Область изобретения
Данное изобретение относится к новым селективным антагонистам α1b-адренергического рецептора и к применению этих и других селективных антагонистов α1b-адренергического рецептора для получения лекарственных препаратов для лечения сексуальной дисфункции человека. Кроме того, изобретение относится к фармацевтическим композициям, содержащим селективные антагонисты α1b-адренергического рецептора и, необязательно, содержащим также простагландины, прямые дилататоры, или ингибиторы 5 цГМФ(cGMP)-фосфодиэстеразы. Наконец, изобретение относится к способу идентификации соединений, полезных для лечения пациентов, страдающих сексуальной дисфункцией.
Предпосылки к созданию изобретения
Сексуальная дисфункция у мужчин и женщин возникает в результате различных механизмов. У мужчин импотенция представляет собой неспособность достигнуть и поддерживать эрекцию, достаточную для полового сношения. Эрекция достигается как результат притока крови в пещеристые тела пениса, что приводит к гиперемии пещеристых тел и последующей эрекции пениса. По оценке, 30 миллионов американских мужчин в некоторой степени испытывают эректильную дисфункцию, преобладание которой увеличивается с возрастом.
Причины импотенции можно разделить на две подкатегории:
1) органическую и 2) психологическую. Органические аспекты импотенции вызываются лежащим в основе сосудистым заболеванием, таким как заболевание, связанное с гипертонией, сахарным диабетом и прописанными лекарствами. Около половины всех случаев импотенции имеют сосудистую природу. Поскольку физиологический процесс эрекции инициируется увеличением кровотока через артерии пениса и сбросом крови в сосудистые пространства пещеристых тел, эректильная дисфункция может проистекать из неспособности артерий пениса расширяться, что подавляет, таким образом, кровоток в эректильную ткань.
Симпатические пути играют основную роль в невральном контроле эрекции пениса. Общепринято, что в состоянии детумесценции выделение норадреналина (НА (NA)), действующего на постсинаптические α1-антагонисты на кавернозных артериях и на пещеристых телах, вносит вклад в поддержание гладкой мышцы пениса в сокращенном состоянии. Напротив, внутрикавернозная инъекция α1-антагонистов, подобных феноксибензамину, фентоламину и моксизилиту, приводит к тумесценции и эрекции.
Недавно было сообщение об эрекционном отклике на трансуретральный празозин у мужчин, а также о расслабляющем влиянии данного антагониста на выделенные ткани и сосуды пениса у особей человека, собаки и крысы мужского пола.
У женщин сексуальный отклик начинается со стимуляции, которая вызывает гиперемию сосудов и приводит к смазыванию влагалища при подготовке к введению пениса. Смазывание происходит вследствие образования выделения, которое, наряду с генитальной гиперемией, образует так называемую оргазменную основу, предшествующую оргазму. Короче говоря, женская сексуальная дисфункция может происходить из-за помех на различных стадиях полового сношения и может относиться либо к органическим, либо к функциональным причинам, либо к ним обеим.
Некоторые причины включают в себя стресс, беспокойство, депрессию, усталость, межличностные конфликты между партнерами или попросту старение, могут приводить к неспособности к гиперемическому отклику, подавляя, таким образом, нормальное смазывание влагалища. В этом состоянии женщины могут быть неспособны к достижению нормального сексуального отклика без соответствующего лечения. Недавно было доказано, что как вагинальная сосудистая гиперемия, так и клиторальная эрекция зависят от увеличенного кровотока. Более того, как сообщалось и для сексуального органа у самцов, было показано, что местная инъекция во влагалище α1-адренергических антагонистов, таких как фентоламин, способна увеличить кровоток и внутривлагалищное давление вплоть до уровней, сопоставимых с уровнями, которых достигают стимуляцией тазового нерва. Эти данные ясно указывают на то, что норадреналин играет важную роль в поддержании мягкости рассматриваемого органа также и в женском сексуальном тракте.
Таким образом, важно идентифицировать новые продукты, обладающие антагонистической активностью по отношению к α1-адренорецепторам, которые могут быть полезны для стимуляции сосудистого расширения артерий в стенках влагалища и клиторе, улучшая при этом смазывание и способствуя продолжению полового акта.
Фармакологические, биохимические исследования и исследования связывания радиолигандов показали наличие трех различных подтипов α1-рецепторов с высоким сродством к празозину, а именно α1А-(α1а-), α1В-(α1b) и α1D-(α1d-), при этом индексы со строчными буквами используют для рекомбинантных рецепторов, а индексы с прописными буквами - для рецепторов в нативных тканях. В функциональных исследованиях были также идентифицированы α1-рецепоры с низким сродством к празозину и названы α1L-.
Некоторые исследования показали наличие данных подтипов α1-адренергических рецепторов в кавернозных тканях животных и человека. Методом гибридизации in situ со специальными олигонуклеотидными зондами и методами иммунного анализа показано, что кавернозные ткани тела человека и крысы экспрессируют все три подтипа клонированных α1-адренергических рецепторов.
С другой стороны, функциональные исследования тканей, относящихся к мужскому половому органу человека, являются спорными, и предполагается участие всех трех подтипов клонированных α1-АДР, или что подтип α1L-АДР является основным медиатором HA-вызванного сокращения в данной ткани. Напротив, о сосудах влагалища до сих пор ничего не известно.
Фармакологическое доказательство однозначного наличия вполне определенного подтипа(пов) α1-адренергических рецепторов в ткани пениса или влагалища представляло бы большое достижение в области лечения мужской или женской сексуальной дисфункции, предоставляя возможность использования селективных α1-антагонистов.
α1-Антагонисты, используемые в настоящее время для лечения преимущественно мужской импотенции, оказывают нежелательные побочные эффекты, такие как приапизм, болезненная эрекция чрезмерной продолжительности, которая может привести к фиброзу кавернозной ткани. Другими побочными эффектами являются боль в пенисе и гипотония.
Цель настоящего изобретения состоит в удовлетворении осознанной необходимости в селективных α1-антагонистах, не подвергающих импотентную мужскую особь побочным эффектам известных методов лечения, в особенности сердечно-сосудистого типа.
Краткое описание сущности изобретения
Данное изобретение направлено на лечение сексуальной дисфункции. С этой целью изобретение связано с применением соединений, имеющих общую формулу I
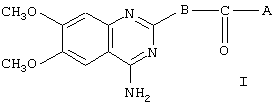
где А представляет собой 2-фурил, замещенный 2-фурил, 2-тетрагидрофурил, замещенную алкокси-, или замещенную феноксиалкильную группу, а В представляет собой одну из следующих групп формулы B1, B2 или В3:
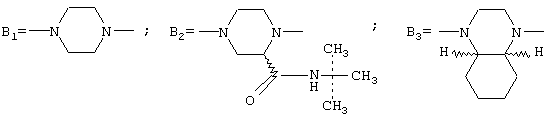 с условием, что если В является группой B1, то А представляет собой замещенную феноксиалкильную группу,
с условием, что если В является группой B1, то А представляет собой замещенную феноксиалкильную группу,
или любого энантиомера, диастереоизомера или фармацевтически приемлемой соли такого соединения,
для получения лекарственного препарата для лечения сексуальной дисфункции. Данный лекарственный препарат может также содержать простагландин, прямой вазодилататор или ингибитор 5 цГМФ фосфодиэстеразы.
Некоторые из соединений I являются новыми. Соответственно, в изобретении представлены также соединения общей формулы II
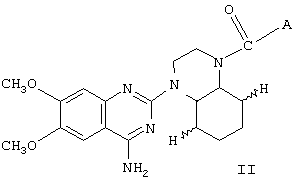
где А представляет собой 2-тетрагидрофурил, замещенный 2-фурил, замещенную алкокси- или замещенную феноксиалкильную группу,
и энантиомеры, диастереоизомеры и фармацевтически приемлемые соли таких соединений.
Фармацевтические композиции, содержащие соединение II или любой энантиомер, диастереоизомер, или фармацевтически приемлемую соль такого соединения и фармацевтически приемлемый разбавитель или носитель, также включены в данное изобретение. Включены в него также и фармацевтические композиции, содержащие соединение I или любой энантиомер, диастереоизомер, или фармацевтически приемлемую соль такого соединения и простагландин, прямой вазодилататор, или ингибитор 5 цГМФ фосфодиэстеразы и фармацевтически приемлемый разбавитель или носитель.
В другом аспекте изобретение связано с использованием соединения, которое
(a) связывается с α1b-адренергическими рецепторами млекопитающих, со сродством по меньшей мере около 10-8 М, и
(b) связывается с α1b-адренергическими рецепторами млекопитающих, со сродством, по меньшей мере в 10 раз большим, чем сродство, с которым данное соединение связывается с α1а- или α1d- или α1L-адренергическими рецепторами млекопитающих для получения лекарственного препарата для лечения сексуальной дисфункции.
В следующем аспекте изобретение связано со способом идентификации соединения, полезного для лечения сексуальной дисфункции. Данный способ включает в себя стадии
(a) индивидуального определения аффинности связывания контрольных соединений с α1b-адренергическим рецептором млекопитающих и α1a- или α1d-адренергическим рецептором методами связывания радиоактивного рецептора,
(b) определения аффинности в отношении α1L-адренергического рецептора путем противодействия эффекту сокращения на α1-адренергических рецепторах на выбранной ткани млекопитающего, и
(c) иденитификации таких соединений, которые
(1) связываются с α1b-адренергическим рецептором с аффинностью по меньшей мере около 10-8 М, и
(2) связываются с α1b-адренергическим рецептором с аффинностью по меньшей мере в 10 раз большей, чем аффинность связывания данного соединения с α1a-, или α1d-, или α1L-адренергическими рецепторами.
