Предшествующий уровень техники
Калиевые каналы являются трансмембранными белками, которые повсеместно экспрессированы в клетках млекопитающих и представляют собой одну из наиболее обширных и разнообразных групп ионных каналов с точки зрения строения молекул. Калиевые каналы играют ключевую роль в регуляции потенциала клеточных мембран и модуляции возбудимости клеток. Калиевые каналы в значительной степени регулируются потенциалом, клеточным метаболизмом, концентрацией ионов кальция и рецептор-опосредованными процессами [Cook, N.S., Trends in Pharmacol. Sciences (1988), 9, 21; и Quast, U., и др., Trends in Pharmacol. Sciences (1989), 10, 431]. Кальций-активируемые калиевые каналы (Кса) являются многообразной группой ионных каналов, деятельность которых зависит от внутриклеточной концентрации кальция. Деятельность Кса-каналов регулируется внутриклеточной концентрацией ионов кальция, мембранным потенциалом и фосфорилированием. На основе их одноканальной проводимости в симметричных калиевых растворах Кса-каналы делят на три подкласса: высокой проводимости (также обозначаемые в литературе как “ВК” или “Maxi-K”), имеющие проводимость более 150 пикосименсов (“pS”); промежуточной проводимости, имеющие проводимость приблизительно 50-150 pS; и низкой проводимости, имеющие проводимость менее 50 pS. Кальций-активируемые калиевые каналы высокой проводимости представлены во многих возбудимых клетках, включая нейроны, клетки сердца и различные виды гладкомышечных клеток [Singer, J. и др., Pflugers Archiv. (1987) 408, 98; Baro, L и др., Pflugers Archiv. (1989) 414 (Suppl. 1), S168; и Ahmed, F и др., Br. J. Pharmacol. (1984) 83, 227].
Ионы калия играют ведущую роль в регуляции мембранного потенциала покоя в большинстве возбудимых клеток и поддерживают трансмембранный потенциал, приблизительно равный равновесному калиевому потенциалу (“Ек”) - около -90 милливольт (“мВ”). Было показано, что открытие калиевых каналов сдвигает мембранный потенциал клетки в сторону Ек, вызывая гиперполяризацию клетки [Cook, N.S., Trends in Pharmacol. Sciences (1988), 9, 21]. Гиперполяризованные клетки менее чувствительны к потенциально опасным, повреждающим деполяризующим стимулам. ВК-каналы, которые регулируются как потенциалом, так и внутриклеточной концентрацией ионов кальция, ограничивают деполяризацию и вход кальция в клетку и могут быть особенно эффективны в блокировании повреждающих стимулов. Следовательно, гиперполяризация клеток посредством открытия ВК-каналов может защищать нервные клетки, также как и другие типы клеток, например, кардиомиоциты [Xu, W., Liu, Y., Wang, S., McDonald, Т., Van Eyk, I.E., Sidor,. А., и O′Rourke, B. (2002) Cytoprotective Role Of Ca2+-activated K+ Channels in the Cardiac Inner Mitochondrial Membrane. Science 298, 1029-1033].
Описано множество синтетических и природных соединений, вызывающих открытие ВК каналов. Особый интерес представляют производные 4-арил-3-гидроксихинолин-2-она, раскрытые, например, в US 5,892,045, опубл. 06.04.1999 г., в US 5,922,735, опубл. 13.07.1999 г., в US 6,353,119, опубл. 05.03.2002 г.
Несмотря на указанные выше достижения в уровне техники, ставшие возможными благодаря использованию производных 4-арил-3-гидроксихинолин-2-она, дальнейшие успехи в изучении класса соединений, способных к модуляции калиевых каналов, в частности кальций-активируемых калиевых каналов высокой проводимости, весьма желательны. Следует ожидать, что такие соединения будут применимы в лечении состояний, являющихся результатом дисфункции поляризации и проводимости клеточной мембраны.
Раскрытие изобретения
В соответствии с настоящим изобретением представлены атропоизомеры производных 3-замещенного-4-арилхинолин-2-она. Атропоизомеры являются соединениями, имеющими следующую общую формулу:
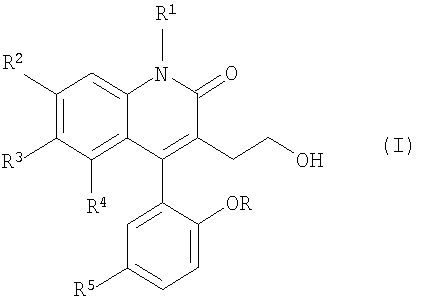
где R, R1, R2, R3, R4 и R5 представляют собой заместители как описано ниже, или их нетоксичную фармацевтически приемлемую соль, сольват или пролекарство; характеризующиеся тем, что соединения атропоизомерно обогащены, и предпочтительно, являются по существу чистыми по отношению к одному атропоизомеру.
Достоинство данного изобретения состоит в том, что теперь возможны стабильные атропоизомеры производных 3-замещенного-4-арилхинолин-2-она. В соответствии с данным изобретением довольно неожиданным оказалось то, что эти атропоизомеры не претерпевают взаимопревращения даже в течение длительных периодов, например 30 дней и более. Как результат этого, фармацевтические композиции могут быть подобраны таким образом, чтобы обогатить атропоизомером наиболее эффективно для лечения в выбранных условиях.
Настоящее изобретение также позволяет получать фармацевтические композиции, включающие атропоизомеры производных 3-замещенного-4-арилхинолин-2-она и осуществлять способы лечения состояний, чувствительных к проводимости калиевых каналов, таких как, например, ишемия, апокалипсический удар, судороги, эпилепсия, астма, синдром раздраженного кишечника, мигрень, черепно-мозговая травма, повреждение спинного мозга, сексуальное расстройство и недержание мочи.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг.1 показывает структурное представление двух атропоизомеров по настоящему изобретению.
Фиг.2 представляет собой ВЭЖХ хроматограмму рацемической смеси атропоизомеров по данному изобретению.
Фиг.3 представляет собой ВЭЖХ хроматограмму атропоизомера по данному изобретению.
Фиг.4 представляет собой ВЭЖХ хроматограмму атропоизомера по данному изобретению.
Фиг.5 представляет собой ВЭЖХ хроматограмму рацемической смеси атропоизомеров по данному изобретению.
Фиг.6 представляет собой ВЭЖХ хроматограмму атропоизомера по данному изобретению.
Фиг.7 представляет собой ВЭЖХ хроматограмму атропоизомера по данному изобретению.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Используемые здесь стереохимические определения и положения в общем соответствуют McGraw-Hill Dictionary of Chemical Terms, S. P. Parker, Ed., McGraw-Hill Book Company, New York (1984) и Stereochemistry of Organic Compounds, Eliel, E. и Wilen, S., John Wiley & Sons, Inc., New York (1994). Многие органические соединения существуют в оптически активных формах, т.е. они способны вращать плоскость плоскополяризованного света. В описании оптически активного соединения префиксы D и L или R и S используются для обозначения абсолютной конфигурации молекулы относительно ее хирального центра (центров). Префиксы д и 1 или (+) и (-) используются для обозначения знака вращения соединением плоскополяризованного света, где (-) или 1 означает, что соединение является левовращающим, а (+) или д означает, что соединение является правовращающим. Для данной химической структуры эти соединения, называемые стереоизомерами, идентичны за исключением того, что они являются зеркальными отражениями друг друга. Определенный стереоизомер зеркальной пары также может быть отнесен к энантиомерам, и смесь таких изомеров зачастую именуется энантиомерной смесью.
Термины "рацемическая смесь" и "рацемат" относятся к эквимолярной смеси двух энантиомерных единиц, не обладающей оптической активностью. Ко всему прочему, здесь используются определения "рацемическая смесь" и "рацемат", подразумевающие эквимолярные смеси двух атропоизомеров.
Термин "хиральный" относится к молекулам, которые обладают свойством неналожения на своего зеркального партнера, в то время как определение "ахиральный" относится к молекулам, способным налагаться на своего зеркального партнера.
Термин "стереоизомеры" относятся к соединениям, которые обладают одинаковым химическим строением, но различаются в отношении расположения атомов или групп в пространстве.
Термин "диастереомер" относится к стереоизомеру, который не является энантимером, например стереоизомер с двумя или более центрами хиральности, у которого молекулы не являются зеркальными отражениями друг друга. Диастереомеры обладают различными физическими свойствами, например точки плавления, точки кипения, спектральные свойства и реакционноспособность. Смеси диастереомеров можно разделять аналитическими методами, имеющими высокое разрешение, такими как электрофорез и хроматография.
Термин "энантиомеры" относится к двум стереоизомерам соединения, которые являются неналагаемыми зеркальными образами друг друга.
Термин "атропоизомер" относится к стереоизомеру, получающемуся из-за ограничения вращения вокруг одинарной связи, где барьер вращения достаточно велик для того, чтобы препятствовать выделению изомерных единиц. Наиболее типично, когда вращение вокруг одинарной связи в молекуле прекращается, или существенно затормаживается, в результате стерических взаимодействий с другими частями молекулы и заместители на обоих концах одинарной связи являются несимметричными.
Термин "атропоизомерно обогащенный" означает, что соединение, т.е. смесь атропоизомеров, содержит большую пропорцию или процент одного из атропоизомеров соединения, по отношению к другому атропоизомеру, т.е. больше чем 50 молярных %.
Термин "практически чистый" означает, что соединение, т.е. смесь атропоизомеров, содержит, по крайней мере 90 молярных %, предпочтительно, по крайней мере 95 молярных %, а еще более предпочтительно по крайней мере 99 молярных % одного атропоизомера.
