Область, к которой относится изобретение
Данное изобретение относится к новым производным 3-замещенного 4-арилхинолин-2-она, которые являются модуляторами активируемых кальцием калиевых каналов (ВК) с высокой проводимостью, и, следовательно, пригодны для защиты нервных клеток и для защиты от болезней, возникающих вследствие дисфункции поляризации клеточных мембран и проводимости.
Данное изобретение предусматривает также способ лечения при помощи производных замещенного хинолин-2-она и фармацевтические композиции на их основе.
Предпосылки создания изобретения
Калиевые каналы играют ключевую роль в регулировании потенциала клеточной мембраны и модуляции возбуждаемости клеток. Калиевые каналы в значительной степени регулируются амплитудой, метаболизмом клеток, процессами, опосредованными кальцием и рецепторами [Cook, N. S., Trends in Pharmacol. Sciences (1988), 9, 21, и Quast U., et al., Trends in Pharmacol. Sciences (1989), 10, 431]. Активируемые кальцием калиевые (Кca) каналы представляют собой другую группу ионных каналов, активность которых зависит от внутриклеточных ионов кальция. Активность Кca каналов регулируется потенциалом внутриклеточной [Са2+] мембраны и фосфорилированием. На основе проводимостей единичных каналов в симметричных растворах К+ Кса каналы подразделяются на три подкласса: с высокй проводимостью (ВК)>150 пс; с промежуточной проводимостью 50-150 пс; с низкой проводимостью <50 пс. Активируемые кальцием калиевые каналы с высокой проводимостью (Maxi-K или ВК) имеются во многих возбуждающихся клетках, включая нейроны, кардиальные клетки и различные типы клеток гладкой мышцы [Singer, et al., Pflugess Archiv. (1987), 408, 98; Baro, I., et al., Pflugess Archiv. (1989) 414 (Suppl. 1), S168, и Ahmed F. et al., Br.J.Pharmacol. (1984) 83, 227].
Ионы калия играют главную роль в регулировании остаточного потенциала мембраны в большинстве возбуждаемых клеток и поддерживают трансмембранное напряжение на уровне потенциала равновесия К+ (Ек), равного примерно 90 мВ. Было установлено, что открывание калиевых каналов сдвигает потенциал клеточной мембраны в сторону равновесия ионов калия (Ек), в результате чего возникает гиперполяризация клетки [Cook, N.S., Trends in Pharmacol. Sciences (1988), 9, 21]. Гиперполяризованные клетки показывают уменьшенный ответ на потенциально вредные деполяризующие раздражители. Каналы ВК, которые регулируются как напряжением, так и внутриклеточными ионами Са2+, ограничивают деполяризацию и поступление кальция и могут частично эффективно блокировать вредные раздражители. Следовательно, гиперполяризация клеток через открывание каналов ВК может привести к защите нервных клеток.
Ряд синтетических и природных соединений со способностью открывать ВК известен. Пирон, экстрагированный из овса посевного avena sativa, был описан как открыватель каналов ВК при использовании метода двухслойных липидов (заявка WO 93/08800, опубликованная 13 мая 1993 г). При проведении очень ограниченного числа электрофизиологических экспериментов было установлено, что 6-бром-8-(метиламино)имидазо[1,2-а]пиразин-2-карбонитрил (SCA-40) является открывателем каналов ВК [Laurent, F. et al., Br. J. Pharmacol. (1993) 108, 622-626]. Было установлено, что флаваноид Phloretin увеличивает возможность открывания активированных Са2+ калиевых каналов в миелинизованных нервных волокнах Xenopus laevis при использовании внешних пэтчей [Koh, D-S, et al., Neuroscience Lett. (1994) 165, 167-170].
В заявке ЕР-А-477819, опубликованной 4 января 1992 года, и в соответствующем патенте США 5200422, выданном 6 апреля 1993 года Olesen и др., описаны различные производные бензимидазола в качестве открывателей каналов ВК при использовании опытов с единичными каналами и фиксацией потенциала всей клетки в клетках гладкой мышцы аорты. Последующие исследования были описаны Olesen et al. в European J. Pharmacol., 251, 53-59(1994).
P.Hewawasam et al. в патенте США 5565483, выданном 15 октября 1996 года, описали ряд замещенных оксиндолов, обладающих способностью открывать ВК каналы. Sit et al. в заявке WO 98/23273, опубликованной 4 июня 1998 года, и в соответствующем патенте США 5892042, выданном 6 апреля 1999 года, раскрыли группу производных 4-арил-3-гидроксихинолин-2-она, a Hewawasam et al. в заявке WO 99/09983, опубликованной 4 марта 1999 года, описали группу производных 4-арил-3-аминохинолин-2-она, являющихся открывателями каналов ВК и производными для лечения нарушений, чувствительных к открыванию калиевых каналов.
E.S.Hamanaka в патенте США 5565472, выданном 15 октября 1996 года, описал ряд 4-арил-3-(гетероарилуреидо)-1,2-дигидро-2-оксохинолина, являющихся ингибиторами ациклоэнзима А; холестеролацилтрансферазы и пригодных в качестве гиполипидемических и антиатеросклеротических агентов.
Целью данного изобретения является создание новых соединений, которые способны модулировать калиевые каналы, в частности активированные кальцием калиевые каналы с высокой проводимостью (ВК), и которые будут пригодны при лечении болезней, возникающих вследствие дисфункции поляризации и проводимости клеточной мембраны.
Сущность изобретения
Настоящее изобретение предусматривает новые производные 3-замещенного 4-арилхинолин-2-она, имеющие общую формулу
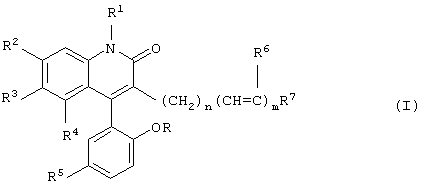
где R, R1, R2, R3, R4, R5, R6 и R7 определены ниже,
и их фармацевтически приемлемые нетоксичные соли, которые являются открывателями активированных кальцием калиевых каналов с высокой проводимостью, также известных как Maxi-K или ВК каналы.
Детальное описание изобретения
Данное изобретение предусматривает новые производные 3-замещенного 4-арилхинолин-2-она, которые являются сильнодействующими открывателями активированных кальцием калиевых каналов с высокой проводимостью (каналы ВК) и имеют формулу
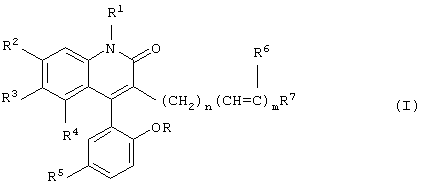
где R и R1 каждый независимо обозначает водород или метил;
R2, R3 и R4 каждый независимо обозначает водород или трифторметил при условии, что R2, R3 и R4 не являются одновременно водородом;
R5 обозначает бром или хлор;
R6 обозначает водород или фтор;
n обозначает целое число от 0 до 6;
m обозначает целое число 0 или 1 и
R7 обозначает СН3, CRR1OH, CHO, C=NOH, СОСН3 или арил, возможно замещенный одним или двумя заместителями, выбранными из группы, состоящей из гидрокси, метокси, амино и ацетиламино,
или их нетоксичные фармацевтически приемлемые соли.
Термин “нетоксичная фармацевтически приемлемая соль”, используемый в описании и в формуле изобретения, включает нетоксичные соли присоединения к неорганическим основаниям. Подходящие неорганические основания, такие как основания щелочных и щелочноземельных металлов, включают основания металлов, таких как натрий, калий, магний, кальций и т.п. Если иное не оговорено, термин “галоген”, используемый в описании и в формуле изобретения, включает бром, хлор, йод и фтор, а термин “галоидное соединение” включает бромид, хлорид и йодид.
Некоторые соединения по изобретению могут существовать в виде несольватируемых форм и в виде сольватируемых форм, включая гидраты, такие как моногидрат, дигидрат, гемигидрат, тригидрат, тетрагидрат и т.п. Продукты могут быть истинными сольватами, а в других случаях продукты могут просто удерживать случайный растворитель или быть смесью сольвата и некоторого количества случайного растворителя. Специалистам в данной области очевидно, что сольватированные формы эквивалентны несольватированным и входят в объем данного изобретения. Некоторые соединения формулы I могут существовать в двух таутомерных формах. Специалистам очевидно, что когда R1 обозначает водород у атома азота, смежного по отношению к атому углерода карбонильной группы, хинолиновое кольцо может существовать в енольной форме. Оба енольных таутомера соединений формулы I входят в объем данного изобретения.
Предпочтительные для использования соединения включают соединения формулы I, перечисленные ниже:
4-(5-хлор-2-метоксифенил)-3-(гидроксиметил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-метоксифенил)-3-(гидроксиметил)-7-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-метоксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)-3-хинолинкарбоксальдегид;
4-(5-хлор-2-метоксифенил)-3-(3-гидрокси-1-пропенил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-метоксифенил)-3-(3-гидроксипропил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-(гидроксиметил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-(3-гидрокси-1-пропенил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-(3-гидроксипропил)-6-(трифторметил)-2(1Н)-хинолинон;
(Е)-4-(5-хлор-2-гидроксифенил)-3-(2-фтор-3-гидрокси-1-пропенил)-6-(трифторметил)-2(1Н)-хинолинон;
(Z)-4-(5-хлор-2-гидроксифенил)-3-(2-фтор-3-гидрокси-1-пропенил)-6-(трифторметил)-2(1Н)-хинолинон;
(Е)-4-(5-хлор-2-метоксифенил)-3-(2-фтор-3-гидрокси-1-пропенил)-6-(трифторметил)-2(1Н)-хинолинон;
(Z)-4-(5-хлор-2-метоксифенил)-3-(2-фтор-3-гидрокси-1-пропенил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-(2-гидроксиэтил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-метоксифенил)-3-(2-гидроксиэтил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-метоксифенил)-3-(4-метоксифенил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-метоксифенил)-3-[(4-метоксифенил)метил]-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-метоксифенил)-3-(4-аминофенил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-(3,4-диметоксифенил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-(2,4-дигидроксифенил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-(4-гидроксифенил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-[(4-гидроксифенил)метил]-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-(4-ацетамидофенил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-(4-аминофенил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-[2-(4-гидроксифенил)этил]-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-метил-6-(трифторметил)-2(1Н)-хинолинон;
4-[4-(5-хлор-2-гидроксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)хинолин-3-ил]-3-бутен-2-он;
4-(5-хлор-2-гидроксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)-3-хинолинкарбоксальдегидоксим;
4-(5-хлор-2-метоксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)-3-хинолинкарбоксальдегидоксим и
4-(5-хлор-2-гидроксифенил)-3-(2-гидрокси-2-метилпропил)-6-(трифторметил)-2(1Н)-хинолинон.
Соединения формулы I могут быть получены различными способами, такими как описанные в примерах и показанные на схемах реакции, приведенных в конкретных примерах, модификации этих описанных способов очевидны для специалистов в данной области.
Реакционные схемы 1-11 иллюстрируют примеры общих методик получения промежуточных соединений и способ получения соединений по изобретению. Специалистам в данной области очевидно, что подходящая замена как соединений, так и описанных способов также входит в объем данного изобретения.
Реакционная схема 1
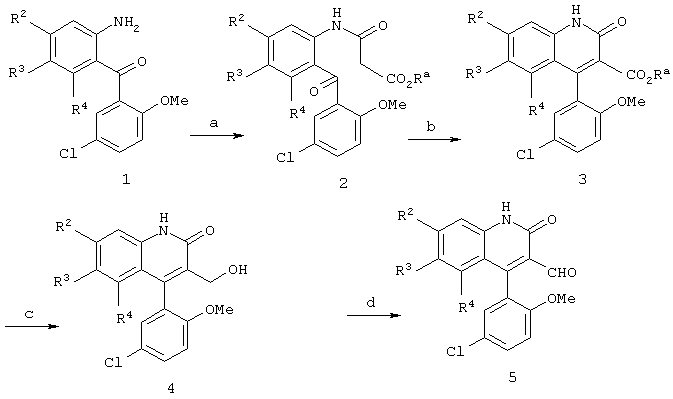
(a) ClC(O)CH2CO2Ra, пиридин, CH2Cl2, 0°C - комнатная температура;
(b) KO+Bu, ТГФ, нагревание с обратным холодильником;
(c) Dibal-H, ТГФ-гексаны, -78°С - комнатная температура;
(d) MnO2, CH2Cl2.
Получение 2(1Н)-хинолинонов формул 4 и 5 показано на реакционной схеме 1. Как показано на этой схеме, ацилирование соединения формулы I ацилхлоридом привело к получению амида формулы 2, где Ra обозначает водород или С1-4алкил, который может циклизоваться и дегидратироваться с получением хинолинона формулы 3 при обработке основанием, таким как трет-бутоксид калия, в инертном органическом растворителе. Обработка эфира формулы 3 восстановителем, таким как диизобутилалюминийгидрид, приводит к образованию первичного спирта формулы 4, который может быть затем окислен окислителем, таким как диоксид марганца, с получением желаемого альдегида формулы 5.
Реакционная схема 2
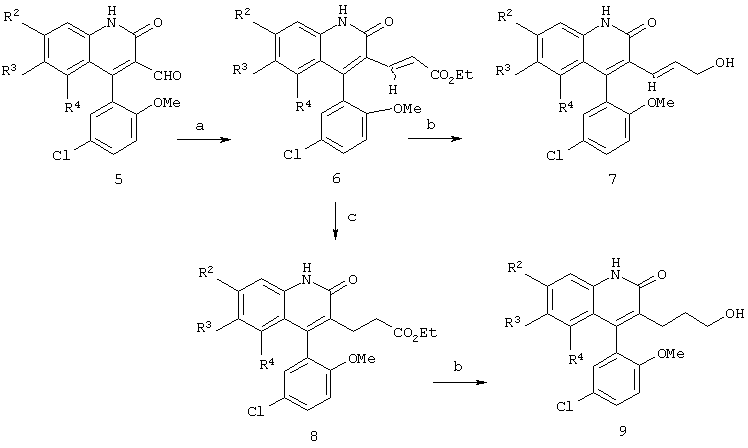
(a) EtOC(O)CH2P(O)(OEt)2, NaH, ДМФ;
(b) Dibal-H, ТГФ,-78°С;
(c) РtO2, ЕtOН-НСl, Н2 (60 ф/дюйм2=413,7 кПа).
Как показано на реакционной схеме 2, гомологизация альдегида формулы 5 может быть легко осуществлена фосфонатом с получением ненасыщенного эфира формулы 6 в виде смеси (Е)- и (Z)-изомеров, которые затем разделяют методом хроматографии на колонке. Восстановление эфира формулы 6 можно осуществить восстановителем, таким как диизобутилалюминийгидрид, с образованием соответствующего аллилового спирта формулы 7. Кроме того, когда желательно получить соединение формулы 9, эфир формулы 6 селективно восстанавливают в условиях гидрирования для восстановления двойной связи и получения насыщенного эфира формулы 8. Обработка эфира формулы 8 в условиях, похожих на условия восстановления эфира формулы 6, приводит к получению соответствующего спирта формулы 9.
Реакционная схема 3
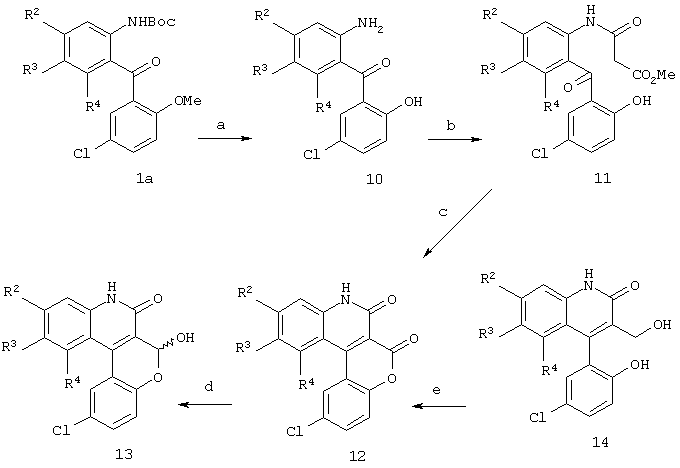
(a) ВВr3, CH2Cl2, -78 -0°С;
(b) СlС(O)СН2СO2Ме, пиридин, СН2Сl2;
(c) KO+Bu, ТГФ, нагревание с обратным холодильником;
(d) Dibal-H, ТГФ, -78°С;
(e) Dibal-H, СН2Cl2 -78°С.
На реакционной схеме 3 показано, что бутилоксикарбонильная (ВОС) и метильная группы могут быть удалены одновременно путем обработки соединения формулы 1а трибромидом бора (ВВr3) с получением анилина формулы 10. Ацилирование анилина формулы 10 позволило получить соответствующий амид формулы 11, который легко циклизуется и дегидратируется в основных условиях при помощи трет-бутоксида калия с образованием лактона формулы 12. Частичное восстановление лактона диизобутилалюминийгидридом в ТГФ привело к получению промежуточного лактола формулы 13. Кроме того, было установлено, что при замене ТГФ метиленхлоридом лактон формулы 12 можно восстановить при помощи диизобутилалюминийгидрида с получением желаемого спирта формулы 14.
Реакционная схема 4
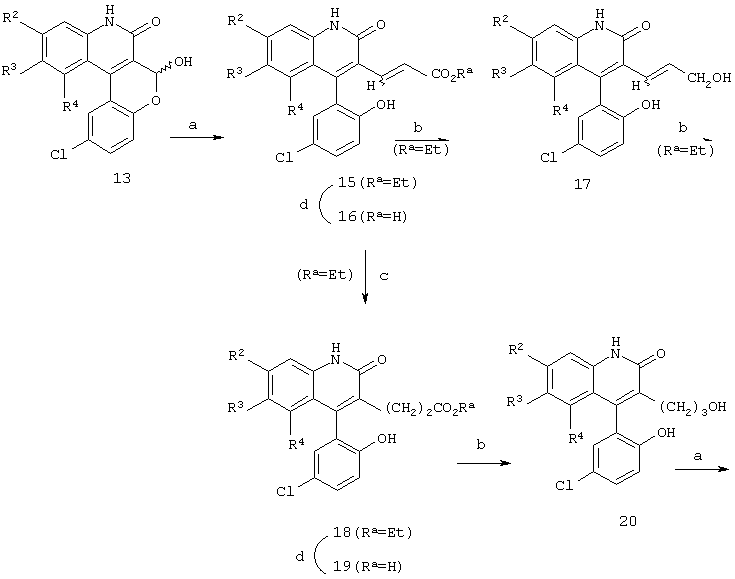
(a) EtOC(O)CH2P(O)(ORa)2, NaH, ДМФ;
(b) Dibal-H, ТГФ - гексаны, -78°С - комнатная температура;
(c) PtO2, ЕtOН-НСl, Н2 (60 ф/дюйм2=413,7 кПа);
(d) NaOH, EtOH, комнатная температура.
Когда нужно получить соединения формул 17 и 20, промежуточный лактол формулы 13 можно обработать, как показано на схеме 4, фосфонатом с получением ненасыщенного эфира формулы 15, затем, если желательно, осуществить омыление с получением ненасыщенной кислоты формулы 16. Кроме того, гидрирование двойной связи соединения формулы 15 приводит к образованию эфира формулы 18, который можно или подвергнуть омылению с получением кислоты формулы 19, или восстановить алюмийгидридом с получением спирта формулы 20.
