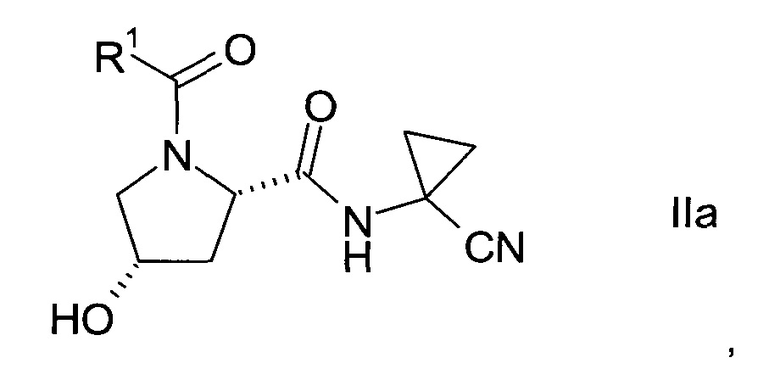
Изобретение относится к способу получения производных пролина формулы:
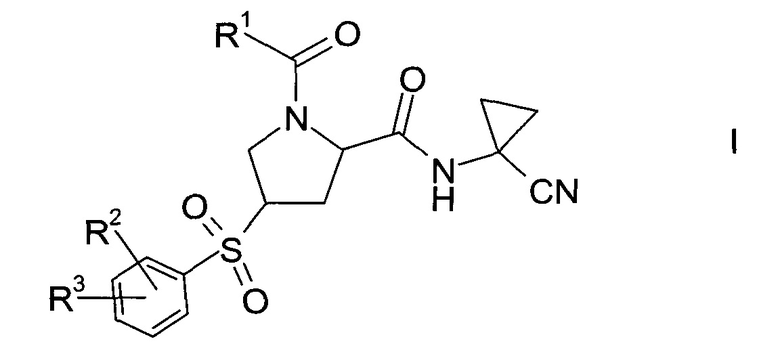
где,
R1 выбран из числа C1-7-алкилов или

где R4 выбран из группы, содержащей C1-7-алкил, галоген-С1-7-алкил, или фенил, возможно, замещенный галогеном.
R2 выбран из группы, содержащей галоген или галоген-С1-7-алкил, и
R3 выбран из группы, содержащей водород, галоген, галоген-С1-7-алкил, С1-7-алкокси, галоген-С1-7-алкокси, или 5- или 6-членный гетероцикл, содержащий один или два атома азота, причем цикл может быть замещен C1-7-алкилом или галогеном.
Пролиновые производные формулы 1 представляют собой предпочтительные ингибиторы цистеиновой протеазы катепсина S и, таким образом, полезны для лечения метаболических заболеваний, таких как диабет, атеросклероз, аневризма брюшной аорты, заболеваний периферических артерий и диабетическая невропатия (РСТ Publ. WO 2010/121918).
Целью настоящего изобретения является разработка масштабируемого способа изготовления соединений формулы I.
Цель может быть достигнута при использовании способа по настоящему изобретению, который включает следующие этапы:
а) трансформация спирта формулы II
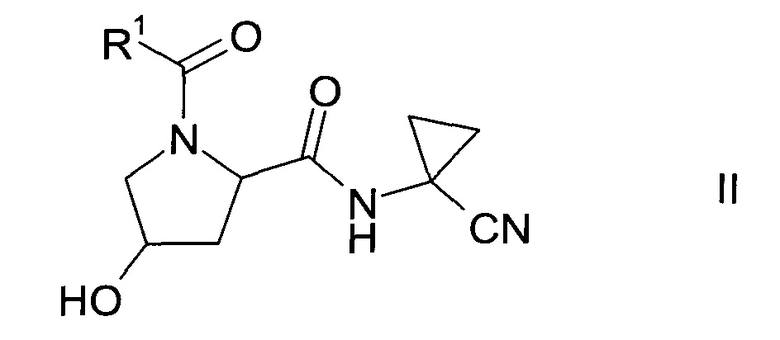
где значение R1 описано выше, в сульфонат формулы III
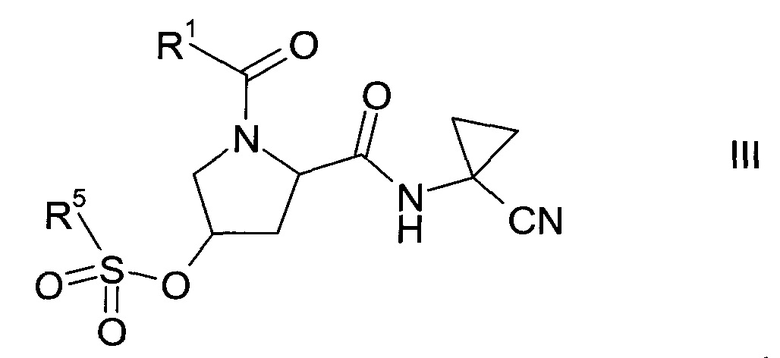
где значение R1 описано выше, а R5 представляет собой C1-7-алкил, галоген-С1-7-алкил или фенил, возможно, замещенный C1-7-алкилом, нитрогруппой или бромом;
b) взаимодействие сульфоната формулы III с тио-соединением формулы IV
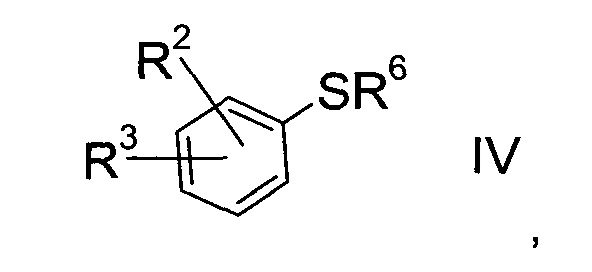
где значение R2 и R3 описаны выше, а R6 представляет собой водород или защитную группу, с получением тиоэфира формулы V.
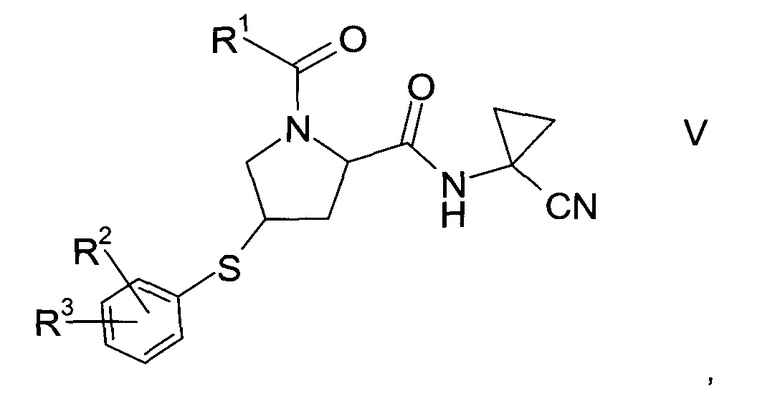
где значения R1, R2 и R3 описаны выше, и
с) окисление тиоэфира формулы V с образованием производного пролина формулы I, где значения R1, R2 и R3 описаны выше.
Для иллюстрации и определения назначения и объема различных терминов, применяемых при описании настоящего изобретения, вводятся следующие определения.
Термин "C1-7-алкил", один или совместно с другими группами, означает разветвленный или линейный одновалентный насыщенный алифатический углеводородный радикал, содержащий от одного до семи атомов углерода, в особенности, от одного до четырех атомов углерода. Этот термин раскрывается примерами радикалов, такими как метил, этил, н-пропил, изопропил, м-бутил, втор-бутил, трет-бутил, пентил, гексил или гептил и их изомеры.
Термин "С1-9-алкил", один или совместно с другими группами, означает разветвленный или линейный одновалентный насыщенный алифатический углеводородный радикал, содержащий от одного до девяти атомов углерода, такой как метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, пентил, циклопентил, гексил, циклогексил, гептил, октил, нонил и их изомеры.
Термин "галоген-С1-7-алкил" означает замещенный галогеном C1-7-алкильный радикал, где значение термина "галоген" раскрыто далее. Конкретные "галоген-С1-7-алкильные" радикалы представляют собой фторированные C1-7-алкильные радикалы, такие как CF3, CH2CF3, СН(CF3)2, СН(СН3)(CF3), C4F9, в особенности CF3.
Термин "С1-7-алкокси" означает разветвленный или линейный одновалентный насыщенный алифатический углеводородный радикал, содержащий от одного до семи атомов углерода, предпочтительно, от 1 до 4 атомов углерода, связанных с атомом кислорода. Примеры "алкокси" включают метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, пентокси, гексилокси или гептокси. Особые используемые здесь алкокси-группы рассматриваются здесь на примерах.
Термин "моно- или ди-(С1-7-алкил)-амино", один или совместно с другими группами, означает один или два разветвленных или прямых одновалентных насыщенных алифатических углеводородных радикалов, содержащих от одного до семи атомов углерода, в особенности, от одного до четырех атомов углерода, связанных с атомом азота. Дополнительно, в случае "ди-(С1-7-алкил)-амино", два C1-7-алкильных радикала могут быть соединены вместе с образованием насыщенного гетероцикла, содержащего атом азота. Примерами "моно- или ди-(С1-7-алкил)-амино" являются метиламино, диметиламино, этиламино, диэтиламино, пирролидинил, этилметиламино, этилпропиламино или пиперидинил.
Термин "галоген-С1-7-алкокси" означает замещенный галогеном радикал C1-7-алкокси, где значение термина "галоген" раскрыто далее. Конкретные "галоген-С1-7-алкокси" радикалы включают фторированные C1-7-алкоксильные радикалы, такие как OCF3, OCH2CF3, OCH(CF3)2, ОСН(СН3)(CF3), OCF4F9, в особенности, OCH2CF3 или ОСН(СН3)(CF3).
Термин "5- или 6-членный гетероцикл, содержащий один или два атома азота" относится к возможно замещенному 5- или 6-членному гетероарильному радикалу, содержащему один или два атома азота и выбранному из группы, содержащей пиридил, пиридазинил, пиримидинил, пиразинил, пирролил, пиразолил или имидазолил, в особенности, пиридинил или пиразолил. Подходящим заместителем является C1-7-алкильная группа, где особенно широко используется метильная группа, или атом галогена, причем особенно широко используется хлор.
Термин "арил" означает радикал фенила, возможно, связанный с одним - тремя заместителями, выбранными из числа C1-7-алкила, C1-7-алкокси и моно- или ди-(С1-7-алкил)-амино.
Термин "галоген" означает атом фтора, хлора, брома или иода.
Получение спирта формулы II:
Спирт формулы II
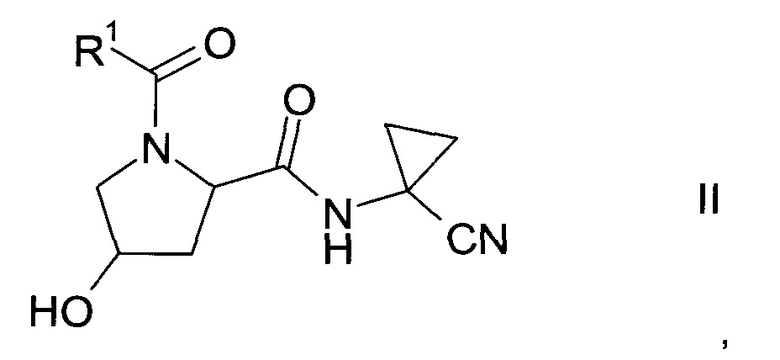
где значение R1 описано выше, можно получить либо
а1) путем реакции гидроксипролинового эфира формулы VI
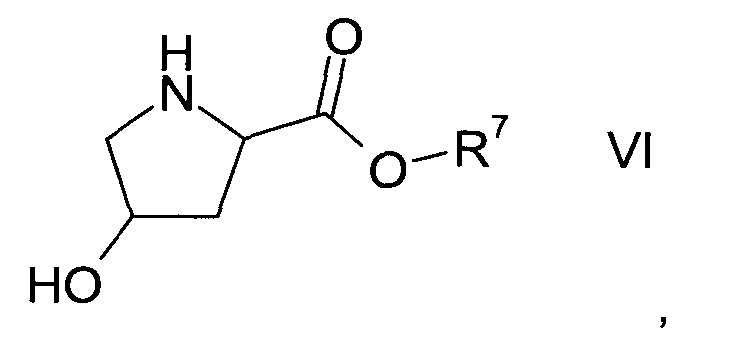
где R7 представляет собой C1-7-алкил,
с карбонильным соединением формулы VII
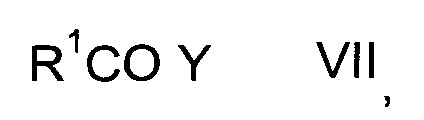
где значение R1 описано выше, a Y представляет собой галоген или ОН, с получением карбонилпролинового эфира формулы IX
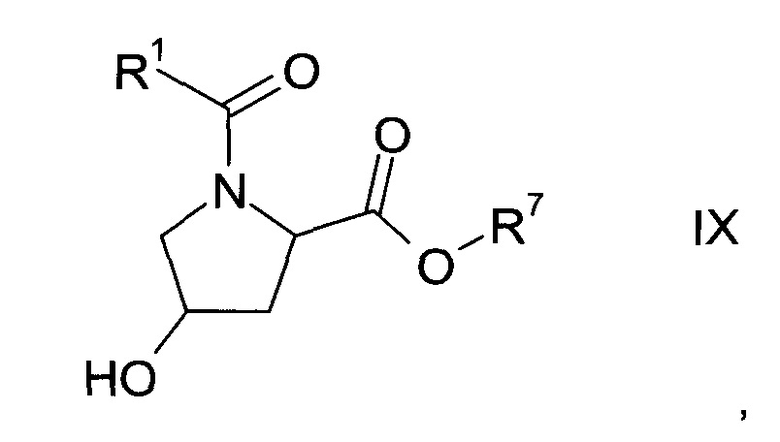
где R1 и R7 описаны выше;
b1) с последующим образованием сульфоната формулы Х
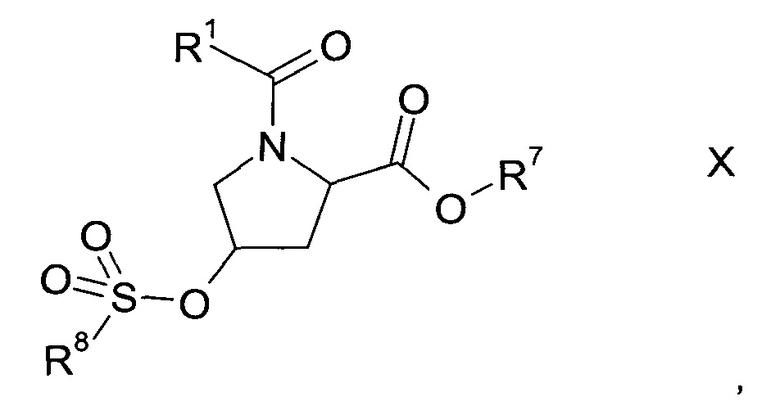
где R1 и R7 описаны выше, a R8 представляет собой C1-7-алкил, возможно, замещенный галогеном или фенил, который возможно замещен C1-7-алкилом, нитро или бром, и
с1) превращением сульфоната формулы Х в присутствии аминоциклопропан карбонитрила формулы XI
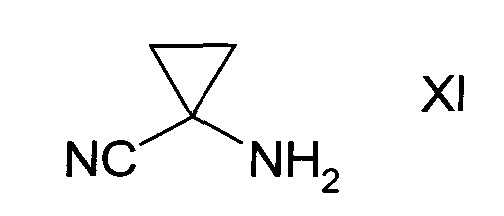
в спирт формулы II
или
а2) путем трансформирования соли сульфоната формулы XII
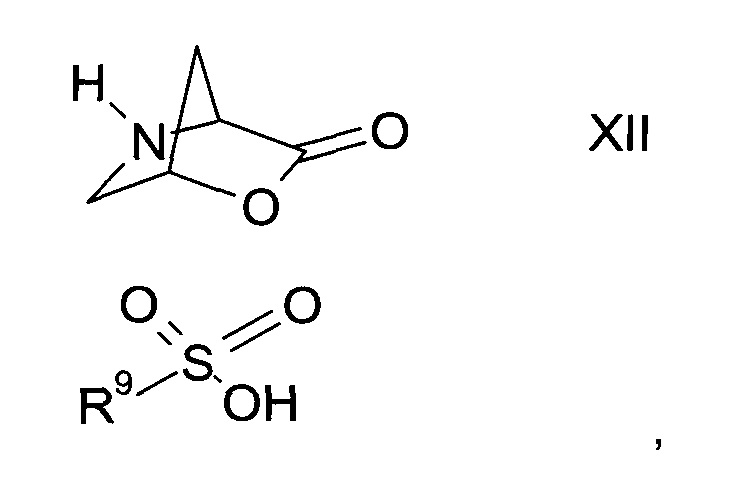
где R9 представляет собой C1-7-алкил или фенил, возможно, замещенный C1-7-алкилом, в амид формулы XIII
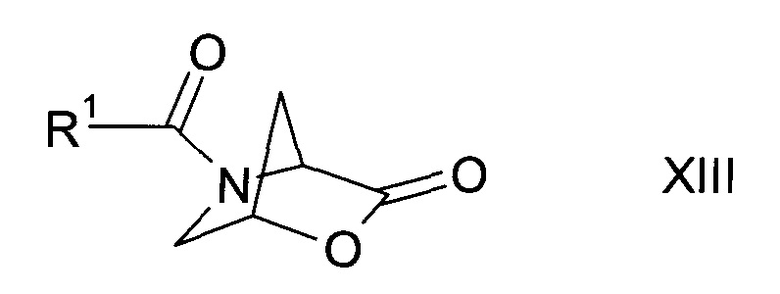
где R1 описан выше, с последующим
b2) превращением амида формулы XIII в присутствии аминоциклопропанкарбонитрила формулы XI
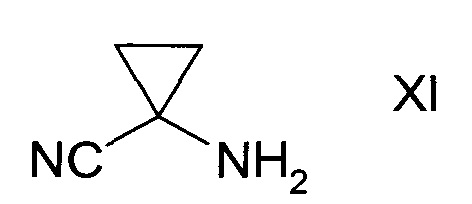
в спирт формулы II.
В соответствии с конкретным воплощением, спирт формулы II, полученный при выполнении описанной выше последовательности этапов а1)-с1) или а2)-b2), представляет собой хиральный изомер формулы
где значение группы R1 описано выше.
Этап а1):
На этапе а1) осуществляется взаимодействие гидроксипролинового эфира формулы VI с карбонильным соединением формулы VII с образованием карбонилпролинового эфира формулы IX.
Эта реакция может проходить в соответствии с принципами, известными опытному специалисту по получению амидов.
Карбонильное соединение формулы VII может представлять собой карбонилхлорид или карбоновую кислоту.
При осуществлении реакции присоединения карбоновой кислоты, как правило, используются распространенные агенты присоединения, используемые для образования амидной связи, например, кратко описанные в журнале Chemical Reviews, том 111 (2011), страницы 6557-6602; например, такими агентами могут быть DCC или DIC.
В соответствии с конкретным воплощением, используемые карбонилхлориды, как правило, готовят in situ из соответствующей карбоновой кислоты с применением распространенного галогенирующего агента, такого как оксалилхлорид или тионилхлорид.
В соответствии с конкретным воплощением настоящего изобретения, гидроксипролиновый эфир формулы VI представляет собой коммерчески доступный гидроксипролиновый метиловый эфир гидрохлорид.
Реакцию, как правило, проводят в инертном органическом растворителе, таком как дихлорметан, тетрагидрофуран или толуол при температурах от -10°C до 25°C.
Удобно, если присутствует подходящий третичный амин, такой как триэтиламин или диизопропилэтиламин.
Карбониловый пролиновый эфир формулы IX может либо быть выделен способами, известными специалисту, либо, предпочтительно, сохранен в растворе и перенесен на следующий этап реакции b1).
Этап b1):
На этапе b1) получают сульфонат формулы X.
Как правило, при этом можно использовать распространенный сульфонирующий агент, такой как метансульфонил хлорид, бензол сульфонил хлорид или п-толуол сульфонил хлорид.
Соответственно, группа R8 конкретно представляет собой метил, фенил или п-толил, более конкретно, метил.
Реакцию преимущественно проводят в той же среде, что была не предыдущем этапе, при температурах от -10°C до 40°C.
Сульфонат формулы Х можно выделить известными специалисту способами, такими как экстракция из реакционной смеси с использованием подходящих растворителей, таких как дихлорметан, и последующей кристаллизацией, например, в изобутил ацетате.
Этап с1):
На этапе с1) осуществляют преобразование сульфоната формулы Х в присутствии аминоциклопропан карбонитрила формулы XI в спирт формулы II.
Реакция включает первоначальный гидролиз сложноэфирной группы, который удобно проводить с неорганическим водным основанием, таким как водный раствор гидроксида щелочного металла, например, гидроксида натрия, гидроксида калия или гидроксида лития, при температуре от 0°C до 30°C, с последующим промежуточным образованием лактона.
Аминоциклопропана карбонитрил формулы XI может реагировать в виде свободного основания, также как и в виде подходящей соли, в особенности, гидрохлорида. Гидрохлорид является наиболее подходящей солью.
Аминолиз лактона с помощью соли аминоциклопропана карбонитрила, такой как гидрохлорид, ускоряется при добавлении, по меньшей мере, стехиометрических количеств соли щелочного металла и алкил- или арилкарбоксилата MR10COO, где М=Li, Na, К или Cs, в частности Na, a R10=C1-9-алкил или арил. В особенности, для ускорения реакции можно использовать 2-этилгексаноат.
Альтернативно, аминолиз лактона с использованием соли аминоциклопропан карбонитрила, такой как гидрохлорид, ускоряется при добавлении суб-стехиометрических количеств алкил- или арилкарбоксилата щелочного металла MR10COO, где М и R10 описаны выше, особенно, натрия 2-этилгексаноата, совместно со стехиометрическим количеством подходящего основания, такого как триэтиламин.
Если используется свободное основание аминоциклопропан карбонитрил, аминолиз лактона ускоряется при добавлении стехиометрических или суб-стехиометрических количеств алкил- или арилкарбоксилата щелочного металла MR10COO, где М и R10 описаны выше, особенно 2-этилгексаноата натрия, или при добавлении стехиометрических или суб-стехиометрических количеств алкил- или арилкарбоновой кислоты R10COOH, где группа R10 определена выше, особенно 2-этилгексановой кислоты.
В систему можно добавить органический растворитель, такой как тетрагидрофуран, реакцию выполняют при температуре от 40 до 130°C, в особенности, от 50 до 70°C.
Выделение продукта из реакционной смеси можно проводить в соответствии со способами, известными специалисту в уровне техники, например, путем экстракции с подходящим растворителем, таким как этилацетат, и последующей кристаллизацией полученного продукта также из подходящего органического растворителя или смеси растворителей, например, этилацетат/гептан.
Альтернативно, спирт формулы II можно синтезировать следующим образом:
Этап а2)
На этапе а2) происходит превращение сульфонатной соли формулы XII в амид формулы XIII.
Подходящими сульфонатными солями формулы XII являются либо метан сульфонат (R9= метил), либо п-толуол сульфонат (R9= п-толил).
Эти сульфонаты можно синтезировать известными в литературе способами, например, приведенными в журнале Journal of Organic Chemistry, том 71, страницы 7133-7145.
Амид получают в соответствии с принципами, известными специалисту в уровне техники, и в соответствии с описанием этапа а1).
Выделение продукта из реакционной смеси можно проводить в соответствии со способами, известными специалисту в уровне техники, например, путем экстракции с подходящим растворителем, таким как дихлорметан, и последующей кристаллизацией полученного продукта также из подходящего органического растворителя или смеси растворителей, например, этилацетат/гептан.
Этап b2)
На этапе b2) происходит превращение амида формулы XIII в спирт формулы II в присутствии аминоциклопропан карбонитрила формулы XI.
Аминоциклопропан карбонитрил формулы XI может вступать в реакцию в виде свободного основания или подходящей соли, особенно, гидрохлорида. Наиболее подходящей солевой формой является гидрохлорид.
Аминолиз лактона с использованием соли аминоциклопропан карбонитрила, такой как гидрохлорид, ускоряется при добавлении по меньшей мере стехиометрических количеств алкил- или арилкарбоксилата щелочного металла MR10COO, где М=Li, Na, К или Cs, в особенности, Na, a R10 представляет собой C1-9-алкил или арил. В частности, для ускорения реакции можно использовать 2-этилгексаноат натрия.