Термин "практически свободный" означает, что соединение содержит менее чем 10 молярных %, предпочтительно менее чем 5 молярных % и более предпочтительно 1 молярных % одного атропоизомера.
Термины "взаимопревращение" и "взаимопревращаться" означают превращение атропоизомерно обогащенного соединения в рацемическую смесь.
Термины "стабильный" и "стабильность" означают, что атропоизомерно обогащенное соединение подвергается взаимному превращению при комнатной температуре, т.е. 25°С, в растворенной форме, например раствор в этаноле 3 милиграмм на милилитр ("мг/мл"), в течение по крайней мере около 1 дня, предпочтительно по крайней мере 10 дней и более, предпочтительно по крайней мере около 30 дней. При испытании стабильности по данному изобретению могут быть использованы растворители, отличные от этанола, например 2-пропанол, смесь спиртовых растворителей с сорастворителями, например толуолом. Когда соединения по данному изобретению находятся в твердой форме, т.е. не растворены или диспергированы в жидкости, они зачастую даже более устойчивы к взаимопревращению, чем когда находятся в жидкой форме.
Термин "фармацевтически приемлемая соль" подразумевает под собой нетоксичные соли, синтезированные обычными химическими методами из родственного соединения, которое содержит основный или кислотный фрагмент. Обычно такие соли могут быть получены реакцией свободных кислой или основной форм этих соединений со стехиометрическим количеством подходящих основания или кислоты в воде или в органическом растворителе, или в смеси их обоих; обычно предпочтительна неводная среда, такая как эфир, этилацетат, этанол, изопропанол или ацетонитрил. Перечень подходящих солей находится в Remington′c Pharmaceutical Sciences, 18th ed., Mack Publishing Company, Easton, PA, 1990, p.1445. Подходящие неорганические основания, такие как основания щелочных и щелочноземельных металлов, включают катионы металла, такие как натрий, калий, магний, кальций и им подобные. Соединения по настоящему изобретению используются в форме свободного основания или кислоты или, вместо этого, в форме фармацевтически приемлемой соли. Все формы включены в объем изобретения.
Термин "галоген" в том значении, как он используется здесь и в формуле изобретения, подразумевает под собой фтор, бром, хлор и йод, в то время как термин "галогенид" подразумевает под собой фторидный, бромидный, хлоридный и йодидный анион.
Термин "терапевтически эффективное количество" означает общее количество каждого активного компонента, которое является достаточным для того, чтобы показать значительное облегчение для пациента, т.е. излечение острых состояний, характеризуемых деблокаторами кальций-активированных К+ каналов высокой проводимости или увеличением скорости излечения таких состояний. При применении к индивидуальному активному ингредиенту, вводимому отдельно, определение относится к такому ингредиенту самому по себе. При применении к комбинации определение относится к суммарным количествам активных ингредиентов, которые приводят к терапевтическому эффекту, вводится ли эта комбинация последовательно или одновременно.
Термин "соединения по изобретению" и равные ему выражения означают, что они охватывают соединения Формулы I и включают в себя пролекарства, фармацевтически приемлемые соли и сольваты, например гидраты. Схожим образом, ссылка на промежуточные соединения, заявляются или нет они сами по себе, охватывает их соли и сольваты, где это позволяется. Ссылки на соединения Формулы I также включают соединения Формулы II и III.
Термин "производное" означает химически модифицированное соединение, где под модификацией подразумевается стандартная процедура для специалиста, такое как сложный эфир или амид кислоты, защитные группы, такие как бензильная группа для спирта или тиола, и трет-бутоксикарбонильная группа для амина.
Термин "сольват" означает физическое объединение соединения данного изобретения с одной или более молекулами растворителя. Это физическое объединение включает в себя образование водородных связей. В определенных случаях сольват может быть выделен, например, когда одна или несколько молекул растворителя включены в кристаллическую решетку твердого кристалла. "Сольват" окружает и фазу раствора, и выделяемые сольваты. Типичные сольваты включают в себя гидраты, этанолаты, метанолаты и тому подобные.
Термин "пациент" включает в себя и человека, и других млекопитающих.
Термин "фармацевтическая композиция" означает композицию, содержащую соединение по изобретению в комбинации с по меньшей мере одним дополнительным фармацевтическим носителем, т.е. активирующая добавка, эксципиент или связующее, такие как растворители, консервирующие агенты, наполнители, агенты, регулирующие поток, измельчающие агенты, увлажняющие агенты, эмульгирующие агенты, суспендирующие агенты, подсластители, ароматизаторы, парфюмерные композиции, антибактериальные агенты, противогрибковые агенты, смазывающие агенты и диспергирующие агенты, в зависимости от способа ввода и форм дозировки. Например, могут быть использованы ингредиенты, приведенные в Remington′c Pharmaceutical Sciences, 18th ed., Mack Publishing Company, Easton, PA (1999).
Словосочетание "фармацевтически приемлемые" здесь применяется по отношению к тем соединениям, материалам, композициям и/или формам дозировки, которые являются пригодными в медицине для применения в контакте с тканями человеческого организма и животных без существенной токсичности, раздражения, аллергического отклика или другой проблемы, или соразмерного осложнения с разумным соотношением риск/успех.
Термин "фармацевтически приемлемые пролекарства", как оно используется здесь, означает те пролекарства из соединений, пригодных по данному изобретению, которые являются пригодными в медицине для применения в контакте с тканями человеческого организма и низших животных без чрезмерной токсичности, раздражения, аллергического отклика и тому подобного, соразмерного с разумным соотношением риск/успех, и эффективными при намеченном применении, а также и цвиттерионные формы, где это возможно, из соединений изобретения.
Термин "пролекарства", как здесь используется, подразумевает под собой любые ковалентно связанные носители, которые высвобождают активное родственное лекарство данного изобретения in vivo, когда такое пролекарство вводят пациенту. Поскольку, как это известно, пролекарства увеличивают многочисленные желательные качества фармацевтических препаратов (т.е. растворимость, биодоступность, производство и т.д.), соединения данного изобретения могут доставляться в форме пролекарства. Так, специалист в данной области будет ожидать, что настоящее изобретение включает пролекарства заявленных соединений, способы их доставки и содержащие их композиции. Пролекарства настоящего изобретения получают модификацией функциональных групп в соединениях таким способом, чтобы модификации отщеплялись, обычными способами in vivo, с получением исходного соединения. Превращение in vivo может происходить, например, в результате некоторых метаболических процессов, таких как химический или ферментный гидролиз карбонового, фосфорного или сульфатного эфира, или восстановление или окисление подходящей группы. Пролекарства включают соединения настоящего изобретения, в которых гидрокси-, амино- или сульфгидрильная группа связана с любой группой так, чтобы пролекарство настоящего изобретения при введении пациенту расщеплялось с получением свободной гидроксильной, свободной амино- или свободной сульфгидрильной группы, соответственно. Функциональные группы, которые могут быть быстро трансформированы, метаболическим разделением in vivo образуют класс групп, реакционных с карбоновой группой настоящего изобретения. Они включают, без ограничений, такие группы, как алканоил (такие как ацетил, пропионил, бутирил и им подобные), незамещенный и замещенный ароил (такой как бензоил и замещенный бензоил), алкоксикарбонил (такой как этоксикарбонил), триалкилсилил (такой как триметил-триэтилсилил), моноэфиры, полученные с дикарбоновыми кислотами (такие как сукцинил), и им подобные. Благодаря легкости, с которой метаболически отщепляемые группы соединений, пригодных в соответствии с изобретением, отщепляются in vivo, соединения с такими группами могут выступать в качестве пролекарств. Соединения с метаболически отщепляемыми группами имеют преимущество в том, что они могут проявлять улучшенную биодоступность в результате улучшенной растворимости и/или скорости всасывания, получая родственное соединение благодаря наличию метаболически отщепляемой группы. Подробное обсуждение пролекарств приведено в следующих источниках: Design of Prodrugs, H. Bundgaard, ed., Elsevier, 1985; Methods in Enzymology, K.Widder и др., Ed., Academic Press, 42, p.309-396, 1985; A Textbook of Drug Design and Development, Krogsgaard-Larsen'and H. Bundgaard, ed., Chapter 5; "Design and Applications of Prodrugs" p.113-191, 1991; Advanced Drug Delivery Reviews, H-Bundgard, 8, p.1-38, 1992; Journal of Pharmaceutical Sciences, 77, p.285, 1988; Chem. Pharm. Bull., N. Nakeya и др., 32, p.692, 1984; Pro-drugs as Novel Delivery Systems. T, Higuchi and V.Stella, Vol.14 of the A.C.S. Symposium Series, and Bioreversible Carriers in Drug Design, Edward B. Roche, ed., American Pharmaceutical Association and Pergamon Press, 1987.
Термин "лечение" относится к: (i) предупреждению заболевания, расстройства или состояния из встречающихся у пациента, который может быть предрасположен к заболеванию, расстройству или состоянию, и/или состояния, которое пока еще не диагностировано; (ii) сдерживанию заболевания, расстройства или состояния, т.е. удержанию его развития; и (iii) облегчению заболевания, расстройства или состояния, т.е. вызыванию регрессии заболевания, расстройства и/или состояния.