Реакционная схема 5
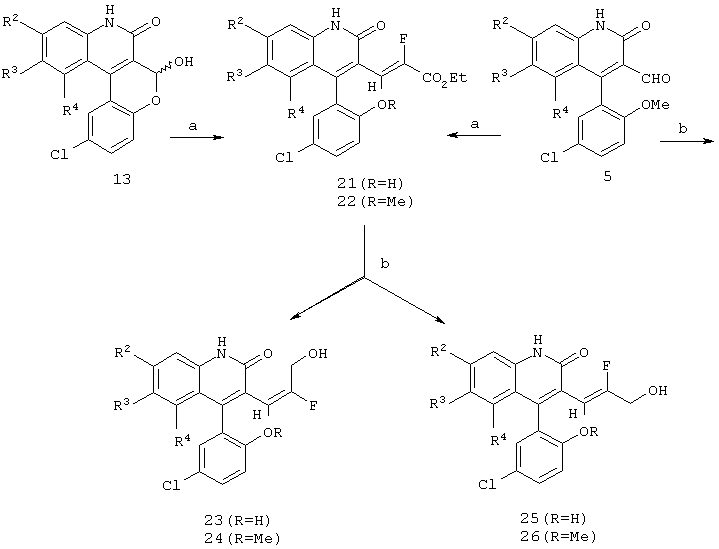
(a) EtOC(O)CHFP(O)(OEt)2, NaH, ДМФ;
(b) Dibal-H, CH2Cl2 -78°C - комнатная температура.
На реакционной схеме 5 показана гомологизация промежуточного лактола формулы 13 фторфосфонатом, как показано на стадии (а) реакционной схемы, что приводит к получению ненасыщенного α -фторированного эфира формулы 21 в виде смеси (Е)-и (Z)-изомеров. Техническую смесь эфиров формулы 21 можно восстановить алюминийгидридом и поученную смесь спиртов разделить хроматографией на колонке с получением (Е)-олефина формулы 23 и (Z)-олефина формулы 25. Подобным образом альдегид формулы 5, который содержит метоксигруппу, можно превратить в соответствующие олефины формул 24 и 26.
Реакционная схема 6
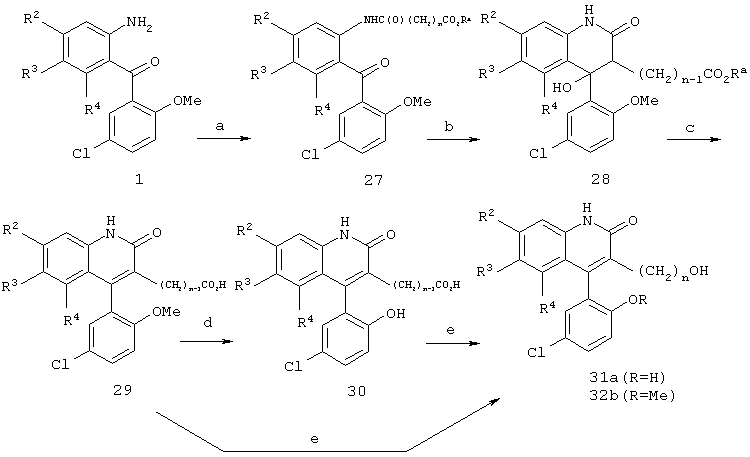
(a) ClC(O)(CH2)nCO2Ra, пиридин, СН2Cl2;
(b) KHMDS, ТГФ, -78°С;
(c) 35% HBr - АсОН, толуол, 80-90°С;
(d) пиридин·НСl,180-200°С;
(e) ВН3·3Ме2, ТГФ, -10°С - комнатная температура.
Получение соединений формул 31а и 31b показано на реакционной схеме 6. Ацилирование анилина формулы 1 хлорангидридом кислоты приводит к образованию соответствующего амида. Амид формулы 27 можно подвергнуть циклизации в основных условиях с получением дегидрогидроксихинолинона формулы 28, который затем можно дегидратировать и деэтерифицировать в кислых условиях, например, при помощи НВr/АсОН или п-TsOH, с получением хинолинона формулы 29. Если желательно, удаление метоксигруппы можно осуществить гидроксихлоридом пиридина при повышенных температурах с получением соответствующего фенола формулы 30. Восстановление кислоты формулы 30 приводит к получению спирта формулы 31а в виде фенола. Кроме того, если нужен метиловый эфир фенола, осуществляют прямое восстановление карбоновой кислоты формулы 29 бораном, что приводит к получению соответствующего спирта формулы 31b.
Реакционная схема 7
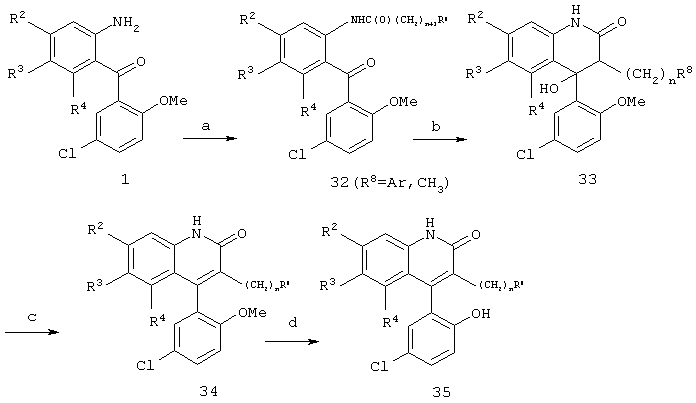
(a) ClC(O)(CH2)n+1R8, пиридин, CH2Cl2;
(b) KHMDS, ТГФ, -78°C;
(c) H+
(d) пиридин · HCl, 180-200°C.
Соединения формул 34 и 35, где n=0-6 и R8 обозначает С1-4алкил или арил, возможно замещенный одним или двумя заместителями, выбранными из группы, состоящей из галогена, гидрокси, метокси, амино, ацетиламино и трифторметила, могут быть получены способом, аналогичным показанному на реакционной схеме 6. Так, реакционная схема 7 иллюстрирует ацилирование соединения формулы 1 с последующими циклизацией и дегидратированием с получением 3-замещенного хинолина формулы 34 в виде метилового эфира. Деметилирование соединения формулы 34 с помощью гидроксихлорида пиридина при повышенных температурах приводит к образованию фенола формулы 35.
Реакционная схема 8
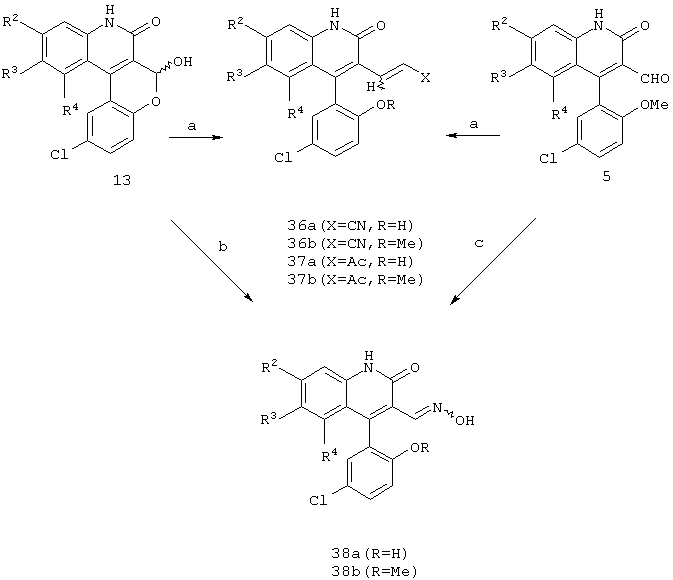
(a) (МеО)2Р(O)СН2Х, NaH, ДМФ;
(b) NH2OH· HCl, Et3N, ТГФ;
(c) NH2OH· HCl, NaOAc, EtOH.
Как показано на реакционной схеме 8, гомологизация промежуточного лактола формулы 13 цианфосфонатом или фосфонацетатом приводит к образованию соответствующего ненасыщенного нитрила формулы 36а или ацетата формулы 37а соответственно. Подобным образом метиловые эфиры формул 36b и 37b могут быть синтезированы из альдегида формулы 5 обработкой или цианфосфонатом или фосфонацетатом соответственно. Оксим формулы 38а может быть получен из промежуточного лактола формулы 13 путем обработки лактола гидроксиламином. Точно так же метиловый эфир формулы 38b может быть получен из альдегида формулы 5.
Реакционная схема 9
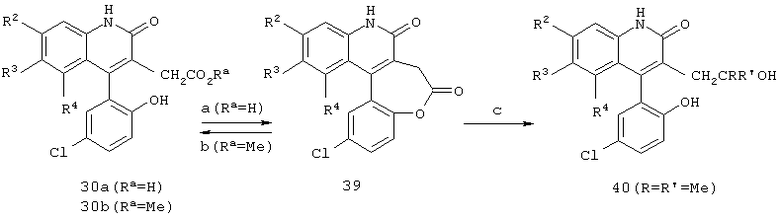
(a) кат. п-TsOH, толуол, нагрев с обратным холодильником;
(b) МеОН, силикагель;
(c) RR1Li, ТГФ, -78°С.
Реакционная схема 9 иллюстрирует образование лактона формулы 39, когда оксикислота формулы 30а обрабатывается каталитическим количеством кислоты в среде толуола при нагревании с обратным холодильником. После очистки лактона формулы 39 на силикагеле с использованием метанола в качестве одного из элюентов, можно превратить лактон в эфир формулы 30b. Если желательно получить замещенный спирт формулы 40, лактон формулы 39 обрабатывают избытком литиевого реагента, такого как метиллитий, с получением дизамещенного спирта формулы 40 или эквивалентным количеством этого реагента с получением монозамещенного спирта.
Реакционная схема 10
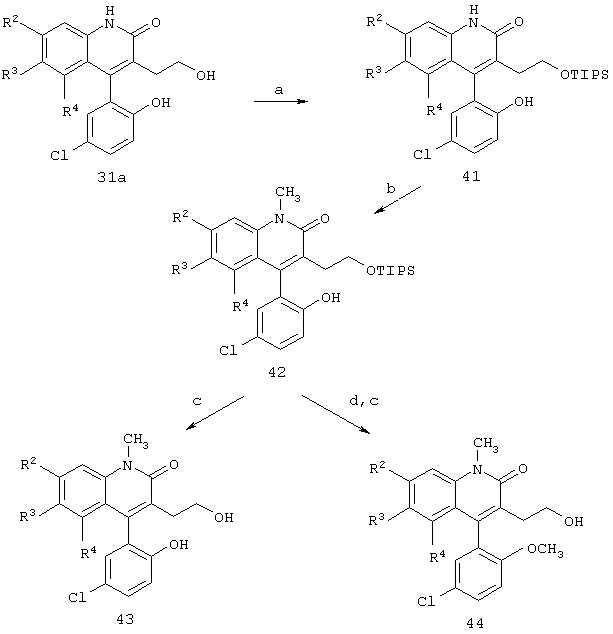
(a) TlPSCl, имидазол, ДМФ;
(b) н-BuLi, СН3I, ТГФ;
(c) ТВАF, ТГФ;
(d) К2СО3, (СН3О)2SO2, ацетон.
Получение N-метильных соединений формул 43 и 44 отражено на реакционной схеме 10. Силилирование спирта формулы 31а триизопропилсилил(ТIРS)хлоридом приводит к получению силилзащищенного эфира формулы 41. N-Алкилирование алкилгалогенидом, таким как метилиодид, позволяет получить соединение формулы 42, которое может быть десилилировано фторидом, стадия (с), с получением соединения формулы 43. Если желательно получить метилированный фенол, соединение формулы 41 обрабатывают диметилсульфатом с последующим десилилированием с получением диметильного аналога формулы 44.
Реакционная схема 11
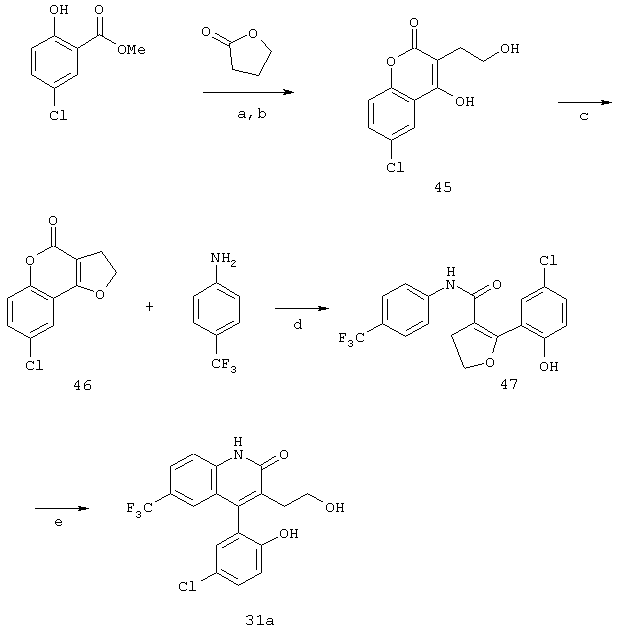
(a) LiHMDS/ТГФ, -78°С ТО, комн. темп.;
(b) 12NHCl;
(c) п-TSA, толуол, нагрев с обратным холодильником;
(d) LiHMDS;
(e) уф, МеОН.
Получение соединения формулы 31а осуществляют, как показано на реакционной схеме 11. Кумарин формулы 45 получают конденсацией γ -бутиролактона с метиловым эфиром хлорсалициловой кислотой, потом его циклизуют при помощи кислоты с получением бензопиран-4-она формулы 46. Обработка соединения 46 п-трифторметиланилином, как показано на стадии (d), приводит к получению дигидрофурана формулы 47, который затем подвергают фотоциклизации в инертном растворителе с получением соединения формулы 31а.
Согласно предпочтительному варианту изобретения соединения формулы I имеют формулу

где R и R1 каждый независимо обозначают водород или метил; R2, R3 и R4, каждый независимо, обозначают водород, галоген, нитро или трифторметил, при условии, что R2, R3 и R4 не являются все одновременно водородом; R5 обозначает бром, хлор или нитро; R6 обозначает водород или фтор; n обозначает целое число от 0 до 6; m обозначает целое число 0 или 1; R7 обозначает СН3, -CRR1OH, CHO, O=NOH, -СОСН3 или арил, возможно замещенный одним или двумя заместителями, выбранными из группы, состоящей из галогена, гидрокси, метокси, амино, ацетиламино и трифторметила, или их нетоксичные фармацевтически приемлемые соли.
Согласно еще одному предпочтительному варианту изобретения соединения являются такими, у которых R и R1 каждый независимо обозначает водород или метил; R2, R3 и R4, каждый независимо, обозначают водород, хлор, нитро или трифторметил при условии, что R2, R3 и R4 не являются все водородом; R5 обозначает хлор; R6 обозначает водород или фтор; n равно 0, 1 или 2; m равно 0 или 1 и R7 обозначает -СН3, -СН2ОН, СНО, -C=NOH, -COCH3 или арил, возможно замещенный галогеном, гидрокси, метокси, амино, ацетиламино или трифторметилом, или их нетоксичными фармацевтически приемлемыми солями.
Согласно еще одному более предпочтительному варианту изобретения соединения формулы I включают соединения, у которых R обозначает водород или метил; R1 и R4 обозначают водород; R2 и R3 каждый независимо обозначает трифторметил; R5 обозначает хлор; R6 обозначает водород; n равно 0, 1 или 2; m равно 0 или 1 и R7 обозначает -СН2ОН или арил, возможно замещенный галогеном, гидрокси, метокси, амино, ацетиламино или трифторметилом, или их нетоксичные фармацевтически приемлемые соли.
Согласно другому аспекту изобретение предусматривает способ лечения или профилактики расстройств, опосредованных открыванием активированных кальцием калиевых каналов с высокой проводимостью (ВК каналов) у млекопитающего, нуждающегося в таком лечении или профилактике, который включает введение указанному млекопитающему терапевтически эффективного количества соединения формулы I или его нетоксичной фармацевтически приемлемой соли. Предпочтительно использовать соединения формулы I при лечении ишемии, удара, конвульсий, астмы, синдрома раздраженной кишки, мигрени, травматического повреждения мозга, недержания мочи и сексуальных расстройств как у мужчин (нарушение эрекции, например, обусловленное сахарным диабетом, повреждением спинного мозга, радикальной простатэктомией, психогенной этиологией или любой другой причиной), так и у женщин за счет улучшения притока крови к половым органам, особенно к corpus cavemosum, и других расстройств, чувствительных к действию ВК каналов.
Согласно еще одному аспекту данное изобретение предусматривает фармацевтические композиции, содержащие, по меньшей мере, одно соединение формулы I в сочетании с фармацевтическим адъювантом, носителем или разбавителем.
Биологическая активность
Калиевые (К+) каналы представляют собой структурно и функционально различающиеся группы селективных для К+ белков, которые имеются в клетках, играя основную роль в регулировании целого ряда ключевых функций клетки [Rudy, В., Neuroscience, 25: 729-749 (1988)]. Будучи распределены широко как класс, К+ каналы распределены по-разному как индивидуальные члены этого класса или как группы [Gehlert, D.R., et al., Neuroscience, 52: 191-205 (1993)]. Вообще активация К+ каналов в клетке, особенно в возбудимых клетках, таких как нейроны или клетки мышц, приводит к гиперполяризации клеточной мембраны или, в случае деполяризованных клеток, к повторной поляризации. В добавление к действию в качестве вольт-клампа эндогенной мембраны К+ каналы могут отвечать на важные события в клетках, такие как изменения внутриклеточной концентрации АТФ или внутриклеточной концентрации кальция (Са2+). Центральная роль К+ каналов в регулировании многочисленных функций клетки делает их особенно важными мишенями для терапевтического воздействия [Cook, N.S., Potassium channels: Structure, classification, function and therapeutic potential. Ellis Horwood. Chinchester (1990)]. Один класс К+ -каналов, активированных Са2+ К+ - каналов с высокой проводимостью (Maxi-K или ВК каналов) регулируется трансмембранным потенциалом, внутриклеточным Са2+ и другими различными факторами, такими как степень фосфорилирования белка каналов [Latorre, R., et al., Ann. Rev. Physiol., 51: 385-399 (1989)]. Высокая проводимость единичного канала (обычно >150 пС) и высокая степень специфичности для К+ ВК каналов свидетельствуют, что небольшое число каналов может оказывать глубокое влияние на проводимость мембран и возбудимость клеток. Кроме того, возрастание вероятности открывания с увеличением внутриклеточного Са2+ показывает, что ВК каналы участвуют в модуляции зависимых от Са2+ явлений, таких как секреция и сокращение мышц [Asano, M., et al., J. Pharmacol. Exp. Ther., 267: 1277-1285 (1993)].
Открыватели ВК каналов проявляют свое действие на клетки путем увеличения вероятности открывания этих каналов [МсКау, M.С., et al., J. Neurophysiol. 71: 1873-1882 (1994); Olesen, S.-P., Exp. Opin. Invest. Drugs, 3: 1181-1188 (1994)]. Это возрастание открывания отдельных ВК каналов приводит к гиперполяризации клеточных мембран, особенно в деполяризованных клетках, вызванной значительным увеличением проводимости всей клетки, опосредованной ВК.