Подробное описание сущности изобретения
В соединениях I группа В2 предпочтительно имеет следующую стереохимию
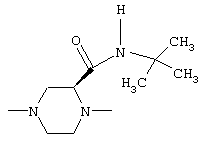
а группа В3 предпочтительно имеет цис-стереохимию, причем соединительные атомы водорода имеют одинаковую ориентацию
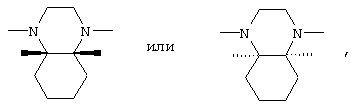
при этом первое предпочтительно. Кроме того, в соединениях I предпочтительно замещенные феноксиалкильные группы А имеют формулу
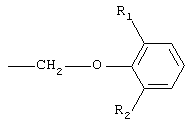
где R1 представляет собой линейную или разветвленную алкильную цепь, содержащую от 1 до 5 атомов углерода, a R2 представляет собой алкокси-группу, содержащую от 1 до 4 атомов углерода; при этом наиболее предпочтительной замещенной феноксиалкильной группой является 6-изопропил-2-метоксифеноксиметильная группа.
Наиболее предпочтительные соединения I включают в себя:
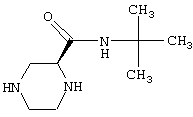
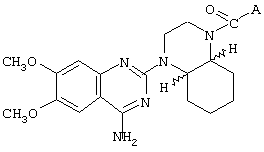
- 4-амино-6,7-диметокси-2-[4-[(2-метокси-6-изопропилфенокси-ацетил)-1-пиперазинил]хиназолин (соединение А),
- 4-амино-6,7-диметокси-2-[(4aR,8aS)-4-(2-фуроил)-цис-октагидро-1-хиноксалинил]хиназолин (соединение В) и
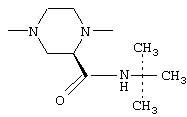
- 4-амино-6,7-диметокси-2-[3(S)-3-(трет-бутилкарбамоил)-4-(2-фуроил)-1-пиперазинил]хиназолин (соединение С).
В соединениях II октагидрохиноксалиновое кольцо предпочтительно имеет (4aR,8aS) конфигурацию. Предпочтительные замещенные феноксиалкильные группы А являются теми же, что и группы, предпочтительные для соединений I. Замещенный алкокси подходящим образом является бензилокси, а замещенный 2-фурил предпочтительно представляет собой 5-метил-2-фурил. В частности, предпочтительны следующие соединения II:
- 4-амино-6,7-диметокси-2-[(±)-4-(2-метокси-6-изопропилфенокси-ацетил)-цис-октагидро-1-хиноксалинил]хиназолин,
- 4-амино-6,7-диметокси-2-[(±)-4-(5-метил-2-фуроил)-цис-октагидро-1-хиноксалинил] хиназолин,
- 4-амино-6,7-диметокси-2-[(±)-4-(2-тетрагидрофуроил)-цис-октагидро-1-хиноксалинил] хиназолин, и
- 4-амино-6,7-диметокси-2-[(±)-4-бензилоксикарбонил-цис-октагидро-1-хиноксалинил]хиназолин.
Способы получения производных хиназолина формулы I описаны в следующих ссылках: WO 95/25726; Giardina D. еt al., J. Med. Chem. 39, 4602-7 (1996); WO 97/11698.
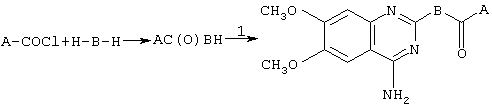
Синтез соединений формулы I можно провести по следующей схеме:
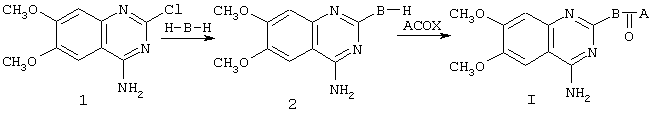
Исходное соединение 1 коммерчески доступно (например, от Lancaster Synthesis Ltd, Eastgate, White Lund, Morecambe, Lancashire, LA3 3DY, England), или иначе может быть получено, как описано у Althuis et al., J. Med. Chem. 20, 146-149 (1977). Амины Н-В-Н могут быть либо в виде рацематов, либо в гомохиральной форме, где это уместно, и могут быть коммерчески доступными, например пиперазин, или могут быть получены описанными в литературе способам. Например, амин
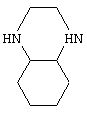
можно получить, как описано у Brill et al., J. Org. Chem. 28, 1135-1138 (1963), или путем стереоселективного синтеза, описанного Brill et al., J. Org. Chem. 29, 579-581 (1964), а амин
можно получить на основе из 2-пиразинкарбоновой кислоты при амидировании с последующим восстановлением и разделением, как описано в Tetr. Lett. 35, 673-676 (1994). Данную реакцию проводят при 150-200°С без растворителя или в присутствии подходящего полярного растворителя, такого как изоамиловый спирт, или нормальный бутиловый спирт, при температуре кипения.
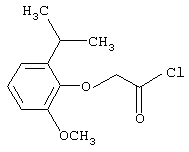
Промежуточные соединения АСОХ коммерчески доступны или могут быть получены, когда А=феноксиалкил, исходя из соответствующего фенольного производного, путем реакции с эфиром галогеналкилкислоты с последующим гидролизом и хлорированием, способами, которые известны специалисту в данной области и описаны в примере 1 для
Конденсацию для получения I можно осуществить путем реакции 2 с АСОХ, где Х представляет собой атом галогена (например, хлора), в хлорсодержащем растворителе, таком как хлороформ или хлористый метилен, или в апротонном полярном растворителе, таком как диметилформамид, в присутствии основания, такого как триэтиламин или диизопропиламин, при температуре от 0°С до 40°С. Альтернативно, когда Х представляет собой гидроксильную группу, конденсацию можно провести в хлорсодержащем или в апротонном полярном растворителе, таком как приведенные выше, в присутствии конденсирующего агента, такого как дициклогексилкарбодиимид, и промотирующего агента, такого как 4-диметиламинопиридин, при температуре от 0°С до 40°С, или другой эквивалентной.
Альтернативно, можно использовать следующую схему:
Подходящие ацилхлориды вводят во взаимодействие с соединениями НВН в полярных растворителях, таких как диметилформамид, ацетон или ацетонитрил, необязательно в присутствии основания, такого как карбонат калия, или цезия, или триэтиламин, при 20-100°С.
После этого промежуточные соединения АС(О)ВН вводят во взаимодействие с соединением 1, получая соединения I. Данное алкилирование можно провести в полярном протонном растворителе, таком как изоамиловый спирт и нормальный бутиловый спирт, или в апротонном растворителе, таком как диметилформамид, при температуре от 60°С до кипения.
Энантиомеры соединений I, в которых В является В2, можно получить, исходя из подходящих НВ2Н энантиомеров, которые получают путем образования соли рацемата с оптически активной кислотой, такой как (S)-10-камфорсульфокислота в подходящем растворителе или смеси растворителей, с последующим разделением диастереомерных солей путем перекристаллизации. Аналогичным образом, энантиомеры соединения I, в которых В является В3, можно получить путем образования соли рацемического промежуточного соединения 2 с подходящей оптически активной кислотой с последующим разделением диастереомеров.
Скрининг соединений-кандидатов для идентификации соединений, которые полезны для воплощения изобретения на практике, включает в себя измерение специфической активности связывания этих соединений с различными нейронными α1-адренергическими рецепторами (таким как α1a-, α1b- и α1d-подтипы, согласно способу Testa et al., Pharmacol. Comm. 6:79-86, 1995), которое можно осуществить с использованием большого числа способов, хорошо известных в данной области, таких как конкурентное связывание с нативными или клонированными рецепторами.
Обычно используют биологический источник, например, α1b-адренергических рецепторов, в котором данный рецептор присутствует в достаточно высокой концентрации, так чтобы определить связывание меченого лиганда было легко. Этот источник может содержать ткань или жидкость млекопитающего (либо in situ, либо после выделения из млекопитающего), или культуру клеток ткани. Рецептор-мишень можно экспрессировать либо из эндогенного (нативного) гена, либо из трансфектированного кодирующего рецептора рекомбинантного гена. Например, печень крысы является богатым (нативным) источником α1B -адренергических рецепторов (Taddei et al., Life Sci. 53: PL177-PL181, 1993). Альтернативно, кДНК α1b-адренергического рецептора хомяка можно временно экпрессировать в клетках COS-7 в культуре (Cottecchi S. et al., Proc. Natl. Acad. Sci. USA 85: 7159-7163, 1988), а кДНК α1b-адренергических рецепторов человека можно экпрессировать в клетках СНО (яичник китайского хомячка) в культуре (Testa et al., Pharmacol. Comm. 6:79-86, 1995).
Кроме того, кДНК α1a- и α1d-адренергических рецепторов человека была экспрессирована в клетках СНО (Testa et al., Pharmacol. Comm. 6:79-86, 1995), тогда как клоны адренергического рецептора α1a быка (ранее α1c) (Schwin et al., J. Biol. Chem. 265:8183-8189, 1990) и α1d крысы (Lomasney et al., J. Biol. Chem. 266: 6365-6369, 1991) были временно экспрессированы в COS-7 клетках и могли быть использованы для достижения селективности для α1b-адренергического рецептора с помощью техники связывания радиорецептора.
После этого определяли способность тестируемых соединений конкурировать с соответствующим меченным лигандом за связывание с рецептором и рассчитывали константу связывания (Ki), используя уравнение Ченга и Прусова (Cheng et al., Biochem Pharmacol. 22: 3099-3108, 1973) или эквивалентный метод расчета, хорошо известный в данной области. Подробное описание дано в примере 8 далее.
С другой стороны, для определения аффинности соединений в отношении подтипа α1L-адренергических рецепторов не существует никаких методов связывания радиорецепторов, даже несмотря на то, что этот подтип можно изучать функциональными способами в различных тканях, таких как брыжеечная или сонная артерии кролика, выводы сосудов и малая брыжеечная артерии крысы, предстательная железа человека (для обзора смотри Doherty J.R. Eur. J. Pharmacol. 361: 1-15, 1998), а также аорта кролика, предварительно обработанная хлорэтилклонидином (Testa et al., J. Pharmacol. Exp. Ther. 281: 1284-1294, 1997).
В данном подходе определяют способность тестируемых соединений подавлять вызванное НА сокращение сосудов и оценивают константу диссоциации (Кb) (Arunlakshana et al., J. Pharmacol. Chemoter. 14: 45-58, 1959), или применяют эквивалентный расчетный метод, хорошо известный в данной области. Подробное описание приведено в примере 9 далее.