Производные 3-замещенного-4-арилхинолин-2-она настоящего изобретения являются атропоизомерно насыщенными соединениями, имеющими формулу
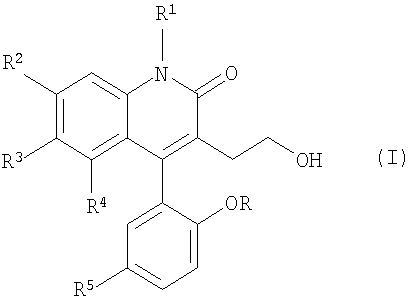
где R и R1 каждый независимо является водородом или метилом;
R2, R3 и R4 каждый независимо является водородом, галогеном, нитро- или трифторметилом при условии, что R2, R3 и R4 не все являются водородом; и их нетоксичная фармацевтически приемлемая соль, сольват или пролекарство.
В первом предпочтительном аспекте изобретения R1 представляет собой водород. В другом предпочтительном аспекте изобретения R1 представляет собой метил. В другом еще более предпочтительном аспекте изобретения R, R2 и R4 представляют собой водород, R3 представляет собой трифторметил и R5 представляет собой хлор.
Один атропоизомер по изобретению может быть представлен формулой
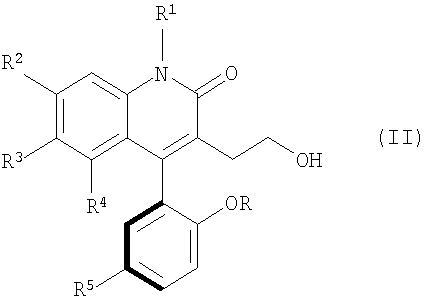
Для целей описания настоящего изобретения вышеуказанная структура произвольным образом отнесена здесь к (-)атропоизомеру или атропоизомеру (А). Жирная часть фенильного кольца в 4 положении хинолинона обозначает частичное вращение фенильного кольца из плоскости, в которой размещен хинолинон.
Другой атропоизомер по изобретению может быть представлен формулой
Для целей описания настоящего изобретения вышеуказанная структура произвольным образом отнесена здесь к (+)атропоизомеру или атропоизомеру (В).
Предпочтительные соединения для использования в способе данного изобретения включают соединения Формулы I, перечисленные ниже:
(1) 4-(5-хлор-2-гидроксифенил)-1-метил-3-(2-гидроксиэтил)-6-(трифторметил)-2(1Н)-хинолинон;
(2) 4-(5-хлор-2-гидроксифенил)-1-метил-3-(2-гидроксиэтил)-7-(трифторметил)-2(1Н)-хинолинон;
(3) 4-(5-хлор-2-гидроксифенил)-3-(2-гидроксиэтил)-6-(трифторметил)-2(1Н)-хинолинон и
(4) 4-(5-хлор-2-гидроксифенил)-3-(2-гидроксиэтил)-7-(трифторметил)-2(1Н)-хинолинон.
Соединения Формулы I могут быть получены различными способами, известными специалисту в данной области, такими как, например, которые раскрыты в US 6,184,231, 06.02.2001 и 6,353,119, 05.03.2002.
Следующая схема реакции иллюстрирует типичные общие методики для получения промежуточных соединений и способы получения соединений Формулы I по данному изобретению. Также специалисту ясно, что подходящая замена раскрываемых здесь материалов и методов даст примеры, проиллюстрированные ниже, и примеры, охваченные объемом изобретения.
Схема реакции 1
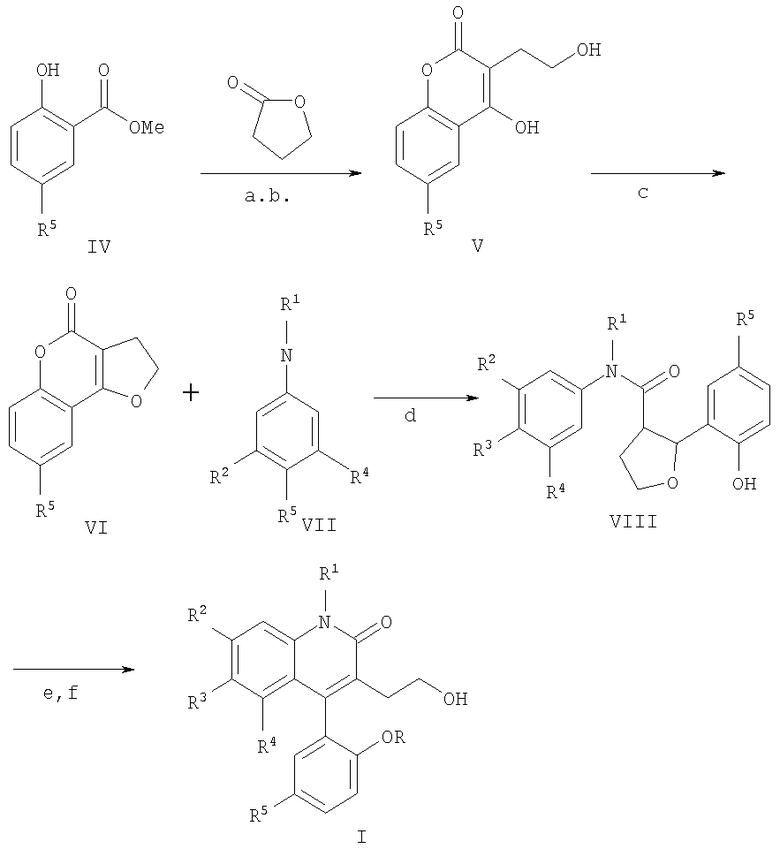
(a) LiHMDS/ТГФ (лития гексаметилдисилазид/тетрагидрофолат), от -78°С до комнатной температуры
(b) 12NHCl
(c) pTSA (пара-толуолсульфонамид), толуол, кипячение с обратным холодильником
(d) LiHMDS
(e) (СН3О)2SO2, К2СО3
(f) hν, MeOH
Приготовление рацемической смеси соединений Формулы I может быть осуществлено посредством реакций, иллюстрированных Схемой реакций 1. Кумариновое соединение Формулы V предпочтительно получают путем конденсации γ-бутиролактона с метиловым эфиром замещенной салициловой кислоты Формулы IV, которая затем легко циклизуется в присутствии каталитического количества кислоты до образования бензопиран-4-она Формулы VI. Обработка соединения 6 замещенным анилином Формулы VII, как показано на стадии (d), приводит к образованию дигидрофурана Формулы VIII, который затем может метилироваться метилирующим агентом, таким как диметилсульфат. Дигидрофуран Формулы VIII затем подвергается фотохимической циклизации в инертном органическом растворителе с получением требуемого соединения Формулы I.
Предпочтительная схема реакций для приготовления соединений Формулы I изложена ниже:
Схема реакций 2
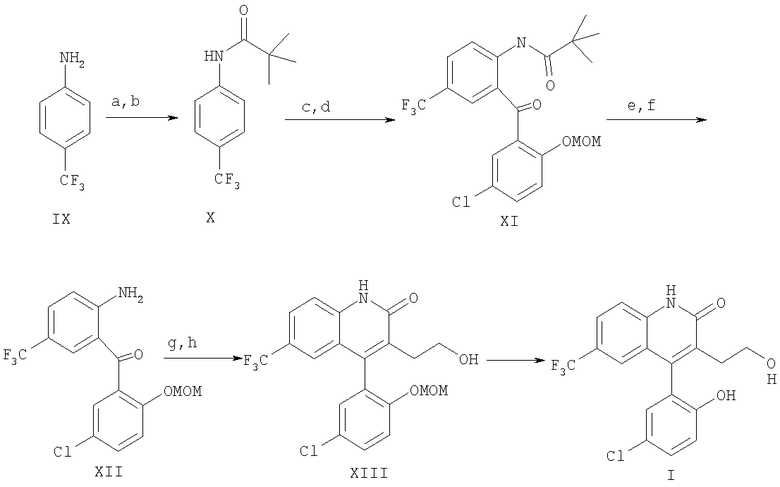
a) МеСООМе, ТГФ
b) NaHMDS, 0 - комнатная температура, 2 часа
c) n-BuLi, -40-0°C
д) от -78 до -40°С, 2 часа
e) NaOH, EtOH,
f) 40°C, 1.5 ч
g)
 , LiHMDS
, LiHMDS
h) 0 - комнатная температура, 1 час, затем H2О, комнатная температура, 2 часа
i) i-PrOH, HCl
j) 45°C, 3 ч
Другая предпочтительная схема реакций для приготовления соединений Формулы 1 изложена ниже:
Схема реакций 3
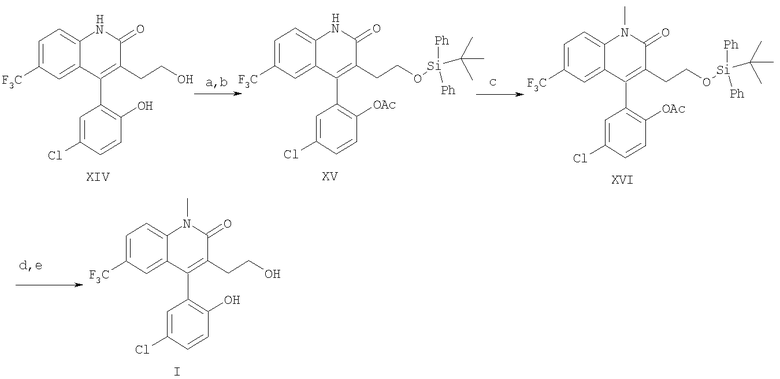
а)
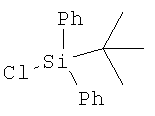 , DMF (диметилформамид), имидазол, комнатная температура, 3 ч
, DMF (диметилформамид), имидазол, комнатная температура, 3 ч
b) Ас2О, Et3N, CH2Cl2, 4 ч
c) MeI, ацетон, К2СО3, комнатная температура
д) LiOH.H2O, EtOH, 18 ч, комнатная температура
е) n-Bu4NF, ТГФ, 36 ч
В соответствии с настоящим изобретением продукт химической реакции, содержащий (-) атропоизомер и (+) атропоизомер, может быть разделен для получения обогащенной фракции, предпочтительно без примеси в отношении одного из атропоизомеров. Желательно, чтобы фракция, содержащая требуемый атропоизомер, например (-) атропоизомер, не содержала бы существенных количеств другого атропоизомера, например (+) атропоизомера. Предпочтительно, предусматривается наличие двух фракций: первая фракция обогащена и предпочтительно чистая в отношении (-) атропоизомера, а вторая фракция обогащена и предпочтительно практически чистая в отношении (+) атропоизомера.
