Область техники
Описан улучшенный способ получения фениламида 5-(4-фторфенил)-1-[2-((2R,4R)-4-гидрокси-6-оксотетрагидропиран-2-ил)этил]-2-изопропил-4-фенил-1Н-пиррол-3-карбоновой кислоты путем синтеза, в котором метилцианоацетат превращают в желаемый продукт в восемь или менее стадий, а также описаны другие ценные промежуточные соединения, применяемые в данном способе.
Уровень техники
Фениламид 5-(4-фторфенил)-1-[2-((2R,4R)-4-гидрокси-6-оксотетрагидропиран-2-ил)этил]-2-изопропил-4-фенил-1Н-пиррол-3-карбоновой кислоты представляет собой ценное промежуточное соединение, применяемое в синтезе Lipitor® (аторвастатина кальция), известного под химическим названием тригидрат кальциевой соли (2:1) [R-(R*,R*)]-2-(4-фторфенил)-β ,δ -дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)карбонил]-1Н-пиррол-1-гептановой кислоты. Указанное выше соединение полезно в качестве ингибитора редуктазы фермента 3-гидрокси-3-метилглутарил-кофермента А (редуктазы HMG-CoA) и, таким образом, полезно в качестве гиполипидемического и/или гипохолестеринемического агента.
В патенте США №4681893, который включен в данное описание посредством ссылки, описаны некоторые транс-6-[2-(3- или 4-карбоксамидо-замещенные-пиррол-1-ил)алкил]-4-гидроксипиран-2-оны, включая транс(±)-5-(4-фтрофенил)-2-(1-метилэтил)-N,4-дифенил-1-[(2-тетрагидро-4-гидрокси-6-оксо-2Н-пиран-2-ил)этил]-1Н-пиррол-3-карбоксамид.
В патенте США №5273995, который включен в данное описание посредством ссылки, описан энантиомер, имеющий (R,R) форму кислоты с разомкнутым кольцом транс-5-(4-фторфенил)-2-(1-метилэтил)-N,4-дифенил-1-[(2-тетрагидро-4-гидрокси-6-оксо-2Н-пиран-2-ил)этил]-1Н-пиррол-3-карбоксамид, т.е. [R-(R*,R*)]-2-(4-фторфенил)-β ,δ -дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)карбонил]-1Н-пиррол-1-гептановая кислота.
В патентах США №№5003080; 5097045; 5103024; 5124482; 5149837; 5155251; 5216174; 5245047; 5248793; 5280126; 5397792; 5342952; 5298627; 5446054; 5470981; 5489690; 5489691; 5510488; 5998633 и 6087511, которые включены в данное описание посредством ссылок, описаны различные способы и ключевые промежуточные соединения для получения аторвастатина.
Кристаллические формы аторвастатина кальция описаны в патентах США №№5969156 и 6121461, которые включены в данное описание посредством ссылок.
Способ синтеза с получением фениламида 5-(4-фторфенил)-1-[2-((2R,4R)-4-гидрокси-6-оксотетрагидропиран-2-ил)этил]-2-изопропил-4-фенил-1Н-пиррол-3-карбоновой кислоты описан в патенте США №5273995.
Асимметричное восстановление β -кетоэфиров, а также β -дикетонов, представляет собой превращение, хорошо известное в органическом синтезе. Однако сложность данных реакций возрастает в случае 1,3,5-трикарбонильных систем, причем в результате часто получают плохой выход и плохую стереоселективность. Фактически, исследования, проведенные Saburi (Tetrahedron, 1997, 1993; 49) и Carpentier (Eur. J. Org. Chem. 1999; 3421) независимо показали, что диастерео- и/или энантиоселективность при асимметричном гидрировании дикетоэфира является низкой или средней. Более того, тот факт, что способы известного уровня техники требуют гидрирования при высоком давлении и значительного времени реакции, делает такие способы непрактичными и неподходящими для промышленных производственных процессов.
Однако было неожиданно обнаружено, что сложные эфиры диола данного изобретения, сложные эфиры (R)-7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-дигидроксигептановой кислоты, могут быть получены непосредственно из соответствующих 1,3,5-трикарбонильных предшественников с высокой стереоселективностью посредством легкой и эффективной реакции асимметрического гидрирования, катализируемой рутением, с применением хиральных нерацемических дифосфиновых лигандов в присутствии вторичных активирующих агентов, таких как протоновые кислоты.
Объектом данного изобретения является краткий и эффективный способ получения фениламида 5-(4-фторфенил)-1-[2-((2R,4R)-4-гидрокси-6-оксотетрагидропиран-2-ил)этил]-2-изопропил-4-фенил-1Н-пиррол-3-карбоновой кислоты. Данный способ позволяет избежать применения дорогих хиральных исходных веществ (этилового эфира (R)-4-циано-3-гидрокси-масляной кислоты) и низкотемпературного диастереоселективного восстановления борана. Более того, ключевая стадия конденсации Паал-Кнорра, общая для настоящего способа и способов известного уровня техники, улучшена посредством значительного снижения времени реакции.
Таким образом, способ данного изобретения имеет значительные преимущества по сравнению со способами известного уровня техники и применим для синтеза в промышленных масштабах.
Краткое описание изобретения
Следовательно, первый аспект данного изобретения включает улучшенный способ получения соединения формулы (13)
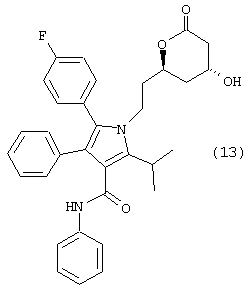
который включает:
Стадию (а) взаимодействия соединения формулы (1)
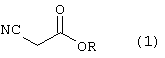
где R является алкилом, арилом, арилалкилом или гетероарилом, в растворителе, с соединением формулы (2)

где R1 является –XR, где
Х является О,
S или
Se, или R1 является
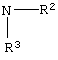 где R2 или R3 независимо являются
где R2 или R3 независимо являются
алкилом,
циклоалкилом,
арилалкилом или
арилом, или
R2 и R3 вместе являются
-(СН2)4-,
-(СН2)5-,
-(CH(R4)-CH2)3-,
-(CH(R4)-CH2)4-,
-(CH(R4)-(CH2)2-CH(R4))-,
-(CH(R4)-(CH2)3-CH(R4))-,
-CH2-CH2-A-CH2-CH2-,
-CH(R4)-CH2-A-CH2-CH2-,
-CH(R4)-CH2-A-CH2-CH(R4)-
где R4 является алкилом, имеющим 1-4 атома углерода, А является О, S или N, и R, такой как определено выше, с получением соединения формулы (3)
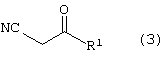
где R1, такой как определено выше;
Стадию (b) взаимодействия соединения формулы (3) с водородом в присутствии катализатора и сильной кислоты, в растворителе, с получением соединения формулы (4)
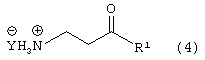
где Y является Cl, Br, TsO, MsO или HSO4 и R1, такой как определено выше;
Стадию (с) взаимодействия соединения формулы (4) с основанием в растворителе с последующим добавлением соединения формулы (5)

где R, такой как определено выше, в растворителе, с получением соединения формулы (6)
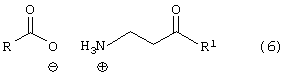
где R и R1 такие, как определено выше;
Стадию (d) взаимодействия соединения формулы (6) с соединением формулы (7)
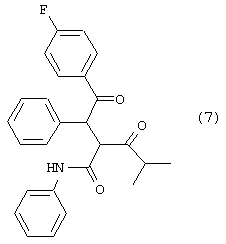
в растворителе с удалением воды с получением соединения формулы (8)
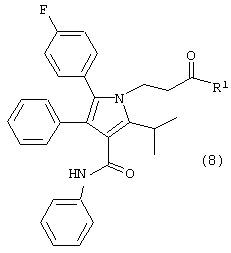
где R1, такой как определено выше;
Стадию (е) взаимодействия соединения формулы (8) с соединением формулы (9)
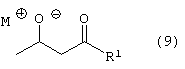
где М является натрием, литием, калием, цинком, магнием, медью, кальцием или алюминием и R1, такой как определено выше, в растворителе, в присутствии сильного основания, с получением соединения формулы (10)
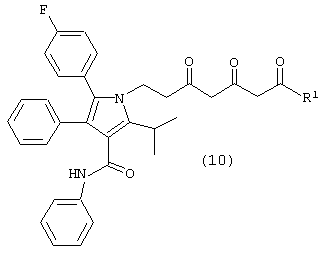
где R1, такой как определено выше;
Стадию (f) взаимодействия соединения формулы (10) с водородом в присутствии катализатора, в растворителе, в присутствии кислоты, с получением соединения формулы (11)
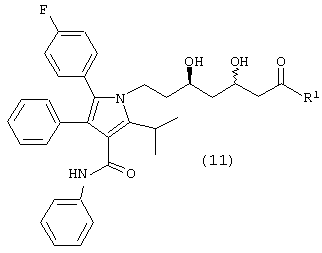
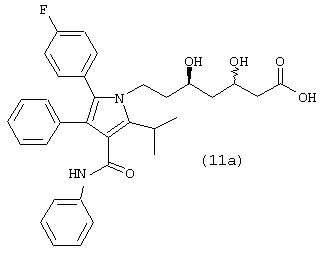
где R1, такой как определено выше, или соединения формулы (11а)
Стадию (g) взаимодействия соединения формулы (11b)

где R1a является ОН, -XR, где
Х является О,
S или
Se, или R1а является
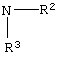 где R2 или R3 независимо являются
где R2 или R3 независимо являются
алкилом,
циклоалкилом,
арилалкилом или
арилом, или
R2 и R3 вместе являются
-(СН2)4-,
-(СН2)5-,
-(CH(R4)-CH2)3-,
-(CH(R4)-CH2)4-,
-(CH(R4)-(CH2)2-CH(R4))-,
-(CH(R4)-(CH2)3-CH(R4))-,
-CH2-CH2-A-CH2-CH2-,
-CH(R4)-CH2-A-CH2-CH2-,
-CH(R4)-CH2-A-CH2-CH(R4)-
где R4 является алкилом, имеющим 1-4 атома углерода, А является О, S или N, и R, такой как определено выше, в растворителе, в присутствии кислоты, с последующим взаимодействием с основанием, ацилирующим агентом и катализатором ацилирования, в растворителе, с получением соединения формулы (12)
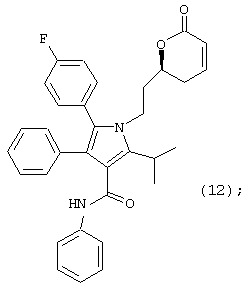
и;
Стадию (h) взаимодействия соединения формулы (12) с НО-М в спирте формулы (17) или (17b)
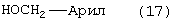 или
или 
где М является натрием, литием, калием, цинком, магнием, медью, кальцием или алюминием; или с соединением формулы (16) или (16b)
 или
или 
где М, такой как определено выше для спирта формулы (17) или (17b), где арил или аллил в соединениях формулы (16) или (16b) и (17) или (17b) является одинаковым, в растворителе, с последующим добавлением водорода в присутствии катализатора и кислоты, с получением соединения формулы (13).
Второй аспект данного изобретения включает улучшенный способ получения соединения формулы (8).
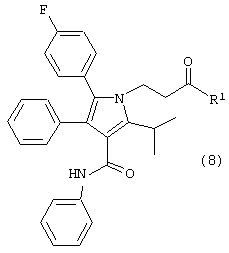
где R1, такой как определено выше, который включает:
взаимодействие соединения формулы (4)
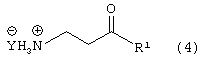
где Y является Cl, Br, TsO, MsO или HSO4 и R1, такой как определено выше, с соединением формулы (20)

где R и М такие, как определено выше, с соединением (7)
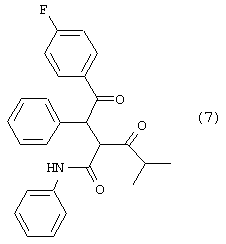
в растворителе с удалением воды, с получением соединения формулы (8).
Третий аспект данного изобретения включает улучшенный способ получения соединения формулы (13)
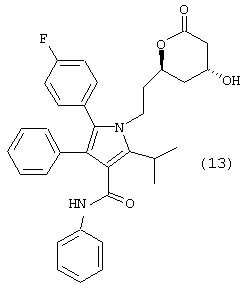
который включает:
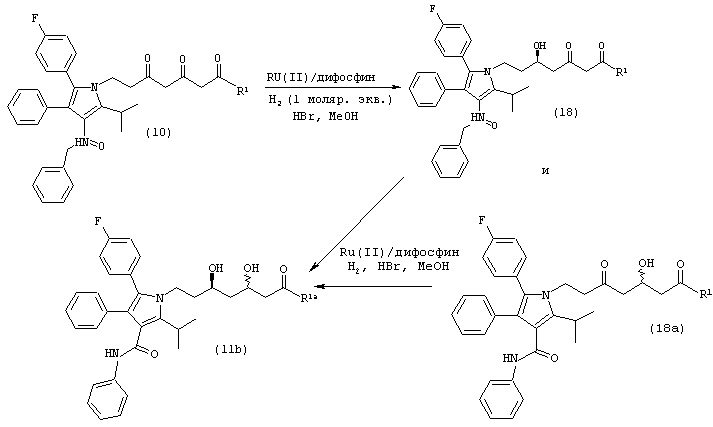
Стадию (а) взаимодействия соединения формулы (11) с ацеталем формулы (15)
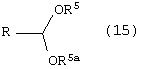
где R5 и R5а одинаковые или различные и независимо являются метилом, этилом или –(CH2)n-, где n является целым числом от 2 до 4 и R, такой как определено выше, в растворителе, в присутствии кислоты, с последующим добавлением альдегида, соответствующего предыдущему ацеталю, в присутствии основания, с получением соединения формулы (14)
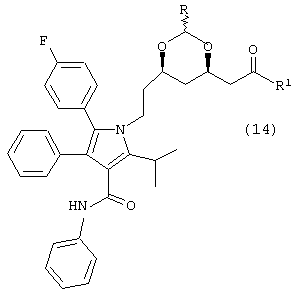
где R1 и R такие, как определено выше;
Стадию (b) взаимодействия соединения формулы (14) в нуклеофильном растворителе, в присутствии кислоты, или необязательного взаимодействия с водородом в присутствии катализатора и кислоты в растворителе, с получением соединения формулы (13); и
Стадию (с) альтернативного взаимодействия соединения формулы (11) или (11а) в ненуклеофильном растворителе, в присутствии кислоты, с получением соединения формулы (13).
Четвертый аспект данного изобретения включает способ получения соединения формулы (11b)
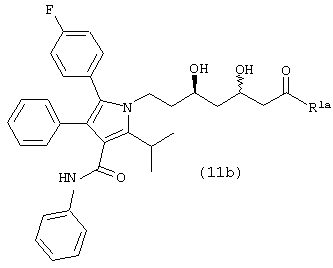
где R1a является ОН, -XR, где
Х является О,
S или
Se, или R1а является
 где R2 или R3 независимо являются
где R2 или R3 независимо являются
алкилом,
циклоалкилом,
арилалкилом или
арилом, или
R2 и R3 вместе являются
-(СН2)4-,
-(СН2)5-,
-(CH(R4)-CH2)3-,
-(CH(R4)-CH2)4-,
-(CH(R4)-(CH2)2-CH(R4))-,
-(CH(R4)-(CH2)3-CH(R4))-,
-CH2-CH2-A-CH2-CH2-,
-CH(R4)-CH2-A-CH2-CH2-,
-CH(R4)-CH2-A-CH2-CH(R4)-
где R4 является алкилом, имеющим 1-4 атома углерода, А является О, S или N, и R является алкилом, арилом, арилалкилом или гетероарилом, который включает:
Стадию (а) взаимодействия соединения формулы (10)
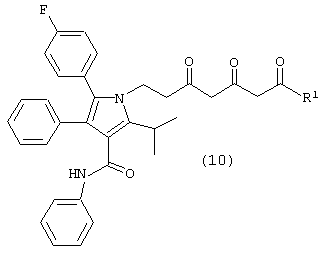
где R1, такой как определено выше, с одним молем водорода, в присутствии катализатора, в растворителе, в присутствии кислоты, с получением соединений формул (18) и/или (18а)
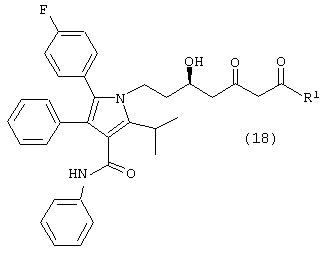
и