Как обсуждалось выше, соединения, полезные для воплощения данного изобретения на практике, связываются с α1b-адренергическим рецептором с Ki, составляющей по меньшей мере 10-8 М, и имеют аффинность в отношении к α1a-, α1b- и α1L-адренергических рецепторов, по меньшей мере в 10 раз меньше. Как только определили, что какое-либо соединение обладает вышеупомянутыми характеристиками, его фармакологическую активность можно доказать с использованием одной или более модельных систем на животных для изучения эрекции самца. Полезные модельные системы на животных включают в себя повышение внутрикавернозного давления у анастезированных крыс и/или собак.
В таких способах соединения вводят в пещеристое тело и измеряют развившееся внутрикавернозное давление одновременно с давлением крови. Эффективность данных соединений лучше определять путем оценки соотношения между внутрикавернозным и кровяным давлением, которые строго коррелированы. Таким образом получают индекс активности, выражаемый в виде процентной величины и отражающий процент ВКД (ICP) по отношению к давлению крови, который может достигать максимального значения 100%. Такие способы подробно описаны в примерах 10 и 11 далее.
Как определено с использованием вышеуказанного в моделях in vivo, полезные соединения вызывают значительное увеличение, по сравнению с контролем носителя, отношения ВКД/ДК при местном введении в дозе 10-1000 мкг, причем давление крови снижается более чем на 20% (30% только при самой большой дозировке). Модель для измерения эффектов продуктов изобретения на влагалищное и клиторальное давление описана в примере 12.
Терапевтические применения
Данное изобретение включает в себя фармацевтические составы, содержащие перечисленные выше антагонисты α1a-адренергического рецептора для лечения мужской и женской сексуальной дисфункции, в частности сексуальной дисфункции вследствие сосудистой природы.
Не желая быть связанными теорией, невральный симпатический контроль поддерживает пенис и стенки влагалища, а также клитор, в их мягком состояниии, и противодействие эффекту симпатических медиаторов в этих тканях с помощью селективных антагонистов α1b-адренергического рецептора позволяет преодолеть этот негативный контроль, с расслаблением гладкой мышцы пениса и расширением кавернозных артерий у мужчин и сосудистой гиперемии у женщин. В результате у мужчин возрастает кровоток в трабекулярные полости пещеристых тел, вызывая в пенисе гиперемию (тумесценцию). Расширение трабекулярных стенок к белочной оболочке сдавливает подоболочные венулы и препятствует оттоку крови, приводя к продолжительной тумесценции, то есть эрекции. У женщин сжатие сосудов позволяет произойти смазыванию влагалища, таким образом, удовлетворительной сексуальной активности.
“Эффективное количество” соединения для лечения сексуальной дисфункции представляет собой количество, приводящее к измеримому улучшению эрекции. У мужчин дополнительным параметром является продолжительность эрекции, тогда как у женщин эффективное количество представляет собой такое количество, которое приводит к измеримому количеству кровотока в клиторе и стенке влагалища.
Эффективное количество для лечения сексуальной дисфункции можно определить экспериментально при использовании способов, известных в данной области, таких как установление матрицы дозировок и частот и сопоставление группы экспериментальных единиц или субъектов в каждой точке данной матрицы. Точное количество, предназначенное для введения пациенту, может изменяться в зависимости от состояния и тяжести расстройства и физического состояния пациента. Измеримое улучшение любого симптома или параметра может определить врач, специализирующийся в данной области, или оно может быть сообщено врачу пациентом. Клинически значительное улучшение определяют как улучшение, заметное пациенту и/или врачу.
Предпочтительно соединения согласно изобретению применяют в сочетании с приемлемым фармакологическим носителем до введения. Такие композиции содержат терапевтически эффективное количество соединения согласно изобретению и фармацевтически приемлемый носитель или эксципиент. Например, когда введение осуществляют путем инъекции, готовят водный раствор, применимый при внутрикавернозной инъекции в пенис. В этом случае носители включают в себя, не ограничиваясь ими, воду, физиологический раствор, забуференный физиологический раствор, соли, глицерин и этанол, либо по отдельности, либо в сочетании. Кроме того, к данным композициям можно добавить нераздражающий консервант, например, такой как хлорид бензалкония.
В случае внутримочеточникового, подкожного или местного введения фармацевтический носитель включает в себя, не ограничиваясь ими, гели, такие как минеральные гели, мази, кремы, растворы, спреи, порошки, пены и липосомные составы. Носитель является водорастворимым, нераздражающим и неповышающим чувствительность кожи. В предпочтительном способе воплощения носитель для введения данного типа имеет полумягкую кремоподобную консистенцию. Этого можно достичь при использовании гидрогеля, такого как гидроксипропилметилцеллюлоза.
Для внутривлагалищного введения в виде влагалищного душа фармацевтические носители включают в себя, не ограничиваясь ими, воду, физиологический раствор, забуференный физиологический раствор, соли, глицерин и этанол, либо по отдельности, либо в сочетании. Более того, к данным композициям можно добавить нераздражающий консервант, включающий в себя, например, хлорид бензалкония.
Носители для введения в виде крема или влагалищного суппозитория включают в себя, но не ограничиваются ими, пропиленгликоль, гидрированный ланолин, масло сладкого миндаля, полигликолевые эфиры жирных кислот, цетиловый спирт, глицерилмоностеарат, эдетат натрия, триглицериды жирных кислот, желатин, глицерин, двуокись титана, парабены.
Фармацевтические композиции согласно настоящему изобретению могут необязательно включать в себя прочие активные агенты, которые повышают или дополняют улучшающие половой акт эффекты соединений данного изобретения. Такие активные агенты включают в себя, не ограничиваясь ими, простагландины, например простагландин Е2; прямые вазодилататоры, например папаверин; и ингибиторы фосфодиэстеразы V типа, например, 1-{[3-(4,7-дигидро-1-метил-7-оксо-3-пропил-1Н-пиразоло[3,4-d]-пиримидин-5-ил-4-этоксифенил]-4-метилпиперазин, известный так же как сильденафил. Данные соединения дополняют прямое действие соединений согласно изобретению в осуществлении требуемых улучшающих эффектов. Использование любого соединения согласно изобретению совместно с сильденафилом может, кроме того, позволить снизить дозу последнего, что сводит к минимуму нежелательные побочные эффекты при введении этого сочетания перорально или внутривенно, или также одним из способов, обсуждаемых в следующих параграфах.
Предпочтительно соединения данного изобретения вводят одним из следующих способов. Соединения согласно изобретению можно вводить путем инъекции, при этом соединения согласно изобретению растворяют в физиологическом растворе в концентрации, изменяющейся в интервале от 0,2 до 20 мг/мл. Объем 0,5 мл вводят внутрикавернозно. В другом примере предпочтительного способа соединения согласно изобретению составляют в минеральном геле, который затем применяют снаружи во внутримочеточниковый катетер. Дозировка соединений данного изобретения находится в интервале от 1 до 10 процентов от массы применяемого геля. Катетер вставляют в мочеиспускательный канал с целью введения соединений согласно изобретению внутрь мочеточника и осуществления расширения сосудов, необходимого для эрекции.
Путем инъекции можно вводить любое количество вышеописанных соединений, которое эффективно для облегчения сексуальной дисфункции человека. Для разовой дозы используют интервал примерно от около 0,1 до 10 мг/дозу. Предпочтительно в разовой дозе используют примерно от 0,3 мг/дозу до 3 мг/дозу.
В случае влагалищного душа концентрация может изменяться в интервале от 0,2% до 5%, тогда как для влагалищного крема концентрация может изменяться в интервале от 1% до 10%. Количество, которое можно ввести при помощи влагалищного суппозитория, может изменяться в интервале от 1 до 100 мг.
Изобретение иллюстрировано следующими примерами и таблицами и фигурами, на которые имеются ссылки в примерах.
Пример 1
Гидрохлорид 4-амино-6,7-диметокси-2-[4-(2-метокси-6-изо-пропилфеноксиацетил)-1-пиперазинил]хиназолина
(I: А=2-метокси-6-изопропилфеноксиметил, В=В1) (соединение А)
2-Метокси-6-изопропилфеноксиуксусная кислота (1А)
Раствор 11,1 мл этилбромацетата в 10 мл толуола прибавляли по каплям при комнатной температуре в течение примерно 15 минут к смеси 20 г NaOH, 30 мл Н2O, 1,1 г хлорида триэтилбензиламмония, 8,4 г 2-изопропил-6-метоксифенола (полученного по Johnson et al., Tetrahedron, 38, 1397-1404 (1982)) и 40 мл толуола. Смесь энергично перемешивали при той же температуре в течение 2 ч, а затем в течение 2 ч при 60-65°С и в течение 6,5 ч при кипении. Во время этой последней стадии прибавляли раствор 6 мл этилбромацетата в 10 мл толуола. В конце смесь разбавляли 250 мл Н2О. Водную фазу отделяли и обрабатывали концентрированной НСl; эмульгированный осадок экстрагировали Et2O (3×50 мл) и промывали органическую фазу водой. Следующую экстракцию проводили 40 мл 20 %-ного Nа2СО3 и обрабатывали этот слабощелочной раствор концентрированной НСl и экстрагировали Et2O (3×40 мл). Экстракты объединяли и выпаривали растворитель, получая 8 г (72%) указанного в заголовке соединения; т.кип. 190°С/0,7 мм рт.ст.
Гидрохлорид 4-амино-6,7-диметокси-2-[4-(2-метокси-6-изо-пропилфеноксиацетил)-1-пиперазинил]хиназолина
К кипящему раствору 6 г промежуточного соединения IА в 30 мл ССl4 прибавляли по каплям 3,6 мл SOCl2 и перемешивали смесь при кипении 2 ч. Маслянистый остаток, полученный при упаривании реакционной смеси, растворяли в 26 мл СНСl3 и прибавляли этот раствор по каплям в течение 30 минут к перемешиваемому раствору 7,75 г 4-амино-6,7-диметокси-2-(1-пиперазинил)хиназолина и 4,1 г Et3N в 50 мл ДМФА. После перемешивания в течение 2 ч растворители выпаривали досуха. Остаток растворяли в 250 мл СНСl3. Данный раствор промывали 2,5%-ным NаНСО3, а затем H2O и, наконец, упаривали досуха. Очистку производили методом колоночной хроматографии с использованием СНСl3/МеОН 100:3 в качестве элюирующей смеси. Остаток суспендировали в 100 мл кипящего этанола, а затем добавляли этанольный НСl в небольшом избытке до растворения. После охлаждения гидрохлоридную соль выделяли отсасыванием и перекристаллизовывали из этанола, получая 6,4 г (45%) продукта; т.пл. 252-254°С.