Конкретный метод разделения смеси атропоизомеров не имеет значения для данного изобретения. Может быть использован любой подходящий метод, известный специалисту в данной области, например образование ионных, диастереомерных солей (или связанных комплексов) с хиральными соединениями и разделение путем фракционной кристаллизации или другими методами, образование диастереомерных соединений с хиральными дериватирующими реагентами, разделение диастереомеров и конверсия в чистые атропоизомеры, прямое разделение атропоизомеров в хиральных условиях на множестве матриц, включая суперкритическую хроматографию и ферментный гидролиз (см., например, Stereochemistry of Carbon Compounds, supra; Lochmuller, C.H., (1975) J. Chromatogr., 113: (3) 283-302).
Например, диастереомерные соли могут быть получены посредством реакции энантиомерно чистых хиральных оснований, таких как, например, бруцин, квинин, эфедрин, стрихнин, α-метил-β-фенилэтиламин (амфетамин) и т.п. с соединениями изобретения, обладающими кислотными свойствами, такие как карбоновая кислота и сульфоновая кислота. Диастереомерные соли могут быть разделены фракционной кристаллизацией или ионной хроматографией. Для разделения соединений изобретения, проявляющих аминные свойства, к образованию диастереомерных солей может привести добавление хиральных карбоновых или сульфоновых кислот, таких как, например, камфорсульфоновая кислота, винная кислота, миндальная кислота или молочная кислота.
В качестве альтернативы рацемическая смесь соединений с целью разделения может быть включена в реакцию с одним энантиомером хирального соединения для образования диастереомерной пары. Например, диастереомерные соединения могут быть получены посредством реакции между соединениями изобретения с энантиомерно чистыми хиральными дериватирующими реагентами, такими как, например, ментиловые производные, с последующим разделением диастереомеров и гидролизом для получения свободного, атропоизомерически обогащенного соединения. Предпочтительный метод установления оптической чистоты предполагает получение хиральных сложных эфиров рацемической смеси, таких как ментиловый эфир или эфир Мошера, например, α-метокси-α-(трифторметил)фенил ацетат [Jacob III. (1982) J. Org. Chem. 47:4165], и анализ спектра ядерного магнитного резонанса (“ЯМР”) на наличие двух атропоизомерных диастереомеров.
Другой альтернативой является разделение рацемической смеси двух атропоизомеров посредством хроматографии с применением хиральной неподвижной фазы см., например, Chiral Liquid Chromatography; W.J.Lough, Ed. Chapman and Hall, New York, (1989); Okamoto, "Optical resolution of dihydropyridine enantiomers by high-performance liquid chromatography using phenylcarbamates of polysaccharides as a chiral stationary phase", J. of Chromatogr. 513:375-378, (1990). Атропоизомеры соединений изобретения могут быть разделены и выделены посредством хроматографии в хиральной неподвижной фазе, например, с использованием Chiralpak AD колонки, например, с использованием мобильной фазы, содержащей: (а) 2-пропанол; и (b) гексан, содержащий в малой пропорции (0,1-0,15%) либо трифторуксусную кислоту (“ТФК”), либо диэтиламин (“ДЭА”).
В качестве альтернативы первичная спиртовая группа в соединениях изобретения может быть этерифицирована хиральной карбоновой кислотой с образованием хиральных сложных эфиров, которые могут быть подвержены диастереоселективному гидролизу ферментом, например эстеразой.
Термин параметров атропоизомеров может быть выполнен любым подходящим методом, известным специалисту в данной области. Примеры подходящих методов включают ЯМР-спектроскопию, например протонный ЯМР или С13 ЯМР, инфракрасную спектроскопию, ультрафиолетовую спектроскопию, физические измерения, например установление температуры плавления, рентгеновская кристаллография и хиральная хроматография для энантиомерной чистоты. Как правило, атропоизомеры могут быть распознаны при помощи методов, используемых для распознавания других хиральных молекул с асимметричными атомами углерода, таких как оптическое вращение и циркулярный дихроизм.
Довольно неожиданно в соответствии с настоящим изобретением было установлено, что разделенные атропоизомеры стабильны. В результате обеспечиваются обогащенные соединения, предпочтительно практически без примеси в отношении требуемого атропоизомера, которые устойчивы к превращению в рацемическую смесь. Возможность обеспечить стабильные атропоизомеры может быть полезной. Например атропоизомер, который более активен в лечении некоторых состояний, может быть назначен в дозировке, существенно меньшей, чем дозировка рацемической смеси, имеющей такую же эффективность, например, наполовину меньшей. В качестве альтернативы введение атропоизомера в той же дозировке, что и рацемическая смесь, может оказаться более эффективным.
Некоторые соединения настоящего изобретения могут существовать в несольватированных формах так же, как и в сольватированных, включая гидратированные формы, такие как моногидрат, дигидрат, гемигидрат, тригидрат, тетрагидрат и подобные им. Продукты могут быть истинными сольватами, в то же время в других случаях они могут лишь содержать остатки растворителя или быть смесью сольвата и небольшого остаточного количества растворителя. Специалисты в данной области должны принять во внимание тот факт, что сольватированные формы равноценны несольватированным формам и включены в рамки настоящего изобретения. Предполагается, что некоторые соединения Формулы I могут существовать в двух таутомерных формах. Специалисты в данной области должны принять во внимание тот факт, что в случае, когда R1 является водородом, связанным с атомом азота, расположенным рядом с карбонильным атомом углерода, хинолиновое кольцо может существовать в енольной форме. Предполагается, что оба енольных таутомера соединений Формулы I включены в рамки настоящего изобретения.
Вещества, открывающие ВК-каналы, реализуют свои клеточные эффекты путем повышения вероятности открытия этих каналов [Gribkoff, V.K., и др., Neuroscientist, 7:166-177 (2001); Gribkoff, V.K. и др., Adv. Phannacol., 37:319-348 (1997); McKay, М.С., и др., J. NeurophysioL, 71: 1873-1882 (1994); и Olesen, S.-P., Exp. Opin. Invest. Drugs, 3: 1181-1188 (1994)]. Такой прирост в частоте открывания отдельных ВК-каналов суммарно приводит к гиперполяризации клеточных мембран, в особенности у деполяризованных клеток, вызванной значительным возрастанием ВК-опосредованной проводимости клетки. Гиперполяризация в свою очередь снижает возбудимость нервных и мышечных клеток и уменьшает вероятность открытия потенциалзависимых кальций-ионных каналов, эффективно снижая внутриклеточные концентрации этих потенциально вредоносных катионов.
Способность соединений по изобретению открывать ВК-каналы и увеличивать ВК-опосредованные потоки ионов калия из клетки наружу была определена в условиях фиксации потенциала путем определения их способности увеличивать поток, опосредованный клональными ВК млекопитающих (mSlo или hSlo), гетерологично экспрессированными на ооцитах Ксенопуса [Butler, А., и др., Science, 261: 221-224 (1993); Dworetzky, S.L, и др., Mol. Brain Res., 27: 189-193 (1994); Gribkoff, V.K., и др., Mol. Pharmacol., 50:206-217 (1996)]. Эти два ВК-канала представляют собой структурно практически одинаковые гомологичные белки, и, как доказано в наших исследованиях, фармакологически неразличимы. Для отделения ВК-потока от естественного (фонового, не-ВК) потока был использован ибериотоксин (IBTX) - специфический сильнодействующий токсин, блокирующий ВК-каналы [Galvez, А., и др., J. Biol. Chem., 265: 11083-11090 (1990)] в субмаксимальной концентрации 50-100 наномоль/литр (“нМ/л”). Относительный вклад ВК канал-опосредованного потока в общий поток наружу клетки был определен путем вычитания потока, оставшегося в присутствии IBTX (не-ВК потока), из потоков, полученных в других экспериментальных условиях (контроль, лекарство и промывание). [Gribkoff, V.K. и др., Mol. Pharmacol., 50:206-217 (1996)]. Было установлено, что при исследуемых концентрациях соединения оказывают незначительное действие на не-ВК естественные потоки в ооцитах. Все соединения были протестированы в 5-10 ооцитах при показанной концентрации 10 микромоль/литр (“µМ”); эффект выбранных соединений изобретения на ВК-поток выражался в процентах от контрольного IBTX-чувствительного потока на единичном трансмембранном потенциале (+140 мВ) и приведен в Таблице 1. Записи выполнены с использованием стандартной двухэлектродной методики фиксации потенциала [Stuhmer, W., и др., Methods in Enzymology, Vol. 207: 319-339 (1992)]; применялась пошаговая деполяризация продолжительностью 500-750 миллисекунд (“мс”) от поддерживаемого потенциала -60 мВ вплоть до заключительного потенциала +140 мВ с шагом 20 мВ. В Таблице 2 приведены данные поток-потенциал (I-V) для двух соединений изобретения, примененных на ооцитах в концентрации 10 µМ. Экспериментальная среда (модифицированный раствор Барта) состояла из следующих веществ (концентрация указана в ммоль/литр): NaCl (88), NaHCO3 (2.4), KCl (1.0), HEPES (10), MgSO4 (0.82), Са(NO3)2 (0.33), CaCl2 (0.41); pH 7.5.