Способность соединений, описанных в данной заявке, открывать ВК каналы и повышать количество выходящих из клетки К+, опосредованных ВК, оценивали в условиях вольт-клампа путем определения их способности увеличивать количество ионов, выходящих из клеток человека, клонированных, опосредованных ВК (mSlo или hSlo), экспрессированных гетерологично в ооцитах Xenopus [Butler, A., et al., Science, 261:221-224 (1993), и Dworetzky, S.I., et al., Mol. Brain Res., 27: 189-193 (1994)]. Две используемые ВК модели представляли собой почти идентичные по структуре гомологичные белки, оказавшиеся, как было доказано, фармакологически идентичными в наших опытах. Для изолирования потока ВК от естественного (основного, не-ВК) потока был использован в супермаксимальной концентрации (50 нМ) специфичный и сильнодействующий токсин ибериотоксин (IBTX), блокирующий ВК каналы [Galvez, A., et al., J. Biol. Chem., 265: 11083-11090 (1990)]. Относительный вклад потока ионов в ВК каналах в общий поток ионов из клетки определяли путем вычитания потока, остающегося в присутствии IBTX (не-ВК потока) из профилей потока, полученных во всех других условиях эксперимента (контрольные образцы, лекарство и промывка). Было установлено, что при испытанной концентрации соединения не оказывают влияния на не-ВК естественные потоки в ооцитах. Все соединения испытывали в, по меньшей мере, 5 ооцитах при единственной концентрации, равной 20 мкМ; влияние выбранных соединений формулы I на поток ионов в ВК выражен в процентах от потока ионов, чувствительного к IBTX (контроль), данные приведены в таблице. Результаты записывались при использовании стандартного метода вольт-клампа с двумя электродами [Stuhmer, W., et al., Methods in Enzymology, Vol.207:309-339 (1992)]. Методика вольт-клампа состояла из стадий деполяризации продолжительностью 500-750 мс от потенциала покоя, равного -60 мВ, до +140 мВ со стадиями в 20 мВ. Среда (модифицированный раствор Барта) содержала (в мМ): NaCl (88), НаНСО3 (2,4), КСl (1,0), HEPES (10), MgSO4 (0,82), Са(NО3)2 (0,33), СаСl2 (0,41); рН 7,5.

Для определения способности этих соединений снижать потерю клеток вследствие нейронной ишемии использовали стандартную модель грызуна с постоянной фокальной ишемией, включающей окклюзию в средней церебральной артерии у спонтанно гипертензивной крысы (модель с окклюзией средней церебральной артерии (МСАО)) [Tamura, A., et al., Journal of Cerebral Blood Flow and Metabolism, Volume 1, 53-60 (1981)]. Выбранные соединения испытывали на модели с фокальным ударом, включающей перманентную МСАО у спонтанно гипертензивной крысы. Эта процедура приводит к обширному неокортикальному инфаркту, объем которого измеряют путем исключения витального красителя в последовательных срезах ткани мозга через 24 ч после МСАО. В данном опыте соединения вводили внутривенно через 2 ч после окклюзии. Например, в этой модели соединение по примеру 21 снижало объем кортикального инфаркта примерно на 25% при введении единичного болюса (0,003 мг/кг) через 2 ч после окклюзии средней церебральной артерии по сравнению с контрольньм опытом при введении носителя (2% ДМСО, 98% пропиленгликоля).
In vivo модель для определения эректильной функции подробно описана в научной литературе [Rehman, J., Chenven, E., Brink, P., Peterson, В., Wolcott, В., Wen, Y.P., MelmanA., Christ, G.: Diminished neurogenic but not pharmacological elections in the 2-3-month experimentally diabetic F-344 rat. Am. J. Physiol. 272: H1960-H1971 (1997)]. Вкратце, крыс (250-600 г) подвергали анестезии при помощи пентобарбитала натрия, вскрывали брюшную полость и идентифицировали кавернозный нерв. В правый corpus cavemosum для измерения внутрикавернозного давления (IСР) помещали катетер. Второй катетер вводили для измерения кровяного давления в сонную артерию. Через катетер, помещенный в яремную вену, вводили испытуемое соединение (0,1, 0,3 и 1 мг/кг, внутривенно) или носитель (ПЭГ 400).
Контрольное внутрикавернозное давление определяли при электростимуляции кавернозного нерва при помощи биполярных стимулирующих электродов (20 Гц, пульс 0,22 мс). Амплитуда раздражителей (0,2-20 мА) регулировалась для получения субмаксимального внутрикавернозного давления (обычно 0,2 или 0,5 мА). Затем, используя постоянное значение амплитуды раздражителей, получали ряд контрольных значений внутрикавернозного давления. Затем вводили испытуемое соединение или носитель (200 мкл, внутривенный болюс) и вновь стимулировали кавернозный нерв для того, чтобы провести измерение кавернозного давления в различные промежутки времени после введения лекарства. Если начальные значения ICP были нестабильными (“пиковая” реакция) или если имели место вариации величины контрольной реакции в зависимости от времени, таких животных исключали. Животные также исключались, если контрольные значения ICP/BP (ВР - кровяное давление) находились за пределами интервала 0,3-0,6. Для оценки статистики осуществляли повторяющиеся измерения ANOVA.
Соединение по примеру 20 (0,1-1 мг/кг) вызывало усиление реакции ICP/BP, вызванной субмаксимальной стимуляцией кавернозного нерва. Значительное увеличение отношения ICP/BP наблюдалось при дозах испытуемого соединения 0,1-1 мг/кг.
Результаты вышеописанных биологических испытаний показывают, что соединения по изобретению являются сильнодействующими открывателями активированных кальцием К+ каналов с высокой проводимостью (Maxi-K или ВК каналы). Таким образом, соединения по изобретению пригодны для лечения нарушений у людей, возникающих вследствие дисфункции поляризации и проводимости клеточной мембраны и предпочтительно показаны для лечения ишемии, удара, конвульсий, эпилепсии, астмы, синдрома раздраженной кишки, мигрени, травматического повреждения мозга, повреждения спинного мозга, сексуального расстройства, недержания мочи и особенно нарушений эрекции у мужчин, других нарушений, чувствительных к активности ВК каналов.
Согласно другому аспекту данное изобретение включает фармацевтические композиции, содержащие, по меньшей мере, одно соединение формулы I в сочетании с фармацевтическим адъювантом, носителем или разбавителем.
Согласно еще одному аспекту настоящее изобретение относится к способу лечения или профилактики нарушений, реагирующих на открывание калиевых каналов, у млекопитающего, нуждающегося в таком лечении или профилактике, который включает введение указанному млекопитающему терапевтически эффективного количества соединения формулы I или его нетоксичной фармацевтически приемлемой соли.
При терапевтическом применении фармакологически активные соединения формулы I обычно вводят в виде фармацевтической композиции, включающей в качестве активного вещества, по меньшей мере, одно такое соединение в сочетании с твердым или жидким фармацевтически приемлемым носителем и, возможно, с фармацевтически приемлемыми адъювантами и эксципиентами при использовании стандартных и обычных методик.
Фармацевтические композиции включают подходящие дозированные формы для орального, парентерального (включая подкожное, внутримышечное, внутрикожное и внутривенное), бронхиального или назального введения. Так, если используют твердый носитель, препарат может быть в виде таблеток помещен в твердую желатиновую капсулу в порошкообразном или гранулированном виде или может быть в виде пастилки или леденца. Твердый носитель может содержать обычные эксципиенты, такие как связующие, наполнители, смазочные агенты для таблеток, дезинтегранты, смачиватели и т.п. Таблетка, если желательно, может быть покрыта обычными методами пленкой. Если используют жидкий носитель, препарат может быть в виде сиропа, эмульсии, мягкой желатиновой капсулы, стерильного раствора для инъекций, водной или неводной жидкой суспензии или может быть сухим продуктом для восстановления водой или другим подходящим носителем перед употреблением. Жидкие препараты могут содержать обычные добавки, такие как суспендирующие агенты, эмульгаторы, смачиватели, неводные носители (включая съедобные масла), стабилизаторы, а также ароматизаторы и/или красители. Для парентерального введения носитель обычно представляет собой стерильную воду, по меньшей мере, в большей части, хотя также можно использовать солевые растворы, растворы глюкозы. Суспензии для инъекций также могут быть использованы, в этом случае применяются обычные суспендирующие агенты. В парентеральные дозированные формы могут быть также добавлены обычные стабилизаторы, буферные агенты и т.п. Особенно полезно введение соединения формулы I непосредственно в парентеральные препараты. Фармацевтические композиции готовят обычными методами, подходящими для получения желательного препарата, содержащего соответствующие количества активного вещества, а именно соединения формулы I по изобретению. См., например, Remington’s Pharmaceutical Sciences Mack Publishig Company, Easton, PA, 17th edition, 1985.
Доза соединений формулы I для получения терапевтического эффекта зависит не только от таких факторов, как возраст, вес или пол пациента и метод введения, но также от желаемой степени активности калиевого канала и активности конкретного соединения, используемого для лечения конкретных нарушения или болезни. Доза конкретного соединения может быть введена в виде единичной дозированной формы, и эта единичная дозированная форма подбирается специалистом в соответствии с относительным уровнем активности. Решение о конкретной дозе (и схеме введения в день) находится в компетенции врача, и они могут меняться в зависимости от конкретных обстоятельств для получения нужного терапевтического эффекта.
Подходящая доза соединения формулы I или фармацевтической композиции на его основе для млекопитающего, включая человека, страдающего и могущего пострадать от любого описанного здесь нарушения, составляет в расчете на активное вещество от примерно 0,01 мкг/кг до 10 мг/кг веса пациента, предпочтительно от примерно 0,1 мкг/кг до 5 мг/кг веса пациента при пероральном введении. При парентеральном введении доза может составлять от 0,1 мкг/кг до 1 мг/кг веса пациента в случае внутривенного введения. Активное вещество предпочтительно вводить в равных дозах от одного до четырех раз в день. Однако обычно вводят небольшую дозу и постепенно увеличивают ее, пока не определят оптимальную дозу для данного пациента.
Соединения по изобретению можно вводить в отдельности или в сочетании с другими подходящими терапевтическими агентами, подходящими для лечения сексуального расстройства, такими как ингибиторы cGMP PDE и особенно ингибиторы cGMP PDE V, например силденафил. Примерами терапевтических агентов являются ингибиторы PDE V, выбранные из имидазохиназолинов (см. WO 98/08848), карбазолов (см. WO 97/03675, WO 97/03985 и WO 95/19978), имидазопуринонов (см. WO 97/19947), бензимидазолов (см. WO 97/24334), пиразолохинолинов (см. патент США 5488055), производных антраниловой кислоты (см. WO 95/18097), конденсированных гетероциклов (см. WO 98/07430) и тиенопиримидинов (см. DE 19632423).
Вышеуказанные терапевтические агенты при применении в сочетании с соединениями по изобретению могут вводиться в количествах, указанных, например, в Physician’s Desk Reference (PDR), или в других количествах, определенных специалистом.
Однако следует иметь в виду, что количество соединения, вводимого в действительности, определяется врачом, исходя из релевантных обстоятельств, включая вид болезни, выбор вводимого соединения, метод введения, возраст, вес и восприимчивость пациента и серьезность симптомов пациента.
Нижеследующие примеры приведены для иллюстрации изобретения и не ограничивают его, в объем данного изобретения входят также многие модификации изобретения.
Описание конкретных примеров
В нижеследующих примерах все значения температур указаны в градусах Цельсия. Точки плавления были определены на капиллярном приборе Gallenkamp для определения точек плавления. Спектры протонного магнитного резонанса (1Н ЯМР) записывались на спектрометре Bruker AC 300. Все спектры измерялись в указанных растворителях и химические сдвиги указаны в единицах δ в слабом поле по сравнению с внутренним стандартом тетраметилсиланом (TMS), константы спинового взаимодействия указаны в герцах (Гц). Сигналы протонов обозначены следующим образом: s синглет; d дублет; t триплет; q квартет; m мультиплег; br уширенный пик; dd, дублет дублета; bd, уширенный дублет; dt, дублет триплета; bs, уширенный синглет; dq, дублет квартета. Инфракрасные (ИК) спектры были измерены на спектрофотометре Perkin Elmer 781 при длине волны от 4000 до 400 см-1, калиброванном на длину волны 1601 см-1, характеризующую поглощение полистирольной пленки, с использованием бромида калия (КВr), спектральные характеристики указаны в см-1. Масс-спектры низкого разрешения (МС) и кажущийся молекулярный вес (МН+) или (М-Н)- определяли на приборе Finnigan TSQ 7000. Элементный анализ указан в процентах по весу. Если в конкретных примерах не указано иное, в названии указаны соединения, у которых R2 и R4 обозначают Н.
Пример 1.
Метиловый эфир 4-(5-хлор-2-метоксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)-3-хинолинкарбоновой кислоты (3, R3=СF3, Ra=СН3).
Стадия А. 1,1-Диметилэтиловый эфир N-[2-[(5-хлор-2-метоксифенил)карбонил]-4-(трифторметил)фенил]аминокарбоновой кислоты.
Беспримесную смесь 4-аминобензотрифторида (35 г, 0,218 моль) и (Вос)2О (52,4 г, 0,24 моль) при перемешивании нагревали при 80°С в течении 2-3 час до тех пор, пока не прекратится выделение СО2. Затем смеси дают охладиться и методом роторного испарения удаляют трет-ВuОН. Полученное белое твердое вещество перекристаллизовывают из смеси гексаны/простой эфир с получением белых иголок (50,6 г, 89%) N-(трет-бутоксикарбонил)-4-аминобензотрифторида.
К охлажденному (-78°С) перемешиваемому раствору N-Boc-4-аминобензотрифторида (26,2 г, 0,1 моль) в сухом ТГФ (130 мл) в атмосфере аргона в течение 20 минут добавляют трет-ВuLi (130 мл, 0,22 мол, 1,7 М в циклогексане). Полученный раствор желтого цвета нагревали до (-45) -(-40)°С и выдерживали в течение 2 часов. Полученную густую суспензию дианиона желтого цвета охлаждали до (-78)°С и быстро добавляли беспримесный сухой метил-5-хлор-2-метоксибензоат. Полученный раствор желто-коричневого цвета нагревали до (-40)°С и выдерживали в течение 1 часа. Реакционную смесь разбавляли эфиром (200 мл) и обрывали реакцию 1 N НСl (250 мл), затем давали смеси нагреться до комнатной температуры. Органический слой отделяли, промывали водой, рассолом и затем сушили (Na2SO4). Выпаривание растворителей приводит к получению твердого вещества светло-желтого цвета (49,9 г), которое растирали с эфиром, получая 31,9 г соединения, указанного в названии т.пл. 148-150° C. ИK (KBr, cм-1): 3280, 1725, 1640, 1530, 1320, 1250, 1150.
1Н ЯМР (300 МГц, ДМСО-d6): δ 1,41 (9H, s), 3,58 (3H, s), 7,19 (1H, d, J=8,9 Гц), 7,49 (1Н, d, J=2,7 Гц), 7,58 (1H, d, J=2,6 Гц), 7,60 (1H, dd, J=8,9 и 2,7 Гц), 7,93 (1H, dd, J=8,7 и 1,9 Гц), 8,12 (1H, s), 8,15 (1H, m), 10,35 (1H, s). МС m/e 430 (МН+).
Вычислено для C20H19ClF3NO4, %: С 55,88; H 4,45; N 3,25.
Найдено, %: С 55,51; H 4,38; N 3,26.
Стадия В. 1-[2-Амино-5-(трифторметил)фенил]-1’-(5-хлор-2-метоксифенил)метанон (1, R3=СF3).
К перемешиваемому раствору 1,1-диметилэтилового эфира N-[2-[5-хлор-2-метоксифенил)карбонил]-4-(трифторметил)фенил]аминокарбоновой кислоты (19 г, 0,044 моль) в этаноле (300 мл), добавляли 3 N НСl. Полученную суспензию нагревали с обратным холодильником в течение 3 часов. Протекание гидролиза контролировали методом ТСХ. Реакционную смесь охлаждали и выливали в холодную воду (500 мл). Полученный продукт экстрагировали эфиром (2× 200 мл) и соединенные эфирные экстракты промывали водой, рассолом и затем сушили (Na2SO4). Выпаривание эфира привело к получению вязкого масла золотисто-желтого цвета, которое после выдержки в течение ночи затвердело с образованием твердого вещества бежевого цвета (14,6 г, 100%). т.пл. 90-92°С. ИК (КВr, см-1): 3340, 3470, 1640, 1320, 1240, 1150, 1025.
1Н ЯМР (300 МГц, ДМСО-d6): δ 3,68 (3H, s), 6,97 (1H, d, J=8,8 Гц), 7,19 (1H, d, J=8,9 Гц), 7,26 (1H, d, J=1,1 Гц), 7,36 (1H, d, J=2,7 Гц), 7,53 (2H, m), 7,92 (2H, brd.s). MC m/e 330 (МН+).
Вычислено для С15Н11СlF3NО2, %: С 54,64; H 3,36; N 4,25.
Найдено, %: С 54,65; H 3,37; N 4,16.
Стадия С. Метиловый эфир 3-[[2-[(5-хлор-2-метоксифенил)карбонил]-4-(трифторметил)фенил]амино]-3-оксопропановой кислоты (2, R3=СF3, Ra=СН2).
Раствор метилмалонилхлорида (1,3 мл, 12 ммоль) в безводном CH2Cl2 (10 мл) добавляли по каплям в атмосфере азота в перемешиваемый охлажденный (0°С) раствор 1-[2-амино-5-(трифторметил)фенил]-1’-(5-хлор-2-метоксифенил)метанона, полученного на стадии В (3,3 г, 10 ммоль), и безводного пиридина (0,97 мл, 12 ммоль) в безводном CH2Cl2 (30 мл). Полученной смеси давали нагреться до комнатной температуры и выдерживали в течение 1 часа. Реакционную смесь охлаждали до (0°С) и затем обрывали реакцию 1 N HCl (1 мл). Органический слой отделяли и последовательно промывали насыщенным NаНСО3, водой, рассолом и затем высушивали (MgSO4). Выпаривание СН2Сl2 приводит к получению твердого вещества бежевого цвета (4,28 г), которое истирали с эфиром, получая соединение, указанное в названии, в виде твердого вещества бледно-желтого цвета (3,98 г, 93%), т.пл. 138-140°С.
ИК (КВr, см-1): 1120, 1314, 1530,1644, 1712,1738.
1Н ЯМР (300 МГц, CDCl3): δ 3,57 (2H, s), 3,66 (3H, s), 3,81 (3H, s), 6,92 (1H, d, J=8,8 Гц), 7,38 (1H, d, J=2,6 Гц), 7,47 (1H, dd, J=8,8 и 2,6 Гц), 7,65 (1H, s), 7,75 (1H, d, J=8,9 Гц), 8,84 (1H, d, J=8,8 Гц), 11,91 (1H, brd. s). MC m/e 428 (М-Н)-.
Стадия D: Метиловый эфир 4-(5-хлор-2-метоксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)-3-хинолинкарбоновой кислоты (3, R3=СF3, R3=СН3).
трет-Бутоксид калия (0,63 г, 5,6 ммоль) добавляли к перемешиваемому раствору метилового эфира 3-[[2-[(5-хлор-2-метоксифенил)карбонил]-4-(трифторметил)фенил]амино]-3-оксопропановой кислоты, полученного на стадии С (2,0 г, 4,65 ммоль), в безводном ТГФ (30 мл) в атмосфере азота. Полученную смесь нагревали с обратным холодильником в течение 30 мин. Реакционной смеси давали охладиться, разбавляли ее эфиром (30 мл) и затем подкисляли 1 N НСl (20 мл). Органический слой отделяли, промывали рассолом и затем сушили (Na2SO4). Выпаривание растворителей привело к получению твердого вещества бежевого цвета (1,94 г), которое перекристаллизовывали из смеси EtOAc/гексаны, получая соединение, указанное в названии, в виде белого твердого вещества (1,82 г, 95%), т.пл. 214-216°С. ИК (КВr, см-1): 1128, 1256,1322, 1662, 1742.