Пример 2
Гидрохлорид 4-амино-6,7-диметокси-2-[(4aR,8aS)-4-(2-фуроил)-цис-октагидро-1-хиноксалинил]хиназолина
(I: А=2-фурил, В=В3) (соединение В)
(±)-(2-Фуроил)-цис-октагидрохиноксалин (2А)
Прибавляли по каплям 1,44 г 48 %-ной бромистоводородной кислоты к раствору 3,85 г цис-октагидрохиноксалина (полученного, как описано Brill et al., J. Org. Chem. 28, 1135-1138 (1963)) в 26 мл этанола и 4 мл Н2O при перемешивании при 40-45°С. К полученному раствору прибавляли по каплям 1,16 г 2-фуроилхлорида в течение 15 минут и продолжали перемешивание в течение 3 ч при 80°С. Раствор концентрировали до малого объема, разбавляли водой и экстрагировали хлороформом. Остаток, полученный после выпаривания растворителя, очищали флэш-хроматографией при элюировании смесью петролейный эфир:этилацетат:метанол:28%-ный водный аммиак 8:6:2:0,2, получая 2,35 г (40%) требуемого соединения. Т.пл. 178°С разл.
(±)-1-(2-Фуроил)-цис-октагидрохиноксалин (2В)
Раствор 2,35 г вышеуказанного промежуточного соединения 2А в 22 мл метанола обрабатывали раствором 1,54 г (S)-(+)-миндальной кислоты в 22 мл метанола. Смесь упаривали досуха, получая остаток, который кристаллизовали, растворяя твердое вещество в 265 мл горячего этилацетата, а затем уменьшая объем при упаривании примерно до 130 мл. Выпавший осадок перекристаллизовывали еще шесть раз из того же растворителя, получая 0,4 г соли (+)-манделата; т.пл. 188-190°С, [α]
Гидрохдорид 4-амино-6,7-диметокси-2-[(4aR,8aS)-4-(2-фуроил)-цис-октагидро-1-хиноксалинил]хиназолина
Смесь 0,21 г указанного выше промежуточного соединения 2В, 0,18 г 4-амино-2-хлор-6,7-диметоксихиназолина и 0,2 г N,N-диизопропилэтиламина в 13 мл изоамилового спирта нагревали при кипении в течение 72 ч. После охлаждения смесь оставляли при 0°С на ночь. После этого твердое вещество выделяли, растирали с холодным 2N NaOH, фильтровали, промывали водой и переводили в соль гидрохлорида. При кристаллизации из Ме-OH/15% EtOH получали 0,06 г указанного в заголовке соединения. Т.пл. 262-264°С, [α]
Пример 3
4-Амино-6,7-диметокси-2-[(3S)-3-(трет-бутидкарбамоил)-4-(2-фуроид)-1-пиперазинил]хиназолин
(I: А=2-фурил, B=B2) (соединение С)
4-Амино-6,7-диметокси-2-[(3S)-3-(трет-бутилкарбамоил)-1-пиперазинил]хиназолин (3А)
Смесь 0,36 г 4-амино-2-хлор-6,7-диметоксихиназолина, 1,08 г (S)-N-третбутил-2-пиперазинкарбоксамида бис-(1S)-(+)-10-камфорсульфоната, полученного, как описано в патенте США 5700364, и 0,94 мл диизопропилэтиламина в 10 мл изоамилового спирта нагревали при кипении в течение 9 ч. После охлаждения до комнатной температуры растворитель выпаривали в вакууме и прибавляли к остатку 50 мл хлористого метилена. Смесь промывали водой (3×20 мл), 5%-ным водным Nа2СО3 (30 мл), водой (3×20 мл), сушили (Na2SO4) и упаривали досуха. Остаток очищали флэш-хроматографией при элюировании смесью хлороформ:2N метанольный аммиак 100:3, получая 0,173 г (30%) требуемого соединения.
1H ЯМР (СDСl3, δ): 1,35 (с, 9Н, С(СН3)3), 1,65-2,10 (м, 1Н, пиперазин NH), 2,30-3,40 (м, 5Н, пиперазин CHs), 3,95 (м, 6Н, ОСН3), 4,45 (д, 1Н, пиперазин СН), 4,68 (дд, 1Н, пиперазин СН), 6,00-6,45 (м, 2Н, NH2), 6,85-7,10 (м, 2Н, CONH и хиназолин Н8), 7, 18 (с, 1Н, хиназолин Н5).
4-Амино-6,7-диметокси-2-[(3S)-3-(третбутидкарбамоил)-4-(2-фуроил)-1-пиперазинил]хиназолин
Смесь 0,243 г указанного выше промежуточного соединения 3А, 0,17 г диизопропилэтиламина, 0,08 мл 2-фуроилхлорида в 10 мл хлористого метилена перемешивали при комнатной температуре в течение 10 ч. Раствор разбавляли хлористым метиленом (10 мл), промывали водой (4×10 мл), 2N NaOH (4×10 мл) и водой (4×10 мл), сушили (Na2SO4) и упаривали досуха. Остаток очищали флэш-хроматографией при элюировании смесью петролейный эфир:этилацетат 100:2, получая 0,2 г (68%) требуемого соединения в виде твердого вещества цвета слоновой кости.
1Н ЯМР (СDСl3, δ): 1,14 (с, 9Н, С(СН3)3), 3,08-3,22 (м, 1Н, пиперазин СН), 3,27-3,47 (м, 2Н, пиперазин CHs), 3,77 (с, 3Н, ОСН3), 3,81 (с, 3Н, ОСН3), 4,10-4,25 (м, 1Н, пиперазин СН), 4,35-4,50 (м, 1Н, пиперазин СН), 4,82-4,98 (м, 2Н, пиперазин CHs), 6,61-6,66 (м, 1Н, фуран Н4), 6,69 (с, 1Н, фуран Н3), 6,95-7,18 (м, 3Н, CONH и NH2), 7,42 (с, 1Н, хиназолин Н8), 7,55 (с, 1Н, хиназолин Н5), 7,85 (с, 1Н, фуран Н5).
Пример 4
Гидрохлорид 4-амино-6,7-диметокси-2-[(±)-4-(2-метокси-6-изопропилфеноксиацетил)-цис-октагидро-1-хиноксадинил]хиназолина·2,5Н2О
(I: А=2-метокси-6-изопропилфеноксиметил, В=В3)
Дигидрохлорид 4-амино-6,7-диметокси-2-[(±)-цис-октагидро-1-хиноксалинил]хиназолина·2,5Н2О (4А)
Смесь 7,85 г 4-амино-2-хлор-6,7-диметоксихиназолина, 13,3 г триэтиламина, 0,4 г диметиламинопиридина, 11,5 г цис-декагидрохиноксалина и 80 мл изоамилового спирта перемешивали при кипении в течение 72 ч. После охлаждения до комнатной температуры растворитель выпаривали досуха и очищали остаток флэш-хроматографией при элюировании смесью петролейный эфир:этилацетат:метанол: 28%-ный гидроксид аммония 8:6:2:0,2. Полученный остаток переводили в соль гидрохлорида и кристаллизовали из смеси изопропанол:метанол 1:1, получая 14,7 г (73%) требуемого соединения; т.пл. 290-295°С.
Гидрохдорид 4-амино-6,7-диметокси-2-[4-хлорацетил-(±)-цис-октагидро-1-хиноксалинил]хиназолина (4В)
К смеси 0,5 г указанного выше промежуточного соединения 4А и 0,21 г диизопропилэтиламина в 15 мл хлористого метилена при перемешивании прибавляли по каплям раствор 0,26 г хлор-ацетилхлорида в 6 мл хлористого метилена в течение 15 минут при 0°С. После 4 ч перемешивания при комнатной температуре и 72 ч стояния в холодильнике твердое вещество выделяли отсасыванием и очищали кристаллизацией из хлороформа, получая 0, 12 г (33%) требуемого продукта; т.пл. >270°С.
1Н ЯМР (СDСl3, δ): 1,30-2,35 (м, 8Н, октагидрохиноксалин CHs позиции 5, 6, 7 и 8), 3,70-4,18 (м, 10Н, октагидрохиноксалин CHs позиции 2 и 3 и 2 ОСН3), 4,20-4,36 (м, 1Н, октагидрохиноксалин Н4а), 4,47 (с, 2Н, CH2Cl), 4,60-4,78 (м, 1Н, октагидрохиноксалин Н8а), 7,48 (с, 1Н, хинолин Н8), 7,75 (с, 1Н, хинолин Н5), 8,66 (шир, 1Н, NH), 8,90 (шир, 1Н, NH), 11,95 (шир, 1Н, NH).
Гидрохлорид 4-амино-6,7-диметокси-2-[(±)-4-(2-метокси-6-изопропилфеноксиацетил)-цис-октагидро-1-хиноксалинил]-хиназолина·2,5 H2O
К раствору 0,16 г 2-изопропил-6-метоксифенола в 5 мл этанола при перемешивании прибавляли 10 мл свежеприготовленного 0,095 М раствора EtONa и продолжали перемешивание 0,5 ч при комнатной температуре. Полученный раствор прибавляли по каплям в течение 15 минут к перемешиваемому раствору 0,2 г указанного выше промежуточного соединения 4В в 50 мл этанола в атмосфере азота. Данную смесь перемешивали в течение 5 ч при комнатной температуре, а затем кипятили 20 часов. Остаток, полученный при выпаривании растворителя, переводили в соль гидрохлорида и кристаллизовали из изопропанола, получая 0,64 г (21%) указанного в заголовке соединения; т.пл. 208-209°С.