Соединения настоящего изобретения применимы для лечения состояний, являющихся результатом дисфункции поляризации и проводимости клеточной мембраны и предпочтительно показаны при ишемии, инсульте, судорогах, эпилепсии, астме, синдроме раздраженной кишки, мигрени, черепно-мозговых травмах, повреждениях спинного мозга, сексуальных расстройствах, недержании мочи и в особенности при эректильной дисфункции у мужчин, а также при других расстройствах, связанных с работой ВК-каналов. Таким образом, с одной стороны настоящее изобретение обеспечивает метод лечения или профилактики состояний, связанных с открытием калиевых каналов, у страдающих от этого пациентов, который включает введение пациенту терапевтически эффективного количества соединения Формулы I или фармацевтически приемлемой соли, его сольвата или пролекарства. С другой стороны, настоящее изобретение обеспечивает способ лечения или предупреждения расстройств, которые опосредованы открытием кальций-активируемых калиевых каналов высокой проводимости, который включает введение пациенту терапевтически эффективного количества соединения Формулы I или фармацевтически приемлемой соли, его сольвата или пролекарства. Соединения Формулы I предпочтительно показаны при ишемии, инсульте, судорогах, астме, синдроме раздраженной кишки, мигрени, черепно-мозговых травмах, недержании мочи и сексуальных дисфункциях как у мужчин (эректильной дисфункции, например, обусловленной сахарным диабетом, повреждением спинного мозга, радикальной простатэктомией, психогенной этиологии или любой другой природы), так и у женщин за счет улучшения кровоснабжения гениталий, в особенности кавернозных тел, и при других расстройствах, связанных с работой ВК-каналов.
С другой стороны, данное изобретение обеспечивает фармацевтические композиции, состоящие по крайней мере из одного соединения Формулы I и носителя.
Фармацевтические композиции включают подходящие лекарственные формы для перорального, парентерального (включая подкожное, внутримышечное, внутрикожное и внутривенное), внутрибронхиального и интраназального применения. Таким образом, если применяется твердый носитель, препарат может быть таблетирован, помещен в твердую желатиновую капсулу, быть в виде порошка или пилюли, или в виде разной формы таблеток. Твердый носитель может содержать обычные наполнители, такие как связывающие агенты, инертные наполнители, смазывающие вещества, дезинтегрирующие агенты, увлажняющие агенты и т.п. Таблетка может, при необходимости, быть покрыта оболочкой по обычной методике. Если применяется жидкий носитель, препарат может быть в виде сиропа, эмульсии, мягких желатиновых капсул, стерильного раствора для инъекций, водной или неводной жидкой суспензии или может быть в виде сухого вещества для растворения в воде или в другом подходящем растворителе перед использованием. Жидкие препараты могут содержать обычные добавки, такие как суспендирующие агенты, эмульсификаторы, увлажняющие агенты, неводные растворители (включая пищевые масла), консерванты, а также ароматизаторы и красители. Для парентерального введения растворитель обычно состоит из стерильной воды, по крайней мере, большей частью, хотя растворы солей, глюкозы и подобные им также могут применяться. Также могут использоваться суспензии для инъекций, в этом случае могут применяться обычные суспендирующие агенты. Обычные консерванты, буферные агенты и подобные им также могут быть добавлены в лекарственные формы для парентерального применения. Особенно эффективно применение соединений Формулы I непосредственно в формах для парентерального введения. Фармацевтические композиции приготавливаются по традиционным методикам, подходящим для нужного препарата, содержащего подходящее количество активного вещества, которое является соединением Формулы I согласно данному изобретению.
Доза соединений Формулы I, необходимая для достижения терапевтического эффекта, будет зависеть не только от таких факторов, как возраст, вес, пол пациента и способ введения, но также и от требуемого уровня активности калиевых каналов и эффективности конкретного соединения, применяемого в отношении конкретного проявления болезни. Также предполагается, что конкретное соединение может быть назначено в дозированной форме, которая будет подбираться специалистом в этой области в зависимости от уровня активности процесса. Конкретную дозу (и кратность приема) определяет лечащий врач, который может скорректировать дозу путем титрования в конкретных условиях для получения желаемого терапевтического эффекта.
Подходящей дозой соединения Формулы I или его фармацевтической композиции для млекопитающих, включая человека, страдающего или предположительно страдающего от любого из указанных здесь состояний, является количество активного вещества от приблизительно 0,01 микрограмм на килограмм (мкг/кг) до 50 миллиграмм на килограмм (мг/кг) массы тела и предпочтительно от примерно 0,1 мкг/кг до 5 мг/кг массы тела при приеме внутрь. Для парентерального введения доза может быть в пределах от 0,1 мкг/кг до 1 мг/кг массы тела при внутривенном введении. Активное вещество предпочтительно назначают в одинаковых дозах от 1 до 4 раз в день. Однако, обычно назначают маленькие дозы, которые затем постепенно повышают до того, как будет установлена оптимальная доза.
Соединения настоящего изобретения могут применяться отдельно или в комбинации с другими подходящими терапевтическими агентами, используемыми в лечении дисфункций поляризации и проводимости клеточных мембран, например сексуальной дисфункции, такими как ингибиторы циклического гуаминмонофосфата, фосфордиэстеразы (“цГМФ, ФДЭ”) и в особенности ингибиторы цГМФ ФДЭ V типа, такие как силденафил. Характерными терапевтическими агентами являются следующие ингибиторы ФДЭ V типа: имидазоквиназолины (см. WO 98/08848), карбазолы (см. WO 97/03675, WO 97/03985 и WO 95/19978), имидазопуриноны (см. WO 97/19947), бензимидазолы (см. WO 97/24334), пиразолохинолины (см. US 5,488,055), производные антраниловой кислоты (см. WO 95/18097), конденсированные гетероциклы (см. WO 98/07430) и тиенопиримидины (см. DE 19632423). Гидрохлорид алосетрона может комбинироваться с соединениями настоящего изобретения для лечения синдрома раздраженной кишки (см., например, US 5,360,800 и 6,284,770).
Вышеуказанные терапевтические агенты, при использовании в комбинации с соединениями настоящего изобретения, могут применяться в дозировках, указанных в the Physician′c Desk Reference (PDR) или в иных, указанных специалистом в данной области.
Однако необходимо понимать, что фактически дозировка применяемого соединения будет определяться лечащим врачом в зависимости от определяющих обстоятельств, таких как причина назначения препарата, выбранное соединение, выбранный путь введения, возраст, вес, ответ пациента на лечение, а также тяжесть симптомов болезни.
ПРИМЕРЫ
Следующие примеры даны для иллюстрации и не должны быть истолкованы как ограниченные объемом формулы изобретения, которая следует ниже.
В нижеследующих примерах все температуры даны в градусах стоградусной шкалы. Точки плавления, измеренные на капиллярном приборе для измерения температур плавления Gallenkamp, являются неисправленными. Спектры протонного магнитного резонанса (1Н ЯМР) были записаны на приборе Bruker AC 300 или 500 МегаГерцовом ("МГц") спектрометре. Все спектры устанавливались в указанных растворителях и химические сдвиги отсчитывались в диапазоне 8 единиц в нижних полях от внутреннего стандарта тетраметилсилана (ТМС), а константы спин-спинового взаимодействия отсчитывались в Герцах (Гц). Примеры расщепления обозначены как следует далее: с, синглет; д, дублет; т, триплет; кв, квартет; м, мультиплет; уш, уширенный пик; дд, дублет дублетов; ушд, уширенный дублет; дт, дублет триплетов; ушс, уширенный синглет; дкв, дублет квартетов. Инфракрасные (ИК) спектры с использованием бромида калия (KBr) были записаны на спектрометре Perkin Elmer 781 в диапазоне от 4000 до 400 см1, калиброваны по поглощению полистирольной пленки при 1601 см1 и записаны в обратных сантиметрах (см1). Масс-спектры низкого разрешения (МС) и истинный молекулярный вес (МН+) или (М-Н)- определяли на приборе Finnigan TSQ 7000. ГЖХ-МС-анализ был проведен на приборе Shimadzu с использованием YMC CIS колонки (4.6×50 мм), применяя 4 или 8 мин линейный градиент при концентрациях от 0 до 100% растворителя В в А (растворитель А: 10% метанол, 90% вода, 0.1% ТФУК; растворитель В: 90% метанол, 10% вода, 0.1% ТФУК) с УФ-детектором при 220 нм. Результаты элементного анализа отсчитывались в виде весовых процентов. Если особо не указывается в специальных включениях, R2 и R4 представляют собой Н в описательной части примеров.
Пример 1
4-(5-Хлор-2-гидроксифенил)-3-(2-гидроксиэтил)-6(трифторометил)-2(1Н)-хинолинон
Вышеуказанное соединение получают как описано ниже.