1Н ЯМР (300 МГц, CDCl3): δ 3,68 (3H, s), 3,70 (3H, s), 6,97 (1H, d, J=8,8 Гц), 7,20 (1H, d, J=2,5 Гц), 7,36 (1H, s), 7,44 (1H, dd, J=8,8 и 2,5 Гц), 7,53 (1H, d, J=8,6 Гц), 7,73 (1H, d, J=8,6 Гц), 12,43 (1H, brd. s). MC m/e 412 (МН+).
Вычислено для С19Н13СlF3NO4, %: С 55,42; H 3,18; N 3,40.
Найдено, %: С 55,27; H 2,94; N 3,30.
Пример 2.
4-(5-Хлор-2-метоксифенил)-3-(гидроксиметил)-6-(трифторметил)-2(1Н)-хинолинон (4, R3=СF3).
Раствор диизобутилалюминийгидрида (2,16 мл 1 М раствора в смеси гексанов, 2,16 ммоль) добавляли по каплям к охлажденному (-78°С) перемешиваемому раствору соединения по примеру 1 (0,22 г, 0,54 ммоль) в безводном ТГФ (10 мл). Смеси давали нагреться до комнатной температуры и перемешивали в течение 2-3 час. Реакционную смесь охлаждали на ледяной бане и затем медленно обрывали реакцию путем добавления по каплям 1 N HCl (10 мл). Реакционную смесь разбавляли ЕtOАс (20 мл), органический слой отделяли, промывали водой, рассолом и затем сушили (MgSO4). Выпаривание растворителей приводило к получению белого твердого вещества (263 мг), которое растирали с эфиром и получали соединение, указанное в заголовке, в виде белого твердого вещества (178 мг, 86%), т.пл. 232-235°С. ИК (KBr, см-1): 1126, 1264, 1322, 1654, 3442.
1Н ЯМР (300 МГц, CDCl3): δ 3,70 (3H, s), 4,41 (1H, d, J=12,5 Гц), 4,53 (1H, d, J=12,5 Гц), 7,01 (1H, d, J=8,8 Гц), 7,15 (1H, d, J=2,6 Гц), 7,33 (1H, s), 7,47 (1H, dd, J=8,8 и 2,6 Гц), 7,52 (1H, d, J=8,6 Гц), 7,71 (1H, d, J=8,6 Гц), 12,33 (1H, brd.s). MC m/e 384 (МН+).
Вычислено для С18Н13СlF3NО3, %: С 56,34; H 3,41; N 3,65.
Найдено, %: С 55,72; H 3,44; N 3,55.
Пример 3.
4-(5-Хлор-2-метоксифенил)-3-(гидроксиметил)-7-(трифторметил)-2(1Н)-хинолинон (4, R3=R4=H, R2=СF3).
По методике, описанной в примерах 1 и 2, получают соединение, указанное в заголовке, т.пл. 174-176°С; MC m/e 384 (МН+).
1Н ЯМР (300 МГц, ДМСО-d6): δ 3,64 (3Н, s), 3,88 (1H, d, J=11,0 Гц), 4,31 (1H, d, J=11,0 Гц), 4,70 (1H, brd.s), 7,05 (1H, d, J=8,4 Гц), 7,23 (1H, d, J=8,9 Гц), 7,29 (1H, d, J=2,4 Гц), 7,36 (1H, d, J=8,4 Гц), 7,55 (1H, dd, J=8,7 и 2,4 Гц), 7,65 (1H, s), 12,23 (1Н, s).
Пример 4.
4-(5-Хлор-2-метоксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)-3-хинолинкарбоксальдегид (5, R3=СF3).
К перемешиваемому раствору соединения по примеру 2 (384 мг, 1 ммоль) в безводном CH2Cl2 (10 мл) добавляли двуокись марганца (0,44 г, 5 ммоль). Полученную суспензию перемешивали в атмосфере азота в течение ночи. Добавляли снова МnО2 (0,44 г, 5 ммоль) и продолжали перемешивать суспензию до завершения окисления (2-3 дня). Суспензию фильтровали через слой целита, промывали дополнительно метиленхлоридом. После выпаривания растворителя получали соединение, указанное в заголовке, в виде бледно-желтого твердого вещества (206 мг, 54%), т.пл. 238-240°С. ИК (КВr, см-1): 1120, 1268, 1320, 1678, 1707.
1Н ЯМР (300 МГц, CDCl3): δ 3,71 (3H, s), 7,01 (1H, d, J=8,9 Гц), 7,10 (1H, d, J=2,6 Гц), 7,48 (1H, m), 7,51 (1H, d, J=2,6 Гц), 7,59 (1H, d, J=8,6 Гц), 7,82 (1H, dd, J=8,7 и 1,8 Гц), 10,29 (1H, s), 12,53 (1H, brd. s). МС m/e 380 (М-Н)-.
Пример 5.
Этиловый эфир (Е)-3-[4-(5-хлор-2-метоксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)-3-хинолинил]-2-пропеновой кислоты (6, R3=CF3).
К перемешиваемой охлажденной суспензии NaH (84 мг, 2,1 ммоль, 60% в минеральном масле) в безводном ДМФ (2 мл) в атмосфере азота добавляли по каплям раствор триэтилфосфонацетата (0,43 г, 1,95 ммоль) в ДМФ (1 мл). Смесь перемешивали 30 минут и затем добавляли беспримесный 4-(5-хлор-2-метоксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)-3-хинолинкарбоксальдегид (0,60 г, 1,62 ммоль). Полученной смеси давали нагреться до комнатной температуры и перемешивали в течение 3 часов. После этого ТСХ показала отсутствие альдегида и образование изомерной смеси сложных эфиров. Реакционную смесь охлаждали на ледяной бане и обрывали реакцию 1 N НСl. Полученный продукт экстрагировали 1:1 смесью эфир/EtOAc, промывали насыщенным NаНСО3, водой, рассолом и затем высушивали (Na2SO4). Выпаривание растворителей привело к получению твердого вещества бежевого цвета (0,765 г), которые перекристаллизовывали из смеси EtOAc/гексаны, с получением соединения, указанного в заголовке, в виде чистого транс (Е)-изомера (0,497 г). Концентрация маточной жидкости с последующим растиранием с эфиром позволила получить еще 123 мг эфира в виде смеси изомеров. Общий выход очищенных эфиров составлял 0,62 г (86%). Характеристики полученного соединения: т. пл. 270-273°С. ИК (КВr, см-1): 1126, 1284, 1322, 1664, 1713.
1Н ЯМР (300 МГц, СDСl3): δ 1,28 (3H, t, J=7,1 Гц) 3,70 (3H, s), 4,20 (2H, q, J=7,1 Гц), 7,04 (1H, d, J=8,9 Гц), 7,11 (1H, d, J=2,6 Гц), 7,24-7,33 (2H, m), 7,43 (1H, s), 7,48-7,52 (2H, m), 7,76 (1H, d, J=8,6 Гц), 12,02 (1H, brd. s). МС m/e 450 (М-Н)-.
Пример 6.
(Е)-4-(5-Хлор-2-метоксифенил)-3-(3-гидрокси-1-пропенил)-6-(трифторметил)-2(1Н)-хинолинон (7, R3=СF3).
К перемешиваемому охлажденному (-78°С) раствору эфира по примеру 5 (0,3 г, 0,66 ммоль) в безводном ТГФ (9 мл) по каплям в атмосфере азота добавляли раствор Dibal-H в смеси гексанов (3 ммоль, 3 мл 1 М раствора). Смеси давали нагреться до комнатной температуры и перемешивали в течение 2 ч. Реакционную смесь охлаждали на ледяной бане и затем медленно обрывали реакцию 1 N НСl (10 мл). Добавляли этилацетат (30 мл), разделяли слои, промывали водой, рассолом и сушили (MgSO4). Выпаривание растворителей привело к получению твердого вещества белого цвета (0,29 г), после растирания с эфиром получали соединение, указанное в заголовке, в виде спирта (234 мг), т.пл. 266-268°С.
1Н ЯМР (300 МГц, ДМСО-d6): δ 3,67 (3H, s), 3,96 (2H, m), 4,76 (1 H, t, J=5,3 Гц), 6,11 (1H, d, J=15,7 Гц), 7,01 (1H, s), 7,18 (1H, dt, J=15,7 и 4,6 Гц), 7,27-7,31 (2H, m), 7,52 (1H, d, J=8,6 Гц), 7,60 (1H, dd, J=8,9 и 2,7 Гц), 7,79 (1H, dd, J=8,6 и 1,8 Гц), 12,36 (1H, s). МС m/e 408 (М-Н)-.
Вычислено для С20Н15ClF3NО3, %: С 58,62; H 3,69; N 3,42.
Найдено, %: С 58,50; H 3,74; N 3,35.
Пример 7.
Этиловый эфир 4-(5-хлор-2-метоксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)-3-хинолинпропановой кислоты (8, R3=СF3).
К раствору эфира, полученного по примеру 5, в этаноле и безводном НСl в колбе Раrr при встряхивании в атмосфере азота добавляли РtO2 (5-10 вес.%). Полученную суспензию гидрировали под давлением 60 ф/дюйм2 (413,7 кПа) в течение ночи. Катализатор отфильтровывали и фильтрат выпаривали в роторном испарителе, получая соединение, указанное в заголовке, т.пл. 193-195°С.
1Н ЯМР (300 МГц, СDСl3): δ 1,16 (3H, m), 2,43 (2H, m), 2,65 (2H, m), 3,65 (3H, s), 4,01 (2Н, m), 6,96-7,01 (2H, m), 7,14 (1H, s), 7,25 (2H, s), 7,40-7,43 (2H, m), 7,60 (1H, brd. s). МС m/e 452 (М-Н)-.
Пример 8.
4-(5-Хлор-2-метоксифенил)-3-(3-гидроксипропил)-6-(трифторметил)-2(1Н)-хинолинон (9, R3=СF3).
По методике, описанной в примере 6, восстанавливают соединение по примеру 7, получая соединение, указанное в заголовке: т.пл. 200-202°С.
1Н ЯМР (300 МГц, CDCl3): δ 1,60 (2H, m), 2,20 (3H, brd. m), 3,50 (2H, m), 3,63 (3H, s), 6,97 (1H, d, J=8,8 Гц), 7,04 (1H, d, J=2,6 Гц), 7,16 (1H, s), 7,19 (1H, s), 7,41 (1H, dd, J=8,8 и 2,6 Гц), 7,49 (1H, d, J=8,3 Гц), 7,65 (1H, J=7,36 Гц), 12,45 (1H, brd. s). МС m/e 412 (МН)+.
Пример 9.
4-(5-Хлор-2-гидроксифенил)-3-(гидроксиметил)-6-(трифторметил)-2(1Н)-хинолинон (14, R3=СF3).
Стадия А. [2-Амино-5-(трифторметил)фенил](5-хлор-2-гидроксифенил)метанон (10, R3-=CF3).
К охлажденному раствору (-78°С) 1,1-диметилэтилового эфира N-[2-[5-хлор-2-метоксифенил)карбонил]-4-(трифторметил)фенил]аминокарбоновой кислоты, полученного по примеру 1 на стадии А (7,0 г, 21,2 ммоль), в метиленхлориде (60 мл) по каплям добавляли 1,0 М раствор ВВr3 в метиленхлориде (46,7 мл, 46,7 ммоль). Полученному раствору красного цвета давали нагреться до комнатной температуры и перемешивали в течение ночи. Реакцию обрывали насыщенным раствором NаНСО3. Органический слой отделяли, промывали водой, рассолом и затем сушили (MgSO4). Выпаривание растворителя привело к получению твердого вещества желто-красного цвета, которое после перекристаллизации из смеси СН2Сl2/гексаны стало желтьм (6,58 г, 98%).
Стадия В. Метиловый эфир 3-[[2-[5-хлор-2-гидроксифенил)карбонил]-4-(трифторметил)фенил]амино]-3-оксипропановой кислоты (11, R3=СF3).
К раствору[2-амино-5-(трифторметил)фенил](5-хлор-2-гидроксифенил)метанона (0,5 г, 1,58 ммоль) и пиридина (0,25 мл, 3,17 ммоль) в метиленхлориде (15 мл) при 0°С по каплям добавляли раствор метилмалонилхлорида (0,34 мл, 3,17 ммоль) в метиленхлориде (10 мл). Затем реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 2 час. Реакционную смесь подкисляли 1 N НСl и отделяли органический слой. Затем его промывали дважды насыщенным NaHCO3, водой, рассолом и сушили (MgSO4). Выпаривание растворителя позволило получить указанное в заголовке соединение в виде желтоватого масла.
Стадия С. 2-Хлор-6,8-дигидро-11-(трифторметил)-7Н-[1]бензопирано[3,4-с]хинолин-6,7-дион (12, R3=СF3).
Сырой метиловый эфир 3-[[2-[5-хлор-2-гидроксифенил)карбонил]-4-(трифторметил)фенил]амино]-3-оксопропановой кислоты, полученный на стадии В, растворяли в ТГФ (15 мл) и добавляли трет-бутоксид калия (1 М в ТГФ, 1,74 ммоль, 1,74 мл). Реакционную смесь нагревали в течение 15 минут с обратным холодильником. Затем ее подкисляли 1 N НСl и отделяли органический слой. Органический слой промывали водой, рассолом и сушили (MgSO4). Выпаривание растворителя позволило получить твердое вещество желтого цвета, которое растирали со смесью этилацетат/гексаны, получая указанное в заголовке соединение в виде твердого вещества желтого цвета (0,48 г, 83%), т.пл.>250°С. МС m/е 366 (МН+).
Вычислено для С17H7ClF3NО3·0,5 Н2О, %: С 54,49; H 2,15; N 3,74.
Найдено, %: С 54,10; H 1,85; N 3.63.
1H ЯМР (ДМСО-d6): δ 7,48 (d, J=8,8 Гц, 1Н), 7,55 (d, J=8,5 Гц, 1Н), 7,76 (dd, J=8,9 и 2,2 Гц, 1Н), 7,98 (d, J=8,7 Гц, 1H), 8,10 (d, J=2,1 Гц, 1Н), 8,42 (s, 1Н). ИК (KBr, см-1): 3479, 3074, 1761, 1652, 1630, 1577, 1368, 1325, 1141.
Стадия D: 4-(5-Хлор-2-гидроксифенил)-3-(гидроксиметил)-6-(трифторметил)-2(1Н)-хинолинон (14, R3=СF3).
К охлаждаемой суспензии (-78°С) 2-хлор-6,8-дигидро-11-(трифторметил)-7Н-[1]бензопиран[3,4-с]хинолин-6,7-диона, полученного на стадии С (1,0 г, 2,73 ммоль), в метиленхлориде (20 мл) по каплям добавляли раствор Dibal-H (I M в метиленхлориде, 13,7 мл, 13,7 ммоль). Реакционную смесь нагревали до 0°С и выдерживали в течение 3 час. Реакционную смесь подкисляли 1 N НСl и дважды экстрагировали этилацетатом. Органический слой отделяли и промывали водой, рассолом и затем высушивали (MgSO4). После выпаривания растворителя и перекристаллизации сырого продукта из смеси этилацетат/гексаны получали соединение, указанное в заголовке, в виде белого твердого вещества (0,7 г, 69%), т.пл.>250°С. МС m/е 368 (М-Н-).
Вычислено для С17Н11СlF3NО3, %: С 55,23; H 3,00; N 3,79.
Найдено, %: С 56,59; H 4,02; N 3,36.
1Н ЯМР (ДМСО-d6): δ 3,90 (dd, J=10,9 Гц, 5,3 Гц, 1Н), 4,36 (dd, J=10,9 Гц, 5,6 Гц, 1 Н), 4,70 (t, J=5,4 Гц, 1H), 7,03 (d, J=8,7 Гц, IH), 7,17 (s, 1Н), 7,26 (d, J=2,7 Гц, 1Н), 7,39 (dd, J=8,7 Гц, 2,7 Гц, 1Н), 7,53 (d, J=8,6 Гц, 1Н), 7,81 (dd, J=8,9 Гц, 1,9 Гц, 1Н), 9,95 (s, 1H), 12,31 (s, 1H).
Пример 10.
Этиловый эфир 3-[4-(5-хлор-2-гидроксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)-3-хинолинил]-2-пропеновой кислоты (15, R3=СF3).
Стадия А. 2-Хлор-6,8-дигидро-6-гидрокси-11-(трифторметил)-7Н-[1]бензопиран[3,4-с]хинолин-7-он (13, R3=СF3).
К охлажденному (-78°С) раствору 2-хлор-6,8-дигидро-11-(трифторметил)-7Н-[1]бензопиран[3,4-с]хинолин-6,7-диона, полученного в примере 9 на стадии С, (1,15 г, 3,15 ммоль) в ТГФ (30 мл) по каплям добавляют раствор Dibal-H (1 М в ТГФ, 15,7 мл, 15,7 ммоль). Реакционную смесь выдерживают при -78°С в течение 4 час. Реакционную смесь подкисляли 1 N НСl и дважды экстрагировали этилацетатом. Органический слой отделяли и промывали водой, рассолом и затем высушивали (MgSO4). Выпаривание растворителя с последующим растиранием сырого продукта с этилацетатом позволило получить указанное в заголовке соединение в виде твердого вещества белого цвета (0,9 г, 78%), т.пл.>260°С. МС m/е 366 (М-Н)-.
Вычислено для С17H9ClF3NО3·0,25 Н2О, %: С 54,86; H 2,57; N 3,76.
Найдено, %: С 54,92; H 2,92; N 3,46.
1Н ЯМР (ДМСО-d6): δ 6,40 (d, J=6,2 Гц, 1Н), 7,31 (d, J=8,7 Гц, 1Н), 7,55-7,63 (т, 3Н), 7,94 (d, J=8,7 Гц, 1H), 8,07 (d, J=2,4 Гц, 1H), 8,38 (s, 1Н), 12,44 (s, 1Н). ИК (КВr, см-1): 3300, 1669, 1631, 1605, 1575, 1326, 1279, 1133.
Стадия В. Этиловый эфир 3-[4-(5-хлор-2-гидроксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)-3-хинолинил]-2-пропеновой кислоты (15, R3=СF3).
К охлажденной суспензии (0°С) NaH (60% в минеральном масле, 41 мг, 1,0 ммоль) в ДМФ (3 мл) по каплям добавляли триэтилфосфонацетат (0,1 мл, 0,5 ммоль). Реакционную смесь перемешивали при 0°С в течение 0,5 час и затем добавляли раствор 2-хлор-6,8-дигидро-6-гидрокси-11-(трифторметил)-7Н-[1]бензопиран[3,4-с]хинолин-7-она, полученного в примере 10 на стадии А (0,15 г, 0,41 ммоль), в ДМФ (5 мл). Смеси красного цвета давали нагреться до комнатной температуры и перемешивали в течение 4 часов. Реакцию медленно обрывали 1 N раствором НСl и экстрагировали этилацетатом. Органический слой отделяли и промывали насыщенным раствором NаНСО3, водой, рассолом и сушили (MgSO4). Выпаривание растворителя с последующей перекристаллизацией из смеси этилацетат/гексаны позволило получить соединение, указанное в заголовке, в виде белого твердого вещества (127 мг, 72%), т.пл. 262-268°С (разл.). МС m/е 436 (М-Н-).
Вычислено для С21Н15ClF3NO4, %: С 57,61; H 3,45; N 3,20.
Найдено, %: С 57,31; H 3,46; N 3,15.