Пример 5
Гидрохлорид 4-амино-6,7-диметокси-2-[(±)-4-(5-метил-2-фуроил)-цис-октагидро-1-хиноксадинил]хиназолина·2,5H2O
(I: А=5-метил-2-фурил, В=В3)
5-Метил-2-фуроилхлорил (5А)
Раствор 0,31 г SOCl2 в 2 мл бензола прибавляли по каплям при 0°С в атмосфере азота к раствору 0,22 г 5-метил-фуранкарбоновой кислоты, полученной способом, описанным Robert et al., Eur. J. Med. Chem. 30, 915-924 (1995), в 5 мл бензола. Смесь перемешивали при 80°С в течение 1 ч, а затем избыток SOCl2 отгоняли. Остаток (0,24 г, 97% от теоретического) использовали на следующей стадии без дополнительной очистки.
Гидрохлорид 4-амино-6,7-диметокси-2-[(±)-4-(5-метил-2-фуроил)-цис-октагидро-1-хиноксалинил]хиназолина·2,5H2O
К раствору 0,56 г указанного выше промежуточного соединения 4А и 0,25 г триэтиламина в 10 мл хлористого метилена при перемешивании прибавляли по каплям раствор 0,24 г указанного выше промежуточного соединения 5А в 5 мл хлористого метилена при 0°С. Смесь перемешивали при комнатной температуре в течение 3 часов, а затем выдерживали при 0-4°С в течение ночи. Осадок собирали отсасыванием и очищали флэш-хроматографией при элюировании смесью петролейный эфир:этилацетат:метанол:28%-ный гидроксид аммония 8:8:2:0,2. Чистое основание переводили в соль гидрохлорида и кристаллизовали из изопропанола, получая 0,2 г (27%) указанного в заголовке соединения; т.пл. 268-270°С.
Пример 6
Гидрохлорид 4-амино-6,7-диметокси-2-[(±)-4-(2-тетрагидро-фуроил)-цис-октагидро-1-хиноксалинил]хиназолина·2,5H2O
(I: А=2-тетрагидрофурил, В=В3)
2-Тетрагидрофуроидхлорил (6А)
Смесь 0,22 г 2-тетрагидро-2-фуранкарбоновой кислоты и 0,5 мл SOCl2 в 10 мл бензола перемешивали при 80°С в течение 1 ч. Избыток SOCl2 и бензола отгоняли, получая 0,25 г маслянистого остатка, который посчитали чистым на 80% и использовали на следующей стадии без дополнительной очистки.
Гидрохлорид 4-амино-6,7-диметокси-2-[(±)-4-(2-тетрагидро-фуроил)-цис-октагидро-1-хиноксалинил]хиназолина·2,5Н2О
Данное соединение получали способом, описанным в примере 5, но с использованием промежуточного соединения 6А вместо промежуточного соединения 5А и с использованием смеси петролейный эфир:этилацетат:метанол:14%-ный гидроксид аммония 8:6:2:0,1 в качестве элюента для флэш-хроматографии. Чистое основание переводили в соль гидрохлорида и кристаллизовали из этанола, получая 21% указанного в заголовке соединения; т.пл. 220-223°С.
Пример 7
Гидрохлорид 4-амино-6,7-диметокси-2-[(±)-4-бензилокси-карбонил-цис-октагидро-1-хиноксалинил]хиназолина·0,75Н2O
(I: А=бензилокси, В=В3)
Данное соединение получали способом, описанным в примере 5, но с использованием бензилоксикарбонилхлорида вместо промежуточного соединения 5А и с использованием смеси петролейный эфир:этилацетат:метанол:14%-ный гидроксид аммония 8:5:0,6:0,025 в качестве элюента для флэш-хроматографии.
Чистое основание переводили в соль гидрохлорида и кристаллизовали из этанола, получая 14% указанного в заголовке соединения; т.пл. 243-245°С.
Пример 8
Анализ связывания радиоактивного лиганда с клонированными α1- адренорецепторами.
Связывание [3Н] празозина с адренорецепторами α1a-быка, α1b-хомяка и α1d-крысы проводили в мембранах клеток COS-7 (эпителиальные клетки почки обезьяны CV-1), кратковременно экспрессирующих адренорецепторы α1a-быка, α1b-хомяка и α1d-крысы.
Создание и трансфекцию индивидуальных α1-адренорецепторов проводили, как описано ранее (Schwinn et аl., J. Biol. Chem. 265: 8183-8189, 1990; Cotecchia S. et al., Proc. Natl. Acad. Sci. USA 85: 7159-7163, 1988; Lomasney et al., J. Biol. Chem. 266: 6365-6369, 1991).
Мембраны клеток COS-7 (35, 35 и 70 мкг белка/образец для α1b, α1a и α1d соответственно) инкубировали в 50 мМ Tris, pH 7,4, содержащего 10 мкМ паргилина и 0,1% аскорбиновой кислоты, с 1,1 нМ [3H] празозина в конечном объеме 0,22 мл в течение 30 минут при 25°С, в отсутствие или в присутствии конкурирующих лекарственных средств (1 пМ-10 мкМ). Неспецифическое связывание было отмечено в присутствии 100 мкМ фентоламина. Инкубирование прекращали при добавлении ледяного Tris-буфера и быстро фильтровали через предварительно обработанные 0,2%-ным полиэтиленимином фильтры Whatman GF/B или Schleicher & Schuell GF52.
Связывание с клонированными подтипами человеческих α1-адренорецепторов проводили в мембранах клеток СНО (клетки яичника китайского хомяка), трансфицированных методом электропорации ДНК, экспрессирующей ген, кодирующий каждый подтип α1-адренорецептора. Клонирование и устойчивую экспрессию гена α1-адренорецептора человека проводили, как описано ранее (Testa et al., Pharmacol. Comm. 6: 79-86, 1995). Мембраны клеток СНО (30 мкг белка) инкубировали в 50 мл Tris, pH 7,4, с 0,2 нМ [3Н] празозина в конечном объеме 1,02 мл в течение 30 минут при 25°С, в отсутствие или в присутствии конкурирующих лекарственных средств (1 нМ-10 мкМ). Неспецифическое связывание было отмечено в присутствии 10 мкМ фентоламина. Инкубирование прекращали при добавлении ледяного Tris-буфера и быстро фильтровали через предварительно обработанные 0,2%-ным полиэтиленимином фильтры Whatman GF/B или Schleicher & Schuell GF52.
Ингибирование специфического связывания радиоактивного лиганда контрольными лекарственными средствами анализировали для оценки величины IC50 с использованием программы нелинейного сглаживания кривой (De Lean et al., A.J. Physiol. 235: E97-E102, 1978). Величину IC50 переводили в константу связывания (Ki) по уравнению Ченга и Прусова (Cheng et al., Biochem, Pharmacol. 22: 3099-3108, 1973). Данные выражены в виде среднего занчения Ki.
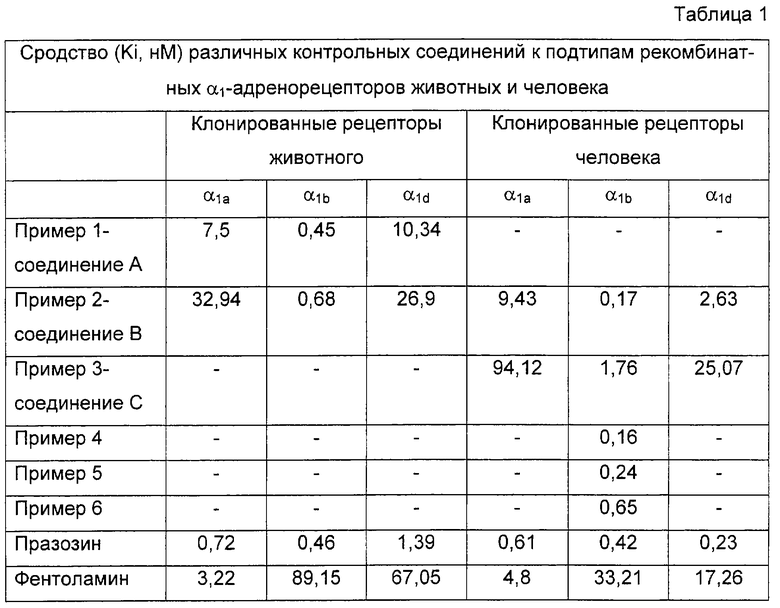
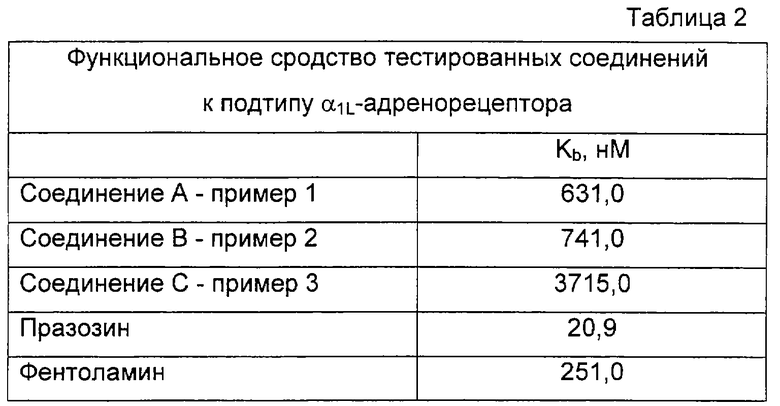
Соединения примеров с 1 по 7 проявляют требуемую действенность по отношению к α1b-адренорецептору, при этом значения их Ki превышают 1×10-8 M (Таблица 1). Соединение А, соединение В и соединение С были также селективны по отношению к α1b-адренорецептору, при этом их сродство к другим α1-подтипам по меньшей мере в 10 раз ниже.