где R1, такой как определено выше; и
Стадию (b) взаимодействия либо соединения формулы (18), либо (18а) с водородом в присутствии катализатора, в растворителе, в присутствии кислоты, с получением соединения формулы (11b).
Пятый аспект данного изобретения включает соединение формулы (6)
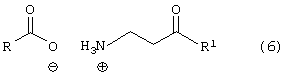
где R является алкилом, арилом, арилалкилом или гетероарилом, и
R1 является XR, где
Х является О,
S или
Se, или R1 является
 где R2 или R3 независимо являются
где R2 или R3 независимо являются
алкилом,
циклоалкилом,
арилалкилом или
арилом, или
R2 и R3 вместе являются
-(СН2)4-,
-(СН2)5-,
-(CH(R4)-CH2)3-,
-(CH(R4)-CH2)4-,
-(CH(R4)-(CH2)2-CH(R4))-,
-(CH(R4)-(CH2)3-CH(R4))-,
-CH2-CH2-A-CH2-CH2-,
-CH(R4)-CH2-A-CH2-CH2-,
-CH(R4)-CH2-A-CH2-CH(R4)-
где R4 является алкилом, имеющим 1-4 атома углерода, А является О, S или N, и R, такой как определено выше.
Особенно предпочтительным является соединение формулы (6), в которой R является PhCH2- или (CH3)3-C- и R1 является
Более предпочтительным является соединение формулы (6), в которой R является PhCH2- и R1 является

Шестой аспект данного изобретения включает соединение формулы (8)
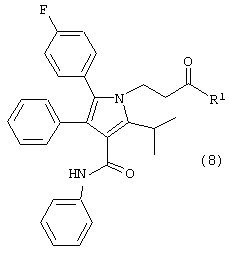
где R1, такой как определено выше.
Особенно предпочтительным является соединение формулы (8), в котором R1 является
Седьмой аспект данного изобретения включает соединение формулы (10) или его фармацевтически приемлемую соль
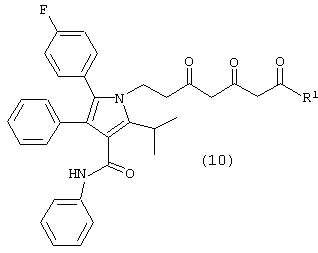
где R1, такой как определено выше.
Особенно предпочтительным является соединение формулы (10), в которой R1 является -O-третичным бутилом, -O-изопропилом, -O-этилом, -O-метилом,
 или –NMe2.
или –NMe2.
Восьмой аспект данного изобретения включает соединение формулы (12)
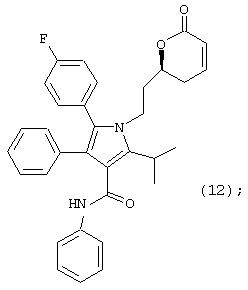
Девятый аспект данного изобретения включает соединение формулы (18) или его фармацевтически приемлемую соль
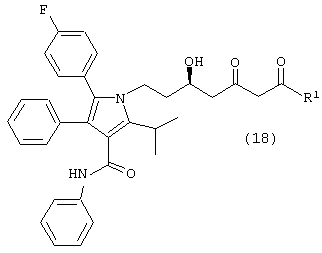
где R1, такой как определено выше.
Особенно предпочтительным является соединение формулы (18), в которой R1 является –О-третичным бутилом, -О-изопропилом, -О-этилом, -О-метилом,
 или –NMe2.
или –NMe2.
Десятый аспект данного изобретения включает соединение формулы (18а) или его фармацевтически приемлемую соль
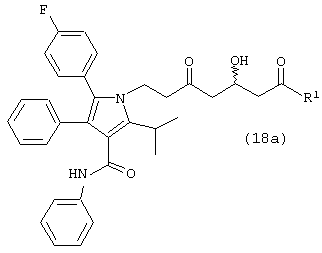
где R1, такой как определено выше.
Особенно предпочтительным является соединение формулы (18а), в которой R1 является –О-третичным бутилом, -О-изопропилом, -О-этилом, -О-метилом,
 или –NMe2.
или –NMe2.
Подробное описание изобретения
Термин “алкил” означает прямой или разветвленный углеводородный радикал, имеющий от 1 до 8 атомов углерода, и включает, например, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, н-гексил, н-гептил, н-октил и подобные.
“Алкокси” и “тиоалкокси” включают О-алкил или S-алкил, имеющий от 1 до 6 атомов углерода, как определено выше для “алкила”.
Термин “циклоалкил” означает насыщенное углеводородное кольцо, имеющее 3-8 атомов углерода, и включает, например, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил и подобные.
Термин “арил” означает ароматический радикал, который представляет собой фенильную группу, фенилалкильную группу, фенильную группу, замещенную 1-4 заместителями, выбранными из алкила, определенного выше, алкокси, определенного выше, тиоалкокси, определенного выше, галогена, трифторметила, диалкиламино, определенного выше для алкила, нитро, циано, 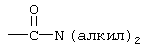 , такой как определено выше для алкила, -(CH2)
, такой как определено выше для алкила, -(CH2)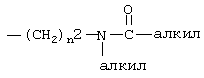 , такой как определено выше для алкила и n2.
, такой как определено выше для алкила и n2.
Термин “аллил” означает углеводородный радикал, имеющий 3-8 атомов углерода, содержащий двойную связь между атомами углерода 2 и 3, незамещенный или замещенный 1-3 заместителями у атомов углерода, имеющих двойную связь, выбранными из алкила или арила, таких как определено выше, и включает, например, пропенил, 2-бутенил, циннамил и подобные.
Термин “арилалкил” означает ароматический радикал, присоединенный к алкильному радикалу, где арил и алкил, такие как определено выше, например, бензил, фенетил, 3-фенилпропил, (4-хлорфенил)метил и подобные.
“Щелочной металл” представляет собой металл группы IA периодической таблицы и включает, например, литий, натрий, калий и подобные.
“Щелочноземельный металл” представляет собой металл группы IIA периодической таблицы и включает, например, кальций, барий, стронций, магний и подобные.
Термин “гетероарил” означает 5- и 6-членный гетероароматический радикал, который может быть необязательно сконденсирован с бензольным кольцом, содержащим 1-3 гетероатома, выбранных из N, О и S, и включает, например, гетероароматический радикал, являющийся 2- или 3-тиенилом, 2- или 3-фуранилом, 2- или 3-пирролилом, 2-, 3- или 4-пиридинилом, 2-пиразинилом, 2-, 4- или 5-пиримидинилом, 3- или 4-пиридазинилом, 1Н-индол-6-илом, 1Н-индол-5-илом, 1Н-бензимидазол-6-илом, 1Н-бензимидазол-5-илом, 2-, 4- или 5-тиазолилом, 3-, 4- или 5-изотиазолилом, 2-, 4- или 5-имидазолилом, 3-, 4- или 5-пиразолилом или 2- или 5-тиадиазолилом, и может быть необязательно замещен заместителем, выбранным из алкила, такого как определено выше, алкокси, такого как определено выше, тиоалкокси, такого как определено выше, галогена, трифторметила, диалкиламино, такого как определено выше для алкила, нитро, циано, 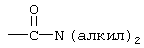 , такого как определено выше для алкила, -(CH2)
, такого как определено выше для алкила, -(CH2)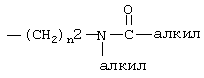 , такого как определено выше для алкила и n2.
, такого как определено выше для алкила и n2.
Фармацевтически приемлемые кислотно-аддитивные соли соединений данного изобретения включают соли, полученные из неорганических кислот, таких как хлористоводородная, азотная, фосфорная, серная, бромистоводородная, йодистоводородная, фтористоводородная, фосфористая и подобные, а также соли, полученные из нетоксичных органических кислот, таких как алифатические моно- и дикарбоновые кислоты, фенилзамещенные алкановые кислоты, гидроксиалкановые кислоты, алкандикислоты, ароматические кислоты, алифатические и ароматические сульфокислоты и т.д. Такие соли включают сульфат, пиросульфат, бисульфат, сульфит, бисульфит, нитрат, фосфат, моногидрофосфат, дигидрофосфат, метафосфат, пирофосфат, хлорид, бромид, йодид, ацетат, трифторацетат, пропионат, каприлат, изобутират, оксалат, малонат, сукцинат, суберат, себакат, фумарат, малеат, манделат,
бензоат, хлорбензоат, метилбензоат, динитробензоат, фталат, бензолсульфонат, толуолсульфонат, фенилацетат, цитрат, лактат, малеат, тартрат, метансульфонат и подобные.
Также включены соли аминокислот, такие как аргинат и подобные, и глюконат, галактуронат (см., например, Berge S. M. et al., “Pharmaceutical Salts” J. of Pharma. Sci., 1977;66:1).
Кислотно-аддитивные соли указанных основных соединений получают взаимодействием свободного основания с достаточным количеством желаемой кислоты с получением соли обычным способом. Свободное основание может быть регенерировано взаимодействием соли с основанием и выделением свободного основания обычным способом. Свободное основание несколько отличается от его соответствующей соли определенными физическими свойствами, такими как растворимость в полярных растворителях, но в другом соли эквивалентны их соответствующим свободным основаниям в целях данного изобретения.
Фармацевтически приемлемые основно-аддитивные соли получают с металлами или аминами, такими как щелочные и щелочноземельные металлы или органические амины. Примеры металлов, применяемых в качестве катионов, включают натрий, калий, магний, кальций и подобные. Примеры подходящих аминов включают N,N’-дибензилэтилендиамин, хлорпрокаин, холин, диэтаноламин, дициклогексиламин, этилендиамин, N-метилглюкамин и прокаин (см., например, Berge S.M. et al., “Pharmaceutical Salts” J. of Pharma. Sci., 1977; 66:1).
Основно-аддитивные соли указанных кислых соединений получают взаимодействием свободной кислоты с достаточным количеством желаемого основания с получением соли обычным способом. Свободная кислота может быть восстановлена взаимодействием соли с кислотой и выделением свободной кислоты обычным способом. Свободная кислота несколько отличается от ее соответствующей соли определенными физическими свойствами, такими как растворимость в полярных растворителях, но в другом соли эквивалентны их соответствующим свободным кислотам в целях данного изобретения.
Кроме того, соединения данного изобретения могут существовать как в не сольватированных формах, так и в сольватированных формах, включая гидратированные формы. В общем, сольватированные формы, включая гидратированные формы, эквивалентны несольватированным формам и включены в объем данного изобретения.
Представленный ниже список включает аббревиатуры и буквенные аббревиатуры, применяемые в схемах и тексте:
Способ данного изобретения в первом аспекте представляет собой новый, улучшенный, экономичный и промышленно применимый способ получения соединения формулы (13)
Способ данного изобретения в первом аспекте представлен на схеме 1. Таким образом, соединение формулы (1), где R является алкилом, арилом, арилалкилом или гетероарилом, подвергают взаимодействию с соединением формулы (2), где R1 является –XR, где
Х является О,
S или
Se, или R1 является
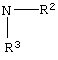 где R2 или R3 независимо являются
где R2 или R3 независимо являются
алкилом,
циклоалкилом,
арилалкилом или
арилом, или
R2 и R3 вместе являются
-(СН2)4-,
-(СН2)5-,
-(CH(R4)-CH2)3-,
-(CH(R4)-CH2)4-,
-(CH(R4)-(CH2)2-CH(R4))-,
-(CH(R4)-(CH2)3-CH(R4))-,
-CH2-CH2-A-CH2-CH2-,
-CH(R4)-CH2-A-CH2-CH2-,
-CH(R4)-CH2-A-CH2-CH(R4)-
где R4 является алкилом, имеющим 1-4 атома углерода, А является О, S или N, и R, такой как определено выше, в растворителе, таком как, например, метил-трет-бутиловый эфир и подобные, с получением соединения формулы (3), где R1, такой как определено выше. Предпочтительно, реакцию проводят с соединением формулы (2), в которой R1-Н является морфолином, в метил-трет-бутиловом эфире.
Соединение формулы (3) подвергают взаимодействию с водородом в присутствии катализатора, такого как, например, Pt/C, Pd/C, в присутствии кислоты, такой как, например, сильная кислота, например, хлористоводородная кислота, бромистоводородная кислота, п-толуолсульфокислота, метансульфокислота, серная кислота и подобные (необязательно восстановление проводят с применением Sponge Ni/NH4OH, гидридов металлов и подобных, с получением свободного основания соединения формулы (4)) в растворителе, таком как, например, метанол, этанол и подобные, с получением соединения формулы (4), в которой Y является Cl, Br, TsO, MsO или HSO4, и R1, такой как определено выше.
Предпочтительно, реакцию проводят в присутствии Pt/C, хлористоводородной кислоты и водорода в метаноле.