1Н ЯМР (ДМСО-d6): δ 1,18 (t, J=7,1 Гц, 3Н), 4,09 (q, J=7,0 Гц, 1Н), 7,08 (d, J=8,8 Гц, 1Н), 7,16 (d, J=15,7 Гц, 1H), 7,20 (s, 1Н), 7,29 (d, J=2,7 Гц, 1Н), 7,34 (d, J=15,7 Гц, 1Н), 7,48 (dd, J=8,7 Гц и 2,7 Гц, 1Н), 7,57 (d, J=8,6 Гц, 1Н), 7,90 (dd, J=8,8 Гц и 1,8 Гц, 1 Н), 10,1 (s, 1H), 12,6 (s, 1Н). ИК (КВr, см-1): 3225, 1683, 1662, 1626, 1323, 1301, 1115.
Пример 11.
3-[4-(5-Хлор-2-гидроксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)-3-хинолинил]-2-пропеновая кислота (16, R3=СF3).
К раствору соединения по примеру 10 (40 мг, 0,09 ммоль) в EtOH (2 мл) добавляли 10 N NaOH (1 мл) и перемешивали при комнатной температуре в течение ночи. Реакционную смесь подкисляли 1 N HCl и отделяли осажденное твердое вещество желтого цвета, указанное в заголовке (34 мг, выход 91%), т.пл. 258-261° С (разл.). МС m/е 408 (М-Н)-.
Вычислено для C19H11ClF3NO4·0,5H2O, %: С 52,79; H 3,15; N 3,24.
Найдено, %: С 52,93; H 2,82; N 3,10.
1Н ЯМР (ДМСО-d6): δ 7,07-7,13 (m, 2Н), 7,18 (s, 1H), 7,25-7,30 (m, 2Н), 7,47 (dd, J=8,7 Гц и 2,7 Гц, 1 Н), 7,57 (d, J=8,7 Гц, 1H), 7,89 (dd, J=8,7 Гц и 1,7 Гц, 1Н), 10,11 (s, 1Н), 12,37 (s, br, 1H), 12,57 (s, 1Н). ИК (КВr, см-1): 3144, 2996, 1676, 1628, 1323, 1270, 1252, 1130.
Пример 12.
4-(5-Хлор-2-гидроксифенил)-3-(3-гидрокси-1-пропенил)-6-(трифторметил)-2(1Н)-хинолинон (17, R3=СF3).
К охлажденной (-78°С) суспензии соединения по примеру 10 (0,2 г, 0,46 ммоль) в метиленхлориде (10 мл) по каплям добавляли раствор Dibal-H (1 М в метиленхлориде, 2,3 мл, 2,3 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 4 часов. Реакционную смесь подкисляли 1 N НСl и дважды экстрагировали этилацетатом. Органический слой отделяли и промывали водой, рассолом и высушивали (MgSO4). Выпаривание растворителя с последующей перекристаллизацией из смеси этилацетат/гексаны позволило получить соединение, указанное в заголовке, в виде твердого вещества белого цвета (0,14 г, 77%), т.пл. 203-209°С (разл.). МС m/е 394 (М-Н)-.
Вычислено для С19H13ClF3NО3·0,5 Н2О, %: С 56,38; H 3,49; N 3,46.
Найдено, %: С 56.35; H 3,72; N 3,29.
1Н ЯМР (ДМСО-d6): δ 3,97 (dt, J=1,7 Гц и 4,9 Гц, 2Н), 4,77 (t, J=5,3 Гц, 1Н), 6,16 (dd, J=15,8 Гц и 1,9 Гц, 1 Н), 7,05 (d, J=8,7 Гц, 1Н), 7,10 (s, 1Н), 7,16 (d, J=2,7 Гц, 1Н), 7,21 (dt, J=15,7 Гц и 4,7 Гц, 1Н), 7,41 (dd, J=8,7 Гц и 2,7 Гц, 1Н), 7,51 (d, J=8,5 Гц, 1Н), 7,78 (dd, J=8,7 Гц и 1,8 Гц, 1Н), 9,90 (s, 1Н), 12,32 (s, 1H). ИК (КВr, см-1): 3286, 1656, 1641, 1322, 1294, 1169, 1120, 1075.
Пример 13.
Этиловый эфир 4-(5-хлор-2-гидроксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)-3-хинолинпропановой кислоты (18, R3=СF3).
К раствору соединения по примеру 10 (0,4 г, 0,91 ммоль) в этаноле (20 мл) добавляли РtO2 (3 8 мг) и 3 капли 1 N HCl. Смесь гидрировали в аппарате Parr под давлением 50 ф/дюйм2 (413,7 кПа) в течение ночи. Катализатор отделяли фильтрованием через целит и промывали этанолом. Фильтрат выпаривали досуха и белый остаток подвергали флэш-хроматрографии (силикагель, 2:1 этилацетат/гексаны) с получением соединения, указанного в заголовке, в виде твердого вещества белого цвета (0,29 г, 72%), т.пл. 241-245°С (разл.). МС m/е 438 (М-Н)-.
Вычислено для C21H17ClF3NO4, %: С 57,35; H 3,90; N 3,18.
Найдено, %: С 57,27; H 4,02; N 2,99.
1Н ЯМР (СD3ОD): δ 1,18 (t, J=7,1 Гц, 3Н), 2,49-2,55 (m, 2H), 2,69-2,75 (m, 2H), 4,05 (q, J=7,1 Гц, 2H), 7,02 (d, J=8,7 Гц, 1H), 7,13 (d, J=2,6 Гц, 1H), 7,26 (s, 1H), 7,38 (dd, J=8,7 Гц и 2,6 Гц, 1Н), 7,51 (d, J=8,6 Гц, 1H), 7,73 (dd, J=8,6 Гц и 1,7 Гц, 1Н). ИК (КВr, см-1): 3353, 1698, 1656, 1626, 1376, 1311, 1270, 1167, 1128, 1074.
Пример 14.
4-(5-Хлор-2-гидроксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)-3-хинолинпропановая кислота (19, R3=СF3).
К раствору соединения по примеру 13 (38 мг, 0,087 ммоль) в ЕtOН (2 мл) добавляли 10 N NaOH (1 мл) и перемешивали смесь при комнатной температуре в течение ночи. Реакционную смесь подкисляли 1 N НС1 и отделяли осадок белого цвета, представляющий собой соединение, указанное в заголовке (30 мг, 84%), т.пл. 255-258°С. МС m/е 410 (М-Н)-.
Вычислено для С19H13ClF3NO4·0,5 Н2О, %: С 54,24; H 3,35; N 3,33.
Найдено, %: С 54,10; H 3,10; N 3,28.
1Н ЯМР (ДМСО-d6): δ 2,32-2,37 (m, 2H), 2,47-2,51 (m, 2H), 7,04-7,07 (m, 2H), 7,25 (d, J=2,6 Гц, 1H), 7,40 (dd, J=8,7 Гц и 2,7 Гц, 1Н), 7,52 (d, J=8,6 Гц, 1H), 7,79 (dd, J=8,7 Гц и 1,9 Гц, 1H), 9,98 (s, 1H), 12,10 (s, br, 1H), 12,31 (s, 1H). ИК (КВr, см-1): 3283, 3155, 1714, 1626, 1560, 1405, 1275, 1194, 1167, 1132.
Пример 15.
4-(5-Хлор-2-гидроксифенил)-3-(3-гидроксипропил)-6-(трифторметил)-2(1Н)-хинолинон (20, R3=СF3).
К охлажденной суспензии (-78°С) соединения по примеру 13 (0,2 г, 0,45 ммоль) в метиленхлориде (10 мл) по каплям добавляли раствор Dibal-H (1 M в метиленхлориде, 3,7 мл, 3,7 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 4 час. Реакционную смесь затем подкисляли 1 N НС1 и дважды экстрагировали этилацетатом. Органический слой отделяли и промывали водой, рассолом и сушили (MgSO4). Выпаривание растворителя с последующей перекристаллизацией из смеси этилацетат/гексаны привело к получению соединения, указанного в заголовке, в виде белого твердого вещества (145 мг, 80%), т.пл. 257-259°С (разл.). МС m/е 398 (МН+).
Вычислено для С19Н15СlF3NО3·0,67 ЕtOАс, %: С 57,00; H 4,49; N 3,07.
Найдено, %: С 57,17; H 4,62; N 2,88.
1H ЯМР (ДМСО-d6): δ 1,5 (m, 2Н), 2,3 (m, 2Н), 3,25 (m, 2Н), 4,35 (m, 1Н), 7,02-7,07 (m, 2Н), 7,21 (d, J=2,5 Гц, 1Н), 7,39 (dd, J=8,7 Гц, 2,7 Гц, 1Н), 7,51 (d, J=8,5 Гц, 1Н), 7,77 (d, J=8,5 Гц, 1H), 9,90 (s, 1Н), 12,24 (s, 1H). ИК (KBr, см-1): 3315, 1654, 1624, 1569, 1324, 1273, 1125, 1073.
Примеры 16 и 17.
(Е)- и (Z)-4-(5-Хлор-2-гидроксифенил)-3-(2-фтор-3-гидрокси-1-пропенил)-6-(трифторметил)-2(1Н)-хинолинон (23, R3=СF3) и (25, R3=СF3).
Стадия А. К охлажденной суспедзии (0°С) NaH (60% минеральное масло, 68 мг, 1,7 ммоль) в ДМФ (5 мл) добавляли триэтил-2-фтор-2-фосфонацетат (0,165 мл, 0,82 ммоль). Полученную смесь перемешивали при 0°С в течение 0,5 час и затем добавляли раствор 2-хлор-6,8-дигидро-6-гидрокси-11-(трифторметил)-7Н-[1]бензопиран[3,4-с]хинолин-7-она, полученного на стадии А в примере 10 (0,25 г, 0,68 ммоль) в ДМФ (5 мл). Реакционной смеси красного цвета давали нагреться до комнатной температуры и перемешивали в течение 4 часов. Реакцию прерывали 1 N HCl и затем смесь экстрагировали этилацетатом. Органический слой отделяли и промывали насыщенным раствором NаНСО3, водой, рассолом и затем высушивали (MgSO4). Выпаривание растворителя позволяет получить смесь эфиров формулы (21) (E:Z=1:2,5, 224 мг, 72%) в виде бесцветного масла.
Стадия В. К охлажденной суспензии (-78°С) сырой смеси эфиров со стадии А (210 мг, 0,46 ммоль) в метиленхлориде (10 мл) добавляли по каплям раствор Dibal-H (1 M в метиленхлориде, 3,3 мл, 3,3 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. Затем реакционную смесь подкисляли 1 N НС1 и экстрагировали дважды этилацетатом. Органический слой отделяли и промывали водой, рассолом и затем высушивали (MgSO4). Сырые изомерные спирты очищали хроматографией на колонке (силикагель, 2:1, этилацетат/гексаны) с получением индивидуального Е-изомера (Е)-4-(5-хлор-2-гидроксифенил)-3-(2-фтор-3-гидрокси-1-пропенил)-6-(трифторметил)-2(1Н)-хинолинона, пример 16 (23, R3=СF3) и Z-изомера (Z)-4-(5-хлор-2-гидроксифенил)-3-(2-фтор-3-гидрокси-1-пропенил)-6-(трифторметил)-2(1Н)-хинолинона, пример 17 (25, R3=СF3). Физические характеристики (Е)- и (Z)-изомеров описаны ниже.
Пример 16. (Е)-4-(5-Хлор-2-гидроксифенил)-3-(2-фтор-3-гидрокси-1-пропенил)-6-(трифторметил)-2(1Н)-хинолинона (23, R3=CF3).
Т.пл. 215-218°С (разл.). МС m/е 412 (М-Н)-.
Вычислено для С19Н12СlF3NО3·0,33 Н2О, %: С 54,37; H 3,04; N 3,34.
Найдено, %: С 54,72; H 3,11; N 3,18.
1Н ЯМР (ДМСО-d6): δ 3,64 (m, 1Н), 3,83 (m, 1H), 4,99 (m, 1Н), 5,61 (d, J=19,6 Гц, 1Н), 7,03 (d, J=8,7 Гц, 1H), 7,20-7,23 (m, 2Н), 7,38 (dd, J=8,7 Гц, 2,7 Гц, 1H), 7,54 (d, J=8,7 Гц, 1H). 7,84 (d, J=8,9 Гц, 1H), 10,09 (s, 1Н), 12,42 (s, 1H).
Пример 17. (Z)-4-(5-Хлор-2-гидроксифенил)-3-(2-фтор-3-гидрокси-1-пропенил)-6-(трифторметил)-2(1Н)-хинолинона (25, R3=СF3).
Т.пл. 242-245°С (разл.). МС m/е 412 (М-Н)-.
Вычислено для С19Н12СlF3NО3·0,33 Н2О, %: С 54,37; H 3,04; N 3,34.
Найдено, %: С 54,62; H 3,27; N 3,11.
1Н ЯМР (ДМСО-d6): δ 3,81-3,86 (m, 2H), 5,35 (t, J=5,9 Гц, 1Н), 5,57 (d, J=40,3 Гц, 1Н), 7,00 (d, J=8,7 Гц, 1Н), 7,12 (d, J=2,6 Гц, 1H), 7,21 (s, 1Н), 7,34 (dd, J=8,7 Гц, 2,7 Гц, 1Н), 7,51 (d, J=8,6 Гц, 1H), 7,81 (dd, J=8,6 Гц, 1,6 Гц, 1Н), 9,90 (s, 1Н), 12,32 (s, 1H).
Примеры 18 и 19.
По методикам, описанным в примерах 16 и 17, получают соединения по примеру 18 (24, R3=СF3) (Е-изомер) и примеру 19 (26, R3=СF3) (Z-изомер) из соединения формулы (5), полученного в примере 4, как показано на реакционной схеме 5. Физические характеристики (Е)- и (Z)-изомеров описаны ниже.
Пример 18. (Е)-4-(5-Хлор-2-метоксифенил)-3-(2-фтор-3-гидрокси-1-пропенил)-6-(трифторметил)-2(1Н)-хинолинон (24, R3=СF3).
Т.пл. 180-182°С. МС m/е 426 (М-Н)-.
1Н ЯМР (СDСl3): δ 3,68 (3Н, s), 4,10 (2H, d, J=24,0 Гц), 5,54 (1H, d, J=18,0 Гц), 7,01 (1H, d, J=8,9 Гц), 7,04 (1H, d, J=2,4 Гц), 7,32 (1H, s), 7,46 (1H, dd, J=8,9 Гц и 2,3 Гц), 7,52 (1H, d, J=8,5 Гц), 7,73 (1H, d, J=7,6 Гц), 11,65 (1H, brd. s).
Пример 19. (Z)-4-(5-Хлор-2-метоксифенил)-3-(2-фтор-3-гидрокси-1-пропенил)-6-(трифторметил)-2(1Н)-хинолинон (26, R3=СF3).
Т.пл. 230-232°С (разл.). МС m/е 426 (М-Н)-.
1Н ЯМР (СDСl3): δ 3,69 (3Н, s), 4,09 (2H, d, J=10,7 Гц), 5,70 (1H, d, J=38,1 Гц), 6,98 (1H, d, J=8,8 Гц), 7,09 (1H, s), 7,35 (1H, s), 7,41 (1H, d, J=8,8 Гц), 7,50 (1H, m), 7,67 (1H, brd. s), 11,75 (1H, brd.s).
Пример 20.
4-(5-Хлор-2-гидроксифенил)-3-(2-гидроксиэтил)-6-(трифторметил)-2(1Н)-хинолинон (31a, R3=СF3, n=2).
Стадия А. Метиловый эфир 4-[[2-[5-хлор-2-метоксифенил)карбонил]-4-(трифторметил)фенил]амино]-4-оксобутановой кислоты (27, R3=CF3, Rа=СН3, n=2).
Беспримесный 3-карбометоксипропионилхлорид (4,8 мл, 0,039 моль) добавляли к перемешиваемому охлажденному (0°С) раствору аминобензофенона, 1-[2-амино-5-(трифторметил)фенил]-1’-(5-хлор-2-метоксифенил)метанона, полученного в примере 1, на стадии В (7,0 г, 0,021 моль) и безводного пиридина (4,8 мл, 0,059 моль) в безводном CH2Cl2 (80 мл). Полученной смеси давали нагреться до комнатной температуры и выдерживают в течение 12 часов. Реакционную смесь подкисляли 1 N НСl (50 мл). Органический слой отделяли и последовательно промывали насыщенным NаНСО3, водой, рассолом и затем сушили (MgSO4). Выпаривание CH2Cl2 и растирание полученного остатка позволило получить 7,71 г (82%) указанного в заголовке амида.
1Н ЯМР (300, CDCl3): δ 2,7 (4Н, m), 3,06 (3Н, s), 3,63 (3Н, s), 6,85 (1Н, d, J=8,9 Гц), 7,16 (1Н, s), 7,30 (1Н, d, J=2,6 Гц), 7,39 (1Н, dd, J=8,8 Гц и 2,6 Гц), 7,57 (1Н, s), 7,66 (1Н, dd, J=8,9 Гц и 1,9 Гц), 8,79 (1Н, d, J=8,8 Гц). МС m/e 444 (МН+).
Стадия В. Метиловый эфир 4-(5-хлор-2-метоксифенил)-4-гидрокси-1,2,3,4-тетрагидро-2-оксо-6-(трифторметил)-3-хинолинуксусной кислоты (28, R3=СF3, Ra=СН3, n=2).
Раствор бис(триметилсилил)амида калия (0,5 М в толуоле, 57 мл, 28,5 ммоль) добавляли к перемешиваемому охлажденному (-78°С) раствору метилового эфира 4-[[2-[5-хлор-2-метоксифенил)карбонил]-4-(трифторметил)фенил]амино]-4-оксобутановой кислоты, полученного на стадии А (4,05 г, 9,1 ммоль), в безводном ТГФ (25 мл) и выдерживали при -78°С в течение 3 часов. Подкисление 1 N НСl и последующая экстракция EtOAc позволила получить сырое, указанное в заголовке соединение (4,05 г, 100%).
Стадия С. 4-(5-Хлор-2-метоксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)-3-хинолинуксусная кислота (29, R3=СF3, n=2).
Перемешиваемую суспензию сырого метилового эфира 4-(5-хлор-2-метоксифенил)-4-гидрокси-1,2,3,4-тетрагидро-2-оксо-6-(трифторметил)-3-хинолинуксусной кислоты, полученного на стадии В (2 г, 4,5 ммоль), в толуоле (25 мл) обрабатывали раствором 35% НВr в уксусной кислоте (5 мл). Полученную смесь нагревали при 85°С в течение ночи. Реакционную смесь выпаривали досуха и распределяли остаток между водой и EtOAc. Экстракт в EtOAc промывали рассолом, высушивали (MgSO4) и затем выпаривали, получая соединение, указанное в заголовке (1,45 г, 78%).
Стадия D. 4-(5-Хлор-2-гидроксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)-3-хинолинуксусная кислота (30, R3=СF3, n=2).
Беспримесную смесь сырой 4-(5-хлор-2-метоксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)-3-хинолинуксусной кислоты, полученной на стадии С (1,45 г, 3,5 ммоль), и пиридинийгидрохлорида (5 г, 43,3 ммоль) нагревали при 185°С в течение 3 часов. Реакционной смеси давали охладиться, добавляли 1 N НСl и осуществляли экстракцию EtOAc с получением соединения, указанного в заголовке (1,20 г, 86%), т.пл. 158-160°С.
1Н ЯМР (300 МГц, CD3OD): δ 3,21 (1H, d, J=16,7 Гц), 3,60 (1H, d, J=16,7 Гц), 7,0 (1Н, d, J=8,8 Гц), 7,13 (1H, d, J=2,6 Гц), 7,31 (1H, m), 7,37 (1H, dd, J=8,8 Гц, 2,6 Гц), 7,52 (1H, d, J=8,6 Гц), 7,75 (1H, d, J=8,6 Гц). МС m/e 398 (МН+).
