х представляет собой атом водорода или галогена или (С1-С3)алкильную, (С1-С3)алкоксильную, трифторметильную или фенилметоксигруппу;
R1 представляет собой атом водорода или (С1-С3)алкильную, циклопропильную или фенилметильную группу,
R2 представляет собой либо (С1-С3)алкильную группу, возможно замещенную метоксигруппой, либо фенил(С1-С3)алкильную группу, возможно замещенную по фенильному кольцу атомом галогена или метильной или метоксигруппой, либо циклогексилметильную группу, либо тиенилметильную группу, либо пиридинилметильную группу, либо фенильную группу, возможно замещенную одним или более чем одним атомом галогена или (С1-С3) алкильной или (С1-С3)алкоксильной группой, либо пиридинильную группу, либо 5-метил-1,2-оксазолильную группу, либо 5-метил-1,3,4-тиадиазолильную группу, либо нафтильную группу;
R3 и R4, независимо один от другого, каждый представляет собой атом водорода, (С1-С3)алкильную группу, 2-метоксиэтильную группу, гидрокси(С2-С4) алкильную группу, карбокси (С1-С3)алкильную группу, (С1-С3) алкоксикарбонил (С1-С3)алкильную группу или фенил(С1-С3)алкильную группу, или же совместно с атомом азота, который их несет, образуют либо пирролидинильную группу, возможно замещенную гидроксильной, пирролидинильную группу, возможно замещенную гидроксильной, этоксильной, метоксикарбонильной или метоксиметильной группой, либо пиперидинильную группу, либо морфолинильную группу, либо 4-метилпиперазинильную группу, либо азетидинильную группу, либо тиазолидинильную группу, а связь между атомами углерода в положениях 3 и 4 является простой или двойной.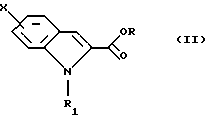
в которой X и R1 такие, как определено в п. 1, a R представляет собой (С1-С3)алкильную группу, подвергают взаимодействию с этилпируватом для получения диэфира общей формулы (IV)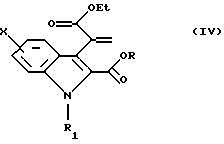
последний затем обрабатывают амином общей формулы R2NH2, в которой R2 такой, как определено в п. 1, для получения сложного эфира общей формулы (V)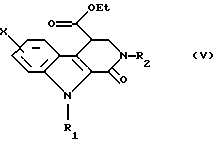
который превращают в соответствующую кислоту общей формулы (VI)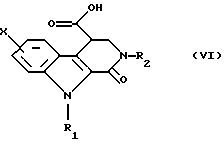
путем гидролиза, затем эту кислоту превращают в первичный, вторичный или третичный амид общей формулы (I')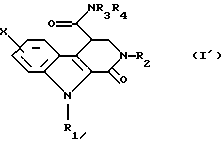
путем взаимодействия с амином общей формулы НNR3R4, в которой R3 и R4 такие, как определено в п. 1, либо через имидазолид, полученный путем взаимодействия с N, N'-карбонилдиимидазолом или через хлорангидрид, и, наконец, если желательно получить соединение, в котором связь между 3 и 4 положениями является двойной связью, соединение общей формулы (I') окисляют при помощи 2,3-дихлор-5,6-дицианоциклогекса-2,5-диен-1,4-диона или 3,4,5,6-тетрахлорциклогекса-3,5-диен-1,2-диона, для получения соответствующего соединения общей формулы (I")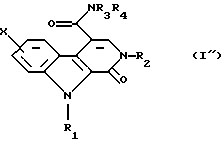
10. Способ получения соединения по п. 1, отличающийся тем, что соединение общей формулы (VIII)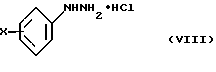
в которой Х такой, как определено в п. 1, подвергают взаимодействию с 2-кетоглутаровой кислотой, а затем обрабатывают в кислой спиртовой среде для получения диэфира общей формулы (IX)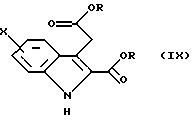
в которой R представляет собой (С1-С3)алкильную группу, затем, по желанию, осуществляют реакцию алкилирования для получения соединения общей формулы (X)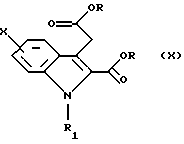
в которой R1 представляет собой (С1-С3)алкильную группу, затем последнее подвергают превращению в присутствии диметилацеталя диметилформамида для получения соединения общей формулы (XI)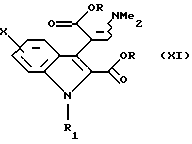
или альтернативно, по желанию, соединение общей формулы (IX) непосредственно превращают в соединение общей формулы (XI), в которой R1 представляет собой метильную группу, в присутствии диметилацеталя диметилформамида, а затем последнее обрабатывают амином общей формулы H2NR2, в которой R2 такой, как определено в п. 1, для получения сложного эфира общей формулы (V)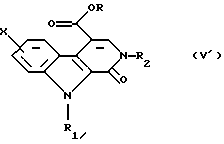
последний затем превращают в соответствующую кислоту общей формулы (VI')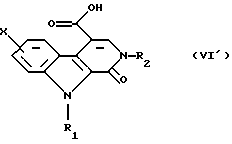
и, наконец, эту кислоту превращают в первичный, вторичный или третичный амид общей формулы (I")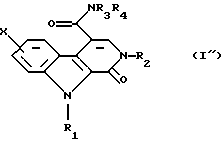
либо через имидазолид, полученный путем взаимодействия с N, N'-карбонилдиимидазолом, либо через хлорангидрид.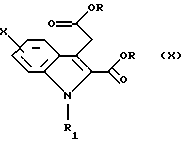
в которой Х и R1 такие, как определено в п. 1, гидролизуют для получения двухосновной кислоты общей формулы (XII)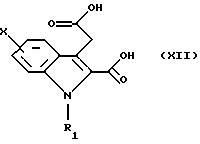
которую превращают в ангидрид для получения соединения общей формулы (XIII)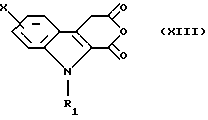
затем последнее подвергают превращению путем взаимодействия с амином общей формулы НNR3R4, в которой R3 и R4 такие, как определено в п. 1, для получения соединения общей формулы (XIV)
которое превращают в сложный эфир общей формулы (XV)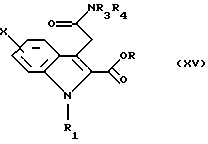
затем последний подвергают обработке в присутствии диметилацеталя диметилформамида для получения соединения общей формулы (XVI)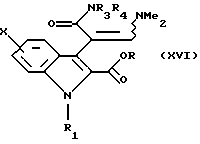
и, наконец, последнее подвергают взаимодействию с амином общей формулы R2NН2, в которой R2 такой, как определено в п. 1, для получения соединения общей формулы (I")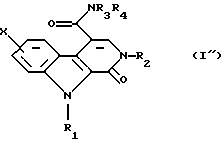
и, наконец, по желанию, вторичный амид общей формулы (I") превращают в третичный амид путем реакции алкилирования.
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| US 5206377 А, 27.04.1993 | |||
| Способ получения производных @ -карболина | 1984 |
|
SU1376946A3 |