Соединение формулы (4) подвергают взаимодействию с основанием, таким как, например, метоксид натрия и подобные, в растворителе, таком как, например, тетрагидрофуран, толуол, метил-трет-бутиловый эфир и подобные, и в спирте, таком как, например, изопропанол, этанол, метанол и подобные, с получением свободного основания, с последующим взаимодействием с соединением формулы (5), где R, такой как определено выше, в растворителе, таком как, например, изопропанол, тетрагидрофуран и подобные, с получением соединения формулы (6), в котором R, такой как определено выше. Необязательно, свободное основание соединения формулы (4) может быть подвергнуто взаимодействию с соединением формулы (5) с получением соединения формулы (6). Предпочтительно реакцию проводят с метоксидом натрия в метил-трет-бутиловом эфире и метаноле с получением свободного основания с последующим взаимодействием с фенилацетатом в тетрагидрофуране.
Соединение формулы (6) подвергают взаимодействию с соединением формулы (7) в растворителе, таком как, например, протонный, апротонный, полярный или неполярный растворитель, например, тетрагидрофуран и подобные, с удалением воды с помощью химического высушивающего агента, такого как, например, молекулярные сита и подобные, или с помощью водяной ловушки Дина-Старка, или с применением азеотропной дистилляции с подходящим растворителем, таким как, например, толуол и подобные, с получением соединения формулы (8), где R1, такой как определено выше. Предпочтительно, реакцию проводят с активированными 3А молекулярными ситами в тетрагидрофуране.
Соединение формулы (8) подвергают взаимодействию с соединением формулы (9), где М является натрием, литием, калием, цинком, магнием, медью, кальцием или алюминием, и R1, такой как определено выше, в растворителе, таком как, например, нереакционноспособный апротонный растворитель, например, тетрагидрофуран, толуол и подобные, в присутствии сильного основания, такого как, например, н-бутиллитий, литий или гексаметилдисилазид калия, диизопропиламид лития и подобные, с получением соединения формулы (10), в которой R1, такой как определено выше. Предпочтительно реакцию проводят с соединением формулы (9), в котором М является натрием, где основанием является н-бутиллитий и растворителем является тетрагидрофуран.
Карбонилы соединения формулы (10) на схеме 1 показаны в кето форме. Однако соединение формулы (10) может претерпевать “кето-енольную” таутомерию и, таким образом, может существовать в нескольких таутомерных формах, которые включены в объем данного изобретения.
Соединение формулы (10) обрабатывают водородом в присутствии катализатора, такого как, например, хиральный нерацемический комплекс рутений (II)-дифосфин. Например, комплекс предшественника рутениевого катализатора, такого как олигомер [дихлор(1,5-циклооктадиен)]рутения (II) и хиральный дифосфиновый лиганд, такой как [(R)-(+)-2,2’-бис(дифенилфосфино)-1,1’-бинафтил]. Однако в данной реакции восстановления может использоваться любой хиральный нерацемический комплекс рутений (II)/дифосфин. Например, предшественники катализатора на основе рутения (II) включают димер [дибром(1,5-дициклооктадиен)] рутения (II), комплекс [бис(2-металлил)циклоокта-1,5-диен]рутения (II) и димер [дихлор(п-цимен)]рутения (II) и подобные. Примеры эффективных хиральных дифосфиновых лигандов включают
2,2’-бис(ди-п-толилфосфино)-1,1’-бинафтил,
2-дифенилфосфинометил-4-дифенилфосфино-1-трет-бутоксикарбонилпирролидин,
производные трицикло[8.2.2.24,7]гексадека-4,6,10,12,13,15-гексан-5,11-диилбис(дифенилфосфина),
4,4’-бидибензофуран-3,3’-диилбис(дифенилфосфин),
6,6’-диметокси[1,1’-бифенил]-2,2’-диил]бисдифенилфосфин,
[5,5’-дихлор-6,6’-диметокси[1,1’-бифенил]-2,2’-диил]бисдифенилфосфин и
производные 1,2-бис(2,5-диметилфосфолано) и подобные,
в растворителе, таком как, например, метанол, этанол, изопропанол и подобные, необязательно в присутствии сорастворителя, например, дихлорметана, тетрагидрофурана, толуола и подобного, в присутствии кислоты, такой как, например, хлористоводородная кислота, бромистоводородная кислота, Dowex® ионообменная смола и подобной, с получением соединения формулы (11) или соединения формулы (11а), где R1, такой как определено
выше. Предпочтительно, реакцию проводят с димером дихлор(п-цимен)рутения (II) и [(R)-(+)-5,5’-дихлор-6,6’-диметокси[1,1’-бифенил]-2,2’-диил]бисдифенилфосфина в метаноле в присутствии хлористоводородной кислоты.
Соединение формулы (11b), в котором R1a является ОН, –XR, где
Х является О,
S или
Se, или R1а является
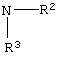 где R2 или R3 независимо являются
где R2 или R3 независимо являются
алкилом,
циклоалкилом,
арилалкилом или
арилом, или
R2 и R3 вместе являются
-(СН2)4-,
-(СН2)5-,
-(CH(R4)-CH2)3-,
-(CH(R4)-CH2)4-,
-(CH(R4)-(CH2)2-CH(R4))-,
-(CH(R4)-(CH2)3-CH(R4))-,
-CH2-CH2-A-CH2-CH2-,
-CH(R4)-CH2-A-CH2-CH2-,
-CH(R4)-CH2-A-CH2-CH(R4)-
где R4 является алкилом, имеющим 1-4 атома углерода, А является О, S или N, и R является алкилом, арилалкилом или гетероарилом, подвергают взаимодействию с кислотой, такой как, например, п-толуолсульфокислота, камфорсульфокислота, серная кислота, хлористый водород и подобные, в ненуклеофильном растворителе, таком как, например, толуол, ацетонитрил, дихлорметан, метил-трет-бутиловый эфир и подобные, с последующим взаимодействием с основанием, таким как, например, триэтиламин, пиридин, диизопропилэтиламин и подобные, и с ацилирующим агентом, таким как, например, уксусный ангидрид, бензоилхлорид, бензилхлорформиат и подобные, в присутствии 4-диметиламинопиридина, с получением соединения формулы (12). Предпочтительно, реакцию проводят в толуоле в присутствии п-толуолсульфокислоты, с последующей обработкой триэтиламином, уксусным ангидридом и 4-диметиламинопиридином в толуоле.
Соединение формулы (12) подвергают взаимодействию с НО-М в спирте формулы (17) или (17b), где М является натрием, литием, калием, цинком, магнием, медью, кальцием или алюминием, или с соединением формулы (16) или (16b), где М, такой как определено выше, в спирте формулы (17) или (17b), где арил или аллил в соединениях формул (16) или (16b) и (17) или (17b) являются одинаковыми, в необязательном сорастворителе, таком как, например, ненуклеофильный растворитель, например, ацетон, тетрагидрофуран, 1,2-диметоксиэтан и подобные, с последующим добавлением водорода в присутствии катализатора, такого как, например, Pd(OH)2/C, Pd/C, Pd/Al2O3 и подобного, в присутствии кислоты, такой как, например, хлористоводородная кислота, бромистоводородная кислота, серная кислота и подобные, с получением соединения формулы (13). Предпочтительно, реакцию проводят с гидроксидом натрия в бензиловом спирте с последующим гидрированием в присутствии Pd(OH)2/C и серной кислоты.
Способ данного изобретения во втором аспекте представлен на схеме 2. Таким образом, соединение формулы (4), полученное как описано на схеме 1, подвергают взаимодействию с соединением формулы (20), где R и М такие, как определено выше, и соединением формулы (7), с удалением воды с помощью химического высушивающего агента, такого как, например, молекулярные сита и подобные, или с помощью водяной ловушки Дина-Старка или с применением азеотропной дистилляции с подходящим растворителем, таким как, например, тетрагидрофуран, толуол и подобные, с получением соединения формулы (8), где R1, такой как определено выше. Предпочтительно реакцию проводят с соединением формулы (20), в которой R является PhCH2 и М является натрием, в присутствии активированных 3А молекулярных сит в тетрагидрофуране.
Способ данного изобретения в третьем аспекте представлен на схеме 3. Таким образом, соединение формулы (11) подвергают взаимодействию с ацеталем формулы (15), где R5 и R5а одинаковые или различные независимо являются метилом, этилом или –(CH2)n, где n является целым числом от 2 до 4, и R, такой как определено выше, в присутствии кислоты, такой как, например, хлористоводородная кислота, п-толуолсульфонат пиридиния, п-толуолсульфокислота и подобные, в растворителе, таком как, например, толуол, дихлорметан, метил-трет-бутиловый эфир и подобные, с последующим добавлением альдегида, соответствующего предыдущему ацеталю формулы (15), в присутствии сильного основания, такого как, например, ненуклеофильное основание, например, третичный бутоксид калия, бис(триметилсилил)амид калия, 1,8-диазабицикло[5.4.0]ундец-7-ен и подобные, с получением соединения формулы (14), в которой R1 и R такие, как определено выше. Предпочтительно, реакцию проводят в диметилацетале бензальдегида в толуоле в присутствии п-толуолсульфокислоты с последующим добавлением бензальдегида и третичного бутоксида калия в тетрагидрофуране.
Соединение формулы (14) подвергают взаимодействию с водородом в присутствии катализатора, такого как, например, палладий на углероде или платина на углероде и подобные, в присутствии кислоты, такой как, например, хлористоводородная кислота и подобные, в растворителе, таком как, например, толуол, тетрагидрофуран, метил-трет-бутиловый эфир, этилацетат и подобные, и в спирте, таком как, например, метанол, этанол и подобные, с получением соединения формулы (13). Предпочтительно, реакцию проводят в толуоле в присутствии платины на углероде, в присутствии метанола, в присутствии хлористоводородной кислоты.
Необязательно соединение формулы (14) подвергают взаимодействию с кислотой, такой как, например, хлористоводородная кислота, п-толуолсульфонат пиридиния, п-толуолсульфокислота и подобные, в растворителе, таком как, например, толуол, дихлорметан, метил-трет-бутиловый эфир и подобные, с получением соединения формулы (13). Предпочтительно реакцию проводят в метиленхлориде в присутствии п-толуолсульфокислоты.
Альтернативно, соединение формулы (11) подвергают взаимодействию с кислотой, такой как, например, хлористоводородная кислота, бромистоводородная кислота, п-толуолсульфокислота и подобные, в ненуклеофильном растворителе, таком как, например, толуол, ацетонитрил, метил-трет-бутиловый эфир, тетрагидрофуран и подобные, с получением соединения формулы (13). Предпочтительно реакцию проводят в толуоле в присутствии п-толуолсульфокислоты.
Способ данного изобретения в четвертом аспекте представлен на схеме 4. Таким образом, соединение формулы (1), где R1, такой как определено выше, подвергают взаимодействию с одним молярным эквивалентом водорода в присутствии катализатора, используя методику, описанную выше для превращения соединения формулы (10) в соединение формулы (11) с получением либо соединения формулы (18), либо соединения формулы (18а), где R1, такой как определено выше, или их смеси. Смесь соединений формулы (18) и (18а) может быть разделена обычными методами, такими как, например, хроматография и подобные. Предпочтительно смесь соединений формул (18) и (18а) разделяют ВЭЖХ.
Соединение формулы (18) или (18а) или их смесь подвергают взаимодействию с водородом в присутствии катализатора, как описано для получения соединения формулы (11), с получением соединения формулы (11b), где R1a, такой как описано выше. Предпочтительно, реакцию проводят с применением, по крайней мере, одного молярного эквивалента водорода.
Соединение формулы (13) может быть превращено в аторвастатинкальций (19) с применением методик, описанных в патентах США №№5273995 и 5969156.
Следующие не ограничивающие примеры иллюстрируют предпочтительные способы получения соединений данного изобретения.
СХЕМА 1
СХЕМА 2