Стадия А - 3-(2-Гидроксиэтил)-4-гидрокси-6-хлоркумарин
К раствору γ-бутиролактона (15.5 г, 178.0 ммоль) в ТГФ (100 мл) при -78°С добавляли 1.0 М раствор L1HMDS (356 мл, 356 ммоль) в ТГФ и полученную смесь перемешивали при -78°С в течение 1.5 часов. Добавляли раствор метилового эфира 5-хлорсалициловой кислоты (16.6 г, 98% чистота, 89.0 ммоль) в ТГФ (95 мл). После перемешивания в течение 1 часа при 0°С смесь нагревали до комнатной температуры в течение ночи до полного завершения реакции. После охлаждения до 0°С медленно добавляли конц. HCl (12 N, 150 мл) до достижения значения рН 1. Реакционную смесь затем перемешивали до тех пор, пока ВЭЖХ анализ не указал на отсутствие кето-эфирных промежуточных соединений. К смеси добавляли 400 мл CH2Cl2 и 300 мл H2O; органическую фазу отделяли, а водный слой экстрагировали CH2Cl2 (100 мл). Органические слои соединяли и высушивали над безводным Na2SO4, растворитель затем удаляли под уменьшенным давлением, что приводит к получению твердого соединения. К раствору твердого соединения ТГФ (290 мл) добавляли гептан (165 мл) для того, чтобы продукт закристаллизовался. После охлаждения до 0-5°С в течение примерно 3 часов продукт выделяли фильтрацией и промывали гептаном. После высушивания в вакууме суммарно было получено 13.9 г (66% выход) заглавного продукта в виде бесцветных кристаллов, т.пл. 185-186°С; МС м/е 240;
1H ЯМР (ДМСО-d6, 300 МГц) δ 7.84 (д, 1Н, J=2.4 Гц), 7.61 (дд, 1Н, J=2.4, 8.8 Гц), 7.38 (д, 1Н, J=8.8 Гц), 3.56 (т, 2Н, J=6.6 Гц), 2.73 (т, 2Н, J=6.6 Гц); 13С ЯМР (ДМСО-d6, 75 МГц) δ 162.6, 159.9, 150.5, 131.4, 127.9, 122.4,118.2, 117.8, 103.2, 59.4, 27.6; ИК (см-1) 3247.2, 2945.1, 2458.6, 1664.9, 1623.9, 1572.7, 1311.5, 1378.1, 1070.8, 825.0.
Расчет для С11Н9O4Cl: С, 54.90; Н, 3.77; Cl, 14.73.
Найдено: С, 54.79; Н, 3.70; Cl, 14.76.
Стадия В - 2,3-Дигидро-8-хлор-4Н-фуробензопиран-4-он
К раствору 3-(2-гидроксиэтил)-4-гидрокси-6-хлоркумарина (Пример 1) (8 г, 33.3 ммоль) в толуоле (360 мл) при комнатной температуре добавляли п-ТСК (0.95 г, 5.0 ммоль) и полученный раствор кипятили с обратным холодильником, удаляя воду с использованием насадки Дина-Старка. Реакционную смесь охлаждали до комнатной температуры и дважды промывали насыщенным водным раствором бикарбоната натрия. Толуол отгоняли при атмосферном давлении до остаточного объема 32 мл. После охлаждения до 70°С продукт начинает кристаллизоваться. Суспензию с кристаллами выдерживали при 55-65°С в течение 30 минут, затем охлаждали до 0-5°С. Продукт выделяли фильтрацией, промывали холодным толуолом и высушивали в вакууме. Суммарно было получено 5.5 г (74% выход) заглавного соединения в виде бесцветных кристаллов, т.пл. 144-146°С; МС м/е 223;
1H ЯМР (CDCl3, 300 МГц) δ 7.58 (д, 1Н, J=2.5 Гц), 7.49 (дд. 1Н, J=2.3, 8.8 Гц), 7.30 (д, 1Н, J=8.9 Гц), 4.90 (т, 2Н, J=9.3 Гц), 3.21 (т, 2Н, J=9.5 Гц); 13С ЯМР (CDCl3, 75 М Гц) δ 166.4, 160.3, 153.4, 132.6, 129.6, 122.4, 118.6, 113.8, 103.6, 74.9, 27.1; ИК (см-1) 3073.1, 2975.8, 1721.2, 1644.4, 1490.8, 1403.7, 1270.6, 1111.8, 1040.1.
Расчет для С11Н7O3Cl: С, 59.35; Н, 3.17; Cl, 15.92.
Найдено: С, 59.13; Н, 3.16; Cl, 15.93.
Стадия С - 4-(4′-Трифторометилфенилкарбоксамид)-5-(2-гидрокси-5-хлор)-2,3-дигидрофуран
К раствору 2,3-дигидро-8-хлор-4Н-фуробензопиран-4-она (Пример 2) (1.02 г, 4.58 ммоль) и 4-(трифторометил)анилина (0.74 г, 4.58 ммоль) в ТГФ (50 мл) при -15°С добавляли LiHMDS (10.5 мл, 10.5 ммоль, l.0 M раствор в ТГФ). Прозрачный красный раствор перемешивали при -15°С М до тех пор, пока ВЭЖХ анализ не указал, что осталось <1% исходного материала (приблизительно 30 минут). Реакционную смесь резко охлаждали прибавлением водного раствора NaH2PO4 (50 мл, 10 вес.% в Н2О). После прибавления трет-бутилметилового эфира (25 мл) слои разделяли и жирную органическую фазу последовательно промывали NaH2PO4 (50 мл, 10 вес.% в Н2О) и насыщенным солевым раствором. После высушивания Na2SO4 раствор концентрировали и получали заглавное соединение в виде прозрачного оранжевого масла (1.76 г, 100% выход), которое закристаллизовывалось при охлаждении. Прибавление дихлорметана (20 мл) дало белые кристаллы, которые выделяли фильтрацией, промывали дихлорметаном (10 мл) и высушивали с получением 1,6 г заглавного соединения (90% выход), т.пл. 180-180.5°С; МС м/е 384 (М+Н)+;
1Н ЯМР (ДМСО-d6, 300 МГц) δ 9.76 (с, 1Н), 9.34 (с, 1Н), 7.76 (д, 2Н, J=8.5 Гц), 7.60 (д, 2Н, J=8.7 Гц), 7.26 (с, 1Н), 7.24 (дд, 1Н, J=2.2, 7.0 Гц), 6.83 (дд, 1Н, J=2.4, 7.1), 4.52 (т, 2Н, J=9.6 Гц), 3.16 (т, 2Н, J=9.6 Гц); 13С ЯМР (ДМСО-d6, 75 МГц) δ 165.5, 159.7, 155.9, 144.7, 132.0, 131.3, 127.3, 123.7, 121.7, 121.2, 119.5, 110.1, 71.5, 32.9; ИК (см-1) 3303.6, 2950.2, 1654.6, 1608.5, 1531.7, 1408.8, 1326.9, 1116.9, 1065.7, 840.4.
Стадия D - 4-(5-Хлор-2-гидроксифенил)-3-(2-гидроксиэтил)-6-(трифторометил)-2(1Н)-хинолинон
Раствор 4-(4′-трифторометилфенилкарбоксамид)-5-(2-гидрокси-5-хлор)-2,3-дигидрофурана, полученного в примере 3 (1.76 г, 4.58 ммоль), в МеОН (500 мл) продували азотом и облучали 450 Вт лампой Hanovia при 30-40°С до тех пор, пока ВЭЖХ анализ не указал, что осталось <1% соединения (Пример 3). Затем упаривали в вакууме и полученное масло растворяли в дихлорметане (50 мл). Кристаллы образовывались после перемешивания в течение одного часа при комнатной температуре. После охлаждения суспензии до 0°С кристаллы выделяли фильтрацией и высушивали. Суммарно было получено 5.5 г (74% выход) заглавного соединения. Суммарно было получено 0.54 г (30% выход) заглавного соединения в виде твердых кристаллов с чистотой согласно ВЭЖХ 97%. т.пл. 253-255°С; МС м/е 384 (М+Н)+.
1H ЯМР (ДМСО-d6, 300 МГц) δ 12.27 (с, 1Н), 9.91 (с, 1Н), 7.79 (д, 1Н, J=8.3 Гц), 7.53 (д, 1Н, J=8.5 Гц), 7.42 (дд, 1Н, J=2.4, 8.6 Гц), 7.26 (д, 1Н, J=2.4 Гц), 7.08 (с, 1Н), 7.06 (д, 1Н, J=8.9 Гц), 4.60 (м, 1Н), 3.44 (м, 2Н), 2.50 (м, 2Н); 13С ЯМР (ДМСО-d6, 75 МГц) δ 163.7, 155.1, 145.9, 141.7, 132.6, 131.5, 131.3, 127.8, 127.4, 125.5, 124.5, 123.5, 121.0, 119.3, 117.9, 60.7, 33.9.
Соединение, полученное в Примере 1, представляет собой рацемическую смесь (-) атропоизомера и (+)атропоизомера, которые структурно представлены на фиг.1. Соединение из Примера 1 стало объектом хиральной хроматографии. Хроматограмма рацемической смеси, а также индивидуальных атропоизомеров представлена на фиг.1, 2, 3 и 4, соответственно.
ПРИМЕР 2
4-(5-Хлор-2-гидроксифенил)-1-метил-3-(2-гидроксиэтил)-6-трифторометил)-2(1Н)-хинолинон
Вышеуказанное соединение получают как описано ниже.