Альтернативно, аминолиз лактона с использованием соли аминоциклопропан карбонитрила, такой как гидрохлорид, ускоряется при добавлении суб-стехиометрических количеств алкил- или арилкарбоксилата щелочного металла MR10COO, где М и R10 описаны выше, особенно, 2-этилгексаноата натрия, совместно со стехиометрическим количеством подходящего основания, такого как триэтиламин.
Если используется свободное основание аминоциклопропан карбонитрил, аминолиз лактона ускоряется при добавлении стехиометрических или суб-стехиометрических количеств алкил- или арилкарбоксилата щелочного металла MR10COO, где М и R10 описаны выше, особенно, 2-этилгексаноата натрия, или при добавлении стехиометрических или суб-стехиометрических количеств алкил- или арилкарбоновой кислоты R10COOH, где группа R10 определена выше, особенно 2-этилгексановой кислоты.
Обычно, реакцию проводят в полярных растворителях, таких как тетрагидрофуран, дихлорметан, вода или их смеси, или без растворителя (мягких условиях), при температуре от 40 до 130°C, в особенности от 50 до 70°C.
Выделение продукта из реакционной смеси можно проводить в соответствии со способами, известными специалисту в уровне техники, например, путем экстракции с подходящим растворителем, таким как этилацетат, и последующей кристаллизацией полученного продукта также из подходящего органического растворителя или смеси растворителей, например, этилацетат/гептан.
Спирт формулы II
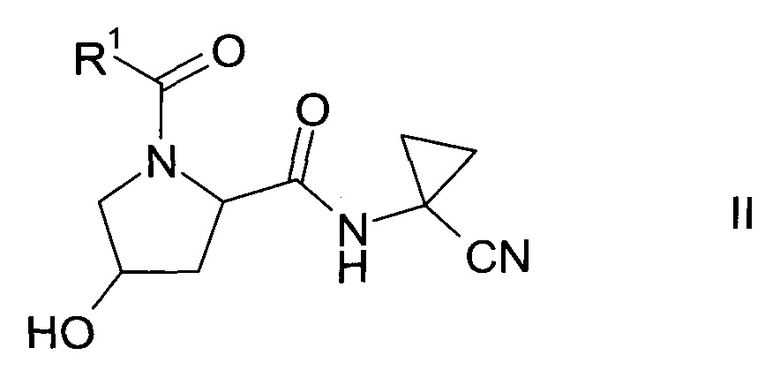
где R1 описан выше, неизвестен в уровне техники и, таким образом, является особым воплощением настоящего изобретения.
В соответствии с еще одним особым воплощением, группа R1 представляет собой
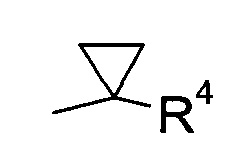
где R4 выбран из группы, содержащей C1-7-алкил, галоген-С1-7-алкил или фенил, возможно, замещенный галогеном.
Более конкретно, R4 представляет собой метил или трифторметил.
В соответствии с еще одним особым воплощением, спирт формулы II представляет собой хиральный изомер формулы
где значение группы R1 описано выше.
Получение тио-соединения формулы IV:
Тио-соединение формулы IV, где R6=Н
Тио-соединение формулы IV
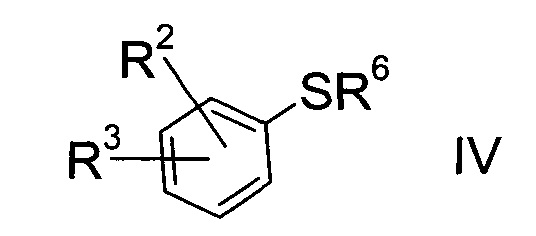
где R2 и R3 определены выше, a R6 представляет собой водород, можно изготовить следующим образом:
а3) снятие защиты с соединения формулы XX
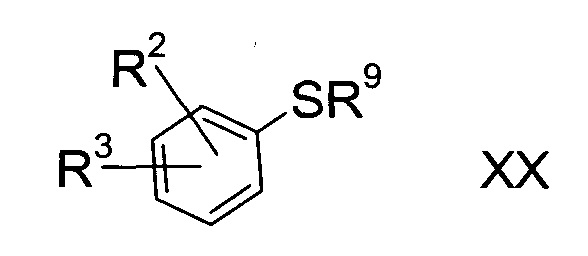
где R2 и R3 определены выше, а R9 представляет собой третичную алкильную группу формулы
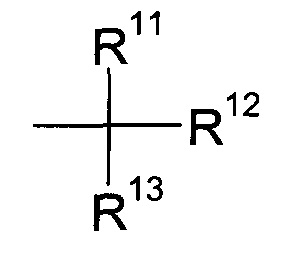
где R11, R12 и R13 независимо друг от друга представляют собой C1-7-алкил с помощью кислоты;
или
b3) снятие защиты с соединения формулы XX
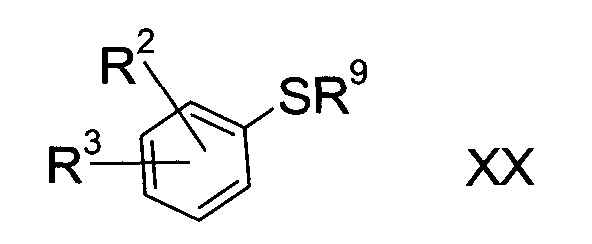
где R2 и R3 определены выше, а R9 представляет собой тритил, с помощью кислоты в присутствии восстановителя;
c3) литиирование галогенированного соединения формулы XXI
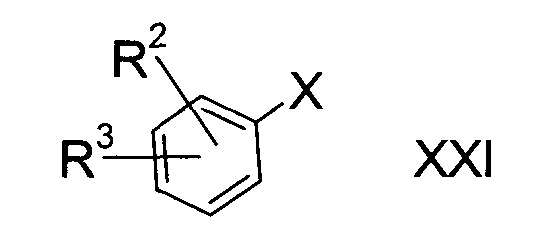
где R2 и R3 определены выше, а Х представляет собой атом галогена, с последующей обработкой продукта серой;
или
d3) взаимодействие галогенированного соединения формулы XXI
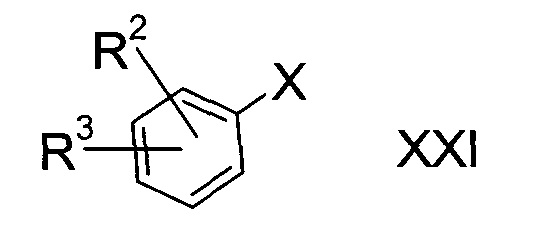
где R2 и R3 определены выше, а Х представляет собой атом галогена с реактивом Гриньяра с последующей обработкой продукта серой;
Этап a3)
Вариант способа a3) предполагает снятие защиты с соединения формулы XX, где R9 представляет собой третичную алкильную группу, действием кислоты.
В особенности, группа R9 соответствует формуле:
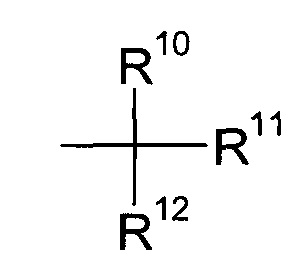
где R10, R11 и R12 независимо друг от друга представляют собой C1-7-алкил и, более конкретно, C1-4-алкил. Еще более конкретно, R9 представляет собой трет-бутил.
Соединения формулы XX либо доступны коммерчески, либо можно получить способами, знакомыми специалисту в уровне техники.
Соединения формулы XX, где R9= трет-бутил, можно приготовить реакцией соответствующего фторированного предшественника с 2-метил-2-пропан-тиолом в присутствии алкоксида щелочи.
Подходящие кислоты можно выбрать из числа водных минеральных кислот, таких как соляная кислота, или из числа органических кислот, таких как трифторуксусная кислота.
При использовании органической кислоты может присутствовать органический растворитель, такой как дихлорметан.
Условия реакции и способы выделения тио-соединений формулы IV зависят от используемой кислоты, но их адаптация может соответствовать методикам, известным специалисту в уровне техники.
Этап b3)
Вариант способа b3) предполагает снятие защиты с соединения формулы XX, где R9 представляет собой тритил, действием трифторуксусной кислоты в присутствии восстановителя.
Подходящим восстановителем является триэтилсилан.
Может использоваться органический растворитель, такой какдихлорметан.
Условия реакции и выделения тио-соединения формулы IV могут соответствовать методикам, известным специалисту в уровне техники.
Этап c3)
Вариант способа c3) включает литиирование галогенированного соединения формулы XXI с последующей обработкой серой.
Подходящие агенты для литиирования могут быть выбраны из числа коммерчески доступных агентов, таких как бутил литий.
Литиирование, как правило, проводят в присутствии органического растворителя, такого как толуол, при температуре от -80 до -20°C.
Удобно использовать также хелатор, такой как диэтиловый эфир или ди-н-пропиловый эфир.
Последующая обработка серой может проводиться при температуре от -80 до -40°C, как правило, в той же реакционной среде, что используется и для литиирования.
Способы выделения тио-соединения формулы IV могут соответствовать методикам, известным в уровне техники.
Этап d3)
Вариант способа d3) предполагает реакцию галогенированного соединения формулы XXI с реактивом Гриньяра с последующей обработкой серой.
Х представляет собой, в особенности, бром.
Подходящие реактивы Гриньяра можно выбрать из числа коммерчески доступных агентов, таких как изопропил магний хлорид или изопропил магний хлорид/литий хлорид в тетрагидрофуране.
Как правило, реакция Гриньяра проходит в присутствии органического растворителя, в котором реактив Гриньяра продается, например, в тетрагидрофуране.
Удобно удерживать температуру реакции в диапазоне от 0 до 40°C, особенно около комнатной температуры.
Последующая обработка серой может проводиться при температуре от -20 до 20°C, как правило, в той же реакционной среде, что используется и реакции Гриньяра.
Выделение тио-соединения формулы IV может соответствовать методикам, известным в уровне техники.
Тио-соединение формулы IV с R6= защитная группа
Подходящая защитная группа может быть выбрана из числа следующих: 2-карбамоилэтил, C1-7-алкоксикарбонилэтил-, а также моно или ди-C1-7-алкиламинокарбонилэтил.
Показано, что особенно удобно, если R6= 2-карбамоилэтил.
Эти соединения можно приготовить по следующей далее схеме. Х представляет собой атом галогена, в частности, брома, группы R2 и R3 соответствуют приведенному выше определению.
Схема 1:
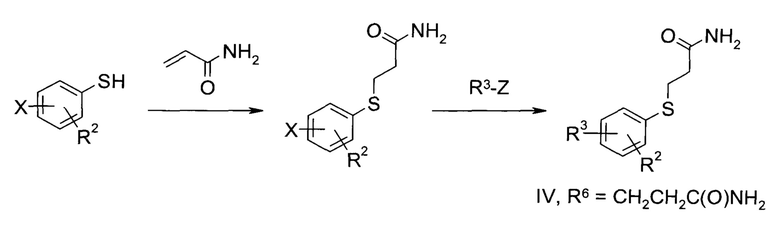
2-Карбамоилэтильную защитную группу можно ввести путем реакции соответствующего тиофенола с акриламидом в присутствии каталитических (или стехиометрических) количеств подходящего основания, например, тетрабората натрия, в подходящем полярном растворителе, таком как метанол, вода или их смесь. В случае других замещенных пропионамидных или сложноэфирных защитных групп реагирующий акриламид заменяют соответствующим образом. С полученным замещенным тиофенолом можно затем работать способами, известными специалисту в уровне техники. Например, чтобы заменить атом брома 5- или 6-членным гетероциклом R3, определенным выше, необходимо провести реакцию с подходящим реагентом R3-Z, где Z=В(ОН)2, В(ОМе)2, B(OEt)2, В(OiPr)2,_4,4,5,5-тетраметил-[1,3,2]-диоксаборил)-, Sn(н-Bu)3, MgX, ZnX или Si(OEt)3, в особенности, В(ОН)2 или 4,4,5,5-тетраметил-[1,3,2]-диоксаборил)-. В конкретных случаях, реакцию проводят в присутствии каталитического количества подходящего комплекса переходного металла, например, [1,1'-бис(дифенилфосфино)ферроцен]-ди-хлор-палладий(II) или тетракис(трифенилфосфин)палладий(0), стехиометрического количества подходящего основания, например, карбоната калия, карбоната натрия или фосфата натрия, в подходящем растворителе, таком как, например, диметилформамид, диметилацетамид, толуол, тетрагидрофуран, трет-бутанол, N-метилпирролидон или диоксан, возможно и предпочтительно, в смеси с водой, при повышенной температуре от 40 до 140°C, предпочтительно от 50 до 70°C.
Кроме того, описанные выше реагенты R3-Z, в особенности, Z=4,4,5,5-тетраметил-[1,3,2]-диоксаборил)-, можно получить in situ, начиная от соответствующих R3-X, где Х представляет собой атом галогена, в особенности, брома или иода, путем их реакции с 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксабороланом) в присутствии соответствующего основания, например, ацетата натрия или калия, и каталитического количества подходящего комплекса палладия, такого как [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II), в подходящем растворителе, например, диметилформамиде, при температуре от 60 до 120°C, в особенности, от 70 до 90°C, с последующей проводимой в той же емкости реакцией с защищенным галотиофенолом, в особенности, используя похожий, более конкретно, тот же растворитель и комплекс палладия.
Этап а)
На этапе а) осуществляется превращение спирта формулы II в сульфонат формулы III.
В соответствии с конкретным воплощением, используется хиральный изомер спирта формулы II, структура которого соответствует формуле
где значение R1 определено выше.
Сульфонирование проводят способами, известными специалисту в уровне техники, применяя распространенные коммерчески доступные агенты сульфонирования. Конкретными агентами сульфонирования являются бензосульфонил хлорид или метансульфонил хлорид.
Как правило, в системе присутствует третичный амин, такой как триэтиламин. Реакцию можно ускорить добавлением подходящего основания Льюиса, например 4-(диметиламино)пиридина.
Как правило, реакцию проводят в органическом растворителе, таком как тетрагидрофуран, при температурах от -10 до 40°C.
Выделение сульфоната формулы III можно проводить способами, известными специалисту в уровне техники.
Сульфонат формулы III
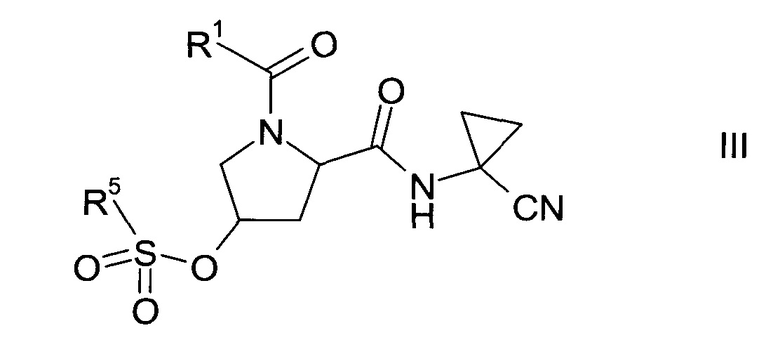
где значение группы R1 описано выше, a R5 представляет собой C1-7-алкил, галоген-С1-7-алкил или фенил, возможно, замещенный C1-7-алкилом, нитрогруппой или бромом, не известен в уровне техники и, таким образом, является особым воплощением настоящего изобретения.
В соответствии с еще одним особым воплощением, R1 представляет собой
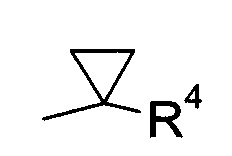
где R4 выбрана из группы, содержащей C1-7-алкил, галоген-С1-7-алкил или фенил, возможно, замещенный галогеном, а R5 представляет собой C1-7-алкил или фенил, возможно, замещенный C1-7-алкилом.
Более конкретно, R4 представляет собой метил или трифторметил, а R5 представляет собой метил или фенил.
В соответствии с другим особым воплощением, сульфонат формулы III представляет собой хиральный изомер формулы:
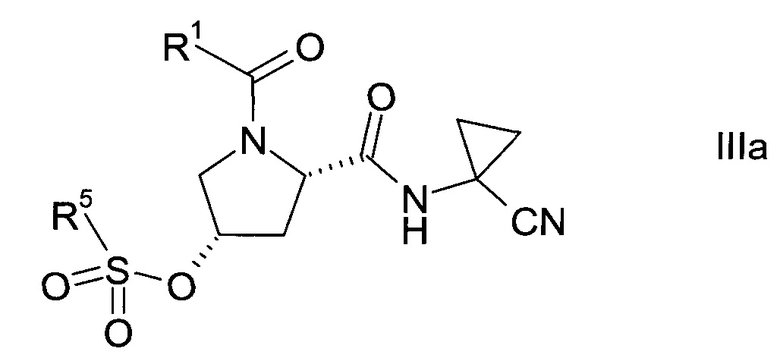
где R1 и R5 описаны выше.
Этап b)
На этапе b) сульфонат формулы III реагирует с тио-соединением формулы IV с образованием тиоэфира формулы V.
Реакцию удобно проводить в присутствии основания, такого как алкоголят или карбонат щелочного металла.
Более конкретно, используется трет-бутилат лития, натрия или калия.
Удобно использовать органический растворитель, такой как тетрагидрофуран, диметилацетамид или их смеси.
Температура реакции может быть выбрана в диапазоне от 10 до 90°C.
Тиоэфир формулы V можно выделить из реакционной смеси в соответствии со способами, известными специалисту в уровне техники, например, путем экстракции с подходящим растворителем, таким как этилацетат или трет-бутилметиловый эфир. Дальнейшую очистку можно проводить кристаллизацией полученного продукта из подходящего органического растворителя, например, толуола, н-гептана, 2-бутанола или их смеси.
Тиоэфир формулы V
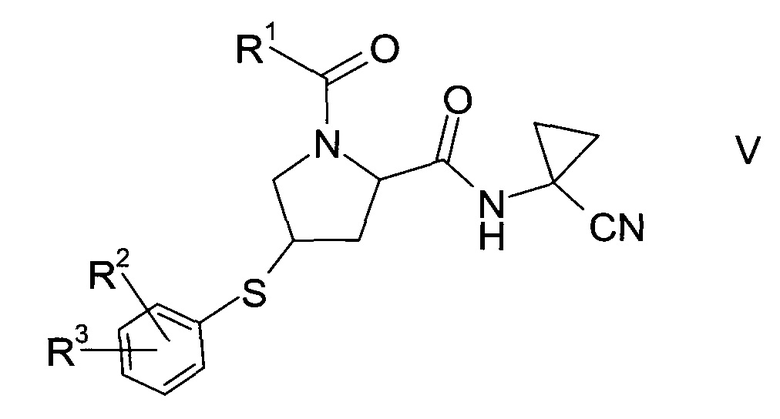
где R1, R2 и R3 описаны выше, неизвестен в уровне техники и, таким образом, представляет собой особое воплощение настоящего изобретения.
В соответствии с еще одним особым воплощением, R1 представляет собой
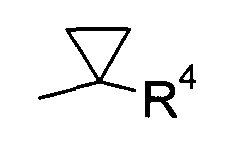
где R4 выбран из группы, содержащей C1-7-алкил, галоген-С1-7-алкил или фенил, возможно, замещенный галогеном.
R2 представляет собой галоген или галоген-С1-7-алкил, и
R3 представляет собой галоген-С1-7-алкокси или 5- или 6-членный гетероцикл, содержащий один или два атома азота, причем цикл может быть замещен C1-7-алкилом или галогеном.
Более конкретно,
R4 представляет собой метил или трифторметил
R2 представляет собой трифторметил или хлор, и
R3 представляет собой 2,2,2-трифторэтокси, 2-метилпирид-4-ил, 1-метил-1Н-пиразол-4-ил или 2,2,2-трифтор-1-метилэтокси.
В соответствии с еще одним особым воплощением, тиоэфир формулы V представляет собой хиральный изомер формулы
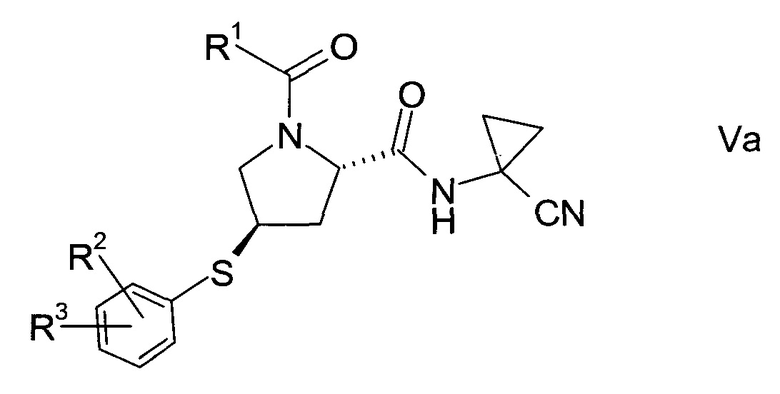
еще более конкретно,
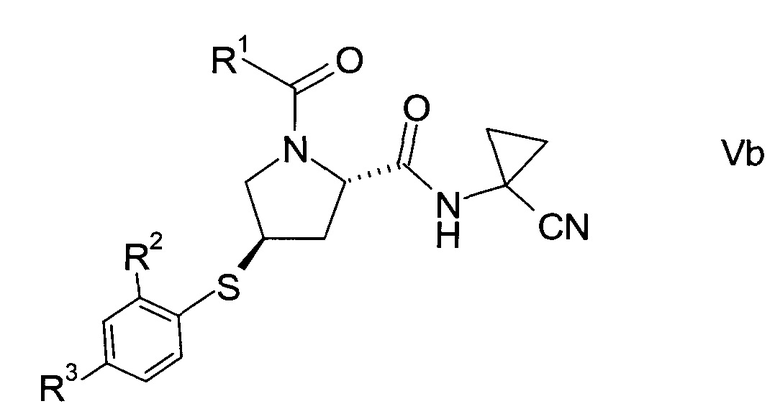
где значения R1, R2 и R3 определены выше.
Этап с)
На этапе с) тиоэфир формулы V окисляется с образованием пролинового производного формулы I.
Окисление может проводиться с использованием коммерчески доступных окислителей, таких как пероксомоносульфат калия, доступный, например, в виде тройной соли "оксон" (Oxone®), или гексагидрат монопероксофталаата магния.
Удобно использовать полярный органический растворитель, такой как ацетонитрил. Дополнительно можно также добавлять воду или водные растворы неорганических кислот, таких как серная или фосфорная кислота.
Температура реакции может быть выбрана в диапазоне от 0 до 60°C.
Пролиновое производное формулы 1 можно выделить из реакционной смеси в соответствии со способами, известными специалисту в уровне техники, например, путем экстракции с подходящим растворителем, таким как этилацетат. Дальнейшую очистку можно проводить кристаллизацией полученного продукта из подходящего растворителя, например, ацетона, изопропанола, воды или их смеси.
В соответствии с конкретным воплощением настоящего изобретения, пролиновые производные формулы 1 представляют собой хиральные изомеры формулы

еще более конкретно, формулы
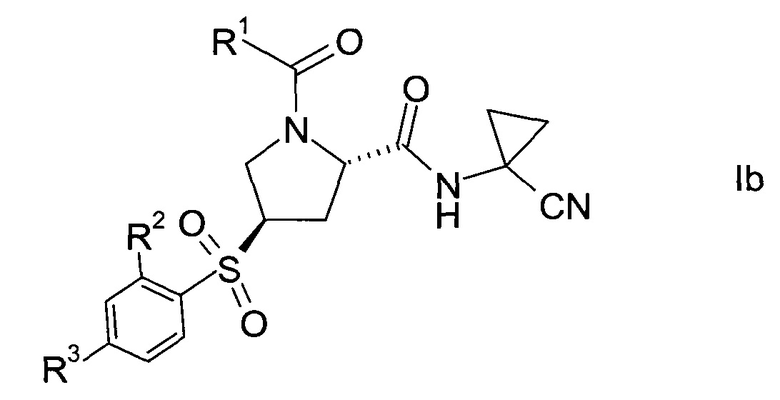
где значения R1, R2 и R3 определены выше.
В соответствии с еще одним конкретным воплощением,
R1 представляет собой
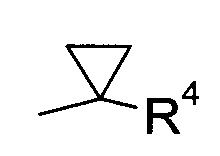
где R4 выбран из группы, содержащей C1-7-алкил, галоген-С1-7-алкил или фенил, возможно, замещенный галогеном,
R2 представляет собой галоген или галоген-С1-7-алкил и
R3 представляет собой галоген-С1-7-алкокси или 5- или 6-членный гетероцикл, содержащий один или два атома азота, причем цикл может быть замещен C1-7-алкилом или галогеном
Более конкретно,
R4 представляет собой метил или трифторметил,
R2 представляет собой трифторметил или хлор, и
R3 представляет собой 2,2,2-трифторэтокси, 2-метилпирид-4-ил, 1-метил-1H-пиразол-4-ил или 2,2,2-трифтор-1-метилэтокси.