СХЕМА 3
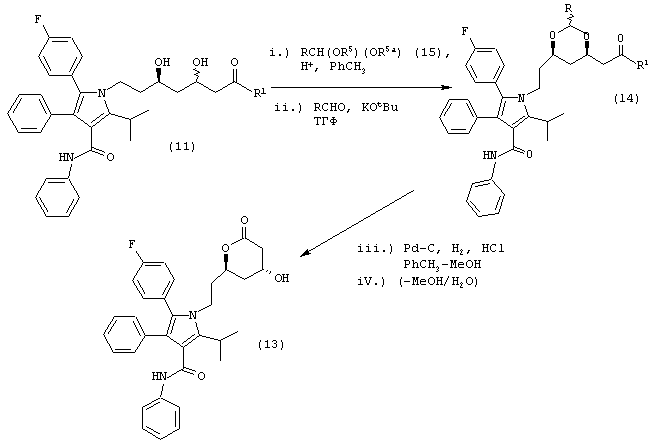
СХЕМА 4
Пример 1
Фениламид 5-(4-фторфенил)-1-[2-((2R,4R)-4-гидрокси-6-оксотетрагидропиран-2-ил)этил]-2-изопропил-4-фенил-1Н-пиррол-3-карбоновой кислоты
Стадия 1: Морфолин-4-ил-3-оксопропионитрил
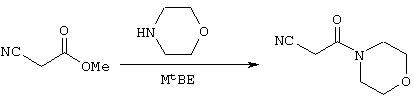
В реактор с инертным азотом, оборудованный обратным холодильником, входом для азота и механической мешалкой, загружают морфолин (1,2 моль), метилцианоацетат (1,0 моль) и MtBE (52 мл). Гомогенный раствор нагревают до примерно 55° С и перемешивают при этой температуре в течение 12-18 часов. MtBE (33 мл) добавляют в течение примерно 15 минут и раствор медленно охлаждают до температуры ниже 50° С, при которой становится очевидным зарождение центров кристаллизации. Добавляют еще MtBE (66 мл) в течение более 1 часа. В течение этого времени реакционную смесь охлаждают до температуры, близкой к температуре окружающей среды. После завершения добавления MtBE реакционную смесь охлаждают при перемешивании до примерно 0° С. Полученный осадок собирают фильтрованием и осадок на фильтре промывают дополнительным количеством MtBE (примерно 40 мл). Твердое вещество сушат в вакууме при температуре примерно 45° С с получением 3-морфолин-4-ил-3-оксопропионитрила (139 г). Данный продукт используют на последующих стадиях без дальнейшей очистки.
m/z (APCI(m+1)) 154,9; вычислено для С7Н10N2O2 154,07
Стадия 2: 3-Амино-1-морфолин-4-илпропан-1-он; гидрохлорид
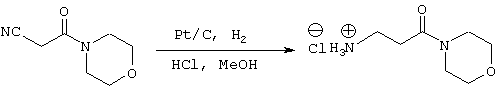
В реактор с инертным азотом загружают 5% Pt-C (43 г; 58% увлажненный водой) с последующим добавлением 3-морфолин-4-ил-3-оксопропионитрила (2,8 моль). Добавляют раствор MeOH (3,4 л) и 12н HCl (3,08 моль) с такой скоростью, чтобы поддерживать внутреннюю температуру на уровне примерно 25° С. Сосуд и его содержимое дегазируют тремя продувками N2 при давлении (50 psi). Атмосферу заменяют водородом тремя продувками Н2 при давлении (50 psi) и реакционную смесь энергично перемешивают при температуре примерно 25° С, поддерживая давление водорода (50 psi) в течение примерно 24 часов. Давление Н2 снижают и заменяют N2. Реакционную смесь пропускают через фильтровальный агент, который далее промывают МеОН (500 мл). Реакционную смесь концентрируют в вакууме до объема примерно 1,4 л и добавляют IPA (2,2 л). Реакционную смесь охлаждают до 0° С и фильтруют. Осадок на фильтре промывают MtBE (500 мл) и сушат в вакууме при температуре примерно 30° С с получением гидрохлорида 3-амино-1-морфолин-4-илпропан-1-она в виде белого твердого вещества (439 г). Данный продукт используют на следующих стадиях без дальнейшей очистки.
1Н ЯМР (400 МГц, ДМСО) δ 2,72 (т, 2Н, J=6,78), 2,96 (т, 2Н, J=6,77), 3,83-3,44 (м, 2Н), 3,52-3,58 (м, 2Н), 8,08 (шс, 3Н).
13С ЯМР (100 МГц, ДМСО) δ 168,4, 65,9, 45,1, 41,45, 35,1, 29,6.
Свободное основание: m/z (APCI(m+1)) 159,2; вычислено для С7Н14N2O2 158,11.
Стадия 3: 3-Амино-1-морфолин-4-илпропан-1-он; соединение с фенилуксусной кислотой
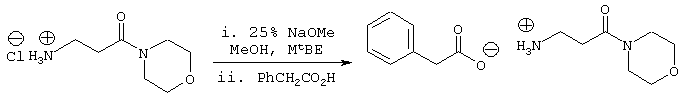
В реактор загружают гидрохлорид 3-амино-1-морфолин-4-илпропан-1-он (765 ммоль). Добавляют МеОН (380 мл) и смесь энергично перемешивают при комнатной температуре в течение примерно 10 минут. Добавляют MtBE (380 мл) и полученную суспензию охлаждают до –10° С, медленно добавляют 25% (об/об) раствор NaOMe в МеОН (765 ммоль) через капельную воронку с такой скоростью, чтобы сохранять внутреннюю температуру на уровне примерно –10° С. Полученную суспензию энергично перемешивают в атмосфере N2 и нагревают до температуры 0° С. Твердые вещества удаляют фильтрованием, промывая дополнительным количеством MtBE (50 мл). Растворитель удаляют в вакууме с получением свободного основания в виде неочищенного масла, которое помещают в MtBE (600 мл). Смесь охлаждают энергичным перемешиванием при температуре примерно 0° С, медленно добавляя фенилуксусную кислоту (765 ммоль) в виде раствора в MtBE (300 мл). После завершения добавления реакционную смесь перемешивают в течение еще 10 минут, в течение которых продукт осаждается из раствора. Твердые вещества собирают фильтрованием, промывая дополнительным количеством MtBE (100 мл), и сушат в вакууме при температуре ≤ 40° С с получением 3-амино-1-морфолин-4-илпропан-1-она, соединения с фенилуксусной кислотой (191 г). Данный продукт используют на последующих стадиях без дальнейшей очистки, он может быть повторно осажден из MtBE.
1Н ЯМР (400 МГц, ДМСО) δ 2,55 (т, 2Н, J=6,78), 2,86 (т, 2Н, J=6,78), 3,62 (т, 2Н), 3,42 (т, 2Н), 6,22 (шс, 3Н), 7,25-7,12 (м, 5Н).
13С ЯМР (100 МГц, ДМСО) δ 174,2, 169,0, 138,2, 129,2, 127,8, 125,5, 66,0, 45,2, 44,4, 41,4, 35,7, 31,6.
Стадия 4: Фениламид 5-(4-фторфенил)-2-изопропил-1-(3-морфолин-4-ил-3-оксопропил)-4-фенил-1Н-пиррол-3-карбоновой кислоты
Способ А
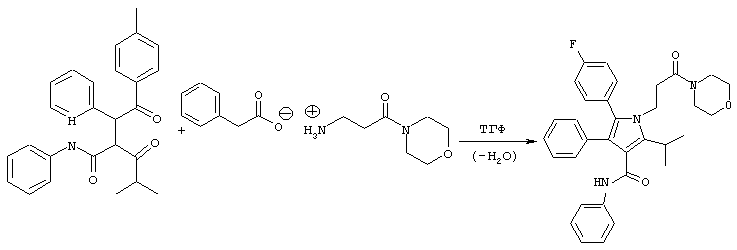
В реактор с инертным азотом, оборудованный подходящим обратным холодильником и экстрактором Сокслета, содержащим свеже-активированные 3А молекулярные сита (4-8 меш; 97,2 г), загружают 3-амино-1-морфолин-4-илпропан-1-он, соединение с фенилуксусной кислотой (765 ммоль) и фениламид 2-[2-(4-фторфенил)-2-оксо-1-фенилэтил]-4-метил-3-оксопентановой кислоты (450 ммоль). Добавляют ТГФ (360 мл) и полученный раствор энергично перемешивают, нагревая при температуре кипения с обратным холодильником в течение примерно 24 часов, в течение которых продукт начинает осаждаться. Добавляют полунасыщенный водный NaHCO3 (100 мл) и реакционную смесь охлаждают при постоянном перемешивании до температуры примерно 0° С. Добавляют MtBE (100 мл) и твердые вещества собирают фильтрованием. Твердое вещество промывают дистиллированной водой (100 мл) и MtBE (2× 100 мл), собирают и сушат в вакууме при температуре ≤ 50° С с получением фениламида 5-(4-фторфенил)-2-изопропил-1-(3-морфолин-4-ил-3-оксопропил)-4-фенил-1Н-пиррол-3-карбоновой кислоты в виде белого твердого вещества (194 г). Данный продукт используют на последующих стадиях без дальнейшей очистки.
m/z (APCI(m-1)) 538,2; (APCI(m+1)) 540,2; вычислено для
С33Н34FN3O3 539,26.
Способ В
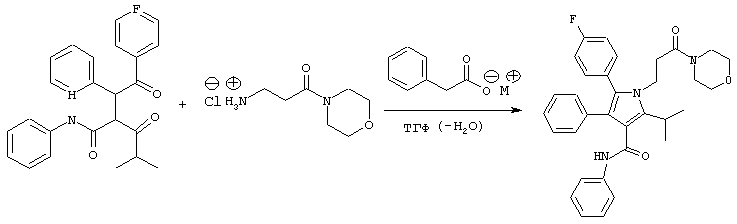
В реактор с инертным азотом, оборудованный подходящим обратным холодильником и экстрактором сокслет, содержащим свеже-активированные 3А молекулярные сита (4-8 меш; 36 г), загружают гидрохлорид 3-амино-1-морфолин-4-илпропан-1-она (170 ммоль), натриевую соль фенилуксусной кислоты (170 ммоль) и фениламид 2-[2-(4-фторфенил)-2-оксо-1-фенилэтил]-4-метил-3-оксопентановой кислоты (100 ммоль). Добавляют ТГФ (150 мл) и полученный раствор энергично перемешивают, нагревая при температуре кипения с обратным холодильником в течение примерно 24 часов, в течение которых продукт начинает осаждаться. Медленно добавляют водный NaHCO3 (100 мл) и реакционную смесь охлаждают при постоянном перемешивании до температуры примерно 0° С. Добавляют MtBE (100 мл) и твердые вещества собирают фильтрованием. Твердое вещество промывают дистиллированной водой (15 мл) и MtBE (2× 15 мл), собирают и сушат в вакууме при температуре ≤ 50° С с получением фениламида 5-(4-фторфенил)-2-изопропил-1-(3-морфолин-4-ил-3-оксопропил)-4-фенил-1Н-пиррол-3-карбоновой кислоты в виде белого твердого вещества (42,1 г). Данный продукт используют на последующих стадиях без дальнейшей очистки.
m/z (APCI(m-1)) 538,2; (APCI(m+1)) 540,2; вычислено для С33Н34FN3O3 539,26.
Способ С
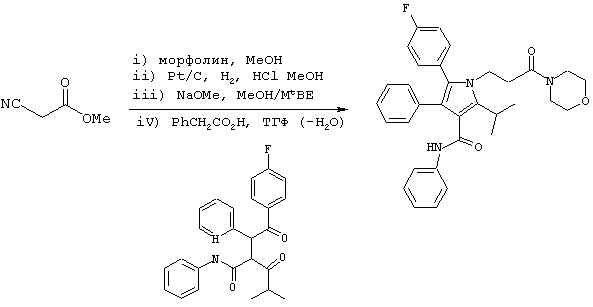
В реактор с инертным азотом, оборудованный обратным холодильником, входом для азота и механической мешалкой загружают морфолин (1,2 моль), метилцианоацетатом (1,0 моль) и MtBE (52 мл). Гомогенный раствор нагревают до примерно 55° С и перемешивают при данной температуре в течение 12-18 часов. MtBE (33 мл) добавляют в течение примерно 15 минут и раствор медленно охлаждают до температуры ниже 50° С, при которой становится очевидным зарождение центров кристаллизации. Добавляют еще MtBE (66 мл) в течение более 1 часа. В течение этого времени реакционную смесь охлаждают до температуры, близкой к температуре окружающей среды. После завершения добавления MtBE реакционную смесь охлаждают при перемешивании до примерно 0° С. Полученный осадок собирают фильтрованием и осадок на фильтре промывают дополнительным количеством MtBE (примерно 40 мл).
Неочищенный 3-морфолин-4-ил-3-оксопропионитрил помещают в МеОН (2 л) и переносят в реактор с инертным азотом под давлением, который загружен 5% Pt-C (55 г; 58% увлажненный водой). Добавляют HCl (12н; 1,1 моль) с такой скоростью, чтобы поддерживать внутреннюю температуру на уровне примерно 25° С. Сосуд и его содержимое дегазируют тремя продувками N2 при давлении (50 psi). Атмосферу заменяют водородом тремя продувками Н2 при давлении (50 psi) и реакционную смесь энергично перемешивают при температуре примерно 25° С, поддерживая давление водорода (50 psi) в течение примерно 24 часов. Давление Н2 снижают и заменяют N2. Реакционную смесь пропускают через фильтровальный агент, который затем промывают МеОН (500 мл). Реакционную смесь концентрируют до увлажненного МеОН твердого вещества, которое повторно суспендируют в IPA (100 мл). Суспензию охлаждают до 0° С и фильтруют. Осадок на фильтре промывают холодным (0° С) IPA и повторно суспендируют в МеОН (500 мл) и MtBE (500 мл). Суспензию охлаждают при перемешивании до –10° С, по каплям добавляют 25% (об/об) раствор NaOMe в МеОН (1 моль) с такой скоростью, чтобы сохранять внутреннюю температуру ≤ –5° С. Полученную суспензию фильтруют с получением прозрачного раствора свободного основания. Растворитель удаляют в вакууме с получением неочищенного масла, которое помещают в ТГФ (450 мл) и охлаждают до примерно 0оС. Полученный раствор переносят в реактор с инертным азотом, который содержит фенилуксусную кислоту (1,0 моль) и фениламид 2-[2-(4-фторфенил)-2-оксо-1-фенилэтил]-4-метил-3-оксопентановой кислоты (590 ммоль). Реактор, оборудованный подходящим обратным холодильником и экстрактором Сокслета, содержащим свеже-активированные 3А молекулярные сита (4-8 меш; 125 г). Полученный раствор энергично перемешивают при кипячении с обратным холодильником в атмосфере N2 в течение примерно 24 часов, в течение которых продукт начинает осаждаться. Добавляют полунасыщенный водный NaHCO3 (130 мл) и реакционную смесь охлаждают при постоянном перемешивании до температуры примерно 0° С. Добавляют MtBE (130 мл) и твердые вещества собирают фильтрованием. Твердое вещество промывают дистиллированной водой (130 мл) и MtBE (2× 130 мл), собирают и сушат в вакууме при температуре ≤ 50° С с получением фениламида 5-(4-фторфенил)-2-изопропил-1-(3-морфолин-4-ил-3-оксопропил)-4-фенил-1Н-пиррол-3-карбоновой кислоты в виде белого твердого вещества (223 г). Данный продукт используют на последующих стадиях без дальнейшей очистки.
m/z (APCI(m-1)) 538,2; (APCI(m+1)) 540,2; вычислено для С33Н34FN3O3 539,26.
Стадия 5: этиловый эфир 7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-диоксогептановой кислоты
Способ А
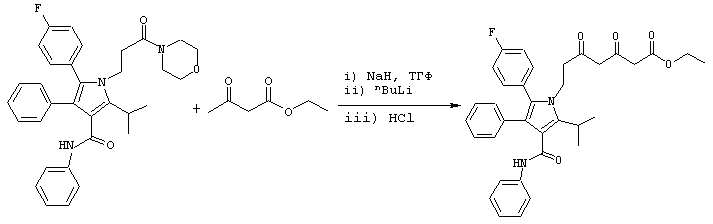