Стадия А: - 2-{3-[2-(трет-бутилдифенил-силанилокси)-этил]-2-оксо-6-трифторометил-1,2-дигидро-хинолин-4-ил}-4-хлорфениловый эфир уксусной кислоты
К раствору соединения из Примера 1 (5.00 г, 13.0 ммоль) в сухом ДМФА (100 мл) добавляли трет-бутилдифенилсилил хлорид (10.74 г, 39.1 ммоль) и имидазол (2.66 г, 39.1 ммоль) при температуре окружающей среды под аргоновой подушкой. Реакционную смесь перемешивали в течение 3 часов и разбавляли этилацетатом (300 мл). Органический слой промывали водой (2×100 мл), насыщенным раствором хлорида натрия (100 мл) и высушивали. Растворитель затем упаривали и получали силиловый эфир (6.61 г, 10.63 ммоль, 81%). К раствору силилового эфира в сухом дихлорметане (200 мл) добавляли Ас2О (5.43 г, 53.2 ммоль) и Et3N (5.38 г, 53.2 ммоль) при комнатной температуре под аргоновой подушкой. Реакционную смесь перемешивали в течение 4 часов и разбавляли этилацетатом (200 мл). Органический слой промывали водой (2×100 мл), насыщенным раствором хлорида натрия (100 мл) и высушивали. Растворитель затем вновь упаривали. Очистка проводилась колоночной хроматографией (SiO2) с использованием в качестве элюента гексана и этилацетата (от 95:5 до 80:20), что дало соединение Стадии А (6.82 г, 96%, время удержания, "Rt"=2.3 минуты при C18 жидкостной хроматографии, "C18 LC" колонка) в виде белого твердого соединения. MS [М+Н]=664. 1H ЯМР (CD3OD) δ 7.74 (д, J=7.6 Гц, 1Н), 7.60 (д, J=6.2 Гц, 1Н), 7.52 (м, 5Н), 7.37 (м, 2Н), 7.31 (м, 5Н), 7.23 (с, 1Н), 7.17 (с, 1Н), 3.94 (м, 1Н), 3.80 (м, 1Н), 2.89 (м, 1Н), 2.72 (м, 1Н), 1.76 (с, 3Н), 0.97 (с, 9Н).
Стадия В: 2-{3-[2-(трет-бутилдифенил-силанилокси)-этил]-1-метил-2-оксо-6-трифторметил-1,2-дигидро-хинолин-4-ил}-4-хлорфениловый эфир уксусной кислоты
Суспензию продукта стадии А (6.82 г, 10.3 ммоль), метилйодида (4.37 г, 30.8 ммоль) и К2СО3 (3.05 г, 30.8 ммоль) в ацетоне (200 мл) при температуре окружающей среды перемешивали в течение 18 часов. В конце разбавляли этилацетатом (200 мл), промывали водой (100 мл), насыщенным раствором хлорида натрия (100 мл) и высушивали. Хроматография градиентом элюирования (SiO2) гексаном, этилацетатом (90:10 до 70:30) приводила к получению соединения Стадии В в качестве основного продукта (5.52 г, 8.1 ммоль, 79%, Rt=2.3 минут C18 LC) в виде белого твердого вещества. MS [M+H]=678. 1H ЯМР (CD3OD) δ 7.82 (д, J=8.1 Гц, 1Н), 7.73 (д, J=8.9 Гц, 1Н), 7.52 (д, J=9.2 Гц, 1Н), 7.48 (м, 4Н), 7.34 (м, 3Н), 7.21 (м, 6Н), 3.91 (м, 1Н), 3.76 (м, 4Н), 2.88 (м, 1Н), 2.70 (м, 1Н), 1.71 (с, 3Н), 0.94 (с, 9Н). Побочный продукт характеризовался как O-метильное производное со следующими характеристиками. MS [M+H]=678. 1H ЯМР (CD3OD) δ 7.82 (д, J=8.1 Гц, 1Н), 7.95 (д, J=5.1 Гц, 1Н), 7.52 (д, J=9.2 Гц, 1Н), 7.48 (м, 4Н), 7.34 (м, 3Н), 7.21 (м, 6Н), 3.91 (м, 1Н), 3.76 (м, 4Н), 2.88 (м, 1Н), 2.70 (м, 1Н), 1.71 (с, 3Н), 0.94 (с, 9Н). Соотношение N-метильного производного к O-метильному производному составляло приблизительно 9:1.
Стадия С: 4-(5-Хлор-2-гидроксифенил)-1-метил-3-(2-гидроксиэтил)-6-трифторметил)-2(1Н)-хинолинон
Эфир ацетилсилила (760 мг, 1.12 ммоль) перемешивали с LiOKH2O (320 мг) в EtOH (10 мл) в течение 18 часов при температуре окружающей среды. В конце летучие растворители удаляли в вакууме. Остаток растворяли в ТГФ (5 мл) и обрабатывали тера-n-бутиламмонийфторидом 91М в ТГФ, 3 эквивалента. После перемешивания в течение 18 часов добавляли еще 3 эквивалента и перемешивали еще 18 часов. ТГФ упаривали, разделяли между EtOAc и водой. Органический слой сушили (Na2SO4), упаривали в вакууме. Остаток очищали колоночной хроматографией на силикагеле с помощью EtOAc:СН2Cl2 (3:2) в качестве элюента. Фракции, содержащие нужное соединение, объединяли и упаривали в вакууме с получением 290 мг (выход 65%) нужного продукта. Перекристаллизация из EtOAc и гексана приводила к получению 152 мг N-метильного производного. 1H ЯМР (500 МГц, ДМСО-d6): δ 2.56 (2Н, м), 3.40 (2Н, м), 3.75 (3Н, с), 4.57 (1Н, м), 7.05 (1Н, д, J=8.8). 7.14 (1Н, с), 7.25 (1Н, д, J=2.6), 7.41 (1Н, дд, J=8.7, 2.7), 7.77 (1Н, д, J=8.8), 7.89 (1Н, дд, J=8.8, 2.0), 9.91 (1Н, с); MS м/е 398 (МН+). Соединение Примера 2 подвергали хиральной хроматографии. Хроматографии рацемической смеси и индивидуальных атропоизомеров представлены на фиг.5, 6 и 7 соответственно.
Пример 3
Разделение атропоизомеров Примера 1
(-)4-(5-Хлор-2-гидрокси-фенил)-3-(2-гидрокси-этил)-6-трифторметил-1H-хинолин2-он
(+)4-(5-Хлор-2-гидрокси-фенил)-3-(2-гидрокси-этил)-6-трифторметил-1Н-хинолин2-он
Раствор 1 (~50 мг) в изо-PrOH: гексан (1:1,2 мл) переносили четырьмя порциями на колонку ChiralPak AD, 21×250 мм, с размером частиц 10 мкм. Элюирование изо-PrOH: гексан (1:19) осуществляли при скорости потока 10 мл/мин. Детектор UVmax при 234 нм применяли. Фракция, содержащая первый по ходу изомер, изомер А, (tR=46.1 мин) при упаривании приводила к получению 20 мг, тогда как фракция, содержащая последний пик (tR=48.3 мин) при упаривании приводила к получению 21 мг изомера В. Хроматограмма рацемата показана на фиг.2.
Характеристика Атропоизомера А (-): 1H ЯМР и 13С ЯМР спектры отличны от спектров, приведенных для рацемата Примера 1. Оптическое вращение изомера А определяли в МеОН, [α]22 D (meOH)=-8.8°.
Характеристика Атропоизомера В (+): 1H ЯМР и 13С ЯМР спектры отличны от спектров, приведенных для рацемата Примера 1. Оптическое вращение изомера В определяли в МеОН, [α]22 D (meOH)=+9.6°.
Разделенные атропоизомеры подвергали ВЭЖХ с обратной фазой, как описано выше, относительно рацемата. Хроматограммы для такого атропоизомера представлены на фиг.3 и 4.
Пример 4
Стабильность Атропоизомеров Примера 3
Исследования хиральной ВЭЖХ: Этанольные растворы (~3 мг/мл) двух атропоизомеров Примера 1 хранили при комнатной температуре и исследовали с помощью хиральной ВЭЖХ, как описано выше, на колонке Chiralpak AD для рацемизации. Два соединения, как было показано, являются стабильными даже через месяц при температуре окружающей среды. Заметной рацемизации не наблюдалось.
Пример 5
Разделение Атропоизомеров из Примера 2
(-)4-(5-Хлор-2-гидрокси-фенил)4-метил-3-(2-гидрокси-этил)-6-трифторметил-1Н-хинолин2-он
(+)4-(5-Хлор-2-гидрокси-фенил)-1-метил-3-(2-гидрокси-этил)-6-трифторметил-1H-хинолин2-он
Рацемат разделяли, как описано в Примере 3, на колонке Chiralpak AD, 4.6×250 мм, 10 мкм с помощью 2-пропанол: гексантрифторуксусной кислоты (1600:399:1). Первый по ходу изомер (tR=8.5 мин) представлял собой изомер А и более медленный (tR=19.0 мин) изомер В. Хроматограмма рацемата показана на фиг.5.
Характеристика Изомера A (-): 1Н ЯМР спектры отличны от спектров, приведенных для рацемата Примера 2. Оптическое вращение изомера А определяли в МеОН, [α]22 D (МеОН)=-6.9°.
Характеристика Изомера В (+):1H ЯМР спектры отличны от спектров, приведенных для рацемата Примера 2. Оптическое вращение изомера В определяли в МеОН, [α]22 D (МеОН)=+5.0°.
Разделенные атропоизомеры подвергали ВЭЖХ с обратной фазой, как описано выше, относительно рацемата. Хроматограммы для такого атропоизомера представлены на фиг.6 и 7.