Пример 9
Функциональное сродство к α1L-адренергическим рецепторам
Функциональную α1-антагонистическую активность контрольных соединений против вызванных норадреналином сокращений аорты кролика, предварительно обработанной хлорэтилклонидином (рецептор (α1L), оценивали по методу Testa (Testa et al., J. Pharmacol. Exp. Ther. 281: 1284-1293, 1997). Взрослых самцов новозеландских кроликов убивали путем цервикального вывиха. Аорту извлекали, помещали в буфер Кребса-Хенслейта (Krebs-Hensleit) и отсекали прилегающую ткань. Из каждой аорты готовили кольца (8 колец на аорту, примерно 4-5 мм шириной) и суспендировали в 20 мл банях для органов, содержащих гидрокарбонатный буфер Кребса следующего состава (мМ): NaCl 112,0, КСl 5,0, CaCl2 2,5, КН2РО4 1,0, MgSO4 1,2, NаНСО3 12,0 и глюкозу 11,1, находящийся в равновесии при 37°С с 95% О2: 5% СО2. К данному буферу прибавляли десметилимипрамин (0,1 мкМ) и кортикостерон (1 мкМ) для блокирования нейронного и экстранейронного поглощения НА, (±)-пропранолол (1 мкМ) для блокирования β адренорецепторов и йохимбин (0,1 мкМ) для блокирования α2-адренорецепторов. Ткани подвергали действию пассивного груза в 2 кг, и измеряли полученное давление с использованием изометрических датчиков (Basile 7003).
Препаратам давали прийти в равновесие в течение 60 минут, а затем в течение каждых 30 минут трижды добавляли 10 мкМ НА. После этого кольца аорты инкубировали с алкилирующим агентом хлорэтилклонидином (5×10-5 М) в течение 30 минут, а затем экстенсивно промывали три раза (в течение 0,5 ч) перед построением кривой концентрации НА-отклик. После вымывания НА и повторного уравновешивания ткани (45 минут) прибавляли тестируемое лекарственное средство, и через 10 минут строили вторую кривую кумулятивной концентрации НА-ответ. Концентрацию каждого антагониста испытывали с использованием 2-3 колец аорты от разных кроликов.
Соотношения доз (то есть соотношение между концентрациями норадреналина, необходимыми для получения половины максимального ответа в присутствии или в отсутствие тестируемого антагониста) рассчитывали при каждой концентрации соединений. Для оценки константы сродства Кb построили график зависимости логарифма соотношений этих доз - 1 от логарифма концентраций соединения (график Шилда (Schild.) ). При использовании только одной или двух концентраций тестированного соединения кажущуюся константу Кb вычисляли по формуле: Кb=[В]/(соотношение дозы - 1), где В представляет собой концентрацию антагониста.
Тестированные соединения проявили селективность в отношении α1b-адренорецептора, в отличие от α1L-адренорецептора. Их функциональное сродство к данному рецептору фактически оказалось по меньшей мере в 10 раз ниже, чем сродство к α1b- подтипу (таблица 2).
Пример 10
Регистрация внутрикавернозного и кровяного давления у крыс
Оценку эректильных свойств различных соединений, испытываемых на крысах, проводили по способу Giuliano et al. (Giuliano et al., J. Urol. 150: 519-524, 1993).
Крыс анастезировали путем внутрибрюшинного введения уретана (1,5 г/кг в стерильном физиологическом растворе) и помещали на гомотермическое одеяло. Их температуру поддерживали при 37°С. Крыс подвергали трахеотомии для облегчения самопроизвольного дыхания и для предотвращения вдыхания слюны. Для записи среднего давления крови (ДК (ВР), мм рт.ст.) в сонную артерию помещали катетер, заполненный гепаринизированным физиологическим раствором (25 МЕ/мл). Пенис выбривали и открывали пещеристые тела. В одно из пещеристых тел вставляли иглу из нержавеющей стали 25-размера для регистрации внутрикавернозного давления (ВКД в мм рт.ст.). Эту иглу соединяли с катетером, заполненным гепаринизированным физиологическим раствором (25 МЕ/мл). Катетеры давления соединяли с датчиками давления (модель 750, Elcomatic Ltd, Глазго, Великобритания).
После 10-минутного периода покоя внутрикавернозно вводили раствор соединения (50 мкл/инъекция). Затем каждые десять минут тем же путем вводили увеличивающиеся дозы одного соединения. Каждой крысе делали пять инъекций (один растворитель плюс четыре кумулятивных дозы), и выбирали пять крыс для изучения одного соединения. Для каждой инъекции и для каждого соединения определяли среднее значение ДК, усреднившееся за десять минут после инъекции. Регистрировали также максимальное значение ВКД, достигаемое во время десятиминутного периода после инъекции. В данных опытах все тестированные соединения растворяли и разбавляли растворителем пропиленгликоль - Соренсен. Значения ВКД и ДК представляли как среднее ± с.о. (систематическая ошибка) среднего, или процентная вариация (± с.о.) базисных значений. Соотношения (ВКД/ДК)·100, соответствующие проценту ДК, достигнутому ВКД, рассчитывали с использованием значения пика влияния на ВКД на среднее давление крови, наблюдаемое в течение 10 минут после инъекции соединений, и представляли как среднее ± с.о. среднего.
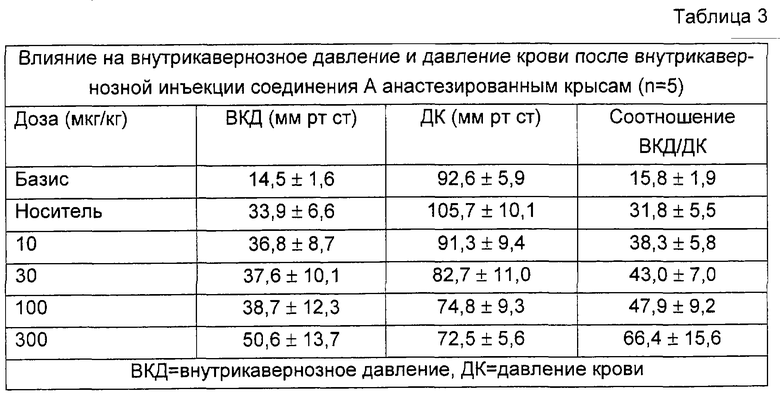
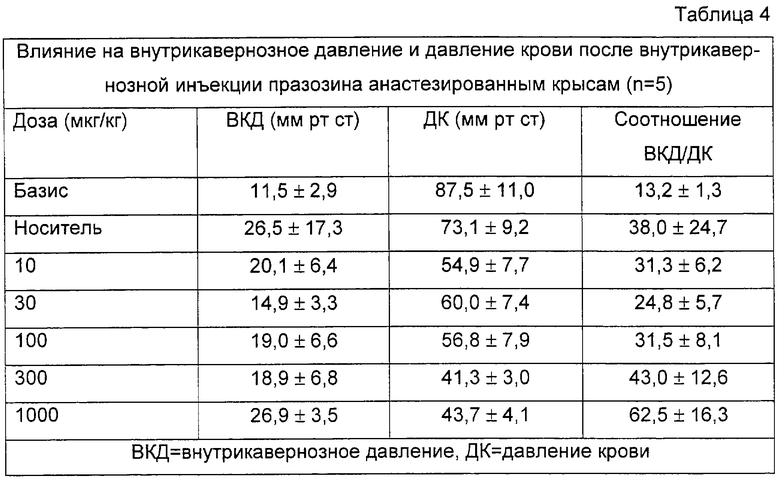
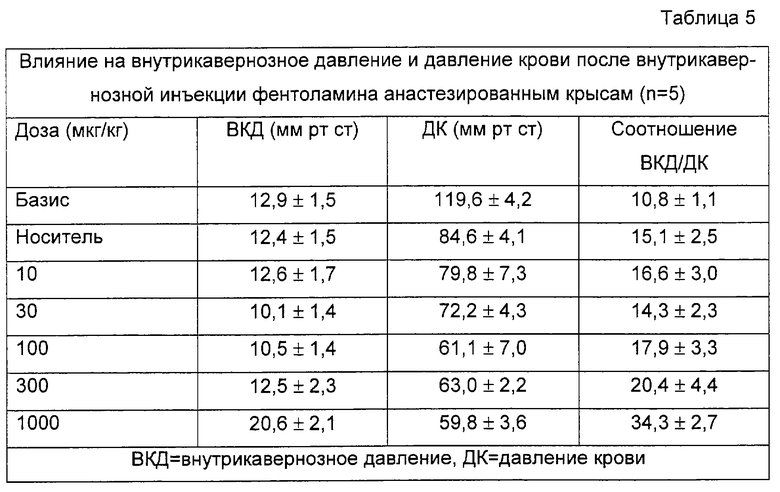
Эффекты соединения А, празозина и фентоламина суммированы в таблицах 3-5. Соединение А повышает ВКД, в зависимости от дозы (от 33,9 мм рт.ст. после инъекции носителя до 50,6 мм рт.ст.) и немного снижает ДК (примерно 30%). Повышение ВКД длится несколько минут, но никогда не перекрывает десятиминутный период скрининга (данные не приведены). Празозин оказался неспособным вызвать какое-либо повышение ВКД и снижал давление крови на 41% в промежутке между инъекцией растворителя и после инъекции 1000 мкг/кг. Фентоламин не изменял ВКД вплоть до 300 мкг/кг. После инъекции 1000 мкг/кг он вызывал продолжительное повышение внутрикавернозного давления, которое длилось несколько минут (не больше 10 минут); при такой дозе фентоламин снижал ДК на 30%.