В сухой реактор с инертным азотом загружают гидрид натрия (300 ммоль). Добавляют безводный ТГФ (150 мл) и полученную смесь охлаждают в атмосфере азота до примерно –20° С. Добавляют этилацетоацетат (307 ммоль) с такой скоростью, чтобы поддерживать внутреннюю температуру реакции на уровне ≤ -10° С. После добавления промывают ТГФ (30 мл) и полученный раствор перемешивают в течение приблизительно 45 минут при ≤ -10оС. Температуру понижают до примерно –18° С. Добавляют 10,0 М раствор н-BuLi в гексане (300 ммоль) с такой скоростью, чтобы поддерживать внутреннюю температуру реакции на уровне ≤ -4° С. После добавления промывают ТГФ (30 мл) и полученный оранжевый раствор перемешивают в течение примерно 90 минут при температуре ≤ -4° С. Температуру понижают до примерно –25° С. К раствору диенолята добавляют фениламид 5-(4-фторфенил)-2-изопропил-1-(3-морфолин-4-ил-3-оксопропил)-4-фенил-1Н-пиррол-3-карбоновой кислоты (74 ммоль), полученную суспензию перемешивают при примерно –23° С в течение 20 часов. Реакционную смесь гасят в смеси 18% водной HCl (898 ммоль) и MtBE (20 мл) с такой скоростью, чтобы поддерживать внутреннюю температуру реакции на уровне ≤ -2° С. Реактор и переносящую систему промывают ТГФ (30 мл) и переносят в реакционную смесь. Двухфазный раствор нагревают до примерно 20° С при перемешивании. Смесь переносят в делительную воронку и фазы разделяют. Органический слой промывают водой (33 мл) и насыщенным водным NaCl (33 мл). Все водные слои повторно экстрагируют MtBE (40 мл). Два органических слоя объединяют и концентрируют в вакууме до неочищенного масла, поддерживая внутреннюю температуру продукта на уровне ≤ 60° С. К маслу добавляют EtOH (24 мл) и смесь снова концентрируют в вакууме. К полученному маслу сразу же добавляют EtOH (330 мл) и воду (33 мл) и раствор продукта выстаивают при температуре ≤ 10° С в течение примерно 14 часов. Полученное твердое вещество собирают, промывают холодным 20% водным EtOH (100 мл) и сушат в вакууме с получением этилового эфира 7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-диоксогептановой кислоты (35,6 г) в виде белого твердого вещества. Данный продукт используют на последующих стадиях без дальнейшей очистки, или, необязательно, он может быть повторно осажден из IPA/Н2О.
HRMS m/z (ESI(m-1)) 581,2463; вычислено для С35Н35FN2O5 582,2530.
По методике, аналогично стадии 5, способ А, заменяя этилацетоацетат соответствующим сложным эфиром или амидом ацетоуксусной кислоты, получают следующие соединения:
Трет-бутиловый эфир 7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-диоксогептановой кислоты.
HRMS m/z (ESI(m-1)) 609,2772; APCI (m+1) 611,3; вычислено для С37Н39FN2O5 610,2843.
Изопропиловый эфир 7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-диоксогептановой кислоты.
m/z (PCI(m+1)) 597; вычислено для С36Н37FN2O5 596,27.
Метиловый эфир 7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-диоксогептановой кислоты.
m/z (PCI(m+1)) 569; вычислено для С34Н33FN2O5 568,24.
Морфолиноамид 7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-диоксогептановой кислоты.
HRMS m/z (ESI(m-1)) 622,2715; вычислено для С37Н38FN3O5 623,2795.
N,N-диметиламид 7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-диоксогептановой кислоты.
m/z (PCI(m+1)) 582; вычислено для С35Н36FN3O4 581,27.
Способ В
Трет-бутиловый эфир 7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-диоксогептановой кислоты
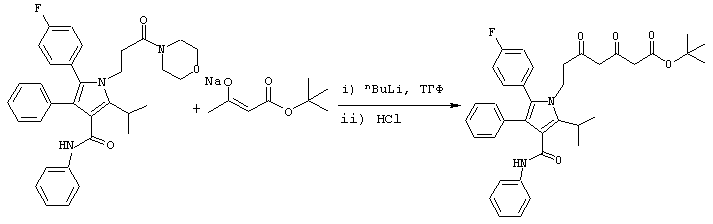
В реактор с инертным азотом загружают натриевую соль трет-бутилацетоацетата (100 ммоль). Добавляют безводный толуол (71,5 мл) и ТГФ (8,2 мл, 101 ммоль) и полученный раствор охлаждают при положительном давлении азота до температуры примерно –10° С. Добавляют 10М раствор н-BuLi в гексане (104 ммоль) с такой скоростью, чтобы поддерживать внутреннюю температуру реакции на уровне ≤ 1° С. После завершения добавления полученный раствор перемешивают еще дополнительно 20-30 минут и температуру понижают до примерно –6° С. Одновременно фениламид 5-(4-фторфенил)-2-изопропил-1-(3-морфолин-4-ил-3-оксопропил)-4-фенил-1Н-пиррол-3-карбоновой кислоты (25 ммоль) загружают во второй реактор с инертным азотом. Добавляют безводный ТГФ (50 мл) при комнатной температуре и полученную суспензию охлаждают до примерно –10° С и перемешивают 15-90 минут. Раствор диенолята добавляют в суспензию морфолинамида с такой скоростью, чтобы поддерживать внутреннюю температуру реакции на уровне примерно –5° С. После добавления суспензию перемешивают при температуре примерно –5° С в течение ≥ 2 часов. При энергичном перемешивании добавляют воду (35 мл) с такой скоростью, чтобы сохранять внутреннюю температуру реакции на уровне ≤ 0° С. Добавляют концентрированную 37% хлористоводородную кислоту (19,0 мл, 229 ммоль) с такой скоростью, чтобы поддерживать внутреннюю температуру реакции на уровне ≤ 0° С. Двухслойную реакционную смесь дистиллируют в вакууме, удаляя >50% органических растворителей. Дистилляцию останавливают и нижний водный слой удаляют. Добавляют воду (55 мл) и дистилляцию в вакууме продолжают до тех пор, пока основная часть органических растворителей не будет удалена. [Примечание: Предпочтительно слить и заменить водный слой до начала вакуумной дистилляции.] Добавляют IPA (100 мл) с последующим добавлением воды (100 мл). Смесь перемешивают в течение ≥ 6 часов, в течение которых продукт затвердевает. Твердое вещество собирают фильтрованием и осадок на фильтре промывают предварительно полученной смесью 1:1 IPA:Н2О. Полученное твердое вещество сушат в вакууме при температуре 35оС с получением трет-бутилового эфира 7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-диоксогептановой кислоты (14,1 г) в виде белого твердого вещества. Продукт используют на последующих стадиях без дальнейшей очистки или, необязательно, он может быть повторно осажден из толуола.
HRMS m/z (ESI(m-1)) 609,2772; APCI (M+1) 611,3; вычислено для С37Н39FN2O5 610,2843.
По методике, аналогично стадии 5, способ В, заменяя натриевую соль трет-бутилацетоацетата соответствующей натриевой солью сложного эфира или амида ацетоуксусной кислоты, получают следующие соединения:
Этиловый эфир 7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-диоксогептановой кислоты.
HRMS m/z (ESI(m-1)) 581,2463; вычислено для С35Н35FN2O5 582,2530.
Изопропиловый эфир 7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-диоксогептановой кислоты.
m/z (PCI(m+1)) 597; вычислено для С36Н37FN2O5 596,27.
Метиловый эфир 7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-диоксогептановой кислоты.
m/z (PCI(m+1)) 569; вычислено для С34Н33FN2O5 568,24.
Морфолиноамид 7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-диоксогептановой кислоты.
HRMS m/z (ESI(m-1)) 622,2715; вычислено для С37Н38FN3O5 623,2795.
N,N-диметиламид 7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-диоксогептановой кислоты.
m/z (PCI(m+1)) 582; вычислено для С35Н36FN3O4 581,27.
Стадия 6: метиловый эфир (5R)-7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-дигидроксигептановой кислоты
Способ А
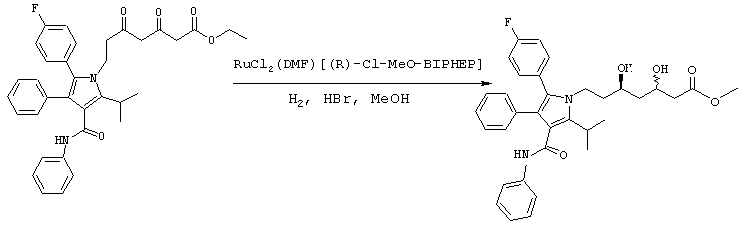
В реактор с инертным азотом загружают этиловый эфир 7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-диоксогептановой кислоты (100,0 ммоль) и МеОН (250 мл). Полученную суспензию нагревают при перемешивании при температуре примерно 55оС с получением гомогенного раствора. Сосуд и его содержимое дегазируют тремя продувками аргона при давлении 50 psi. При постоянном потоке аргона добавляют 1 М HBr в метаноле (7,0 ммоль) и катализатор RuCl2(ДМФ)n[(R)-Cl-MeO-BIPHEP)] (0,5 ммоль) и реактор продувают дополнительным количеством аргона при давлении 50 psi. Атмосферу заменяют водородом тремя продувками при давлении 50 psi. Реакционную смесь энергично перемешивают при температуре 65° С при поддерживаемом давлении водорода (50 psi) до тех пор, пока не прекратится поглощение водорода. Реакционную смесь охлаждают до температуры окружающей среды, давление водорода снижают и заменяют азотом. Неочищенный МеОН раствор метилового эфира (5R)-7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-дигидроксигептановой кислоты используют на следующих стадиях без очистки или, необязательно, он может быть выделен флэш-хроматографией на колонке с силикагелем, элюируя смесью этилацетат-гексан.
ВЭЖХ анализ (YMC ODS AQ S5; 1 мл/мин; 30° С; 254 нм: СН3CN/H2O, 60:40 (0-22 мин) до 100:0 (27-37 мин) до 60:40) показал соотношение син:анти изомеров 1:1,5. Хиральный ВЭЖХ анализ (Chiralcel OD-H колонка; 5% EtOH:гексаны; tR(3R,5R)=23,1 мин/tR(3R,5S)=18,0 мин/tR(3S,5S)=24,8 мин/tR(3S,5R)=19,9 мин) показал энантиомерный избыток при С-5 ≥ 98%, преимущественно (R) конфигурации. m/z (PCI(m+1)) 573; вычислено для С34Н37FN2O5 572,27.
По методике, аналогично стадии 6, способ А, используя раствор соответствующего спирта вместо МеОН, получают следующие соединения, например:
Этиловый эфир (5R)-7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-дигидроксигептановой кислоты
m/z (PCI(m+1)) 587; вычислено для С35Н39FN2O5 586,28.
Хиральный ВЭЖХ анализ (Chiralcel OD-H колонка; 5% EtOH:гексаны; tR(3R,5R)=17,6 мин/tR(3R,5S)=14,7 мин/tR(3S,5S)=20,9 мин/tR(3S,5R)=15,9 мин) показал энантиомерный избыток при С-5 ≥ 98%, преимущественно (R) конфигурации.
Изопропиловый эфир (5R)-7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-дигидроксигептановой кислоты
m/z (PCI(m+1)) 601; вычислено для С36Н41FN2O5 600,30.
По методике, аналогично стадии 6, способ А, используя соответствующий сложный эфир или амид со стадии 5 в ненуклеофильном/некоординирующем растворителе (например,
толуоле) вместо МеОН и уксусную кислоту вместо HBr можно избежать трансэтерификации и получить следующие соединения, например:
Трет-бутиловый эфир (5R)-7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-дигидроксигептановой кислоты
m/z (APCI(m+1)) 615,3; вычислено для С37Н43FN2O5 614,32.
Морфолиноамид (5R)-7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-дигидроксигептановой кислоты
m/z (APCI(m+1+НСО2Н)) 672,3; вычислено для С37Н42FN3O5 627,31.
N,N-диметиламид (5R)-7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-дигидроксигептановой кислоты
m/z (APCI(m+1)) 586; вычислено для С35Н40FN3O4 585,30.
По методике, аналогично стадии 6, способ А, используя альтернативные Ru(II)-хиральные дифосфиновые комплексы вместо RuCl2(ДМФ)n[(R)-Cl-MeO-BIPHEP)] в качестве катализатора гидрирования получают идентичные продукты с различными энантиомерными избытками при С-5. Например, при восстановлении этилового эфира 7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-диоксогептановой кислоты до метилового эфира (5R)-7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-дигидроксигептановой кислоты происходит следующее:
RuCl2(ДМФ)n[(R)-(+)-BINAP] комплекс дает продукт с 90% эи (преимущественно (R) конфигурацию) при С-5, что определено хиральным ВЭЖХ анализом.
RuCl2(ДМФ)n[(R)-(+)-pTol-BINAP] комплекс дает продукт с 91% эи (преимущественно (R) конфигурацию) при С-5, что определено хиральным ВЭЖХ анализом.
RuCl2(ДМФ)n[(R)-(+)-C4-TunaPhos] комплекс дает продукт с 93% эи (преимущественно (R) конфигурацию) при С-5, что определено хиральным ВЭЖХ анализом.
RuCl2(ДМФ)n[(R)-(+)-C2-TunaPhos] комплекс дает продукт с 98% эи (преимущественно (R) конфигурацию) при С-5, что определено хиральным ВЭЖХ анализом.