Примеры:
Общая часть:
Все растворители и реагенты были приобретены у коммерческих поставщиков и использованы в том виде, в каком приобретены. Как правило, реакции сопровождались анализом по методу ТСХ (ТСХ плашки F254, "Мерк"), ЖХ (жидкостная хроматография) или ГХ (газовая хроматография). Протонные спектры ЯМР записывали на приборе "Брукер" (Bruker) 300, 400 или 600 МГц, химические сдвиги (δ, м.д.) записывались относительно триметилсилана в качестве внутреннего стандарта в следующем формате: химический сдвиг в м.д. (форма пика, константа сопряжения, если применимо, интеграл). Используются следующие сокращения ЯМР: s, синглет; d, дублет; t, триплет; q, квадруплет; quint, квинтет; sext, секстет; hept, гептет; m, мультиплет; br, уширение. Чистоту анализировали методом ВЭЖХ в обращенной фазе. Элементный анализ проводили на приборе Agilent 1100 & 1200 (производство "Аджилент"). Элементный анализ проводили в компании "Солвиас АГ" (Solvias AG, Маттенштрассе, почтовый ящик, СН-4002 Базель, Швейцария). Колоночную хроматографию проводили на силикагеле 60 (32-60 меш, 60 Å) или на заполненных колонках (Isolute Flash Si). Масс-спектры записывали на спектрометре Agilent 6520 QTOF при следующих одновременно задаваемых условиях (множественный режим, multimode): ESI (электронно-ионная ионизация) и APCI (химическая ионизация при атмосферном давлении), а также на приборе Agilent 5975 в режиме EI (электронная ионизация) с детекцией либо положительно (pos.), либо отрицательно (neg.) заряженных ионов. Если не указано иное, регистрировались положительно заряженные ионы.
Получение спирта:
А1. Получение (1-циано-циклопропил)-амида (2S,4S)-4-гидрокси-1-(1-трифторметил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты
а.) (1S,4S)-3-Оксо-2-окса-5-азония-бицикло[2.2.1]гептан метансульфонат
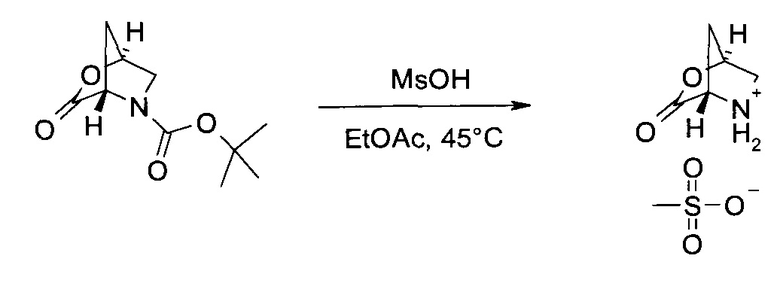
Трет-бутиловый эфир (1S,4S)-3-оксо-2-окса-5-аза-бицикло[2.2.1]гептан-5-карбоновой кислоты (100,0 г, 469 ммоль) растворили в этилацетате (970 мл) и при температуре 45°C добавили метансульфоновую кислоту (43,5 мл, 659 ммоль). Смесь 16 часов перемешивали при 45°C. Суспензию охладили до комнатной температуры, профильтровали, осадок промыли этилацетатом (240 мл) и высушили в вакууме в получением заявленного соединения в виде белого кристаллического вещества (94,2 г, 96%). MS (El, neg): m/z=113 [катион - Hf, 69 [катион - Н - CO2]+, 68 [катион - Н - НСО2]+. 1H ЯМР (ДМСО-d6, 600 МГц): δ 2,12 (dd, J=1,2 Гц, 12,0 Гц, 1Н), 2,33 (s, 3H), 2,59 (d, J=12,0 Гц, 1Н), 3,33 и 3,50 (АВХ, JAB=12,0 Гц, JAX=1,9 Гц, JBX=0 Гц, каждый 1Н), 4,58 (s, 1Н), 5,41 (s, 1Н), 9.74 (br s, 2H).
b) (15,45)-5-(1-Трифторметил-циклопропанкарбонил)-2-окса-5-аза-бицикло[2.2.1]гептан-3-он
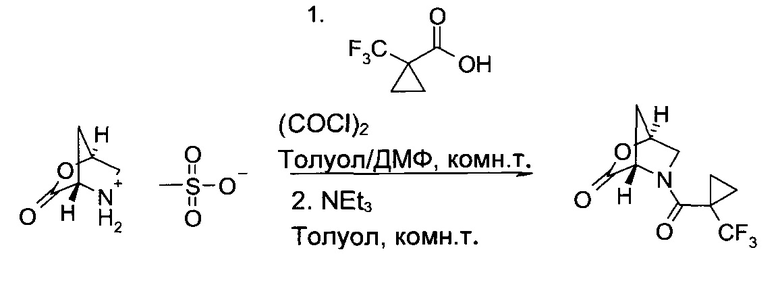
1-Трифторметил-циклопропанкарбоновую кислоту (167.0 г, 1084 ммоль) суспендировали в толуоле (500 мл), после чего добавили диметилформамид (3.6 мл, 47 ммоль). Смесь охладили на водяной бане до 2°C, после чего по каплям в течение 25 минут добавили раствор оксалил хлорида (90 мл, 1037 ммоль) в толуоле (167 мл). После этого смесь перемешивали еще 30 минут и затем 4 часа при комнатной температуре. Затем ее снова охладили до 0°C (сухой лед/метанольная баня) и медленно добавили метансульфонат (1S,4S)-3-оксо-2-окса-5-азония-бицикло[2.2.1]гептана (200 г, 956 ммоль), тетрагидрофуран (330 мл) и триэтиламин (500 мл, 3,59 моль), удерживая температуру ниже 5°C. Следует отметить, что, особенно после добавления 50% триэтиламина, реакция становится сильно экзотермической, и важно обеспечивать достаточное охлаждение системы. После этого смесь 20 часов перемешивали при комнатной температуре, вылили в водный раствор лимонной кислоты (10% в воде, 1,6 л), и фазы разделили. Водную фазу проэкстрагировали этилацетатом (3×500 мл). Объединенные органические экстракты трижды промыли концентрированным соляным раствором (500 мл), высушили над сульфатом натрия и сконцентрировали в вакууме. Неочищенный продукт (245 г, коричневое масло) растворили в дихлорметане (330 мл), после чего добавили этилацетат (130 мл) и гептан (660 мл), а дихлорметан осторожно удалили перегонкой в вакууме. Продукт начал кристаллизоваться. Суспензию охладили до 2°C (ледяная баня) и перемешивали один час, затем отфильтровали. Осадок промыли смесью этилацетат/гептан 1:9 (об./об., 300 мл) и высушили в вакууме, получив указанное в заголовке соединение в виде светло-коричневого порошка (219 г, 92%). 1H ЯМР (CDCl3, 400 МГц): (51,17-1,25 (m, 1H), 1,30 (dd, J=5,3 Гц, 8,3 Гц, 1H), 1,37-1,46 (m, 2H), 2,13 и 2,37 (АВ, JAB=10,7 Гц, все 1Н), 3,63 и 3,73 (AB, JAB=12,1 Гц, все 1Н), 4,99 (s,1H), 5,21 (s, 1H).
с) (13,45)-5-(1-Метил-циклопропанкарбонил)-2-окса-5-аза-бицикло[2.2.1]гептан-3-он
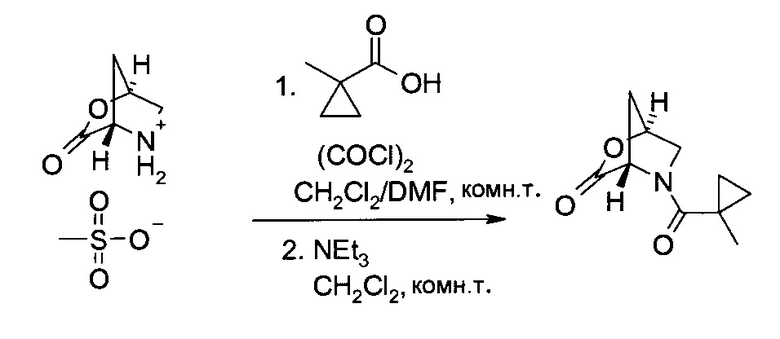
1-Метил-циклопропанкарбоновую кислоту (56,4 г, 552 ммоль) растворили в дихлорметане (365 мл), и добавили диметилформамид (405 мкл, 5,2 ммоль). Смесь охладили до 2°C, после чего по каплям добавили оксалил хлорид (70,8 г, 547 ммоль). Системе позволили нагреться и 90 минут перемешивали при комнатной температуре. После этого ее добавили к суспензии метансульфоната (15,45)-3-оксо-2-окса-5-азония-бицикло[2.2.1]гептана (110 г, 526 ммоль) в дихлорметане (400 мл). Полученную суспензию охладили до 2°C, и к ней медленно добавили триэтиламин (256 мл, 1,84 моль) (экзотермический процесс). После 70-минутного перемешивания при комнатной температуре в систему добавили раствор лимонной кислоты (81,0 г, 421 ммоль) в воде (550 мл) при 2°C. После разделения системы на фазы водную фазу проэкстрагировали дихлорметаном (300 мл). Объединенные органические экстракты промыли водой (400 мл) и сконцентрировали в вакууме до объема, приблизительно, 500 мл. Добавили этилацетат (330 мл), и остаточный дихлорметан удалили перегонкой в вакууме (внутренняя температура 40°C). Затем добавили еще этилацетата (50 мл), внутренняя температура возросла до 50°C, после чего медленно добавили гептан (800 мл). Кристаллизация началась после добавления около 300 мл гептана. Суспензию перемешивали 12 часов при комнатной температуре, затем профильтровали. Кристаллы промыли холодным гептаном (400 мл) и высушили в вакууме при 40°C, получив указанное в заголовке соединение в виде бесцветных кристаллов (88,45 г, 86%). Т.пл. 101-102°C. MS (ESI & APCI): m/z=196,1 [М+Н]+. 1H ЯМР (CDCl3, 600 МГц); δ 0,60-0,67 (m, 2H), 0,87-0,91 (m, 1H), 1,13-1,17 (m, 1H), 1,39 (s, 3H), 2,04 (dd, J=1,2 Гц, 10,9 Гц, 1H), 2,32 (d, J=10,8 Гц, 1H), 3,59 и 3,69 (АВ, JAB=11,5 Гц, каждый 1H), 4,97 (s, 1H), 5,18 (s, 1H).
d) (1-Циано-циклопропил)-амид (2S,4S)-4-гидрокси-1-(1-трифторметил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты
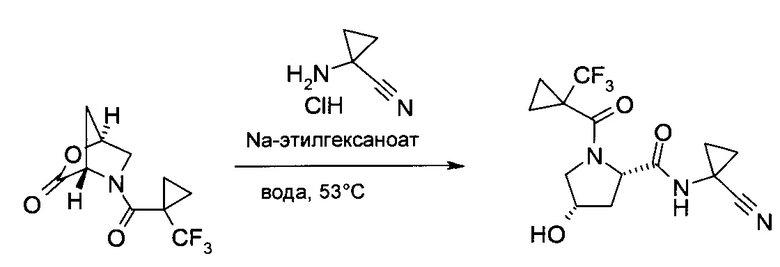
В воде (1,32 л) растворили (1S,4S)-5-(1-трифторметил-циклопропанкарбонил)-2-окса-5-аза-бицикло[2.2.1]гептан-3-он (220 г, 883 ммоль), 1-амино-циклопропанкарбонитрил гидрохлорид (140 г, 1,18 моль) и 2-этилгексаноат натрия (97%, 230 г, 1,34 моль). Смесь 20 часов перемешивали при 53°C. После охлаждения до комнатной температуры в систему добавили тетрагидрофуран (880 мл), после чего закислили концентрированной соляной кислотой (37 масс. %, 47 мл) и добавили хлорид натрия (440 г). После экстракции этилацетатом (1×1,4 L, 3×550 мл) объединенные органические экстракты высушили над сульфатом натрия и сконцентрировали в вакууме. По достижении объема, приблизительно, 1,5 л и при добавлении кристаллов затравки продукт начал кристаллизоваться. Объем суспензии дополнительно снизили до, приблизительно, 500 мл и охладили до 2°C (ледяная баня). После перемешивания 60 минут кристаллы отфильтровали, промыли смесью этилацетат/гептан 1:1 (об./об., 600 мл) и гептаном (300 мл) и высушили в вакууме с получением заявленного соединения в виде грязно-белых кристаллов (255,0 г, 87%), 1H ЯМР (CDCl3, 400МГц): δ 1,18-1,29 (m, 4H), 1,30-1,42 (m, 2H), 1,50-1,59 (m, 2H), 2,17-2,26 (m, 1H), 2,29 (d, J=14,5 Гц, 1Н), 3,73 и 3,96 (АВХ, JAB=11,8 Гц, JAX=4,3 Гц, JBX=0 Гц, все 1H), 4,43-4,53 (m, 2H), 4,81 (br d, J=8,3 Гц, 1H), 7,73 (s, 1H).
A2. Получение (1-циано-циклопропил)-амида (2S,4S)-4-гидрокси-1-(1-трифторметил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты
а) Метиловый эфир (2S,4R)-4-метансульфонилокси-1-(1-трифторметил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты
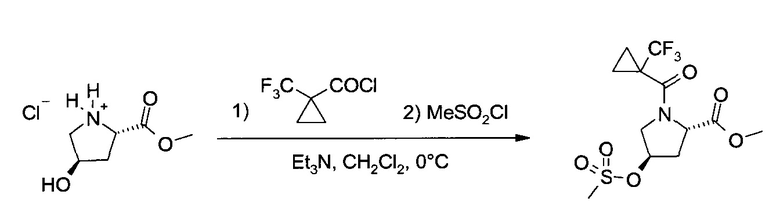
К суспензии 1-трифторметилциклопропан-1-карбоновой кислоты (40,4 г, 263 ммоль) и ДМФ (0,4 мл, 5,25 ммоль) в дихлорметане (50 мл) при перемешивании добавили оксалил хлорид (36,7 г, 289 ммоль), добавление и перемешивание проводили 2 часа при комнатной температуре (наблюдается бурное выделение газа!). После дополнительного перемешивания в течение еще получаса (выделение газа прекратилось) чистый раствор хлорангидрида перенесли в 100 мл капельную воронку и из нее в течение получаса при энергичном перемешивании добавили при 0°C к суспензии гидрохлорида гидроксипролин метилового эфира (45,4 г, 250 ммоль) и триэтиламина (101 г, 1000 ммоль) в дихлорметане (950 мл). После дополнительного перемешивания еще 1 час при 0°C в течение получаса добавили метансульфонил хлорид (31,5 г, 275 ммоль), перемешивание при 0°C продолжали еще полчаса, и затем гидролизовали холодную суспензию 1 M HCl (500 мл, pH=1). Нагрев систему до комнатной температуры, органический слой промыли 5% соляным раствором (500 мл), и проэкстрагировали дихлорметаном (250 мл) два водных слоя. Объединенные органические слои высушили (Na2SO4) и упарили (35-45°C/≥10 мбар), получив бежевый кристаллический неочищенный продукт (91,4 г), который растворили в изопропилацетате (360 мл) при ~70°C. Кристаллизация, начавшаяся при охлаждении и внесении затравки, была завершена при перемешивании при комнатной температуре с последующим добавлением по каплям гептана (540 мл) и перемешиванием 4 часа при -20°C. Фильтрация и промывка холодной системой изопропилацетат: гептан 2:3 позволила после сушки (10 мбар/55°C/4 ч) получить продукт (83,2 г, 92.6%) в виде грязно-белого кристаллического порошка, т.пл. 123-124°C. 
b) (1-Циано-циклопропил)-амид (2S,4S)-4-гидрокси-1-(1-трифторметил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты
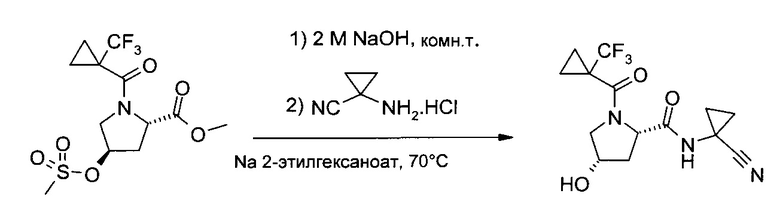
К метиловому эфиру (2S,4R)-4-метансульфонилокси-1-(1-трифторметил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты (71,9 г, 200 ммоль) при перемешивании и охлаждении на льду разом добавили 2,0 M NaOH (120,0 мл, 240,0 ммоль). После снятия с ледяной бани белую суспензию один час перемешивали при комнатной температуре. Нейтрализацию проводили добавлением 2,0 M HCl (20,0 мл, 40,0 ммоль; pH ~7), а затем разом добавили 1-аминоциклопропанкарбонитрил гидрохлорид (23,7 г 200 ммоль) и 2-этилгексаноат натрия (37,7 г, 220 ммоль), затем двухфазную реакционную смесь 22 часа перемешивали при 70°C и охладили до ~35°C. В систему добавили дихлорметан (100 мл) и NaCl (16 г) и продолжили перемешивание до растворения NaCl (около 15 минут). Подкислив раствор 25% HCl (~12 мл, pH ~1), смесь проэкстрагировали дихлорметаном (3×200 мл), и все три органических слоя по отдельности промыли 5% NaHCO3 (40 мл pH ~8). Объединенные органические слои высушили (Na2SO4), профильтровали и упарили (35-50°C/≥5 мбар), получив бежевый кристаллический осадок (88,5 г), который растворили в изобутилацетате (500 мл) при ~110°C. Кристаллизация, начавшаяся при охлаждении и внесении затравки, была завершена при перемешивании при комнатной температуре в течение часа и при -20°C в течение 4 часов. Фильтрация и промывка холодным изобутилацетатом с последующей сушкой (10 мбар/55°C/4 ч) позволила получить заявленные продукт (47,7 г, 72,0%) в виде грязно-белого кристаллического порошка, т.пл. 156-157°C. 
A3. Получение (1-циано-циклопропил)-амида (2S,4S)-4-гидрокси-1-(1-метил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты
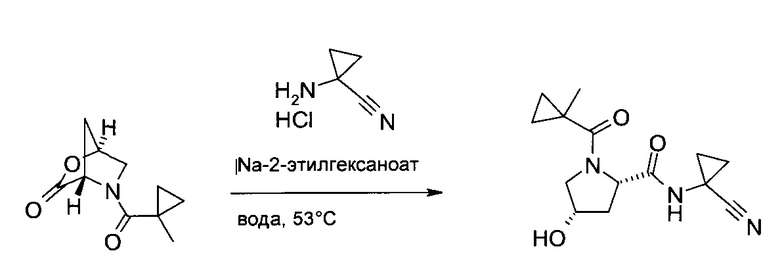
(1S,4S)-5-(1-Метил-циклопропанкарбонил)-2-окса-5-аза-бицикло[2.2.1]гептан-3-он (100 г, 512 ммоль), 1-амино-циклопропанкарбонитрил гидрохлорид (62,0 г, 523 ммоль) и 2-этилгексаноат натрия (97%, 96,5 г, 563 ммоль) растворили в воде (500 мл). Смесь 16 часов перемешивали при 50°C. После охлаждения до комнатной температуры добавили дихлорметан (500 мл), смесь подкислили соляной кислотой (25 масс. %, 13,8 мл, 106 ммоль), и добавили хлорид натрия (120 г). Смесь перемешивали 1 час, фазы разделили, водный слой проэкстрагировали дихлорметаном (3×330 мл), и объединенные органические экстракты сконцентрировали в вакууме до объема, приблизительно, 1,1 л. В систему добавили этилацетат (1,1 л), смесь сконцентрировали в вакууме до объема, приблизительно, 1 л, после чего в течение 30 минут в систему добавили 1 л гептана. Полученную суспензию 2 часа перемешивали при 2°C, профильтровали, осадок промыли холодной системой этилацетат/гептан 1:1 (об./об., 340 мл) и гептаном (340 мл), после чего высушили в вакууме, получив указанное в заголовке соединение в виде желтых кристаллов (129,4 г, 90%). MS (ESI & APCI): m/z=278,1 [M+H]+. 1H ЯМР (CDCl3, 600 МГц): δ 0,60-0,65 (m, 2H), 0,89-0,92 (m, 1H), 0,95-0,98 (m, 1H), 1,20-1,26 (m, 2H), 1,33 (s, 3H), 1,50-1,57 (m, 2H), 2,13 (ddd, J=5,0 Гц, 9,0 Гц, 14,1 Гц, 1H), 2,39 (br d, J=14,3 Гц, 1H), 3,76 (dd, J=4,3 Гц, 11,6 Гц, 1H), 3,85 (d, J=11,6 Гц, 1H), 4,48-4,53 (m, 1H), 4,57 (d, J=9,0 Гц, 1H), 4,70 (br s, 1H), 7,92 (br s, 1H).
А4. Получение (1-циано-циклопропил)-амида (2S,4S)-4-гидрокси-1-(1-метил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты
а) 1-Метил-циклопропанкарбонил хлорид
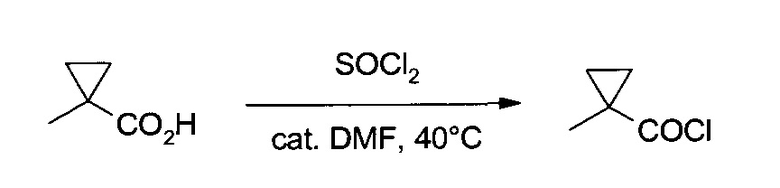
К 1-метил-циклопропанкарбоновой кислоте (100,1 г, 1000 ммоль) и ДМФ (0,37 г, 5,0 ммоль) осторожно при перемешивании и при температуре ~40°C добавили тионилхлорид (125,0 г, 1050 ммоль), добавление проводили в течение 1 часа. Систему еще один час перемешивали при 40°C, после чего образовавшийся неочищенный продукт (122,3 г) очистили перегонкой на колонке Вигро, получив указанное в заголовке соединение (114,8 г, 96,8%) в виде желто-коричневой жидкости, т.пл. 129-130°C/~1000 мбар. 1H ЯМР (CDCl3, 400 МГц) δ 0,98 (m, 2Н), 1,40 (s, 3H), 1,59 (m, 2H).
b) Метиловый эфир (2S,4R)-4-метансульфонилокси-1-(1-метил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты
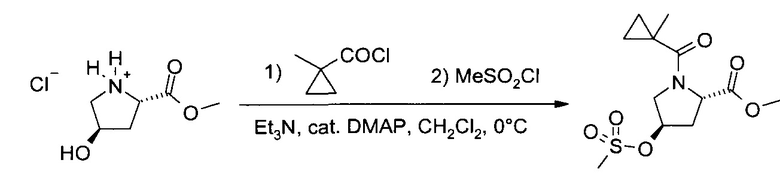
К суспензии гидрохлорида гидроксипролинметилового эфира (36,3 г, 200 мол), триэтиламина (65,8 г, 650 ммоль) и диметиламинопиридина (1,25 г, 10 ммоль) в дихлорметане (800 мл) при 0°C добавили 1-метил-циклопропанкарбонил хлорид (24,9 г, 210 ммоль), процесс проводили полчаса. После дополнительного перемешивания еще в течение 2 часов, в систему в течение получаса добавили метансульфонил хлорид (28,64 г, 250 ммоль; примечание 7), после чего перемешивали еще один час при 0°C. Холодную реакционную смесь перенесли в делительную воронку и промыли 1М HCl (400 мл) и 10% концентрированным соляным раствором (400 мл). Водные слои экстрагировали дихлорметаном (400 мл), после чего комбинированные органические слои высушили над Na2SO4. После фильтрования и упаривания растворителя (35-45°C/≥10 мбар) удалось получить неочищенный кристаллический продукт (63,8 г), который растворили в изобутилацетате (250 мл) при ~70°C. После внесения затравки при ~50°C кристаллизацию завершили, охладив смесь до комнатной температуры и перемешивая всю ночь при -20°C. Фильтрование и промывка холодным изобутилацетатом дали после высушивания (50°C/10 мбар/3 ч) указанное в заголовке соединение (57,5 г, 94,2%) в виде белого кристаллического порошка, т.пл. 102-103°C. 