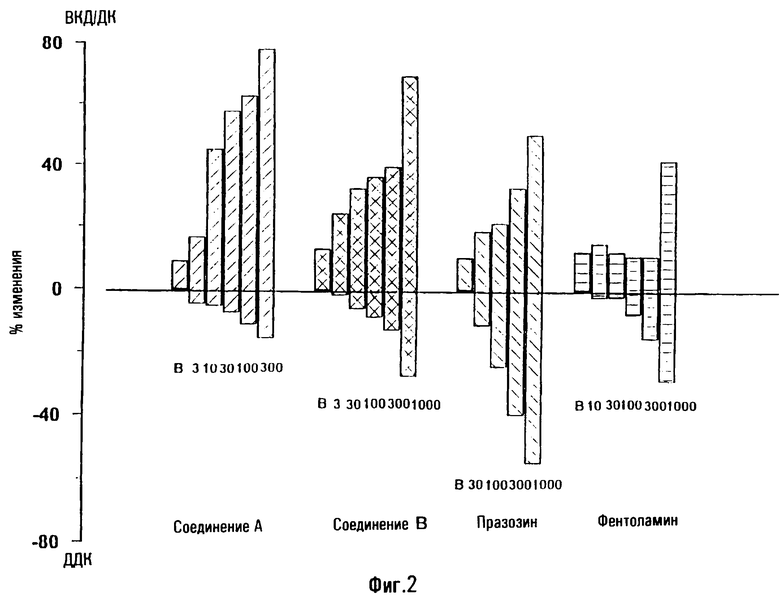
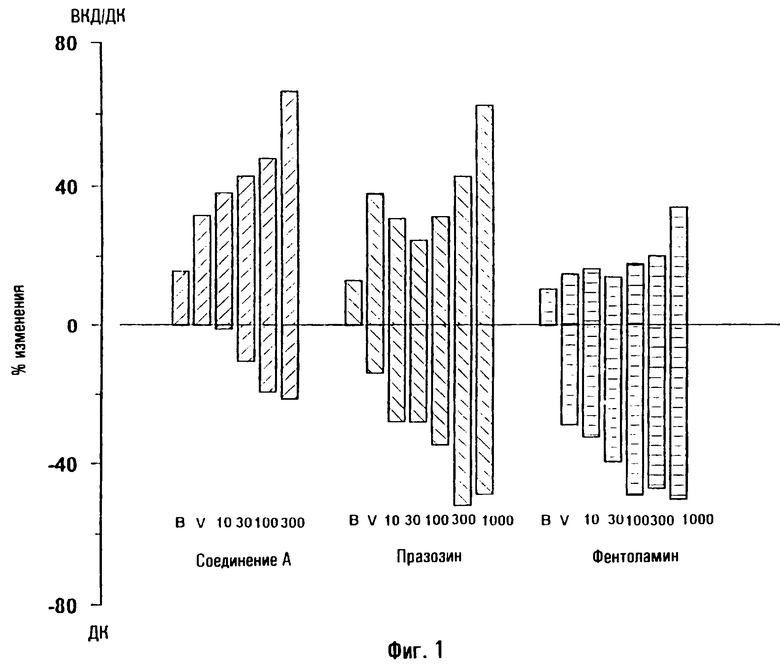
На фигуре 1 показано влияние носителя и различных доз тестированных соединений на отношения ВКД/ДК, соответствующее проценту от давления крови (ДК), достигаемому внутрикавернозным давлением (ВКД(IСР)), после внутрикавернозной инъекции у крыс. Данные представляют собой средние значения данного отношения. Базис: первый столбик; носитель: 2ой столбик; различные тестируемые дозы: остальные столбики (соединение А: 10, 30, 100 и 300 мкг; празозин и фентоламин: 10, 30, 100, 300 и 1000 мкг). Кроме того, показано процентное уменьшение среднего ДК, оцененное по сравнению с базисными значениями, приведенными в таблицах 3-5. Соединение А повышает отношение ВКД/ДК в зависимости от дозы. Увеличения больше 40% были получены на фоне снижения давления крови, не превышавшего 20%. Напротив, повышение ВКД/ДК, вызванное празозином и фентоламином, слабо зависело от дозы, и оба соединения вызывали в одинаковых дозах заметную гипотонию, причем снижение давления крови было равно или превышало 40%.
Соединение А повышало соотношение от 31,8 после инъекции растворителя до 66,4 после инъекции 300 мкг/кг соединения. Повышение, полученное при введении самой большой дозы празозина, отражает лишь снижение давления крови, поскольку ВКД не повышается вовсе. Фентоламин вызывает слабое повышение только при самой большой дозе.
Пример 11
Регистрация внутрикавернозного и кровяного давления у собак
Оценку эректильных свойств на собаках проводили по методу Carati (Carati et al., J. Physiol. 384: 525-538, 1987), с некоторыми изменениями.
Самцов гончей собаки анастезировали пентобарбиталом натрия (в.в. (внутривенно) Nembutal, 35 мг/кг для индукции и 4 мг/кг для поддержания) и интубировали внутритрахеальной трубкой с манжетой для облегчения свободной вентиляции. Коллатераль левой бедренной вены канюлировали РЕ катетером для вливания анестезирующего средства. За соматическим ДК наблюдали с помощью датчика давления Mikro-tip 6F (Miliar Instruments), введенного в дугу аорты через правую общую сонную артерию. ВКД измеряли при помощи иглы 20-размера, помещенной в левое или правое пещеристое тело, и ту же иглу использовали для внутрикавернозной инъекции лекарственных средств. Иглу присоединяли к катетеру, заполненному гепаринизированным физиологическим раствором (25 МЕ/мл). Сигнал давления инициировали ВМ 614/2 усилителями на многоканальном полиграфе. Предназначенные для тестирования соединения вводили путем внутрикавернозной инъекции в объеме 0,5 мл, и после каждой инъекции иглу промывали 0,5 мл физиологического раствора. Наполнители для растворения лекарственных средств испытывали перед первой дозой каждого лекарственного средства. Соединения вводили кумулятивным путем с 30-минутным интервалом между дозами. ВКД (мм рт.ст.) измеряли при пиковом эффекте после введения соединений. Продолжительность тумесценции (ПТ (DT), минут) определяли от начала повышения ВКД от его базисного значения (мм рт.ст.) до возвращения к основной линии. Систолическое давление крови и диастолическое давление крови (мм рт.ст.) измеряли при пиковом эффекте после введения лекарственных средств, для того, чтобы оценить эффекты соединений на ДК независимо от эффектов на ВКД. Кроме того, систолическое давление крови определяли во время максимального значения ВКД после внутрикавернозной инъекции, для оценки отношений ВКД/ДК.
В данных экспериментах соединение А, соединение В и фентоламин (1 или 3 мг/мл) растворяли в 10% (об/об) N,N-диметилформамиде и затем растворяли в деионизированной Н2O. Празозин растворяли в деионизированной H2О. Эти данные представляли как среднее ± с.о. среднего, или процентную вариацию (± с.о.) базисных значений.
Результаты внутрикавернозного введения лекарственных средств анастезированным собакам приведены в таблицах 6-9. Носители, использованные для растворения лекарственных средств, испытывали перед каждой дозой каждого соединения, и они не оказывали влияния ни на внутрикавернозное давление, ни на соматическое давление крови (данные не показаны).
Все тестированные лекарственные средства вызывают повышение внутрикавернозного давления (ВКД). Соединение А повышает ВКД в зависимости от дозы (по сравнению с базисными значениями ВКД) от 12 мм рт.ст. при 3 мкг/кг до 96,5 мм рт.ст. при самой большой дозе (300 мкг/кг). Продолжительность повышения внутрикаверноэного давления (ПТ (DT)) также зависела от дозы и длилась по меньшей мере 27 минут при самой большой дозе.
Соединение вызывало слабую зависящую от дозы гипотонию (рассчитано по диастолическому давлению крови) от -10 до -19 мм рт.ст.. Соединение В повышало ВКД зависящим от дозы образом от 13,7 мм рт.ст. (при 3 мкг/кг) до 73,3 мм рт.ст. (1000 мкг/кг) и вызывало гипотонию лишь при самой большой дозе (-31 мм рт.ст. по диастолическому давлению крови). ПТ длится примерно 40 минут при самой большой дозе. Празозин и фентоламин повышают ВКД при дозах, вызывающих продолжительную гипотонию. Кроме того, повышение ВКД, наблюдаемое после инъекции этих стандартных соединений, не зависело от дозы. Празозин при 1000 мкг вызывал повышение ВКД только на 36 мм рт.ст. и снижал диастолическое давление крови на 71 мм рт.ст. Аналогичным образом, фентоламин (при той же дозе) повышал ВКД на 43 мм рт.ст., но снижал диастолическое давление крови на 37 мм рт.ст..
На фигуре 2 показаны эффекты носителя и различных доз тестированных соединений на соотношение ВКД/ДК, соответствующее проценту от давления крови (ДК), достигаемому внутрикавернозным давлением (ВКД) после внутрикавернозной инъекции собакам. Данные представляют собой средние значения соотношения. Базис: первый столбик; различные исследованные дозы: остальные столбики (соединение А: 3, 10, 30, 100 и 300 мкг; соединение В: 3, 30, 100, 300 и 1000 мкг; празозин: 30, 100, 300 и 1000 мкг; фентоламин: 10, 30, 100, 300 и 1000 мкг). Кроме того, приведены процентные снижения оцененного DBP по сравнению с базисными значениями, приведенными в таблицах 6-9.
Соединение А повышает соотношение ВКД/ДК зависимым от дозы образом. Повышение более 80% было получено при снижениях давления крови, не превышающих 20%. Похожие результаты получили после введения соединения В. Напротив, повышения ВКД/ДК, вызванные празозином, были меньше повышений, полученных после введения соединений А и В, и данное стандартное соединение вызвало значительную гипотонию. Фентоламин повышает соотношение ВКД/ДК только после введения самой большой дозы, что вызывает существенную гипотонию.