Стадия Е. 4-(5-Хлор-2-гидроксифенил)-3-(2-гидроксиэтил)-6-(трифторметил)-2(1Н)-хинолинон (31а, R3=СF3, n=2).
Раствор комплекса боран-метилсульфид (2 М в ТГФ, 125 мл, 0,25 моль) в течение 20 минут по каплям добавляли в атмосфере азота к перемешиваемому охлажденному (-10°С) раствору 4-(5-хлор-2-гидроксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)-3-хинолинуксусной кислоты, полученной на стадии D (20 г, 0,05 моль) в безводном ТГФ (125 мл). Полученной прозрачной реакционной смеси давали нагреться при комнатной температуре и продолжали перемешивание в течение 2-3 дней (ЖХВР показывает отсутствие исходного вещества). Реакционную смесь охлаждали на ледяной бане и затем обрывали реакцию добавлением по каплям 1 N NaOH (125 мл) до щелочной реакции и затем смесь подкисляли 1 N НСl. Добавляли эфир (250 мл) и разделяли слои, промывали водой, рассолом и затем подвергали сушке (Na2SO4). Выпаривание растворителей привело к получению твердого вещества коричневого цвета (21,4 г), которое перекристаллизовывали из EtOAc-МеОН, получая 2,6 г чистого твердого вещества белого цвета (первую порцию). Растирание концентрированной маточной жидкости вместе с эфиром позволило получить 8,7 г белого твердого вещества (вторая порция). Вторую порцию перекристаллизовывали из ЕtOАс-МеОН и соединяли с первой порцией, получая 11,1 г указанного в заголовке соединения в виде твердого вещества белого цвета, т.пл. 255-256°С.
1Н ЯМР (300 МГц, СD3ОD): δ 2,73 (2 Н, m), 3,64 (2Н, t, J=7,4 Гц), 7,0 (1Н, d, J=8,8 Гц), 7,15 (1Н, d, J=2,6 Гц), 7,23 (1H, broad s), 7,36 (1H, dd, J=8,8 Гц, 2,6 Гц), 7,48 (1H, d, J=8,6 Гц), 7,71 (1 H, dd, J=8,6 и 1,8 Гц). МС m/e 384 (МН+).
Вычислено для С18Н13СlF3NО3, %: С 56,34; H 3,41; N 3,65.
Найдено, %: С 56,18; H 3,58; N 3,48.
Пример 21.
4-(5-Хлор-2-метоксифенил)-3-(2-гидроксиэтил)-6-(трифторметил)-2(1Н)-хинолинон (31b, R3=CF3, n=2).
По методике примера 20, стадия Е, из соединения, полученного на стадии С примера 20, получают соединение, указанное в заголовке.
Т.пл. 219-221° С.
1Н ЯМР (300 МГц, CDCl3): δ 7,71 (1H, d, J=8,4 Гц), 7,53-7,46 (2Н, m), 7,23 (1H, s), 7,11 (1H, d, J=2,7 Гц), 7,02 (1H, d, J=8,7 Гц), 3,79 (m, 2H), 3,70 (s, 1H), 3,79 (t, 2H). МС m/e 397 (МН+).
Вычислено для С19Н15СlF3NО3, %: С 57,37; H 3,80; N 3,52.
Найдено, %: С 57,31; H 3,94; N 3,38.
Примеры 22 и 23.
Общая методика получения соединений формул (34) и (35)
Стадия А. Ацилирование аминобензофенона формулы (1).
Беспримесный ацилхлорид (1,2 экв.) добавляли к перемешиваемому охлажденному (0°С) раствору аминобензофенона формулы (1) (1 экв.) и безводного пиридина (1,3 экв.) в безводном CH2Cl2. Полученной смеси давали нагреться до комнатной температуры и выдерживали 2-3 часа. Реакционную смесь подкисляли 1 N НСl, разделяли слои и органический слой промывали насыщенным раствором NаНСО3, водой, рассолом и затем высушивали (MgSO4). Выпаривание CH2Cl2 привело к получению соответствующего амида общей формулы (32).
Стадия В. Циклизация амида формулы (32).
Раствор бис(триметилсилил)амида калия (0,5 М в толуоле, 3 экв.) добавляли к перемешиваемому охлажденному (-78°С) раствору амида формулы (32) (1 экв.) в безводном ТГФ и выдерживали при -78°С в течение 3 час. Подкисление 1 N НСl и последующая экстракция EtOAc привели к получению хинолина формулы (33).
Стадия С. Дегидратирование хинолина формулы (33).
Перемешиваемую суспензию сырого соединения формулы (33) в толуоле обрабатывали раствором 35% НВr в уксусной кислоте. Полученную смесь нагревали при 85°С в течение ночи. Реакционную смесь выпаривали досуха и остаток распределяли между водой и EtOAc. Экстракт в EtOAc промывали рассолом и высушивали (MgSO4), а затем выпаривали растворитель с получением хинолинона формулы (34).
Стадия D. Деметилирование соединения формулы (34).
Беспримесную смесь сырого хинолина формулы (34) (1 экв.) и пиридинийхлорида (5 экв.) нагревали при 185°С в течение 3 часов. Реакционной смеси давали охладиться, добавляли 1 N НСl и затем осуществляли экстракцию при помощи EtOAc с получением соответствующего гидроксисоединения формулы (35).
Пример 22.
4-(5-Хлор-2-метоксифенил)-3-(4-метоксифенил)-6-(трифторметил)-2(1Н)-хинолинон (34а, R3=СF3, R8=4-метоксифенил, n=0).
Т.пл. 130-135°С.
1H ЯМР (300 МГц, СDСl3): δ 7,62 (1H, d, J=8,7 Гц), 7,36-7,25 (3Н, m), 7,10 (2Н, d, J=8,7 Гц), 6,93 (1H, d, J=2,7 Гц), 6,81 (1H, d, J=8,7 Гц), 6,77 (2Н, d, J=8,7 Гц), 3,77 (s, 3Н), 3,62 (s, 3H).
MC m/e 459 (МН+).
Вычислено для С24Н17СlF3NО3, %: С 62,69; H 3,73; N 3,05.
Найдено, %: С 62,74; H 3,92; N 2,89.
Пример 23.
4-(5-Хлор-2-метоксифенил)-3-[(4-метоксифенил)метил]-6-(трифторметил)-2(1Н)-хинолинон (34b, R3=CF3, R8=4-метоксифенил, n=1).
Т.пл. 110-114°С.
1Н ЯМР (300 МГц, СDСl3): δ 7,62 (1H, d, J=8,7 Гц), 7,47-7,43 (1H, dd, J=2,7 Гц и 8,7 Гц), 7,29 (1H, d, J=2,7 Гц), 7,23 (1H, s), 7,00-6,92 (3Н, m), 6,70 (1H, d, J=8,7 Гц), 3,78 (dd, 2H), 3,72 (s, 3Н), 3,57 (s, 3H). МС m/e 473 (MH+).
Пример 24.
4-(5-Хлор-2-метоксифенил)-3-(4-нитрофенил)-6-(трифторметил)-2(1Н)-хинолинон (34c, R3=CF3, R8=4-нитрофенил, n=0).
Т.пл. 218-222°С.
1Н ЯМР (300 МГц, СDСl3): δ 8,12 (2Н, d, J=8,7 Гц), 7,72 (1H, d, J=8,7 Гц), 7,43-7,36 (4Н, m), 7,33-7,29 (dd, 1H, J=2,7 и 8,7 Гц), 6,95 (1H, d, J=2,7 Гц), 6,82 (1H, d, J=8,7 Гц). МС m/e 474 (МН+).
Вычислено для С23Н14ClF3N2O4, %: С 58,18; H 2,97; N 5,90.
Найдено, %: С 57,70; H 3,20; N 5,65.
Пример 25.
4-(5-Хлор-2-метоксифенил)-3-(4-аминофенил)-6-(трифторметил)-2(1Н)-хинолинон (34d, R3=СF3, R8=4-аминофенил, n=0).
Т.пл. 287°С.
1Н ЯМР (300 МГц, СDСl3): δ 7,62 (1H, d, J=8,4 Гц), 7,44 (1H, d, J=8,4 Гц), 7,31 (s, 1H), 7,27-7,23 (m, 4H), 6,94-6,79 (dd, 1H, J=3,6 и 8,7 Гц), 6,82 (s, 1H), 6,52 (d, 1H, J=8,7 Гц). МС m/e 444 (МН+).
Вычислено для С23H16ClF3NО2, %: С 62,10; H 3,63; N 6,30.
Найдено, %: С 61,89; H 3,81; N 6,06.
Пример 26.
4-(5-Хлор-2-метоксифенил)-3-метил-6-(трифторметил)-2(1Н)-хинолинон (34е, n=0, R8=Me, R3=СF3).
Раствор соответствующего соединения формулы (33) (5,63 ммоль) 33% НВr в АсОН (38,3 ммоль) и 10 мл АсОН нагревали до 75°С в течение 3 часов. Раствор охлаждали до комнатной температуры и реакцию обрывали Н2О (50 мл), затем перемешивали смесь в течение 12 часов. Осадок отфильтровывали, промывали водой и сушили в вакууме. Твердое вещество бледно-коричневого цвета перекристаллизовывали из смеси этилацетат/гексан, получая указанное в заголовке соединение в виде твердого вещества белого цвета (0,550 г, выход 27%).
1Н ЯМР (300 МГц, СDСl3): δ 2,04 (s, 3Н), 3,72 (s, 3Н), 7,03 (d, 1H, J=9,0 Гц), 7,12 (s, 1H), 7,44 (m, 3Н), 7,65 (d, 1H, J=8,4 Гц), 10,93 (brs, 1H). МС m/e 368 (MH+).
Вычислено для С18Н13СlF3NО2·0,33 Н2О, %: С 58,79; H 3,56; N 3,81.
Найдено, %: С 58,89; H 3,82; N 3,53.
Пример 27.
4-(5-Хлор-2-гидроксифенил)-3-(3,4-диметоксифенил)-6-(трифторметил)-2(1Н)-хинолинон (35а, R3=CF3, R8=3,4-диметоксифенил, n=0).
Т.пл.140-142°С.
1Н ЯМР (300 МГц, СDСl3): δ 7,65 (1H, d, J=8,7 Гц), 7,37 (1H, d, J=8,7 Гц), 7,35 (1H, s), 7,29-7,25 (1H, dd, J=2,7 Гц и 8,7 Гц), 6,96 (1H, d, J=2,4 Гц), 6,87-6,76 (2Н, m), 6,63 (1H, d, J=1,8 Гц), 3,85 (s, 3Н), 3,69 (s, 3Н), 3,62 (s, 3Н). МС m/e 489 (MH+).
Вычислено для С25Н19ClF3NO4, %: С 61,30; H 3,91; N 2,86.
Найдено, %: С 61,42; H 3,89; N 2,75.
Пример 28.
4-(5-Хлор-2-гидроксифенил)-3-(2,4-дигидроксифенил)-6-(трифторметил)-2(1Н)-хинолинон (35b, R3=CF3, R8=2,4-дигидроксифенил, n=0).
Т.пл. 295°С (разл.).
1Н ЯМР (300 МГц, СD3ОD): δ 7,73 (1Н, d, J=8,7 Гц), 7,49 (1Н, d, J=8,7 Гц), 7,31-7,27 (2Н, dd, J=2,7 Гц и 8,7 Гц), 7,03-7,00 (2Н, m), 6,89 (1Н, d, J=8,7 Гц), 6,53-6,49 (2Н, m). МС m/e 462 (МН+).
Вычислено для С22Н13СlF3NО4, %: С 59,01; H 2,93; N 3,13.
Найдено, %: С 58,38; H 3,15; N 2,96.
Пример 29.
4-(5-Хлор-2-гидроксифенил)-3-(4-гидроксифенил)-6-(трифторметил)-2(1Н)-хинолинон (35с, R3=СF3, R8=4-гидроксифенил, n=0).
Т.пл. 220-240°С.
1Н ЯМР (300 МГц, СDСl3): δ 7,53 (1Н, d, J=8,7 Гц), 7,20 (1Н, d, J=8,7 Гц), 7,24-7,17 (2Н, m), 6,97-6,88 (4Н, dd, J=8,7 Гц и 1,9Гц), 6,81 (1Н, d, J=8,7 Гц), 6,77 (2Н, d, J=8,7 Гц), 3,77 (s, 3Н), 3,62 (s, 3Н). МС m/e 459 (МН+).
Пример 30.
4-(5-Хлор-2-гидроксифенил)-3-[(4-гидроксифенил)метил]-6-(трифторметил)-2(1Н)-хинолинон (35d, R3=СF3, R8=4-гидроксифенил, n=1).
Т.пл. 242-250°С.
1Н ЯМР (300 МГц, CD3OD): δ 7,73 (1Н, d, J=8,7 Гц), 7,70 (1Н, d, J=8,7 Гц), 7,50 (1H, d, J=8,7 Гц), 7,42-7,27 (2Н, m), 6,70-6,87 (2Н, m), 6,82 (2Н, d, J=8,7 Гц), 6,56 (d, 2H, J=8,1 Гц), 3,91-3,51 (2H, dd, J=13,8 Гц и 14,7 Гц). МС m/e 445 (МН+).
Вычислено для C23H15ClF3NO3·0,5 Н2О, %: С 60,68; H 3,52; N 3,08.
Найдено, %: С 60,71; H 3,91; N 2,82.
Пример 31.
4-(5-Хлор-2-гидроксифенил)-3-(4-ацетамидофенил)-6-(трифторметил)-2(1Н)-хинолинон (35е, R3=СF3, R8=4-ацетамидофенил, n=0).
Т.пл. 240-260°С.
1Н ЯМР (300 МГц, СD3ОD): δ 7,77 (1Н, d, J=8,7 Гц), 7,55 (1Н, d, J=8,7 Гц), 7,48-7,38 (m, 3Н), 7,19-7,15 (m, 3Н), 6,87-6,83 (m, 2H), 2,09 (s, 3Н). МС m/e 472 (МН+).
Вычислено для C24H16ClF3N3O3, %: С 56,59; H 3,93; N 5,50.
Найдено, %: С 57,21; H 3,73; N 5,28.
Пример 32.
4-(5-Хлор-2-гидроксифенил)-3-(4-аминофенил)-6-(трифторметил)-2(1Н)-хинолинон (35f, R3=СF3, R8=4-аминофенил, n=0).
Т.пл. 222-224°С.
1Н ЯМР (300 МГц, СD3OD): δ 7,74 (1Н, d, J=8,4 Гц), 7,53 (1H, d, J=8,4 Гц), 7,36 (s, 1H), 7,15 (dd, 1H, J=2,7 и Гц), 6,99 (2H, d, J=8,4 Гц), 6,85-6,81 (m, 2H), 6,58 (1H, d, J=8,4 Гц). МС m/e 446,8 (МН+).
Вычислено для С22Н14СlF3N2O3, %: С 59,14; H 3,16; N 6,27.
Найдено, %: С 60,27; H 3,52; N 6,32.
Пример 33.
4-(5-Хлор-2-гидроксифенил)-3-[2-(4-гидроксифенил)этил]-6-(трифторметил)-2(1Н)-хинолинон (35g, R3=СF3, R8=4-гидроксифенил, n=2).
Т.пл.205-207°С.
1Н ЯМР (300 МГц, СD3ОD): δ 7,73-7,69 (1Н, dd, J=1,8 Гц и 8,7 Гц), 7,52 (1Н, d, J=8,7 Гц), 7,38-7,35 (dd, IH, J=2,7 Гц и 6,2 Гц), 7,23 (1H, s), 7,01 (d, 1H, J=8,7 Гц), 6,78-6,60 (2Н, dd, J=8,7 и 7,3 Гц), 2,68 (4Н, m). МС m/e 459 (МН+).
Вычислено для С24Н17СlF3NО3·1,5 H2O, %: С 59,16; H 4,11; N 2,88.
Найдено, %: С 58,71; H 3,78; N 2,86.
Пример 34.
4-(5-Хлор-2-гидроксифенил)-3-метил-6-(трифторметил)-2(1Н)-хинолинон (35h, n=0, R8=Me, R3=СF3).
MC m/z: 352 (MH-). ИК (KBr): 3183, 1655,1321, 1263,1122 см-1.
1Н ЯМР (ДМСО-d6): δ 1,86 (3Н, s), 7,04 (1H, d, J=8,8 Гц), 7,12 (1H, s), 7,22 (1H, d, J=2,6 Гц), 7,39 (1H, dd, J=2,6 Гц, 8,7 Гц), 7,52 (1H, d, J=8,6 Гц), 7,78 (1H, d, J=8,7 Гц), 9,92 (1H, s), 12,26 (1H, s).
Вычислено для C17H11ClF3NO2·0,5 Н2О, %: С 56,26; H 3,33; N 3,86.
Найдено, %: С 56,57; H 3,16; N 3.81.
Пример 35.
3-[4-(5-Хлор-2-гидроксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)хинолин-3-ил]-акрилонитрил (36а, R=Н, Х=CN, R3=СF3).
К охлажденной (0°С) суспензии NaH (60% минеральное масло, 33 мг, 0,82 ммоль) в ДМФ (5 мл) по каплям добавляли диэтилцианометилфосфонат (63 мкл, 0,39 ммоль). Реакционную смесь перемешивали при 0°С в течение 0,5 часа и добавляли раствор 2-хлор-6,8-дигидро-6-гидрокси-11-(трифторметил)-7Н-[1]бензопиран[3,4-с]хинолин-7-она, полученного на стадии А в примере 10 (120 мг, 0,33 ммоль), в ДМФ (5 мл). Полученной смеси, окрашенной в красный цвет, давали нагреться до комнатной температуры и перемешивали в течение 2 ч. Реакцию обрывали 1 N НСl и затем смесь экстрагировали этилацетатом. Органический слой отделяли и промывали насыщенным NаНСО3, водой, рассолом и затем высушивали (MgSО4). Выпаривание растворителя привело к получению желтоватого масла, которое затем очищали хроматографией на колонке (силикагель, 1:1 этилацетат/гексаны) с получением указанного в заголовке соединения в виде твердого вещества желтого цвета (71 мг, 56%).
Т.пл.>265°С. МС m/е: 389 (М-Н)-.
Вычислено для С19Н10СlF3N2О2, %: С 58,40; H 2,58; N 7,17.
Найдено, %: С 58,16; H 2,81; N 6,87.
1Н ЯМР (ДМСО-d6): δ 6,85 (1Н, d, J=16,4 Гц), 7,10 (1Н, d, J=8,8 Гц), 7,21 (1Н, s), 7,23 (1H, d, J=16,4 Гц), 7,30 (1H, d, J=2,7 Гц), 7,49 (1H, dd, J=8,8 Гц, 2,7 Гц), 7,59 (1H, d, J=8,6 Гц), 7,93 (1H, dd, J=8,7 Гц, 1,7 Гц), 10,19 (1H, s), 12,70 (1H, s). ИК (KBr, см-1): 3333, 2224, 1656, 1625, 1585, 1321, 1265, 1118, 1073.
Пример 36.
3-[4-(5-Хлор-2-метоксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)хинолин-3-ил]акрилонитрил (36b, R=Me, X=CN, R3=СF3).
Соединение, указанное в заголовке, получали из соединения формулы (5), полученного в примере 4, по методике, описанной в примере 35.
Т.пл.>250°С. МС m/е: 403 (М-Н)-.
1H ЯМР (ДМСО-d6): δ 3,71 (3Н, s), 6,92 (1H, d, J=16,3 Гц), 7,08 (2Н, m), 7,31 (1H, s), 7,32 (1H, d, J=16,3 Гц), 7,54 (2Н, m), 7,81 (1H, dd, J=8,5 Гц и 1,6 Гц), 12,41 (1H, brd s). ИК (KBr, см-1): 2216, 1665, 1321, 1127.