с) (1-Циано-циклопропил)-амид (2S,4S)-4-гидрокси-1-(1-метил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты
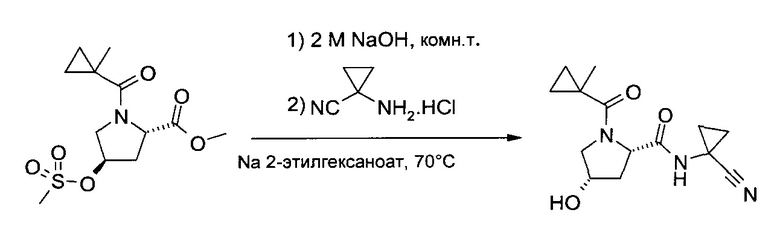
К 2 M NaOH (60,0 мл, 120 ммоль) при 0°C разом добавили метиловый эфир (2S,4R)-4-метансульфонилокси-1-(1-метил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты (30,54 г, 100 ммоль). Ледяную баню убрали, и белую суспензию в течение часа нагрели до комнатной температуре. В систему добавили 2 M HCl (10,0 мл, 20 ммоль), после чего разом ввели 1-аминоциклопропанкарбонитрил гидрохлорид (11,86 г, 100 ммоль) и 2-этилгексаноат натрия (18,28 г, 110 ммоль); затем двухфазную реакционную смесь в течение 21 часа перемешивали при 70°C. Охладив ее до ~35°C, добавили дихлорметан (50 мл) и NaCl (8,0 г), перемешивание продолжали до растворения NaCl. Смесь закислили 25% HCl (8 мл) и перенесли в делительную воронку, где экстрагировали дихлорметаном (4×150 мл). Все четыре органических слоя по отдельности промыли 5% NaHCO3 (20 мл). Комбинированные органические слои высушили (Na2SO4), профильтровали и упарили до сухого состояния (40°C/≥5 мбар), получив бежевый кристаллический осадок (38,5 г), который растворили в дихлорметане (200 мл). Замену растворителя на этилацетат провели на роторном испарителе постепенным добавлением этилацетата (350 мл) при 60-80°C/950 мбар и в то же самое время отгонкой дихлорметана. Кристаллическую суспензию охладили до комнатной температуры и в течение ночи перемешивали при -20°C. Фильтрование и промывка холодным этилацетатом позволила после сушки (50°C/10 мбар/4 ч) получить указанное в заголовке соединение (20,8 г, 75,0%) в виде белого кристаллического порошка, т.пл. 159,5-160,5°C. 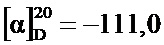
В. Получение тио-соединения:
В1. Получение 2-хлор-4-((S)-2,2,2-трифтор-1-метил-этокси)-бензотиола
а) 2-Хлор-4-фтор-1-тритилсульфанил-бензол
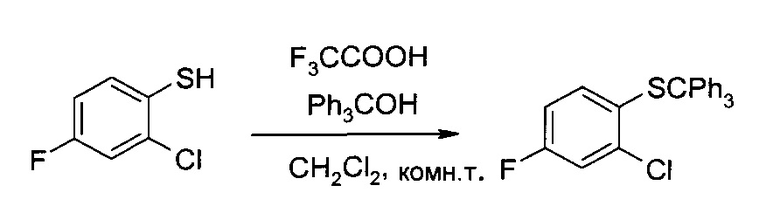
2-Хлор-4-фтор-бензотиол (250 г, 1,54 моль) растворили в дихлорметане (1,25 л), после чего туда при комнатной температуре добавили трифторуксусную кислоту (120 мл, 1,57 моль) и затем трифенилметанол (400 г, 1,54 моль) (при добавлении трифенилметанола необходимо проводить охлаждение на ледяной бане). Смесь полтора часа перемешивали при комнатной температуре, сконцентрировали и высушили в вакууме, получив указанное в заголовке соединение в виде желтого твердого вещества (622 г, 99% чистоты по методу ВЭЖХ, 99%), которое на следующем этапе использовалось без дополнительной очистки. 1H ЯМР (CDCl3, 400 МГц): δ 6,55 (ddd, J=2,9 Гц, 8,0 Гц, 8,9 Гц, 1H), 6,91 (dd, J=6,2 Гц, 8,9 Гц, 1H), 6,97 (dd, J=2,9 Гц, 8,6 Гц, 1H), 7,20-7,27 (m, 9H), 7,34-7,39 (m, 6H).
b) 2-Хлор-4-((S)-2,2,2-трифтор-1-метил-этокси)-1-трибутилсульфанил-бензол
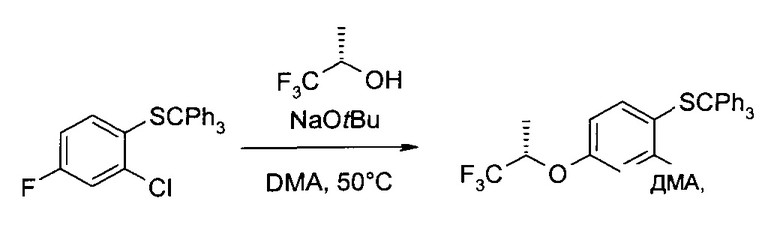
(5)-1,1,1-Трифтор-пропан-2-ол (100 г, 877 ммоль) растворили в диметилацетамиде (600 мл), и охладили раствор до 2°C (ледяная баня). При этой температуре добавили раствор трет-бутоксида натрия (77,5 г, 790 ммоль) в диметилацетамиде (100 мл), и смесь перемешивали 15 минут. Затем в систему добавили раствор 2-хлор-4-фтор-1-тритилсульфанил-бензола (200 г, 494 ммоль) в диметилацетамиде (100 мл) при комнатной температуре, и перемешивали ее 3 ч при 50°C. После этого содержимое реакционного сосуда вылили в смесь соляного раствора (200 мл), льда (1,2 кг) и воды (2,0 л), и проэкстрагировали трет-бутил-метиловым эфиром (2×1000 мл). Объединенные органические экстракты промыли соляным раствором/водой 1:1 (об./об., 300 мл) и сконцентрировали в вакууме с получением неочищенного продукта в виде желтого вязкого масла. Масло затем растворили в этаноле (1,6 л), после чего при комнатной температуре медленно добавили воду (240 мл). Полученную суспензию перемешивали 14 часов, охладили до 2°C (ледяная баня) и перемешивали еще 2 часа, затем профильтровали. Твердый осадок промыли этанолом/водой 4:1 (об./об., 500 мл) и высушили в вакууме, получив указанное в заголовке соединение в виде белых кристаллов (238 г, 97%). 1H ЯМР (CDCl3, 400 МГц): δ 1,43 (d, J=6,5 Гц, 3H), 4,50 (qq, J=6,2 Гц, 6,2 Гц, 1Н), 6,44 (dd, J=2,8 Гц, 8,7 Гц, 1H), 6,84 (d,J=2,7 Гц, 1Н), 6,90 (d, J=8,6 Гц, 1Н), 7,18-7,27 (m, 9H), 7,33-7,39 (m, 6H).
с) 2-Хлор-4-((S)-2,2,2-трифтор-1-метил-этокси)-бензотиол
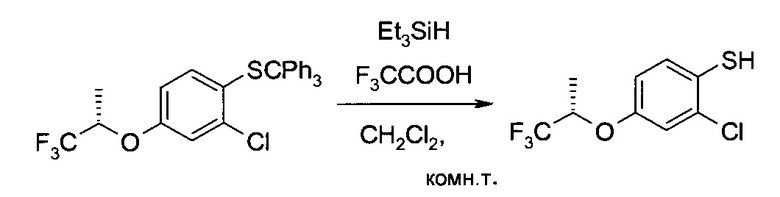
2-Хлор-4-((S)-2,2,2-трифтор-1-метил-этокси)-1-тритилсульфанил-бензол (220 г, 441 ммоль) растворили в дихлорметане (800 мл), после чего при комнатной температуре добавили трифторуксусную кислоту (165 мл, 2,15 моль) и затем триэтилсилан (110 мл, 674 ммоль, необходимо охлаждение на ледяной бане). После получасового перемешивания при комнатной температуре смесь сконцентрировали в вакууме. К остатку добавили раствор гидроксида калия (2М в воде, 1,1 л), и перемешивали суспензию 15 минут. Профильтровав смесь, твердый осадок промыли водой. Объединенный фильтрат закислили до pH<2 добавлением соляной кислоты (25% в воде, 330 мл) и проэкстрагировали трет-бутилметиловым эфиром (3×500 мл). Объединенные органические экстракты промыли водным раствором гидрокарбоната калия (1М, 330 мл), высушили над сульфатом натрия и сконцентрировали в вакууме. Мутное масло обработали гептаном (80 мл) и профильтровали. Оставшийся твердый осадок еще раз промыли гептаном (20 мл). Объединенный фильтрат сконцентрировали и высушили в вакууме с получением заявленного соединения в виде бесцветной жидкости (105 г, 93%). 1H ЯМР (CDCl3, 400 МГц): δ 1,49 (dd, J=6,5 Гц, 0,5 Гц, 3H), 3,75 (s, 1H), 4,55 (qq, J=6,2 Гц, 6,2 Гц, 1H), 6,79 (dd, J=3,0 Гц, 8,9 Гц, 1H), 7,03 (d, J=2,7 Гц, 1H), 7,29 (d, J=8,6 Гц, 1H).
В2. Получение 4-(2,2,2-трифтор-этокси)-2-трифторметил-бензотиола
а) 4-Фтор-2-трифторметил-1-тритилсульфанил-бензол
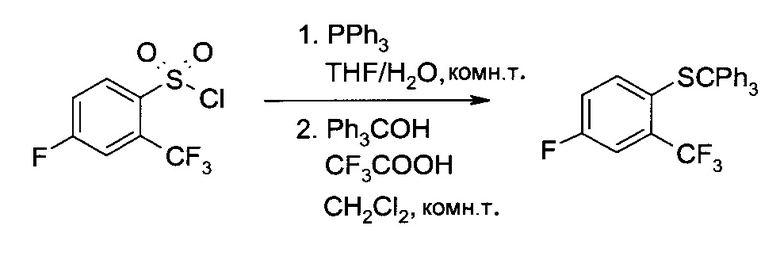
Трифенилфосфин (126,2 г, 481 ммоль) растворили в тетрагидрофуране (155 мл), после чего туда при 22°C в течение получаса добавили раствор 4-фтор-2-трифторметил-бензосульфонил хлорида (40,0 г, 152 ммоль) в тетрагидрофуране (80 мл). Желтую суспензию 15 минут перемешивали при комнатной температуре, после чего добавили воды (48 мл), и полученный чистый раствор перемешивали еще 20 минут. В систему добавили концентрированный раствор гидроксида натрия (32 масс. % в воде, 34 мл, 367 ммоль), затем добавили еще воды (260 мл), тетрагидрофуран полностью отогнали в вакууме, и полученную водную суспензию профильтровали. Отфильтрованные осадки (Трифенилфосфин и трифенилфосфиноксид) тщательно промыли водой (360 мл), объединенные фильтраты закислили добавлением водного раствора соляной кислоты (25 масс. % в воде, 40,0 мл, 307 ммоль), проэкстрагировали дихлорметаном (1×200 мл, 1×60 мл), и полученные объединенные органические экстракты (около 260 мл) использовали на следующем этапе без дополнительной обработки.
Трифенилметанол (39,7 г, 149,4 ммоль) растворили в растворе неочищенного 4-фтор-2-трифторметил-бензотиола в дихлорметане (около 260 мл), полученном на предыдущем этапе, после чего туда при комнатной температуре добавили раствор трифторуксусной кислоты (21,2 г, 186 ммоль) в дихлорметане (20 мл). Перемешав в течение 15 минут, смесь подщелочили, последовательно добавив воду (20 мл), концентрированный водный раствор гидроксида натрия (32 масс. %, 26,9 г, 215 ммоль) и снова воду (240 мл). Фазы разделили, водный слой проэкстрагировали дихлорметаном (100 мл), и объединенные органические экстракты сконцентрировали в вакууме при 50°C, приблизительно, до объема 330 мл. В систему при дистилляции непрерывно добавляли этанол (400 мл), пока общий объем не перестал меняться (замена растворителя). Раствору (около 300 мл) позволили остыть до комнатной температуры, кристаллизация началась при 42°C. Суспензию 18 часов перемешивали при комнатной температуре и 1 час при 0°C. После фильтрования осадок промыли холодным этанолом (100 мл) и высушили в вакууме при 40°C, получив указанное в заголовке соединение в виде белых кристаллов (52,5 г, 79%). MS (El): m/z=243 [CPh3]+. 1H ЯМР (CDCl3, 600 МГц): δ 6,73 (ddd, J=2,8 Гц, 8,3 Гц, 8,3 Гц, 1H), 7,04 (dd, J=5,4 Гц, 8,8 Гц, 1H), 7,18-7,25 (m, 10H), 7,35-7,37 (m, 6H). Аналитический расчет для C26H18F4S: С, 71,22; Н, 4,14; S, 7,31; F, 17,33. Обнаружено: С, 71,16; Н, 4,26; S, 7,15; F, 17,09.
b) 4-(2,2,2-Трифтор-этокси)-2-трифторметил-1-тритилсульфанил-бензол
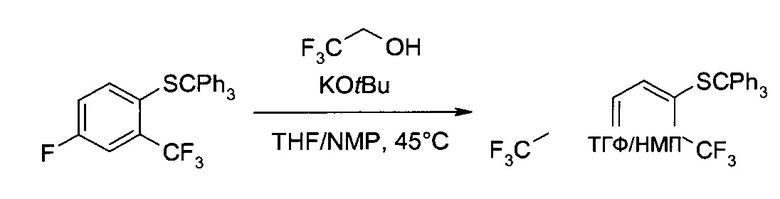
Калия трет-бутоксид (98 масс. %, 39,17 г, 342,1 ммоль) суспендировали в тетрагидрофуране (190 мл), после чего при комнатной температуре добавили раствор трифторэтанола (35,65 г, 356,4 ммоль) в тетрагидрофуране (28 мл) (экзотермическая реакция). Смесь перемешивали 15 минут, после чего добавили раствор 4-фтор-2-трифторметил-1-тритилсульфанил-бензола (125 г, 285 ммоль) в 1-метил-2-пирролидоне (240 мл) и тетрагидрофуран (290 мл). Полученный коричневый раствор полчаса перемешивали при комнатной температуре, а затем еще 2 часа при внутренней температуре 45°C. После этого в систему добавили воду (720 мл), концентрированный солевой раствор (125 мл) и трет-бутилметиловый эфир (720 мл), а затем разделили фазы. Органический слой промыли раствором хлорида натрия (59,3 г, 1015 ммоль) в воде (380 мл) и сконцентрировали в вакууме при температуре 40°C, приблизительно, до объема 400 мл. Раствор разбавили этанолом (300 мл), и в процессе дистилляции непрерывно добавляли еще этанола (480 мл), пока объем не перестал меняться, остановившись на уровне, приблизительно, 700 мл (замена растворителя). Полученную суспензию 14 часов перемешивали при комнатной температуре и 1 час при 0°C, а затем добавили воду (140 мл), и суспензию еще перемешивали 1,5 часа при 0°C. После фильтрования осадок промыли холодной смесью этанол/вода 5:1 (об./об., 288 мл) и высушили в вакууме с получением заявленного соединения в виде мелких желтых кристаллов (140,9 г, 95%). MS (El): m/z=243 [Cph3]+. 1Н ЯМР (CDCl3, 600 МГц): δ 4,26 (q, J=8,0 Гц, 2Н), 6,61 (dd, J=2,9 Гц, 8,8 Гц, 1Н), 7,04 (d, J=8,8 Гц, 1Н), 7,07 (d, J=2,9 Гц, 1Н), 7,18-7,25 (m, 9H), 7,33-7,37 (m, 6H).
с) 4-(2,2,2-Трифтор-этокси)-2-трифторметил-бензотиол
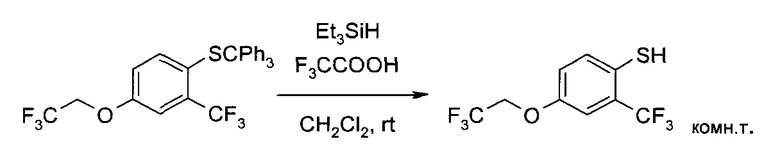
4-(2,2,2-Трифтор-этокси)-2-трифторметил-1-тритилсульфанил-бензол (100 г, 193 ммоль) растворили в дихлорметане (600 мл), и при комнатной температуре добавили трифторуксусную кислоту (44,9 г, 386 ммоль), а затем раствор триэтилсилана (24,9 г, 214 ммоль) в дихлорметане (75 мл) при 18°C (охлаждение на ледяной бане). Желтую смесь перемешивали 3 часа при комнатной температуре. Затем добавили воду (600 мл) и дихлорметан отогнали в вакууме при интенсивном перемешивании. Добавили трет-бутилметиловый эфир (600 мл), и полученную двухфазную смесь защелочили до pH 12 добавлением водного раствора гидроксида натрия (32 масс. %, 54 мл, 583 ммоль). Фазы разделили, водный слой проэкстрагировали трет-бутилметиловым эфиром (400 мл), закислили соляной кислотой (25 масс. % в воде, 35 мл, 268 ммоль) до pH 3 и экстрагировали трет-бутилметиловым эфиром (600 мл). Органический экстракт промыли раствором гидрокарбоната натрия (16,1 г, 193 ммоль) в воде (500 мл) и водой (500 мл) и сконцентрировали в вакууме с получением заявленного соединения в виде светло-желтой жидкости (49,7 г, 90%). MS (ESI & APCI, neg): m/z=275,0 [M-H]+. 1H ЯМР (CDCl3, 600 МГц): δ 3,66 (q, J=2,5 Гц, 1H), 4,36 (q, J=8,0 Гц, 2Н), 6,99 (dd, J=2,9 Гц, 8,6 Гц, 1H), 7,23 (d, J=2,8 Гц, 1H), 7,39 (d, J=8,6 Гц, 1Н).
B3. Получение 4-(2,2,2-трифтор-этокси)-2-трифторметил-бензотиола
а1) 1-Бром-4-(2,2.2-трифтор-этокси)-2-трифторметил-бензол
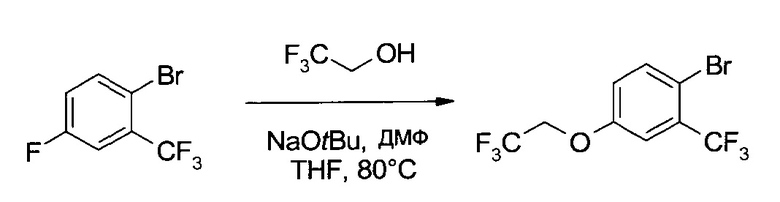
К 2,4 М раствору трет-бутоксида натрия в ТГФ (156,3 мл, 375 ммоль, Chemetall) добавили ДМФ (38,5 мл, 500 ммоль) и затем 2,2,2-трифторэтанол (41,27 г, 413 ммоль). После добавления еще 2-бром-5-фторбензотрифторида (60,8 г, 250 ммоль) реакционную смесь нагрели до кипения (с обратным холодильником) и 7 часов перемешивали при ~80°C. Охладив смесь до 25°C, добавили ТБМЭ (800 мл), после чего смесь промыли 1М раствором HCl (400 мл), 5% NaHCO3 (400 мл) и 10% соляным раствором (400 мл). Органический слой высушили (Na2SO4), профильтровали и упарили до сухого состояния (60°C/≥5 мбар), получив неочищенное указанное в заголовке соединение (80,0 г, 99,1%) в виде желтого масла, использовавшегося на следующем этапе без очистки. 1H ЯМР (CDCl3, 400 МГц) δ 4,38 (q, J=7,9 Гц, 2Н), 6,98 (dd, J1=8,6 Гц, J2=2,7 Гц, 1H), 7,28 (d, J=2,7 Гц, 1H), 7,65 (d, J=8,9 Гц, 1H).
b1) 4-(2,2,2-Трифтор-этокси)-2-трифторметил-бензотиол
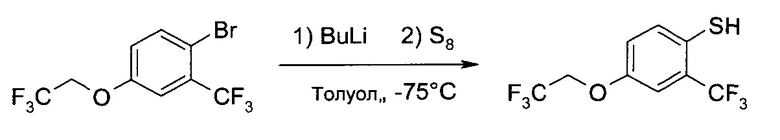
К раствору 1-бром-4-(2,2,2-трифтор-этокси)-2-трифторметил-бензола (80,8 г, 250 ммоль) в толуоле (1000 мл) добавили диэтиловый эфир (52 мл, 500 ммоль). Охладив систему до -75°C, при этой температуре в течение 30 минут при перемешивании добавили 2,5 М раствор бутиллития в толуоле (105 мл, 263 ммоль) и перемешивали при этой же температуре еще 30 минут. После этого при -75°C добавили порошок серы (8,8 г, 275 ммоль), и перемешивание продолжали 7 часов. Холодную желтую суспензию вылили при перемешивании в смесь толуола (1000 мл) и 0,5М NaOH (1000 мл). После интенсивного перемешивания 5 минут два слоя разделили, и водный слой проэкстрагировали толуолом (500 мл). Затем водный слой охладили до ~10°C, закислили 5М HCl (~150 мл) и проэкстрагировали дихлорметаном (1000 мл). Дихлорметановый слой промыли 10% соляным раствором (1000 мл), высушили над Na2SO4, профильтровали и упарили до сухого состояния (60°C/≥5 мбар), получив неочищенный желтый маслянистый продукт (58,2 г). Очистка перегонкой позволила получить указанное в заголовке соединение (55,1 г, 79,8%) в виде желто-коричневого масла, т.кип. 84-86°C/2.3 мбар. 1H ЯМР (CDCl3, 400 МГц) δ 3,66 (q, J=2,4 Гц, 1Н), 4,36 (q, J=8,1 Гц, 2Н), 6,98 (dd, J1=8,6 Гц, J2=2,7 Гц, 1H), 7,23 (d, J=2,7 Гц, 1H), 7,39 (d, J=8,6 Гц, 1Н).
b2) 4-(2,2,2-Трифтор-этокси)-2-трифторметил-бензотиол
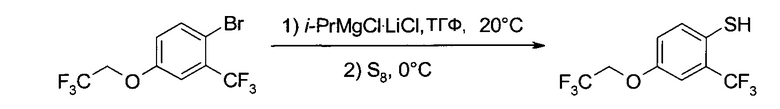
К раствору 1-бром-4-(2,2,2-трифтор-этокси)-2-трифторметил-бензола (16,2 г, 50 ммоль) в ТГФ (200 мл) при перемешивании и температуре 20°C добавили 1,3 М раствор изопропилмагний хлорида/литий хлорида (1:1) в ТГФ (46,2 мл≅45,2 г, 60 ммоль Turbo-Grignard (Турбо-Гриньяр), производство "Чеметалл" (Chemetall)); добавление проводили в течение 30 минут. После двухчасового перемешивания при 20°C чистый желтый раствор охладили до 0°C и туда разом добавили порошок серы (1,84 г, 57,5 ммоль). Перемешивание при 0°C продолжали 2 часа, после чего при интенсивном перемешивании смесь реакционную смесь гидролизовали 0,5 М HCl (200 мл) и дважды проэкстрагировали ТМБЭ (200 мл и 100 мл). Два органических слоя промыли 0,5 М NaOH (200 мл), слой NaOH закислили 6 М HCl (20 мл) при охлаждении на льду и проэкстрагировали ТБМЭ (200 мл). Слой ТБМЭ промыли 10% соляным раствором (100 мл), высушили (Na2SO4), профильтровали и упарили (≤60°C/≥5 мбар), получив неочищенное указанное в заголовке соединение (11,4 г) в виде желтого масла, которое очистили перегонкой по методу Kugelrohr (11,1 г, 80,4%), т.кип.≈90°C/2 мбар. 1H ЯМР (CDCl3, 400 МГц) δ 3,66 (q, J=2,4 Гц, 1Н), 4,36 (q, J=8,1 Гц, 2Н), 6,98 (dd, J1=8,6 Гц, J2=2,7 Гц, 1H), 7,23 (d, J=2,7 Гц, 1H), 7,39 (d, J=8,6 Гц, 1H).