RuCl2(ДМФ)n[(S)-(-)-MeO-BIPHEP] комплекс дает продукт с 95% эи (преимущественно (S) конфигурацию) при С-5, что определено хиральным ВЭЖХ анализом.
RuCl2[(R)-(+)-Cl-MeO-BIPHEP](NEt3)n комплекс дает продукт с ≥ 98% эи (преимущественно (R) конфигурацию) при С-5, что определено хиральным ВЭЖХ анализом.
RuCl2[(R)-(+)-BINAP](NEt3)n комплекс дает продукт с 91% эи (преимущественно (R) конфигурацию) при С-5, что определено хиральным ВЭЖХ анализом.
RuCl2[(R)-(+)-pTol-BINAP](NEt3)n комплекс дает продукт с 91% эи (преимущественно (R) конфигурацию) при С-5, что определено хиральным ВЭЖХ анализом.
[Ru(TFA)2((R)-(+)-Cl-MeO-BIPHEP)]n комплекс дает продукт с ≥ 98% эи (преимущественно (R) конфигурацию) при С-5, что определено хиральным ВЭЖХ анализом.
[Ru(TFA)2((R)-(+)-BINAP)]n комплекс дает продукт с 90% эи (преимущественно (R) конфигурацию) при С-5, что определено хиральным ВЭЖХ анализом.
Способ В
В реактор с инертным азотом загружают димер хлорида бензолрутения (II) (11 мг) и (R)-(+)-C2-TunaPhos (26 мг). Реактор продувают N2 при давлении и добавляют через шприц МеОН, продутый N2. Полученную смесь тщательно продувают N2 и перемешивают при температуре 25оС в течение 30 минут. В реактор через шприц добавляют раствор трет-бутилового эфира 7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-диоксогептановой кислоты (0,5 г) и МеОН, продувом N2 (4,5 мл), и полученную смесь перемешивают в атмосфере N2 при температуре 60оС в течение 30 минут. Раствор перемешивают при температуре 60оС при поддерживаемом давлении Н2 60 psi в течение 22 часов. Реакционную смесь охлаждают до температуры окружающей среды и повторно продувают N2. Неочищенный МеОН раствор метилового эфира (5R)-7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-дигидроксигептановой кислоты используют на следующих стадиях без очистки или, необязательно, он может быть выделен флэш-хроматографией на колонке с силикагелем, элюируя смесью этилацетат-гексан.
ВЭЖХ анализ (YMC ODS AQ S5; 1 мл/мин; 30оС; 254 нм: СН3CN/H2O, 60:40 (0-22 мин) до 100:0 (27-37 мин) до 60:40) показал соотношение син:анти изомеров 1:1,4.
Хиральный ВЭЖХ анализ (Chiralcel OD-H колонка; 5% EtOH:гексан; tR(3R,5R)=23,1 мин/tR(3R,5S)=18,0 мин/tR(3S,5S)=24,8 мин/tR(3S,5R)=19,9 мин) показал энантиомерный избыток при С-5 ≥ 97%, преимущественно (R) конфигурации.
m/z (PCI(m+1)) 573; вычислено для С34Н37FN2O5 572,27.
Стадия 7: фениламид 5-(4-фторфенил)-2-изопропил-1-[2-((S)-6-оксо-3,6-дигидро-2Н-пиран-2-илэтил]-4-фенил-1Н-пиррол-3-карбоновой кислоты
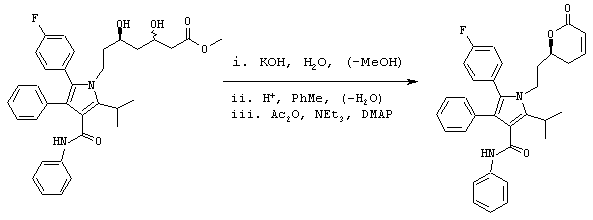
Подходящий реактор с инертным азотом загружают КОН (110,0 ммоль) и водой (300 мл). К быстро перемешиваемому раствору добавляют неочищенный раствор метилового эфира (5R)-7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-дигидроксигептановой кислоты со стадии 6 (примерно 100 ммоль/>98% эи) в МеОН (250 мл). Смесь нагревают в атмосфере азота до внутренней температуры примерно 85° С. Одновременно МеОН удаляют дистилляцией. Полученную реакционную смесь охлаждают до температуры 45° С и промывают MtBE (2× 150 мл). Фазы MtBE разделяют и удаляют. К водной фазе с температурой 45° С добавляют толуол (125 мл) с последующим медленным добавлением 6н HCl (20 мл). Двухфазную смесь перемешивают в течение 10 минут и слои разделяют. Водную фазу экстрагируют второй порцией толуола (125 мл) и удаляют. Объединенные органические фазы нагревают до температуры кипения с обратным холодильником в атмосфере азота. В это время собирают 130 мл дистиллята и удаляют. Полученный раствор охлаждают до примерно 60° С и последовательно добавляют NEt3 (140 ммоль), DMAP (2,0 ммоль) и Ac2O (70,0 ммоль) с такой скоростью, чтобы сохранять внутреннюю температуру реакции на уровне от 55° С до 65° С. Раствор перемешивают в течение примерно 1,5 часов при температуре 60° С. Смесь охлаждают до температуры 50° С и медленно добавляют 1н HCl (100 мл). Двухфазную смесь перемешивают в течение 10 минут, фазы разделяют и водную фазу удаляют. Органическую фазу промывают второй порцией 1н HCl (100 мл) и водой (100 мл), поддерживая температуру 45° С-55° С. Раствор в толуоле разбавляют Bu2O (200 мл) и полученный раствор медленно охлаждают до температуры 0° С при постоянном перемешивании. Полученное твердое вещество собирают на фильтровальной воронке и сушат в вакууме с получением фениламида 5-(4-фторфенил)-2-изопропил-1-[2-((S)-6-оксо-3,6-дигидро-2Н-пиран-2-илэтил]-4-фенил-1Н-пиррол-3-карбоновой кислоты в виде белого/беловатого твердого вещества (34,4 г). Данный продукт используют на последующих стадиях без дальнейшей очистки или, необязательно, он может быть повторно осажден из IPA/Н2О.
m/z (PCI(m+1)) 523; вычислено для С33Н31FN2O3 522,23.
Хиральный ВЭЖХ анализ (Chiralpack AD колонка; 1 мл/мин; 30° С; 254 нм; 10% IPA:гексан; tR(R)=18 мин/tR(S)=16 мин) показал энантиомерный избыток >98%, преимущественно (R) конфигурацию.
Стадия 8: фениламид 5-(4-фторфенил)-1-[2-((2R,4R)-4-гидрокси-6-оксотетрагидропиран-2-ил)этил]-2-изопропил-4-фенил-1Н-пиррол-3-карбоновой кислоты
Способ А
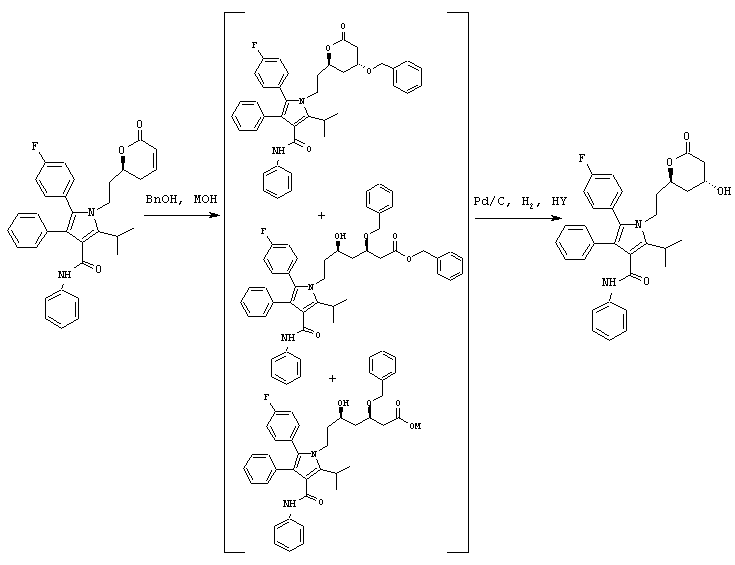
В продутый аргоном реактор загружают фениламид 5-(4-фторфенил)-2-изопропил-1-[2-((S)-6-оксо-3,6-дигидро-2Н-пиран-2-илэтил]-4-фенил-1Н-пиррол-3-карбоновой кислоты (0,020 моль/>99% эи) и бензиловый спирт (52 мл). Реакционную смесь охлаждают до температуры –10° С и добавляют NaOH (0,040 моль). После перемешивания в течение 19 часов при температуре –10° С реакцию гасят 37% HCl (0,042 моль) и разбавляют водой (25 мл) и толуолом (25 мл). После нагревания смеси до температуры окружающей среды нижний водный слой удаляют. Верхний органический слой объединяют с 20% Pd(OH)2/C (1,0 г) и H2SO4 (0,01 моль) и гидрируют при давлении водорода 50 psi при температуре 50° С в течение 16 часов. Реакционную смесь нагревают до температуры 80° С и фильтруют через диатомовую землю. Реактор и катализатор промывают горячим толуолом (10 мл). Нижний водный слой удаляют. Верхний органический слой промывают теплым раствором водной HCl (0,16 г, 37% HCl в 25 мл горячей воды) и нагревают до температуры кипения с обратным холодильником в течение 2,5 часов в атмосфере аргона с одновременным азеотропным удалением воды. Реакционную смесь охлаждают до температуры 65° С и вносят затравку фениламида 5-(4-фторфенил)-1-[2-((2R,4R)-4-гидрокси-6-оксотетрагидропиран-2-ил)этил]-2-изопропил-4-фенил-1Н-пиррол-3-карбоновой кислоты. Через 2 часа реакционную смесь медленно охлаждают до температуры окружающей среды. Полученную суспензию охлаждают до температуры примерно 0оС. Продукт собирают и промывают холодным толуолом (25 мл). Полученное твердое вещество растворяют в горячем толуоле (95 мл), охлаждают до температуры 65° С и выстаивают в течение 2 часов. Реакционную смесь медленно охлаждают до температуры окружающей среды и затем охлаждают до температуры 0° С. Продукт собирают, промывают холодным толуолом (25 мл) и сушат в вакууме при температуре 70° С в течение ночи с получением фениламида 5-(4-фторфенил)-1-[2-((2R,4R)-4-гидрокси-6-оксотетрагидропиран-2-илэтил]-2-изопропил-4-фенил-1Н-пиррол-3-карбоновой кислоты (8,4 г) в виде белого твердого вещества.
ВЭЖХ анализ (YMC ODS AQ S5; 1 мл/мин; 30° С; 254 нм; СН3CN/H2O, 60:40 (0-22 мин) до 100:0 (27-37 мин) до 60:40) показал соотношение син:анти изомеров >99:1.
Хиральный ВЭЖХ анализ (Chiralcel OF; 60° С; 254 нм; 20% IPA:гексан; tR(3R,5R)=26 мин/tR(3R,5S)=59 мин/tR(3S,5S)=33 мин/tR(3S,5R)=37 мин) показал энантиомерный избыток при С-5 >99%, преимущественно (R) конфигурации.
m/z (PCI(m+1)) 541; вычислено для С33Н33FN2O4 540,24.
По методике, аналогично стадии 8, способ А, замещенные производные бензилового спирта (например, п-метоксибензиловый спирт) могут использоваться вместо бензилового спирта с получением соответствующих соединений.
Способ В
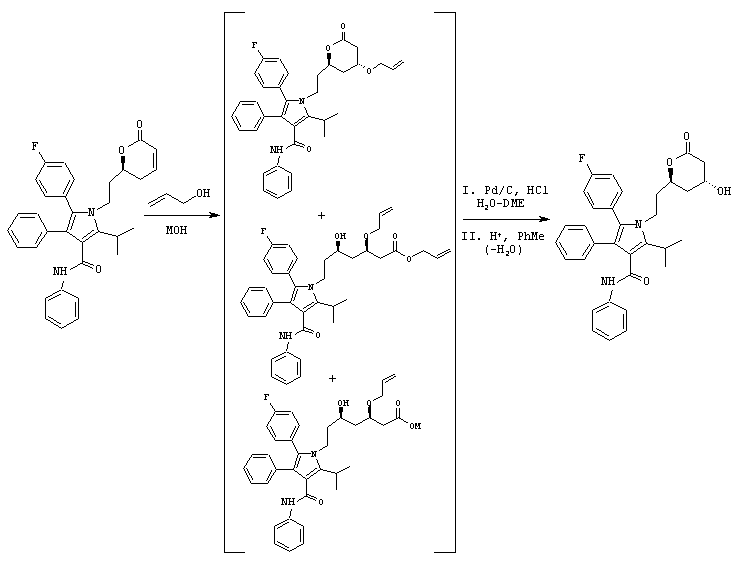
В продутый аргоном реактор загружают фениламид 5-(4-фторфенил)-2-изопропил-1-[2-((S)-6-оксо-3,6-дигидро-2Н-пиран-2-илэтил]-4-фенил-1Н-пиррол-3-карбоновой кислоты (19,1 ммоль/>99% эи) и аллиловый спирт (50 мл). Реакционную смесь охлаждают до температуры –5° С и добавляют LiOH (38,2 ммоль). После перемешивания в течение 1 часа при температуре –5° С реакцию гасят 37% HCl (42 ммоль) и толуолом (125 мл). После нагревания смеси до температуры окружающей среды реакционную смесь концентрируют до объема примерно 75 мл. Добавляют толуол (50 мл) и реакционную смесь концентрируют дистилляцией до неочищенного масла, которое затвердевает при выстаивании. Неочищенный остаток помещают в ДМЭ (340 мл). К полученному раствору добавляют деионизированную воду (20 мл), п-толуолсульфокислоту (2,25 г) и 5% Pd/C (11 г, 50% увлажненный водой). Полученную смесь нагревают до температуры 45° С в атмосфере N2 в течение 1,5 часов и при температуре окружающей среды в течение еще 16 часов. Раствор пропускают через вспомогательную фильтрующую присадку для удаления катализатора и растворитель удаляют в вакууме. Остаток помещают в толуол (50 мл). Добавляют воду (75 мл) и КОН (950 мг), реакционную смесь нагревают до температуры 65° С и слои разделяют. Водную фазу промывают толуолом (25 мл) при температуре 65° С и объединенные слои толуола удаляют. К водной фазе добавляют толуол (50 мл) с последующим добавлением 6н HCl (3,8 мл). Смесь энергично перемешивают при температуре 65° С в течение 5 минут и фазы разделяют. Фазу толуола нагревают до температуры кипения с обратным холодильником в течение 2,5 часов с одновременным азеотропным удалением воды. Реакционную смесь охлаждают до температуры 65° С и вносят затравку фениламида 5-(4-фторфенил)-1-[2-((2R,4R)-4-гидрокси-6-оксотетрагидропиран-2-ил)этил]-2-изопропил-4-фенил-1Н-пиррол-3-карбоновой кислоты. Через 2 часа реакционную смесь медленно охлаждают до температуры окружающей среды. Полученную суспензию охлаждают до температуры примерно 0° С. Продукт собирают и промывают холодным толуолом (25 мл).
Полученное твердое вещество растворяют в горячем толуоле (95 мл), охлаждают до температуры 65° С и выстаивают в течение 2 часов. Реакционную смесь медленно охлаждают до температуры окружающей среды и затем охлаждают до 0° С. Продукт собирают, промывают холодным толуолом (25 мл) и сушат в вакууме при температуре 70° С в течение ночи с получением фениламида 5-(4-фторфенил)-1-[2-((2R,4R)-4-гидрокси-6-оксотетрагидропиран-2-илэтил]-2-изопропил-4-фенил-1Н-пиррол-3-карбоновой кислоты в виде белого твердого вещества.
ВЭЖХ анализ (YMC ODS AQ S5; 1 мл/мин; 30оС; 254 нм; СН3CN/H2O, 60:40 (0-22 мин) до 100:0 (27-37 мин) до 60:40) показал соотношение син:анти изомеров >99:1.
Хиральный ВЭЖХ анализ (Chiralcel OF; 60° С; 254 нм; 20% IPA:гексаны; tR(3R,5R)=26 мин/tR(3R,5S)=59 мин/tR(3S,5S)=33 мин/tR(3S,5R)=37 мин) показал энантиомерный избыток при С-5 >99%, преимущественно (R) конфигурации.
m/z (PCI(m+1)) 541; вычислено для С33Н33FN2O4 540,24.
По методике, аналогично стадии 8, способ В, замещенные производные аллилового спирта (например, кротиловый спирт) могут использоваться вместо аллилового спирта с получением соответствующих соединений.
Способ С