Пример 37(а).
4-[4-(5-Хлор-2-гидроксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)хинолин-3-ил]-3-бутен-2-он (37а, R=Н, Х=Ac, R3=СF3).
Указанное в заголовке соединение получали из соединения 13, полученного на стадии А в примере 10, по методике, описанной в примере 35.
Т.пл. 186-188°С. МС m/е: 406 (М-Н)-.
ИК (КВr, см-1): 3185, 1656,1629,1322,1284, 1169,1125, 1076.
Вычислено для С20Н13СlF3NО3, %: С 58,91; H 3,21; N 3,43.
Найдено, %: С 58,64; H 3,05; N 3,23.
Пример 37(b).
4-[4-(5-Хлор-2-метоксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)хинолин-3-ил]-3-бутен-2-он (37b, R=Me, X=Ac, R3=CF3).
Соединение, указанное в заголовке, получали из соединения 5, полученного в примере 4, по методике, описанной в примере 5.
Т.пл. 232-234°С. МС m/е: 422 (МН+).
ИК (KBr, см-1): 2844, 1686, 1625, 1656, 1588, 1320.
Вычислено для С21Н15СlF3NО3, %: С 59,80; H 3,58; N 3,32.
Найдено, %: С 59,60; H 3,56; N 3,22.
Пример 38.
4-(5-Хлор-2-гидроксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)-3-хинолин-карбоксальдегидоксим (38а, R=Н, R3=СF3).
К суспензии 2-хлор-6,8-дигидро-6-гидрокси-11-(трифторметил)-7Н-[1]бензопиран[3,4-с]хинолин-7-она, полученного на стадии А в примере 10 (41 мг, 0,11 ммоль), в ТГФ (10 мл) добавляли гидрохлорид гидроксиламина (9,3 мг, 0,13 ммоль) и триэтиламин (0,038 мл, 0,28 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Выпаривали ТГФ и добавляли воду. Собирали осадок светло-желтого цвета и сушили его на воздухе, получая указанное в заголовке соединение (36 мг, 85%), т.пл. 195-198°С (разл.).
МС m/е: 383 (МН+).
Вычислено для С17Н10СlF3NО3, %: С 53,35; H 2,63; N 7,32.
Найдено, %: С 53,18; H 4,55; N 6.87.
1Н ЯМР (ДМСО-d6): δ 6,98 (d, J=8,8 Гц, 1Н), 7,19-720 (m, 2Н), 7,34 (m, 1Н), 7,54 (d, J=8,6 Гц, 1Н), 7,85 (m, 1H), 7,95 (s, 1H), 9,87 (s, br, 1H), 11,30 (s, 1H), 12,46 (s, br, 1H). ИК (KBr, см-1): 3247, 1661, 1629, 1322, 1265, 1168, 1121, 1076.
Пример 39.
4-(5-Хлор-2-метоксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)-3-хинолинкарбоксальдегидоксим (38b, R=Me, R3=СF3).
Перемешиваемую суспензию соединения, полученного в примере 4 (80 мг, 0,21 ммоль), Na2OH· HCl (18 мг, 0,25 ммоль) и безводного NaOAc (20 мг, 0,25 ммоль) в абсолютном этаноле (2 мл) нагревали с обратным холодильником в течение 1 часа. Этанол выпаривали в роторном испарителе и распределяли остаток между EtOAc и водой. Слой в EtOAc отделяли и промывали водой, рассолом и затем высушивали (Na2SO4). Выпаривание EtOAc с последующим растиранием сырого продукта с простым эфиром привело к получению указанного в заголовке соединения в виде твердого вещества белого цвета (56 мг), т.пл. 255-258°С. ИК (КВr, см-1): 3207, 1669, 1323, 1267, 1122.
1Н ЯМР (ДМСО-d6): δ 3,66 (3Н, s), 7,08 (1Н, s), 7,22 (1Н, d, J=8,9 Гц), 7,28 (1Н, d, J=2,6 Гц), 7,53 (2Н, dd, J=8,9 и 2,8 Гц), 7,85 (1Н, dd, J=8,7 и 1,7 Гц), 7,97 (1Н, s), 11,29 (1Н, s), 12,50 (1H, brd s). МС 397 (MH+).
Пример 40.
Метиловый эфир 4-(5-хлор-2-гидроксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)-3-хинолинуксусной кислоты (30b, Ra=Me, R3=СF3).
Стадия А. 2-Хлор-7,9-дигидро-12-(трифторметил)-[1]бензоксепино[4,5-с]хинолин-6,8-дион (39, R3=СF3).
Перемешиваемую смесь карбоновой кислоты формулы 30а (Ra=Н, R3=СF3) (1,20 г, 3,0 ммоль) и каталитического количества п-TsOH нагревали с обратным холодильником в толуоле в течение 4 часов. Растворитель удаляли в роторном испарителе. Остаток обрабатывали насыщенным раствором бикарбоната натрия и экстрагировали EtOAc. Органические экстракты высушивали над MgSO4 и концентрировали с получением соединения, указанного в заголовке (783 мг, 69%).
1Н ЯМР (300 МГц, ДМСО-d6): δ 3,23 (1Н, d, J=13,3 Гц), 4,26 (1Н, d, J=13,3 Гц), 7,52 (1Н, d, J=8,8 Гц), 7,60 (1Н, m), 7,76 (1H, dd, J=8,8 и 2,6 Гц), 7,87 (1H, d, J=2,6 Гц), 7,92 (2Н, m), 12,62 (1H, brd). МС m/e: 380 (МН+).
Стадия В. Метиловый эфир 4-(5-хлор-2-гидроксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)-3-хинолинуксусной кислоты (30b, Ra=Me, R8=CF3).
После очистки лактона 2-хлор-7,9-дигидро-12-(трифторметил)-[1]бензоксепино[4,5-с]-хинолин-6,8-диона, полученного на стадии А, хроматографией на колонке с силикагелем с использованием смеси CH2Cl2 - МеОН в качестве элюента был получен эфир, указанный в заголовке.
1Н ЯМР (300 МГц, ДМСО-d6): δ 3,17 (1Н, d, J=16,4 Гц), 3,47 (1Н, d, J=16,4 Гц), 3,53 (3Н, s), 7,05 (1H, d, J=8,7 Гц), 7,11 (1Н, d, J=2,7 Гц), 7,18 (1Н, m), 7,42 (1H, dd, J=8,7 и 2,7 Гц), 7,55 (1H, d, J=8,5 Гц), 7,84 (1H, dd, J=8,7 и 2,7 Гц), 10,0 (1H, s), 12,4 (1H, brd s). МС m/e: 412 (МН+).
Пример 41.
4-(5-Хлор-2-гидроксифенил)-3-(2-гидрокси-2-метилпропил)-6-(трифторметил)-2(1Н)-хинолинон (40, R=R1=Me, R3=СF3).
Раствор метиллития (1М в ТГФ, 1,6 мл, 1,6 ммоль) добавляли к охлажденному (-78°С) перемешиваемому раствору 2-хлор-7,9-дигидро-12-(трифторметил)-[1]бензоксепино[4,5-с]хинолин-6,8-диона, полученного в примере 40, стадия А (16 мг, 0,3 ммоль), в безводном ТГФ (3 мл) в атмосфере азота. После перемешивания в течение 1 часа при -78°С удаляли охлаждающую баню и продолжали перемешивать в течение 16 часов. Реакцию обрывали 1 N НСl и осуществляли экстракцию EtOAc. Сырой продукт очищали флэш-хроматографией (силикагель, 19:1 СН2Сl2/МеОН) с получением указанного в заголовке соединения (21 мг) в виде твердого вещества бежевого цвета.
МС m/z: 412 (МН+).
1Н ЯМР (ДМСО-d6): δ 0,95 (6Н, s), 2,6 (1H, dd), 7,0 (1H, d), 7,1 (1H, s), 7,2 (1H, s), 7,39 (1H, d), 7,55 (1H, d), 7,8 (1H, d), 9,95 (1H, s), 12,5 (1H, s).
Пример 42.
4-(5-Хлор-2-гидроксифенил)-1-метил-3-(2-гидроксиэтил)-6-(трифторметил)-2(1Н)-хинолинон (43, R3=СF3).
Стадия А. 4-(5-Хлор-2-гидроксифенил)-3-(2-триизопропилсилилоксиэтил)-6-(трифторметил)-2(1Н)-хинолинон (41, R3=СF3).
Беспримесный триизопропилсилилхлорид (0,293 мл, 1,37 ммоль, 1,05 экв.) добавляли к перемешиваемому раствору соединения по примеру 2 и имидазола (0,134 г, 1,97 ммоль, 1,5 экв.) в безводном ДМФ (10 мл). После выдержки при комнатной температуре в течение 12 часов полученную смесь выливали в водный 1 N раствор НСl (50 мл). Водный слой экстрагировали этилацетатом (3× 50 мл). Соединенные органические слои промывали рассолом (25 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме. Сырой остаток очищали на хроматографической колонке с силикагелем (3:1-2:1 гексан/этилацетат) с получением 0,357 г прозрачного вязкого масла (выход 50%). Аналитический образец соединения, указанного в заголовке, получают кристаллизацией из смеси этилацетат/гексан, т.пл. 209-210°С.
1H ЯМР (300 МГц, ДМСО-d6): δ 0,91 (21Н, s), 2,58 (2Н, m), 3,63 (2Н, m), 7,04 (1Н, d, J=8,8 Гц), 7,06 (1Н, s), 7,23 (1Н, d, J=2,6 Гц), 7,40 (1Н, dd, J=8,7 и 2,3 Гц), 7,51 (1Н, d, J=8,6 Гц), 7,78 (1H, dd, J=8,6 и 1,7 Гц), 9,92 (1H, s), 12,26 (1H, s). МС m/e: 540 (МН+).
Стадия В. 4-(5-Хлор-2-гидроксифенил)-1-метил-3-(2-триизопропилсилилоксиэтил)-6-(трифторметил)-2(1Н)-хинолинон (42, R3=СF3).
К перемешиваемому раствору хинолинона со стадии А (0,244 г, 0,453 ммоль) в ТГФ при -78°С добавляли н-бутиллитий (0,593 мл, 0,950 ммоль, 2,1 экв., 1,6 М/гексан). Через 15 минут добавляли беспримесный иодметан и давали полученной смеси нагреться до комнатной температуры. После перемешивания в течение 12 часов реакцию обрывали водной 1 N НСl (10 мл) и полученную смесь выливали в воду (50 мл). Осуществляли экстракцию водного слоя этилацетатом (3× 50 мл). Соединенные органические слои промывали рассолом (25 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме. Сырой остаток очищали на колонке с силикагелем (4:1-3:1 гексан/этилацетат) с получением 0,202 г соединения, указанного в заголовке в виде твердого вещества белого цвета (выход 81%).
1Н ЯМР (300 МГц, ДМСО-d6): δ 0,92 (21Н, s), 2,63 (2Н, m), 3,66 (2Н, t, J=7,4 Гц), 3,75 (3Н, s), 7,05 (1Н, d, J=8,8 Гц), 7,15 (1Н, s), 7,24 (1Н, d, J=2,7 Гц), 7,41 (1Н, dd, J=8,7 и 2,6 Гц), 7,78 (1Н, d, J=8,9 Гц), 7,91 (1Н, dd, J=9,0 и 1,9 Гц), 9,95 (1Н, s).
Стадия С. 4-(5-Хлор-2-гидроксифенил)-1-метил-3-(2-гидроксиэтил)-6-(трифторметил)-2(1Н)-хинолинон (43, R3=СF3).
Тетрабутиламмонийфторид (0,380 мл, 0,308 ммоль, 2 экв., 1,0 М/ТГФ) добавляли к перемешиваемому раствору хинолинона со стадии В (0,105 г, 0,190 ммоль) в ТГФ (10 мл). Через 12 часов сырую реакционную смесь выливали в воду (50 мл). Водный слой экстрагировали этилацетатом (3× 50 мл). Соединенные органические слои промывали рассолом (25 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме. Сырой остаток очищали методом хроматографии на колонке (силикагель, 2,5% метанол/хлороформ) с получением 74 мг твердого вещества белого цвета. Это вещество перекристаллизовывали из смеси этилацетат/гексан и получали 0,064 г (выход 86%) указанного в заголовке соединения, т.пл. 229-230°С.
1Н ЯМР (300 МГц, ДМСО-d6): δ 2,56 (2Н, m), 3,40 (2Н, m), 3,75 (3Н, s), 4,57 (1Н, m), 7,05 (1Н, d, J=8,8 Гц), 7,14 (1Н, s), 7,25 (1Н, d, J=2,6 Гц), 7,41 (1Н, dd, J=8,7 и 2,7 Гц), 7,77 (1Н, d, J=8,8 Гц), 7,89 (1Н, dd, J=8,8 и 2,0 Гц), 9,91 (1Н, s). МС m/e: 398 (МН+).
Вычислено для С19Н15СlF3NО3, %: С 57,37; H 3,80; N 3,52.
Найдено, %: С 57,69; H 3,88; N 3,28.
Пример 43.
4-(5-Хлор-2-метоксифенил)-1-метил-3-(2-гидроксиэтил)-6-(трифторметил)-2(1Н)-хинолинон (44, R3=СF3).
Смесь хинолинона, полученного в примере 42, стадия В (0,202 г, 0,365 ммоль), диметилсульфата (0,038 мл, 0,402 ммоль, 1,1 экв.) и карбоната калия (0,056 г, 0,402 ммоль, 1,1 экв.) в ацетоне нагревали с обратным холодильником в течение 3 часов. Полученную смесь выливали в воду (25 мл). Затем осуществляли экстракцию водного слоя этилацетатом (3× 50 мл). Соединенные органические слои промывали рассолом (25 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме. Сырой силилированный хинолинон (0,228 г) использовали в последующей реакции без очистки или определения его характеристик. К перемешиваемому раствору силилированного хинолинона (0,228 г, 0,401 ммоль) в ТГФ (10 мл) добавляли тетрабутиламмонийфторид (0,802 мл, 0,802 ммоль, 2 экв., 1,0 М/ТГФ). Через 12 часов сырую реакционную смесь выливали в воду (50 мл). Водный слой экстрагировали этилацетатом (3× 50 мл). Соединенные органические слои промывали рассолом (25 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме. Сырой остаток очищали с использованием хроматографии на колонке (силикагель, 1:1-1:2 гексан/этилацетат) с получением 0,148 г твердого вещества белого цвета. Это вещество перекристаллизовывали из смеси этилацетат/гексан, получая 0,139 г (выход 84%) указанного в заголовке соединения, т.пл. 175-176°С.
1Н ЯМР (300 МГц, ДМСО-d6): δ 2,48 (2Н, m), 3,37 (2Н, m), 3,67 (3Н, s), 3,75 (3Н, s), 4,57 (1Н, t, 5,4), 7,06 (1H, s), 7,29 (1H, d, J=9,0 Гц), 7,35 (1H, d, J=1,3 Гц), 7,61 (1H, dd, J=8,8 и 2,7 Гц), 7,77 (1H, d, J=8,8 Гц), 7,89 (1H, dd, J=8,8 и 1,9 Гц). МС m/e: 412 (МН+).
Вычислено для С20Н17СlF3NО3, %: С 58,33; H 4,16; N 3,40.
Найдено, %: С 58,30; H 4,07; N 3,18.
Пример 44.
4-(5-Хлор-2-гидроксифенил)-1-метил-3-(2-гидроксиэтил)-6-(трифторметил)-2(1Н)-хинолинон (43, R3=СF3).
Стадия А. 4-(5-Хлор-2-гидроксифенил)-3-(2-триизопропилсилилоксиэтил)-6-(трифторметил)-2(1Н)-хинолинон (41, R3=СF3).
Беспримесный триизопропилсилилхлорид (0,293 мл, 1,37 ммоль, 1,05 экв.) добавляли к перемешиваемому раствору кинолона 31а, полученного в примере 20, и имидазола (0,134 г, 1,97 ммоль, 1,5 экв.) в безводном ДМФ (10 мл). После выдержки в течение 12 часов при комнатной температуре полученную смесь выливали в водную 1 N HCl (50 мл). Водный слой экстрагировали этилацетатом (3× 50 мл). Соединенные органические слои промывали рассолом (25 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме. Сырой остаток очищали на колонке с силикагелем (3:1-2:1 гексан/этилацетат) с получением 0,357 г прозрачного вязкого масла (выход 50%). Аналитический образец получали кристаллизацией из смеси этилацетат/гексан, т. пл. 209-210°С.
1Н ЯМР (300 МГц, ДМСО-d6): δ 0,91 (21Н, s), 2,58 (2Н, m), 3,63 (2Н, m), 7,04 (1Н, d, J=8,8 Гц), 7,06 (1Н, s), 7,23 (1Н, d, J=2,6 Гц), 7,40 (1Н, dd, J=8,7 и 2,3 Гц), 7,51 (1Н, d, J=8,6 Гц), 7,78 (1Н, dd, J=8,6 и 1,7 Гц), 9,92 (1Н, s), 12,26 (1H, s). МС m/e: 540 (MH+).
Стадия В. 4-(5-Хлор-2-гидроксифенил)-1-метил-3-(2-триизопропилсилилоксиэтил)-6-(трифторметил)-2(1Н)-хинолинон (42, R3=СF3).
н-Бутиллитий (0,593 мл, 0,950 ммоль, 2,1 экв., 1,6 М/гексан) добавляли к перемешиваемому раствору хинолинона 41 (0,244 г, 0,453 ммоль), полученного на стадии А, в ТГФ при -78°С. Через 15 минут добавляли беспримесный йодметан и давали полученной смеси нагреться до комнатной температуры. После перемешивания в течение 12 часов реакцию обрывали водной 1 N НСl (10 мл) и выливали в воду (50 мл). Водный слой экстрагировали этилацетатом (3× 50 мл). Соединенные органические слои промывали рассолом (25 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме. Сырой остаток очищали, используя колонку с силикагелем (4:1-3:1 гексан/этилацетат) с получением 0,202 г указанного в заголовке соединения в виде твердого вещества белого цвета (выход 81%).
1Н ЯМР (300 МГц, ДМСО-d6): δ 0,92 (21Н, s), 2,63 (2Н, m), 3,66 (2Н, t, J=7,4 Гц), 3,75 (3Н, s), 7,05 (1H, d, J=8,8 Гц), 7,15 (1H, s), 7,24 (1H, d, J=2,7 Гц), 7,41 (1H, dd, J=8,7 и 2,6 Гц), 7,78 (1H, d, J=8,9 Гц), 7,91 (1H, dd, J=9,0 и 1,9 Гц), 9,95 (1H, s).
Стадия С. 4-(5-Хлор-2-гидроксифенил)-1-метил-3-(2-гидроксиэтил)-6-(трифторметил)-2(1Н)-хинолинон (43, R3=СF3).
Тетрабутиламмонийфторид (0,380 мл, 0,308 ммоль, 2 экв., 1,0 М/ТГФ) добавляли к перемешиваемому раствору хинолинона (0,105 г, 0,190 ммоль), полученного на стадии В, в ТГФ (10 мл). Через 12 часов сырую реакционную смесь выливали в воду (50 мл). Водный слой экстрагировали этилацетатом (3× 50 мл). Соединенные органические слои промывали рассолом (25 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме. Сырой остаток очищали на хроматографической колонке (силикагель, 2,5% метанол/хлороформ), получая 74 мг твердого вещества белого цвета. Это вещество перекристаллизовывали из смеси этилацетат/гексан с получением 0,064 г (выход 86%) указанного в заголовке соединения, т.пл. 229-230°С.