В4. Получение 2-хлор-4-((S)-2,2,2-трифтор-1-метил-этокси)-бензотиола
а) 1-Бром-2-хлор-4-((S)-2,2,2-трифтор-1-метил-этокси)-бензол
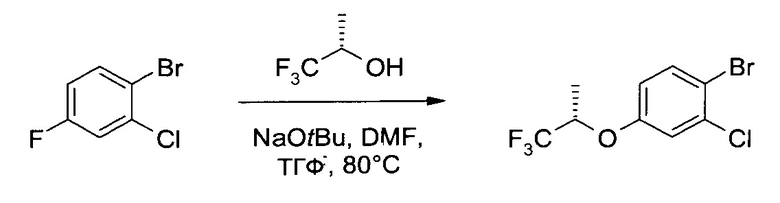
К 2,4 М раствору трет-бутоксида натрия в ТГФ (156,3 мл, 375 ммоль) при перемешивании добавили (S)-1,1,1-трифтор-пропан-2-ол (47,1 г, 413 ммоль), ДМФ (77,0 мл, 1000 ммоль) и 1-бром-2-хлор-4-фтор-бензол (52,4 г, 250 ммоль). Реакционную смесь нагрели до кипения (с обратным холодильником) и 19 часов перемешивали при ~80°. После охлаждения до комнатной температуры добавили ТБМЭ (1000 мл), и смесь промыли 1М HCl (500 мл), 5% NaHCO3 (500 мл) и 10% соляным раствором (400 мл). Водные слои проэкстрагировали ТВМЕ (400 мл), а органические слои высушили (Na2SO4), профильтровали и упарили до сухого состояния (≤60°C/≥5 мбар), получив 75,8 г неочищенного заявленного соединения. Дистилляция на колонке Вигре позволила получить бесцветное масло (71,5 г, 94,2%), т.кип.~70°C/0,1 мбар. 1Н ЯМР (CDCl3, 400 МГц) δ 1,50 (dd, J1=6,4 Гц, J2=0,5 Гц, 3H), 4,59 (hept, J=6,2 Гц, 1Н), 6,76 (dd, J1=8,9 Гц, J2=3,0 Гц, 1H), 7,09 (d, J=2,7 Гц, 1H), 7,52 (d, J=8,9 Гц, 1H).
b) 2-Хлор-4-((S)-2,2,2-трифтор-1-метил-этокси)-бензотиол
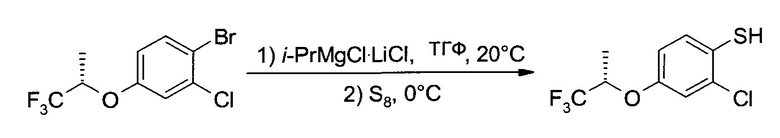
К раствору 1-бром-2-хлор-4-((S)-2,2,2-трифтор-1-метил-этокси)-бензола (15,2 г, 50 ммоль) в ТГФ (200 мл) при перемешивании и 20°C добавили 1,3 М раствор изопропилмагний хлорида/литий хлорида (1:1) в ТГФ (50,0 мл≅49,0 г, 65 ммоль Turbo-Grignard (Турбо-Гриньяр), "Чеметалл"); добавление проводили в течение 30 минут. После двухчасового дополнительного перемешивания при 20°C желтый раствор охладили до -5°C и туда разом добавили серу (1,92 г, 60 ммоль). После перемешивания при 0°C в течение 2 часов ледяную баню убрали, и гидролизовали реакционную смесь 1М HCl (125 мл). Смесь дважды проэкстрагировали ТМБЭ (200 мл и 100 мл), и промыли органические слои 1 М NaOH (125 мл). Слой NaOH выделили, охладили до ~10°C и закислили 6 М HCl (25 мл). После экстракции ТМБЭ (200 мл) и промывки 10% соляным раствором (100 мл), органический слой высушили (Na2SO4), профильтровали и упарили (≤50°C/≥5 мбар), получив (10,1 г, 78,7%) неочищенного заявленного соединения в виде желтого масла. 1H ЯМР (CDCl3, 400 МГц) δ 1,49 (dd, J1=6,5 Гц, J2=0,5 Гц, 3H), 3,75 (s, 1H), 4,55 (hept, J=6,2 Гц, 1Н), 6,79 (dxd, J1=8,9 Гц, J2=3,0 Гц, 1Н), 7,03 (d, J=2,7 Гц, 1H), 7,29 (d, J=8,6 Гц, 1H).
В5. Получение 3-[4-(2-метил-пиридин-4-ил)-2-трифторметил-фенилсульфанил]-пропионамида
а) 4-Бром-2-трифторметил-бензотиол
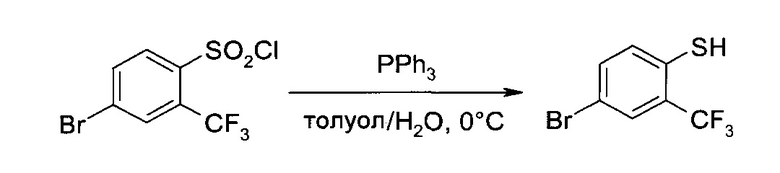
4-Бром-2-трифторметил-бензосульфонил хлорид (375 г, 1,16 моль) растворили в толуоле (1,5 л), и добавили туда раствор трифенилфосфина (994 г, 3,79 моль) в толуоле (1,5 л) при температуре 5-10°C, добавление проводили 45 минут. Желтую суспензию перемешивали 30 минут при 0-5°C, после чего добавили воду (360 мл) при температуре 5-12°C (сильно экзотермическая реакция), и полученную бесцветную суспензию 20 минут перемешивали при комнатной температуре. После фильтрации осадок промыли толуолом (1 л). Объединенные органические слои проэкстрагировали раствором гидроксида калия (1М в воде, 2,8 л). В процессе экстракции образовались три слоя. Верхний слой выбросили, остальные два промыли толуолом (1 л). Водную фазу закислили до pH 3-4 добавлением лимонной кислоты (280 г, 1,46 моль). После введения в систему н-гептана (1 л) осадок отфильтровали и промыли н-гептаном (500 мл). Объединенные слои фильтрата разделили, и водную фазу проэкстрагировали н-гептаном (1,5 л). Объединенные органические экстракты высушили над сульфатом натрия. После этого в систему добавили силикагель (250 г), полученную кашицу перемешивали 10 минут при комнатной температуре, профильтровали и отфильтрованный силикагель промыли н-гептаном (1 л). Объединенный фильтрат сконцентрировали и высушили в вакууме при 45°C, получив 291,2 г (98%) заявленного соединения в виде бесцветной жидкости, которую без дополнительной очистки использовали на следующем этапе. MS (El): m/z=256,9, 254,9 [M+H]+. 1Н ЯМР (CDCl3, 400 МГц): δ 3,76 (q, J=2,8 Гц, 1Н), 7,26 (d, J=8,3 Гц, 1Н), 7,48 (dd, J=2,0 Гц, 8,5 Гц, 1Н), 7,75 (d, J=1,9 Гц, 1Н).
b) 3-(4-Бром-2-трифторметил-фенилсульфанил)-пропионамид
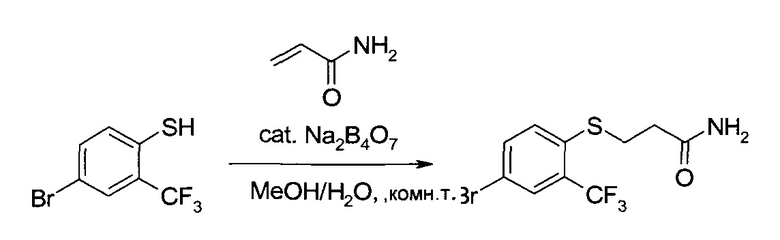
4-Бром-2-трифторметил-бензотиол (259,2 г, 1,01 моль) растворили в метаноле (1,3 л) и воде (2,6 л), после чего при комнатной температуре добавили акриламид (130 г, 1,82 моль) и затем тетраборат натрия (25,9 г, 129 ммоль). Суспензию перемешивали 40 часов. После фильтрования твердый осадок промыли водой (2,6 л) и н-гептаном (2,6 л), а затем высушили в вакууме, получив указанное в заголовке соединение в виде белого порошка (325,6 г, 98%). MS (El): m/z=330,0, 328,0 [M+Н]+. 1H ЯМР (d6-ДМСО, 400 МГц): δ 2,40 (t, J=7,2 Гц, 2Н), 3,24 (t, J=7,2 Гц, 2Н), 6,92 (bs, 1H), 7,37 (bs, 1H), 7,60 (d, J=8,3 Гц, 1Н), 7,82-7,88 (m, 2Н).
c1) 3-[4-(2-Метил-пиридин-4-ил)-2-трифторметил-фенилсульфанил]-пропионамид
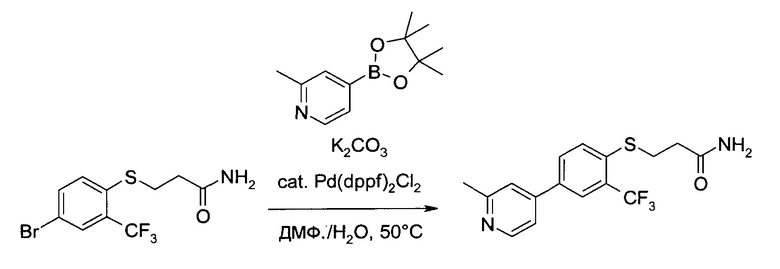
3-(4-Бром-2-трифторметил-фенилсульфанил)-пропионамид (300 г, 914 ммоль) растворили в N,N-диметилформамиде (3,0 л) в присутствии карбоната калия (300 г, 2,17 моль). После этого в систему добавили пинаколовый эфир 2-метилпиридин-4-борной кислоты (285 г, 1,3 моль) и воду (240 мл). Раствор дегазировали, и в него добавили [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (30 г, 41 ммоль). Смесь перемешивали 20 часов при 50°C. Охладив ее до комнатной температуры, смесь вылили в ледяную воду (5°C, 5 л) и проэкстрагировали этилацетатом (1×3,5 л, 2х1,5 л). Объединенные органические экстракты промыли соляным раствором/водой (1:1 об./об., 750 мл) и соляным раствором (750 мл), а затем добавили метанол (600 мл), и высушили смесь над сульфатом натрия. В систему добавили 400 г силикагеля, кашицу отфильтровали и прокомн.т. этилацетатом/метанолом (9:1 об./об., 1,5 л). Объединенные фильтраты сконцентрировали в вакууме. Остаток ресуспендировали в толуоле (600 мл) и н-гептане (300 мл), после чего 5 минут перемешивали при 60°C и 1 час при комнатной температуре. Осадок отфильтровали, промыли системой толуол/н-гептан (4:1 об./об., 300 мл) и н-гептаном (300 мл), а затем высушили в вакууме, получил 230,3 г коричневого кристаллического вещества. Кристаллы дополнительно очистили растиранием в системе изопропанол/гептан (1:1 об./об., 600 мл) 30 минут при комнатной температуре и 30 минут при температуре 0-4°C (ледяная баня). Осадок отфильтровали, промыли системой н-гептан/изопропанол (4:1 об./об., 300 мл) и высушили в вакууме при 65°C, получив указанное в заголовке соединение в виде светло-коричневого кристаллического твердого вещества (215,7 г, 69%). MS (El): m/z=341,1 [M+Н]+. 1Н ЯМР (d6-ДМСО, 400 МГц): δ 2,48 (t, J=7,2 Гц, 2Н), 2,54 (s, 3H), 3,32 (t, J=7,2 Гц, 2Н), 6,95 (bs, 1H), 7,41 (bs, 1H), 7,58 (dd, J=1,6 Гц, 5,4 Гц, 1H), 7,68 (s, 1H), 7,77 (d, J=9,1 Гц, 1H), 8,02-8,08 (m, 2Н), 8,52 (d, J=5,1 Гц, 1H).
c2) 3-[4-(2-Метил-пиридин-4-ил)-2-трифторметил-фенилсульфанил]-пропионамид (один реакционный сосуд с бромпиколином)

4-Бром-2-метилпиридин (6,0 г, 34,9 ммоль) растворили в диметилформамиде (60 мл), и при комнатной температуре добавили в систему ацетат калия (10,0 г, 102 ммоль) и 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (10 г, 39,4 ммоль). Раствор дегазировали, и в него добавили [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (400 мг, 547 мкмоль). Коричневую реакционную смесь перемешивали 22 часа при 80°C. После охлаждения до комнатной температуры добавили еще 3-(4-бром-2-трифторметил-фенилсульфанил)-пропионамид (8,0 г, 24,4 ммоль), калия карбонат (8,0 г, 57,9 ммоль), диметилформамид (20 мл), и воду (16 мл). Смесь дегазировали, перемешивали 30 минут при комнатной температуре, после чего добавили еще одну порцию [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (400 мг, 547 мкмоль). Полученную коричневую реакционную смесь перемешивали 20 часов при 60°C. После охлаждения до комнатной температуры ее вылили в воду (150 мл) и проэкстрагировали этилацетатом (1×80 мл, 2×40 мл). Объединенные органические экстракты промыли системой соляной раствор/вода (1:1 об./об., 20 мл) и соляным раствором (20 мл), добавили метанол (16 мл), высушили смесь над сульфатом натрия. В смесь добавили силикагель (16 г), кашицу профильтровали и промыли этилацетатом/метанолом (9:1 об./об., 40 мл). Объединенные фильтраты сконцентрировали в вакууме. Остаток обработали системой толуол/н-гептан (1:1 об./об., 24 мл) и перемешивали полученную суспензию 30 минут при комнатной температуре. После фильтрации остаток промыли толуолом/н-гептаном (4:1 об./об., 10 мл) и н-гептаном (10 мл) и высушили в вакууме, получив указанное в заголовке соединение в виде коричневых кристаллов (5,7 г, 71%). MS (El): m/z=341,1 [M+Н]+. 1H ЯМР (d6-ДМСО, 400 МГц) δ 2,48 (t, J=7,2 Гц, 2Н), 2,54 (s, 3H), 3,32 (t, J=7,2 Гц, 2Н), 6,95 (bs, 1Н), 7,41 (bs, 1H), 7,58 (dd, J=1,6 Гц, 5,4 Гц, 1Н), 7,68 (s, 1H), 7,77 (d, J=9,1 Гц, 1H), 8,02-8,08 (m, 2Н), 8,52 (d,J=5,1 Гц, 1H).
В6. Получение 3-[4-(1-метил-1Н-пиразол-4-ил)-2-трифторметил-фенилсульфанил]-пропионамида
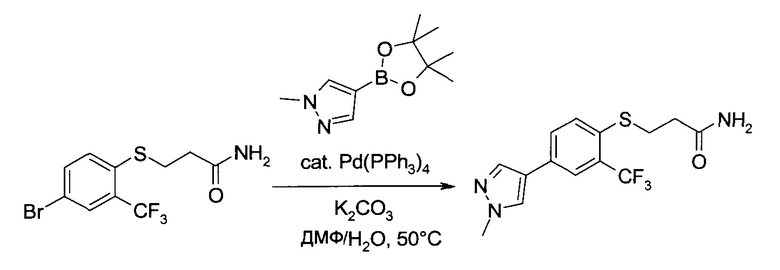
3-(4-Бром-2-трифторметил-фенилсульфанил)-пропионамид (160 г, 488 ммоль) в атмосфере Ar растворили в N,N-диметилформамиде (1,5 л), после чего в систему добавили 1-метил-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-пиразоле (143 г, 687 ммоль), карбонат калия (160 г, 1,16 моль) и воду (130 мл). Раствор дегазировали и добавили тетракис(трифенил-фосфин)палладий(0) (24,0 г, 20,8 ммоль). Смесь 16 часов перемешивали при 50°C. Охладив до комнатной температуры, ее вылили в воду (5 мл) и проэкстрагировали этилацетатом (1×3 л, 2×1 л). Объединенные органические экстракты промыли соляным раствором (1 л), объем в вакууме уменьшили, приблизительно, до 2 л, и высушили смесь над сульфатом натрия. Полученный фильтрат еще раз упарили в вакууме, продолжая процесс, пока не начал образовываться осадок и не образовалась гомогенная кашица. После этого в систему порциями в процессе ее перегонки начали добавлять -трет-бутилметиловый эфир (1 л), поддерживая общий объем постоянным (замена растворителя). Суспензию охладили до 5°C (ледяная баня) и профильтровали. Осадок промыли трет-бутилметиловым эфиром (500 мл) и высушили в вакууме, получив 141 г указанного в заголовке соединения в виде коричневых кристаллов (88%). MS (ESI & APCI): m/z=330,1 [M+H]+. 1H ЯМР (CDCl3, 600 МГц): δ 2,54 (t, J=7,4 Гц, 2H), 3,27 (t, J=7,4 Гц, 2H), 5,39 (bs, 1H), 5,52 (bs, 1H), 7,56 (s, 1H), 7,57 (s, 1H), 7,66 (s, 1H), 7,73 (bs, 1H), 7,77 (d, J=0,7 Гц, 1H).
В7. Получение гидрохлорида 4-(2-метил-пиридин-4-ил)-2-трифторметил-бензотиола
а) 4-(4-Фтор-3-трифторметил-фенил)-2-метил-пиридин

К суспензии 4-фтор-3-(трифторметил)фенилборной кислоты (42,6 г, 205 ммоль) в толуоле (200 мл) при перемешивании добавили 4-бром-2-метилпиридин (34,4 г, 200 ммоль) и 2 М водный раствор карбоната калия (200 мл). После введения в систему комплекса 1,1'-бис(дифенилфосфино)-ферроцен-палладий(II)дихлорида и дихлорметана (81,7 мг, 0.1 ммоль), полученную двухфазную желтоватую реакционную смесь перемешивали при кипячении с обратным холодильником (88°C) в течение 23 часов. Образовавшуюся коричневатую реакционную смесь охладили до комнатной температуры и проэкстрагировали толуолом (200 мл). Промыв слой толуола 10% соляным раствором (200 мл), его высушили над Na2SO4 (50 г) и при перемешивании 30 минут обрабатывали древесным углем (2 г). Фильтрование и упаривание (50°C/≥10 мбар) позволили получить неочищенное указанное в заголовке соединение (50,7 г, 99,4%) в виде грязно-белого кристаллического остатка, который использовался на следующем этапе без очистки. 1H ЯМР (CDCl3, 400 МГц) δ 2,64 (s, 3H), 7,25-7,36 (m, 3H), 7,76-7,82 (m, 1H), 7,84 (dd, J1=6,7 Гц, J2=2,4 Гц, 1H), 8,58 (d, J=5,0 Гц, 1H). ESI-MS (m/z) [M+H]+ 256,3 (100).
b) 4-(4-трет-Бутилсульфанил-3-трифторметил-фенил)-2-метил-пиридин
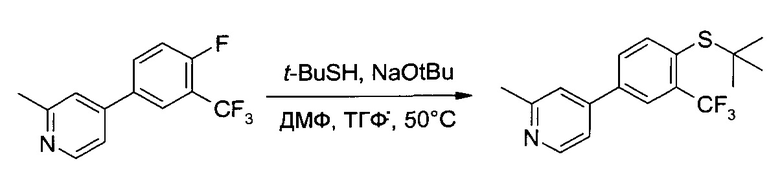
К раствору 4-(4-фтор-3-трифторметил-фенил)-2-метил-пиридина (51,0 г, ~200 ммоль) в ТГФ (100 мл) осторожно при комнатной температуре добавили 2-метил-2-пропантиол (23,5 г, 260 ммоль) и ДМФ (29,2 г, 400 ммоль). В течение 50 минут в систему вводили 25% раствор трет-бутоксид натрия в ТГФ (96,1 г=106 мл, 250 ммоль, производство Chemetall), полученную бежевую суспензию 17 часов перемешивали при 50°C. Коричневатую суспензию перенесли на делительную воронку, наполнили ТБМЭ (500 мл) и промыли водой (500 мл) и 10% соляным раствором (500 мл). Два водных слоя проэкстрагировали ТБМЭ (300 мл) и высушили комбинированные органические слои (Na2SO4). Фильтрование и упаривание (45°C/≥10 мбар) позволили получить неочищенное указанное в заголовке соединение (65,5 г, 100,6%) в виде коричневого масла, которое использовалось на следующем этапе без очистки. 1H ЯМР (CDCl3, 400 МГц) δ 1,39 (s, 9H), 2,65 (s, 3H), 7,33 (d, J=5,1 Гц, 1H), 7,39 (s, 1H), 7,73 (dd, J1=8,1 Гц, J2=1,9 Гц, 1H), 7,83 (d, J=8,1 Гц, 1H), 7,96 (d, J=1,9, 1H), 8,60 (d, J=5,4, 1H). ESI-MS (m/z) [М+НГ 326 (100).
с) 4-(2-Метил-пиридин-4-ил)-2-трифторметил-бензотиола гидрохлорид
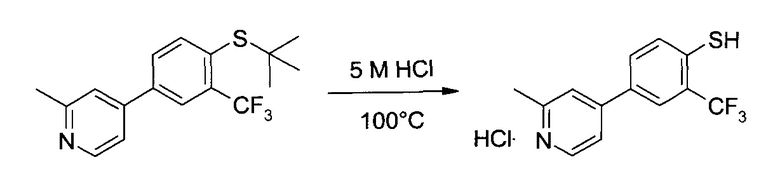
4-(4-трет-бутилсульфанил-3-трифторметил-фенил)-2-метил-пиридин (65,1 г, ~200 ммоль) растворили в 5 М HCl (800 мл), полученный желтоватый раствор перемешивали в условиях кипячения с обратным холодильником при 100°C в течение 22 часов. После охлаждения до комнатной температуры в течение, приблизительно, одного часа, и перемешивании при этой температуре еще полчаса, бежевую суспензию профильтровали, и промыли отфильтрованный осадок деионизированной водой (400 мл) и ацетоном (200 мл), а затем высушили (50°C/≥10 мбар/24 ч) с получением 57,4 г (93,9%) указанного в заголовке соединения в виде грязно-белого кристаллического порошка, т.пл.>270°C (разложение). 1H ЯМР (CDCl3+2 капли ТФК, 400 МГц) δ 2,96 (s, 3H), 4,06 (q, J=3,3 Гц, 1H), 7,63 (d, J=8,3 Гц, 1H), 7,74 (dd, J,=8,3 Гц, J2=2,1 Гц, 1H), 7,84 (d, J=1,9 Гц, 1H), 7,93 (dd, J1=6,3 Гц, J2=1,9 Гц, 1Н), 7,97 (d, J=1,9, 1H), 8,79 (d, J=6,2 Гц, 1H), 9,96 (s, 14H, TFA), 15,18 (brs, 1H). ESI-MS (m/z) [M-HCl]- 270 (100).
B8. Получение 4-(1-метил-1H-пиразол-4-ил)-2-трифторметил-бензотиола
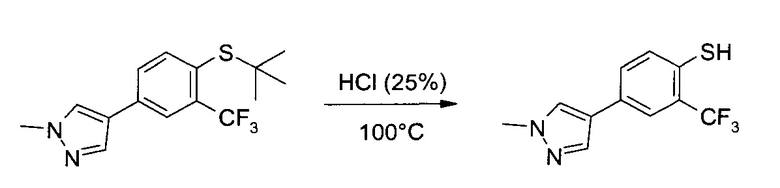
4-(4-трет-Бутилсульфанил-3-трифторметил-фенил)-1-метил-1Н-пиразол (40,0 г, 127 ммоль) суспендировали в соляной кислоте (25 масс. %, 780 мл) и перемешали при кипячении с обратным холодильником при 100°C. Часть соляной кислоты отогнали (около 80 мл). После того, как, по данным ВЭЖХ, реакция завершилась, смесь охладили до 85°C, добавили воду (240 мл), и дополнительно охладили смесь до 20°C. pH раствора довели до 4,0 добавлением гидроксида натрия (32% масс. %, около 480 мл) в воде (960 мл). Водную фазу проэкстрагировали 2-метилтетрагидрофураном (2×320 мл), объединенные органические экстракты промыли раствором хлорида натрия (40,0 г) в воде (400 мл) и сконцентрировали в вакууме. Остаток заново растворили в тетрагидрофуране (260 мл), сконцентрировали в вакууме, еще раз растворили в тетрагидрофуране (110 мл) и профильтровали, чтобы удалить неорганические соли. Осадок промыли тетрагидрофураном (30 мл) и сконцентрировали в вакууме объединенный фильтрат, получив указанное в заголовке соединение в виде светло-коричневого твердого вещества (32,4 г, 98,6%), которое без дополнительной очистки использовалось на следующем этапе.