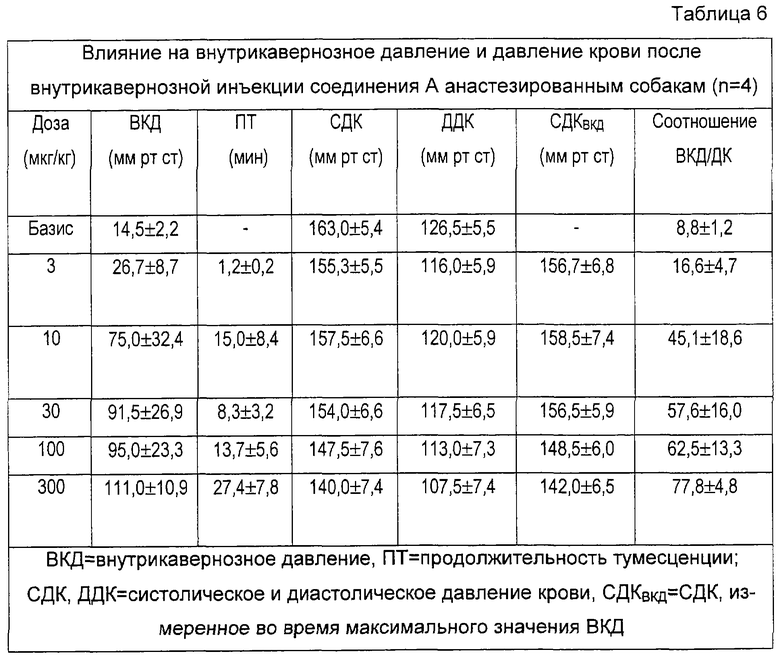
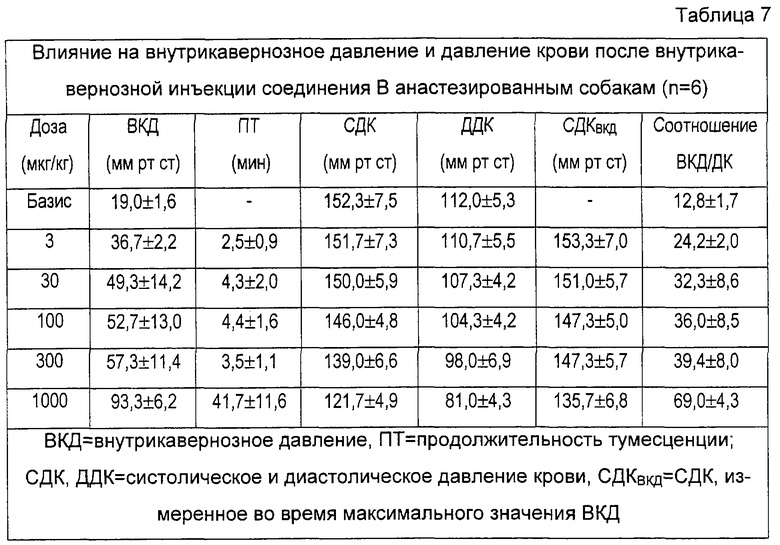
Результаты примеров 10 и 11 показывают полезность селективных α1b-антагонистов для лечения эректильной дисфункции.
Соединение А как у собак, так и у крыс, и соединение В у собак вызывают зависящее от дозы повышение ВКД с очень малыми гипотензивными эффектами. Проэректильную активность таких соединений получали при более низких дозах, чем дозы фентоламина и празозина, и снижение диастолического давления крови было меньше, чем снижение, вызванное стандартными лекарственными средствами.
Фентоламин повышает ВКД у собак и крыс при очень высоких дозах, и его проэректильная активность сопровождается продолжительной гипотонией. Аналогичным образом празозин у собак вызывает повышение ВКД, сопровождающееся сильной гипотонией.
У крыс празозин не повышает внутрикавернозное давление при внутрикавернозном введении, и следовательно, не обладает проэректильными свойствами для данных видов животных.
Кроме того, продолжительность действия, наблюдаемого после инъекции соединения А (и соединения В при самых высоких исследованных дозах) для собак была больше, чем продолжительность действия тестированных стандартных соединений.
Пример 12
Оценка влияния на влагалищное и клиторальное давление у самок кроликов
Способ оценки влияния продуктов данного изобретения на влагалищное и клиторальное давление у самок кроликов представляет собой способ, описанный Park К et. al., Int. J. Import. Res. 9, 27-37 (1997), модифицированный соответствующим образом.
Самок кроликов новозеландской линии анастезировали фенобарбиталом и вводили в сонную артерию катетер для регистрации давления крови. Брюшную и подвздошную артерии, на которые для измерения периферического кровотока помещали электромагнитные датчики кровотока, и ветвь тазового нерва, который раздражает влагалище и клитор, открыли и выделили срединной лапаротомией. Давление в стенке влагалища и клиторе определяли, внедряя иглы (размер 21G), соединенные с датчиком давления, во влагалищное губчатое тело и пещеристые тела клитора соответственно. Тестируемые соединения вводили локально в подэпителиальный слой влагалищной губчатой ткани или вводили внутривенно.
Определяли влияние на влагалищное и клиторальное давление после местного введения и влияние на давление, вызванное электростимуляцией тазового нерва (параметры раздражения: 10 В, 16 Гц, 8 мс).
В указанных выше экспериментальных моделях результаты, полученные с соединениями данного изобретения, показывают их эффективное использование для лечения сексуальной дисфункции на фоне присутствия очень небольших побочных эффектов гипотензивной природы.
Изобретение относится к новым селективным антагонистам α1b-адренергическим рецепторам формулы :
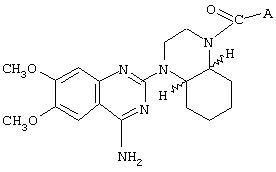
где А представляет собой 2-тетрагидрофурил, 2-фурил, замещенный линейным или разветвленным алкилом, содержащим от 1 до 5 атомов углерода, бензилокси-группу или феноксиалкильную группу, замещенную линейным или разветвленным алкилом, содержащим от 1 до 5 атомов углерода, и/или алкокси-группой, содержащей от 1 до 4 атомов углерода, или энантиомер, диастереоизомер или фармацевтически приемлемая соль такого соединения, а также к фармацевтической композиции на основе этих соединений, обладающий способностью улучшать состояние, связанное с сексуальной дисфункцией. Изобретение относится также к применению известных соединений, аналогичных по своей структуре соединениям формулы I, для получения лекарственных средств для лечения сексуальной дисфункции и фармацевтической композиции на их основе. Технический результат – получение соединений и фармацевтических средств на их основе для лечения сексуальной дисфункции. 4 н. и 15 з.п. ф-лы, 2 ил., 9 табл.
где А представляет собой 2-тетрагидрофурил, 2-фурил, замещенный линейным или разветвленным алкилом, содержащим от 1 до 5 атомов углерода, бензилокси-группу или феноксиалкильную группу, замещенную линейным или разветвленным алкилом, содержащим от 1 до 5 атомов углерода, и/или алкокси-группой, содержащей от 1 до 4 атомов углерода,
или энантиомер, диастереоизомер или фармацевтически приемлемая соль такого соединения.
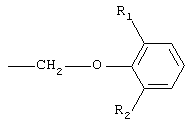
где R1 представляет собой линейную или разветвленную алкильную цепь, содержащую от 1 до 5 атомов углерода;
R2 представляет собой алкоксигруппу, содержащую от 1 до 4 атомов углерода.
4-амино-6,7-диметокси-2-[(±)-4-(2-метокси-6-изопропилфеноксиацетил) -цис-октагидро-1-хиноксалинил] хиназолин,
4-амино-6,7-диметокси-2-[(±)-4-(5-метил-2-фуроил)-цис-октагидро-1-хиноксалинил]хиназолин,
4-амино-6,7-диметокси-2-[(±)-4-(2-тетрагидрофуроил)-цис-октагидро-1-хиноксалинил]хиназолин,
4-амино-6,7-диметокси-2-[(±)-4-бензилоксикарбонил-цис-октагидро-1-хиноксалинил]хиназолин,
или энантиомер, диастереоизомер или фармацевтически приемлемая соль такого соединения.
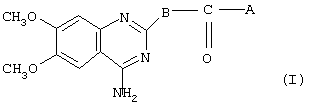
где А представляет собой 2-фурил, 2-тетрагидрофурил, 2-фурил, замещенный линейным или разветвленным алкилом, содержащим от 1 до 5 атомов углерода, бензилоксигруппу или феноксиалкильную группу, замещенную линейным или разветвленным алкилом, содержащим от 1 до 5 атомов углерода, и/или алкоксигруппой, содержащей от 1 до 4 атомов углерода, а В представляет собой одну из следующих групп формулы B1, В2 или В3:
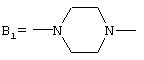
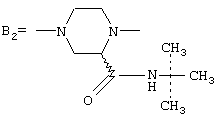
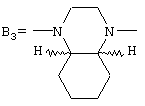
при условии, что (i) если В представляет собой группу B1, то А представляет собой замещенную феноксиалкильную группу, и (ii) если В представляет собой группу В3, то А представляет собой группу 2-фурила,
или энантиомера, диастереоизомера или фармацевтически приемлемой соли такого соединения для получения лекарственного средства для лечения сексуальной дисфункции.
 или
или 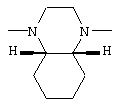
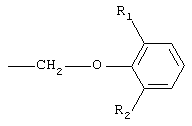
где R1 представляет собой линейную или разветвленную алкильную группу, содержащую от 1 до 5 атомов углерода;
R2 представляет собой алкоксигруппу, содержащую от 1 до 4 атомов углерода.
4-амино-6,7-диметокси-2-[4-(2-метокси-6-изопропилфенокси-ацетил)-1-пиперазинил]хиназолина,
4-амино-6,7-диметокси-2-[(4aR,8aS)-4-(2-фуроил)-цис-октагидро-1-хиноксалинил]хиназолин и
4-амино-6,7-диметокси-2-[(3S)-3-(трет-бутилкарбамоил)-4-(2-фуроил)-1-пиперазинил]хиназолин
или энантиомера, диастереоизомера или фармацевтически приемлемой соли такого соединения.
(a) соединение общей формулы I, как определено в п. 9, или энантиомер, диастереоизомер или фармацевтически приемлемую соль такого соединения, (b) простагландин, прямой вазодилататор или ингибитор 5 цГМФ-фосфодиэстеразы и (c) фармацевтически приемлемый разбавитель или носитель.
| RU 93004423 A, 20.05.1995.RU 2124008 C1, 27.12.1998.Leonardi A | |||
| et al | |||
| J | |||
| of Medical Chemistry | |||
| Металлический водоудерживающий щит висячей системы | 1922 |
|
SU1999A1 |
| Способ уравновешивания движущихся масс поршневых машин | 1925 |
|
SU427A1 |
| Giardin A.D | |||
| et al | |||
| J | |||
| of Medical Chemistry | |||
| Способ изготовления фанеры-переклейки | 1921 |
|
SU1993A1 |
| Регистратор для дел | 1925 |
|
SU690A1 |
| Giardin A.D | |||
| et al | |||
| J | |||
| of Medical Chemistry | |||
| Предохранительное устройство для паровых котлов, работающих на нефти | 1922 |
|
SU1996A1 |
| ВЕРТИКАЛЬНЫЙ ФРЕЗЕРНЫЙ СТАНОК ДЛЯ ОБРАБОТКИ СТЫКОВ В СОЕДИНЯЕМЫХ ПОД УГЛОМ ТРУБАХ | 1925 |
|
SU4602A1 |