1Н ЯМР (300 МГц, ДМСО-d6): δ 2,56 (2Н, m), 3,40 (2Н, m), 3,75 (3Н, s), 4,57 (1H, m), 7,05 (1H, d, J=8,8 Гц), 7,14 (1H, s), 7,25 (1H, d, J=2,6 Гц), 7,41 (1H, dd, J=8,7 и 2,7 Гц), 7,77 (1H, d, J=8,8 Гц), 7,89 (1H, dd, J=8,8 и 2,0 Гц), 9,91 (1H, s). МС m/e: 398 (МН+).
Вычислено для С19Н15СlF3NО3, %: С 57,37; H 3.80; N 3,52.
Найдено, %: С 57,69; H 3,88; N 3,28.
Пример 45.
4-(5-Хлор-2-метоксифенил)-1-метил-3-(2-гидроксиэтил)-6-(трифторметил)-2(1Н)-хинолинон (44, R3=СF3).
Смесь хинолинона 42, полученного в примере 44, стадия В (0,202 г, 0,365 ммоль), диметилсульфата (0,038 мл, 0,402 ммоль, 1,1 экв.) и карбоната калия (0,056 г, 0,402 ммоль, 1,1 экв.) в ацетоне (15 мл) нагревали с обратным холодильником в течение 3 часов. Полученную смесь выливали в воду (25 мл). Водный слой экстрагировали этилацетатом (3× 50 мл). Соединенные органические слои промывали рассолом (25 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме. Сырой хинолинон (0,228 г) использовали для проведения последующей реакции без очистки или определения его характеристик.
Тетрабутиламмонийфторид (0,802 мл, 0,802 ммоль, 2 экв., 1,0 М/ТГФ) добавляли к перемешиваемому раствору хинолинона, полученного выше (0,228 г, 0,401 ммоль) в ТГФ (10 мл). Через 12 часов сырую реакционную смесь выливали в воду (50 мл). Водный слой экстрагировали этилацетатом (3× 50 мл). Соединенные органические слои промывали рассолом (25 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме. Сырой остаток очищали на колонке (силикагель, 1:1-1:2 гексан/этилацетат) с получением 0,148 г белого твердого вещества. Это вещество перекристаллизовывали из смеси этилацетат/гексан, получая 0,139 г (выход 84%) указанного в заголовке соединения, т.пл. 175-176°С.
1H ЯМР (300 МГц, ДМСО-d6): δ 2,48 (2Н, m), 3,37 (2Н, m), 3,67 (3Н, s), 3,75 (3Н, s), 4,57 (1Н, t, 5,4), 7,06 (1Н, s), 7,29 (1H, d, J=9,0 Гц), 7,35 (1H, d, J=1,3 Гц), 7,61 (1H, dd, J=8,8 и 2,7 Гц), 7,77 (1H, d, J=8,8 Гц), 7,89 (1H, dd, J=8,8 и 1,9 Гц). МС m/e: 412 (МН+).
Вычислено для С20Н17СlF3NО3, %: С 58,33; H 4,16; N 3,40.
Найдено, %: С 58,30; H 4,07; N 3,18.
Пример 46. 4-(5-Хлор-2-метоксифенил)-3-метил-6-(трифторметил)-2(1Н)-хинолинон.
Раствор соединения 33 (R3=СF3, R8=Н, n=1) (5,63 ммоль), 33% НВr в АсОН (38,3 ммоль) и 10 мл АсОН нагревали до 75°С в течение 3 часов. Раствор охлаждали до комнатной температуры, обрывали реакцию водой (50 мл) и затем перемешивали в течение 12 часов. Осадок отфильтровывали, промывали Н2О и сушили в вакууме. Вещество светло-коричневого цвета перекристаллизовывали из смеси этилацетат/гексан. Соединение, указанное в заголовке, выделяли в виде белого вещества (0,550 г, выход 27%).
1Н ЯМР (300 МГц, СDСl3): δ 2,04 (s, 3Н), 3,72 (s, 3Н), 7,03 (d, 1H, J=9,0 Гц), 7,12 (s, 1H), 7,44 (m, 3Н), 7,65 (d, 1H, J=8,4 Гц), 10,93 (br s, 1H). МС m/e 368 (MH+).
Вычислено для C18H13ClF3NO2·0,33 Н2О, %: С 58,79; H 3,56; N 3,81.
Найдено, %: С 58,89; H 3,82; N 3,53.
Пример 47.
4-(5-Хлор-2-гидроксифенил)-3-(2-гидроксиэтил)-6-(трифторметил)-2(1Н)-хинолинон (31a, R3=CF3, n=2).
Стадия А. 3-(2-Гидроксиэтил)-4-гидрокси-6-хлоркумарин (45).
К раствору γ -бутиролактона (15,5 г, 178,0 ммоль) в ТГФ (100 мл) при -78°С добавляли 1,0 М раствор LiHMDS в тетрагидрофуране (356 мл, 356 ммоль) и перемешивали полученную смесь при -78°С в течение 1,5 часов. Добавляли раствор метилового эфира 5-хлорсалициловой кислоты (16,6 г, степень чистоты 98%, 89,0 ммоль) в ТГФ (95 мл). После перемешивания в течение 1 часа при 0°С смесь нагревали до комнатной температуры в течение ночи для завершения реакции. После охлаждения до 0°С добавляли концентрированную НСl (12 N, 150 мл) для доведения рН до 1. Реакционный раствор перемешивали до тех пор, пока ЖХВР не показала отсутствие промежуточного кетоэфира. К смеси добавляли 400 мл СН2Сl2 и 300 мл Н2О; отделяли органическую фазу и экстрагировали водный слой CH2Cl2 (100 мл). Органические слои соединяли и сушили над безводным Na2SO4, затем удаляли при уменьшенном давлении растворитель с получением твердого вещества. К раствору твердого вещества в ТГФ (290 мл) добавляли гептан (165 мл) для кристаллизации продукта. После охлаждения до 0-5°С в течение примерно 3 часов продукт выделяли фильтрованием и промывали гептаном. После сушки в вакууме было получено в целом 13,9 г (выход 66%) соединения, указанного в заголовке, в виде белых кристаллов, т.пл. 185-186°С. МС m/z 240.
1Н ЯМР (ДМСО, 300 МГц): δ 7,84 (d, 1Н, J=2,4 Гц), 7,61 (dd, 1H, J=2,4 и 8,8 Гц), 7,38 (d, 1H, J=8,8 Гц), 3,56 (t, 2H, J=6,6 Гц), 2,73 (t, 2H, J=6,6 Гц). 13C ЯМР (ДМСО, 75 МГц): δ 162,6, 159,9, 150,5, 131,4, 127,9, 122,4, 118,2, 117,8, 103,2, 59,4, 27,6. ИК (см-1): 3247,2, 2945,1, 2458,6, 1664,9, 1623,9. 1572,7, 1311,5, 1378,1, 1070,8, 825,0.
Стадия В. 2,3-Дигидро-8-хлор-4Н-фуробензопиран-4-он (46).
К раствору 3-(2-гидроксиэтил)-4-гидрокси-6-хлоркумарина (45) (8 г, 33,3 ммоль) в толуоле (360 мл) при комнатной температуре добавляли п-TSA (0,95 г, 5,0 ммоль) и полученный раствор нагревали с обратным холодильником при удалении воды в ловушке Дина-Старка. Реакционную смесь охлаждали до комнатной температуры и промывали дважды насыщенным раствором бикарбоната натрия. Толуол удаляли отгонкой при атмосферном давлении до получения конечного объема 32 мл. После охлаждения до 70°С продукт начал кристаллизоваться. Суспензию кристаллов выдерживали при 55-65°С в течение 30 минут с последующим охлаждением до 0-5°С. Продукт выделяли фильтрованием, промывали холодным толуолом и высушивали в вакууме. Получали 5,5 г (выход 74%) соединения, указанного в заголовке, в виде белых кристаллов, т.пл. 144-146°С. МС m/z 223 (М+Н).
1Н ЯМР (СDСl3, 300 МГц): δ 7,58 (d, 1Н, J=2,5 Гц), 7,49 (dd, 1H, J=2,3 и 8,8 Гц), 7,30 (d, 1H, J=8,9 Гц), 4,90 (t, 2H, J=9,3 Гц), 3,21 (t, 2H, J=9,5 Гц). 13С ЯМР (CDCl3, 75 МГц): δ 166,4, 160,3, 153,4, 132,6, 129,6, 122,4, 118,6, 113,8. 103,6, 74,9. 27,1. ИК (см -1): 3073,1, 2975,8, 1721,2, 1644,4, 1490,8, 1403,7, 1270,6, 1111,8, 1040,1.
Стадия С. 4-(4’-Трифторметилфенилкарбоксамид)-5-(2-гидрокси-5-хлор)-2,3-дигидрофуран (47).
К раствору 2,3-дигидро-8-хлор-4Н-фуробензопиран-4-она (46) (1,02 г, 4,58 ммоль) и 4-(трифторметил)анилина (0,74 г, 4,58 ммоль) в ТГФ (50 мл) при -15°С добавляли LiHMDS (10,5 мл, 10,5 ммоль, 1,0 М раствор в ТГФ). Прозрачный раствор красного цвета перемешивали при -15°С до тех пор, пока ЖХВР не показала менее 1% оставшегося соединения (46) (примерно 30 минут). Реакцию обрывали путем добавления водного раствора NaH2PO4 (50 мл, 10 вес.% в H2O). После добавления МТВЕ (25 мл) слои разделяли и обогащенную органическую фазу промывали последовательно NaH2PO4 (50 мл, 10 вес.% в Н2О) и насыщенным рассолом. После сушки над Na2SO4 раствор концентрировали с получением указанного в заголовке соединения в виде прозрачного оранжевого масла (1,76 г, выход 100%), которое кристаллизовалось после охлаждения. Добавление дихлорметана (20 мл) приводило к получению белых кристаллов, которые выделяли путем фильтрования, промывали дихлорметаном (10 мл) и сушили с получением 1,6 г соединения, указанного в заголовке (90% выход), т.пл. 180-180,5°С. МС m/z 384 (М+Н).
1Н ЯМР (ДМСО, 300 МГц): δ 9,76 (s, 1H), 9,34 (s, 1H), 7,76 (d, 2H, J=8,5 Гц,), 7,60 (d, 2H, J=8,7 Гц), 7,26 (s, 1H), 7,24 (dd, 1H, J=2,2 и 7,0 Гц), 6,83 (dd, 1H, J=2,4 и 7,1 Гц), 4,52 (t, 2H, J=9,6 Гц), 3,16 (t, 2H, J=9,6 Гц). 13С ЯМР (ДМСО, 75 МГц): δ 165,5, 159,7, 155,9, 144,7, 132,0, 131,3, 127,3, 123,7, 121,7, 121,2, 119,5, 110,1, 71,5, 32,9; ИК (см-1): 3303,6, 2950,2, 1654,6, 1608,5, 1531,7, 1408,8,1326,9, 1116,9,1065,7, 840,4.
Стадия D. 4-(5-Хлор-2-гидроксифенил)-3-(2-гидроксиэтил)-6-(трифторметил)-2(1Н)-хинолинон (31a, R3=СF3, n=2).
Раствор 4-(4’-трифторметилфенилкарбоксамид)-5-(2-гидрокси-5-хлор)-2,3-дигидрофурана (47), полученного на стадии С, (1,76 г, 4,58 ммоль), в МеОН (500 мл) промывали азотом и облучали лампой Hanovia мощностью 450 Вт при 30-40°С С до тех пор, пока ЖХВР не показала наличие менее 1% оставшегося соединения (47). Затем МеОН концентрировали в вакууме и растворяли в дихлорметане (50 мл) полученное масло. Кристаллы образовались после перемешивания в течение 1 часа при комнатной температуре. После охлаждения суспензии до 0°С кристаллы выделяли путем фильтрования и подвергали их сушке. Было получено 0,54 г (выход 30%) соединения, указанного в заголовке, в виде кристаллического твердого вещества со степенью чистоты (ЖХВР), равной 97%, физические характеристики которого идентичны характеристикам соединения по примеру 20.
| название | год | авторы | номер документа |
|---|---|---|---|
| ДИФЕНИЛЬНЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 1997 |
|
RU2175319C2 |
| 3-ЗАМЕЩЕННЫЕ ОКСИНДОЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ КАЛИЙНЫХ КАНАЛОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ | 1996 |
|
RU2165925C2 |
| СИММЕТРИЧНЫЕ И НЕСИММЕТРИЧНЫЕ ПРОИЗВОДНЫЕ ДИФЕНИЛМОЧЕВИНЫ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПОДАВЛЕНИЯ РОСТА ОПУХОЛЕВЫХ КЛЕТОК, ОПОСРЕДОВАННОГО КИНАЗОЙ RAF | 1998 |
|
RU2247109C9 |
| ПРОИЗВОДНЫЕ ГЕТЕРОАРЕНКАРБОКСАМИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРИМЕНЕНИЕ | 2003 |
|
RU2320656C2 |
| АНТАГОНИСТЫ РЕЦЕПТОРА FLT3 | 2015 |
|
RU2710928C2 |
| НОВЫЕ АНТАГОНИСТЫ 5-НТ2 | 2016 |
|
RU2706229C2 |
| ЗАМЕЩЕННЫЕ ФЕНИЛНАФТАЛИНЫ В КАЧЕСТВЕ ЭСТРОГЕННЫХ АГЕНТОВ | 2002 |
|
RU2314283C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 1-АЦИЛ-4-ФЕНИЛСУЛЬФОНИЛПРОЛИНАМИДА И НОВЫЕ ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2012 |
|
RU2615997C2 |
| ПРОИЗВОДНЫЕ ИНДОЛИН-2-ОНА В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗ | 2013 |
|
RU2627706C2 |
| АТРОПОИЗОМЕРЫ ПРОИЗВОДНЫХ 3-ЗАМЕЩЕННОГО-4-АРИЛХИНОЛИН-2-ОНА | 2003 |
|
RU2352563C2 |
Изобретение относится к новым производным 3-замещенного 4-арилхинолин-2-она формулы
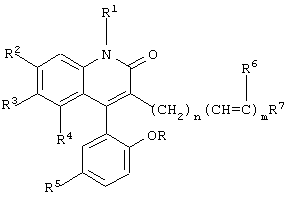
где R и R1 каждый независимо обозначает водород или метил; R2, R3 и R4 каждый независимо обозначает водород или трифторметил при условии, что R2, R3 и R4 не являются одновременно водородом; R5 обозначает бром или хлор; R6 обозначает водород или фтор; n обозначает целое число от 0 до 6; m обозначает целое число 0 или 1 и R7 обозначает СН3, CRR1OH, СНО, C=NOH, СОСН3 или арил, возможно замещенный одним или двумя заместителями, выбранными из группы, состоящей из гидрокси, метокси, амино и ацетиламино, или их нетоксичным фармацевтически приемлемым солям. Технический результат – получение новых соединений, которые являются модуляторами активированных кальцием К+-каналов с высокой проводимостью и пригодны для лечения расстройств, реагирующих на открывание калиевых каналов. 1 н. и 7 з.п. ф-лы, 1 табл.
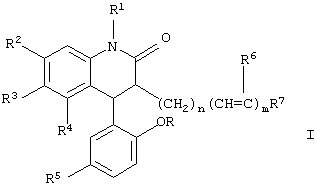
где R и R1 каждый независимо обозначает водород или метил;
R2, R3 и R4 каждый независимо обозначает водород или трифторметил при условии, что R2, R3 и R4 не являются одновременно водородом;
R5 обозначает бром или хлор;
R6 обозначает водород или фтор;
n обозначает целое число от 0 до 6;
m обозначает целое число 0 или 1;
R7 обозначает СН3, CRR1OH, СНО, C=NOH, СОСН3 или арил, возможно замещенный одним или двумя заместителями, выбранными из группы, состоящей из гидрокси, метокси, амино и ацетиламино,
или их нетоксичные фармацевтически приемлемые соли.
где R обозначает водород или метил;
R2, R3 и R4 каждый независимо обозначает водород или трифторметил при условии, что R2, R3 и R4 не являются одновременно водородом;
R5 обозначает хлор;
R6 обозначает водород или фтор;
n обозначает целое число от 0 до 3;
m обозначает целое число, равное 0 или 1;
R7 обозначает СНО, C=NOH, или арил, возможно замещенный одним или двумя заместителями, выбранными из группы, включающей гидрокси, метокси, амино и ацетиламино,
или его нетоксичная фармацевтически приемлемая соль.
4-(5-хлор-2-метоксифенил)-3-(гидроксиметил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-метоксифенил)-3-(гидроксиметил)-7-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-метоксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)-3-хинолинкарбоксальдегид;
4-(5-хлор-2-метоксифенил)-3-(3-гидрокси-1-пропенил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-метоксифенил)-3-(3-гидроксипропил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-(гидроксиметил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-(3-гидрокси-1-пропенил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-(3-гидроксипропил)-6-(трифторметил)-2(1Н)-хинолинон;
(Е)-4-(5-хлор-2-гидроксифенил)-3-(2-фтор-3-гидрокси-1-пропенил)-6-(трифторметил)-2(1Н)-хинолинон;
(Z)-4-(5-хлор-2-гидроксифенил)-3-(2-фтор-3-гидрокси-1-пропенил)-6-(трифторметил)-2(1Н)-хинолинон;
(Е)-4-(5-хлор-2-метоксифенил)-3-(2-фтор-3-гидрокси-1-пропенил)-6-(трифторметил)-2(1Н)-хинолинон;
(Z)-4-(5-хлор-2-метоксифенил)-3-(2-фтор-3-гидрокси-1-пропенил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-(2-гидроксиэтил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-метоксифенил)-3-(2-гидроксиэтил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-метоксифенил)-3-(4-метоксифенил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-метоксифенил)-3-[(4-метоксифенил)метил]-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-метоксифенил)-3-(4-аминофенил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-(3,4-диметоксифенил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-(2,4-дигидроксифенил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-(4-гидроксифенил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-[(4-гидроксифенил)метил]-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-(4-ацетамидофенил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-(4-аминофенил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-[2-(4-гидроксифенил)этил]-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-метил-6-(трифторметил)-2(1Н)-хинолинон;
4-[4-(5-хлор-2-гидроксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)хинолин-3-ил]-3-бутен-2-он;
4-(5-хлор-2-гидроксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)-3-хинолинкарбоксальдегидоксим;
4-(5-хлор-2-метоксифенил)-1,2-дигидро-2-оксо-6-(трифторметил)-3-хинолинкарбоксальдегидоксим;
4-(5-хлор-2-гидроксифенил)-3-(2-гидрокси-2-метилпропил)-6-(трифторметил)-2(1Н)-хинолинон.
4-(5-хлор-2-метоксифенил)-3-(3-гидрокси-1-пропенил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-метоксифенил)-3-(3-гидроксипропил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-(гидроксиметил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-(3-гидрокси-1-пропенил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-(3-гидроксипропил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-(2-гидроксиэтил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-метоксифенил)-3-(2-гидроксиэтил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-(4-гидроксифенил)-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-[(4-гидроксифенил)метил]-6-(трифторметил)-2(1Н)-хинолинон;
4-(5-хлор-2-гидроксифенил)-3-(4-аминофенил)-6-(трифторметил)-2(1Н)-хинолинон.
| WO 9823273 A1, 04.06.1998 | |||
| US 5565483 A, 15.10.1996 | |||
| RU 2001910 C1, 30.10.1993 | |||
| RU 96111002 A, 27.09.1998. |