С. Получение продукта
С1. Получение (1-циано-циклопропил)-амида (2S,4R)-4-[2-хлор-4-((8)-2,2,2-трифтор-1-метил-этокси)-бензосульфонил]-1-(1-трифторметил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты
а) (3S,5S)-5-(1-Циано-циклопропилкарбамоил)-1-(1-трифторметил-циклопропанкарбонил)-пирролидин-3-иловый эфир бензолсульфоновой кислоты
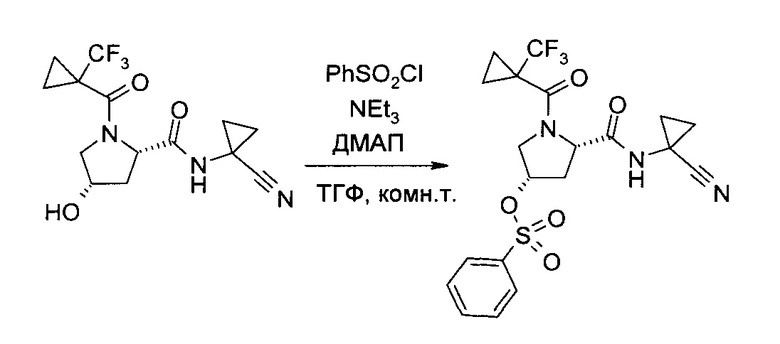
(1-Циано-циклопропил)-амид (2S,4S)-4-гидрокси-1-(1-трифторметил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты (100,0 г, 301,8 ммоль) растворили в тетрагидрофуране (500 мл). Смесь охладили до 2°C (ледяная баня), после чего последовательно добавили бензосульфонил хлорид (99%, 48 мл, 370,5 ммоль), 4-(диметиламино)пиридин (98%, 2,0 г, 16,0 ммоль) и триэтиламин (75,0 мл, 539 ммоль), и смесь перемешивали 15 минут. Реакционной смеси позволили нагреться до комнатной температуры и перемешивали 20 часов. После охлаждения до 2°C (ледяная баня), добавили воду (150 мл) и метанол (350 мл). Тетрагидрофуран осторожно отогнали в вакууме (около 500 мл), после чего медленно добавили воду (500 мл). После введения 300 мл воды добавлением в систему затравки индуцировали кристаллизацию. Полученную суспензию перемешивали 30 минут при 2°C (ледяная баня), а затем профильтровали. Твердый осадок промыли системой метанол/вода 1:2 (об./об., 300 мл) и гептан (300 мл), и высушили в вакууме, получив указанное в заголовке соединение в виде грязно-белых кристаллов (140,8 г, 99%). 1H ЯМР (CDCl3, 400 МГц): δ 1,06-1,27 (m, 4Н), 1,28-1,41 (m, 2Н), 1,44-1,54 (m, 2H), 2,26 (ddd, J=5,9 Гц, 9,4 Гц, 14,2 Гц, 1Н), 2,59 (ddd, J=3,8 Гц, 3,8 Гц, 14,2 Гц, 1Н), 3,90 и 4,03 (ABX, JAB=12,5 Гц, JAX=4,0 Гц, JBX=5,2 Гц, каждый 1Н), 4,57 (br d, J=5,1 Гц, 1Н), 5,02-5,09 (m, 1H), 7,08 (br s, 1H), 7,61 (t, J=7,8 Гц, 2Н), 7,71 (t, J=7,5 Гц, 1H), 7,95 (d, J=7,2 Гц, 2H).
b) (1-Циано-циклопропил)-амид (2S,4R)-4-[2-хлор-4-((8)-2,2,2-трифтор-1-метил-этокси)-бензосульфонил]-1-(1-трифторметил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты
Этап 1: (1-Циано-циклопропил)-амид (2S,4R)-4-[2-хлор-4-((8)-2,2,2-трифтор-1-метил-этокси)-фенилсульфанил]-1-(1-трифторметил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты

Эфир бензолсульфоновой кислоты и (33,55)-5-(1-циано-циклопропилкарбамоил)-1-(1-трифторметил-циклопропанкарбонил)-пирролидин-3-ила (225 г, 477 ммоль) растворили в диметилацетамиде (1,125 л) и добавили карбонат калия (166,5 г, 1,193 моль). При комнатной температуре медленно добавили раствор 2-хлор-4-((S)-2,2,2-трифтор-1-метил-этокси)-бензотиола (142 г, 553,2 ммоль) в тетрагидрофуране (135 мл), поддерживая внутреннюю температуру ниже 29°C (необходимо охлаждение на ледяной бане). Смесь перемешивали при комнатной температуре 5,5 часов. После добавления льда (700 г) и воды (2 л) смесь проэкстрагировали трет-бутилметиловым эфиром (1×1,5 л, 3×750 мл). Объединенные органические экстракты промыли соляным раствором (450 мл) и сконцентрировали в вакууме, получив указанное в заголовке соединение в виде коричневого вязкого масла (300,4 г). Неочищенный продукт (содержащий диметилацетамид) без дополнительной очистки использовался на следующем этапе.
Этап 2: (1-Циано-циклопропил)-амид (2S,4R)-4-[2-хлор-4-((S)-2,2,2-трифтор-1-метил-этокси)-бензосульфонил]-1-(1-трифторметил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты
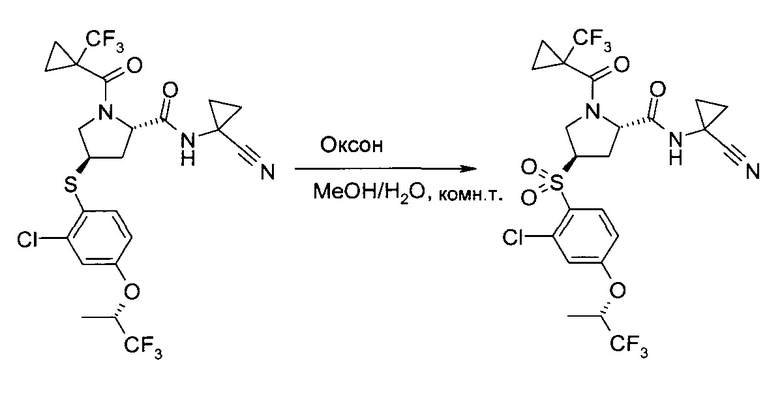
В метаноле (1,225 л) и воде (385 мл) суспендировали оксон (1,5 кг, 2,44 моль), после чего при температуре реакции 10-18°C (необходимо охлаждение на ледяной бане) добавили раствор (1-циано-циклопропил)-амида (2S,4R)-4-[2-хлор-4-((3)-2,2,2-трифтор-1-метил-этокси)-фенилсульфанил]-1-(1-трифторметил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты (неочищенный продукт, содержащий диметилацетамид, 90,5% чистота по весу, 300,4 г, 477,2 ммоль) в метаноле (800 мл). Смесь перемешивали при комнатной температуре 20 часов. Суспензию профильтровали, и оставшийся твердый осадок промыли метанолом (900 мл). В фильтрат добавили воду (900 мл), и метанол отогнали в вакууме. Полученный раствор проэкстрагировали трет-бутилметиловым эфиром (2×900 мл). Объединенные органические экстракты промыли раствором метабисульфита натрия (40,0 г, 206 ммоль) в воде (450 мл), раствором гидрокарбоната калия (1М в воде, 450 мл) и соляным раствором (450 мл). После сушки над сульфатом натрия добавили 300 г силикагеля. Полученную суспензию профильтровали, и оставшийся силикагель промыли трет-бутилметиловым эфиром (900 мл). Объединенные фильтраты сконцентрировали в вакууме и подвергли азеотропной перегонке с метанолом (2×500 мл). Неочищенный продукт (белая пена, 270 г), растворили в метаноле (450 мл) и добавили к 4 л воды при энергичном перемешивании. Суспензию 18 часов перемешивали при комнатной температуре, профильтровали, и промыли раствор водой (900 мл) и гептаном (900 мл). После сушки в вакууме указанное в заголовке соединение получили в виде аморфного белого твердого вещества (259 г, 97,6% чистота по методу ВЭЖХ, 88% за обе ступени вместе). MS (ESI & APCI): m/z=602,1 [М+Н]+, 619,1 [М+NH4]+. 1Н ЯМР (CDCl3, 400 МГц): δ 1,11-1,21 (m, 2H), 1,30-1,43 (m, 1H), 1,35-1,40 (m, 1H), 1,42-1,47 (m, 1H), 1,49-1,56 (m, 3H), 1,57 (d, J=7,1 Гц, 3H), 2,16-2,23 (m, 1H), 2,86 (ddd, J=5,6 Гц, 8,3 Гц, 14,2 Гц, 1H), 3,85 (dd, J=7,5 Гц, 13,6 Гц, 1H), 4,34-4,39 (m, 1H), 4,72 (brd, J=13,3 Гц, 1Н), 4,76-4,84 (m, 2H), 7,02 (dd, J=2,5 Гц, 9,0 Гц, 1H), 7,18 (d, J=2,5 Гц, 1H), 7,60 (s, 1H), 8,00 (d,J=8,9 Гц, 1H).
C2. Получение (1-циано-циклопропил)-амида (2S,4R)-1-(1-метил-циклопропанкарбонил)-4-[4-(2,2,2-трифтор-этокси)-2-трифторметил-бензосульфонил]-пирролидин-2-карбоновой кислоты
а1) Эфир бензолсульфоновой кислота и (3S,5S)-5-(1-циано-циклопропилкарбамоил)-1-(1-метил-циклопропанкарбонил)-пирролидин-3-ила
(1-Циано-циклопропил)-амид (2S,4S)-4-гидрокси-1-(1-метил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты (62,0 г, 224 ммоль) и 4-(диметиламино)пиридин (98%, 1,5 г, 12,0 ммоль) растворили в дихлорметане (370 мл), после чего при комнатной температуре в систему добавили бензосульфонил хлорид (31,1 мл, 242 ммоль) и затем триэтиламин (49,8 мл, 358 ммоль), и смесь перемешивали 16 часов. После этого ее разбавили водой (140 мл) и закислили добавлением соляной кислоты (25 масс. %, 21,4 мл). Фазы разделили и промыли органический слой водой (160 мл). Объединенные водные фазы проэкстрагировали дихлорметаном (160 мл), объединенные органические экстракты сконцентрировали в вакууме до объема 430 мл. Растворитель заменили непрерывной перегонкой в вакууме (внутренняя температура 40°C, 670-170 мбар) на этанол (добавлено 670 мл), при этом общий объем сохранялся постоянным (430 мл). Полученному раствору позволили остыть до комнатной температуры и начала кристаллизацию введением затравки (около 10 мг кристаллов). После начала кристаллизации суспензию при комнатной температуре перемешивали 30 минут, в течение еще 50 минут вводили гептан (430 мл), и после этого продолжали перемешивание еще 12 часов. Затем суспензию профильтровали, осадок промыли системой гептан/этанол 2:1 (об./об., 321 мл) и гептаном (107 мл) и высушили в вакууме при 40°C, получив указанное в заголовке соединение в виде белого кристаллического твердого вещества (80,0 г, 85%). MS (ESI & APCI): m/z=418,1 [M+H]+. +H ЯМР (CDCl3, 600 МГц): δ 0,59-0,64 (m, 1H), 0,65-0,69 (m, 1H), 0,80-0,90 (m, 1H), 0,99-1,05 (m, 1H), 1,12-1,21 (m, 2H), 1,30 (s, 3H), 1,47-1,53 (m, 2H), 2,15 (ddd, J=5,8 Гц, 9,2 Гц, 14,6 Гц, 1H), 2,70 (br d, J=14,0 Гц, 1H), 3,83 (dd, J=3,8 Гц, 12,3 Гц, 1H), 4,08 (dd, J=5,3 Гц, 12,4 Гц, 1H), 4,55 (br s, 1H), 5,04-5,08 (m, 1H), 7,49 (br s, 1H), 7,61 (t, J=7,8 Гц, 2Н), 7,70 (t, J=7,5 Гц, 1H), 7,96 (d, J=7,6 Гц, 2H).
b1) (1-Циано-циклопропил)-амид (2S,4R)-1-(1-метил-циклопропанкарбонил)-4-[4-(2,2,2-трифтор-этокси)-2-трифторметил-фенилсульфанил]-пирролидин-2-карбоновой кислоты
Раствор 4-(2,2,2-трифтор-этокси)-2-трифторметил-бензотиола (56,1 г, 196 ммоль) в тетрагидрофуране (93 мл) при комнатной температуре добавили к суспензии трет-бутоксида калия (22,8 г, 203 ммоль) в тетрагидрофуране (370 мл). Оранжево-коричневый прозрачный раствор 20 минут перемешивали, после чего добавили раствор эфира бензолсульфоновой кислоты и (3S,5S)-5-(1-циано-циклопропилкарбамоил)-1-(1-метил-циклопропанкарбонил)-пирролидин-3-ила (74,4 г, 178 ммоль) в диметилацетамиде (240 мл). Смесь перемешивали 15 минут при комнатной температуре. В систему добавили воду (550 мл) и этилацетат (500 мл), и разделили фазы. Водный слой проэкстрагировали этилацетатом (500 мл), объединенные органические экстракты промыли раствором хлорида натрия (124 г) в воде (1,1 л) (2×600 мл) и сконцентрировали в вакууме до объема около 260 мл. После этого добавили толуол (600 мл), смесь еще раз сконцентрировали в вакууме до объема, приблизительно, 260 мл, и добавили еще одну порцию толуола (240 мл). Теплую смесь (внутренняя температура 45°C) профильтровали, осадок промыли толуолом (120 мл) и позволили объединенному фильтрату медленно остыть (в течение 60 минут) до комнатной температуры. Кристаллизация началась при внутренней температуре 26°C. Суспензию один час перемешивали при 22°C, затем в течение 45 минут добавили гептан (620 мл), и суспензию перемешивали еще 17 часов при комнатной температуре, после чего профильтровали. Осадок промыли системой толуол/гептан 1:1 (об./об., 240 мл) и гептаном (120 мл) и высушили в вакууме при 40°C, получив указанное в заголовке соединение в виде грязно-белого кристаллического вещества (87,7 г, 96,1% чистота, 88%). MS (ESI & APCI): m/z=536,1 [M+Н]+. 1H ЯМР (CDCl3, 600 МГц): δ 0,58-0,62 (m, 1H), 0,64-0,68 (m, 1H), 0,89-0,93 (m, 1H), 1,04-1,09 (m, 1H), 1,16-1,22 (m, 2H), 1,31 (s, 3H), 1,45-1,55 (m, 2H), 1,88-1,94 (m, 1H), 2,79-2,85 (m, 1H), 3,83-3,88 (m, 1H), 3,91-3,98 (m, 2H), 4,41 (q, J=7,9 Гц, 2H), 4,61 (dd, J=4,7 Гц, 8,0 Гц, 1H), 7,12 (dd, J=2,9 Гц, 8,6 Гц, 1H), 7,31 (d, J=2,8 Гц, 1H), 7,61 (d, J=8,6 Гц, 1H), 8,04 (br s, 1H).
b2) (1-Циано-циклопропил)-амид (2S,4R)-1-(1-метил-циклопропанкарбонил)-4-[4-(2,2,2-трифтор-этокси)-2-трифторметил-фенилсульфанил]-пирролидин-2-карбоновой кислоты
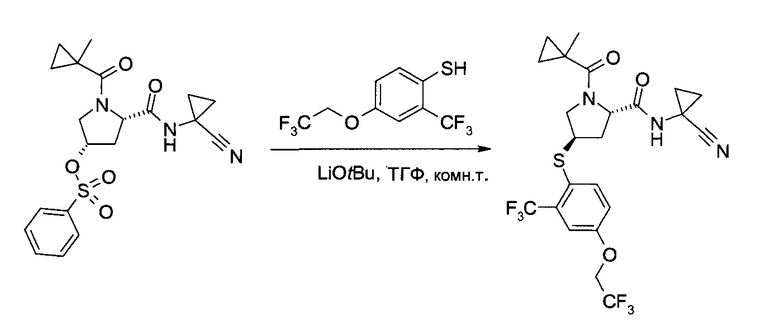
К суспензии 4-(2,2,2-трифтор-этокси)-2-трифторметил-бензотиола (30,4 г, 110 ммоль) и (3S,5S)-5-(1-циано-циклопропилкарбамоил)-1-(1-метил-цикло-пропанкарбонил)-пирролидин-3-ил бензолсульфоната (41,7 г, 100 ммоль) в ТГФ (50 мл) при перемешивании и комнатной температуре в течение 40 минут добавили 20% раствор трет-бутоксида лития в ТГФ (44,0 г≅49,5 мл, 110 ммоль). После перемешивания 18 часов при комнатной температуре реакционную смесь гидролизовали деионизированной водой (400 мл) и дважды проэкстрагировали этилацетатом (400 мл & 200 мл). Оба органических слоя промыли 10% соляным раствором (200 мл), объединили, высушили над Na2SO4 и профильтровали. Упаривание растворителя и осторожная сушка (45°C/≥10 мбар) позволили получить 56,3 г неочищенного продукта в виде объемной грязно-белой пены, которую растворили в 250 мл толуола при 50°C. Кристаллизация, начавшаяся в процессе охлаждения, завершилась после часового перемешивания смеси при комнатной температуре при одновременно прибавлении 200 мл гептана по капле (примечание 11) и последующего перемешивания в течение 20 часов при комнатной температуре. Фильтрование и сушка (50°C/10 мбар/3 дня для удаления толуола до концентрации ~1%) привели к получению 51,2 г (95,7%) указанного в заголовке соединения в виде грязно-белого кристаллического порошка, т.пл. 66-76°C. 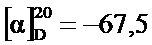
c1) (1-Циано-циклопропил)-амид (2S,4R)-1-(1-метил-циклопропанкарбонил)-4-[4-(2,2,2-трифтор-этокси)-2-трифторметил-бензосульфонил]-пирролидин-2-карбоновой кислоты
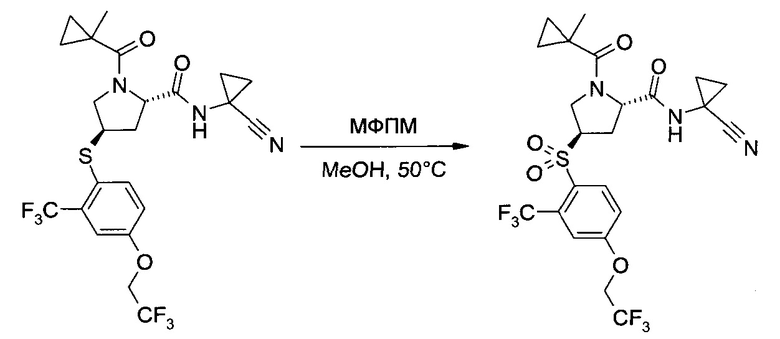
К раствору гексагидрата монопероксофталаата магния (11,8 г, 20 ммоль, 84% по данным анализа) в метаноле (100 мл) при перемешивании добавили раствор (1-циано-циклопропил)-амида (2S,4R)-4-[2-хлор-4-(2,2,2-трифтор-этокси)-фенилсульфанил]-1-(1-метил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты (10,7 г, 20 ммоль) в метаноле (30 мл), процесс проводили в течение 15 минут. После перемешивания при 50°C в течение 8 часов разом добавили еще гексагидрата монопероксофталаата магния (13,0 г, 22 ммоль) и перемешивали белую суспензию еще 16 часов. Реакционную смесь охладили до комнатной температуры и разрушили избыток монопероксофталаата магния добавлением по каплям 39% раствора бисульфита натрия (~9 мл). После удаления большей части метанола на роторном испарителе (45°C/≥80 мбар) к белой кашице добавили дихлорметан (100 мл), и осторожно нейтрализовали смесь, доведя pH до 7 путем добавления 1 М NaOH (~95 мл) при перемешивании. Органический слой промыли 5% NaHCO3 (50 мл) и оба водных слоя проэкстрагировали дихлорметаном (50 мл). Комбинированные органические слои высушили (Na2SO4), профильтровали и упарили (40°C/≥10 мбар), получив белую объемную пену (11,51 г), которую растворили в изопропаноле (50 мл) при ~60°C. При перемешивании в течение 30 минут добавили деионизированную воду (100 мл), и полученную белую суспензию 3 часа перемешивали при комнатной температуре. Смесь профильтровали, промыли системой изопропанол-вода 1:2 (~20 мл) и высушили (50°C/10 мбар/4 часа), получив указанное в заголовке соединение (10,6 г, 93,3%) в виде белого кристаллического порошка, т.пл. 125,5-129,5°C. 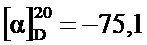
c2) (1-Циано-циклопропил)-амид (2S,4R)-1-(1-метил-циклопропанкарбонил)-4-[4-(2,2,2-трифтор-этокси)-2-трифторметил-бензосульфонил]-пирролидин-2-карбоновой кислоты
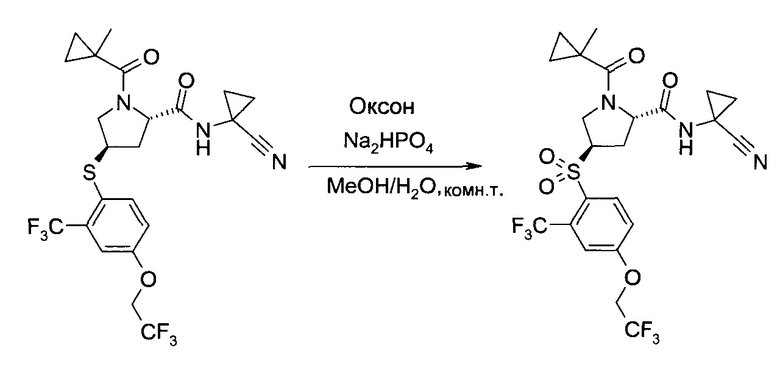
В метаноле (570 мл) и воде (810 мл) суспендировали оксон (264,6 г, 430 ммоль) и гидрофосфат натрия (457,2 г, 2,72 моль), после чего при комнатной температуре в течение 30 минут добавили раствор (1-циано-циклопропил)-амида (2S,4R)-1-(1-метил-циклопропанкарбонил)-4-[4-(2,2,2-трифтор-этокси)-2-трифторметил-фенилсульфанил]-пирролидин-2-карбоновой кислоты в метаноле (600 мл). Смесь перемешивали 90 часов при комнатной температуре. Суспензию нагрели до 40°C, профильтровали, и профильтрованную твердую фракцию промыли метанолом (1000 мл). Объединенный фильтрат сконцентрировали в вакууме до объема 1,1 л и добавили дихлорметан (900 мл). После разделения фаз водный слой проэкстрагировали дихлорметаном (450 мл), объединенные органические экстракты промыли раствором тиосульфата натрия (26,6 г, 168 ммоль) в воде (900 мл) и водой (900 мл), профильтровали и сконцентрировали в вакууме, приблизительно, до объема 220 мл. После этого в систему добавили изопропанол (900 мл) и раствор сконцентрировали в вакууме, приблизительно, до объема 500 мл. При 50°C добавили воду, и раствору позволили остыть до комнатной температуры (начало кристаллизации). Медленно добавили в систему еще воды (500 мл), и кристаллическую суспензию перемешивали 5 часов при комнатной температуре. После этого суспензию профильтровали, осадок промыли системой вода/изопропанол 2:1 (об./об., 270 мл) и водой (270 мл) и высушили в вакууме при 50°C, получив указанное в заголовке соединение в виде бесцветных кристаллов (87,0 г, 91%). т.пл. 126,0-127,0°C. MS (ESI & APCI): m/z=568,1 [М+Н]+. 1H ЯМР (CDCl3, 600 МГц): δ 0,62-0,66 (m, 1H), 0,73-0,78 (m, 1H), 1,07-1,14 (m, 2H), 1,15-1,21 (m, 2H), 1,36 (s, 3H), 1,46-1,54 (m, 2H), 2,21 (ddd, J=5,4 Гц, 8,4 Гц, 13,8 Гц, 1H), 2,71-2,77 (m, 1H), 3,89 (dd, J=7,5 Гц, 12,4 Гц, 1H), 4,13-4,19 (m, 1H),4,51 (q, J=7,7 Гц, 2H), 4,69 (dd, J=3,8 Гц, 12,4 Гц, 1H), 4,75 (dd, J=5,1 Гц, 8,3 Гц, 1H), 7,25 (dd, J=2,7 Гц, 8,9 Гц, 1H), 7,53 (d, J=2,6 Гц, 1H), 7,97 (br s, 1H), 8,19 (d, J=8,8 Гц, 1H). Анализ согласно расчету C23H23F6N3O5S: С, 48,68; Н, 4,08; N, 7,40; S, 5,65; F, 20,09. Обнаружено: С, 48,59; Н, 4,05; N, 7,53; S, 5,76; F, 20,08.