Операция А
В реактор с инертным азотом загружают трет-бутиловый эфир (5R)-7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-дигидроксигептановой кислоты (10,0 ммоль), диметилацеталь бензальдегида (44,0 ммоль), толуол (40 мл) и моногидрат п-толуолсульфокислоты (1,0 ммоль). Реакционную смесь энергично перемешивают в вакууме в течение примерно 20 часов или до завершения реакции, определяемого ВЭЖХ анализом аликвоты. Раствор охлаждают в атмосфере азота до температуры примерно –50° С, добавляют 1М раствор KOtBu в ТГФ (9,0 ммоль) тремя равными порциями, разделенными промежутками 30-45 минут. Полученный раствор перемешивают еще дополнительно 12-14 часов при температуре 0° С. Реакцию гасят медленным добавлением 1н HCl (10 мл). Полученную двухфазную смесь нагревают до температуры примерно 15° С и переносят в делительную воронку, где водную фазу удаляют. Органическую фазу промывают насыщенным водным NaCl (100 мл), сушат над безводным MgSO4 (25 г), фильтруют и концентрируют в вакууме до неочищенного масла. Данный продукт используют в дальнейших стадиях без очистки или, необязательно, он может быть повторно осажден из смеси простой эфир/гексаны.
m/z (APCI(m+1)) 703,4; вычислено для С44Н47FN2O5 702,35.
По методике, аналогично стадии 8, способ С, операция А, используя соответствующий сложный эфир со стадии 6 вместо трет-бутилового эфира (5R)-7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-дигидроксигептановой кислоты получают следующие соединения, например:
метиловый эфир ((4R,6R)-6-{2-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-этил}-2-фенил[1,3]диоксан-4-ил)уксусной кислоты
этиловый эфир ((4R,6R)-6-{2-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-этил}-2-фенил[1,3]диоксан-4-ил)уксусной кислоты
изопропиловый эфир ((4R,6R)-6-{2-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-этил}-2-фенил[1,3]диоксан-4-ил)уксусной кислоты
Операция В
В реактор, содержащий инертный азот под давлением, загружают трет-бутиловый эфир ((4R,6R)-6-{2-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]этил}-2-фенил[1,3]диоксан-4-ил)уксусной кислоты из операции А (5,0 г), 5% Pd/C (0,45 г; 50% Н2О увлажненный), 2н HCl в МеОН (1,9 мл), толуол (11 мл) и МеОН (3,1 мл). Реактор и его содержимое дегазируют двумя циклами частичного откачивания газа и созданием повышенного давления азота (25 мм рт. ст. и 50 psi, соответственно). Атмосферу заменяют водородом тремя циклами частичного откачивания газа и созданием повышенного давления водорода (25 мм рт. ст. и 50 psi, соответственно). Реакционную смесь энергично перемешивают при температуре 40° С при положительном давлении Н2 (примерно 50 psi) в течение примерно 2,5 часов. Реакционную смесь охлаждают до температуры окружающей среды и давление водорода сбрасывают и заменяют азотом. Реакционную смесь пропускают через фильтрующий агент для удаления катализатора, тщательно промывая МеОН (2× 5 мл). К полученному раствору добавляют КОН (0,6 г) в воде (25 мл). Реакционную смесь энергично перемешивают в атмосфере азота и нагревают до внутренней температуры реакции примерно 90° С, удаляя МеОН дистилляцией. Двухфазную смесь охлаждают до температуры 70° С и верхнюю фазу толуола отделяют и удаляют. Водную фазу промывают второй порцией толуола (10 мл) при температуре 70° С. Органическую фазу также отделяют и удаляют. К водной фазе добавляют толуол (10 мл) с последующим медленным добавлением 2н HCl (5 мл). Двухфазную смесь перемешивают в течение 10 минут и слои разделяют. Водную фазу экстрагируют второй порцией толуола (10 мл) и удаляют. Объединенные органические фазы нагревают до температуры кипения с обратным холодильником с водной ловушкой Дина-Старка в течение 2,5 часов в атмосфере аргона. Реакционную смесь охлаждают до температуры 65° С и вносят затравку фениламида 5-(4-фторфенил)-1-[2-((2R,4R)-4-гидрокси-6-оксотетрагидропиран-2-ил)этил]-2-изопропил-4-фенил-1Н-пиррол-3-карбоновой кислоты. Через 2 часа реакционную смесь медленно охлаждают до температуры окружающей среды. Полученную суспензию охлаждают до температуры примерно 0° С. Продукт собирают и промывают холодным толуолом (5 мл). Полученное твердое вещество растворяют в горячем толуоле (20 мл), охлаждают до температуры 65° С и выстаивают в течение 2 часов. Реакционную смесь медленно охлаждают до температуры окружающей среды и затем охлаждают до температуры 0° С. Продукт собирают, промывают холодным толуолом (5 мл) и сушат в вакууме при температуре 70° С в течение ночи с получением фениламида 5-(4-фторфенил)-1-[2-((2R,4R)-4-гидрокси-6-оксотетрагидропиран-2-илэтил]-2-изопропил-4-фенил-1Н-пиррол-3-карбоновой кислоты в виде белого твердого вещества.
m/z (PCI(m+1)) 541; вычислено для С33Н33FN2O4 540,24.
Способ D
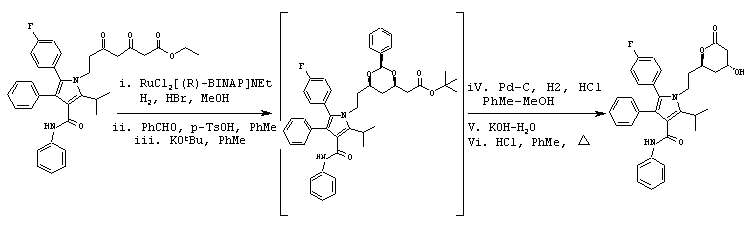
В реактор с инертным азотом под давлением загружают этиловый эфир 7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-дигидроксигептановой кислоты (100,0 ммоль) и EtOH (250 мл). Полученную суспензию нагревают при перемешивании до температуры примерно 55° С с получением гомогенного раствора. Сосуд и его содержимое дегазируют тремя продувками аргона при давлении 50 psi. При постоянном потоке аргона добавляют 1М HBr в метаноле (7,0 ммоль) и катализатор RuCl2[(R)-BINAP)]NEt3 (0,5 ммоль) и реактор продувают дополнительным количеством аргона при давлении 50 psi. Атмосферу заменяют водородом тремя продувками при давлении 50 psi. Реакционную смесь энергично перемешивают при температуре 65° С при поддерживаемом давлении водорода (50 psi) до тех пор, пока не прекратится поглощение Н2. Реакционную смесь охлаждают до температуры примерно 50° С, давление водорода снижают и заменяют азотом. Неочищенный EtOH раствор метилового эфира (5R)-7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-дигидроксигептановой кислоты разбавляют толуолом (250 мл). К полученному раствору добавляют бензальдегид (150 ммоль) и моногидрат п-TsOH (5 ммоль). Реакционную смесь нагревают до температуры реактора 110° С, удаляя EtOH и воду путем образования азеотропов с толуолом. Раствор охлаждают в атмосфере азота до температуры примерно –5° С, добавляют 1М раствор KOtBu в ТГФ (90 ммоль) тремя равными порциями, разделенными промежутками 30-45 минут. Полученный раствор перемешивают еще дополнительно 12-14 часов при 0° С. Реакцию гасят медленным добавлением 1н HCl (100 мл). Полученную двухфазную смесь нагревают до температуры примерно 15° С и переносят в делительную воронку, где водную фазу удаляют. Органическую фазу промывают насыщенным водным NaCl (25 мл), сушат над безводным MgSO4 (5 г), фильтруют и концентрируют в вакууме до неочищенного масла, которое помещают в МеОН (200 мл). Раствор переносят в реактор с инертным азотом под давлением, содержащий 5% Pd/C (5 г; 50% увлажненный водой). Добавляют концентрированную HCl (2 мл) и реакционную смесь перемешивают при поддерживаемом давлении Н2 (50 psi) в течение примерно 3 часов при температуре 50° С. Реакционную смесь охлаждают до температуры окружающей среды, Н2 заменяют N2 и катализатор удаляют фильтрованием. Полученный раствор метилового эфира (3R,5R)-7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-дигидроксигептановой кислоты переносят в реактор с инертным азотом, в который загружены КОН (110,0 ммоль) и воды (300 мл). Реакционную смесь нагревают в атмосфере азота до внутренней температуры реакции примерно 85° С. Одновременно удаляют МеОН дистилляцией. Полученную реакционную смесь охлаждают до температуры 45° С и промывают MtBE (2× 150 мл). Фазы MtBE отделяют и удаляют. К водной фазе при температуре 45° С добавляют толуол (125 мл) с последующим медленным добавлением 6н HCl (20 мл). Двухфазную смесь перемешивают в течение 10 минут и слои разделяют. Водную фазу экстрагируют второй порцией толуола (125 мл) и удаляют. Объединенные органические фазы нагревают до температуры кипения с обратным холодильником с водной ловушкой Дина-Старка в течение 2,5 часов в атмосфере аргона. Реакционную смесь охлаждают до температуры 65° С и вносят затравку фениламида 5-(4-фторфенил)-1-[2-((2R,4R)-4-гидрокси-6-оксотетрагидропиран-2-ил)этил]-2-изопропил-4-фенил-1Н-пиррол-3-карбоновой кислоты. Через 2 часа реакционную смесь медленно охлаждают до температуры окружающей среды. Полученную суспензию охлаждают до примерно 0° С. Продукт собирают и промывают холодным толуолом (100 мл). Полученное твердое вещество растворяют в горячем толуоле (350 мл), охлаждают до температуры 65° С и выстаивают в течение 2 часов. Реакционную смесь медленно охлаждают до температуры окружающей среды и затем охлаждают до 0° С. Продукт собирают, промывают холодным толуолом (100 мл) и сушат в вакууме при температуре 70° С в течение ночи с получением фениламида 5-(4-фторфенил)-1-[2-((2R,4R)-4-гидрокси-6-оксотетрагидропиран-2-илэтил]-2-изопропил-4-фенил-1Н-пиррол-3-карбоновой кислоты в виде белого твердого вещества.
m/z (PCI(m+1)) 541; вычислено для С33Н33FN2O4 540,24.
Стадия 9: кальциевая соль (R,R)-7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-дигидроксигептановой кислоты
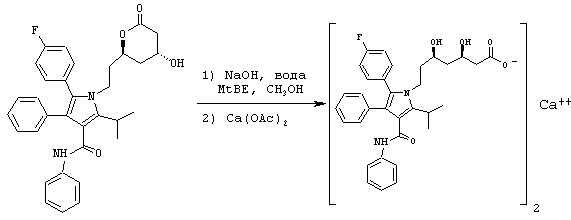
В продутый аргоном реактор загружают фениламид 5-(4-фторфенил)-1-[2-((2R,4R)-4-гидрокси-6-оксотетрагидропиран-2-илэтил]-2-изопропил-4-фенил-1Н-пиррол-3-карбоновой кислоты (14,8 ммоль), MtBE (45 мл) и МеОН (20 мл). Добавляют раствор NaOH (15,2 ммоль) в воде (103 мл) и реакционную смесь нагревают до температуры 52° С. После нагревания в течение примерно 1 часа реакционную смесь охлаждают до температуры 34° С и слои разделяют. Верхний органический слой удаляют. Нижний водный слой промывают MtBE (33 мл) при температуре примерно 33° С. Нижний водный слой разбавляют MtBE (2 мл) и нагревают до температуры 52° С в атмосфере аргона. Добавляют теплый раствор Ca(OAc)2·H2O (7,5 ммоль) в воде (44 мл) в течение примерно 2 часов. Через примерно 5 минут после начала добавления Ca(OAc)2 в реакционную смесь вносят затравку суспензии кальциевой соли (R,R)-7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-дигидроксигептановой кислоты (0,08 ммоль) в воде (1,2 мл) и метаноле (0,4 мл). После завершения добавления Ca(OAc)2 реакционную смесь выстаивают в течение примерно 15 минут при температуре 52° С и охлаждают до температуры 20° С. Продукт собирают, промывают последовательно раствором водного метанола 2:1 (48 мл) и водой (49 мл). После сушки в вакууме при температуре 70° С получают кальциевую соль (R,R)-7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-дигидроксигептановой кислоты (8,7 г) в виде белого твердого вещества. Аналитические спецификации полученного продукта соответствуют значениям, представленным в известном уровне техники.
Получение катализаторов
Пример А
RuCl2(ДМФ)n[(R)-(+)-Cl-MeO-BIPHEP] комплекс
Подходящую реакционную колбу загружают ДМФ (17,5 мл). Колбу и ее содержимое дегазируют двумя циклами частичного откачивания газа и созданием повышенного давления азота (25 мм рт. ст. и 10 psi, соответственно). Избыточное давление азота сбрасывают и сразу же добавляют димер хлорида бензолрутения(II) (0,50 ммоль) и (R)-(+)-Cl-MeO-BIPHEP (1,10 ммоль). Колбу и ее содержимое снова дегазируют двумя циклами частичного откачивания газа и созданием повышенного давления азота (25 мм рт. ст. и 10 psi, соответственно). Избыточное давление азота сбрасывают и реактор нагревают до температуры примерно 100° С в течение 10 минут. Полученный раствор охлаждают до температуры ≤ 50° С и растворитель удаляют в вакууме с получением RuCl2(ДМФ)n[(R)-(+)-Cl-MeO-BIPHEP] в виде ржаво-коричневого твердого вещества. Неочищенный комплекс используют непосредственно в последующих реакциях без очистки или получают четкие характеристики или, необязательно, хранят в инертной атмосфере для дальнейшего использования.
По методике, аналогично примеру А, используя соответствующие хиральные лиганды дифосфина вместо (R)-(+)-Cl-MeO-BIPHEP получают следующие комплексы, например:
RuCl2(ДМФ)n[(R)-(+)-BINAP]n комплекс
RuCl2(ДМФ)n[(R)-(+)-pTol-BINAP]n комплекс
RuCl2(ДМФ)n[(R)-(+)-C4-TunaPhos]n комплекс
RuCl2(ДМФ)n[(R)-(+)-C2-TunaPhos]n комплекс
RuCl2(ДМФ)n[(S)-(-)-MeO-BIPHEP]n комплекс
Пример В
RuCl2[(R)-(+)-BINAP] (NEt3)n комплекс
В подходящий реактор под давлением инертного азота загружают димер дихлор(1,5-циклооктадиен)рутения (II) (0,15 ммоль) и (R)-(+)-BINAP (0,32 ммоль). Добавляют толуол (8,0 мл) с последующим добавлением триэтиламина (4,5 ммоль). Реактор и его содержимое дегазируют двумя циклами частичного откачивания газа и созданием повышенного давления азота (25 мм рт. ст. и 10 psi, соответственно). Избыточное давление азота сбрасывают, реактор герметизируют и нагревают до примерно 140° С, при которой его выдерживают в течение примерно 4 часов. Полученный прозрачный красный раствор охлаждают до температуры ≤ 40° С и растворитель удаляют в вакууме с получением RuCl2[(R)-(+)-BINAP](NEt3)n комплекса в виде ржаво-коричневого твердого вещества. Неочищенный комплекс используют непосредственно в последующих реакциях без очистки или получают четкие характеристики, или, необязательно, хранят в инертной атмосфере для дальнейшего использования.
По методике, аналогично примеру В, используя соответствующие хиральные лиганды дифосфина вместо (R)-(+)-BINAP получают следующие комплексы, например:
RuCl2[(R)-(+)-Cl-MeO-BIPHEP](NEt3)n комплекс
RuCl2[(R)-(+)-BINAP](NEt3)n комплекс
RuCl2[(R)-(+)-pTol-BINAP](NEt3)n комплекс
Пример С
[Ru(TFA)2((R)-(+)-Cl-MeO-BIPHEP)]n комплекс
Подходящую реакционную колбу загружают ацетоном (50 мл). Колбу и ее содержимое дегазируют двумя циклами частичного откачивания газа и созданием повышенного давления аргона (25 мм рт. ст. и 10 psi, соответственно). Избыточное давление аргона сбрасывают и сразу же добавляют (0,50 ммоль) и (R)-(+)-Cl-MeO-BIPHEP (0,51 ммоль). Колбу и ее содержимое снова дегазируют двумя циклами частичного откачивания газа и созданием повышенного давления аргона (25 мм рт. ст. и 10 psi, соответственно). Избыточное давление аргона снижают и реактор энергично перемешивают при температуре примерно 30° С. Через шприц добавляют трифторуксусную кислоту (1,2 ммоль) и реакционную смесь перемешивают еще 1 час. Растворитель удаляют в вакууме при осторожном пропускании О2, с получением [Ru(TFA)2((R)-(+)-Cl-MeO-BIPHEP)]n комплекса в твердого вещества. Неочищенный комплекс используют непосредственно в последующих реакциях без очистки или получают четкие характеристики, или, необязательно, хранят в инертной атмосфере для дальнейшего использования.
По методике, аналогично примеру С, используя соответствующие хиральные лиганды дифосфина вместо (R)-(+)-Cl-MeO-BIPHEP получают следующие комплексы, например:
[Ru(TFA)2((R)-(+)-MeO-BIPHEP)]n комплекс
[Ru(TFA)2((R)-(+)-BINAP)]n комплекс
[Ru(TFA)2((R)-(+)-p-Tol-BINAP)]n комплекс
Пример 2 получения соединений формулы 18 и 18 (а).
Трет.-бутиловый эфир 7-[2-(4-Фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3-гидрокси-5-оксогептановой кислоты
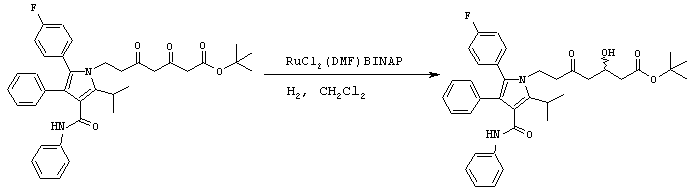
В реактор под давлением с инертным аргоном загружают трет.-бутиловый эфир 7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-диоксогептановой кислоты (14,0 ммол) в 40 мл хлористого метилена. К перемешиваемой суспензии добавляют RuCl2 (DMF) BINAP. Полученную суспензию дегазируют тремя продувками аргона при давлении 50 psi. Сосуд и его содержимое продувают тремя продувками водорода при давлении 50 psi, и смесь нагревают при 55° С с получением гомогенного раствора. Реакционную смесь перемешивают при поддерживаемом давлении водорода (50 psi) до прекращения поглощения водорода. Реакционную смесь охлаждают до температуры окружающей среды и убирают давление водорода. Неочищенный раствор трет.-бутилового эфира 7-[2-(4-Фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3-гидрокси-5-оксогептановой кислоты концентрируют в вакууме и очищают колоночной флеш-хроматографией на силикагеле, элюируя смесями этилацетат-гептан.
ВЭЖХ анализ (YMC ODS AQ S5; 1 мл/мин; 30° C, 254 нм:
CH3CN/H2O, 60:40 (0-5 мин) до 100:0 (15-22 мин) до 60:40 (25 мин); tR = 14,8 мин.
13C ЯMP (СDCl3, 100 МГц) δ 21,5, 25,5, 28,3, 39,9, 41,5, 44,6, 48,5, 64,5, 81,5, 115,2, 115,6, 115,7, 120,5, 122,8, 123,6, 126,7, 127,7, 127,8, 128,3, 128,6, 128,7, 130,3, 132,5, 132,9, 134,3, 138,3, 141,3, 161,1, 163,5, 164,5, 171,4, 205,8.
Пример 3 получения соединения формулы 11b.
Метиловый эфир 7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3,5-дигидроксигептановой кислоты
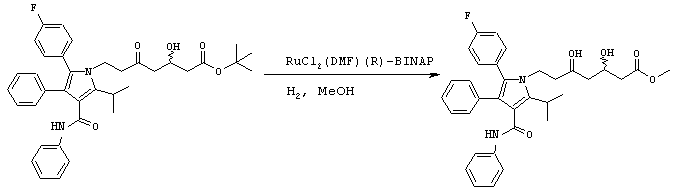
В реактор под давлением с инертным аргоном загружают трет.-бутиловый эфир 7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3-гидрокси-5-оксогептановой кислоты (2,5 ммол) в 9 мл метанола. К перемешиваемой суспензии добавляют RuCl2 (DMF)BINAP и НВr/МеОН (0,1 ммол). Полученную суспензию дегазируют тремя продувками аргона при давлении 60 psi. Сосуд и его содержимое продувают тремя продувками водорода при давлении 60 psi, и смесь нагревают при 65° С с получением гомогенного раствора. Реакционную смесь перемешивают при поддерживаемом давлении водорода (50 psi) до прекращения поглощения водорода. Реакционную смесь охлаждают до температуры окружающей среды и убирают давление водорода. Неочищенный раствор метилового эфира 7-[2-(4-фторфенил)-5-изопропил-3-фенил-4-фенилкарбамоилпиррол-1-ил]-3-гидрокси-5-оксогептановой кислоты концентрируют в вакууме.
ВЭЖХ соотношение син:анти изомеров 1:1,5,
Хиральный ВЭЖХ анализ (Chiralcel OD-H колонка; 5% ЕtOН:гексаны; tR(3R,5R)=23,1 мин/tR(3R,5S)=18,0 мин/tR(3S,5R)=19,9 мин) показал энантиомерный избыток при С-5=87%, преимущественно (R) конфигурации.
Изобретение относится к новому способу получения фениламида 5-(4-фторфенил)-1-[2-((2R,4R)-4-гидрокси-6-оксотетрагидропиран-2-ил)этил]-2-изопропил-4-фенил-1Н-пиррол-3-карбоновой кислоты, который заключается в превращении метилцианоацетата в целевой продукт в восемь или менее стадий. Изобретение также относится к ценным промежуточным продуктам, которые получают в результате осуществления указанных стадий заявленного способа. Фениламид 5-(4-фторфенил)-1-[2-((2R,4R)-4-гидрокси-б-оксотетрагидропиран-2-ил)этил]-2- изопропил-4-фенил-1Н-пиррол-3-карбоновой кислоты является ценным промежуточным продуктом в синтезе аторвастатина кальция, который используется в качестве гиполипидемического и/или гипохолестеринемического агента. Технический результат - данный способ позволяет избежать применение дорогих хиральных исходных веществ и снизить время процесса. 10 н. и 2 з.п. ф-лы.
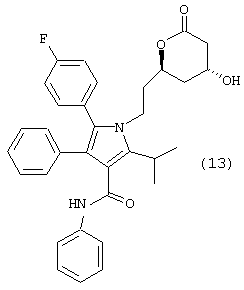
который включает
стадию (а) взаимодействия соединения формулы (1)