Пример 6
Моторика кишки в ответ на стресс, вызванный воздушной струей
Предполагается, что стрессорное влияние окружающей среды играет роль в развитии синдрома раздраженной кишки и других состояний, сопровождающихся нарушением кишечной моторики. На животных моделях было показано, что стрессорное влияние окружающей среды вызывает усиление дефекации, вероятно, как результат усиления двигательной активности кишки. Основываясь на этом, мы проверили гипотезу, что вещества, открывающие maxi-K+ калиевые каналы, настоящего изобретения могут быть эффективны в снижении стресс-индуцированного ускорения кишечной моторики, отмечаемого по выделению каловых комков.
Крысы были разделены случайным образом на 2 группы, которым вводили либо растворитель (полиэтиленгликоль в дозе 400 грамм на моль, 1 мл/кг, интраперитонеально), либо соединение по Примеру 1 в виде рацемата (20 мг/кг). Через 1,5 часа после процедуры каждая крыса была частично иммобилизована в плексиглазовом контейнере и подвергнута стрессорному воздействию струей воздуха. Это осуществлялось путем обдувания каждого из частично иммобилизованных животных струей воздуха в течение 2 минут 3-4 раза с интервалом 8-10 минут. Выделяющийся кал собирали и записывали его сырой вес. Данные были проанализированы путем вычисления среднего веса кала в каждой группе и сравнения полученных средних значений с использованием статистического теста (непарного, двустороннего критерия t; p<0,05).
Средний вес выделенного кала в группе крыс, которым вводили растворитель, составил 0,37 г, а в группе крыс, которым вводили исследуемое соединение 0,03 г. У большинства животных первой группы (9 из 12) было отмечено изменение дефекации на воздействие воздушной струей. Напротив, только у 3 из 11 животных, которым вводили исследуемое соединение, наблюдалась подобная реакция. Средний вес выделенного кала в этой группе был значительно меньше, чем в группе крыс, которым вводили растворитель.
Было установлено, что соединения настоящего изобретения могут значительно ослаблять стресс-индуцированную кишечную моторику крыс.
Результаты вышеизложенных биологических исследований доказывают, что соединения данного изобретения могут быть действенными агентами, открывающими кальций-активируемые калиевые каналы высокой проводимости (Maxi-K или ВК-каналы). Кроме того, было установлено, что атропоизомеры обладают различной активностью.
Хотя данное изобретение описывает конкретные обстоятельства, специалисту в данной области должно быть понятно, что другие вытекающие из них обстоятельства также включены в рамки данной заявки. Например, возможные схемы реакций получения атропоизомеров, отличные от описанных в заявке. Также включено в рамки изобретения применение заместителей, равноценных указанным здесь, например, R, R1, R2, R3, R4, R5. Более того, все документы, цитированные в этой заявке, например патенты и публикации, объединены ссылками.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 1,3,4-ОКСАДИАЗОЛОНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 1999 |
|
RU2202548C2 |
| ФОСФАТНЫЕ ПРОИЗВОДНЫЕ ФТОРОКСИНДОЛОВ И СПОСОБ ЛЕЧЕНИЯ С ИХ ИСПОЛЬЗОВАНИЕМ | 2003 |
|
RU2312857C2 |
| ПРОИЗВОДНЫЕ 3-ЗАМЕЩЕННОГО 4-АРИЛХИНОЛИН-2-ОНА В КАЧЕСТВЕ МОДУЛЯТОРОВ КАЛИЕВЫХ КАНАЛОВ | 1999 |
|
RU2240998C2 |
| ПИРИДОПИРИМИДИНОНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРА N-МЕТИЛ-D-АСПАРТАТА | 2016 |
|
RU2717665C2 |
| СТЕРЕОИЗОМЕРНО ОБОГАЩЕННЫЕ 3-АМИНОКАРБОНИЛЬНЫЕ БИЦИКЛОГЕПТЕНОВЫЕ ПИРИМИДИНДИАМИНЫ И ИХ ПРИМЕНЕНИЯ | 2005 |
|
RU2416604C2 |
| КОНФОРМАЦИОННО ОГРАНИЧЕННЫЕ ИНГИБИТОРЫ PI3K И mTOR | 2014 |
|
RU2669696C2 |
| СОЕДИНЕНИЯ, КОТОРЫЕ УСИЛИВАЮТ РЕЦЕПТОР ГЛУТАМАТА, И ИХ ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2005 |
|
RU2403242C2 |
| 3-ЗАМЕЩЕННЫЕ ОКСИНДОЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ КАЛИЙНЫХ КАНАЛОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ | 1996 |
|
RU2165925C2 |
| ДИФЕНИЛЬНЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 1997 |
|
RU2175319C2 |
| МОДУЛЯТОРЫ СВЯЗАННЫХ С MAS РЕЦЕПТОРОВ G-БЕЛКА X4 И СВЯЗАННЫЕ С НИМИ ПРОДУКТЫ И СПОСОБЫ | 2020 |
|
RU2815715C2 |
Изобретение относится к новым атропоизомерам формулы
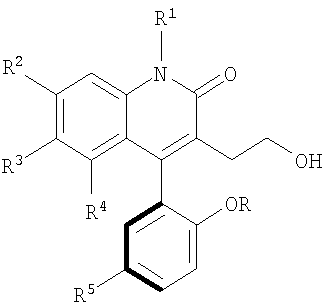
в которой R и R1 каждый независимо представляет собой водород или метил; R2, R3 и R4 каждый независимо представляет собой водород, или трифторметил, при условии, что R2, R3 и R4 все не представляют собой водород; и R5 представляет собой бром, хлор; или к его нетоксичной фармацевтически приемлемой соли, сольвату. Изобретение также относится к фармацевтической композиции, а также к способу лечения. Технический результат - получение новых биологически активных соединений, обладающих свойствами, открывающими кальций активируемые калиевые каналы высокой проводимости. 4 н. и 8 з.п. ф-лы, 2 табл., 7 ил.
1. Атропоизомер формулы
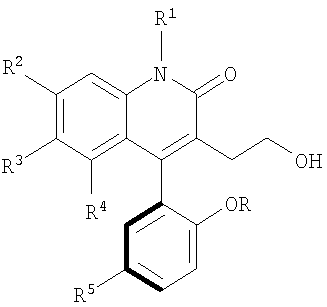
в которой
R и R1 каждый независимо представляет собой водород или метил;
R2, R3 и R4 каждый независимо представляет собой водород или трифторметил, при условии, что R2, R3, и R4 все не представляют собой водород; и
R5 представляет собой бром, хлор; или его нетоксичная фармацевтически приемлемая соль, сольват.
2. Атропоизомер по п.1, по существу свободный от его соответствующего атропоизомера формулы
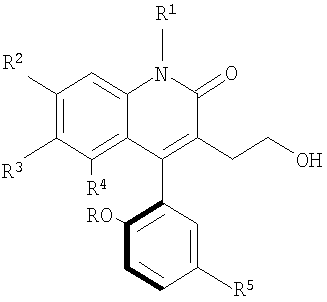
в котором R, R1, R2, R3, R4 и R5 определены в п.1.
3. Атропоизомер по п.1, в котором R1 представляет собой водород.
4. Атропоизомер по п.1, в котором R1 представляет собой метил.
5. Атропоизомер по п.1, который является стабильным по крайней мере 1 день.
6. Атропоизомер формулы
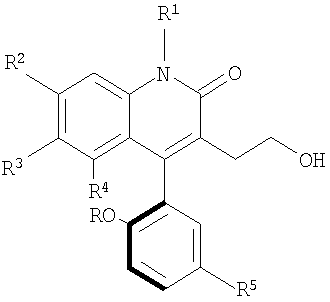
в котором
R и R1 каждый независимо представляет собой водород или метил;
R2, R3 и R4 каждый независимо представляет собой водород, или трифторметил, при условии, что R2, R3 и R4 все не представляют собой водород; и
R5 представляет собой бром, хлор; или его нетоксичная фармацевтически приемлемая соль, сольват.
7. Атропоизомер по п.6, по существу свободный от его соответствующего атропоизомера формулы
в котором R, R1, R2, R3, R4 и R5 определены в п.6.
8. Атропоизомер по п.6, в котором R1 представляет собой водород.
9. Атропоизомер по п.6, в котором R1 представляет собой метил.
10. Атропоизомер по п.6, который является стабильным, по крайней мере, 1 день.
11. Фармацевтическая композиция, обладающая свойствами, открывающими кальций-активируемые калиевые каналы высокой проводимости, включающая соединение по п.1 или 6, и фармацевтически приемлемый носитель.
12. Способ лечения состояния, связанного с открытием активируемых кальцием калиевых каналов высокой проводимости у млекопитающего, который включает введение млекопитающему терапевтически эффективного количества соединения по п.1 или 6.
| US 6184231 В1, 06.02.2001 | |||
| US 6353119 В1, 05.03.2002 | |||
| ДИЭТИЛАМИНОЭТИЛАМИДА 1-ПРОПИЛ-2-ОКСО-4-ГИДРОКСИХИНОЛИН-3-КАРБОНОВОЙ КИСЛОТЫ ГИДРОХЛОРИД, ПРОЯВЛЯЮЩИЙ АНЕСТЕЗИРУЮЩУЮ, ПРОТИВОАРИТМИЧЕСКУЮ, АНТИОКСИДАНТНУЮ, АНТИМИКРОБНУЮ И ФУНГИЦИДНУЮ АКТИВНОСТЬ | 1990 |
|
RU1774624C |
| RU 95113873 А, 27.07.1997. | |||