C3. Получение (1-циано-циклопропил)-амида (2S,4R)-4-[4-(2-метил-пиридин-4-ил)-2-трифторметил-бензосульфонил]-1-(1-трифторметил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты
Этап 1: (1-Циано-циклопропил)-амид (2S,4R)-4-[4-(2-метил-пиридин-4-ил)-2-трифторметил-фенилсульфанил]-1-(1-трифторметил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты
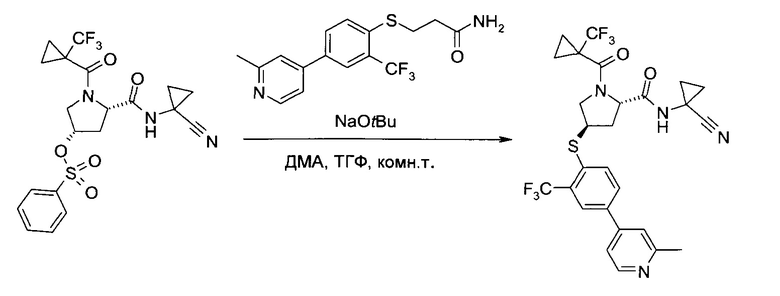
3-[4-(2-Метил-пиридин-4-ил)-2-трифторметил-фенилсульфанил]-пропионамид (200 г, 588 ммоль) растворили в тетрагидрофуране (1,0 л), после чего туда добавили трет-бутоксид натрия (55,5 г, 578 ммоль), и тонкую суспензию перемешивали 2 часа при комнатной температуре. В систему добавили N,N-диметилацетамид (500 мл), и раствор перемешивали еще полтора часа. К нему добавили раствор эфира бензолсульфоновой кислоты и (3S,5S)-5-(1-циано-циклопропилкарбамоил)-1-(1-трифторметил-циклопропанкарбонил)-пирролидин-3-ила (237,5 г, 504 ммоль) в N,N-диметилацетамиде (500 мл), после чего перемешивали прозрачный коричневый раствор в течение 40 часов. Смесь разбавили водой (3 л) и проэкстрагировали трет-бутилметиловым эфиром (1×2 л, 3×1 л). Объединенные органические экстракты промыли водным раствором карбоната натрия (1 М, 1,0 л) и соляным раствором (1,0 л), высушили над сульфатом натрия и сконцентрировали в вакууме, получив указанное в заголовке соединение в виде коричневой смолы (327,8 г). Неочищенный продукт (содержащий диметилацетамид) использовали на следующем этапе без дополнительной очистки. Чтобы охарактеризовать продукт, 4 г образца неочищенного вещества очистили фильтрацией через силикагель (20 г, элюент: этилацетат (200 мл)). 3,3 г очищенного продукта представляли собой грязно-белое твердое вещество. MS (El): m/z=583,1 [М+Н]+. 1H ЯМР (CDCl3, 300 МГц): δ 1,10-1,36 (m, 6Н), 1,49-1,58 (m, 2H), 1,99-2,12 (m, 1H), 2,65 (s, 3H), 2,90-3,02 (m, 1H), 3,92 & 4,02 (ABX, JAB=11,9 Гц, JAX=4,8 Гц, JBX=5,8 Гц, каждый 1H), 4,12-4,22 (m, 1H), 4,71 (dd, J=4,7 Гц, 7,8 Гц, 1H), 7,31 (d, J=5,3 Гц, 1H), 7,37 (br s, 1H), 7,60-7,69 (m, 2H), 7,80 (d, J=8,3 Гц, 1H), 7,93 (s, 1H), 8,60 (d, J=5,1 Гц, 1H).
Этап 2: (1-Циано-циклопропил)-амид (2S,4R)-4-[4-(2-метил-пиридин-4-ил)-2-трифторметил-бензосульфонил]-1-(1-трифторметил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты
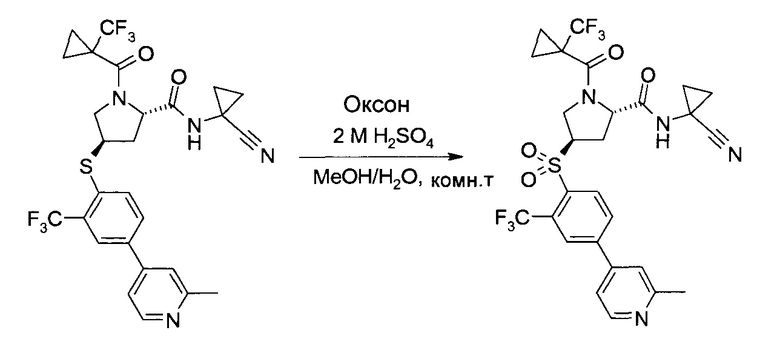
(1-Циано-циклопропил)-амид (2S,4R)-4-[4-(2-метил-пиридин-4-ил)-2-трифторметил-фенилсульфанил]-1-(1-трифторметил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты (исходный продукт, содержащий диметилацетамид, 89,4 масс % чистоты, 327,8 г, 503 ммоль) растворили в метаноле (930 мл) и серной кислоте (1 М в воде, 3,7 л) при 0-5°C (ледяная баня). В смесь разом добавили оксон (980 г, 1590 ммоль) (слегка экзотермическая реакция), после чего перемешивали смесь 20 часов при комнатной температуре. После этого в систему ввели целит (100 г), смесь профильтровали и осадок промыли системой вода/метанол (4:1 об./об., 500 мл). К объединенному фильтрату маленькими порциями добавили метабисульфит натрия (183 г, 965 ммоль) (экзотермическая реакция). Затем добавили этилацетат (1,5 л), и защелочили двухфазную смесь до pH>8, добавив водный раствор аммиака (25 масс %, 1,2 л). После разделения фаз, водный слой проэкстрагировали этилацетатом (2×1,2 л), объединенные органические экстракты промыли водой/соляным раствором (1:1 об./об., 750 мл) и соляным раствором (750 мл), высушили над сульфатом натрия и сконцентрировали в вакууме при 55°C. Неочищенный продукт растворили в системе дихлорметан/метанол (100:3 об./об., 1,45 л) и добавили силикагель (500 г). Кашицу профильтровали и промыли системой дихлорметан/метанол (100:3 об./об., 2,9 л). Объединенные фильтраты сконцентрировали в вакууме при 55°C. Остаток, белую пену (279,2 г), растворили в этаноле (1600 мл) при 70°C, после чего в раствор быстро добавили предварительно нагретую воду (60-65°C, 800 мл) и медленно охладили полученную смесь. Суспензию перемешивали 20 часов при комнатной температуре и профильтровали. Осадок промыли системой этанол/вода (1:1 об./об., 800 мл) и н-гептаном (800 мл), а затем высушили в вакууме при 55°C, получив указанное в заголовке соединение в виде бесцветных кристаллов (228,1 г, 74% за 2 этапа). MS (ESI & APCI): m/z=615,1 [М+H]+. 1Н ЯМР (CDCl3, 600 МГц): δ 1,11-1,22 (m, 2Н), 1,32-1,42 (m, 2H), 1,46-1,57 (m, 4H), 2,24-2,30 (m, 1H), 2,69 (s, 3H), 2,86 (ddd, J=5,7 Гц, 8,0 Гц, 14,1 Гц, 1Н), 3,88 (dd, J=7,1 Гц, 13,3 Гц, 1Н), 4,14-4,19 (m, 1H),4,83(dd, J=1,7 Гц, 13,3 Гц, 1Н), 4,88 (dd, J=5,6 Гц, 8,6 Гц, 1Н), 7,35 (dd, J=1,4 Гц, 5,2 Гц, 1Н), 7,40 (bs, 1Н), 7,63 (bs, 1Н), 8,01 (dd, J=1,8 Гц, 8,2 Гц, 1H), 8,17 (d, J=1,6 Гц, 1Н), 8,29 (d, J=8,3 Гц, 1Н), 8,68 (d, J=5,2 Гц, 1Н).
С4. Получение (1-циано-циклопропил)-амида (2S,4R)-4-[4-(1-метил-1N-пиразол-4-ил)-2-трифторметил-бензосульфонил]-1-(1-трифторметил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты
Вариант 1 процесса:
Этап 1: (1-Циано-циклопропил)-амид (2S,4R)-4-[4-(1-метил-1Н-пиразол-4-ил)-2-трифторметил-фенилсульфанил]-1-(1-трифторметил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты
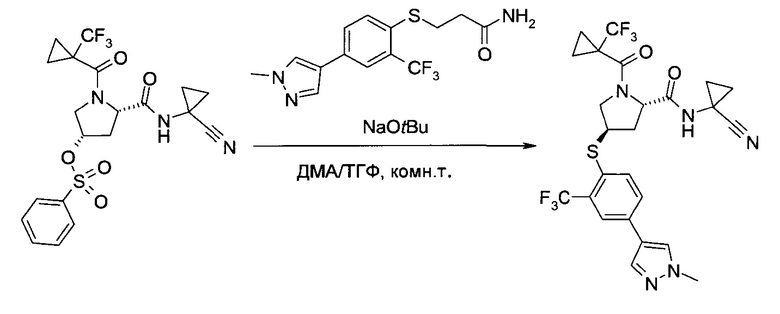
3-[4-(1-Метил-1H-пиразол-4-ил)-2-трифторметил-фенилсульфанил]-пропионамид (16.3 г, 49.6 ммоль) растворили в тетрагидрофуране (80 мл), добавили трет-бутоксид натрия (4,7 г, 48,9 ммоль), и перемешивали тонкую суспензию 2 часа при комнатной температуре. В систему добавили N,N-диметилацетамид (40 мл), и смесь при комнатной температуре перемешивали еще 2,5 часа. К ней добавили раствор эфира бензолсульфоновой кислоты и (3S,5S)-5-(1-циано-циклопропилкарбамоил)-1-(1-трифторметил-циклопропан-карбонил)-пирролидин-3-ила (20 г, 42,4 ммоль) в N,N-диметилацетамиде (40 мл), после чего перемешивали желтую, тонкую суспензию 42 часа при комнатной температуре. Смесь разбавили водой (240 мл) и проэкстрагировали трет-бутилметиловым эфиром (1×160 мл, 3×80 мл). Объединенные органические экстракты промыли водным раствором карбоната натрия (1 M, 80 мл) и соляным раствором (80 мл), высушили над сульфатом натрия и сконцентрировали в вакууме, получив указанное в заголовке соединение в виде светло-коричневой пены (27,1 г). Неочищенный продукт (содержащий диметилацетамид) использовали на следующем этапе без дополнительной очистки. Чтобы охарактеризовать продукт, 2 г образца неочищенного вещества очистили колоночной хроматографией на силикагеле (элюент: градиент этилацетат/гептан 4:1 (об./об.) - этилацетат). Подобным образом получили 1,7 г очищенного вещества. MS (ESI & APCI): m/z=572,1 [М+Н]+, 589,2 [М+NH4]+. 1H ЯМР (CDCl3, 600 МГц): δ 1,12-1,22 (m, 3H), 1,28-1,34 (m, 3H), 1,49-1,54 (m, 2H), 2,02 (ddd, J=5,6 Гц, 8,2 Гц, 13,6 Гц, 1Н), 2,88 (ddd, J=5,7 Гц, 5,7 Гц, 13,7 Гц, 1Н), 3,89-3,99 (m, 2H), 3,97 (s, 3H), 4,04-4,09 (m, 1Н), 4,69 (dd, J=5,2 Гц, 8,2 Гц, 1Н), 7,55 (d, J=8,2 Гц, 1Н), 7,60 (bs, 1Н), 7,62 (dd, J=1,8 Гц, 8,2 Гц, 1Н), 7,69 (s, 1Н), 7,77 (d, J=1,8 Гц, 1Н), 7,79 (s, 1Н).
Этап 2: (1-Циано-циклопропил)-амид (2S,4R)-4-[4-(1-метил-1Н-пиразол-4-ил)-2-трифторметил-бензосульфонил]-1-(1-трифторметил-циклопропанкарбрнил)-пирролидин-2-карбоновой кислоты

В метаноле (45 мл) и воде (16 мл) суспендировали оксон (53,0 г, 86,2 ммоль) и гидрофосфат натрия (13,0 г, 91,4 ммоль), после чего в течение 15 минут добавили раствор (1-циано-циклопропил)-амида (2S,4R)-4-[4-(1-метил-1Н-пиразол-4-ил)-2-трифторметил-фенилсульфанил]-1-(1-трифторметил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты (неочищенный продукт, содержащий диметилацетамид, 89,3 масс. % чистоты, 11,2 г, 17,5 ммоль) в метаноле (30 мл) при внутренней температуре 5-15°C. Ярко-желтую суспензию перемешивали 20 часов при комнатной температуре. Отфильтровав смесь, твердый осадок промыли метанолом (50 мл). К объединенному фильтрату добавили воду (30 мл), после чего в вакууме отогнали метанол. Водный раствор остатка проэкстрагировали этилацетатом (3×20 мл). Объединенные органические экстракты последовательно промыли раствором метабисульфита натрия (1,9 г, 10,0 ммоль) в воде (30 мл), насыщенным водным раствором гидрокарбоната натрия (30 мл) и соляным раствором (30 мл), высушили над сульфатом натрия и сконцентрировали в вакууме. Остаток растворили в системе дихлорметан/метанол (100:3 (об./об.), 50 мл) и профильтровали через слой силикагеля (20 г). Этот слой промыли системой дихлорметан/метанол (100:3 об./об., 100 мл), после чего сконцентрировали объединенный фильтрат в вакууме. Неочищенный продукт в виде желтой пены (9,4 г) растворили в этилацетате (25 мл) и толуоле (94 мл), сконцентрировали в вакууме, приблизительно, до объема 80 мл и полтора часа перемешивали при комнатной температуре. Суспензию профильтровали, и оставшийся твердый осадок промыли толуолом (20 мл) и н-гептаном (20 мл) и высушили в вакууме. Полученное белое кристаллическое вещество (7,15 г) снова растворили в ацетоне (35 мл) и воде (70 мл). В образовавшуюся эмульсию внесли затравку, и 20 часов энергично перемешивали суспензию при комнатной температуре. Осадок после фильтрования промыли системой ацетон/вода (1:4 (об./об.), 20 мл) и н-гептаном (20 мл) и высушили в вакууме, получив указанное в заголовке соединение в виде бесцветных кристаллов (6,03 г, 57% за 2 этапа). MS (ESI & APCI): m/z=604,1 [М+Н]+, 621,1 [М+NH4]+. 1H ЯМР (CDCl3, 600 МГц): δ 1,10-1,22 (m, 2H), 1,30-1,35 (m, 1Н), 1,35-1,41 (m, 1H), 1,45-1,59 (m, 4H), 2,22-2,28 (m, 1H), 2,84 (add, J=5,9 Гц, 8,0 Гц, 14,3 Гц, 1H), 3,84 (dd,J=7,1 Гц, 13,2 Гц, 1H), 4,00 (s, 3H), 4,08-4,14 (m, 1H), 4,81 (d, J=13,3 Гц, 1H), 4,86 (dd, J=5,8 Гц, 8,6 Гц, 1H), 7,65 (bs, 1H), 7,80-7,83 (m, 2H), 7,89 (s, 1H), 7,99 (d, J=1,6 Гц, 1H), 8,12 (d, J=8,4 Гц, 1H).
Вариант 2 процесса:
Этап 1: (1-Циано-циклопропил)-амид (2S,4R)-4-[4-(1-метил-1Н-пиразол-4-ил)-2-трифторметил-фенилсульфанил]-1-(1-трифторметил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты
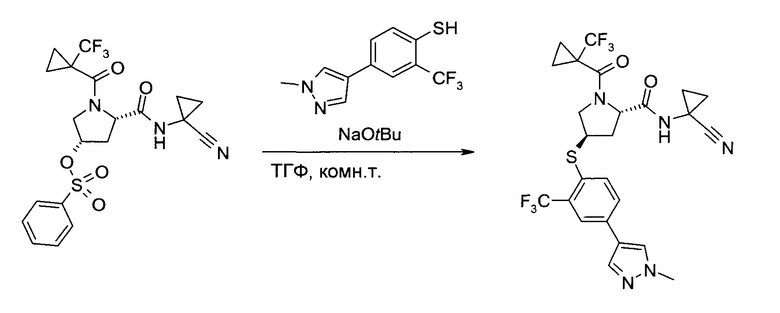
(3S,5S)-5-(1-Циано-циклопропилкарбамоил)-1-(1-трифторметил-цикло-пропанкарбонил)-пирролидин-3-иловый эфир (52,4 г, 111 ммоль) и 4-(1-метил-1H-пиразол-4-ил)-2-трифторметил-бензотиол (32,4 г, 122 ммоль) растворили в тетрагидрофуране (250 мл). При внутренней температуре 20-28°C в течение 20 минут добавили раствор трет-бутоксида натрия в тетрагидрофуране (25 масс. %, 44,8 г, 49,2 мл, 116 ммоль). Смесь 18 часов перемешивали при комнатной температуре. После этого добавили раствор хлорида натрия в воде (10 масс. %, 250 мл) и 2-метилтетрагидрофуран (250 мл). Фазы разделили, органический слой промыли раствором хлорида натрия в воде (10 масс. %, 250 мл) и сконцентрировали в вакууме. Неочищенный продукт растворили в ацетонитриле (250 мл), сконцентрировали в вакууме, снова растворили в ацетонитриле (250 мл) и профильтровали через активированный уголь, чтобы удалить неорганические соли. Отфильтрованное вещество промыли ацетонитрилом (100 мл), и объединенные фильтраты сконцентрировали в вакууме, получив неочищенное указанное в заголовке соединение в виде пены, на следующем этапе оно использовалось без дополнительной очистки (64,0 г, чистота 97,6% по методу ВЭЖХ, 98,3%).
Этап 2: (1-Циано-циклопропил)-амид (2S,4R)-4-[4-(1-метил-1Н-пиразол-4-ил)-2-трифторметил-бензосульфонил]-1-(1-трифторметил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты
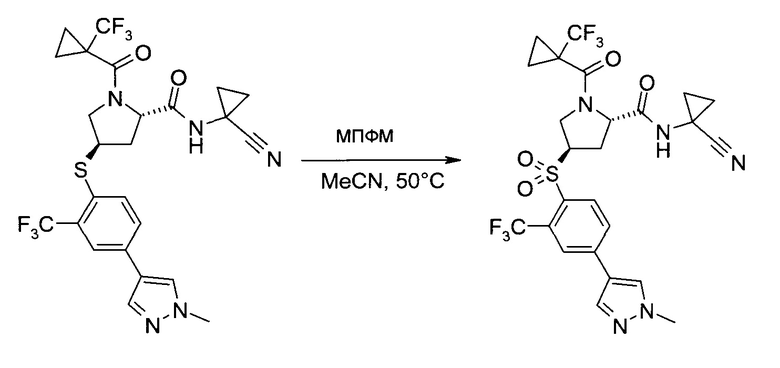
(1-циано-циклопропил)-амид (2S,4R)-4-[4-(1-метил-1Н-пиразол-4-ил)-2-трифторметил-фенилсульфанил]-1-(1-трис(эторметил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты (неочищенный продукт, чистота 97,6%, 64,0 г, 109 ммоль) растворили в ацетонитриле (480 мл) и добавили гексагидрат монопероксофталаата магния (76,0 г, 154 ммоль). Полученную белую суспензию перемешивали 5 часов при 50°C. После этого добавили еще суспензии гексагидрата монопероксофталаата магния (76,0 г, 154 ммоль) в ацетонитриле (150 мл) и перемешивали смесь еще 16 часов при 50°C. Охладив ее затем до 20°C, добавили воду (480 мл) и раствор метабисульфита натрия в воде (40 масс. %, 56,8 мл, 109 ммоль). Смесь защелочили до pH 8, добавлением водного раствора гидроксида натрия (3 М, около 290 мл), проэкстрагировали дихлорметаном (1 л), промыли органическую фазу насыщенным водным раствором гидрокарбоната натрия (1 л) и сконцентрировали в вакууме. Остаток растворили в дихлорметане (300 мл) и профильтровали, удалив твердые примеси. Растворитель в фильтрате заменили на этанол непрерывной перегонкой при поддержании постоянного объема (использовали около 750 мл этанола). Началась кристаллизация требуемого продукта. Полученную суспензию перемешивали 12 часов при 0°C, профильтровали, осадок промыли холодным этанолом (50 мл) и высушили в вакууме при 45°C, получив указанное в заголовке соединение в виде белых кристаллов (50,0 г, 74,9%). MS (ESI & APCI): m/z=604,1 [М+Н]+, 621,1 [М+NH4]+. 1Н ЯМР (CDCl3, 600 МГц): δ 1,10-1,22 (m, 2H), 1,30-1,35 (m, 1H), 1,35-1,41 (m, 1H), 1,45-1,59 (m, 4H), 2,22-2,28 (m, 1H), 2,84 (add, J=5,9 Гц, 8,0 Гц, 14,3 Гц, 1H), 3,84 (dd, J=7,1 Гц, 13,2 Гц, 1Н), 4,00 (s, 3H), 4,08-4,14 (m, 1H), 4,81 (d, J=13,3 Гц, 1H), 4,86 (dd, J=5,8 Гц, 8,6 Гц, 1H), 7,65 (bs, 1H), 7,80-7,83 (m, 2H), 7,89 (s, 1H), 7,99 (d, J=1,6 Гц, 1H), 8,12 (d, J=8,4 Гц, 1H).
С5. Получение (1-циано-цикло-пропил)-амида (2S,4R)-1-(1-метил-циклопропанкарбонил)-4-[4-(2-метил-пиридин-4-ил)-2-трифторметил-бензосульфонил]-пирролидин-2-карбоновой кислоты
Этап 1: (1-Циано-циклопропил)-амид (2S,4R)-1-(1-метил-циклопропанкарбонил)-4-[4-(2-метил-пиридин-4-ил)-2-трифторметил-фенилсульфанил]-пирролидин-2-карбоновой кислоты
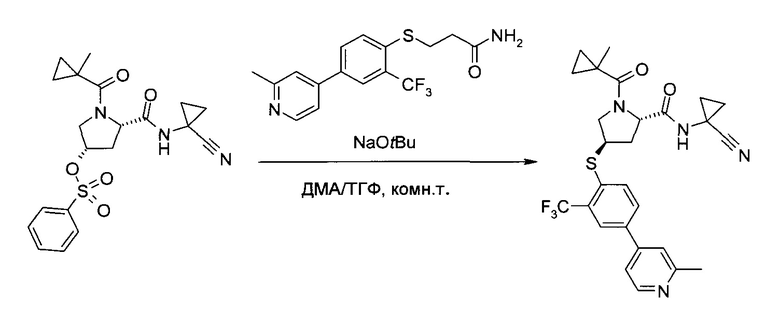
3-[4-(2-Метил-пиридин-4-ил)-2-трифторметил-фенилсульфанил]-пропионамид (15,0 г, 44,1 ммоль) растворили в тетрагидрофуране (75 мл), после чего добавили трет-бутоксид натрия (4,15 г, 43,2 ммоль) и перемешивали тонкую суспензию 2 часа при комнатной температуре. Затем добавили N,N-диметилацетамид (37 мл), и раствор перемешивали еще 2 часа. Добавили раствор эфира бензолсульфоновой кислоты и (35,55)-5-(1-циано-циклопропилкарбамоил)-1-(1-метил-циклопропанкарбонил)-пирролидин-3-ила (15,7 г, 37,7 ммоль) в N,N-диметилацетамиде (37 мл), после чего перемешивали коричневый прозрачный раствор 65 часов при комнатной температуре. После этого смесь разбавили холодной водой (240 мл) и проэкстрагировали трет-бутилметиловым эфиром (1×180 мл, 5×90 мл). The объединенные органические экстракты промыли водным раствором карбоната натрия (1 М, 90 мл) и соляным раствором (90 мл), высушили над сульфатом натрия и сконцентрировали в вакууме, получив указанное в заголовке соединение в виде коричневой смолы (22,05 г). Неочищенный продукт (содержащий диметилацетамид) использовали на следующем этапе без дополнительной очистки. MS (El): m/z=529,1 [М+H]+. 1H ЯМР (CDCl3, 400 МГц): δ 0,56-0,66 (m, 2H), 0,87-0,93 (m, 1H), 1,04-1,09 (m, 1H), 1,18-1,22 (m, 2H), 1,31 (s, 3H), 1,45-1,57 (m, 2H), 1,95-2,04 (m, 1H), 2,65 (s, 3H), 2,95-3,01 (m, 1H), 3,91 & 4,02 (ABX, JAB=11,3 Гц, JAX=5,6 Гц, JBX=6,5 Гц, каждый 1Н), 4,14-4,22 (m, 1H), 4,66 (dd, J=4,3 Гц, 8,1 Гц, 1Н), 7,31 (dd, J=1,5 Гц, 5,3 Гц, 1Н), 7,37 (bs, 1Н), 7,69 & 7,80 (ABX, JAB=8,2 Гц, JAX=О Гц, JBX=2,0 Гц, each 1Н), 7,93 (d, J=1,9 Гц, 1H), 8,06 (bs, 1H), 8,60 (d, J=5,1 Гц, 1H).