где R является алкилом, арилом, арилалкилом или гетероарилом, в растворителе, с соединением формулы (2)

где R1 является -XR, где
Х является О,
S или
Se, или R1 является

где R2 или R3 независимо являются
алкилом,
циклоалкилом,
арилалкилом или
арилом, или
R2 и R3 вместе являются
-(CH2)4-,
-(СН2)5-,
-(СН(R4)-СН2)3-,
-(СH(R4)-CH2)4-,
-(CH(R4)-(CH2)2-CH(R4))-,
-(CH(R4)-(CH2)3-CH(R4))-,
-СН2-СН2-А-СН2-СН2-,
-СН (R4)-СН2-А-СН2-СН2-,
-СН(R4)-CH2-A-CH2-CH(R4)-
где R4 является алкилом, имеющим 1-4 атома углерода, А является О, S или N, и R, такой как определено выше, с получением соединения формулы (3)
где R1, такой как определено выше;
стадию (b) взаимодействия соединения формулы (3) с водородом в присутствии катализатора и сильной кислоты, в растворителе, с получением соединения формулы (4)
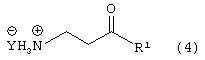
где Y является Cl, Br, TsO, MsO или HSO4 и R1, такой как определено выше;
стадию (с) взаимодействия соединения формулы (4) с основанием в растворителе с последующим добавлением соединения формулы (5)

где R такой, как определено выше, в растворителе, с получением соединения формулы (6)
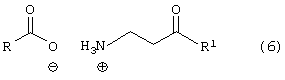
где R и R1 такие, как определено выше;
стадию (d) взаимодействия соединения формулы (6) с соединением формулы (7)
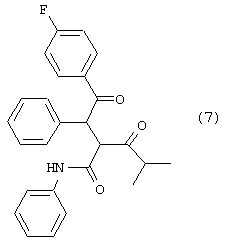
в растворителе с удалением воды с получением соединения формулы (8)
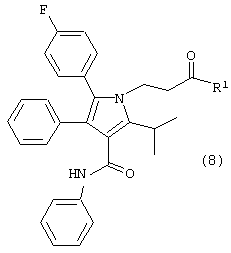
где R1, такой как определено выше;
стадию (е) взаимодействия соединения формулы (8) с соединением формулы (9)

где М является натрием, литием, калием, цинком, магнием, медью, кальцием или алюминием, и R1, такой как определено выше, в растворителе, в присутствии сильного основания, с получением соединения формулы (10)

где R1, такой как определено выше;
стадию (f) взаимодействия соединения формулы (10) с водородом в присутствии катализатора на основе рутения, в растворителе, в присутствии кислоты, с получением соединения формулы (11)
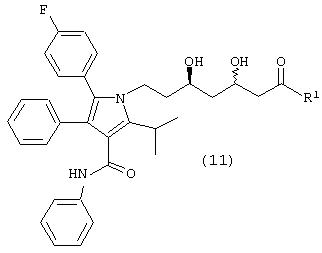
где R1, такой как определено выше, или соединения формулы (11a)
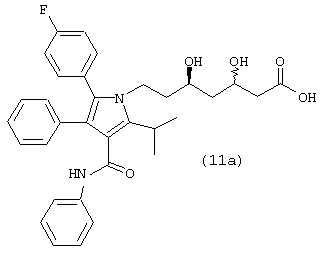
стадию (g) взаимодействия соединения формулы (11b)
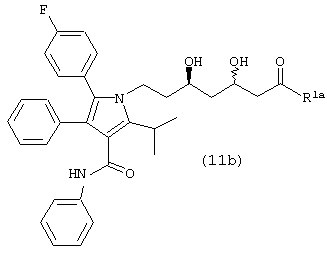
где R1а является ОН, -XR, где
Х является О,
S или
Se, или R1a является
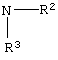
где R2 или R3 независимо являются
алкилом,
циклоалкилом,
арилалкилом или
арилом, или
R2 и R3 вместе являются
-(СН2)4-,
-(СН2)5-,
-(CH(R4)-CH2)3-,
-(СH(R4)-CH2)4-,
-(СН(R4)-(СH2)2-СН(R4))-,
-(CH(R4)-(CH2)3-CH(R4))-,
-СН2-СН2-А-СН2-СН2-,
-СН(R4)-СН2-А-СН2-СН2-,
-СН(R4)-CH2-A-CH2-CH(R4)-
где R4 является алкилом, имеющим 1-4 атома углерода, А является О, S или N, и R является алкилом, арилом, арилалкилом или гетероарилом, в растворителе, в присутствии кислоты, с последующим взаимодействием с основанием, ацилирующим агентом и катализатором ацилирования, в растворителе, с получением соединения формулы (12)

и
стадию (h) взаимодействия соединения формулы (12) с НО-М в спирте формулы (17) или (17b)
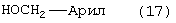 или
или 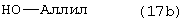
где М является натрием, литием, калием, цинком, магнием, медью, кальцием или алюминием; или с соединением формулы (16) или (16b)
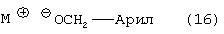 или
или 
где М, такой как определено выше для спирта формулы (17) или (17b), где арил или аллил в соединениях формулы (16) или (16b) и (17) или (17b) являются одинаковыми, в растворителе, с последующим добавлением водорода в присутствии катализатора и кислоты, с получением соединения формулы (13).
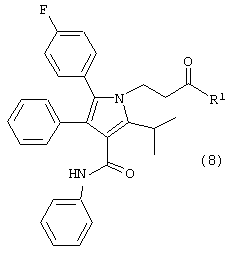
где R1 является -XR, где
Х является О,
S или
Se, или R1 является
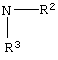
где R2 или R3 независимо являются
алкилом,
циклоалкилом,
арилалкилом или
арилом, или
R2 и R3 вместе являются
-(СН2)4-,
-(СН2)5-,
-(СН(R4)-СH2)3-,
-(CH(R4)-CH2)4-,
-(CH(R4)-(CH2)2-CH(R4))-,
-(CH(R4)-(CH2)3-CH(R4))-,
-СН2-СН2-А-СН2-СН2-,
-СН(R4)-CH2-A-CH2-CH2-,
-СН(R4)-CH2-A-CH2-CH (R4)-
где R4 является алкилом, имеющим 1-4 атома углерода, А является О, S или N, и R является алкилом, арилом, арилалкилом или гетероарилом, который включает
взаимодействие соединения формулы (4)
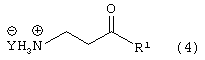
где Y является Cl, Br, TsO, MsO или HSO4 и R1, такой как определено выше, с соединением формулы (20)
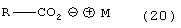
где R, такой как определено выше и М является натрием, литием, калием, цинком, магнием, медью, кальцием или алюминием, с соединением (7)
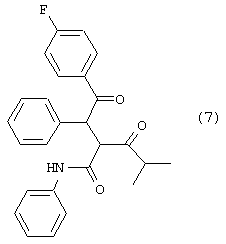
в растворителе с удалением воды, с получением соединения формулы (8).
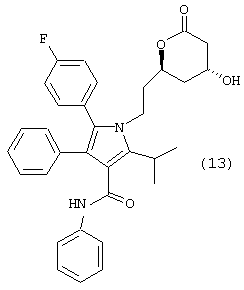
который включает
стадию (а) взаимодействия соединения формулы (11)
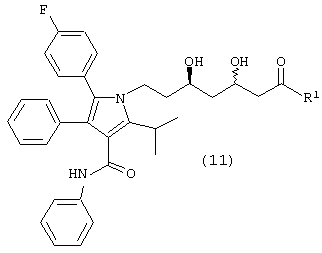
где R1 является -XR, где
X является О,
S или
Se, или R1 является
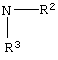
где R2 или R3 независимо являются
алкилом,
циклоалкилом,
арилалкилом или
арилом, или
R2 и R3 вместе являются
-(СН2)4-,
-(СН2)5-,
-(CH(R4)-CH2)3-,
-(СH(R4)-СН2)4-,
-(CH(R4)-(CH2)2-CH(R4))-,
-(СH(R4)-(CH2)3-CH(R4))-,
-СН2-СН2-А-СН2-СН2-,
-СН(R4)-СН2-А-СН2-СН2-,
-СН(R4)-CH2-A-CH2-CH(R4)-
где R4 является алкилом, имеющим 1-4 атома углерода, А является О, S или N, и R является алкилом, арилом, арилалкилом или гетероарилом, с ацеталем формулы (15)
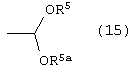
где R5 и R5a одинаковые или различные и независимо являются метилом, этилом или -(СН2)n-, где n является целым числом от 2 до 4 и R, такой как определено выше, в растворителе, в присутствии кислоты, с последующим добавлением альдегида, соответствующего предыдущему ацеталю, в присутствии основания, с получением соединения формулы (14)

где R1 и R такие, как определено выше;
стадию (b) взаимодействия соединения формулы (14) в растворителе, в присутствии кислоты, или необязательного взаимодействия с водородом в присутствии катализатора и кислоты в растворителе, с получением соединения формулы (13); и
стадию (с) альтернативного взаимодействия соединения формулы (11) или (11а)
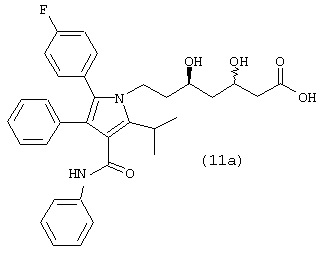
в растворителе, в присутствии основания, с получением соединения формулы (13).
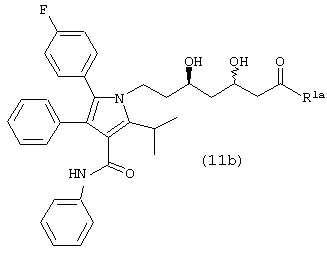
где R1a является ОН, -XR, где
Х является О,
S или
Se, или R1a является
где R2 или R3 независимо являются
алкилом,
циклоалкилом,
арилалкилом или
арилом, или
R2 и R3 вместе являются
-(СН2)4-,
-(СН2)5-,
-(CH(R4)-CH2)3-,
-(СH(R4)-СН2)4-,
-(CH(R4)-(CH2)2-CH(R4))-,
-(СH(R4)-(CH2)3-CH(R4))-,
-СН2-СН2-А-СН2-СН2-,
-СН(R4)-СН2-А-СН2-СН2-,
-СН(R4)-CH2-A-CH2-CH(R4)-
где R4 является алкилом, имеющим 1-4 атома углерода, А является О, S или N, и R является алкилом, арилом, арилалкилом или гетероарилом, который включает
стадию (а) взаимодействия соединения формулы (10)
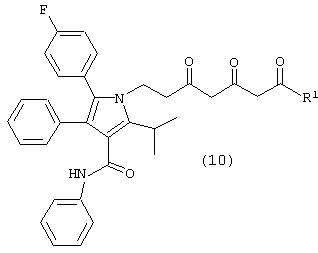
где R1 является XR или R1 является
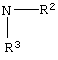
где X, R, R2 и R3 такие, как определено выше, с одним молем водорода, в присутствии катализатора на основе рутения, в растворителе, в присутствии кислоты, с получением соединений формул (18) и/или (18а)
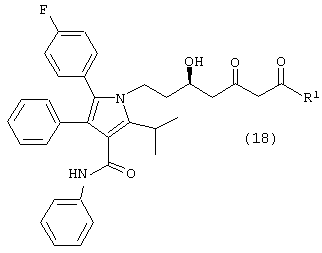
и

где R1, такой как определено выше; и
стадию (b) взаимодействия либо соединения формулы (18), либо (18а) с водородом в присутствии катализатора на основе рутения, в растворителе, в присутствии кислоты, с получением соединения формулы (11b).

где R является PhCH2- или (СН3)3-С- и R1 является
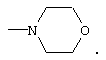
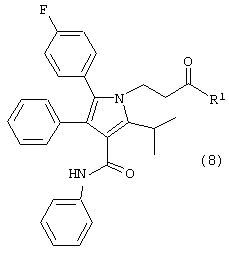
где R1 является

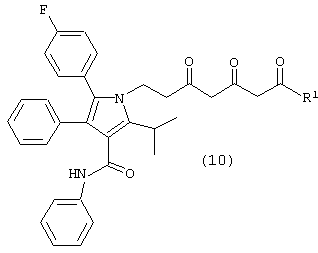
где R1 является -O-третичным бутилом, -О-изопропилом, -O-этилом, -O-метилом,
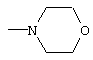 или -NMe2.
или -NMe2.

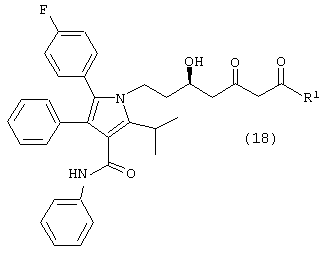
где R1 является O-третичным бутилом, -O-изопропилом, -O-этилом, -O-метилом.
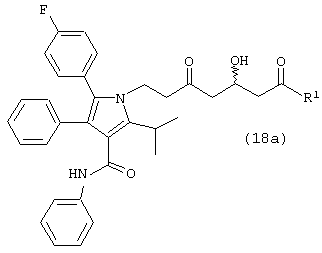
где R1 является O-третичным бутилом, -O-изопропилом, -O-этилом, -O-метилом.
| US 5273995 А, 28.12.1993 | |||
| Кулиса для фотографических трансформаторов и увеличительных аппаратов | 1921 |
|
SU213A1 |
| US 5298627 А, 29.03.1994 | |||
| УСТРОЙСТВО для КОРРЕЛЯЦИОННЫХ ИЗМЕРЕНИЙ | 0 |
|
SU247633A1 |