Этап 2: (1-Циано-циклопропил)-амид (2S,4R)-1-(1-метил-циклопропанкарбонил)-4-[4-(2-метил-пиридин-4-ил)-2-трифторметил-бензосульфонил]-пирролидин-2-карбоновой кислоты
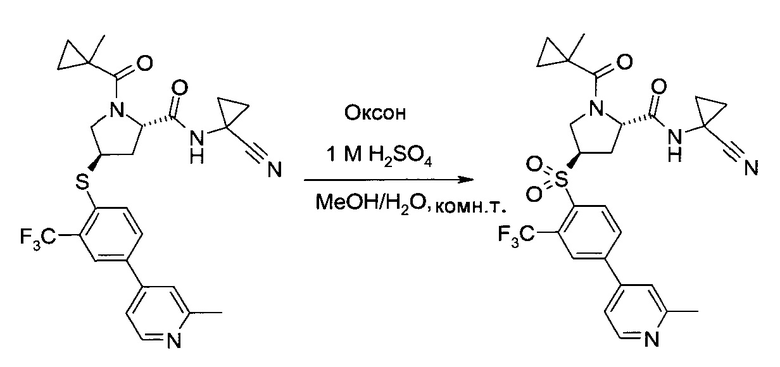
(1-Циано-циклопропил)-амид (2S,4R)-1-(1-метил-циклопропанкарбонил)-4-[4-(2-метил-пиридин-4-ил)-2-трифторметил-фенилсульфанил]-пирролидин-2-карбоновой кислоты (неочищенный продукт, содержащий диметилацетамид, чистота 90,2 масс. %, 22,1 г, 37,7 ммоль) растворили в метаноле (70 мл) и серной кислоте (1 М в воде, 280 мл) при 0-5°C (ледяная баня). В систему разом добавили оксон (73.5 г, 120 ммоль) (слегка экзотермическая реакция), и перемешивали смесь 20 часов при комнатной температуре. После этого добавили целит (7,5 г) и профильтровали смесь. Осадок промыли системой вода/метанол (4:1 об./об., 40 мл). В комбинированный фильтрат небольшими порциями добавили метабисульфит натрия (14,0 г, 73,6 ммоль) (экзотермическая реакция). Затем добавили этилацетат (120 мл) и настроили кислотность двухфазной смеси до pH>8, добавляя водный раствор аммиака (25 масс. %, 90 мл). После разделения фаз водный слой проэкстрагировали этилацетатом (2×120 мл). Объединенные органические экстракты промыли системой вода/соляной раствор (1:1 об./об., 60 мл) и соляной раствор (60 мл), высушили над сульфатом натрия и сконцентрировали в вакууме at 55°C. Неочищенный продукт (21,7 г) растворили в системе дихлорметан/метанол (100:3 об./об., 120 мл). Затем туда добавили силикагель (40 г), полученную кашицу профильтровали и промыли системой дихлорметан/метанол (100:3 об./об., 240 мл). Объединенные фильтраты сконцентрировали в вакууме при 55°C. Остаток, белую пену (18,1 г), растворили в этаноле (95 мл) при 70°C и быстро добавили предварительно нагретую воду (60-65°C, 135 мл). В прозрачный раствор внесли затравку, и медленно охладили полученную смесь. Суспензию 5 часов перемешивали при комнатной температуре и профильтровали. Осадок промыли системой этанол/вода (1:2 об./об., 50 мл) и н-гептаном (50 мл) и высушили в вакууме при 55°C, получив указанное в заголовке соединение в виде тонкого белого порошка (14,1 г, 71% за 2 этапа). MS (ESI & APCI): m/z=561,2 [М+H]+. 1H ЯМР (CDCl3, 600 МГц): δ 0,62-0,67 (m, 1H), 0,74-0,80 (m, 1H), 1,08-1,15 (m, 2H), 1,15-1,21 (m, 2H), 1,38 (s, 3H), 1,46-1,54 (m, 2H), 2,27 (add, J=5,6 Гц, 8,4 Гц, 13,9 Гц, 1H), 2,69 (s, 3H), 2,77 (ddd, J=5,7 Гц, 8,4 Гц, 13,4 Гц, 1H), 3,93 (dd, J=7,6 Гц, 12,4 Гц, 1H), 4,21-4,27 (m, 1H), 4,73 (dd, J=3,9 Гц, 12,4 Гц, 1H), 4,78 (dd, J=5,0 Гц, 8,4 Гц, 1H), 7,36 (dd, J=1,4 Гц, 5,2 Гц, 1H), 7,41 (s, 1H), 7,89 (bs, 1H), 8,01 (dd, J=1,8 Гц, 8,2 Гц, 1Н), 8,17 (d, J=1,5 Гц, 1H), 8,32 (d, J=8,3 Гц, 1H), 8,68 (d, J=5,2 Гц, 1H).
С6. Получение (1-циано-циклопропил)-амида (2S,4R)-4-[4-(2-метил-пиридин-4-ил)-2-трифторметил-фенилсульфанил]-1-(1-трифторметил-циклопропанкарбонил)-пирролидин-2-карбоновой кислоты
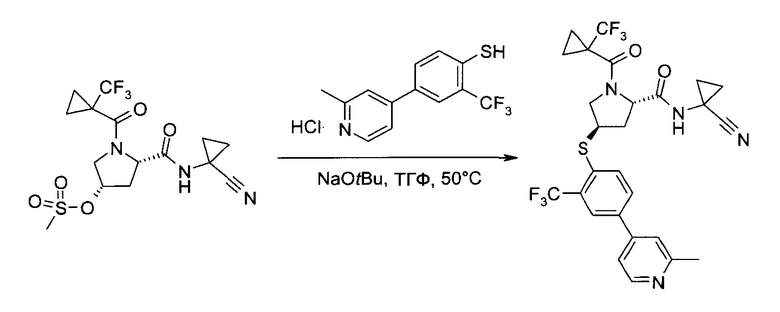
К суспензии эфира метансульфоновой кислоты и (3S,5S)-5-(1-циано-циклопропилкарбамоил)-1-(1-трифторметил-циклопропанкарбонил)-пирролидин-3-ила (20,5 г, 50 ммоль) и 4-(2-метил-пиридин-4-ил)-2-трифторметил-бензотиол гидрохлорида (17,6 г, 57,5 ммоль) в ТГФ (75 мл) при перемешивании и 0°C в течение 30 минут добавили 25% раствор трет-бутоксида натрия в ТГФ (43,2 г, 112,5 ммоль) и перемешивали реакционную смесь 4,5 часа при 50°C. Охладив реакционную смесь до 10°C, ее добавили к дихлорметану (250 мл) и дважды промыли деионизированной водой (2×250 мл). Органический слой высушили (Na2SO4), профильтровали и упарили на роторном испарителе (45°C/≥10 мбар), получив неочищенное указанное в заголовке соединение (30,9 г, 106%) в виде бежевой аморфной пены, которая использовалась на следующем этапе без дополнительной очистки. 1H ЯМР (CDCl3, 400 МГц) δ 1,10-1,35 (m, 6H), 1,47-1,58 (m, 2H), 2,00-2,11 (m, 1H), 2,65 (s, 3H), 2,92-3,01 (m, 1H), 3,92 & 4,02 (ABC, JAB=11,8 Гц, JAC=5,0 Гц, JBC=6,2 Гц, каждый 1H), 4,17 (quint, J≈5,5 Гц, 1H), 4,72 (dd, J1=8,1 Гц, J2=4,8 Гц, 1H), 7,31 (dd, J1=5,1 Гц, J2=1,5 Гц, 1H), 7,37 (s, 1H), 7,62 (s, 1H), 7,66 (d, J=8,3 Гц, 1H), 7,80 (dd, J1=8,1 Гц, J2=1,9 Гц, 1H), 7,93 (d, J=2,1 Гц, 1Н), 8,60 (d, J=5,1 Гц, 1Н). ESI-MS (m/z) [M+H]+ 583 (40).
| название | год | авторы | номер документа |
|---|---|---|---|
| АРИЛСУЛЬФОНИЛМЕТИЛЬНЫЕ ИЛИ АРИЛСУЛЬФОНАМИДНЫЕ ПРОИЗВОДНЫЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ РАССТРОЙСТВ, ВОСПРИИМЧИВЫХ К ЛЕЧЕНИЮ ЛИГАНДАМИ ДОФАМИНОВЫХ D РЕЦЕПТОРОВ, С ИХ ПОМОЩЬЮ | 2005 |
|
RU2442781C2 |
| СПИРО-АМИНОСОЕДИНЕНИЯ, ПРИГОДНЫЕ ДЛЯ ЛЕЧЕНИЯ НАРУШЕНИЙ СНА И ЛЕКАРСТВЕННОГО ПРИВЫКАНИЯ | 2010 |
|
RU2562609C2 |
| МОДУЛЯТОРЫ РЕЦЕПТОРА НМДА И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2621049C2 |
| СОЕДИНЕНИЯ ТИЕНОПИРИМИДИНА | 2013 |
|
RU2637925C2 |
| МОДУЛЯТОРЫ ЭСТРОГЕНОВЫХ РЕЦЕПТОРОВ | 2017 |
|
RU2738646C2 |
| ПРОИЗВОДНЫЕ КАРБОКСИЗАМЕЩЕННЫХ (ГЕТЕРО)АРОМАТИЧЕСКИХ КОЛЕЦ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2016 |
|
RU2733750C2 |
| СОЕДИНЕНИЯ, ИНГИБИРУЮЩИЕ МЕТАЛЛОФЕРМЕНТЫ | 2017 |
|
RU2764666C2 |
| НОВЫЕ ПИРАЗОЛПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ | 2017 |
|
RU2769448C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПИРРОЛИДИНА | 2017 |
|
RU2736498C1 |
| ПРОИЗВОДНЫЕ 5-АМИНО-4, 6-ДИЗАМЕЩЕННОГО ИНДОЛА И 5-АМИНО-4, 6-ДИЗАМЕЩЕННОГО ИНДОЛИНА КАК МОДУЛЯТОРЫ КАЛИЕВЫХ КАНАЛОВ | 2008 |
|
RU2483060C2 |
Изобретение относится к способу получения производных пролина формулы 1, где R1 выбран из C1-7-алкила или радикала формулы II, где R4 выбран из группы, содержащей С1-7-алкил, галоген-С1-7-алкил и фенил, возможно замещенный галогеном; R2 выбран из группы, содержащей галоген или галоген-С1-7-алкил; и R3 выбран из группы, содержащей водород, галоген, галоген-С1-7-алкил, С1-7-алкокси, галоген-С1-7-алкокси или 5- или 6-членный гетероцикл, содержащий один или два атома азота, причем цикл возможно замещен C1-7-алкилом или галогеном, и новым промежуточным соединением, используемым в способе. Производные пролина формулы 1 являются предпочтительными ингибиторами цистеиновой протеазы катепсина S и таким образом полезны при лечении метаболических заболеваний, таких как диабет, атеросклероз, аневризма брюшной аорты, заболевания периферических артерий и диабетическая невропатия. 3 н. и 26 з.п. ф-лы, 1 пр.
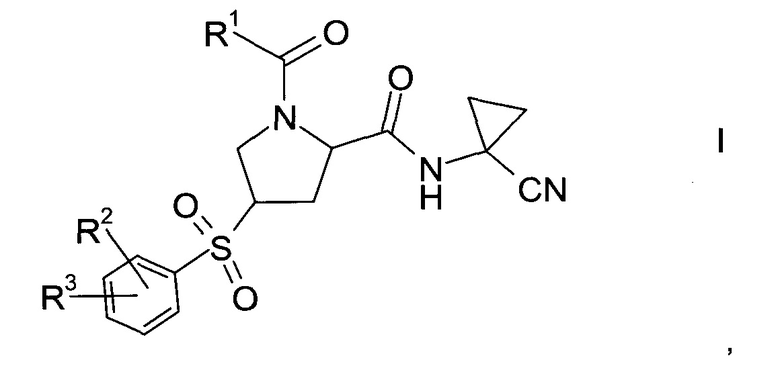
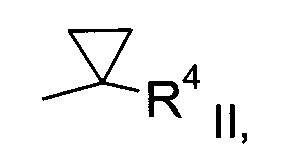
1. Способ получения производных пролина формулы I
где
R1 выбран из C1-7-алкила или
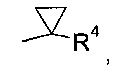
где R4 выбран из С1-7-алкила, галоген-С1-7-алкила или фенила, возможно замещенного галогеном;
R2 выбран из галогена или галоген-С1-7-алкила; и
R3 выбран из водорода, галогена, галоген-С1-7-алкила, C1-7-алкокси, галоген-C1-7-алкокси или 5- или 6-членного гетероцикла, содержащего один или два атома азота, причем цикл необязательно замещен С1-7-алкилом или галогеном;
включающий следующие этапы:
а) трансформация спирта формулы II

где R1 представляет собой такой, как описано выше, в сульфонат формулы III
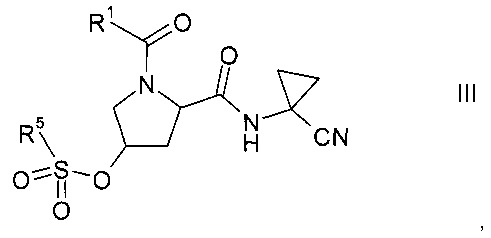
где R1 представляет собой такой, как описано выше, a R5 представляет собой C1-7-алкил, галоген-С1-7-алкил или фенил, возможно замещенный C1-7-алкилом, нитрогруппой или бромом
b) взаимодействие сульфоната формулы III с тио-соединением формулы IV
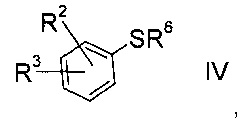
где R2 и R3 представляют собой такие, как описано выше, a R6 представляет собой водород или защитную группу, с получением тиоэфира формулы V
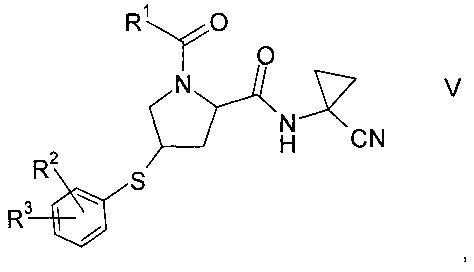
где R1, R2 и R3 представляют собой такие, как описано выше, и
c) окисление тиоэфира формулы V с получением производного пролина формулы I, где R1, R2 и R3 представляют собой такие, как описано выше,
и где способ дополнительно отличается тем, что спирт формулы II получают путем
а1) взаимодействия гидроксипролинового эфира формулы VI
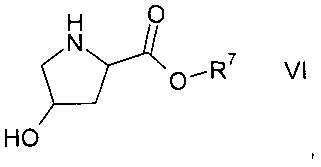
где R7 представляет собой С1-7-алкил,
с карбонильным соединением формулы VII
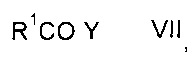
где R1 представляет собой такой, как описано выше, a Y представляет собой галоген или ОН, с получением карбонилпролинового эфира формулы IX,
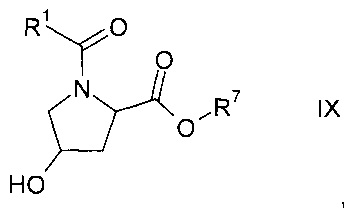
где R1 и R7 представляют собой такие, как описано выше;
b1) последующего образования сульфоната формулы X
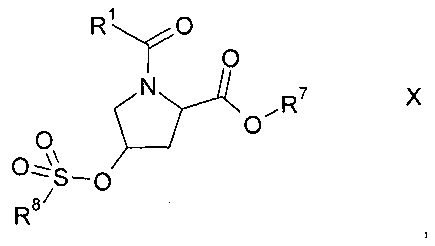
где R1 и R7 представляют собой такие, как описано выше, a R8 представляет собой C1-7-алкил, возможно замещенный галогеном, или фенил, который возможно замещен С1-7-алкилом, нитро или бромо, и
с1) превращения сульфоната формулы X в присутствии аминоциклопропанкарбонитрила формулы XI
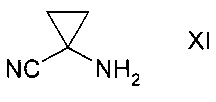
в спирт формулы II.
2. Способ по п. 1, где R1 представляет собой остаток формулы
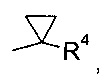
где R4 выбран из С1-7-алкила, галоген-С1-7-алкила или фенила, возможно замещенного галогеном.
3. Способ по п. 1, где
R2 выбран из галогена или галоген-С1-7-алкила; и
R3 выбран из галоген-С1-7-алкокси или 5- или 6-членного гетероцикла, содержащего один или два атома азота, причем цикл возможно замещен С1-7-алкилом или галогеном.
4. Способ по п. 1, где остаток формулы
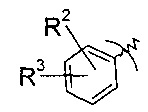
представляет собой

где R2 и R3 представляют собой такие, как описано выше.
5. Способ по п. 1, где производные пролина формулы I представляют собой хиральные изомеры формулы
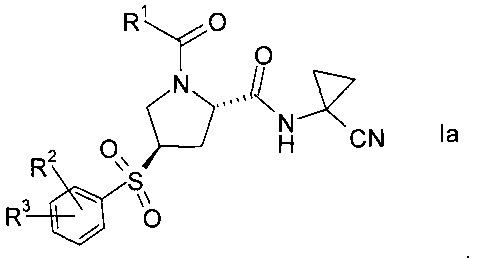
6. Способ по любому из пп. 1-5, где трансформацию на этапе а) осуществляют с использованием сульфонирующего агента в присутствии органического растворителя при температурах от -10°С до 40°С.
7. Способ по любому из пп. 1-5, где взаимодействие на этапе b) осуществляют в присутствии основания в органическом растворителе при температуре от 10°С до 90°С.
8. Способ по любому из пп. 1-5, где окисление на этапе с) осуществляют с использованием окислителя в присутствии органического растворителя при температуре от 0°С до 60°С.
9. Способ по п. 8, где окислитель представляет собой пероксомоносульфат калия или гексагидрат монопероксофталата магния.
10. Способ по п. 1, где спирт формулы II представляет собой хиральный изомер формулы

где R1 представляет собой такой, как описано выше.
11. Способ по п. 1, где взаимодействие на этапе а1) проводят в органическом растворителе при температуре от -10°С до 25°С.
12. Способ по п. 1, где взаимодействие на этапе b1) проводят с использованием сульфонирующего агента в органическом растворителе при температурах от -10°С до 40°С.
13. Способ по п. 1, где взаимодействие на этапе с1) проводят в присутствии соли карбоксилата NaR10COO, где R10=C1-9-алкил или арил, в растворителе при температуре от 40°С до 130°С.
14. Способ по п. 13, где используют 2-этилгексаноат натрия.
15. Способ по п. 1, где тио-соединение формулы IV
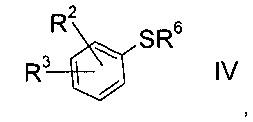
где R2 и R3 представляют собой такие, как определено выше, a R6 представляет собой водород, получают путем
а3) снятия защиты с соединения формулы XX
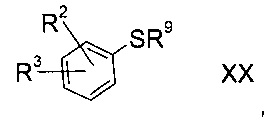
где R2 и R3 представляют собой такие, как определено выше, a R9 представляет собой третичную алкильную группу формулы
где R11, R12 и R13 независимо друг от друга представляют собой С1-7-алкил, с помощью кислоты;
или
b3) снятия защиты с соединения формулы XX
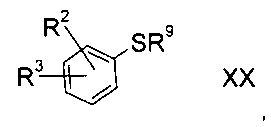
где R2 и R3 представляют собой такие, как определено выше, a R9 представляет собой тритил, с помощью трифторуксусной кислоты в присутствии восстановителя;
с3) литиирования галогенированного соединения формулы XXI
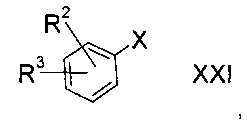
где R2 и R3 представляют собой такие, как определено выше, а X представляет собой атом галогена, с последующей обработкой продукта серой;
или
d3) взаимодействия галогенированного соединения формулы XXI

где R2 и R3 представляют собой такие, как определено выше, а X представляет собой атом галогена, с реактивом Гриньяра с последующей обработкой продукта серой.
16. Способ по п. 15, где группа R9 соединения формулы XX, используемого в реакции этапа а3), представляет собой трет-бутил.
17. Способ по п. 15, где кислоту, используемую в реакции этапа а3), выбирают из водного раствора неорганической кислоты или органической кислоты.
18. Способ по п. 15, где восстановитель, используемый в реакции этапа b3), представляет собой триэтилсилан.
19. Способ по п. 15, где взаимодействие на этапе c3) выполняют с бутиллитием в качестве агента литиирования в органическом растворителе при температуре от -80°С до -20°С.
20. Способ по п. 15, где взаимодействие с серой на этапе с3) выполняют в органическом растворителе при температуре от -80°С до -40°С.
21. Способ по п. 15, где взаимодействие этапа d3) выполняют с реактивом Гриньяра, выбранным из изопропилмагнийхлорида и изопропилмагнийхлорида/литий хлорида в органическом растворителе при температуре от 0°С до 40°С.
22. Способ по п. 15, где взаимодействие с серой на этапе d3) выполняют в органическом растворителе при температуре от -20°С до 20°С.
23. Спирт формулы II
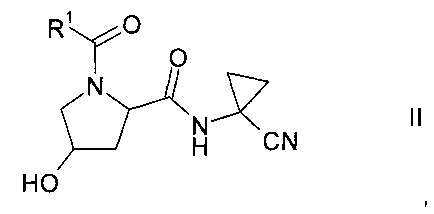
где R1 выбран из С1-7-алкила или
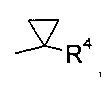
где R4 представляет собой метил или трифторметил.
24. Спирт по п. 23 следующей структуры:
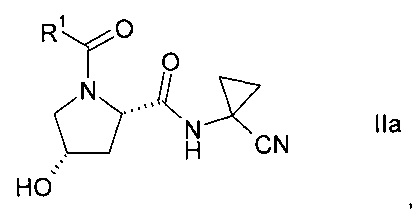
где R1 представляет собой такой, как описано выше.
25. Тиоэфир формулы V

где
R1 выбран из C1-7-алкила или
где R4 выбран из С1-7-алкила, галоген-С1-7-алкила или фенила, возможно замещенного галогеном;
R2 выбран из галогена или галоген-С1-7-алкила; и
R3 выбран из водорода, галогена, галоген-С1-7-алкила, C1-7-алкокси, галоген-C1-7-алкокси или 5- или 6-членного гетероцикла, содержащего один или два атома азота, причем цикл возможно замещен С1-7-алкилом или галогеном.
26. Тиоэфир по п. 25, где
R1 представляет собой

где R4 выбран из С1-7-алкила, галоген-С1-7-алкила или фенила, возможно замещенного галогеном;
R2 представляет собой галоген или галоген-С1-7-алкил; и
R3 представляет собой галоген-С1-7-алкокси или 5- или 6-членный гетероцикл, содержащий один или два атома азота, причем цикл может быть замещен С1-7-алкилом или галогеном.
27. Тиоэфир по любому из пп. 25 и 26, где,
R4 представляет собой метил или трифторметил;
R2 представляет собой трифторметил или хлор; и
R3 представляет собой 2,2,2-трифторэтокси, 2-метилпирид-4-ил, 1-метил-1H-пиразол-4-ил или 2,2,2-трифтор-1-метилэтокси.
28. Тиоэфир по п. 25 структуры
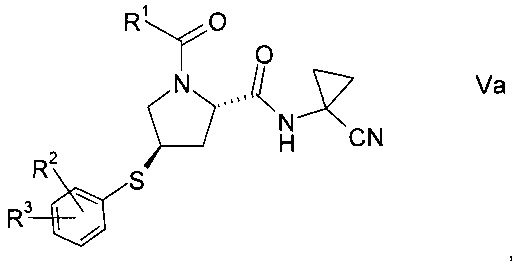
где R1, R2 и R3 представляют собой такие, как описано выше.
29. Тиоэфир по п. 25 структуры
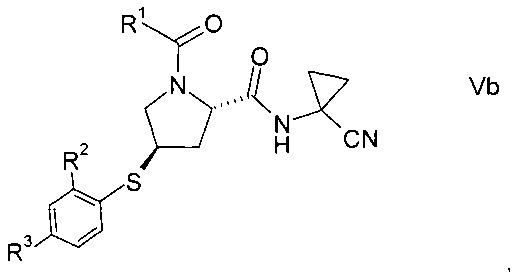
где R1, R2 и R3 представляют собой такие, как описано выше.
| ПРОИЗВОДНЫЕ ПРОЛИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КАТЕПСИНА | 2010 |
|
RU2535479C2 |
| LEO A | |||
| HARDEGGER et al., "Systematic investigation of halogen bonding in protein-ligand interactions", ANGEWANDTE CHEMIE INTERNATIONAL EDITION, 2011, vol | |||
| Устройство для выпрямления многофазного тока | 1923 |
|
SU50A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

